

Abstract
Traumatic axonal injury (TAI) and the associated axonopathy are common consequences of traumatic brain injury (TBI) and contribute to significant neurological morbidity. It has been previously suggested that TAI activates a highly conserved program of axonal self-destruction known as Wallerian degeneration (WD). In the present study, we utilize our well-established impact acceleration model of TBI (IA-TBI) to characterize the pathology of injured myelinated axons in the white matter tracks traversing the ventral, lateral, and dorsal spinal columns in the mouse and assess the effect of Sterile Alpha and TIR Motif Containing 1 (Sarm1) gene knockout on acute and subacute axonal degeneration and myelin pathology. In silver-stained preparations, we found that IA- TBI results in white matter pathology as well as terminal field degeneration across the rostrocaudal axis of the spinal cord. At the ultrastructural level, we found that traumatic axonopathy is associated with diverse types of axonal and myelin pathology, ranging from focal axoskeletal perturbations and focal disruption of the myelin sheath to axonal fragmentation. Several morphological features such as neurofilament compaction, accumulation of organelles and inclusions, axoskeletal flocculation, myelin degeneration and formation of ovoids are similar to profiles encountered in classical examples of WD. Other profiles such as excess myelin figures and inner tongue evaginations are more typical of chronic neuropathies. Stereological analysis of pathological axonal and myelin profiles in the ventral, lateral, and dorsal columns of the lower cervical cord (C6) segments from wild type and Sarm1 KO mice at 3 and 7 days post IA-TBI (n = 32) revealed an up to 90% reduction in the density of pathological profiles in Sarm1 KO mice after IA-TBI. Protection was evident across all white matter tracts assessed, but showed some variability. Finally, Sarm1 deletion ameliorated the activation of microglia associated with TAI. Our findings demonstrate the presence of severe traumatic axonopathy in multiple ascending and descending long tracts after IA-TBI with features consistent with some chronic axonopathies and models of WD and the across-tract protective effect of Sarm1 deletion.
Keywords: Neuropathy, Wallerian degeneration, Neurodegeneration, Myelin, Oligodendrocyte, Microglia, Corticospinal tract, Electron microscopy, Traumatic axonal injury
1. Introduction
Axonal degeneration is ubiquitous in neurological disorders. The best characterized type of axonal breakdown is Wallerian degeneration (WD) (Waller, 1850), a highly conserved program of axonal self-destruction that was classically described in experimental axotomy in the peripheral nervous system as the delayed fragmentation of the axon distal to injury (Conforti et al., 2014). Wallerian degeneration may be also encountered, to various degrees, in diverse disorders including peripheral neuropathy, chemotherapy-induced neuropathy, glaucoma, and perhaps in neurodegenerative diseases of the central nervous system (CNS) (Wang et al., 2002; Beirowski et al., 2008; Howell et al., 2013; Zhu et al., 2013; Geisler et al., 2016; Turkiew et al., 2017). To this list investigators have recently added traumatic axonopathy encountered in models of diffuse brain injury (Henninger et al., 2016; Ziogas and Koliatsos, 2018; Koliatsos and Alexandris, 2019; Marion et al., 2019; Maynard et al., 2020). Initially associated with the depletion of the labile NAD-synthesizing enzyme NMNAT2 (Gilley and Coleman, 2010; Loreto et al., 2020) the molecular understanding of WD has substantially expanded with the recent discovery of its trigger signal, the NADase Sterile Alpha And TIR Motif Containing 1 (SARM1) (Osterloh et al., 2012; Gerdts et al., 2013; Essuman et al., 2017) and upstream players such as the atypical ubiquitin E3 ligase complex of MYCBP2-SKP1-FBXO45 and members of the stress-activated MAPK cascade (Desbois et al., 2018; Summers et al., 2018).
Traumatic axonopathy, an evolving pathology underlying a common type of traumatic brain injury (TBI) known as diffuse or traumatic axonal injury (DAI or TAI) has been the subject of extensive research for some period of time (Adams et al., 1984; Blumbergs et al., 1989; Maxwell et al., 1991; Mittl et al., 1994; Gentleman et al., 1995; Povlishock and Christman, 1995; Maxwell et al., 1997; Povlishock et al., 1997; Lifshitz et al., 2007; Koliatsos et al., 2011; Wang et al., 2011; Johnson et al., 2013; Maxwell et al., 2015). However, it has been only recently demonstrated that axonal degeneration in some cases of TAI may be triggered by SARM1 signaling (Henninger et al., 2016; Ziogas and Koliatsos, 2018; Marion et al., 2019; Maynard et al., 2020).
We have previously shown that, in the impact acceleration TBI (IA-TBI) model of TAI, there is axonal dystrophy and some fragmentation in descending tracts such as corticospinal and reticulospinal and ascending tracts such as the gracilis in the brain stem acutely within 24 h after injury (Ziogas and Koliatsos, 2018). By analyzing swellings in YFP-labeled axons we further showed that this early TAI-related axonopathy is at least partially dependent on SARM1 signaling. In the present study, we extend our previous observations and explore whether TAI in these long tracts triggers WD. To this effect, we explore changes at longer survival times (3–7 days post injury), i.e. the time frame typical of WD, and focus on the upper spinal cord to take advantage of its unique anatomy where several ascending and descending tracts converge in a small region. We examine changes in myelinated axons in the ventral, lateral, and dorsal columns of the cervical spinal cord that contain some of the tracts examined by us in the spinal cord early after TBI (Ziogas and Koliatsos, 2018). We characterize the features of traumatic axonopathy and degeneration at the semithin and ultrastructural level and explore the dependency of such lesion profiles on SARM1.
2. Materials and methods
2.1. Experimental subjects - the impact acceleration TBI model
For the study we used 10 to 14 week-old male C57BL/6 J wild-type mice and transgenic Sarm1 KO mice (Szretter et al., 2009) backcrossed to C57BL/6 J for at least 10 generations (gift from Dr. A. Hoke, Johns Hopkins University). Sarm1 KO mice were generated by homozygous breeding and genotyping was confirmed for each mouse by PCR. Mice were subjected to IA-TBI or sham injury (Table 1) as described with slight modifications (Ziogas and Koliatsos, 2018; Welsbie et al., 2019; Alexandris et al., 2022b). Briefly, mice were anaesthetized with a mixture of isoflurane, oxygen and nitrous oxide, the cranium was exposed, and a 5mm-thick stainless-steel disc was glued onto the skull midway between bregma and lambda sutures. Then, a 50 g weight was dropped from 85 cm on the metal disk, while the mouse was placed on a foam mattress (4–0 spring constant foam, Foam to Size Inc., Ashland, VA), with the body immobilized with tape. The mouse was positioned with the forelimbs on the elevated platform of the foam pad. Immediately after injury the disc was removed and the skull was inspected for skull fractures. The rare animals with fractures (<2%) were excluded.The scalp incision was closed with surgical staples. Sham animals underwent the same procedure omitting the weight drop step. Neurological recovery was assessed by the presence and duration of apnea or irregular breathing (present in 46% of mice, median duration 53 s with IQR 74.5 s) and the revival of the righting reflex (median 210 s with IQR of 210 s). Subjects with apnea/irregular breathing >150 s and time to righting reflex >550 s were excluded (< 5%). With the current lesion settings and based on blood-brain barrier labeling with EZ-Link™ Sulfo-NHS-LC-Biotin, the biomechanical disruption in the CNS associated with impact acceleration appeared to have extended beyond the brainstem to involve the upper cervical cord. While mice with this severity of injury do not typically have overt motor or other behavioral deficits following recovery, they do display substantial deficits in a pellet-reaching task (40–50% reduction in successful attempts; Koliatsos et al., unpublished observations). After recovery, animals were returned to their vivarium with a 12-h light/12-h dark cycle and ad libitum access to food and water. Surgical procedures and injuries were performed under aseptic conditions and all animal handling and postoperative procedures were carried according to protocols approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions (Protocol Number: MO19M458).
Table 1.
Experimental groups and histological procedures.
| Experimental Groups | Injury and Survival | Histological procedures |
|---|---|---|
| 1. Mapping of degenerating fiber tracts with silver staining | 3d post IA-TBI (n = 3 per TBI or sham injury, WT)) | Silver staining (amino-cupric silver technique) of transverse and horizontal spinal cord sections. IBA1 staining for activated microglia. |
| 2. Characterization of spinal cord traumatic axonopathy in WT mice with electron microscopy | Sham (n = 4), 3d post IA-TBI (n = 4), 7d post IA-TBI (n =3) | Electron microscopy in transverse and longitudinal cervical spinal cord sections |
| 3. Characterization of traumatic axonopathy with toluidine-stained semithin sections in WT and SARM1 KO mice | 3d post IA-TBI or sham-injury (n = 8 per genotype), 7d post IA-TBI (n = 8 per genotype) | Stereological quantitation of lower cervical transverse semithin sections (at C6). |
| 4. Assessment of neuroinflammation with Iba1 immunohistochemistry | 7d post IA-TBI or sham injury (n = 6 per genotype), | Iba1 immunostaining and quantitation of % area coverage in the CST (at C6) |
2.2. Silver staining
Mice in experimental group 1 were transcardially perfused with freshly depolymerized and filtered paraformaldehyde in PBS (4% in 0.1 M PBS, pH 7.4). Spinal cords were dissected and postfixed overnight in the same fixative. Tissues were embedded in gelatin, sectioned at the coronal and transverse planes (30 μm) and processed for de Olmos modified amino-cupric silver for degenerating axons (de Olmos et al., 1994) by Neuroscience Associates (Knoxville, TN). Ionized calcium binding adaptor molecule 1 (IBA1) immunohistochemistry for microglia was performed on adjacent sections.
