

Abstract
Conformational diseases, such as Alzheimer, Parkinson and Huntington diseases, are part of a common class of neurological disorders characterized by the aggregation and progressive accumulation of proteins bearing aberrant conformations. Huntington disease (HD) has autosomal dominant inheritance and is caused by mutations leading to an abnormal expansion in the polyglutamine (polyQ) tract of the huntingtin (HTT) protein, leading to the formation of HTT inclusion bodies in neurons of affected patients. Interestingly, recent experimental evidence is challenging the conventional view by which the disease pathogenesis is solely a consequence of the intracellular accumulation of mutant protein aggregates. These studies reveal that transcellular transfer of mutated huntingtin protein is able to seed oligomers involving even the wild-type (WT) forms of the protein. To date, there is still no successful strategy to treat HD. Here, we describe a novel functional role for the HSPB1-p62/SQSTM1 complex, which acts as a cargo loading platform, allowing the unconventional secretion of mutant HTT by extracellular vesicles. HSPB1 interacts preferentially with polyQ-expanded HTT compared with the WT protein and affects its aggregation. Furthermore, HSPB1 levels correlate with the rate of mutant HTT secretion, which is controlled by the activity of the PI3K/AKT/mTOR signalling pathway. Finally, we show that these HTT-containing vesicular structures are biologically active and able to be internalized by recipient cells, therefore providing an additional mechanism to explain the prion-like spreading properties of mutant HTT. These findings might also have implications for the turn-over of other disease-associated, aggregation-prone proteins.
Graphical Abstract
Graphical Abstract.

Introduction
Cell-to-cell communication by extracellular vesicles (EVs) is a growing field of investigation in basic cell biology research (1–6), biomarker discovery (7–10) and therapeutic drug delivery (11–14). EVs are now known to comprise a heterogenous population of vesicles that can differ in size, density, composition as well as biological function. One approach aimed at elucidating the function of such vesicles is to identify their cargo; by identifying the proteins that are found in distinct EV subpopulations and exploiting them for visualization and tracking purposes, one can unveil divergent roles of such vesicles. Thus, in the last two decades, research efforts have sought to understand the mechanism by which EVs form, are secreted, interact with recipient cells and, similarly, how to best harness EVs as delivery vehicles (15–17).
One family of potential cargoes are the small heat shock proteins (sHSPs), a family of 11 proteins, named HSPB1 through HSPB11, some of which are ubiquitously expressed, while others tissue specific (18,19). They are defined by their low molecular weight (ranging from 16 to 28 kDa, as detailed in Supplementary Material, Fig. S1A) and are characterized by a conserved α-crystallin domain (ACD) of 80 amino acids, normally present in the central region of these proteins. This domain is important for the formation of the oligomeric structure (20). The ACD is flanked by a less conserved N-terminal domain and a variable C-terminal extension, which both play an important role in oligomerization (21,22). Some members of the sHSP family such as αA-crystallin, αB-crystallin and Hsp27 form large oligomeric species (23).
Organisms and cells can respond and adapt to various stress conditions by increasing the expression of these proteins. The sHSPs are involved in many cellular physiological and pathological functions such as cell death, cell cycle, protein degradation and maintaining cytoskeletal integrity (24). Among the others, sHSPs can interact with aggregation-prone proteins associated to neurodegenerative diseases, preventing their aggregation (25,26). In this context, Huntington disease (HD), Alzheimer disease (AD), Parkinson disease (PD) and amyotrophic lateral sclerosis (ALS) are some examples of conformational diseases characterized by the progressive accumulation of misfolded protein, formation of protein aggregates and inclusion bodies, which ultimately lead to neuronal toxicity and death. In addition, several chaperones can also undergo secretion and such a phenomenon can impact on and contribute to the pathogenesis of several neurodegenerative disorders (27–31). In particular, HSPB1 has been shown to exert a protective effect by reducing the accumulation of intracellular toxic oligomers and to be released by astrocytes in response to production of amyloid-beta (32,33). Notably, (macro)autophagy has emerged as a critical pathway in regulating the unconventional secretion of several intracellular proteins (34–36). Indeed, we have recently reported how autophagy can contribute to the unconventional secretion of the small heat shock protein HSPB5/CRYAB, by hampering the autophagosome–lysosome heterotypic fusion step, which is critical for the proper turn-over of autophagy substrates, including the PD-associated A53T variant of alpha-synuclein (37).
Our laboratory is interested in understanding the functional interactions between sHSPs and aggregation-prone proteins that are features of neurodegenerative diseases. Huntington disease (HD) is a neurodegenerative disorder caused by the expansion of a CAG repeat (>35) in exon 1 of huntingtin HTT gene, which results in an abnormal polyglutamine tract (38,39). HD is characterized by the deposition of large intracellular protein aggregates, or inclusion bodies, composed of N-terminal fragments of mutant HTT. The accumulation of soluble oligomeric species of HTT can cause neurotoxicity (39–42). The turn-over of mutant HTT is largely dependent on the activity of the autophagic pathway (43). Mutant HTT can be degraded by autophagy through both mTOR-dependent and -independent autophagic pathways. In addition, mutant HTT can interact with the autophagy cargo receptor protein p62/SQSTM1 to initiate autophagosome formation, and such a mechanism promotes autophagic degradation of the mutant HTT protein (43–48).
Importantly, mutant HTT may have the propensity to propagate by a prion-like mechanism (49). For example, Masnata et al. in 2019 have shown that SH-SY5Y cells, THP1-derived macrophages and iGABA human neurons take up toxic HTT-Exon1 fibrils (50). Uptake is accompanied by the aggregation of endogenous HTT. In addition, several reports have shown how mutant HTT can be subjected to inter-cellular spreading by means of EVs, misfolding-associated protein secretion (MAPS) and the formation of nanotubular structures (NTNs) (51–55).
As part of an ongoing screen aimed at identifying novel signalling pathways and components capable of regulating unconventional secretion, we found that HSPB1 is able to interact with p62/SQSTM1 to modulate synergistically its unconventional secretion in an autophagy-independent manner. In addition, both proteins form a platform for the functional disaggregation, sorting and secretion of mutant HTT, via a mechanism that is regulated by the PI3K/AKT/mTOR signalling axis (56–59). Finally, we demonstrate how the pool of HSPB1-loaded EVs are functionally capable of guaranteeing the transcellular spreading of mutant HTT. Importantly, these findings reveal a novel mechanism for the spreading and seeding of protein aggregates, which may have wider implications for and impact the pathobiological mechanisms underlying other neurodegenerative disorders.
Results
Screening for the identification of HSPB family members unconventionally secreted
Small heat shock proteins (HSPBs) are a family of proteins with chaperone-like activity, which can prevent protein aggregation. We have previously reported how the small heat shock protein HSPB5 is secreted by an unconventional secretion pathway that involves multi-vesicular bodies (MVBs). Importantly, we found that HSPB5 secretion was regulated by phosphorylation at serine residue 59, which acts by inhibiting its recruitment to the autophagosomes (37). To identify whether other HSPB family members undergo a similar unconventional secretion, HeLa cells were transiently transfected with V5-HSPB1 to V5-HSPB8 over-expressing constructs. Using SDS-PAGE to analyse the cell lysate and secreted fraction, we found that HSPB1, HSPB4, HSPB5 and, to a lesser extent, HSPB6 were secreted into and readily detectable in the culturing media (Fig. 1A–B).
Figure 1.

Screening for the identification of HSPB family members unconventionally secreted. (A–B) HeLa cells were transiently transfected with constructs encoding the different V5-tagged HSPBs. Twenty four hours after transfection, cells were cultured in DMEM with 1% FBS for 8 h to allow protein secretion. Both secreted fraction and cell lysate were collected and processed by western blot analysis. All the proteins were present in the cell lysate, but only HSPB1, HSPB4 and HSPB5 were detected in the secreted fraction. (C–D) EVs were isolated from conditioned media of HeLa cells transiently transfected with the GFP-CD63 over-expression construct by differential ultracentrifugation. P100 fractions were processed and ultrathin sections (60 nm) prepared as detailed in Materials and Methods section and the grid observed by transmission electron microscopy (C, scale bar: 50 nm). For the immunogold staining of sections, an anti-CD63 specific antibody and 10-nm gold particles were used (D, scale bar: 100 nm). (E–F) HeLa cells were kept in culture in 10-cm dishes for 48 h. In the last 8 h, a secretion assay was performed. Secreted fractions were subjected to ultracentrifugation at 100000xg for 2 h. Pellet (P100) fractions were subjected to nanoparticle tracking analysis. The plots in (E) and (F) report the concentration and particles size average distribution of EVs. (G–H) HeLa cells were placed in DMEM 1% FBS for 8 h in the presence or not of 5-mg brefeldin A, then secreted fraction and cell lysate were collected. Secreted fractions were centrifuged at 100 000×g for 2 h. Pellet (P100) was resuspended in lysis buffer and processed for western blot analysis using an HSPB1 antibody. HSPB1 is present in the P100 fraction and in the cell lysate but not in the S100, also upon brefeldin A treatment, confirming that HSPB1 is secreted in EVs unconventionally. (I–K) Western blot analysis of ATG16L1 WT and KO cells, which are unable to conjugate LC3 and form autophagosomes (E). Parental and ATG16L1 KO HeLa cells were transfected with V5-HSPB1 over-expression construct. After 24 h, cells were placed in DMEM 1% FBS for 8 h, then secreted fraction and cell lysate were collected. Secreted fraction was centrifuged at 100 000 g for 2 h. Pellet (P100) fractions were lysed and processed for western blot analysis using a HSPB1 antibody. HSPB1 is present in the P100 fraction and in the cell lysate but not in the S100, also in ATG16L1 KO Hela cells, suggesting that HSPB1 secretion is autophagy independent. The graphs in (B), (D) and (G) report the quantitative analysis of protein secretion relative to loading control from at least three independent experiments. The y-axis values are shown as the OUT/IN relative ratio and the error bars denote standard deviations. The P-values for the densitometric analyses were determined by Student’s t-test using STATVIEW v4.53 (Abacus Concepts) (n = 3, NS: non-significant).
We first evaluated whether endogenous HSPB1 was contained within EVs (60,61). In particular, we found that endogenous HSPB1, as well as the exosomal markers CD63 and CD9, were present in the P100 fraction representing the EVs isolated by means of differential ultracentrifugation (37) of conditioned media derived from HeLa cells (Supplementary Material, Fig. S1B–D). To confirm the presence of EVs purified with our experimental approach, we decided to further characterize the P100 fractions by electron microscopy and nanoparticle tracking analysis. As reported in Figure 1C, by transmission electron microscopy we could observe in the P100 fractions processed and analysed by electron microscopy a number of heterogeneous vesicular structures (arrowheads in Fig. 1C) of different size. We also performed immunogold labelling on these sections, confirming the presence of the exosomal marker CD63 on at least on some of these structures (Fig. 1D, white arrowhead in the magnified inset). As expected, these structures were non-homogeneous in terms of size and composition. In order to corroborate these observations, we analysed the size distribution of particles in our samples by means of nanoparticle tracking analysis (NTA). Consistent with the electron microscopy analysis, our results showed how the diameter of the particles contained in the P100 fractions ranged from 70 to 200 nm (Fig. 1E–F), with a mean value of 124.6 ± 0.8 nm, which is compatible with the size range of exosomes and EVs (50–140 nm) reported in the literature (1–6). All together, these data confirm that our experimental protocol allows us to successfully isolate EVs derived from conditioned media of cultured cells, in which we could detect both endogenous and over-expressed HSPB1.
In order to provide further evidence that HSPB1 is secreted by unconventional secretion, HeLa cells were transiently transfected with V5-HSPB1 and treated for 8 h with 5 μg/ml brefeldin A (Bref. A), which reversibly blocks the protein transport from the endoplasmic reticulum to the Golgi compartment, without affecting endocytosis or lysosomal function (62). Both secreted fractions and cell lysates were collected and EVs were isolated by ultracentrifugation. HSPB1 secretion was not affected by brefeldin A treatment (Fig. 1G–H), therefore confirming that HSPB1 can be secreted unconventionally.
We next investigated whether the autophagic pathway might be involved in the unconventional secretion of HSPB1 by measuring the amount of HSPB1 secreted in ATG16L1 KO HeLa cells (63), which are unable to produce autophagosomes (Fig. 1I). We did not observe any reduction in HSPB1 secretion compared with the isogenic control cells (Fig. 1J–K). Hence, our results showed that HSPB1 can undergo unconventional secretion. However, differently from what previously reported for the HSPB5 protein (37), such a phenomenon occurs in an autophagy-independent manner.
HSPB1 interacts with p62/SQSTM1 and synergistically supports its unconventional secretion
Despite the lack of functional correlation between the activity of the autophagic pathway and the secretion of HSPB1, it has been previously shown that HSPB1 interacts with the autophagy cargo receptor p62/SQSTM1 (64). Interestingly, the interaction between HSPB1 and p62/SQSTM1 is instrumental for allowing cells some initial steps required for the regulation of autophagosome biogenesis (64). We initially performed a number of complementary experiments to validate the interaction between HSPB1 and p62/SQSTM1 and to evaluate whether the two proteins were able to be secreted together. To this end, HeLa cells were co-transfected with HA-p62/SQSTM1 and V5-HSPB1. Anti V5-tag antibody was used to immunoprecipitate HSPB1. As shown in Figure 2A (top panel), p62/SQSTM1 did specifically interact with HSPB1 (Fig. 2A, lane 3), as no pull-down was observed by using a control antibody (Fig. 2A, lane 4 and Supplementary Material, Fig. S2A). We then performed the reverse immunoprecipitation with an HA antibody to pull down p62/SQSTM1, and once again we were able to validate its interaction with HSPB1 (Fig. 2A, bottom panel, lane 3).
Figure 2.

