

A. Rationale
Nearly all soft materials are based on organic polymer molecules. In other words they are derived from macromolecules constructed around the chemistry of carbon. Yet there are roughly 100 other elements in the periodic table that could in principle provide the building blocks for polymers and for soft materials. A few “inorganic” elements are incorporated into gels and related materials. Examples include silicate, aluminate, and titanate sol-gel substances, but the opportunities for diversification within these systems are relatively limited. The poly(organosiloxanes) (silicones) were the first hybrid inorganic-organic polymers to be discovered and they continue to play a major role in research and technology. Nevertheless, a need exists for additional systems that have the structural diversity found in the field of organic polymers, combined with the attributes of inorganic skeletal elements rather than carbon. It is with this objective in mind that we have devoted several decades to exploring the synthesis and diversification of the polyphosphazenes.
A. What are Polyphosphazenes?
Polyphosphazenes are hybrid inorganic-organic high polymers comprised of a backbone of alternating phosphorus and nitrogen atoms with two (usually organic) side groups linked to each phosphorus. Several hundred poly(organophosphazenes) are known with diverse side groups or combinations of different side groups, together with a range of alternative skeletal architectures that range from linear to cyclolinear, comb, star, dendritic, or block copolymeric.1 Examples are shown in Figure 1. The focus of this review is on linear, block copolymer, and dendritic species.
Figure 1.

Different poly(organophosphazene) architectures. The linear, block copolymers, and dendrimers are the main subject of this article. The other types are still undergoing development.
These polymers are unique in numerous ways, but especially in their mode of synthesis. Classical organic polymers are prepared by the addition polymerization of olefins or vinyl compounds or by various condensation or ring-opening reactions. For nearly all classical polymers the required side groups must be in place on the monomer before polymerization. After polymerization it is often difficult to modify or replace the side groups in classical polymers by substitution chemistry, mainly because these groups are usually chosen specifically for their lack of reactivity during polymerization. For example, it is not possible to chlorinate polyethylene to form poly(vinyl chloride) without extensive polymer chain cleavage, or to replace the chlorine atoms in poly(vinyl chloride) by other groups. Even when a side group modification is theoretically favorable, as in the case of poly(vinyl acetate), 100% hydrolysis to poly(vinyl alcohol) is difficult. Alternatively, any attempt to begin with a monomer that has reactive side groups suitable for later modification can interfere with the mechanism of polymerization and is thus avoided. Methods are being developed to circumvent this problem, for example in the synthesis of comb and brush structures, but this is still not a wide-ranging procedure in the field of classical organic polymers.
Most polyphosphazenes are different from their organic counterparts in the sense that the organic or organometallic side groups are introduced by macromolecular substitution reactions after the polymer chains have been assembled.2–4 The main method for the synthesis of most polyphosphazenes is illustrated in Figure 2. It is a process that involves an initial thermal ring-opening polymerization of the cyclic, totally inorganic precursor, hexachlorocyclotriphosphazene (1) (prepared from PCl5 and ammonium chloride), to high molecular weight poly(dichlorophosphazene) (2), followed by replacement of all the chlorine atoms along each chain using organic or organometallic nucleophiles (4-6). This approach is possible because of the very high reactivity of phosphorus-chlorine bonds. Thus, this is an almost unique process in which the structural diversity of the final polymers is achieved by use of the wide variety of existing nucleophiles (more that 250 at the present count) and combinations of two or more different nucleophiles that have led to the synthesis of more than 700 different polymers. Because different side groups generate different properties, the range of properties and potential uses rivals those of some of the largest classical organic polymer systems.
Figure 2.

Pathways used for the synthesis of most of the known linear-type poly(organophosphazenes)
Although the ring-opening polymerization route to poly(dichlorophosphazene) yields the longest polymer chains (up to 15,000 repeating units), it is not the only method available. Use of a living, room temperature condensation of a chlorophosporanimine (3 in Figures 2 and 3) gives a narrow molecular weight distribution polymer but with shorter polymer chains than the ring-opening route, but allows access to block copolymers, combs, dendrimers, and so on.5–9 Alternative condensation processes have been developed by other research groups that also provide direct access to chloro-substituted medium molecular weight polymers.10,11 All these forms of poly(dichlorophosphazene) can be used as reactive macromolecular intermediates in the same way as the polymers produced by the ring-opening route.
Figure 3.