2.3. Electron microscopy
Mice in experimental groups 2 and 3 were transcardially perfused with 2% paraformaldehyde/2% glutaraldehyde in 50 mM sodium cacodylate, 50 mM phosphate and 3 mM magnesium chloride buffer (pH = 7.4) for 30 min. Tissues were left in situ at room temperature for two hours before dissection of spinal cord in 1 mm blocks at C6 and post- fixation overnight at 4 °C in the same fixative. On a rotator, tissues were washed in 75 mM sodium cacodylate, 75 mM phosphate, 3 mM magnesium chloride (pH =7.4), for 15 min 5 times. To adjust osmolarity, the first two rinsing steps included 2:1 and 1:1 mixture of fixative solution and buffer at 4 °C, respectively. Tissues were then incubated in freshly prepared 2% osmium tetroxide with 1.6% potassium ferrocyanide in wash buffer for 2 h at room temperature in the dark. Tissues were rinsed in 100 mM maleate, 3.5% sucrose buffer (pH = 6.2) for 10 min 3 times. They were then incubated with 2% uranyl acetate in maleate-sucrose buffer for 1 h (dark) followed by step-wise washing steps in maleate buffer, 1:1 maleate buffer with distilled water and finally distilled water for 5 min each. Tissue blocks were dehydrated in a graded ethanol series, embedded in EMbed 812 resin (EMS14120, Electron Microscope Sciences, Hatfield, PA). Semithin (1 μm) and thin (60 nm) sections were sectioned in a Leica UC7 ultramicrotome. Semithin sections were stained with 1% toluidine blue. Thin sections were mounted in square mess copper grids (EMS300-Cu, Electron Microscope Sciences, Hatfield, PA) and stained with uranyl acetate and lead citrate and observed with a Hitachi H7600 transmission electron microscope (Hitachi High- Technologies Corporation, Tokyo, Japan).
2.4. Stereological quantitation of axons and their pathology
Mice in group 3 were perfused and tissues were processed as for group 1. Semithin sections (1 μm) were prepared and stained with toluidine blue. Pathological axonal and myelin profiles (primarily axons with condensed or hydropic axoplasm, collapsed myelin sheaths, excess myelin figures, gross discontinuities in myelin sheath, tomacula and myelin bodies) were counted separately in the ventral, lateral, and dorsal columns on coronal semithin sections through segments C6-C7. The ventral column was defined as the region between the midline and the ventral root axons of the deltoid group of motor neurons; the lateral column was defined as the region between the lateral border of ventral column and the lateral edge of dorsal root entry zone; the dorsal column was defined as the region between the medial edge of dorsal root entry zone and the midline. Only one side of each section was used for counts, with random selection of laterality. For the quantitative assessment of the dorsal corticospinal tract (CST), an index pathway that is well delineated in the dorsal column, the latter was further subdivided into gracilis (Gr) and cuneatus (Cu) fasciculi and the CST based on the characteristic axon calibers of the corresponding axons.
Stereological analysis by an investigator blinded to experimental history was performed using the optical fractionator probe and systematic random sampling, with the aid of a motorized stage Axioplan microscope (Carl Zeiss Inc.) and Stereo Investigator® software (Microbrightfield Inc., Williston, VT). Regions of interest were defined at 10× or 20× objective. For the ventral (VC) and lateral columns (LC), myelin profiles were counted with a 40× objective with a 11.50 × 11.50 μm counting frame, 51.43 × 51.43 μm grid, and sampling fraction of 5%. For the dorsal columns (including the CST), stereological analysis was performed at 100× with a counting frame size of 4 × 4 μm, grid size of 17.89 × 17.89 μm and sampling fraction of 5%.
To quantitate intact corticospinal axons, stereological analysis by an investigator blinded to experimental history was performed at 100×. Axons were counted as apparently intact if they had normal appearance and did not meet any of the criteria for pathological profiles (as above) or display any other alterations, such as thinning or thickening of the myelin, metachromatic staining of the cytoplasm. Axon profiles with these latter milder alterations are reported as “equivocal” profiles.
Statistical analyses were performed using GraphPad Prism 9 (GraphPad Software, La Jolla, Ca, USA; RRID:SCR_002798). Normality of the data was assessed by the Shapiro-Wilk test and statistical significance for differences between genotypes and effect of time was assessed with a two-way ANOVA test (Fdf,df). Post-hoc multiple comparisons (tdf) were adjusted by the Holm-Šídákś method (adjusted p-values are reported unless stated otherwise). Data are presented as means ± standard error of mean. Statistical significance was set at p < 0.05.
2.5. Quantitation and analysis of electron microscopy profiles
For quantitative analysis of particular axonal/myelin profiles (excess myelin figures, inner tongue evaginations), the CST was chosen as an index white matter tract, as it can be easily delineated anatomically at low magnification and has a more uniform distribution of axon calibers. In each EM grid, the random superposition of the sample on the copper grid lines, allows for an unbiased sampling of the CST region. Areas of interest were identified at low magnification (4000×) at the corners and center of each hole (90 × 90 μm) in the copper grid array; and then micrographs were captured at 20,000×(6–8 per case). To avoid bias due to different axon sizes, a stereological counting frame of 6.3 × 6.5 μm was fitted in Fiji (RRID:SCR_002285) and profiles were counted blind to group assignment using the cell counter plugin. For each case (n = 3–4 per group), about 250 axons were assessed. For the estimation of axonal morphometric parameters we used the MyelTracer software by Kaiser et al. (Kaiser et al., 2021). Due to the lognormal distribution of morphological parameters, summary statistics for each case were calculated based on geometric means (Alexandris et al., 2022b).
2.6. Assesment of neuroinflammation
Mice in group 4 were perfused with 4% PFA in PBS. Spinal cord segments corresponding to C6–7 were cryoprotected and sectioned at 50-μm thick sections with a freezing microtome. Immunohistochemistry was performed after blocking in 5% normal goat serum, 0.3% Triton X, in PBS with a rabbit polyclonal antibody against IBA1 (1:200, BIOCARE MEDICAL, # CP 290 A, RRID: AB_10578940) for two days at 4 °C, and a polyclonal goat anti-rabbit antibody conjugated to Alexa Fluor™ Plus 594 (1:300, Thermo Fisher Scientific, # A48284, RRID:AB_2896348) overnight at 4 °C. Z-stack images through the CST were captured at 20× with a Zeiss LSM 880 (two sections per case) and analyzed with Fiji (RRID: SCR_002285). Maximum projections of the IBA1 channel were converted to binary maps by adaptive thresholding after background subtraction and Gaussian blurring, and the % area coverage were estimated for the CST.
3. Results
3.1. Impact acceleration traumatic brain injury causes degeneration in long tracts in ventral, lateral and dorsal spinal columns
To explore the distribution of axonal degeneration in the spinal cord after IA-TBI, we assessed cupric silver-stained preparations (de Olmos et al., 1994) from subjects euthanized 3 days after injury, i.e. a time frame in which injured axons may undergo WD. We found axon degeneration signal in select spinal tracts in the ventral, lateral, and dorsal white matter (Fig. 1) but not in sham-injured mice (Supplementary Fig. 1). With the exception of the CST, gracilis and cuneatus, all of which are clearly and reliably delineated in their characteristic positions in the dorsal funiculus, in most cases it is difficult to know the exact location of these tracts on silver material. In the description that follows, we will refer to non-dorsal tracts based on their correspondence to the map of Watson and Harrison (Watson and Harrison, 2012).
Fig. 1.
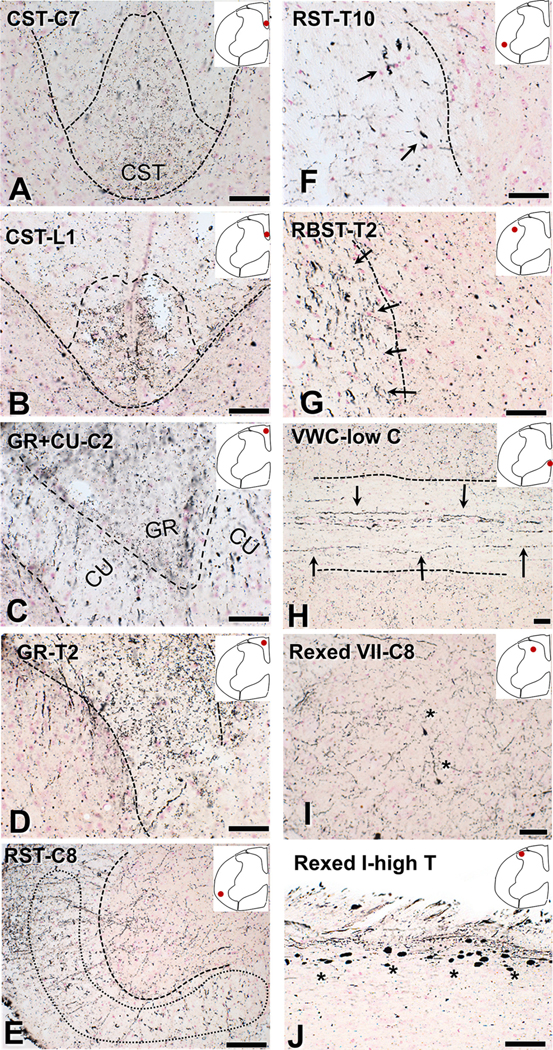
Degeneration of ventral, lateral and dorsal spinal columns after impact acceleration injury revealed with amino-cupric silver at 3 days. With the exception of (E), dotted lines mark white-gray matter border or midline. The anatomical location is indicated in each panel with an inset and spinal segments are denoted with letters and numbers (C, cervical; T, thoracic; L, lumbar). Approximate tract delineation is based on the map of Watson and Harrison (2012). (A-B) Degenerative changes in the corticospinal tract in coronal sections. (C–D). Degenerating fibers in the gracilis and cuneatus tracts in coronal. (E-F) Degenerative changes in large-caliber axons in the ventral and lateral columns roughly corresponding to the reticulospinal tract (arrows). (G) Coronal section through the thoracic cord showing silver precipitation in large fibers corresponding to the rubrospinal tract (arrows). (H) Horizontal section through the ventral white commissure in the lower cervical cord showing large degenerating fibers putatively belonging to the reticulospinal tract (arrows). (I) Fine silver precipitation in neuropil in Rexed VII of a coronal section through the lower cervical cord. (J) Argyrophilic axonal spheroids in the dorsal root entry zone (Rexed I) in a horizontal section through higher thoracic cord. Asterisks indicate pathological axon swellings and bulbs. CST, corticospinal tract; CU, cuneatus; GR, gracilis; RBST, rubrospinal tract; RST, reticulospinal tract; VWC, ventral white commissure. Scale bars: 50 μm.