HSPB1 interacts with p62/SQSTM1 and supports synergistically its unconventional secretion. (A) HeLa cells were transiently transfected with V5-HSPB1-V5 and HA-p62/SQSTM1. Twenty four hours after transfection, cell lysates were prepared and immunoprecipitation was performed using either anti-V5 or anti-HA antibody. HSPB1 and p62/SQSTM1 interaction was detected in both orientations. In addition, an unconjugated control IgG antibody was used to verify the specificity of the interaction (lane 4 of the top and bottom panels). (B–C) HeLa cells were transiently transfected with GFP-HSPB1 and HA-p62/SQSTM1. After 24 h, cells were semi-permeabilized by digitonin treatment, fixed, subjected to immunofluorescence and analysed by confocal microscopy. Magnifications (10×) of the merged channels are shown in the insets. The co-localization analysis was performed using an ImageJ plug-in (JaCoP). The graph shows Mander’s coefficient (fraction of HSPB1-positive structures overlapping with p62-positive structures and reverse, grey bars). Scale bars: 10 μm. (D) HeLa cells were placed in DMEM 1% FBS for 8 h, then secreted fraction and cell lysate were collected. Secreted fraction was centrifuged at 100 000 g for 2 h. Pellet (P100) was resuspended in lysis buffer and processed for western blot analysis using a p62/SQSTM1 antibody. P62/SQSTM1 is present in the P100 fraction and in the cell lysate but not in the S100. (E–F) HeLa cells were transfected with HSPB1-V5, P62-HA or both. After 24 h, the cells were placed in 1% FBS for 8 h to allow protein secretion. Both cell lysate and secreted fraction were collected and analysed. p62/SQSTM1 secretion is increased by HSPB1 over-expression as shown also in the quantification (F). (G–H) HeLa cells were co-transfected with V5-HSPB1 and HA-p62/SQSTM1. Twenty four hours after transfection, cells were washed three times and placed in serum-free DMEM (NO FBS) for the indicated times (from 0 to 24 h). Secreted fractions were collected from 0.5 up to 24 h. Both HSPB1 and p62/SQSTM1 secretion increases over time, but the amount of p62/SQSTM1 is decreased after 24 h. (I–J) Schematic diagram depicting the experimental protocol deployed to assess the dynamic variations of HSPB1 and p62/SQSTM1 secretion upon serum deprivation. HeLa cells were serum starved or left in complete medium. Then cells were placed in 1% FBS or NO FBS for 8 h to allow protein secretion and 1 h before collection 1% FBS was added to the rescue condition. Cell lysates were collected and analysed using the indicated antibodies. AKT phosphorylation is reduced by serum deprivation and is increased again after re-addition of FBS. (K–L) HeLa cells were transiently transfected with V5-HSPB1 and HA-p62/SQSTM1. After 24 h, cells were serum starved or left in complete medium. Then, cells were placed in 1% FBS for 8 h to allow protein secretion, and 1 h before collection, FBS was added back to the rescue condition. p62/SQSTM1 secretion is increased after serum starvation and the secretion is reversible. The graphs in (F), (H) and (L) report the quantitative analysis of HSPB1 and p62/SQSTM1 protein secretion relative to loading control from at least three independent experiments. The y-axis values are shown as the OUT/IN relative ratio and the error bars denote standard deviations. The P-values for the densitometric analyses were determined by factorial ANOVA or Student’s t-test using STATVIEW v4.53 (Abacus Concepts) (n = 3, **P < 0.001; NS: non-significant).
We did also perform co-immunoprecipitation experiments and we were able to validate the interaction between endogenous HSPB1 and p62/SQWSTM1 (Supplementary Material, Fig. S2B–C). To confirm and further understand the spatial relationship between HSPB1 and p62/SQSTM1, we performed a co-localization analysis by immunofluorescence and confocal imaging. HeLa cells were transiently transfected with HA-p62/SQSTM1 and GFP-HSPB1 and then subjected to semi-permeabilization by digitonin treatment, as previously reported (37,65). Co-localization analysis showed a strong co-localization between HSPB1 and p62/SQSTM1 (Mander’s coefficient higher than 0.6, in both ways) in HeLa cells (Fig. 2B–C), therefore reinforcing the co-IP-based interaction data. Such an observation was also confirmed by looking at the co-localization of endogenous HSPB1 and p62/SQSTM1 proteins upon digitonin treatment (Supplementary Material, Fig. S2D–E). In view of the relevance of p62/SQSTM1 for the regulation of both the autophagy and the ubiquitin–proteasome system (UPS) (66), we also asked whether the over-expression of HSPB1 could influence the activity of these pathways. To this end, we performed a panel of experiments to measure the activity of the autophagic pathway. The over-expression of HSPB1 did not show any effect on the LC3-II and p62/SQSTM1 levels in basal conditions or in the presence of bafilomycin A1 (Supplementary Material, Fig. S3A–D). We also looked at the effect of HSPB1 over-expression on the number of aggregates generated by the GFP-Q74HTT exon 1 reporter, which is a well-established autophagy substrate (43). GFP-HTT(Q74) levels were reduced in cells over-expressing HSPB1, albeit to a lesser extent than in those treated with trehalose (Supplementary Material, Fig. S3E), a well-known autophagy enhancer (46). Despite the lack of any direct effect of the HSPB1 over-expression on the activity of the autophagic pathway, the latter effect is entirely consistent with previous reports and can be explained by the key role played by HSPB1 and other small HSPBs in regulating the activity of disaggregation machineries (27,30–32). We also performed a UbiquitinG76V-GFP (degron) assay to assess proteasome activity (66,67) and we could not observe any significant effect deriving from the HSPB1 over-expression (Supplementary Material, Fig. S3F). Hence, we can conclude that the unconventional secretion of over-expressed HSPB1 does not depend on or reflect any substantial change in the activity of the major systems controlling cellular proteostasis, including the autophagic pathway and the UPS.
In view of the interaction between HSPB1and p62/SQSTM1 and of the observations that also endogenous HSPB1 could be detected in the EVs isolated from cells (Supplementary Material, Fig. S1C), we asked whether p62/SQSTM1 itself could undergo secretion. Indeed, we found that endogenous p62/SQSTM1 was present in the P100 fraction, representing the EVs isolated upon differential ultracentrifugation of conditioned media derived from non-transfected HeLa cells (Fig. 2D).
In order to infer about the potential functional correlation between HSPB1 and the secretion of p62/SQSTM1, we performed secretion assays in HeLa cells that were transiently transfected with V5-HSPB1 alone, HA-p62/SQSTM1 alone or in combination. We found that p62/SQSTM1 was present in the secreted fraction and that upon over-expression of HSPB1 the secretion of p62/SQSTM1 was significantly increased, whereas the levels of HSPB1 levels remained unchanged, suggesting the existence of a directional synergism (i.e. HSPB1 on p62/SQSTM1, but not the opposite), in the unconventional secretion of the two proteins (Fig. 2E–F). In view of these data, we decided to look at the general effect deriving from the HSPB1 over-expression on the generation of EVs. To this end, we performed nanoparticle tracking analysis by comparing P100 fractions obtained from either empty vector or HSPB1-transfected cells. Interestingly, the particle profiling did not show any overt effect deriving from the HSPB1 over-expression neither in terms of concentration, nor in terms of size distribution of the analysed particles (Supplementary Material, Fig. S4A–D). Consistent with this, we could not observe any effect of HSPB1 on the secretion rate of the exosomal marker CD63 (Supplementary Material, Fig. S4E–F). All together, these data prompted us to hypothesize that HSPB1, under certain conditions, could play a role in regulating the loading and/or composition of (at least some) of the EVs that can be secreted by cells.
It is known that p62/SQSTM1 serves as a signalling hub in nutrient sensing, activating mTORC1, which regulates protein translation, anabolism, cell growth and autophagy (68,69). In order to evaluate whether serum starvation could regulate secretion of both HSPB1 and p62/SQSTM1, we performed a run-on secretion assay in HeLa cells transiently transfected with HSPB1 and p62/SQSTM1, in the absence of serum (NO FBS). To this end, secreted fractions were collected over a time-course, from 0.5 to 24 hours. Interestingly, we found that the secretion of both HSPB1 and p62/SQSTM1 were increased over time; however, the kinetics of the secretion of the two proteins displayed some remarkable differences. In particular, HSPB1 secretion increased up to 8 h, when it reached a plateau. Conversely, p62/SQSTM1 secretion progressively increased up to 12 h and then decreased at 24 h (Fig. 2G–H), suggesting that its secretion is temporally regulated and reversible. We hypothesized that the amount of p62/SQSTM1 present in the secreted fractions might represent the result of a dynamic equilibrium between secretion and re-uptake of these vesicular carriers (EVs). Hence, we decided to further investigate the modulation of the signalling axis that could regulate this phenomenon. In particular, in order to understand the variations of the dynamic properties induced by serum starvation, we performed the experiments comparing control cells kept in full medium (FM) to serum-starved cells (NO FBS) and cells where the signalling was restored (RESCUE), by adding back the serum (as schematically depicted in Fig. 2I). To monitor the experimental protocol, we looked at the phosphorylation state of PKB/AKT, which is a direct substrate of the PI3K pathway, in response to full medium, starvation and the rescue condition (47). As expected, the phosphorylation of AKT was blunted upon serum deprivation and fully rescued by re-adding serum to cultured cells (Fig. 2J). Notably, the secretion of p62/SQSTM1, but not HSPB1, was strongly increased by serum starvation. Importantly, the re-addition of serum was able to revert the effect on p62/SQSTM1 (Fig. 2K–L). However, such a phenomenon was not due to any change in either the interaction (Supplementary Material, Fig. S5A) or the co-localization between HSPB1 and p62/SQSTM1 (Supplementary Material, Fig. S5B–C), induced by serum starvation. Importantly, we were also able to confirm the effect of serum starvation by looking at the secretion of endogenous p62/SQSTM1, which was detected in the P100 fraction derived from HeLa cells and increased in serum starvation conditions (Supplementary Material, Fig. S5D–E). Finally, we performed an experiment to assess whether the synergistic effect exerted by the over-expression of HSPB1 could still be observed upon serum starvation. Differently from what is reported in steady-state condition, the HSPB1 over-expression does not induce any further increase in the serum-starvation increased secretion of p62/SQSTM1 (Supplementary Material, Fig. S6A–B). Likewise, we could not observe any additional effect deriving from the over-expression of HSPB1 when we looked at the progressive secretion of p62/SQSTM1 in a run-on experiment performed in serum starvation condition (Supplementary Material, Fig. S6C–D), suggesting that the latter acts upstream of HSPB1 and results in a ‘bottle-neck effect’ (i.e. saturating effect) on this phenomenon, at least under these experimental conditions. Hence, our data strongly suggest that the modulation of growth-factor-dependent signalling response, elicited, for instance, upon serum deprivation, can indeed trigger the unconventional secretion of p62/SQSTM1. Nonetheless, the over-expression of the small heat shock protein HSPB is sufficient to synergistically enhance the secretion of p62/SQSTM1 even in steady-state conditions.
Effect of HSPB1 mutants on the unconventional secretion of p62/SQSTM1
HSPB1 contains within the N-terminal domain three serine residues that can undergo phosphorylation, namely Ser15, Ser78 and Ser82 (70). It is known that the combined phosphorylation of these serine residues can influence the oligomerization status of the protein (71) and, as a consequence, modulate the chaperone function of HSPB1 (Supplementary Material, Fig. S7A) (72). Indeed, it has been proposed that the alpha-crystallin domain of HSPB1 is responsible for its ability to form dimers and, in response to variations of the phosphorylation of the N-terminal domain, the protein can further multimerize (72,73). As such, these dynamic transitions between low and high molecular weight isoforms allow HSPB1 to interact with different client proteins and/or to modulate the affinity towards substrate proteins (72). In view of these considerations, we asked whether the phosphorylation/oligomerization state of HSPB1 could be relevant for its own secretion, and for the interaction with p62/SQSTM1 (and, ultimately, for the effect on the secretion of the latter). To this end, HeLa cells were transfected with either HSPB1 WT, HSPB1 3A (S15A/S78A/S82A) mutant or HSPB1 3D (S15D/S78D/S82D) mutant constructs, and we performed cell fractionation experiments followed by isopycnic ultracentrifugation to evaluate the oligomerization status of HSPB1 (71). In particular, cell lysates were loaded on a discontinuous sucrose gradient (5–20%, w/v) and ultracentrifuged at 100 000 g for 24 h at 4°C. Twelve fractions were collected from the top to the bottom of the gradient and the remaining pellet was resuspended in sample buffer. All the samples were subjected to immunoblot analysis. As previously described by Bolhuis and Richter-Landsberg (71), the fraction distribution was different between the WT and 3A/3D-HSPB1 mutants (Supplementary Material, Fig. S7B–C). In particular, the 3A-HSPB1 mutant closely resembled the distribution of the WT protein (with peaks positioned between fractions 9 and 10, plus the presence of a right-side shoulder), whereas the 3D-HSPB1 was shifted towards the left (i.e. lighter) part of the gradient (peak at fraction 7–8 interface), consistent with a lower oligomerization state of the 3D-variant protein (Supplementary Material, Fig. S7B–C). We then evaluated the secretion of both phospho-mutant proteins and found that both 3A-HSPB1 and 3D-HSPB1 were present in P100 fractions at comparable levels (Fig. 3A–B) indicating that, differently from what was previously reported for the HSPB5/CRYAB protein (37), HSPB1 can be conveyed into and secreted by means of vesicular structures independently of its phosphorylation (and therefore oligomerization) state.
Figure 3.

Effect of HSPB1 mutants on the unconventional secretion of p62/SQSTM1. (A–B) HeLa cells were transiently transfected with 3A-HSPB1 and 3D-HSPB1 phosphorylation mutants. After 24 h, cells were placed in DMEM 1% FBS for 8 h and secreted fractions were collected, pre-cleared and centrifuged at 100 000×g for 2 h. Pellet (P100) fractions were lysed and processed for western blot analysis using an anti-FLAG antibody. The graph in (B) reports the quantitative analysis of 3A-HSPB1 and 3D-HSPB1 protein levels contained in the P100 fractions, relative to the inputs, from at least three independent experiments. (C–D) HeLa cells were transiently transfected with HA-p62/SQSTM1 and either 3A-HSPB1 or 3D-HSPB1 expression constructs. After 24 h, cell extracts were collected and 3A-HSPB1, 3D-HDSPB1 (E) or p62/SQSTM1 (F) was immunoprecipitated using the indicated antibodies. Co-immunoprecipitation was then evaluated with specific antibodies, as indicated. 3A-HSPB1 and 3D-HSPB1 are both able to co-immunoprecipitate with p62/SQSTM1, but the phospho-mimetic mutant (3D) shows a higher affinity for p62/SQSTM1 compared with the 3A. (E–F) HeLa cells were co-transfected with p62-HA and either 3A-HSPB1 or 3D-HSPB1. Twenty four hours after transfection, cells were serum starved overnight or left in complete medium. Then, cells were washed and placed in 1% FBS or serum-free DMEM (NO FBS) to allow protein secretion, and 1 h before collection, FBS was added to the rescue condition. Both cell lysates and secreted fractions were collected. 3D-HSPB1 strongly increases p62/SQSTM1 secretion both in steady state and in serum starvation. The graph in (F) and the inset report the quantitative analysis and the best-fit functional curve of HSPB1 and p62/SQSTM1 protein secretion, respectively, relative to loading control from three independent experiments. The y-axis values in the graph are shown as the OUT/IN relative ratio and the error bars denote standard deviations. The P-values for the densitometric analyses were determined by factorial ANOVA or Student’s t-test using STATVIEW v4.53 (Abacus Concepts) (n = 3, *P < 0.05; **P < 0.01; NS: non-significant).