The living cationic condensation polymerization route to poly(dichlorophosphazene). This provides access to block, comb and dendritic copolymers.
The simplest utilization of the macromolecular substitution process is when the nucleophile bears only one nucleophilic site. Most of the polymers produced during the earlier phases of this field fall into this category. However, if the nucleophile bears two or more nucleophilic centers, the secondary functional units may react with two different chains thus leading to crosslinking during macromolecular substitution. Accordingly, secondary reactive sites on the nucleophile, such as hydroxyl, amino, or carboxylic acid units, must be protected before chlorine replacement and then subsequently deprotected by the use of non-aggressive reagents after the side group has been coupled to the phosphazene polymer chain.12
Other routes to poly(organophosphazenes) also exist, including the use of fluorocyclophosphazenes to produce fluorophosphazene polymers for macromolecular substitution reactions (these are preferred for organometallic substitutions).13 Moreover, a few ring-opening polymerizations are known in which cyclic phosphazenes that bear both chlorine or fluorine and organic substituents can be polymerized to yield polymers with both unreactive organic side groups and reactive halogen atoms.14 Condensations of organic-substituted phosphoranimines to give specific organophosphazenes directly are also available. 15–20 These methods are valuable for the synthesis of specific macromolecules that are difficult to prepare by the other procedures. Nevertheless, the most diverse range of polymers has been prepared by the macromolecular substitution route described above, and the polymers described in the following sections were made by this method.
(B). Structure-Property Relationships
The development of routes to new polymers, while interesting from a conceptual viewpoint, is of long-term interest only if new combinations of properties can be generated. The macromolecular substitution approach used for most poly(organophosphazenes) is valuable mainly because of the ease of diverse property tuning that is possible. For example, even when the most common backbone arrangement is retained (essentially linear architecture, broad molecular weight distribution, and average molecular weights above a million) polymers are accessible with properties that range from water-soluble to highly hydrophobic, from water-stable to bioerodible, from colorless to colored, from radiation stable to radiation crosslinkable, from elastomers to rigid solids, or from hydrogels to hydrophobic membranes. Many of these polymers resist combustion. All this is possible by varying the side groups via the macromolecular substitution protocol. Examples of different side groups and their influence on polyphosphazene properties are given in Chart 1, and a few specific polymers (7-16) relevant to this article are shown in Figure 4.
Chart 1.

Properties associated with different side groups in poly(organophosphazenes)
Figure 4.