In the case of CST, we saw clear labeling in the cervical, thoracic and lumbar cord (Fig. 1 A–B). In the case of gracilis, we saw intense labeling in all spinal cord segments. In the case of cuneatus, we found strong labeling in the cervical cord (Fig. C–D). In the ventral white commissure and ventral funiculus, we found cupric silver precipitation in large axons corresponding to the ventral reticulospinal tract, primarily in cervical and thoracic segments (Fig. 1 E–F). Large abnormal axon profiles in the ventral most positions, putatively belonging to the vestibulospinal tract, were present in some cases. We also found strong axon degeneration signal in laterally directed fibers in the ventral white commissure suggestive of crossing spinothalamic axons. We also saw occasional motor axons coursing in the ventral funiculus, especially in the cervical protuberance. In the lateral funiculus, we found silver precipitation in large axons in locations corresponding to the dorsal reticulospinal tract in lumbar, thoracic and cervical segments and large axons in the area of the rubrospinal tract, mostly in cervical and thoracic segments (Fig. 1G).
In the gray matter, we found cupric silver signal in large axons crossing the lower Rexed laminae or midline ventral to the central canal, mostly in cervical and thoracic segments. We also saw signal consistent with terminal degeneration throughout the gray matter (Rexed 1–8) mostly in cervical segments (Fig. 1I). Furthermore, we observed large argyrophilic axon spheroids in dorsal root entry zone, mainly in thoracic segments (Fig. 1J). In many of these locations, axonal pathology corresponded to the presence of deramified IBA1(+) microglial profiles or microglial nodules (Fig. 2).
Fig. 2.
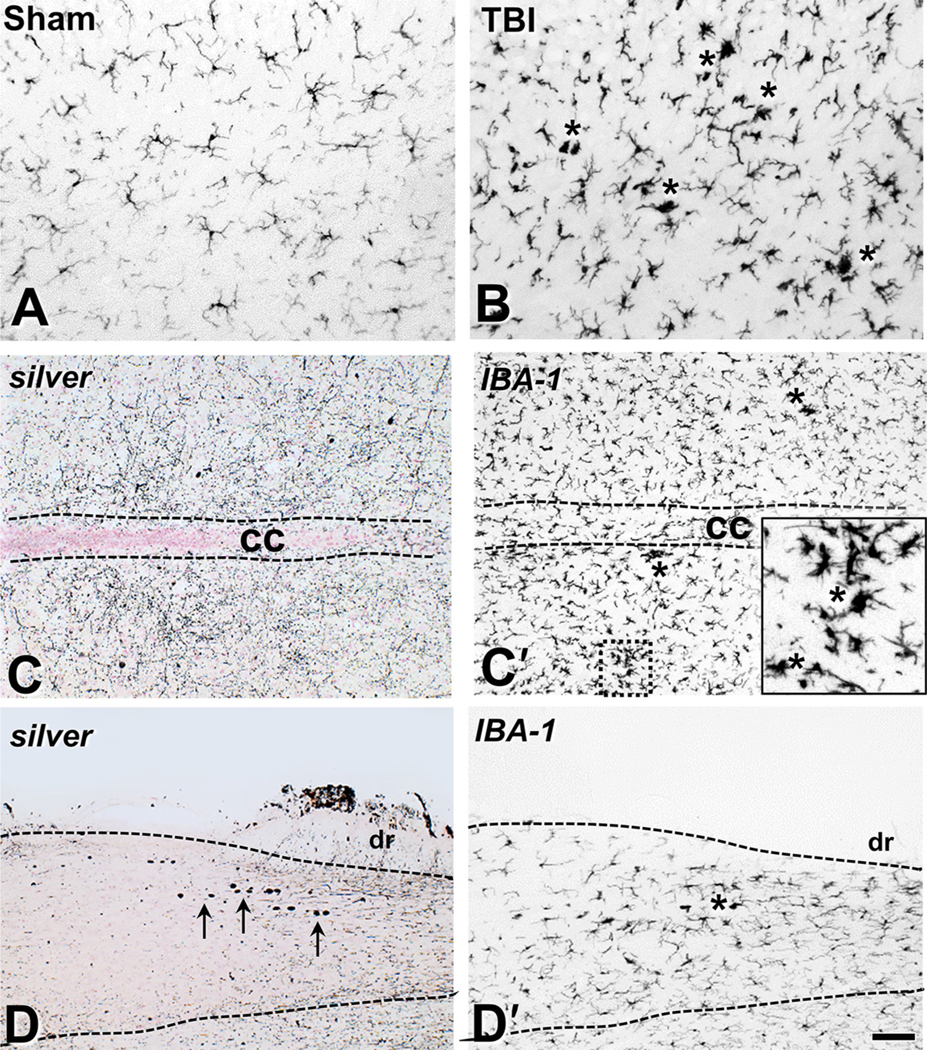
Traumatic brain injury is associated with microglial activation in the spinal cord tracts. (A-B) IBA1 immunohistochemistry labels resting microglia in the mid-cervical ventral column of sham-injured mice (A) and the deramified microglia and microglial nodules (asterisks) 3 days after IA-TBI (B). (C-C′) Adjacent horizontal sections through Rexed VII at a mid-cervical level stained for cupric silver (C) and IBA1 immunoreactivity (C′). Note the widespread fine neuropil silver precipitation in terminals in (C) and extensive microglial activation with the presence of microglial nodules (asterisks and inset) in (C′). (D-D′) Silver (D) and IBA1 (D′) staining of adjacent horizontal sections through Rexed I at an upper thoracic level Arrows in panel (D) indicate the presence of large axonal spheroids in the root entry zone; same area is featured by deramified IBA1(+) profiles in panel (D′). cc, central canal; dr, dorsal root. Scale bars: 100 μm.
3.2. Ultrastructural observations
The ultrastructure of axons in the dorsal and ventral columns showed a remarkable variety of changes including axoplasmic alterations (Figs. 3–4), multifarious changes in myelin sheath and related myelin figures (Figs. 5–6), profiles of axonal fragmentation such as ovoids (Fig. 7), and changes in oligodendrocyte cytoplasmic domains attached to the axon, primarily the inner tongue process (Fig. 8). Pathological changes in the axon, sheath, and axon-myelin spaces do not always occur together in a single axonal segment. Normal axonal profiles could be wrapped in pathological sheaths and, conversely, normal-looking myelin sheaths could be wrapped around abnormal axons.
Fig. 3.

Gross alterations of spinal axons 3 days after impact acceleration injury. Electron micrographs are from sections through the corticospinal tract (A-D, F) or the dorsal columns (E) corresponding to C6–7 segments. (A-B) Transverse axonal profiles showing hydropic changes (asterisks) with dissolution of structure at different degrees, and the presence of distended mitochondria (B). Note the thin myelin sheath in the pathological axon in (B). (C–D) Transversely sectioned profiles with axoplasmic flocculation (C) and advanced condensation (D; asterisks). In both cases, myelin sheath appears normal. (E-F) Accumulation of mitochondria, inclusions and dense bodies in distended segments of longitudinally (E) and transversely (F) sectioned axons (arrowheads). Note the relatively thin myelin sheath in (E) and the absence of myelin sheath in (F). CST, corticospinal tract; DC, dorsal column. Scale bars: A-C, 400 nm; D-E, 500 nm; F, 1 μm.
Fig. 4.

Subtle alterations of spinal cord axons 3 days after impact acceleration injury. Electron micrographs are from sections through the ventral column corresponding to C6–7 segments. (A-B) Regional rarefaction or deformation of microtubule content in ax1 (A, arrowheads; also throughout ax3) and in an axonal protrusion/varicosity of the axon illustrated in (B, arrowheads). Compare with normal axons (e.g. ax2 in A). The myelin sheath of pathological axons is often compact, indistinguishable from that in non-injured axons. (C) Higher magnification of (B), showing localized disruption of the cytoskeleton (arrowheads) and compaction of neurofilaments in the underlying region (asterisks). VC, ventral column. Scale bars; A, C, 400 nm; B, 1 μm.
Fig. 5.

Gross myelin pathology in the form of collapsed myelin or excess myelin profiles. Electron micrographs are from sections through the ventral column in panels (A-B) and the corticospinal tract in panels (C-E), (C6–7 level). (A-B) Collapsed myelin profiles, i.e., myelin sheaths closely apposed to each other with little or no tissue between them, are seen at 3 days (A) and, more commonly, at 7 days (B) post-injury (asterisks). Oli, oligodendrocyte. (C-E) Excess myelin profiles appear as morphologically complex tube-like structures with morphological similarities to collapsed myelin objects. These profiles loop around normal-looking or abnormal axons or oligodendrocyte cytoplasm. Note the ensheathing of normal axonal material (ax) in (C), but also surrounding of tissue with electron density typical of oligodendrocytes (C–D, asterisks). These various tissues may be contained in the same enclosure (C,D). Excess myelin often wraps itself in concentric loops containing normal and abnormal tissue elements in the form of tomacula (E). CST, corticospinal tract; VC, ventral column. Scale bars: A, B, D, E, 500 nm; C, 800 nm.
Fig. 6.

Subtle myelin pathology after traumatic brain injury. Electron micrographs are from sections through the corticospinal tract (A), gracilis/cuneatus (B) and ventral column (C–D) (C6–7 level). (A-B) In longitudinal (A) and transverse (B) sections, loose and more compact myelin loops (ml) form ectopically between intact axons (ax) and apparently intact sheaths, with/without oligodendrocyte cytoplasm (asterisk in A). (C–D) Here the myelin sheath of normal-looking axons is split by a massive accumulation of dense oligodendrocyte cytoplasm (asterisks). In (C) there is also disorderly loose myelin (ml). ac, astrocyte; CST, corticospinal tract; Gr/Cu, gracilis and cuneatus; VC, ventral column. Scale bars: A, 600 nm; B, 1 μm; C, 250 nm; D-E, 500 nm.
Fig. 7.

Ovoid-like formation and axonal fragmentation after traumatic brain injury. Electron micrographs are from sections through the dorsal columns (A-C), and ventral columns (D-E) (C6–7 level). (A) A longitudinally cut axon showing accumulation of mitochondria and dense bodies (see also Fig. 3 E–F) but also periodic segmental constriction of the axon (arrowheads), typical of early Wallerian degeneration in peripheral axons. (B–C) Ovoid formations in longitudinal sections of myelinated axons (arrows) with (C) or without (B) the preservation of portions of hydropic axoplasm (asterisk). In both panels, note the complex arrangement of compact-appearing myelin sheaths. (D-E) Profiles of myelin bodies that may represent a later stage of degeneration at 7 days post- injury. There is loss of periodicity in myelin sheaths. ac, astrocyte; DC, dorsal column; VC, ventral column. Scale bars: A, D, E, 2 μm; B–C, 1 μm.