In order to understand whether the phosphorylation state of HSPB1 could affect its interaction with p62/SQSTM1, we performed co-immunoprecipitation experiments in HeLa cells transiently co-transfected with the phospho-mutant HSPB1 variants, along with HA-p62/SQSTM1. We confirmed that both the 3A- and 3D-HSPB1 variants were able to interact with and to co-immunoprecipitate p62/SQSTM1 (Fig. 3C). However, when we performed the co-immunoprecipitation in reverse (i.e. with an HA antibody to pull down p62/SQSTM1), we were able to appreciate a higher co-immunoprecipitation of the 3D-HSPB1 mutant with p62/SQSTM1 compared with the 3A variant (Fig. 3D). This is consistent with previous studies, reporting that the 3D-HSPB1 variant, in view of its reduced oligomerization ability, can display a higher promiscuity and/or affinity towards some client proteins (74,75). We then decided to test whether such a different affinity of p62/SQSTM1 for the 3A/3D phospho-mimetic variants of HSPB1 could affect its secretion. To this end, HeLa cells were transfected with HA-p62/SQSTM1 in combination with either the 3A-HSPB or 3D-HSPB1 and we performed our standardized secretion assays. Strikingly, the over-expression of the phospho-mimetic protein (3D)-HSPB1 was able to increase, compared with the 3A variant, the secretion of p62/SQSTM1, both at steady state and upon serum starvation/rescue (Fig. 3E–F and inset). Collectively, these data suggest that HSPB1 interacts with p62/SQSTM1 and is able to regulate its unconventional secretion, indicating that the HSPB1-p62/SQSTM1 complex can exert in cis a tangential effect on the cargo loading and secretion step.
The HSPB1 over-expression induces the clearance and secretion of mutant HTT
HSPB1 is able to protect cells from toxicity caused by misfolded proteins, for instance, in the case of disease-associated mutant proteins, such as α-synuclein, the mutated Superoxide Dismutase 1 (SOD1) and HTT, which are able to cause neurodegeneration (76–78). In addition, it has been recently reported how some proteotoxic stress can favour the p62/SQSTM1- and autophagy-dependent clearance of protein aggregates bearing an expanded polyglutamine (polyQ) tract, such as mutant huntingtin (79). We therefore decided to investigate the potential role of the HSPB1-p62/SQSTM1 complex in the turn-over and secretion of aggregation-prone proteins, such as mutant HTT. To this end, HeLa cells were transfected for 48 h with WT-HSPB1 and either the WT N-terminal fragment of huntingtin (1-588, 17 poly-Q, hereafter referred to as WT-HTT) or the expanded mutant huntingtin (1-588, 138 poly-Q, hereafter referred to as mut HTT), as detailed (Fig. 4A). Importantly, we did not observe any overt change in cellular viability induced by the over-expression of either WT- or the mut HTT when compared with non-transfected cells (Supplementary Material, Fig. S8A), ruling out any potential effect deriving from cellular toxicity. In HSPB1 over-expressing cells, we could observe the secretion of both forms of HTT (Fig. 4B); interestingly, the secretion of the mutant protein was much greater than the wild-type (WT) form, suggesting that HSPB1 preferentially regulates the secretion of the aggregation-prone mutant HTT. As an additional control, we performed the Triton X-100 soluble/insoluble fractionation protocol on the intracellular fractions of cells transfected with HA-p62/SQSTM1 and mut HTT (as schematically detailed in Supplementary Material, Figure S8B), a well-described and standardized method to determine protein solubility and aggregation (80). This allowed us to show how, at steady state, the fractionation profile of mut HTT, p62/SQSTM1 and endogenous HSPB1 were different (confirming also the sufficient dynamic range of the analysis we have deployed in this study) and largely unaffected by serum starvation (Supplementary Material, Fig. S8C). In addition, we observed how both the soluble and insoluble levels of mut HTT in the cell lysate were remarkably reduced upon over-expression of HSPB1 (Fig. 4C–E), with a less pronounced effect observed on the levels of WT-HTT (Supplementary Material, Fig. S8D).
Figure 4.

The HSPB1 over-expression induces the clearance and secretion of mutant huntingtin. (A) Schematic representation of HTT WT and mutant (MUT) huntingtin constructs. (B) HeLa cells were transiently transfected with V5-HSPB1 and WT or mut HTT. Cells were placed in 1% FBS for 8 h and cell lysate and secreted fractions were analysed with the indicated antibodies. (C–E) HeLa cells were transfected with mutant HTT with or without V5-HSPB1. Cells were placed in 1% FBS for 8 h and secreted fractions were collected and processed as previously described. Cell lysates were subjected to solubility fractionation, as detailed in Supplementary Material, Figure S5(B) and analysed by western blot. HSPB1 over-expression reduces both the soluble and insoluble fractions and increases HTT mutant secretion. The graph in (D) and (E) report the quantitative analysis of mut HTT protein levels and secretion relative to loading control from three independent experiments. The y-axis values are shown as the OUT/IN relative ratio and the error bars denote standard deviations. (F–G) HeLa cells were transiently transfected with the GFP-HTT Q74 over-expression construct with or without V5-HSPB1 for 24 h. Cells were placed in 1% FBS for 8 h. Cell lysate and secreted fraction were collected and analysed by western blot. The graph in (G) reports the quantitative analysis of GFP-HTT Q74 protein secretion relative to loading control. The y-axis values are shown as the OUT/IN relative ratio and the error bars denote standard deviations. (H–I) HeLa cells were transfected with V5-HSPB1 and WT or mutant huntingtin. Cell lysates were collected and HSPB1 was immunoprecipitated using an anti-V5 antibody. Both WT and mut HTT co-immunoprecipitate with HSPB1. The graph in (I) reports the quantitative analysis of WT and mut HTT co-immunoprecipitated by HSPB1. The y-axis values are shown as the mean values with standard deviations deriving from three independent experiments. The P-values for the densitometric analyses were determined by factorial ANOVA using STATVIEW v4.53 (Abacus Concepts) (n = 3, *P < 0.05; **P < 0.01; NS: non-significant).
Collectively, these data are entirely consistent with previous reports, showing that HSPB1 is indeed capable of reducing the aggregation propensity of several disease-associated mutant proteins such as huntingtin, α-synuclein and SOD1 (30,31,34,76–78) by means of its disaggregation activity (81). Notably, our data do confirm and integrate such a scenario, showing how the over-expression of HSPB1 is capable of preferentially reducing the aggregation of mut HTT and at the same time to favour its secretion. Consistent with this, we observed the same effect of HSPB1 over-expression on the secretion of the GFP-Q74-HTT reporter vector (Fig. 4F–G), whereas the secretion of the control GFP-Q21-HTT reporter remained unchanged (Supplementary Material, Fig. S8E–F). As HSPB1 was able to preferentially increase the secretion of mut HTT, we tested whether this difference was due to a different affinity of HSPB1 towards the mutated form. To this end, we performed co-immunoprecipitation experiments in HeLa cells transiently transfected with HSPB1 and either WT or mut HTT. HSPB1 was immunoprecipitated using an anti-V5 antibody and co-immunoprecipitation was assessed using an anti-Flag antibody to detect HTT. Strikingly, we observed how HSPB1 interacts preferentially with mut HTT (Fig. 4H–I), suggesting that the higher affinity for the mutant protein bearing the expanded polyQ-tract might favour the cargo loading of mut HTT, therefore facilitating its increased secretion observed upon HSPB1 over-expression. Finally, in order to assess the physiological relevance of these observations, we decided to test whether the HSPB1 over-expression could exert any effect on the secretion of endogenous p62/SQSTM1 and huntingtin proteins. To this end, P100 fractions were isolated from HeLa cells transiently transfected with either empty vector or V5-HSPB1 and analysed by western blot. Consistent with the effect observed upon serum starvation (Supplementary Material, Fig. S5D–E), we could observe a significant increase of secreted p62/SQSTM1 in HSPB1-over-expressing cells, compared with control cells (Supplementary Material, Fig. S9A–B). However, such an effect was not observed for the full-length endogenous huntingtin, whose low levels of secretion remained unchanged upon HSPB1 over-expression (Supplementary Material, Fig. S9A–B).
Serum starvation acts upstream of the HSPB1-p62/SQSTM1 complex and triggers the secretion of mutant HTT
HSPB1 contains three serine residues that can be phosphorylated by several kinases (70), particularly mitogen-activated protein kinases associated protein kinases (MAPK/MK2,3). The phosphorylation status regulates the function of an oligomerization form of HSPB1 (71). Notably, p62/SQSTM1 represents one of the major cellular hubs regulating autophagy cargoes selection and loading (44,82). Among others, the functional interaction with ubiquitinated and polyQ-bearing proteins is essential for their degradation (83,84). Since we have shown that serum starvation can modulate p62/SQSTM1 secretion but not HSPB1 secretion, we asked whether the secretion of mut HTT could be regulated by the same signalling pathway. As shown in Figure 5(A) and (B), mut HTT secretion was increased upon serum starvation. However, we performed co-immunoprecipitation experiments in HeLa cells transiently transfected with HSPB1 and mut HTT and we did not observe any variation in the interaction of HSPB1 with mut HTT upon serum starvation (Fig. 5C–D), suggesting that the increased secretion is not consequent to a direct change in the affinity or binding of HSPB1 for the mut HTT. Notably, we could appreciate how the HTT appears as a doublet, with the faster migrating band of the doublet being preferentially secreted upon serum starvation (Fig. 5A). Both WT and mut HTT can undergo a number of different post-translational modifications, including phosphorylation, SUMOylation, ubiquitination, acetylation, proteolytic cleavage and palmitoylation (85). HTT can undergo several proteolytic cleavages by caspases under basal and apoptotic conditions. In particular, it has been reported that HTT can be cleaved by caspase-6 and caspase-3. There are two primary sites of caspase cleavage in HTT that map to locations associated with cellular toxicity, Asp552 and Asp586 (85–88), which reside within the 1-588 N-terminal fragment used in our study (Fig. 4A).
Figure 5.
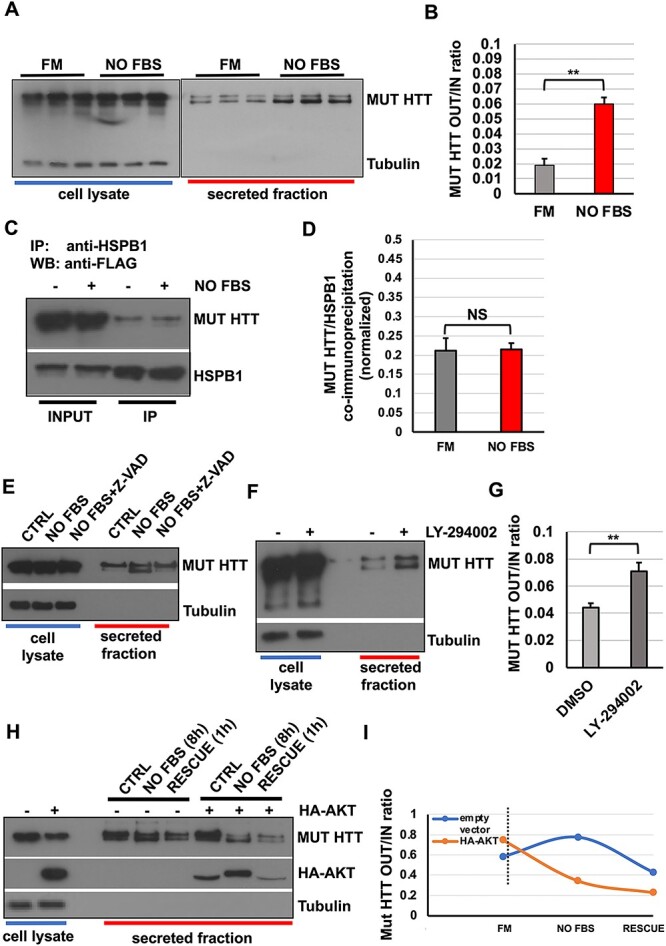
Serum starvation acts upstream of HSPB1-p62/SQSTM1 and triggers the secretion of mutant huntingtin. (A–B) HeLa cells were transiently transfected with mutant HTT. After 24 h, cells were serum starved or left in complete medium. Secreted fractions were collected and analysed by western blot. Serum starvation increases mut HTT secretion. (C–D) HeLa cells were co-transfected with V5-HSPB1 and mut HTT. After 24 h, cells were either kept in full medium or serum starved overnight. Cell lysates were collected and HSPB1 was immunoprecipitated using an anti-V5 antibody. The graph in (D) reports the quantitative analysis of mut HTT co-immunoprecipitated by HSPB1. Serum starvation does not affect the HSPB1/mut HTT interaction. (E) HeLa cells were transiently transfected with mut HTT for 24 h. Cells were left in complete medium or serum starved overnight in the presence or absence of 10-mm Z-VAD, and then secretion assays were performed in the same conditions. Secreted fractions were collected and analysed by western blot. Z-VAD treatment inhibit the proteolytic cleavage of mut HTT, without interfering with its secretion. (F–G) HeLa cells were transiently transfected with mut HTT. After 24 h, cells were treated or not with 10 μm LY-294002 overnight. Then cells were placed in 1% FBS (replenishing the LY-294002) for 8 h and both cell lysates and secreted fraction were collected and analysed. LY-294002 is able to mimic serum starvation and increases mut HTT secretion. (H–I) HeLa cells were transiently transfected with mut HTT with or without HA-AKT. Cells were kept in complete medium or serum starved overnight. Then, cells were placed in 1% FBS to allow protein secretion and 1 h before collection FBS was added to the rescue condition. The over-expression of AKT reduces mut HTT secretion both at steady state and in response to serum starvation/rescue conditions. The P-values for the densitometric analyses were determined by factorial ANOVA or Student’s t-test using STATVIEW v4.53 (Abacus Concepts) (n = 3, **P < 0.01; NS: non-significant).
Based on this evidence, we investigated whether the lower band of mut HTT could be the result of a proteolytic cleavage operated by a caspase. We therefore performed a secretion assay on HeLa cells transfected with mut HTT upon serum starvation, which is likely responsible for inducing (in the long term) the activation of apoptotic cell death-related pathways, because of growth factor deprivation (89), in the absence or presence of Z-VAD, a well-known pan-caspase inhibitor (90). As shown in Figure 5E, caspase inhibition was able to blunt almost completely the appearance of the lower band of mut HTT in the secreted fraction, nonetheless still allowing the secretion and accumulation, compared with both control and serum-starved condition, of the full-length fragment in the secreted fraction. This data therefore confirms that mut HTT is preferentially secreted as a caspase-dependent proteolytic cleavage product in response to serum starvation. However, such a cleavage does not represent a limiting factor, or a required step, for the secretion itself.
Huntingtin protein contains many phosphorylation sites, and it has been reported that phosphorylation status regulates aggregation of the protein and, in turn, neuronal toxicity and cell death (91–94). Importantly, huntingtin is a substrate of PKB/AKT and phosphorylation of huntingtin by AKT is crucial to mediate the neuroprotective effects exerted by IGF-1 (95). We performed a secretion assay to look for mut HTT secretion in the presence or absence of the selective PI3K inhibitor, LY294002. Consistent with the effect exerted by serum starvation, mut HTT secretion was increased in the presence of LY294002 (Fig. 5F–G), therefore confirming that the activity of the PI3K pathway can modulate HTT secretion.