Specific examples of the range of properties accessible by variations in the side groups of polyphosphazenes.
(C). Elastomers
Elastomers are soft materials that play a major role in engineering and biomedicine. Classical elastomers such as natural rubber, polyisobutylene, or silicone rubber are well known. The molecular characteristics that favor elastomer formation are highly flexible polymer chains to allow facile molecular reorientation during impact or stress, cross-linking to avoid permanent shape change caused by unrestricted chain slippage, and in some cases an onset of crystallinity when the material is stretched. Elastomers absorb impact energy or tensile stress by allowing torsional bond motion and limited bond angle distortion, but give back that energy as the chains and side groups return to their preferred higher entropy conformations. Note that an uncross-linked amorphous elastomer may have gum-like properties and be capable of viscous liquid flow at room temperature. Only after the chains have been cross-linked do the true elastomeric properties appear.
However, additional characteristics are needed for uses under certain extreme condition. For example, components that come in contact with aggressive fuels or hydraulic fluids need to resist absorption of these liquids. Other elastomers must absorb impact but rebound slowly after a short time delay (hysteresis). Polymeric elastomers for cardiovascular devices must resist absorption of aqueous fluids or lipids or the deposition of proteins. Moreover, a serious need exists for polymers that are resistant to high energy radiation. Fire resistance is an additional requirement for some high-technology uses. Some of the earliest poly(organophosphazenes) were developed with high performance elastomers in mind.
1. Halogen-Containing Phosphazene Elastomers.
(a). Fluoroalkoxy-derivatives.
The first stable high molecular weight poly(organophosphazene), synthesized by the author and his coworkers in the mid-1960’s,2–4 is a species with trifluoroethoxy side groups (8). This is a non-elastomeric, fire-resistant, semi-crystalline high polymer that forms hydrophobic films, fibers, and surface coatings. Probably more studies have been carried out on this polymer than on any other polyphosphazene21–23 including its investigation as an inert biomedical material.24
It was subsequently shown that the introduction of two different fluoroalkoxy side groups (9)25–28 eliminates the crystallinity by destroying molecular symmetry, and the polymers become elastomers especially when they are cross-linked. Cross-linking is achieved by the incorporation of a third substituent that contains unsaturated organic groups, albeit in small concentrations, followed by free radical cross-coupling reactions. The uniqueness of these elastomers lies in their flexibility at low temperatures (glass transition temperatures in the range of −60°C), their resistance to hydrocarbons and other fluids, and their impact-dampening properties. They were developed and manufactured as high performance elastomers for use in land vehicle and aerospace components and, incidentally, for uses in dental devices29. They have also been examined as cardiovascular materials because of their hydrophobicity and resistance to lipid absorption. The inorganic backbone, which is transparent from the near infrared, through the visible, and to the 220 nm region in the ultraviolet, is more resistant to ultra violet, visible, and high-energy radiation than most organic elastomers, and is also an effective fire retardant. Since that time we have synthesized numerous mixed-substituent polyphosphazene elastomers that bear fluorinated, chlorinated, organosilicon, and other side groups. Examples are shown in Figure 4.
Polyphosphazene elastomers are not restricted to those that contain only fluoroalkoxy side groups. A combination of fluoroalkoxy side units and non-fluorinated groups, provided they are distributed randomly along the chains, will also favor elastomeric or flexible behavior. Examples are macromolecules that have organosilicon units as cosubstituent groups, as shown in structure 10. These elastomers are synthesized in a different way from most other phosphazene polymers, specifically by the prior synthesis of small molecule cyclophosphazene trimers that bear both chlorine and the organosilicon side groups, followed by ring-opening polymerization and replacement of the chlorine atoms by organic groups.30
(b). Chloroalkoxy derivatives.
Recently, a new series of polyphosphazenes has been synthesized with chloroalkoxy side groups,31 including mixed-substituent species with non-chlorinated co-substituents. A typical formula of a mixed substituent polymer is [NP(OCH2CF3)x(OCH2CCl3)y]n. Properties variations are accessible through variations in the ratio of the fluoroalkoxy and chloroalkoxy side groups These too are soft materials, but with leathery rather than elastomeric characteristics, a consequence of the greater bulk of the chlorinated side chains and their influence on reduced chain mobility. Although fluoroalkoxy derivatives are already resistant to combustion, the presence of the chlorinated side groups enhances this property.
2. Halogen-Free Elastomers.
Another series of inherent elastomers has been synthesized in our program by the use of non-halogenated alkoxy side groups such as ethoxy, n-propoxy, n-butoxy, n-pentoxy, and so on.32 Structure 7 in Figure 4 is an example. Elastomeric properties are generated even when only one type of side group is present. In other words, molecular asymmetry or side group positional disorder is not necessary with these side units for the formation of non-crystalline elastomers. These polymers have lower glass transition temperatures than their fluorinated analogues. Some are in the range of −100°C, which is close to the lower theoretical limit for any polymer. This characteristic is attributed to a combination of low torsional barriers within both the backbone and the side groups. Mixed-substituent polyphosphazenes with alkoxy and methoxyethoxyethoxy side chains are also gums and elastomers. As with the fluoroalkoxy derivatives, cross-linking via unsaturated organic side groups turns the initial gums into elastomers. The presence of numerous carbon-hydrogen bonds in the side groups means that cross-linking can also be accomplished by exposure to gamma rays or even by the use of a photosensitizer and ultraviolet light. Similar elastomers are accessible via the crosslinking of polyphosphazenes that bear only oligoethyleneoxy side groups (15), but the interest in these lies more in their utility in hydrogels, organogels, or ion conductors (see later).
A few aryloxyphosphazene polymers also show flexible or elastomeric character, but the glass transition temperatures are near room temperature. Specifically mixed-substituent polymers with both phenoxy and methyl- or ethyl-phenoxy side groups (to avoid crystallinity) are soft materials which, when crosslinked, have been used as non-flammable expanded foam thermal insulating materials.26,27
3. Block and Graft Copolymers.
A third class of soft matter polyphosphazene elastomers is based on block or graft copolymers,33,34 typically diblock species in which one block is a highly flexible polyphosphazene and the second block is a more rigid organic polymer or a flexible polysiloxane Alternatively, a phosphazene block with flexible side chains can be connected to a phosphazene block with rigid, bulky side groups. Triblock copolymers have also been produced. Specific examples are shown in Figure 5.
Figure 5.