Fig. 8.

Oligodendrocyte cytoplasm protrusions into the main axon space seen after traumatic brain injury. Electron micrographs are from sections through the dorsal columns (A,C) and ventral columns (B, D) (C6–7 level). (A) Lobular evaginations of oligodendrocyte cytoplasm. These entities are separated from axons via intact membranes (arrowheads). Some axons have cytoskeletal disorganization (ax1 in A;); compare with a normal axon without evident affiliation with such evaginations (ax2 in A). (B) Cross-sectional configuration of evaginations similar to the ones in (A). (C) Some evaginations contain dark oligodendrocyte cytoplasm (arrowhead) and vacuolar or hyperdense inclusions (asterisk). (D) Axon showing evidence of evagination process. The inner tongue of the oligodendrocyte (oli; shown in orange), intends deeply the axon and partially partitions the axoplasm (Ax; light blue). Note the degenerative flocculation and absence of microtubules in the axoplasm. DC, dorsal column; VC, ventral column. Scale bars: 500 nm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Changes in the axon proper included hydropic (Fig. 3 A–B) and less commonly dense (Fig. 3 C–D) degeneration of the axoplasm at both 3 and 7 days post-injury. The former was associated with dissolution and aggregation (flocculation) of the axoskeleton and sometimes the presence of abnormal mitochondria (Fig. 3 B). Pleiomorphic vesicles, dense bodies, mitochondria, and myelin inclusions were frequently present in enlarged segments of the axon (Fig. 3 E–F). The most common axonal alterations were focal rarefaction of microtubules and compaction of neurofilaments, often appearing side-by-side in the same region (Fig. 4).
Myelin changes were common and extremely diverse, although less elaborate in small-caliber axons. Excess myelin figures and collapsed myelin sheaths with no evident axon content were especially common. These profiles had normal-appearing compact myelin and were more common at 7 compared to 3 days post-injury (Fig. 5). The clearly defined anatomical boundaries of the CST made it amenable site for quantitative analysis which revealed an increase in frequency of excess myelin from 0.38% in sham mice to 2% at 3 days post injury (t6 = 4.9, p = 0.0027) and 2.7% at 7 days (t5 = 6.35, p = 0.0015). Excess myelin figures usually extended over some distance and engaged in complex wrapping formations around normal and pathological axoplasm (Fig. 5 C–E); sometimes they wrapped themselves on already myelinated axons to form tomacula (Fig. 5 E).
We also observed focal alterations in myelin sheaths associated with both normal and abnormal axons (Fig. 6), usually in the form of loose or more compact myelin loops in the space between axon and sheath, with or without associated oligodendrocyte cytoplasm (Fig. 6 C–D). Demyelination was also present: morphometric analysis of myelinated axons in the CST revealed an increase in the corrected g-ratio from 0.68 to 0.74 (t6 = 10.19, p = 0.00005) and 0.73 (t5 = 4.42, p = 0.007) at 3 days and 7 days respectively, due to thinning of the myelin sheath (at day 3 by 20%, t6 = 2.69, p = 0.036) in the absence of significant changes in average axon calibers (t6 = 0.94, p = 0.4 and t5 = 0.45, p = 0.7 respectively). The increase in the g-ratio was observed across all axon calibers at 3 days with some partial recovery for large-caliber axons at day 7 (Supplementary Fig. 2). In addition to the pathological profiles described above, we also observed axon/myelin fragmentation in the form of periodic constriction of degenerating axons (Fig. 7 A) or the presence of ovoids (Fig. 7 B–E).
There were also changes in the oligodendrocyte cytoplasmic domains associated with the axon. The axon-sheath space was increased by 50% at 3 days (t6 = 3.89, p = 0.008) and was occupied either by a uniformly enlarged inner tongue process or by lobular compartments of oligodendrocyte cytoplasm (Fig. 8). The latter formations, previously described as oligodendrocyte or Schwann cell – axon networks or inner tongue evaginations (Bilbao and Schmidt, 2014) were sometimes encountered in large myelinated axons of sham animals, but increased in frequency and complexity after injury (Fig. 8A–D). In injured animals, such evaginations often contained pathological inclusions (e.g. Fig. 8 D). In the CST, a preferred site for quantitative EM analysis for reasons explained above, complex evagination profiles (defined as those containing abnormal inclusions and/or having multiple compartments) were increased by 4.3 times (t6 = 3.233, p = 0.017) compared to sham- injured animals at 3 days post injury, but returned to baseline at 7 days.
3.3. Traumatic axonopathy in the spinal cord depends on SARM1 signaling
We and others have previously shown that Sarm1 deletion can reduce the acute pathological burden associated with traumatic axonopathy (Henninger et al., 2016; Ziogas and Koliatsos, 2018; Marion et al., 2019) in the vicinity of the presumed biomechanical disruption, especially in the brainstem and cervical cord after impact acceleration injury (Ziogas and Koliatsos, 2018). To further assess the role of SARM1 in ongoing axonopathic events beyond the acute phase, we elected to count the density of pathological axonal profiles in the white matter of the lower cervical spinal cord at 3 and 7 days following injury (n = 8 per genotype per time point), on semithin sections using stereological methods (Fig. 9). Pathological profiles counted included axons with condensed axoplasm and myelin pathology such as excess myelin figures, breaks in myelin sheath, excess myelin figures, tomacular formations, and collapsed myelin or free myelin debris (myelin bodies) (Fig. 9 A–C). These profiles correspond to morphologies disclosed with EM (see previous section).
Fig. 9.
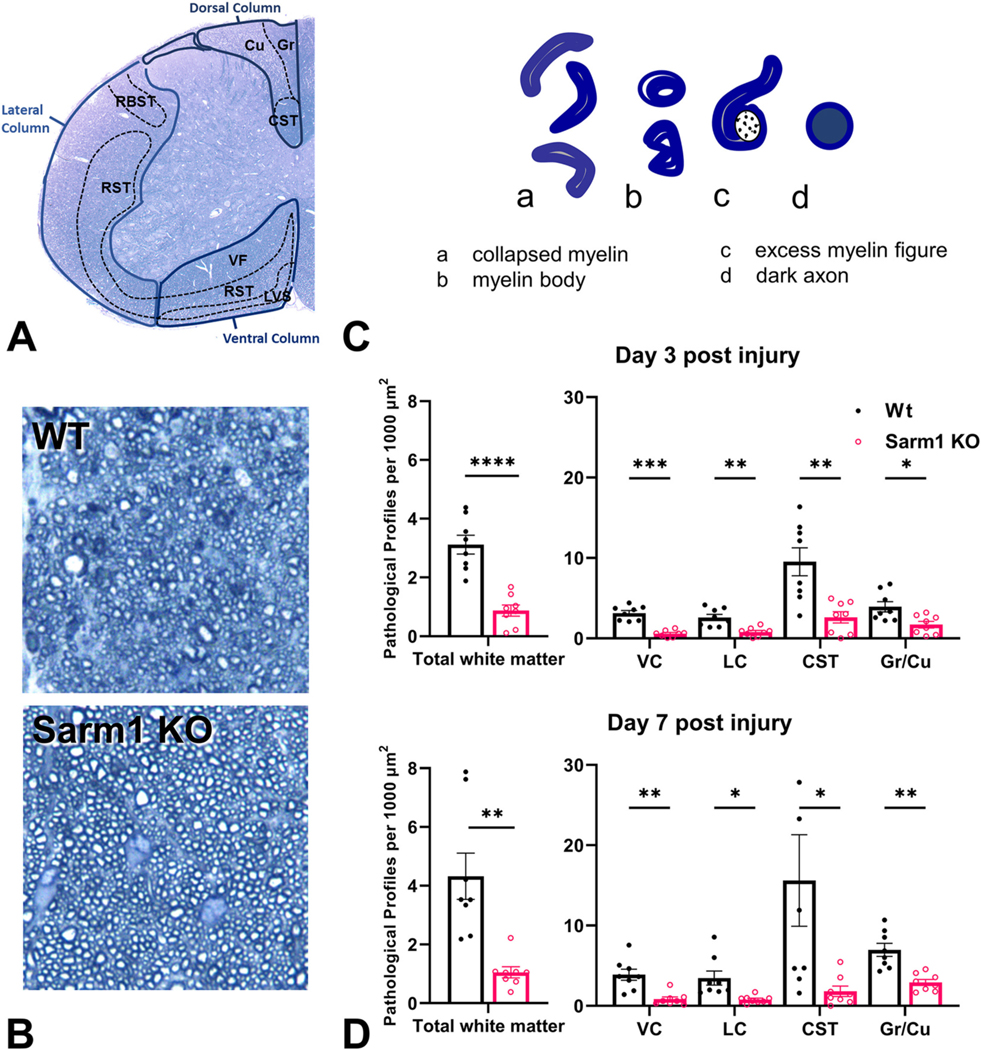
The effects of Sarm1 knockout on pathological axonal degeneration in the spinal cord after impact acceleration traumatic brain injury. (A) Toluidine blue-stained semithin transverse section of mouse cervical spinal cord with annotations of white matter tracts based on Watson and Harrison (2012). White matter tracts counted in this study are delineated with solid blue lines (DC, LC, VC). (B) Representative images from wt and Sarm1 KO mice through the CST 3 days after injury. (C) Sketches of main types of pathological profiles unequivocally visualized on semithin material in this study. (D) Pathological profile densities (number of pathological profiles per mm2) in the lower cervical spinal cord (C6–7) 3 and 7 days after IA-TBI, in WT and SARM1 KO mice. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. CST, corticospinal tract; Cu, Cuneatus; Gr, gracilis; LC, lateral column; lvs, lateral vestibulospinal tract; rbst, rubrospinal tract; rst-c, caudal reticulospinal tract; rst-v, ventral reticulospinal tract; VC, ventral column; vf, ventral funiculus. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
A 2-way between-groups ANOVA was used to examine the main effects and interactions between genotype (wt vs Sarm1 KO) and post- injury survival (3 days vs 7 days post injury) on total white matter pathology. There was a significant effect for genotype (F1,28 = 38.50, p < 0.0001), explaining 54% of the variation, but not for day (3 vs 7 days; F1,28 = 2.42, p = 0.13), or their interaction (F1,28 = 1.33, p = 0.25). Sarm1 deletion afforded robust protection and reduced the total pathological burden on both day 3 and day 7 post-injury by 71 ± 7%; (t28 = 4.59, p < 0.0001), and 75 ± 6% (t8 = 4.39; p = 0.002), respectively (Fig. 9 C–D). Analysis based on total pathology counts (instead of pathology densities) revealed virtually identical effects (not shown).