Next, to test whether the modulation of the PI3K pathway activity could also have an effect in the opposite direction, we deployed the serum starvation and rescue protocol to modulate the nutrient-dependent signalling pathway, previously used for assessing the secretion of p62/SQSTM1 (Fig. 2I), by looking at the effect of AKT over-expression on the secretion of mut HTT. To this end, HeLa cells were transiently transfected with mut HTT, in the presence or absence of HA-AKT over-expression. The secretion assay was performed in the presence or absence of serum and with the re-addition of serum for the rescue condition. As already observed in the case of p62/SQSTM1 (Fig. 3K–L), the secretion of mut HTT, which was induced by starvation, could be promptly reversed by adding back serum. In addition, the AKT over-expression largely hampered the response of mut HTT to both starvation and rescue conditions (Fig. 5H–I). Notably, we could also observe how AKT itself can be secreted by cells at steady state and in response to serum starvation/rescue (Fig. 5H), mirroring the behaviour observed for p62/SQSTM1 (Fig. 3K–L). Together, these data confirm that nutrient and growth factor availability can regulate the unconventional secretion of mutant HTT by means of the PI3K/AKT signalling pathway.
The formation of the HSPB1-p62/SQSTM1-mutant HTT ternary complex is instrumental for the unconventional secretion of mutant HTT
To dissect and better understand the molecular mechanisms linking the unconventional secretion of mutant HTT with the activity of the HSPB1-p62/SQSTM1 complex, we first sought to analyse their reciprocal interactions and to understand whether and how they could be regulated. We found that in HeLa cells, both HSPB1 and p62/SQSTM1 were able to co-immunoprecipitate with mut HTT in the intracellular fraction (Fig. 6A–B). Interestingly, when mut HTT was immunoprecipitated from the secreted fraction, we found that only HSPB1 was co-immunoprecipitated (Fig. 6A). This is consistent with the expectation that the large majority of the mut HTT-p62/SQSTM1 complexes will be engulfed into autophagosomes and delivered to the lysosomal compartment for degradation (44). In addition, when we performed the co-immunoprecipitation experiment in serum starvation condition, we observed a reduction in the amount of p62/SQSTM1 being co-immunoprecipitated by mut HTT (Fig. 6B), suggesting therefore that serum starvation can modulate the intracellular interaction of mut HTT with the components of the described complex and, in turn, influence their secretion. It is worth noting that both p62/SQSTM1 and HTT have been reported to interact with mTOR on the lysosome (68,96), which represents a key hub in controlling nutrient sensing and energy metabolism, as well as autophagy (69). In view of these observations, we examined whether the HSPB1-p62/SQSTM1-mut HTT ternary complex described here could be related to the one containing the mTOR complex. By co-immunoprecipitation and confocal imaging analysis, we could show a significant co-localization between mut HTT with mTOR (Supplementary Material, Fig. S10A–B) and the lysosomal marker LAMP1 (Supplementary Material, Fig. S10C–D), which remained unchanged upon serum starvation. Furthermore, we could observe an increased co-localization between mut HTT and LAMP1 upon serum starvation (Supplementary Material, Fig. S10E–F). Finally, we could also confirm by co-immunoprecipitation experiments the interaction between mTOR and mut HTT, as well as p62/SQSTM1, which was slightly increased upon serum starvation (Supplementary Material, Fig. S10G). However, we could not detect any HSPB1 in these pull-down experiments, suggesting therefore that the HSPB1-p62/SQSTM1-mut HTT ternary complex we have described here is independent and acts differently from the one containing mTOR and localized at the lysosome (68,96).
Figure 6.

The formation of the HSPB1-p62/SQSTM1-MUT HTT ternary complex is instrumental for the unconventional secretion of mutant huntingtin. (A) HeLa cells were transfected with mut HTT, HA-p62/SQSTM1 with or without V5-HSPB1. Twenty four hours after transfection, cells were placed in DMEM 1% FBS and cell lysate and secreted fraction were collected. Mutant HTT was immunoprecipitated using anti-FLAG antibody and Co-IP assessed with the indicated antibodies. Mutant HTT is able to interact with HSPB1 both intracellularly and in the secreted fractions, whereas p62/SQSTM1 interacts with mut HTT only intracellularly. (B) HeLa cells were transiently transfected with mut HTT and p62/SQSTM1. Twenty four hours after transfection, cells were kept in full medium or serum starved overnight, cell lysates were prepared and mut HTT immunoprecipitated with an anti-FLAG antibody. Serum starvation does reduce the interaction between mut HTT and p62/SQSTM1. (C–D) HeLa cells were silenced for scramble CTRL, HSPB1 or p62/SQSTM1 with 100-nm siRNA mix. HSPB1 and p62/SQSTM1 depletion were confirmed using specific antibodies. After 72 h, cells were re-transfected with the same mix, supplemented with 2 μg of FLAG-tagged mut HTT over-expression construct. After 48 h, cell lysates were prepared and mut HTT was immunoprecipitated using an anti-FLAG antibody and co-immunoprecipitation analysis performed with the indicated antibodies. (E–F) HeLa cells were silenced for Ctrl, HSPB1 or p62/SQSTM1 and transfected with mutant HTT as detailed in (C–D). Cells were semi-permeabilized with digitonin immediately before fixation. Indirect immunofluorescence was performed and analysed by confocal microscopy. Co-localization analysis was performed by using ImageJ software. Thirty cells per experimental condition were analysed and mean values with standard deviations are reported in the graph. Scale bar: 10 μm. (G) Schematic representation of the hierarchy dictating the assembly and function of the HSPB1-p62-mut HTT ternary complex. (H–I) HeLa cells were silenced for scramble control, HSPB1 or p62/SQSTM1 with 100-nm siRNA and transfected with 2 μg of FLAG mut HTT, as detailed above. Cell lysate and secreted fraction were collected and analysed by western blot. The P-values for the co-localization and densitometric analyses were determined by Student’s t-test using STATVIEW v4.53 (Abacus Concepts) (n = 3, **P < 0.01; ***P < 0.001).
In order to further characterize the mechanisms underlying the secretion of mutant HTT and its dependency on the HSPB1-p62/SQSTM1 complex, we performed HSPB1 and p62/SQSTM1 depletion experiments using siRNA. HeLa cells were transfected with a siRNA pool (smart pool; Dharmacon) targeting either HSPB1 or p62/SQSTM1 for 5 days. On day 3, mut HTT was transfected in HeLa cells. Depletion of both HSPB1 and p62/SQSTM1 was assessed by western blot (Fig. 6C). First, we used control, HSPB1- and p62/SQSTM1-depleted cells to perform co-immunoprecipitation experiments, by pulling down mut HTT and looking at the interaction with HSPB1 and p62/SQSTM1. We found that the interaction between mut HTT and p62/SQSTM1 was reduced upon depletion of HSPB1. Conversely, there was no effect on the amount of HSPB1 co-immunoprecipitated with mut HTT when p62/SQSTM1 was depleted (Fig. 6D). Likewise, the reduced interaction between mut HTT and p62/SQSTM1 was confirmed by the confocal imaging analysis, showing a reduced co-localization between mut HTT and p62/SQSTM1 upon serum starvation or in HSPB1-depleted cells (Fig. 6E–F). Interestingly, these data suggest a precise hierarchy dictating the formation and the stabilization of this secretory cargo complex, whereby HSPB1 acts upstream of and can interact with p62/SQSTM1, mut HTT or both (Fig. 6G), with the PI3K/AKT pathway being able to regulate upstream its assembly and/or activity. Finally, the same siRNA-based approach was deployed to perform a secretion assay to examine the effect of this complex on mut HTT secretion. The depletion of HSPB1 strongly reduced mut HTT secretion compared with control cells (Fig. 6H–I). Likewise, the p62/SQSTM1 knockdown had a significant, albeit smaller, effect on the secretion of mut HTT. Together, these data confirm how the HSPB1-p62/SQSTM1 functional complex plays a critical role for the secretion of mutant huntingtin and that the activity of the complex can exert a tangential effect on the cargo loading and secretion step.
The over-expression of HSPB1 facilitates the spreading of mutant HTT and is regulated in trans by the PI3K/AKT signalling axis
Recently, it has been shown that mutant huntingtin can be secreted by neuronal cells or spread between neurons (55,56,97,98). We have shown that HSPB1 plays an important role in regulating the disaggregation and secretion of mut HTT.
In view of the role exerted by the HSPB1-p62/SQSTM1 complex in secretion of mutant HTT, we next asked whether HSPB1 could be equally instrumental for its transcellular spreading. To this end, we established a spreading assay in order to understand whether HSPB1 could also regulate the ability of mut HTT-loaded EVs to be up-taken in trans by recipient cells (Supplementary Material, Fig. S11A). Briefly, HeLa cells were transiently transfected with mut HTT with or without HSPB1 for 48 hours (feeder cells). The culturing medium of feeder cells (namely, cells transiently transfected with either mut HTT or mut HTT + HSPB1) was replaced with DMEM supplemented with 1% FBS and kept overnight. The following day, the conditioned medium derived from feeder cells was cleared by centrifugation. We first confirmed that mut HTT and HSPB1 were present in the secreted fraction from feeder cells (Fig. 7A), then transferred on recipient (i.e. non-transfected) cells, that were previously incubated at 4°C for 30 min in order to block and synchronize the uptake. When the conditioned medium was added, the cells were promptly warmed up and shifted back at 37°C for the indicated time points, ranging from 0.5 up to 8 hours. At the end of incubation, both secreted and intracellular fractions of the recipient cells were collected and analysed. Strikingly, both HSPB1 and mut HTT were present in the cell lysate of recipient cells, indicating that both proteins were able to be internalized in a time-dependent manner (Fig. 7B–D). In addition, we could show how the HSPB1 over-expression was able to induce a faster and higher uptake of mut HTT (Fig. 7C–D). In particular, the integral amount of the total mut HTT uptake (AUC, area under the curve) showed a normalized fold increase of 2.851 ± 0.5627, compared with empty vector transfected cells (Fig. 7D), suggesting how HSPB1 is not only able to favour the loading of mut HTT into EVs but might also functionalize these carriers in terms of docking/uptake at the interface with recipient cells. Furthermore, such a phenomenon was also observed in the human SK-N-BE2 neuronal cell line, suggesting that the HSPB1-dependent induced spreading of mut HTT might affect also the inter-neuronal spreading of the protein (Supplementary Material, Fig. S11B–C). Importantly, to confirm that the plateauing in uptake observed with mut HTT was not due to depletion of EVs from the conditioned media, we checked the levels of mut HTT and HSPB1 that remained in the conditioned media that had been applied to the recipient cells (Fig. 7E). Such observation was instrumental to confirm that an abundance of mut HTT and HSPB1 remained in the conditioned media after 8 hours of treatment, indicating that the levels of mut HTT that were internalized represented the maximum internalization rather than an exhaustion of EVs available in the conditioned media.
Figure 7.

The spreading of mutant huntingtin is facilitated by HSPB1 over-expression and is regulated in trans by the PI3K/AKT signalling axis. (A–D) Feeder cells were prepared by transiently transfecting HeLa cells with FLAG-tagged mut HTT, with or without V5-HSPB1. Twenty four hours after transfection, cells were placed in DMEM 1% FBS for 12 h to allow protein secretion. Cell lysate and secreted fractions were collected and analysed (A). In addition, secreted fractions were used as conditioned medium for assessing the EV-mediated spreading of secreted proteins on recipient cells, as detailed in Supplementary Material, Figure S7. HSPB1 over-expression increases the uptake of mut HTT in recipient cells (B). The graph in (C) reports the relative levels of HSPB1, mut HTT alone or in the presence of HSPB1, internalized over time by recipient cells, whereas in (D), the cumulative value (area under curve, AUC) of the total uptake, for each experimental condition, is reported. In particular, for the quantification of the internalized mut HTT, it was taken in consideration and factored in the increased amount of protein present in the conditioned medium derived from HSPB1 over-expressing cells (fold increase: 2.267 ± 0.017) compared with empty vector transfected cells (A). The integral amount of the total mut HTT uptake (AUC, area under the curve) reported in (D) represents a normalized value (fold increase: 2.851 ± 0.5627) compared with empty vector transfected cells. The same approach has been applied to determine the AUC reported in (H) and in the Supplementary Figure S11(C). Histograms indicate mean values with standard deviations from three independent experiments. (E–H) Feeder cells were prepared by transiently co-transfecting HeLa cells with FLAG-tagged mut HTT and V5-HSPB1. Cell lysates and secreted fractions were collected and conditioned medium, prepared as already detailed in (A–B), was used for the spreading experiment on recipient cells. In order to infer about the potential effect in trans of the PI3K/AKT signalling pathway, recipient cells were kept at steady state (Ctrl), transfected with HA-AKT or pre-treated with 10 μM LY-294002, 24 h before receiving the conditioned medium obtained from feeder cells for the indicated time points (0.5 to 8 h). The graph in (G) reports the relative levels of mut HTT internalized over time by recipient cells, whereas in (H) the cumulative value (area under curve, AUC) of the total uptake, for each experimental condition, is reported. Histograms indicate mean values with standard deviations from three independent experiments. The P-values for the densitometric analyses were determined by factorial ANOVA using STATVIEW v4.53 (Abacus Concepts) (n = 3, *P < 0.05; **P < 0.01).
In view of the previous observations linking nutrient availability (i.e. serum starvation) and the activity of the PI3K/AKT signalling axis on the secretion of mut huntingtin (Fig. 5A–B, D–E and F–G), we next went on to assess whether the PI3K/AKT activity could exert an effect, also in trans, on the ability of recipient cells to uptake HSPB1/mut HTT-loaded vesicles. To this end, we performed a spreading assay by dispensing conditioned medium derived from HSPB1-mut HTT co-transfected cells (Fig. 7E) on recipient cells at steady state (control) or in recipient cells where the activity of the PI3K pathway was either up-regulated (by over-expression of AKT) or blunted (by pre-treatment with the PI3K inhibitor LY-294002). Strikingly, we could observe how the AKT over-expression was able to reduce, compared with control cells, the kinetic and the total uptake of both mut HTT and, albeit to a lesser extent, HSPB1. Conversely, in the presence of the PI3K inhibitor LY-294002, recipient cells showed a remarkable increase of the intracellular pool of mut HTT (Fig. 7F–H), indicating the existence of a dynamic equilibrium between the uptake and recycling that these EVs are subjected to by recipient cells. Collectively, the results from starvation and rescue experiments (Fig. 5) and PI3K/AKT manipulation confirm how the effect of HSPB1 and serum starvation on mut HTT secretion is coordinated and 2-fold: on one hand, the activity of the HSPB1-p62/SQSTM1 complex is able to facilitate in cis the cargo loading and secretion step. On the other hand, these EVs are biologically active and can undergo uptake (or re-uptake) and recycling by recipient cells in a PI3K/AKT-dependent manner. In particular, our data indicate that the inward/outward dynamic equilibrium of HSPB1/mut HTT-loaded EVs is regulated also in trans by the PI3K/AKT signalling axis.