We then attempted to assess differences in pathological burden and possible differential effects of SARM1 deletion across white matter tracts separated by location in the ventral, lateral, and dorsal column. Further sub-regional analysis of individual white matter tracts in the ventral and lateral columns is difficult without the use of tracing methods. However, the dorsal column can be further subdivided in the CST which courses as a bundle of densely packed small axons in the lower part of the dorsal column and the ascending fasciculi (combined Cu and Gr) based on their distinct topography and large caliber of axons (Fig. 9A).
The density of pathological axons (pathological profiles per mm2) was highly correlated among different regions (Supplementary Fig. 3). At both 3 and 7 days post injury the CST had the highest density of pathological profiles, perhaps associated with the small calibers and hence higher packing density of axons: on average, pathology in the CST is 4, 3.5 and 2.3 times higher compared to the lateral column (p = 0.033), ventral column (p = 0.009) and the Cu/Gr (p = 0.006). Sarm1 deletion substantially reduced pathology in all regions studied (Fig. 9 D). At 7 days post-injury, there was nearly 90% reduction in pathology in the CST, 75–80% in the ventral and lateral columns, and approximately 60% in the Cu/Gr (Supplemental Fig. 4). One-way ANOVA demonstrated that the effect of Sarm1 deletion was variable among regions (F3,28 = 5.36; p = 0.005); primarily because of the difference between the CST and the Cu/Gr (t28 = 3.92, p = 0.003; Supplementary Fig. 4). Τo further validate that our counts on semithin sections also reflect the status of axons at the ultrastructural level, we counted pathological axonal and myelin profiles on EM material on day-7 animals (n =7) with a focus on the CST that is relatively easy to delineate. We found that Sarm1 deletion leads to reductions in both excess myelin figures (by 81%; t3.8 = 3.0, p = 0.04) and degenerated axon profiles (by 87%; t4.3 = 2.45, one-sided p = 0.03).
To determine whether Sarm1 deletion preserves the number of normal axons, we counted intact myelinated axons in the CST by stereology (Fig. 10). At baseline, wt and Sarm1 KO mice were estimated to have 28,749 ± 3473 and 30,253 ± 3599 axons (t12 = 0.8, p = 0.4), of which 97–8% ± 2% were assessed as apparently intact. In wt mice, IA- TBI leads to a loss of 22% of intact axons by day 7 (t12 = 3.33, p = 0.006) while Sarm1 KO mice show no loss of intact axons (t14 = 0.91, p = 0.4) and significant protection relative to wt mice (t13.9 = 3.8 p = 0.002) (Fig. 10 A). As we used non-complimentary criteria for intact axons and pathological profiles, in injured cases we also identified a population of axons with minor alterations (e.g. myelin thinning or thickening, or axons with mild metachromatic staining of the cytoplasm) that didn’t meet criteria for either group, termed here as equivocal profiles (Fig. 10). Although, there was a significant increase in these profiles compared to sham-injured animals (t6.2 = 2.8, p = 0.03), there was no significant difference in the number of equivocal axons between the two genotypes after injury (Fig. 10).
Fig. 10.

Stereological analysis of apparently intact, pathological and equivocal axonal profiles in the corticospinal tract of wt and Sarm1 KO mice 7 days after injury. Values are expressed relative to the total axon counts of sham animals for each genotype. Sarm1 knockout leads to significant protection of intact axons (t13.9 = 3.8 p = 0.002) and significant reduction in pathological profiles (t8 = 4.39; p = 0.002) compared to injured wt mice.
3.4. Sarm1 deletion ameliorates neuroinflammation associated with traumatic axonopathy
Based on the finding that Sarm1 deletion has a profound effect on the degeneration of axons in the first 7 days after injury, we hypothesized that the reduced pathological burden would be associated with reduced neuroinflammation. To test this hypothesis, we performed immunohistochemistry for the microglial marker IBA1 and assessed changes in the area covered by IBA1 immunoreactivity in the CST after TBI, relative to sham-injured controls, separately for each genotype (Fig. 11). We found that in wt animals, IA-TBI leads to 40% increase in IBA1(+) immunoreactivity (t8 = 2.90, p = 0.018). In contrast, there is no significant change in injured Sarm1 KO mice from baseline.
Fig. 11.

Sarm1 knockout ameliorates TBI-induced microglial reactivity in the corticospinal tract. (A) Representative confocal images of IBA1 immunoreactivity in the CST of wt animals 7 days after TBI and sham injury. (B) Changes in the areal density of IBA1 immunoreactivity in the corticospinal tract of wt and Sarm1 KO mice after TBI compared to sham-injured mice for each genotype. In wt, but not Sarm1 KO mice, there is significant increase in IBA1 immunoreactivity after TBI, t8 = 2.90, p = 0.018 (*).
4. Discussion
Our findings demonstrate the presence of traumatic axonopathy in ascending and descending tracts of the spinal cord in a well-established model of impact-acceleration traumatic brain injury, and the attenuating effect of Sarm1 deletion in axonal degeneration. Besides extending our previous observations on acute traumatic axonopathy in the CST (Ziogas and Koliatsos, 2018) to later time points, we explore morphological features of axonopathy and dependency on SARM1 signaling in additional long-axon tracts, by leveraging the unique advantage of the spinal cord where several ascending and descending tracts conveniently cluster in a small region. This anatomical environment allows the exploration of signatures of traumatic axonopathy across diverse groups of long axons. Our findings show that traumatic axonopathy shares morphological features with peripheral neuropathy and other types of axonopathies, including axonal dystrophy encountered in neurodegenerative diseases of the CNS. Some of these morphological features and the dependency of traumatic axonopathy on SARM1 suggest the involvement of a WD program that can be amenable to genetic and, in the future, pharmacological interventions.
4.1. Morphological features of TAI-associated axonopathy
Traumatic (diffuse) axonal injury and the associated axonopathy is a common neuropathology in various types of TBI across the severity spectrum (Strich, 1956; Adams et al., 1984; Blumbergs et al., 1989; Blumbergs et al., 1994; Mittl et al., 1994; Koliatsos et al., 2011). The study of white matter integrity in various models of TBI including impact acceleration (Foda and Marmarou, 1994; Povlishock et al., 1997; Xu et al., 2016; Ziogas and Koliatsos, 2018; Welsbie et al., 2019) closed head impact (Marion et al., 2018; Marion et al., 2019; Maynard et al., 2020), fluid percussion injury (Lifshitz et al., 2007; Wang et al., 2011; Eakin et al., 2015), optic nerve stretch (Maxwell et al., 1991; Maxwell and Graham, 1997; Maxwell et al., 2015) and blast injury (Koliatsos et al., 2011) has been the focus of several investigations. Previous studies employing electron microscopy in some of these models have revealed some key features of axoplasmic and myelin pathology (Maxwell and Graham, 1997; Povlishock et al., 1997; Wang et al., 2011; Maxwell et al., 2015; Mierzwa et al., 2015; Marion et al., 2018) and, in the present study, we have confirmed and extended several of these observations.
We have previously shown that, in the IA-TBI model of TAI, there is axonal dystrophy and some fragmentation in descending tracts such as corticospinal and reticulospinal and ascending tracts such as the gracilis in the brain stem acutely within 24 h after injury (Ziogas and Koliatsos, 2018). In the current study, we have further characterized the sequelae of the injury on ascending and descending white matter tracts that link the brain stem with the spinal cord, the bulk of which is exactly the same as shown in our previous study (Ziogas and Koliatsos, 2018). The distribution of the biomechanical disruption in lower brain stem-upper cervical cord is explainable by the translational and rotational acceleration of the head onto and away from the foam pad and the relative laxity of the upper body. While we cannot exclude some injury in lower cervical cord, we believe that the bulk of axonopathy is due to secondary degeneration from the above biomechanical disruption.
With respect to pathological changes in the axoplasm and axon proper, at the ultrastructural level, we identified a spectrum of profiles that range from focal perturbations of the axoskeleton to complete axonal dissolution and the formation of ovoids. The first set of observations which involves localized axoskeletal disorganization with loss of microtubules and/or compaction of neurofilaments, is not classically associated with WD, but is common in models of TBI (Jafari et al., 1998). The evolution of these cytoskeletal changes has been better characterized in vitro in response to biomechanical perturbations including axonal stretch or compression (Tang-Schomer et al., 2010; Fournier et al., 2015). The second set of observations includes profiles of axons with evidence of transport arrest (i.e. accumulation of organelles and dense bodies), condensation or dissolution of the cytoskeleton, formation of periodic axon constrictions and ultimately the formation of ovoids. Such profiles have been observed in both models of TAI (Maxwell and Graham, 1997; Povlishock et al., 1997; Beirowski et al., 2010) and in classical WD in models of axotomy in the PNS (Ghabriel and Allt, 1979; Beirowski et al., 2005; Jung et al., 2011; Catenaccio et al., 2017) and CNS (Maxwell et al., 1991; Maxwell and Graham, 1997; Narciso et al., 2001; Beirowski et al., 2010).
Some changes involving the myelin sheath of axons, e.g. separation or loosening of lamellae, have been previously observed in traumatic axonopathy acutely after injury (Maxwell, 2013) and in the course of WD (Ghabriel and Allt, 1979). Accumulation of cytoplasmic material between myelin lamellae has not been reported before; we speculate that it may indicate dysfunction of the myelinic channel system (see below) or degeneration of the oligodendrocyte process. Pathological configurations of normal-looking compact myelin were also very common in our material, as either collapsed myelin or excess myelin figures. Collapsed myelin figures, i.e. myelin sheaths that lack ensheathed axons, are classically encountered in WD (Franson and Ronnevi, 1989; George and Griffin, 1994) and are thought to represent the sheaths of degenerated axons before the end stage of myelin bodies (Franson and Ronnevi, 1989; George and Griffin, 1994; Armstrong et al., 2016). In contrast, excess (or redundant) myelin figures present either as myelin outfoldings away from the ensheathed axon or as multiple myelin wrappings around the axon resulting in focal thickenings (tomacula). In these profiles the axon often appears normal. Excess myelin profiles have been previously characterized in a closed-head impact model of TBI (Marion et al., 2019), as well as in experimental autoimmune encephalomyelitis (Bando et al., 2015). Such profiles are also encountered in chronic neuropathies, including hereditary neuropathy with liability to pressure palsies (also known as tomaculous neuropathy), demyelinating neuropathies, and hereditary motor-sensory neuropathies.(Madrid and Bradley, 1975; Bilbao and Schmidt, 2014) Excess myelin figures are also encountered during development (Cullen and Webster, 1979; Djannatian et al., 2021) and can also be observed after genetic perturbation of the axoglial adhesion and myelination apparatus e.g. with mutations of the myelin-associated glycoprotein, or ablation of Cdc42, Rac1 or N-Wasp genes (Fujita et al., 1998; Thurnherr et al., 2006; Katanov et al., 2020). Whether excess myelin figures are a consequence of a disruption in axoglial signaling due to the circumstances of the injury or an aberrant repair mechanism is not known, but this type of pathological signature suggests the engagement of an active myelination process.
Besides the formation of excess myelin figures, a number of subtle morphological observations in our material may also suggest an active engagement of the ensheathing oligodendrocyte. One piece of evidence is the increase in the size of the inner tongue. This may represent benign cytoplasm edema, degenerative changes, or reflect a repair process initiated by the oligodendrocyte. Another observation is the transient increase in inner tongue evaginations, some of which appear to contain degenerative material. Although inner tongue evaginations are sometimes seen next to paranodes or incisures of large myelinated axons in naïve animals (Spencer and Thomas, 1974), they are more extensive in clinical and experimental neuropathies or in the proximal stumps of transected axons (Spencer and Thomas, 1974; Bilbao and Schmidt, 2014). As far as we know this is the first report of a similar process in traumatic axonopathy. Some of these morphologies may represent a mechanism of selective phagocytosis of pathological axon segments by the ensheathing glia (Spencer and Thomas, 1974; Gatzinsky, 1996; Bilbao and Schmidt, 2014) and, indeed, in our material they were frequently encountered next to focal degenerative changes of the axoplasm.
The morphological features of traumatic axonopathy discussed in the previous paragraphs reveal a wide range of pathological profiles that involve the axonal, myelin sheath and axon-glial domains. Many of these profiles have signatures observed in classical models of WD (axotomy), while there are also several features more typical of TAI-induced axonopathy (e.g. focal axoskeletal and myelin perturbations) or chronic neuropathies (e.g. excess myelin figures and axon-glial networks). The previous changes are not only featured by a remarkable diversity, but also a wide range of pathological severity, some of which may be quite segmental. The variable extent and degree of cytoskeletal perturbations and their apparent presence in both axons that seem to retain viability and in degenerating axons may reflect not only the variable effect of the biomechanical impact of the initial injury on individual axons, but also the variable engagement of WD pathways. Oligodendrocytes appear to be substantially engaged in the axonopathic process, as manifested by evidence of remyelination and also changes in the oligodendrocyte cytoplasm attached to the axon, some of which may reflect the phagocytosis of axonal material. Whether oligodendrocytes may also play an active role in axonal fragmentation, as proposed for Schwann cells during WD in the PNS (Jung et al., 2011; Catenaccio et al., 2017) is a question that deserves further investigation.
4.2. SARM1 dependency of traumatic axonopathy across spinal cord tracts
SARM1 dependency is a key feature in the current molecular understanding of WD that, although not an exclusive property(Simon and Watkins, 2018) appears to be a specific feature of Wallerian breakdown of the axon (Conforti et al., 2014; Gerdts et al., 2016; Hill et al., 2016; Koliatsos and Alexandris, 2019; Coleman and Hoke, 2020). Indeed, in the current study, the presence or absence of the Sarm1 gene explains 50% of the observed variance in axonal and myelin pathology after IA- TBI, and its deletion reduces pathology across white matter tracts, at both 3 and 7 days after injury, by up to 90%. Sarm1 deletion also preserved the integrity of axons (apparently intact axons) to virtually pre- injury levels and suppressed the secondary neuroinflammatory response. These findings are consistent with the protective effect of Sarm1 deletion or the presence of the related Wallerian degeneration slow (WldS) mutation in other models of TBI (Henninger et al., 2016; Yin et al., 2016; Ziogas and Koliatsos, 2018; Marion et al., 2019; Maynard et al., 2020; Bradshaw et al., 2021).
By studying multiple white matter tracts side by side, we have also identified a differential efficacy of Sarm1 deletion, as exemplified by the 1.5 times higher reduction in pathology in the CST compared to the Gr/Cu fasciculi. One potential explanation is that at the plane of our observations, the descending CST contains predominantly distal axon segments with respect the neuronal soma and injury, whereas the ascending Gr/Cu fasciculi contain mostly proximal axon segments, and as such would indicate that SARM1 has a predominate action within the distal axon. However, other explanations may also include differences in the mechanical or vascular composition of the disruption, and differences in axon caliber or vulnerability of the different neuronal populations. Whether a methodological epiphenomenon or an interesting feature in the biological response of different axon tracts to injury, this type of variance should be taken into account when it comes to the design and assessment of therapeutic interventions.
Although SARM1 signaling in the execution of WD is known to involve intrinsic axonal processes and the primary prevention of axonopathy by Sarm1 deletion would therefore be specific to the axon, the observed reduction in pathological burden in our study involves both axoplasmic and myelin abnormalities. This protective effect of Sarm1 deletion on myelin pathology has also been reported in another model of single closed head injury as a reduction in TBI associated excess myelin figures and putative demyelinated axon profiles (Marion et al., 2019; Bradshaw et al., 2021). In light of the apparent lack of a role of SARM1 in oligodendrocytes we hypothesize that, in the context of IA-TBI, most of the observed myelin pathology, particularly collapsed myelin figures, ovoids and myelin bodies, is secondary to injury signals transmitted to the oligodendrocyte from the injured axon or secondary to axonal degeneration and, therefore, amenable to SARM1 deletion. Future studies with neuronal- (or oligodendrocyte-) targeted conditional deletion of Sarm1 may further elucidate the specific role of SARM1 in driving oligodendrocyte pathology after TBI.
Our study has certain limitations that need to be taken into account for the interpretation of our findings. Although the Sarm1 KO mice used in the present study have been backcrossed for >10 generations to the C57BL/6 J strain, recent genetic analysis revealed that they may have genetic variations in neighboring genes belonging to the original 129 strain (Uccellini et al., 2020). Nonetheless, the specific effect of Sarm1 deletion on axon degeneration in vivo has also been confirmed by CRISPR/Cas9 editing (Uccellini et al., 2020) and the expression of dominant negative constructs (Geisler et al., 2019). Moreover, here we used the congenic C57BL/6 J mice as wild type controls, which were of the same background but not littermates with the Sarm1 KO mice. Therefore we have not controlled for litter effects. However, the protective effect of Sarm1 interference on axonal and myelin pathology after TBI has also been demonstrated in experiments comparing Sarm1 KO with wild-type littermates (Bradshaw et al., 2021).
While these results indicate robust protection of Sarm1 deletion against TAI-induced degeneration, and are consistent with suppression of behavioral deficits in a pellet reaching task after IA-TBI in Sarm1 KO mice (Koliatsos et al., unpublished observations), we cannot extrapolate on the long-term effect of the protective effect or its role in preserving neuronal connectivity and function. A couple of recent studies, however, indicate that ablation of Sarm1, may indeed preserve axonal integrity 1.5 months after single closed head injury (Bradshaw et al., 2021) and ameliorate pathology and behavioral deficits 6 months after repeated mild closed head injury (Maynard et al., 2020). Finally, in this study we have not investigated the effect of severity and number of injuries, or that of age and sex, which warrant further assessment.
The dependence of traumatic axonopathy on SARM1 signaling has an important translational value in view of recent progress in the development of pharmacological agents to inhibit SARM1 (Liu et al., 2018; Loring et al., 2020; Hughes et al., 2021; Alexandris et al., 2022a; Bratkowski et al., 2022; Feldman et al., 2022). In contrast to the all-or-none nature of experimental axotomy, the biomechanical disruption of the axon in the context of TAI may be related to varying degrees of damage, from acute disconnection to partial or small axonal perturbations, all of which may trigger WD and breakdown (Koliatsos and Alexandris, 2019). Indeed, in the present study we routinely encountered injured axons with only focal lesions and intact myelin sheaths. In this case, SARM1 inhibition would allow the recovery of otherwise healthy, rescuable axons. Apart from the primary effect of SARM1 inhibition on the survival of injured axons, any reduction of the pathological burden associated with WD may be by itself beneficial due to the amelioration of WD-related neuroinflammation and possible secondary neurotoxicity (Gaudet et al., 2011). These are important issues that should be addressed in future studies.
Supplementary Material
Acknowledgements
We would like to thank Mr. Norman Barker, director of the Pathology Photography and Graphic Arts Laboratory (JHU), for his help in the acquisition of the brightfield micrographs, and Mr. Joseph Belamarich (JHU) for his help with stereology. We would also like to thank Mr. Mike Delannoy (JHU) for his help in developing the EM fixation protocol.
Funding
V.E.K received funding from the National Eye Institute (RO1EY028039), the National Institute of Neurological Disorders and Stroke (R01NS114397), the Department of Defense (W81XWH-14- 0396), and from generous gifts from the Kate Sidran Family foundation. In all cases, the funding agencies were not involved in the acquisition, analysis, interpretation and/or presentation/reporting of data. This work also received support from National Institutes of Health core grant 5P30EY001765.