Discussion
A novel role for the HSPB1-p62/SQSTM1 functional complex in the unconventional secretion of aggregation-prone mutant proteins
In this work, we provide a working model for the HSPB1–p62-SQSTM1-mediated regulation of the unconventional secretion of the HD-associated mutant HTT protein (Fig. 8). As part of an ongoing screen, we have found that HSPB1 is secreted unconventionally by means of EVs in HeLa cells and MEF cell lines. HSPB1 interacts with the autophagy cargo receptor p62/SQSTM1 and regulates synergistically its unconventional secretion. The phospho-mimetic form of the chaperone (3D-HSPB1) was able to increase p62/SQSTM1 secretion, showing a higher affinity compared with the 3A phospho-null form. This can likely be explained because the phosphorylation of HSPB1 favours the recognition of a subset of client proteins dedicated to specific cellular functions, as previously reported (70–72,74).
Figure 8.

Proposed model for the HSPB1/p62-mediated regulation of the unconventional secretion and transcellular spreading of mutant huntingtin. In response to both physiological and pathological conditions, such as variation of nutrient availability or the accumulation of aggregation-prone proteins, cells can activate a number of pathways, including the PI3K/AKT signalling axis, autophagy and unconventional secretion, aimed at counteracting cellular stress (1). The HSPB1/p62 functional complex can act as cargo selection platform, allowing the loading of cellular components, such as mutant HTT and other aggregation-prone proteins, destined to secretion (2). The activity of this complex can allow the generation and the regulated secretion of diverse extracellular vesicles (i.e. EVs and exosomes), which can display a different content and composition (3). These vesicular transport carriers are biologically active and capable of being taken up by recipient cells, contributing to the prion-like transcellular spreading of aggregation-prone proteins. Such a phenomenon can represent an additional pathophysiological mechanism underlying the onset of neurodegenerative diseases, such as Huntington disease (HD) (4).
Our data suggest also that HSPB1 and p62/SQSTM1 form an intracellular complex, acting like a cargo selection platform where, among other potential substrates, aggregation-prone proteins are sorted and sent for either degradation or secretion (Fig. 8). In particular, we propose a working model in which HSPB1 and p62/SQSTM1 interact, generating a platform for the cargo selection and loading of EVs. The activity of this platform can be regulated in response to nutrients and growth-factor-activated cellular signalling, or in response to the accumulation of neurodegenerative disease-associated, aggregation-prone proteins, such as mutant HTT, whose degradation and turn-over has previously been shown to be mediated by the autophagosome–lysosome system (43–48). Our data demonstrate how a significant pool of mutant HTT can be directed towards the HSPB1-p62/SQSTM1 complex, which displays a higher affinity for the mutant protein, bearing the polyQ-expanded tract (namely, Q138, in this work). This is consistent with previous reports showing the critical role exerted by p62/SQSTM1 in recognizing and delivering to the autophagosomes ubiquitinated and aggregation-prone proteins (79,84, 99–101).
The formation of the HSPB1-p62/SQSTM1 ternary complex is instrumental for the loading of mutant HTT, with the assistance of HSPB1, into these vesicular transport structures. Thereby, we propose an additional mechanism that cells can harness to remove aggregation-prone proteins and ultimately minimize cellular toxicity deriving from their progressive accumulation, as observed in several neurodegenerative disorders (102). Indeed, we have shown that HSPB1 over-expression increases the secretion of mutant HTT. Conversely, siRNA-mediated depletion of HSPB1 decreases HTT secretion, indicating that HSPB1 is necessary for this mechanism. Moreover, we have found that both HSPB1 and p62/SQSTM1 can interact with mutant HTT (Q138). However, HSPB1 interacts with HTT both intracellularly and extracellularly, whereas p62/SQSTM1 is able to interact with HTT only inside that cell, suggesting that upon interaction and loading into EVs, HTT is likely secreted along with HSPB1. Nutrient availability and growth-factor-induced signalling can influence this process, as we have shown that serum starvation reduces the HTT interaction with p62/SQSTM1 and promotes its secretion. Consistent with this, treatment with the class I PI3K inhibitor LY-294002 mimics the effect of serum starvation and induces mut HTT secretion, whereas the PKB/AKT over-expression exerts the opposite effect. Finally, it is worth noting how the expression of HSPs, HSPB1 and other members of the small heat shock protein family can be transcriptionally induced by a number of stress conditions and how this can be harnessed and have a beneficial effect for a number of pathological conditions (103–106). For instance, the transcriptional activation of CRYAB/HSPB5, mediated by the inhibition of the glutaminyl cyclase (QPCT) enzyme, has been proved to reduce mutant huntingtin HTT aggregation and toxicity and to rescue the HD phenotype both in in vitro and in vivo disease models (107). Likewise, the transcriptional up-regulation of HSPB8 mediated by colchicine and doxorubicin treatment is able to induce the clearance of ALS-associated mutant forms of TDP-43 (108). Hence, it will be definitely interesting in future studies to test whether the up-regulation of endogenous HSPB1 is sufficient to modulate and if so, to which extent, the activity and/or the selectivity of the functional complex we have described here.
The HSPB1-p62/SQSTM1 functional complex can exert a tangential effect on the cargo recruitment and loading of EVs
In this work, we provide a number of converging observations supporting the idea that the HSPB1-p62/SQSTM1 functional complex can exert in cis a ‘catapult-like’ tangential effect (i.e. the sequential identification, cargo binding/entry and release/egress from the platform we describe here) during the cargo selection and loading steps of EVs: (i) the over-expression of both WT and mutant HSPB1 can synergistically induce the unconventional secretion of p62/SQSTM1, but not of HSPB1 itself; (ii) any variation in the affinity (for instance with the 3D-HSPB1 phosphomimic mutant) and the increased intracellular interaction between HSPB1 and p62/SQSTM1 is not maintained in the secreted fraction; (iii) the increased secretion of both p62/SQSTM1 and mutant HTT, observed upon serum starvation, is not mirrored by an increased interaction with HSPB1, neither at intracellular nor at extracellular level; (iv) the siRNA-based functional data suggest a precise hierarchy dictating the formation and the stabilization of this secretory cargo complex, whereby HSPB1 acts upstream of and can interact independently with either p62/SQSTM1 or mutant HTT, or both. In this scenario, the PI3K/AKT pathway is able to regulate the assembly, stability and/or activity of the HSPB1-p62/SQSTM1 complex. In our working model, the activity of the functional complex could act in a coordinated fashion with both the inward membrane fusion events as well as with the egress from the MVBs, which ultimately allow the regulated release of EVs-containing aggregates of mutant HTT within them. In such a scenario, the over-expression of HSPB1 (Fig. 4) likely allows an increase in the loading and secretion of mut HTT, compared with the WT protein. Such a scenario, along with the higher affinity of the HSPB1-p62/SQSTM1 complex for the expanded-poly-Q mutant protein, could also explain why the over-expression of HSPB1 is not capable of increasing the secretion rate of the full-length endogenous (i.e. ‘wildtype’) huntingtin protein (Supplementary Material, Fig. S9).
The activity of p62/SQSTM1 as an autophagy cargo receptor, widely described in the literature, depends on the state of phosphorylation of the UBA domain. The phosphorylation of the Ser407 residue within the UBA domain involves the monomerization of p62/SQSTM1 and the ability to interact with ubiquitin: once this pathway is activated, p62/SQSTM1-positive structures are recruited and lead to autophagosome-mediated degradation (79,82–84). Nevertheless, our data show how a small but significant fraction of mutant HTT can escape degradation route and be secreted extracellularly with the assistance of HSPB1. It is therefore conceivable that p62/SQSTM1 is capable of pursuing an alternative, albeit complementary, pathway. This allows the cargo to be transported to the unconventional secretion platform described in this study, rather than to the autophagosome. Collectively, this leads us to propose that the HSPB1-p62/SQSTM1 platform can facilitate the transfer of mutant HTT-positive multi-protein complexes into highly dense vesicular structures: future ultrastructural studies of the abovementioned vesicles will be necessary to shed further light on such a hypothesis.
In this regard, it is worth highlighting how the formation of the ternary complex favours the unconventional secretion of mutant HTT, despite the fact these proteins are neither secreted simultaneously, or necessarily together. The evidence for this is 2-fold: immunofluorescence and biochemical studies confirm the interaction between p62/SQSTM1 and mutant HTT and in particular highlight the high co-localization of these proteins in membrane-associated (i.e. digitonin extraction resistant) structures within cells. Nevertheless, our immunoprecipitation studies performed on conditioned media reveal the absence of interaction of p62/SQSTM1 and HSPB1 in the same EVs, whereas the mut HTT-HSPB1 interaction is still maintained. Hence, all together our evidence strongly suggests the existence of a tangential effect operated by the complex, which ultimately allows the generation and differential secretion of HSPB1-mut HTT-loaded exosomes/EVs (Fig. 8).
These data, along with the effect exerted by the HSPB1 over-expression (Fig. 4A–E) suggest that, in the context of the functional complex, HSPB1 is the major factor responsible for the loading and secretion of mutant HTT. Consistent with this, when HSPB1 was depleted with siRNA, HTT secretion was strongly affected (Fig. 6I), suggesting that HSPB1 is necessary for its secretion. Furthermore, this occurs by means of the same subset of vesicular structures, where their interaction is likely preserved (Fig. 6A). Collectively, these data imply that the activity of HSPB1 is critical for allowing the cargo loading and selection of mutant HTT into both autophagosomes (mediated by p62/SQSTM1) and secretory vesicles (EVs). Interestingly, a precise hierarchy dictates the formation and the stabilization of this secretory cargo complex, whereby HSPB1 acts upstream of and can interact with p62/SQSTM1, mut HTT or both (as summarized in Fig. 6H), with the PI3K/AKT pathway being able to regulate upstream its assembly and/or activity.
The HSPB1 can facilitate the transcellular spreading of mutant huntingtin
Finally, HSPB1 has also a positive role in facilitating the spreading of mutant HTT (Figs. 7 and Supplementary Material, Fig. S11). It is worth noting (Fig. 7B–D) how the transcellular transfer of mut HTT shows a time-dependent kinetic variation, namely that, when HSPB1 is over-expressed, the uptake of mut HTT is accelerated. Interestingly, in this condition, a doublet of HTT bands can be readily detected. In this regard, it is conceivable to hypothesize that cleaved mut HTT is preferentially transferred into the EVs (as suggested by the effect of z-VAD, Fig. 6C), as well as internalized by recipient cells. Nevertheless, one could still envisage the occurrence of some in-trans effects, such as the cleavage and/or degradation of the pool of mutant HTT being transferred in recipient cells. Finally, the question remains as to how mutant HTT is secreted and taken up in the absence of HSPB1 over-expression. Here, it should be noted that the kinetics of mut HTT entry may be partly dependent on HSPB1 expressed endogenously by feeder cells.
The EV-based secretion mechanism described here is likely necessary for the disposal of the mutant HTT aggregates, which can be considered, along with other mechanisms regulating exocytosis, an alternative to the canonical degradation route controlled by the autophagosome–lysosome system (102). Hence, one cannot rule out that other proteins resistant to either proteasomal or lysosomal degradation may also undertake this pathway. Furthermore, it should be emphasized that the three-component model proposed here represents only a part of a much more complex pathway likely involving other proteins. Further studies, such as MS-based or siRNA-based screenings, will be necessary to identify other molecular partners involved in the regulation of this mechanism.
Unconventional secretion, complementary to the canonical degradation, plays a main role in the pathophysiology of many neurodegenerative diseases. Notably, we did not observe any significant change in the intracellular levels of mutant huntingtin with none of the experimental perturbations operated, such as serum starvation (Fig. 5A–B) or the PI3K pathway pharmacological inhibition (Fig. 5F–G) capable of impacting the secretion of mutant huntingtin HTT, suggesting that the increased secretion of mutant HTT does not lead to a detectable decrease in intracellular levels of the protein, at least in our experimental set-up (8 hours of secretion). Together, these data suggest that mutant HTT unconventional secretion is unlikely to have a major contribution to its clearance. However, we cannot exclude a possibility that a small rate of mutant huntingtin HTT exocytosis over a prolonged period might eventually have an impact on the clearance of the mutant protein in HD patients. In such scenarios, although it is conceivable to expect that, on one hand, such a mechanism could be advantageous to alleviate the proteostatic stress induced by the intracellular accumulation of protein aggregates, on the other hand this may lead to the transcellular propagation of pathological aggregates. With this in mind, there is a pressing need to further characterize the molecular determinants (i.e. receptors, co-receptors and other membrane components) responsible for the biogenesis and the acquisition of the specific biological properties of these vesicles. By doing so, one could identify novel druggable targets for HD capable of inhibiting specifically the ability of mutant HTT-loaded exosomes/EVs to undergo the cell-to-cell transfer and therefore to limit the toxicity deriving from progressive spreading and accumulation of these protein aggregates within neighbouring cells. Such characterization would also elucidate whether other aggregation-prone proteins are handled in a similar fashion and would have an impact on our understanding of the pathogenesis of and potential therapeutic targets for other neurodegenerative diseases.
Materials and Methods
Antibodies
The following antibodies were used: mouse monoclonal anti-HSP27 antibody (Santa Cruz Biotechnology, USA; #sc13132) and mouse monoclonal anti-p53 (Santa Cruz Biotechnology; #sc126); mouse monoclonal anti-HSP27 antibody (clone G3.1) (ThermoFisher, USA, catalogue #MA3-015), mouse IgG isotype control antibody (ThermoFisher, USA, catalogue #10400C), mouse monoclonal anti-huntingtin antibody (Millipore, USA, #MAB2166), mouse monoclonal anti-CD63 (Santa Cruz Biotechnology, sc-51662),) and mouse monoclonal anti-FLAG antibody (Sigma-Aldrich, Milan, Italy; #F1804); mouse monoclonal anti-HA (Sigma-Aldrich; #H9658, clone HA7); rabbit polyclonal anti-LC3 (Novus Biologicals, USA; #NB100-2220); mouse monoclonal anti-LAMP1 (clone H4A3, obtained from Developmental Studies Hybridoma Bank, University of Iowa); rabbit polyclonal anti-p62 (MBL International, Woburn, MA; #PM045); mouse monoclonal anti-V5 (Santa Cruz, USA; #sc271944); rabbit polyclonal anti-actin (Sigma-Aldrich, Milan, Italy; #A2066); mouse monoclonal anti-α-tubulin antibody (Sigma-Aldrich, Milan, Italy; #T6199); rabbit polyclonal anti-ATG16L1 antibody (MBL Life Sciences, Woburn, MA; #PM040); rabbit polyclonal anti-mTOR (Cell Signaling Technologies, Danvers, MA; #2983); rabbit polyclonal anti-phospho AKT (Cell Signaling Technologies, Danvers, MA; #4060); anti-total AKT (Cell Signaling Technologies, Danvers, MA; #4691); peroxidase conjugated anti-mouse and anti-rabbit IgG (Sigma-Aldrich, Milan, Italy); Texas-Red-conjugated anti-mouse and anti-rabbit IgG, FITC-conjugated goat anti-mouse and anti-rabbit IgG, Cy5-conjugated goat anti-mouse and anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA).