Abbreviations:
- CNS
Central nervous system
- CST
Corticospinal tract
- DC
Dorsal column
- Gr/Cu
Gracilis and cuneatus fasciculi
- IA
Impact acceleration
- LC
Lateral column
- SARM1
Sterile Alpha and TIR Motif Containing 1
- TAI
Traumatic axonal injury
- TBI
Traumatic brain injury
- VC
Ventral column
- WD
Wallerian degeneration
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.expneurol.2022.114252.
Declaration of Competing Interest
None.
Data availability
Data will be made available on request.
References
- Adams JH, Doyle D, Graham DI, Lawrence AE, McLellan DR, 1984. Diffuse axonal injury in head injuries caused by a fall. Lancet 2 (8417–8418), 1420–1422. [DOI] [PubMed] [Google Scholar]
- Alexandris AS, Ryu J, Rajbhandari L, Harlan R, McKenney J, Wang Y, Aja S, Graham D, Venkatesan A, Koliatsos VE, 2022a. Protective effects of NAMPT or MAPK inhibitors and NaR on Wallerian degeneration of mammalian axons. Neurobiol. Dis. 171, 105808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandris AS, Wang Y, Frangakis CE, Lee Y, Ryu J, Alam Z, Koliatsos VE, 2022b. Long-term changes in axon calibers after injury: observations on the mouse corticospinal tract. Int. J. Mol. Sci. 23 (13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RC, Mierzwa AJ, Marion CM, Sullivan GM, 2016. White matter involvement after TBI: clues to axon and myelin repair capacity. Exp. Neurol. 275 (Pt 3), 328–333. [DOI] [PubMed] [Google Scholar]
- Bando Y, Nomura T, Bochimoto H, Murakami K, Tanaka T, Watanabe T, Yoshida S, 2015. Abnormal morphology of myelin and axon pathology in murine models of multiple sclerosis. Neurochem. Int. 81, 16–27. [DOI] [PubMed] [Google Scholar]
- Beirowski B, Adalbert R, Wagner D, Grumme DS, Addicks K, Ribchester RR, Coleman MP, 2005. The progressive nature of Wallerian degeneration in wild-type and slow Wallerian degeneration (WldS) nerves. BMC Neurosci. 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Babetto E, Coleman MP, Martin KR, 2008. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur. J. Neurosci. 28 (6), 1166–1179. [DOI] [PubMed] [Google Scholar]
- Beirowski B, Nogradi A, Babetto E, Garcia-Alias G, Coleman MP, 2010. Mechanisms of axonal spheroid formation in central nervous system Wallerian degeneration. J. Neuropathol. Exp. Neurol. 69 (5), 455–472. [DOI] [PubMed] [Google Scholar]
- Bilbao JM, Schmidt RE, 2014. Biopsy Diagnosis of Peripheral Neuropathy. Springer. [Google Scholar]
- Blumbergs PC, Jones NR, North JB, 1989. Diffuse axonal injury in head trauma. J. Neurol. Neurosurg. Psychiatry 52, 838–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, Mclean AJ, 1994. Staining of amyloid precursor protein to study axonal damage in mild head- injury. Lancet 344 (8929), 1055–1056. [DOI] [PubMed] [Google Scholar]
- Bradshaw DV, Knutsen AK, Korotcov A, Sullivan GM, Radomski KL, Dardzinski BJ, Zi X, McDaniel DP, Armstrong RC, 2021. Genetic inactivation of SARM1 axon degeneration pathway improves outcome trajectory after experimental traumatic brain injury based on pathological, radiological, and functional measures. Acta Neuropathol. Commun. 9 (1), 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratkowski M, Burdett TC, Danao J, Wang X, Mathur P, Gu W, Beckstead JA, Talreja S, Yang YS, Danko G, Park JH, Walton M, Brown SP, Tegley CM, Joseph PRB, Reynolds CH, Sambashivan S, 2022. Uncompetitive, adduct-forming SARM1 inhibitors are neuroprotective in preclinical models of nerve injury and disease. Neuron. In press. [DOI] [PubMed] [Google Scholar]
- Catenaccio A, Llavero Hurtado M, Diaz P, Lamont DJ, Wishart TM, Court FA, 2017. Molecular analysis of axonal-intrinsic and glial-associated co-regulation of axon degeneration. Cell Death Dis. 8 (11), e3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MP, Hoke A, 2020. Programmed axon degeneration: from mouse to mechanism to medicine. Nat. Rev. Neurosci. 21 (4), 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Gilley J, Coleman MP, 2014. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 15 (6), 394–409. [DOI] [PubMed] [Google Scholar]
- Cullen MJ, Webster HD, 1979. Remodelling of optic nerve myelin sheaths and axons during metamorphosis in Xenopus laevis. J. Comp. Neurol. 184 (2), 353–362. [DOI] [PubMed] [Google Scholar]
- de Olmos JS, Beltramino CA, de Olmos De Lorenzo S, 1994. Use of an amino-cupric-silver technique for the detection of early and semiacute neuronal degeneration caused by neurotoxicants, hypoxia, and physical trauma. Neurotoxicol. Teratol. 16 (6), 545–561. [DOI] [PubMed] [Google Scholar]
- Desbois M, Crawley O, Evans PR, Baker ST, Masuho I, Yasuda R, Grill B, 2018. PAM forms an atypical SCF ubiquitin ligase complex that ubiquitinates and degrades NMNAT2. J. Biol. Chem. 293 (36), 13897–13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djannatian M, Weikert U, Safaiyan S, Wrede C, Deichsel C, Kislinger G, Ruhwedel T, Campbell DS, van Ham T, Schmid B, Hegermann J, Mobius W, ¨ Schifferer M, Simons M, 2021. Myelin biogenesis is associated with pathological ultrastructure that is resolved by microglia during development (Pre-Print). bioRxiv. 10.1101/2021.02.02.429485. [DOI] [Google Scholar]
- Eakin K, Rowe RK, Lifshitz J, 2015. Modeling Fluid Percussion Injury: Relevance to Human Traumatic Brain Injury. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Kobeissy FH, Boca Raton: (FL). [PubMed] [Google Scholar]
- Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J, 2017. The SARM1 toll/interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron 93 (6), 1334–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman HC, Merlini E, Guijas C, DeMeester KE, Njomen E, Kozina EM, Yokoyama M, Vinogradova E, Reardon HT, Melillo B, Schreiber SL, Loreto A, Blankman JL, Cravatt BF, 2022. Selective inhibitors of SARM1 targeting an allosteric cysteine in the autoregulatory ARM domain. Proc. Natl. Acad. Sci. U. S. A. 119 (35), e2208457119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foda MAA, Marmarou A, 1994. A new model of diffuse brain injury in rats. 2. Morphological characterization. J. Neurosurg. 80 (2), 301–313. [DOI] [PubMed] [Google Scholar]
- Fournier AJ, Hogan JD, Rajbhandari L, Shrestha S, Venkatesan A, Ramesh KT, 2015. Changes in neurofilament and microtubule distribution following focal axon compression. PLoS One 10 (6), e0131617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franson P, Ronnevi LO, 1989. Myelin breakdown in the posterior funiculus of the kitten after dorsal rhizotomy. A qualitative and quantitative light and electron microscopic study. Anat. Embryol. (Berl.) 180 (3), 273–280. [DOI] [PubMed] [Google Scholar]
- Fujita N, Kemper A, Dupree J, Nakayasu H, Bartsch U, Schachner M, Maeda N, Suzuki K, Popko B, 1998. The cytoplasmic domain of the large myelin-associated glycoprotein isoform is needed for proper CNS but not peripheral nervous system myelination. J. Neurosci. 18 (6), 1970–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatzinsky KP, 1996. Node-paranode regions as local degradative centres in alpha-motor axons. Microsc. Res. Tech. 34 (6), 492–506. [DOI] [PubMed] [Google Scholar]
- Gaudet AD, Popovich PG, Ramer MS, 2011. Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J. Neuroinflammation 8, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Doan RA, Strickland A, Huang X, Milbrandt J, DiAntonio A, 2016. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 139 (Pt 12), 3092–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Huang SX, Strickland A, Doan RA, Summers DW, Mao X, Park J, DiAntonio A, Milbrandt J, 2019. Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J. Exp. Med. 216 (2), 294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman SM, Roberts GW, Gennarelli TA, Maxwell WL, Adams JH, Kerr S, Graham DI, 1995. Axonal injury: a universal consequence of fatal closed head injury? Acta Neuropathol. 89 (6), 537–543. [DOI] [PubMed] [Google Scholar]
- George R, Griffin JW, 1994. Delayed macrophage responses and myelin clearance during Wallerian degeneration in the central nervous system: the dorsal radiculotomy model. Exp. Neurol. 129 (2), 225–236. [DOI] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Sasaki Y, DiAntonio A, Milbrandt J, 2013. Sarm1- mediated axon degeneration requires both SAM and TIR interactions. J. Neurosci. 33 (33), 13569–13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Milbrandt J, DiAntonio A, 2016. Axon self-destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron 89 (3), 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghabriel MN, Allt G, 1979. The role of Schmidt-Lanterman incisures in Wallerian degeneration. II. An electron microscopic study. Acta Neuropathol. 48 (2), 95–103. [DOI] [PubMed] [Google Scholar]
- Gilley J, Coleman MP, 2010. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 8 (1), e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henninger N, Bouley J, Sikoglu EM, An J, Moore CM, King JA, Bowser R, Freeman MR, Brown RH Jr., 2016. Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1. Brain 139 (Pt 4), 1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CS, Coleman MP, Menon DK, 2016. Traumatic axonal injury: mechanisms and translational opportunities. Trends Neurosci. 39 (5), 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Soto I, Libby RT, John SW, 2013. Intrinsic axonal degeneration pathways are critical for glaucomatous damage. Exp. Neurol. 246, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RO, Bosanac T, Mao X, Engber TM, DiAntonio A, Milbrandt J, Devraj R, Krauss R, 2021. Small molecule SARM1 inhibitors recapitulate the SARM1(− /− ) phenotype and allow recovery of a metastable pool of axons fated to degenerate. Cell Rep. 34 (1), 108588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari SS, Nielson M, Graham DI, Maxwell WL, 1998. Axonal cytoskeletal changes after nondisruptive axonal injury. II. Intermediate sized axons. J. Neurotrauma 15 (11), 955–966. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH, 2013. Axonal pathology in traumatic brain injury. Exp. Neurol. 246, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Cai W, Lee HK, Pellegatta M, Shin YK, Jang SY, Suh DJ, Wrabetz L, Feltri ML, Park HT, 2011. Actin polymerization is essential for myelin sheath fragmentation during Wallerian degeneration. J. Neurosci. 31 (6), 2009–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser T, Allen HM, Kwon O, Barak B, Wang J, He Z, Jiang M, Feng G, 2021. MyelTracer: a semi-automated software for myelin g-ratio quantification. eNeuro 8 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katanov C, Novak N, Vainshtein A, Golani O, Dupree JL, Peles E, 2020. N-Wasp regulates oligodendrocyte myelination. J. Neurosci. 40 (32), 6103–6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliatsos VE, Alexandris AS, 2019. Wallerian degeneration as a therapeutic target in traumatic brain injury. Curr. Opin. Neurol. 32 (6), 786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliatsos VE, Cernak I, Xu L, Song Y, Savonenko A, Crain BJ, Eberhart CG, Frangakis CE, Melnikova T, Kim H, Lee D, 2011. A mouse model of blast injury to brain: initial pathological, neuropathological, and behavioral characterization. J. Neuropathol. Exp. Neurol. 70 (5), 399–416. [DOI] [PubMed] [Google Scholar]