Reagents and chemicals
Clarity Western ECL Substrate was from Bio-Rad (California, USA; #170-5061), and Protein A–Sepharose was from Roche (Milan, Italy; #CL4B 17-0780-01). Digitonin, bafilomycin A1, brefeldin-A, rapamycin and trehalose were purchased from Sigma-Aldrich, Milan, Italy. LY-294002 and Z-VAD were purchased from Tocris Biosciences (UK).
Constructs and siRNA reagents
The Frt-V5/HSPB1-HSPB8 expression vectors have been previously described (109). The pFLAG-CMV-HSP27 3A (S15A/S78A/S82A) and 3D (S15D/S78D/S82D) were purchased from ADDGENE (catalogue: #84997 and #85188). The pCDNA3-HA-Akt1 was purchased from ADDGENE (catalogue #73408). The EGFP-CD63 was purchased from ADDGENE (catalogue #62964) (110). The HA-tagged p62/SQSTM1 construct has been previously described (83) and was purchased from ADDGENE (catalogue: #28027). The EGFP-HDQ21 and EGFP-HDQ74 vectors were characterized previously (111). The constructs expressing the WT and mutant FLAG-tagged huntingtin N-terminal fragment (1-588) have been previously described (112). The Ub-G76V/GFP over-expression construct has been previously described (113). The smart pool siRNA against human HSPB1, p62/SQSTM1 and scramble control were purchased from Horizon Discoveries (Cambridge, UK). The construct expressing the GFP-tagged HSPB1 have been described previously (114). The CD9-mEGFP construct was purchased from ADDGENE (catalogue #182864) (115).
Cell culture and transfections
HeLa cells, SK-N-BE2 cells, isogenic WT and ATG16L1 KO HeLa cells were grown in Dulbecco’s modified essential medium (DMEM), containing 10% fetal bovine serum (FBS), 100 U/ml penicillin/streptomycin and 2-mm l-glutamine at 37°C in a humidified atmosphere of 5% CO2 and 95% O2. For the serum starvation experiments, cells were washed twice and incubated in serum-free DMEM, in the presence of l-glutamine. Cells were transfected using X-tremeGENE™ 9 DNA Transfection Reagent (Roche, Milan, Italy) according to the manufacturer’s instructions. For the siRNA-based experiments, cells were transfected with Lipofectamine 2000 (Invitrogen, USA), according to the manufacturer’s instructions.
Western blot analysis
Cells were washed twice with 1× PBS and lysed on ice for 30 min in 1× lysis buffer, containing 150-mm NaCl, 10-mm Tris–HCl pH 6.8, 1-mm EDTA pH 8.0, 10% glycerol and 1% Triton X-100, and supplemented with a protease inhibitor cocktail (cOmplete-mini EDTA-free tablets-Roche) and, when appropriate, with phosphatase inhibitors (Sigma-Aldrich). After centrifugation for 15 min at 14 000 rpm at 4°C, cleared lysates were obtained. Sample proteins were separated using 12% SDS-PAGE gels, followed by their transfer onto PVDF and detection using primary antibodies (1:1000) followed by incubation with HRP mouse or rabbit IgG (1:5000) (GE Healthcare Amersham, Little Chalfont, UK) in PBS containing 5% non-fat dry milk (Bio-Rad Laboratories). In all the experiments, unless otherwise indicated, the volumes of both lysis buffer and conditioned media used were kept constant (namely, 600 μL for the six multi-well format and 1500 μL for the 10-cm dish format) and the same ratio between intracellular and extracellular pool samples was used (1:10 IN/OUT ratio, volume/volume) in order to allow and optimize the detection of even small amounts of secreted proteins.
Isolation of exosomes and EVs by differential ultracentrifugation
For the isolation of EVs, cells were grown in 10-cm dishes and conditioned media obtained and cleared by a first centrifugation at 1500 rpm for 5 min at 4°C to eliminate cellular debris. Equal volumes of cleared conditioned media were subjected to ultracentrifugation at 100 000 g (45 000 rpm) in MLS-50 Swinging-Bucket Rotor (Beckman Coulter, cat. no. 367280) mounted on the Optima MAX-130K Ultracentrifuge (Beckman Coulter) for 2 h at 4°C. The floating (S100) and pellet (P100) fractions were then subjected to further analysis.
Nanoparticle tracking analysis of of extracellular vesicles
For the NTA, the P100 fractions obtained as indicated above were resuspended in 200 μl of sterile 1× PBS. Mean diameter of EVs and the number of particles were measured by using the NanoSight NS300 apparatus (NanoSight; Alfatest, Rome, Italy) equipped with a blue laser (Blue488). Briefly, 10 μl of the P100 fractions was further diluted with 1× PBS to a final volume of 1 ml (dilution factor = 1:100) and loaded into the instrument. The instrument’s software NTA 3.4 Build 3.4.4 was used for the measurement. Instrumental settings for the measurements were camera type: sCMOS; number of frames: 1498.
Analysis of EVs by transmission electron microscopy
For the analysis by electron microscopy, the P100 fractions were fixed with a solution of 4% paraformaldehyde (PFA) and 0.05% glutaraldehyde (GA), prepared in 0.2-M HEPES (pH 7.4) for 30 min (RT), followed by 10 min washes in acetone graded solutions (50 to 100%). Samples were then embedded in resin overnight (RT) and baked for 24 h at 65°C. Ultrathin sections (60 nm) were finally obtained and double stained with 2% uranyl acetate for 20 min and lead citrate for 10 min and collected on a nickel grid. Ten microliters of exosome suspension was used for negative staining. An antibody against CD63 (Santa Cruz Biotechnology, sc51662) and protein A gold (PAG 10 nm) were used for the immunostaining of exosomes. The grids were observed by transmission electron microscopy (TEM). EM images were acquired at 80 kW using an FEI Tecnai-12 electron microscope (ThermoFisher) equipped with a VELETTA CCD digital camera.
Triton X-100 soluble/insoluble fractionation
The soluble/insoluble fractionation protocol was performed as already described (80) and schematically depicted in Supplementary Material, Figure S5(B). Briefly, cells were lysed on ice in 1× buffer B (150 mm NaCl, 10 mm Tris–HCl pH 7.4, 1 mm EDTA pH 8.0) containing 1% Triton X-100. After 30 min, cell lysates were centrifuged at 14 000 rpm for 15 min at 4°C. Supernatants were collected and Laemmli buffer was added. These were considered as Triton X-100 soluble fractions. For the Triton X-100 insoluble fractions, the pellets were resuspended in 1× lysis buffer (containing 150-mm NaCl, 10-mm Tris–HCl pH 6.8, 1-mm EDTA pH 8.0, 10% glycerol, 1% Triton X-100) and kept on ice for 30 min. Finally, Laemmli buffer was added and samples processed for western blot analysis.
Immunoprecipitation and co-immunoprecipitation assays
For immunoprecipitation experiments, cells were lysed in 1× buffer B (150-mm NaCl, 10-mm Tris–HCl pH 7.4, 1-mm EDTA pH 8.0) containing 0.6% CHAPS in the presence of protease inhibitor cocktail. For the isolation of Flag-, V5-, HA-tagged or endogenous proteins, 1–2 mg of the cell extracts was first subjected to a pre-clearing step (1 h in protein A/G-conjugated agarose beads) and then incubated with 5–10 μg of the specific primary antibodies for 16 h at 4°C. Next, 30–40 μl of a 50% Protein-A/G beads slurry solution prepared in the same buffer (Amersham GE Healthcare) was added and incubated for 1 h at 4°C. The beads were then pelleted by centrifugation and washed three times in lysis buffer. The bound protein was eluted by adding 50 μl of 1× Laemmli sample buffer and subjected to SDS-PAGE and western blot analysis as detailed above.
Quantification of poly-Q74 huntingtin aggregate formation
Quantification of GFP/Q74 aggregate formation were assessed as already previously described. Briefly, for each experimental condition, at least 200 EGFP/HDQ74-transfected cells were selected and the number of cells with aggregates analysed and scored by using a fluorescence microscope. The identity of the slides was unavailable to the observer until all slides had been analysed. The experiments were performed in triplicate and repeated at least three times.
UbG76V-GFP reporter degradation assay
HeLa cells were seeded in six-well plates and transfected with the UbG76V-GFP reporter (107), in combination or not with V5-HSPB1 and cultured in full medium for 48 h. To allow the block of the proteasome, HeLa cells were incubated (8 h; 37°C) with either DMSO or with 10-mm MG132. Cells were then placed at 4°C, washed with cold 1× PBS, recovered by trypsinization and fixed in 1× PBS/3.7% PFA for 30 min at room temperature. The amount of GFP-derived fluorescence was finally quantified by flow cytometry on a FACSCalibur apparatus (Becton Dickinson).
Mutant huntingtin transcellular spreading assay
Feeder cells (HeLa) were transiently transfected with mut HTT alone or mut HTT plus WT V5-HSPB1. After 48 h, cells were placed in DMEM supplemented with 1% FBS for 12 h to allow protein secretion. Secreted fractions were collected and centrifuged at 1500 rpm for 5 min to remove any cellular debris. Recipient cells were washed twice with prechilled serum-free DMEM medium supplemented with 10-mm HEPES pH 7.4 and placed at 4°C for 30 min in the same medium to stop endocytic activity and synchronize the uptake. Cells were then washed with pre-warmed medium and equal volumes of conditioned medium were added to each well at different time points, ranging from 0.5 up to 8 h. After 8 h, both conditioned medium and intracellular fractions were collected. For the intracellular fractions, cells were lysed in 150 ml of lysis buffer and analysed by western blot, as detailed above.
Cell fractionation by discontinuous sucrose gradient isopycnic ultracentrifugation
HSPB1 and huntingtin oligomerization status was analysed by isopycnic ultracentrifugation on a discontinuous sucrose gradient (5–20% w/v), as described by Martin and Ames (116), with some slight modifications. Briefly, cytoplasmic extracts were prepared with reassembly buffer (100 mM Tris, 0.5-mm MgSO4, 1-mm EGTA, 2-mm dithiothreitol; pH 6.8), containing protease inhibitors and phosphatase inhibitors (Roche Diagnostics, Mannheim, Germany). Equal amounts of protein were loaded on a 5–20% sucrose gradient and centrifuged at 100 000 g (45 000 rpm) in MLS-50 Swinging-Bucket Rotor (Beckman Coulter, cat. no. 367280) mounted on the Optima MAX-130K Ultracentrifuge (Beckman Coulter) for 24 h at 4°C. Twelve fractions (450 ml each) were collected from the top to the bottom of the gradient. The remaining pellets were resuspended in sample buffer (50 μl) and subjected to immunoblot analysis as above indicated.
Immunofluorescence, confocal microscopy and co-localization analysis
Cells were grown on glass coverslips washed with ice-cold KHM buffer (110-mm KOAc, 20-mm HEPES, pH 7.2, 2-mm MgOAc). Cells were permeabilized with digitonin at 40 μg/ml in KHM buffer for 5 min on ice and fixed in 4% formaldehyde in PBS for 30 min at room temperature. After blocking in PBS with 1% BSA, the cells were incubated with the primary antibodies diluted in PBS–5% BSA for 1 h at room temperature. After washing in 1× PBS, the primary antibodies were detected using anti-Ig secondary antibodies conjugated to Alexa Fluor (Alexa Fluor 488, 568 and 647) (Molecular Probes, Invitrogen). DNA was stained with 250 ng/ml DAPI for 5 min. Confocal images were acquired at 63× magnification (63× NA 1.4 Plan-Apochromat oil-immersion lens) on a LSM710 Meta (Carl Zeiss, Jena, Germany). For co-localization analysis, 20 images containing on average four cells per field were taken for each experimental condition. Each experiment was run at least three independent times. Exposure settings were unchanged throughout acquisition. Images were analysed using the JaCoP plug-in within the ImageJ software. Pearson’s and Mander’s (original, non-threshold) coefficients were used for assessing co-localization. Student’s t-tests were performed to determine statistical significance for correlation and co-localization coefficients. Images shown are single plane projections of representative images using ImageJ and Photoshop software (Adobe).
Statistical analysis
In all the experiments, we determined the significance levels for comparisons between groups by a two-tailed Student’s t-test, repeated measurements or Factorial ANOVA test using STATVIEW v4.53 (Abacus Concepts), where appropriate. Densitometric analysis on the immunoblots was performed using ImageJ software. Experiments were performed at least three times in triplicate. In all the main or supplementary figures, error bars denote standard deviations. The P-values for assessing densitometric analysis on the immunoblots, EGFP-HDQ74 aggregation, co-localization analysis and spreading/uptake assays were determined using Student’s t-test (***P < 0.001; **P < 0.01; *P < 0.05; NS, non-significant).
Supplementary Material
Acknowledgements
The overall project was conceived by M.R., with the contribution of A.F. The experiments were designed by M.R. and performed by R.B., G.S., R.D.M., S.N., E.P., M.D.G., S.V.W., M.G.C., C.C., R.P., M.D.A., A.F. and M.R. The manuscript was written and edited by R.B., A.F. and M.R. All the authors read, revised and edited the manuscript. M.R. is grateful to the Department of Physiology, Development and Neuroscience (PDN, University of Cambridge, UK), for hosting him as a Visiting Academic Fellow. M.R. would like to thank Professor David C. Rubinsztein (CIMR, University of Cambridge, UK), Professor M.A. De Matteis (Telethon Institute for Genetics and Medicine, Pozzuoli, Italy) and Professor Stefano Bonatti (Department of Molecular Medicine and Medical Biotechnologies, University of Naples ‘Federico II’, Italy) for sharing reagents, scientific support and the helpful discussions.
Contributor Information
R Bonavita, Department of Molecular Medicine and Medical Biotechnologies, University of Naples “Federico II”, 80131 Naples, Italy.
G Scerra, Department of Molecular Medicine and Medical Biotechnologies, University of Naples “Federico II”, 80131 Naples, Italy.
R Di Martino, Institute for Endocrinology and Experimental Oncology “G. Salvatore,” National Research Council, 80131 Naples, Italy; Institute of Biochemistry and Cell Biology, National Research Council, 80131 Naples, Italy.
S Nuzzo, IRCCS SYNLAB SDN, 80143 Naples, Italy.
E Polishchuk, Telethon Institute of Genetics and Medicine (TIGEM), 80078 Pozzuoli, Italy.
M Di Gennaro, Department of Molecular Medicine and Medical Biotechnologies, University of Naples “Federico II”, 80131 Naples, Italy.
S V Williams, Department of Physiology, Development and Neuroscience, University of Cambridge, CB2 3DY Cambridge, UK.
M G Caporaso, Department of Molecular Medicine and Medical Biotechnologies, University of Naples “Federico II”, 80131 Naples, Italy.
C Caiazza, Department of Molecular Medicine and Medical Biotechnologies, University of Naples “Federico II”, 80131 Naples, Italy.
R Polishchuk, Telethon Institute of Genetics and Medicine (TIGEM), 80078 Pozzuoli, Italy.
M D’Agostino, Department of Molecular Medicine and Medical Biotechnologies, University of Naples “Federico II”, 80131 Naples, Italy.
A Fleming, Department of Physiology, Development and Neuroscience, University of Cambridge, CB2 3DY Cambridge, UK.
M Renna, Department of Molecular Medicine and Medical Biotechnologies, University of Naples “Federico II”, 80131 Naples, Italy; Department of Physiology, Development and Neuroscience, University of Cambridge, CB2 3DY Cambridge, UK.
Funding
Italian Minister for Research and University (PRIN research grant no.: 20177XJCHX assigned to M.R. and partially by departmental funding to M.R.)
Conflict of Interest statement. None declared.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material. The datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request.
References
- 1. Camussi, G., Deregibus, M.C., Bruno, S., Grange, C., Fonsato, V. and Tetta, C. (2011) Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am. J. Cancer Res., 1, 98–110. [PMC free article] [PubMed] [Google Scholar]
- 2. Del Conde, I., Shrimpton, C.N., Thiagarajan, P. and Lopez, J.A. (2005) Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood, 106, 1604–1611. [DOI] [PubMed] [Google Scholar]
- 3. Gatti, S., Bruno, S., Deregibus, M.C., Sordi, A., Cantaluppi, V., Tetta, C. and Camussi, G. (2011) Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol. Dial. Transplant., 26, 1474–1483. [DOI] [PubMed] [Google Scholar]
- 4. Lai, R.C., Arslan, F., Lee, M.M., Sze, N.S., Choo, A., Chen, T.S., Salto-Tellez, M., Timmers, L., Lee, C.N., El Oakley, R.M. et al. (2010) Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res., 4, 214–222. [DOI] [PubMed] [Google Scholar]
- 5. Segura, E., Nicco, C., Bèrangère, L., Vèron, P., Raposo, G., Batteaux, F., Amigorena, S. and Théry, C. (2005) ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood, 106, 216–223. [DOI] [PubMed] [Google Scholar]
- 6. Viaud, S., Terme, M., Flament, C., Taieb, J., André, F., Novault, S., Escudier, B., Robert, C., Caillat-Zucman, S., Tursz, T. et al. (2009) Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Ra. PLoS One, 4, 4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cazzoli, R., Buttitta, F., Di Nicola, M., Malatesta, S., Marchetti, A., Rom, W.N. and Pass, H.I. (2013) microRNAs derived from circulating exosomes as noninvasive biomarkers for screening and diagnosing lung cancer. J. Thorac. Oncol., 8, 1156–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matsumura, T., Sugimachi, K., Iinuma, H., Takahashi, Y., Kurashige, J., Sawada, G., Ueda, M., Uchi, R., Ueo, H., Takano, Y. et al. (2015) Exosomal microRNA in serum is a novel biomarker of recurrence in human colorectal cancer. Br. J. Cancer, 113, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Melo, S.A., Luecke, L.B., Kahlert, C., Fernandez, A.F., Gammon, S.T., Kaye, J., LeBleu, V.S., Mittendorf, E.A., Weitz, J., Rahbari, N. et al. (2015) Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature, 523, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Michael, A., Bajracharya, S.D., Yuen, P.S.T., Zhou, H., Star, R.A., Illei, G.G. and Alezivos, I. (2010) Exosomes from human saliva as a source of microRNA biomarkers. Oral Dis., 16, 34–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alvarez-Erviti, L., Seow, Y., Yin, H., Betts, C., Lakhal, S. and Wood, M.J.A. (2011) Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol., 29, 341–345. [DOI] [PubMed] [Google Scholar]
- 12. Didiot, M.C., Hall, L.M., Coles, A.H., Haraszti, R.A., Godinho, B.M.D.C., Chase, K., Sapp, E., Socheata, L., Alterman, J.F. and Hassler, M.R. (2016) Exosome-mediated delivery of hydrophobically modified siRNA for huntingtin mRNA silencing. Mol. Ther., 24, 1836–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ohno, S.I., Takanashi, M., Sudo, K., Ueda, S., Ishikawa, A., Matsuyama, N., Fujita, K., Mizutani, T., Ohgi, T., Ochiya, T. et al. (2013) Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther., 21, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tian, Y., Li, S., Song, J., Ji, T., Zhu, M., Anderson, G.J., Wei, J. and Nie, G. (2014) A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials, 35, 2383–2390. [DOI] [PubMed] [Google Scholar]
- 15. Kowal, J., Arras, G., Colombo, M., Jouve, M., Morath, J.P., Primdal-Bengtson, B., Dingli, F., Loew, D., Tkach, M. and Théry, C. (2016) Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA, 113, E968–E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Willms, E., Johansson, H.J., Mäger, I., Lee, Y., Blomberg, K.E., Sadik, M., Alaarg, A., Smith, C.I., Lehtiö, J., El Andaloussi, S. et al. (2016) Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep., 6, 22519. 10.1038/srep22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoshioka, Y., Konishi, Y., Kosaka, N., Katsuda, T., Kato, T. and Ochiya, T. (2013) Comparative marker analysis of extracellular vesicles in different human cancer types. J Extracell. Ves., 2, 20424. 10.3402/jev.v2i0.20424.eCollection2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. The UniProt Consortium (2017) UniProt: the universal protein knowledgebase. Nuc. Ac. Res., 46, 2699 PMID: 29425356. 10.1093/nar/gky092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Penke, B., Bogár, F., Crul, T., Sántha, M., Toth, M.E. and Vígh, L. (2018) Heat shock proteins and autophagy pathways in neuroprotection: from molecular bases to pharmacological interventions. Int. J. Mol. Sci., 19, 325. 10.3390/ijms19010325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giese, K.C. and Vierling, E. (2002) Changes in oligomerization are essential for the chaperone activity of a small heat shock protein in vivo and in vitro. J. Biol. Chem., 277, 46310–46318. [DOI] [PubMed] [Google Scholar]
- 21. Basha, E., O’Neill, H. and Vierling, E. (2012) Small heat shock proteins and α-crystallins: dynamic proteins with flexible functions. Trends Biochem. Sci., 37, 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boelens, W.C. (2020) Structural aspects of the human small heat shock proteins related to their functional activities. Cell Stress Chaperones, 25, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Delbecq, S.P. and Klevit, R.E. (2013) One size does not fit all: the oligomeric states of aB crystallin. FEBS Lett., 587, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Basha, E., Lee, G.J., Breci, L.A., Hausrath, A.C., Buan, N.R., Giese, K.C. and Vierling, E. (2004) The identity of proteins associated with a small heat shock protein during heat stress in vivo indicates that these chaperones protect a wide range of cellular functions. J. Biol. Chem., 279, 7566–7575. [DOI] [PubMed] [Google Scholar]
- 25. Lindner, R.A., Kapur, A. and Carver, J.A. (1997) The interaction of the molecular chaperone, alpha-crystallin, with molten globule states of bovine-lactalbumin. J. Biol. Chem., 272, 27722–27729. [DOI] [PubMed] [Google Scholar]
- 26. Rajaraman, K., Raman, B., Ramakrishna, T. and Rao, C.M. (2001) Interaction of human recombinant alphaA-and alphaB-crystallins with early and late unfolding intermediates of citrate synthase on its thermal denaturation. FEBS Lett., 497, 118–123. [DOI] [PubMed] [Google Scholar]
- 27. Escusa-Toret, S., Vonk, W.I.M. and Frydman, J. (2013) Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat. Cell Biol., 15, 1231–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chaplot, K., Jarvela, T.S. and Lindberg, I. (2020) Secreted chaperones in neurodegeneration. Front. Aging Neurosci., 12, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fagan, A.M. (2015) What does it mean to be ‘amyloid-positive’? Brain, 138, 514–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Muranova, L.K., Ryzhavskaya, A.S., Sudnitsyna, M.V., Shatov, V.M. and Gusev, N.B. (2019) Small heat shock proteins and human neurodegenerative diseases. Biochemistry (Mosc), 84, 1256–1267. [DOI] [PubMed] [Google Scholar]
- 31. Vendredy, L., Adriaenssens, E. and Timmerman, V. (2020) Small heat shock proteins in neurodegenerative diseases. Cell Stress Chaperones, 25, 679–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wilhelmus, M.M.M., Otte-Höller, I., Wesseling, P., de Waal, R.M.W., Boelens, W.C. and Verbeek, M.M. (2006) Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol. Appl. Neurobiol., 32, 119–130. [DOI] [PubMed] [Google Scholar]
- 33. Nafar, F., Williams, J.B. and Mearow, K.M. (2015) Astrocytes release HspB1 in response to amyloid-β exposure in vitro. J. Alzh. Dis., 49, 251–263. [DOI] [PubMed] [Google Scholar]
- 34. Ojha, J., Masilamoni, G., Dunlap, D., Udoff, R.A. and Cashikar, A.G. (2011) Sequestration of toxic oligomers by HspB1 as a cytoprotective mechanism. Mol. Cell. Biol., 31, 3146–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Padmanabhan, S. and Manjithaya, R. (2020) Facets of autophagy based unconventional protein secretion-the road less travelled. Fron. Mol. Biosci., 7, 586483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang, M. and Schekman, R. (2013) Unconventional secretion, unconventional solutions. Science, 340, 622–626. [DOI] [PubMed] [Google Scholar]
- 37. D’Agostino, M., Scerra, G., Cannata Serio, M., Caporaso, M.G., Bonatti, S. and Renna, M. (2019) Unconventional secretion of α-crystallin B requires the autophagic pathway and is controlled by phosphorylation of its serine 59 residue. Sci. Rep., 9, 16892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bates, G.P., Dorsey, R., Gusella, J.F., Hayden, M.R., Kay, C., Leavitt, B.R., Nance, M., Ross, C.A., Scahill, R.I., Wetzel, R. et al. (2015) Huntington disease. Nat. Rev. Dis. Primers, 1, 15005. [DOI] [PubMed] [Google Scholar]
- 39. Ghosh, R. and Tabrizi, S.J. Huntington disease. Handb. Clin. Neurol., 147, 255–278. [DOI] [PubMed] [Google Scholar]
- 40. Arrasate, M., Mitra, S., Schweitzer, E.S., Segal, M.R. and Finkbeiner, S. (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature, 431, 805–810. [DOI] [PubMed] [Google Scholar]
- 41. Nucifora, L.G., Burke, K.A., Feng, X., Arbez, N., Zhu, S., Miller, J., Yang, G., Ratovitski, T., Delannoy, M., Muchowski, P.J. et al. (2012) Identification of novel potentially toxic oligomers formed in vitro from mammalian-derived expanded huntingtin exon-1 protein. J. Biol. Chem., 287, 16017–16028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bäuerlein, F.J.B., Saha, I., Mishra, A., Kalemanov, M., Martinéz-Sánchez, A., Klein, R., Dudanova, I., Hipp, M.S., Hartl, F.U., Baumeister, W. and Fernández-Busnadiego, R. (2017) In situ architecture and cellular interactions of polyQ inclusions. Cell, 171, 179–187. [DOI] [PubMed] [Google Scholar]
- 43. Ravikumar, B., Duden, R. and Rubinsztein, D.C. (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet., 11, 1107–1117. [DOI] [PubMed] [Google Scholar]
- 44. Bento, C.F., Renna, M., Ghislat, G., Puri, C., Ashkenazi, A., Vicinanza, M., Menzies, F.M. and Rubinsztein, D.C. (2016) Mammalian autophagy: how does it work? Annu. Rev. Biochem., 85, 685–713. [DOI] [PubMed] [Google Scholar]
- 45. Corrochano, S., Renna, M., Carter, S., Chrobot, N., Kent, R., Stewart, M., Cooper, J., Brown, S.D.M., Acevedo-Arozena, A. and Rubinsztein, D.C. (2012) Synuclein levels modulate Huntington’s disease in mice. Hum. Mol. Genet., 21, 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Renna, M., Jimenez-Sanchez, M., Sarkar, S. and Rubinsztein, D.C. (2010) Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J. Biol. Chem., 285, 11061–11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Renna, M., Figueira-Bento, C., Fleming, A., Menzies, F.M., Siddiqi, F.H., Ravikumar, B., Puri, C., Garcia-Arencibia, M., Sadiq, O., Corrochano, S. et al. (2013) IGF-1 receptor antagonism inhibits autophagy. Hum. Mol. Genet., 22, 4528–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ehrnhoefer, D.E., Martin, D.D.O., Schmidt, M.E., Qiu, X., Ladha, S., Caron, N.S., Skotte, N.H., Nguyen, Y.T.N., Vaid, K., Southwell, A.L. et al. (2018) Preventing mutant huntingtin proteolysis and intermittent fasting promote autophagy in models of Huntington disease. Acta Neuropathol. Comms., 6, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jucker, M. and Walker, L.C. (2018) Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci., 21, 1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pearce, M.M.P. and Kopito, R.R. (2018) Prion-like characteristics of polyglutamine-containing proteins. Cold Spring Harb. Perspect. Med., 8, a024257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Masnata, M., Sciacca, G., Maxan, A., Bousset, L., Denis, H.L., Lauruol, F., David, L., Saint-Pierre, M., Kordower, J.H., Melki, R. et al. (2019) Demonstration of prion-like properties of mutant huntingtin fibrils in both in vitro and in vivo paradigms. Acta Neuropathol., 137, 981–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abounit, S., Wu, J.W., Duff, K., Vixtoria, G.S. and Zurzolo, C. (2016) Tunneling nanotubes: a possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion, 10, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Costanzo, M., Abounit, S., Marzo, L., Danckaert, A., Chamoun, Z., Roux, P. and Zurzolo, C. (2013) Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J. Cell Sci., 126, 3678–3685. [DOI] [PubMed] [Google Scholar]