- Lifshitz J, Kelley BJ, Povlishock JT, 2007. Perisomatic thalamic axotomy after diffuse traumatic brain injury is associated with atrophy rather than cell death. J. Neuropathol. Exp. Neurol. 66 (3), 218–229. [DOI] [PubMed] [Google Scholar]
- Liu HW, Smith CB, Schmidt MS, Cambronne XA, Cohen MS, Migaud ME, Brenner C, Goodman RH, 2018. Pharmacological bypass of NAD(+) salvage pathway protects neurons from chemotherapy-induced degeneration. Proc. Natl. Acad. Sci. U. S. A. 115 (42), 10654–10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loreto A, Hill CS, Hewitt VL, Orsomando G, Angeletti C, Gilley J, Lucci C, Sanchez-Martinez A, Whitworth AJ, Conforti L, Dajas-Bailador F, Coleman MP, 2020. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol. Dis. 134, 104678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loring HS, Parelkar SS, Mondal S, Thompson PR, 2020. Identification of the first noncompetitive SARM1 inhibitors. Bioorg. Med. Chem. 28 (18), 115644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid R, Bradley WG, 1975. The pathology of neuropathies with focal thickening of the myelin sheath (tomaculous neuropathy): studies on the formation of the abnormal myelin sheath. J. Neurol. Sci. 25 (4), 415–448. [Google Scholar]
- Marion CM, Radomski KL, Cramer NP, Galdzicki Z, Armstrong RC, 2018. Experimental traumatic brain injury identifies distinct early and late phase axonal conduction deficits of white matter pathophysiology, and reveals intervening recovery. J. Neurosci. 38 (41), 8723–8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion CM, McDaniel DP, Armstrong RC, 2019. Sarm1 deletion reduces axon damage, demyelination, and white matter atrophy after experimental traumatic brain injury. Exp. Neurol. 321, 113040. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, 2013. Damage to myelin and oligodendrocytes: a role in chronic outcomes following traumatic brain injury? Brain Sci. 3 (3), 1374–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell WL, Graham DI, 1997. Loss of axonal microtubules and neurofilaments after stretch-injury to guinea pig optic nerve fibers. J. Neurotrauma 14 (9), 603–614. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, Irvine A, Graham JH, Adams TA, Gennarelli R. Tipperman, Sturatis M, 1991. Focal axonal injury: the early axonal response to stretch. J. Neurocytol. 20 (3), 157–164. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, Povlishock JT, Graham DL, 1997. A mechanistic analysis of nondisruptive axonal injury: a review. J. Neurotrauma 14 (7), 419–440. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, Bartlett E, Morgan H, 2015. Wallerian degeneration in the optic nerve stretch-injury model of traumatic brain injury: a stereological analysis. J. Neurotrauma 32 (11), 780–790. [DOI] [PubMed] [Google Scholar]
- Maynard ME, Redell JB, Zhao J, Hood KN, Vita SM, Kobori N, Dash PK, 2020. Sarm1 loss reduces axonal damage and improves cognitive outcome after repetitive mild closed head injury. Exp. Neurol. 327, 113207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierzwa AJ, Marion CM, Sullivan GM, McDaniel DP, Armstrong RC, 2015. Components of myelin damage and repair in the progression of white matter pathology after mild traumatic brain injury. J. Neuropathol. Exp. Neurol. 74 (3), 218–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittl RL, Grossman RI, Hiehle JF, Hurst RW, Kauder DR, Gennarelli TA, Alburger GW, 1994. Prevalence of MR evidence of diffuse axonal injury in patients with mild head injury and normal head CT findings. AJNR Am. J. Neuroradiol. 15 (8), 1583–1589. [PMC free article] [PubMed] [Google Scholar]
- Narciso MS, Hokoc JN, Martinez AM, 2001. Watery and dark axons in Wallerian degeneration of the opossum’s optic nerve: different patterns of cytoskeletal breakdown? An. Acad. Bras. Cienc. 73 (2), 231–243. [DOI] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH Jr., Conforti L, Coleman M, Tessier-Lavigne M, Zuchner S, Freeman MR, 2012. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337 (6093), 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povlishock JT, Christman CW, 1995. The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J. Neurotrauma 12 (4), 555–564. [DOI] [PubMed] [Google Scholar]
- Povlishock JT, Marmarou A, McIntosh T, Trojanowski JQ, Moroi J, 1997. Impact acceleration injury in the rat: evidence for focal axolemmal change and related neurofilament sidearm alteration. J. Neuropathol. Exp. Neurol. 56 (4), 347–359. [PubMed] [Google Scholar]
- Simon DJ, Watkins TA, 2018. Therapeutic opportunities and pitfalls in the treatment of axon degeneration. Curr. Opin. Neurol. 31 (6), 693–701. [DOI] [PubMed] [Google Scholar]
- Spencer PS, Thomas PK, 1974. Ultrastructural studies of the dying-back process. II. The sequestration and removal by Schwann cells and oligodendrocytes of organelles from normal and diseases axons. J. Neurocytol. 3 (6), 763–783. [DOI] [PubMed] [Google Scholar]
- Strich SJ, 1956. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J. Neurol. Neurosurg. Psychiatry 19, 163–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers DW, Milbrandt J, DiAntonio A, 2018. Palmitoylation enables MAPK- dependent proteostasis of axon survival factors. Proc. Natl. Acad. Sci. U. S. A. 115 (37), E8746–E8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szretter KJ, Samuel MA, Gilfillan S, Fuchs A, Colonna M, Diamond MS, 2009. The immune adaptor molecule SARM modulates tumor necrosis factor alpha production and microglia activation in the brainstem and restricts West Nile Virus pathogenesis. J. Virol. 83 (18), 9329–9338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang-Schomer MD, Patel AR, Baas PW, Smith DH, 2010. Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J. 24 (5), 1401–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurnherr T, Benninger Y, Wu X, Chrostek A, Krause SM, Nave K-A, Franklin RJM, Brakebusch C, Suter U, Relvas JB, 2006. Cdc42 and Rac1 signaling are both required for and act synergistically in the correct formation of myelin sheaths in the CNS. J. Neurosci. 26 (40), 10110–10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkiew E, Falconer D, Reed N, Hoke A, 2017. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J. Peripher. Nerv. Syst. 22 (3), 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uccellini MB, Bardina SV, Sánchez-Aparicio MT, White KM, Hou YJ, Lim JK, García-Sastre A, 2020. Passenger mutations confound phenotypes of SARM1- deficient mice. Cell Rep. 31 (1), 107498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller A, 1850. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog and observations of the alterations produced thereby in the structure of their primitive fibers. Philos. Trans. R. Soc. Lond. Ser. B 140, 423–429. [Google Scholar]
- Wang MS, Davis AA, Culver DG, Glass JD, 2002. WldS mice are resistant to paclitaxel (taxol) neuropathy. Ann. Neurol. 52 (4), 442–447. [DOI] [PubMed] [Google Scholar]
- Wang J, Hamm RJ, Povlishock JT, 2011. Traumatic axonal injury in the optic nerve: evidence for axonal swelling, disconnection, dieback, and reorganization. J. Neurotrauma 28 (7), 1185–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson C, Harrison M, 2012. The location of the major ascending and descending spinal cord tracts in all spinal cord segments in the mouse: actual and extrapolated. Anat. Rec. (Hoboken) 295 (10), 1692–1697. [DOI] [PubMed] [Google Scholar]
- Welsbie DS, Ziogas NK, Xu L, Kim BJ, Ge Y, Patel AK, Ryu J, Lehar M, Alexandris AS, Stewart N, Zack DJ, Koliatsos VE, 2019. Targeted disruption of dual leucine zipper kinase and leucine zipper kinase promotes neuronal survival in a model of diffuse traumatic brain injury. Mol. Neurodegener. 14 (1), 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Nguyen JV, Lehar M, Menon A, Rha E, Arena J, Ryu J, Marsh- Armstrong N, Marmarou CR, Koliatsos VE, 2016. Repetitive mild traumatic brain injury with impact acceleration in the mouse: multifocal axonopathy, neuroinflammation, and neurodegeneration in the visual system. Exp. Neurol. 275 (Pt 3), 436–449. [DOI] [PubMed] [Google Scholar]
- Yin TC, Voorhees JR, Genova RM, Davis KC, Madison AM, Britt JK, Cintron- Perez CJ, McDaniel L, Harper MM, Pieper AA, 2016. Acute axonal degeneration drives development of cognitive, motor, and visual deficits after blast- mediated traumatic brain injury in mice. eNeuro 3 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Zhang L, Sasaki Y, Milbrandt J, Gidday JM, 2013. Protection of mouse retinal ganglion cell axons and soma from glaucomatous and ischemic injury by cytoplasmic overexpression of Nmnat1. Invest. Ophthalmol. Vis. Sci. 54 (1), 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziogas NK, Koliatsos VE, 2018. Primary traumatic axonopathy in mice subjected to impact acceleration: a reappraisal of pathology and mechanisms with high-resolution anatomical methods. J. Neurosci. 38 (16), 4031–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.