- 54. Scherzinger, E., Sittler, A., Schweige, R.K., Heiser, V., Lurz, R., Hasenbank, R., Bates, G.P., Lehrach, H. and Wanker, E.E. (1999) Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc. Nat. Acad. USA, 96, 4604–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tang, B.L. (2018) Unconventional secretion and intercellular transfer of mutant huntingtin. Cell, 7, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Trajkovic, K., Jeong, H. and Krainc, D. (2017) Mutant huntingtin is secreted via a late endosomal/lysosomal unconventional secretory pathway. J. Neurosci., 37, 9000–9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim, D.-H., Sarbassov, D.D., Ali, S.M., King, J.E., Latek, R.R., Erdjument-Bromage, H., Tempst, P. and Sabatini, D.M. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell, 110, 163–175. [DOI] [PubMed] [Google Scholar]
- 58. Kim, E., Goraksha-Hicks, P., Li, L., Neufeld, T.P. and Guan, K.L. (2008) Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol., 10, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Saxton, R.A. and Sabatini, D.M. (2017) mTOR signalling in growth, metabolism and disease. Cell, 169, 361–371. [DOI] [PubMed] [Google Scholar]
- 60. De Maio, A., Cauvi, D.M., Capone, R., Bello, I., Egberts, W.V., Arispe, N. and Boelens, W. (2019) The small heat shock proteins, HSPB1 and HSPB5, interact differently with lipid membranes. Cell Stress Chaperones, 24, 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Clayton, A., Turkes, A., Navabi, H., Mason, M.D. and Tabi, Z. (2005) Induction of heat shock proteins in B-cell exosomes. J. Cell Sci., 118, 3631–3638. [DOI] [PubMed] [Google Scholar]
- 62. Kano, F., Sako, Y., Tagaya, M., Yanagida, T. and Murata, M. (2000) Reconstitution of brefeldin A-induced Golgi tubulation and fusion with the endoplasmic reticulum in semi-intact Chinese hamster ovary cells. Mol. Biol. Cell, 11, 3073–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pavel, M., Imarisio, S., Menzies, F.M., Jimenez-Sanchez, M., Siddiqi, F.H., Wu, X., Renna, M., O'Kane, C.J., Crowther, D.C. and Rubinsztein, D.C. (2016) CCT complex restricts neuropathogenic protein aggregation via autophagy. Nat Comms., 7, 13821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Haidar, M., Asselbergh, B., Adriaenssens, E., De Winter, V., Timmermans, J.-P., Auer-Grumbach, M., Juneja, M. and Timmerman, V. (2019) Neuropathy-causing mutations in HSPB1 impair autophagy by disturbing the formation of SQSTM1/p62 bodies. Autophagy, 15, 1051–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Plutner, H., Davidson, H.W., Saraste, J. and Balch, W.E. (1992) Morphological analysis of protein transport from the ER to Golgi membranes in digitonin-permeabilized cells: role of the p58 containing compartment. J. Cell Biol., 119, 1097–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Korolchuk, V.I., Mansilla, A., Menzies, F.M. and Rubinsztein, D.C. (2009) Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell, 33, 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dantuma, N.P. and Lindsten, K. (2010) Stressing the ubiquitin-proteasome system. Cardiovasc. Res., 85, 263–271. [DOI] [PubMed] [Google Scholar]
- 68. Duran, A., Amanchy, R., Linares, J.F., Joshi, J., Abu-Baker, S., Porollo, A., Hansen, M., Moscat, J. and Diaz-Meco, M.T. (2011) p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell, 44, 134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Settembre, C. and Ballabio, A. (2014) Lysosomal adaptation: how the lysosome responds to external cues. Cold Spring Harb. Perspect. Biol., 6, a016907. 10.1101/cshperspect.a016907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kostenko, S. and Moens, U. (2009) Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell. Mol. Life Sci., 66, 3289–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bolhuis, S. and Richter-Landsberg, C. (2010) Effect of proteasome inhibition by MG-132 on HSP27 oligomerization, phosphorylation and aggresome formation in the OLN-93 oligodendroglia cell line. J. Neurochem., 114, 960–971. [DOI] [PubMed] [Google Scholar]
- 72. Arrigo, A.P. (2017) Mammalian HspB1 (Hsp27) is a molecular sensor linked to the physiology and environment of the cell. Cell Stress Chaperones, 22, 517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lambert, H., Charette, S.J., Bernier, A.F., Guimond, A. and Landry, J. (1999) HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J. Biol. Chem., 274, 9378–9385. [DOI] [PubMed] [Google Scholar]
- 74. Arrigo, A.-P. and Gibert, B. (2013) Protein interactomes of three stress inducible small heat shock proteins: HspB1, HspB5 and HspB8. Int. J. Hyperth., 29, 409–422. [DOI] [PubMed] [Google Scholar]
- 75. Arrigo, A.-P. and Gibert, B. (2012) HspB1 dynamic phospho-oligomeric structure dependent interactome as cancer therapeutic target. Curr. Mol. Med., 12, 1151–1163. [DOI] [PubMed] [Google Scholar]
- 76. Wyttenbach, A., Sauvageot, O., Carmichael, J., Diaz-Latoud, C., Arrigo, A.-P. and Rubinsztein, D.C. (2002) Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet., 11, 1137–1151. [DOI] [PubMed] [Google Scholar]
- 77. Zourlidou, A., Payne Smith, M.D. and Latchman, D.S. (2004) HSP27 but not HSP70 has a potent protective effect against α-synuclein-induced cell death in mammalian neuronal cells. J. Neurochem., 88, 1439–1448. [DOI] [PubMed] [Google Scholar]
- 78. An, J.J., Lee, Y.P., Kim, D.W., Sohn, E.J., Jeong, H.J., Kang, H.W., Shin, M.J., Kim, M.J., Ahn, E.H., Jang, S.H. et al. (2009) Transduced HSP27 protein protects neuronal cell death by enhancing FALS-associated SOD1 mutant activity. BMB Rep., 42, 136–141. [DOI] [PubMed] [Google Scholar]
- 79. Lim, J., Lachenmayer, M.L., Wu, S., Liu, W., Kundu, M., Wang, R., Komatsu, M., Oh, Y.J., Zhao, Y. and Yue, Z. (2015) Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet., 11, e1004987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ji, C.H., Kim, H.Y., Lee, M.J., Heo, A.J., Park, D.Y., Lim, S., Shin, S., Ganipisetti, S., Yang, W.S., Jung, C.A. et al. (2022) The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system. Nat Comms., 13, 904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mogk, A., Bukau, B. and Kampinga, H.H. (2018) Cellular handling of protein aggregates by disaggregation machines. Mol. Cell, 69, 214–226. [DOI] [PubMed] [Google Scholar]
- 82. Sanchez-Martin, P., Saito, T. and Komatsu, M. (2019) p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J., 286, 8–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Fan, W., Tang, Z., Chen, D., Moughon, D., Ding, X., Chen, S., Zhu, M. and Zhong, Q. (2010) Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy, 6, 614–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Matsumoto, G., Wada, K., Okuno, M., Kurosawa, M. and Nukina, N. (2011) Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell, 44, 279–289. [DOI] [PubMed] [Google Scholar]
- 85. Wellington, C.L., Singaraja, R., Ellerby, L., Savill, J., Roy, S., Leavitt, B., Cattaneo, E., Hackam, A., Sharp, A., Thornberry, N. et al. (2000) Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J. Biol. Chem., 275, 198310–119838. [DOI] [PubMed] [Google Scholar]
- 86. Wellington, C.L., Ellerby, L.M., Gutekunst, C.-A., Rogers, D., Warby, S., Graham, R.K., Loubser, O., van Raamsdonk, J., Singaraja, R., Yang, Y.-Z. et al. (2002) Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington’s disease. J. Neurosci., 22, 7862–7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fryer, J.D. and Zoghbi, H.Y. (2006) Huntingtin’s critical cleavage. Nat. Neurosci., 9, 1088–1089. [DOI] [PubMed] [Google Scholar]
- 88. Graham, R.K., Deng, Y., Slow, E.J., Haigh, B., Bissada, N., Lu, G., Pearson, J., Shehadeh, J., Bertram, L., Murphy, Z. et al. (2006) Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell, 125, 1179–1191. [DOI] [PubMed] [Google Scholar]
- 89. Brunet, A., Bonni, A., Zigmond, M.J., Lin, M.Z., Juo, P., Hu, L.S., Anderson, M.J., Arden, K.C., Blenis, J. and Greenberg, M.E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell, 96, 857–868. [DOI] [PubMed] [Google Scholar]
- 90. Slee, E.A., Zhu, H., Chow, S.C., MacFarlane, M., Nicholson, D.W. and Cohen, G.M. (1996) Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem. J., 315, 21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Watkin, E.E., Arbez, N., Waldron-Roby, E., O'Meally, R., Ratovitski, T., Cole, R.N. and Ross, C.A. (2014) Phosphorylation of mutant huntingtin at serine 116 modulates neuronal toxicity. PLoS One, 9, e88284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cariulo, C., Azzollini, L., Verani, M., Martufi, P., Boggio, R., Chiki, A., Deguire, S.M., Cherubini, M., Gines, S., Lawrence Marsh, J. et al. (2017) Phosphorylation of huntingtin at residue T3 is decreased in Huntington’s disease and modulates mutant huntingtin protein conformation. Proc. Nat. Acad. USA., 114, E10809–E10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Anne, S.L., Saudou, F. and Humbert, S. (2007) Neurobiology of disease phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. J. Neurosci., 27, 7318–7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bruyère, J., Abada, Y.-S., Vitet, H., Fontaine, G., Deloulme, J.-C., Cès, A., Denarier, E., Pernet-Gallay, K., Andrieux, A., Humbert, S. et al. (2020) Presynaptic APP levels and synaptic homeostasis are regulated by Akt phosphorylation of huntingtin. elife, 9, e56371. 10.7554/eLife.56371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Humbert, S., Bryson, E.A., Cordelieres, F.P., Connors, N.C., Datta, S.R., Finkbeiner, S., Greenberg, M.E. and Saudou, F. (2002) The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves huntingtin phosphorylation by AKT. Dev. Cell, 2, 831–837. [DOI] [PubMed] [Google Scholar]
- 96. Rui, Y.N., Xu, Z., Patel, B., Chen, Z., Chen, D., Tito, A., David, G., Sun, Y., Stimming, E.F., Bellen, H.J. et al. (2015) Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol., 17, 262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pecho-Vrieseling, E., Rieker, C., Fuchs, S., Bleckmann, D., Esposito, M.S., Botta, P., Goldstein, C., Bernhard, M., Galimberti, I., Müller, M. et al. (2014) Transneuronal propagation of mutant huntingtin contributes to non–cell autonomous pathology in neurons. Nat. Neurosci., 17, 1064–1072. [DOI] [PubMed] [Google Scholar]
- 98. Babcock, D.T. and Ganetzky, B. (2015) Transcellular spreading of huntingtin aggregates in the Drosophila brain. Proc. Nat. Acad. USA., 112, E5427–E5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bjørkøy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Overvatn, A., Stenmark, H. and Johansen, T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol., 171, 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Doi, H., Adachi, H., Katsuno, M., Minamiyama, M., Matsumoto, S., Kondo, N., Miyazaki, Y., Iida, M., Tohnai, G., Qiang, Q. et al. (2013) p62/SQSTM1 differentially removes the toxic mutant androgen receptor via autophagy and inclusion formation in a spinal and bulbar muscular atrophy mouse model. J. Neurosci., 33, 7710–7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rogov, V., Dötsch, V., Johansen, T. and Kirkin, V. (2014) Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell, 53, 167–178. [DOI] [PubMed] [Google Scholar]
- 102. Menzies, F.M., Fleming, A., Caricasole, A., Bento, C.F., Andrews, S.P., Ashkenazi, A., Füllgrabe, J., Jackson, A., Jimenez-Sanchez, M., Karabiyik, C. et al. (2017) Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron, 93, 1015–1034. [DOI] [PubMed] [Google Scholar]
- 103. Georgopoulos, C. and Welch, W.J. (1993) Role of the major heat shock proteins as molecular chaperones. Annu. Rev. Cell Biol., 9, 601–634. [DOI] [PubMed] [Google Scholar]
- 104. Wu, C. (1995) Heat shock transcription factors: structure and regulation. Annu. Rev. Cell Dev. Biol., 11, 441–469. [DOI] [PubMed] [Google Scholar]
- 105. Takayama, S., Reed, J.C. and Homma, S. (2003) Heat-shock proteins as regulators of apoptosis. Oncogene, 22, 9041–9047. [DOI] [PubMed] [Google Scholar]
- 106. Sun, X., Ou, Z., Xie, M., Kang, R., Fan, Y., Niu, X., Wang, H., Cao, L. and Tang, D. (2015) HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene, 34, 5617–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jimenez-Sanchez, M., Lam, W., Hannus, M., Sönnichsen, B., Imarisio, S., Fleming, A., Tarditi, A., Menzies, F.M., Dami, T.E., Xu, C. et al. (2015) siRNA screen identifies QPCT as a druggable target for Huntington’s disease. Nat. Chem. Biol., 11, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Crippa, V., D’Agostino, V., Cristofani, R., Rusmini, P., Cicardi, M.E., Messi, E., Loffredo, R., Pancher, M., Piccolella, M., Galbiati, M. et al. (2016) Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci. Rep., 6, 22827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Vos, M.J., Zijlstra, M.P., Kanon, B., van Waarde-Verhagen, M.A., Brunt, E.R., Oosterveld-Hut, H.M., Carra, S., Sibon, O.C. and Kampinga, H.H. (2010) HSPB7 is the most polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum. Mol. Genet., 19, 4677–4693. [DOI] [PubMed] [Google Scholar]
- 110. Rous, B.A., Reaves, B.J., Ihrke, G., Briggs, J.A.G., Gray, S.R., Stephens, D.J., Banting, G. and Luzio, J.P. (2002) Role of adaptor complex AP-3 in targeting wild-type and mutated CD63 to lysosomes. Mol. Biol. Cell, 13, 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Narain, Y., Wyttenbach, A., Rankin, J., Furlong, R.A. and Rubinsztein, D.C. (1999) A molecular investigation of true dominance in Huntington’s disease. J. Med. Genet., 36, 739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Luo, S., Vacher, C., Davies, J.E. and Rubinsztein, D.C. (2005) Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J. Cell Biol., 169, 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Dantuma, N.P., Lindsten, K., Glas, R., Jellne, M. and Masucci, M.G. (2000) Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol., 18, 538–543. [DOI] [PubMed] [Google Scholar]
- 114. Hoffman, L., Jensen, C.C., Yoshigi, M. and Beckerle, M. (2017) Mechanical signals activate p38 MAPK pathway-dependent reinforcement of actin via mechanosensitive HspB1. Mol. Biol. Cell, 28, 2661–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Dunsing, V., Petrich, A. and Chiantia, S. (2021) Multicolor fluorescence fluctuation spectroscopy in living cells via spectral detection. eLife, 10, e69687. 10.7554/eLife.69687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Martin, R.G. and Ames, B.N. (1961) A method for determining the sedimentation behaviour of enzymes: application to protein mixtures. J. Biol. Chem., 236, 1372–1379. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material. The datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request.