Block copolymers derived from polyphosphazenes and classical polymers
The attraction of block copolymers for both research and solving practical problems is that the properties of two entirely different polymers may be combined in one macromolecule. For example, an elastomeric block combined with a rigid block can give materials or membranes that have both strength and impact resistance. A hydrophobic block connected to a hydrophilic block provides a pathway to the formation of micelles in which nano-sized spheres self assemble in water with the hydrophilic blocks in the corona and the hydrophobic blocks in the core, or vice versa in a hydrophobic medium. Such assemblies offer opportunities for micelle use as carrier vehicles for hydrophobic drug delivery or for forming suspensions of dyes or pigments for ink jet printing. Other block copolymers may self assemble in the solid state to give nanostructures such as laminar or gyroid arrangements, or cylindrical micelles Because many polyphosphazenes are fire retardant, the linkage of a polyphosphazene block to a flammable organic polymer block has obvious technological benefits.
A particularly intriguing group of block copolymers synthesized in our program contain a polyphosphazene connected to a poly(organosiloxane) (silicone) block, thus generating hybrids of the two most widely developed inorganic-organic elastomeric systems.33
(D). Hydrogels
Hydrogels are lightly crosslinked modifications of water-soluble polymers that absorb aqueous media to a limit defined by the crosslink density. The higher the crosslink density, the less fluid can be absorbed. Specific gamma-radiation (5–20 Mr) cross-linked, water-stable polyphosphazenes swell by absorbing water but contract by extrusion of water when the temperature, pH, or ion strength is changed.31 This allows their incorporation into devices for the control of enzyme or inorganic catalyst activity, or for membranes or surface coatings with variable permeability.
The molecular features that give rise to water solubility in polyphosphazenes are associated with organic side groups that contain alkyl ether units or alkyl or aryl groups with hydroxyl or carboxylate functional units. For example, polyphosphazenes with oligo-ethyleneoxy side groups, glucose units, or aryloxy groups with hydroxyl or sodium carboxylate functional substituents are all water-soluble. Crosslinking of these can be accomplished by gamma radiation, ultraviolet irradiation, or by the introduction of divalent or trivalent ions that replace the sodium ions in carboxylate derivatives.35−40
The temperature-based responsive behavior of gels derived from polymers such as 15 is attributed to the same phenomena that are found in totally organic polymers that have amphiphilic chains. These species have a lower critical solution temperature (LCST) below which the polymers are soluble in water or the crosslinked gels are fully expanded in aqueous media. Temperature increases above the LCST cause precipitation of the uncrosslinked polymer or contraction of the crosslinked hydrogel by extrusion of water. This behavior is attributed to the presence of both hydrophilic (etheric oxygen) and hydrophobic (alkyl) components in the skeleton or side chains. Below the LCST hydrogen bonding between water and the hydrophilic components of the polymer ensures solubility. Above the LCST the hydrogen bonding, which declines with rising temperature, is insufficient to counteract the hydrophobicity of the alkyl components, and the uncrosslinked polymer precipitates or the gel contracts.
In polyphosphazene hydrogels the LCST can be tuned by changes to the length or degree of branching of the side groups as illustrated in Chart 2.35–40 This opens possibilities for a number of uses, including responsive membranes with permeabilities that change suddenly as the temperature is raised or lowered. For example, reactions catalyzed by enzymes trapped in a membrane can proceed when the gel is in an expanded state, but are interrupted when the enzymes are insulated from substrates by gel contraction.37 Drug delivery through a membrane may be controlled in a similar way. There are many other potential uses for these materials in both medicine41,42 and general technology.
Chart 2.

Lower critical solution temperatures of ethyleneoxy-substituted polyphosphazenes
Gel response to temperature changes is but one of several phenomena that have been explored with polyphosphazene hydrogels. Another is the response to changes in pH where increases in proton concentration tip the balance between solubility and insolublity. For example, polyphosphazenes that bear both oligoethyleneoxy and p-carboxyphenoxy side groups (structure 16) are responsive to pH changes.40 Variations in ion strength, or replacement of one cation in solution by another, also trigger gel expansion or contraction (Figure 7). Thus, pH changes from basic to acidic switch the polymer from soluble to insoluble or change a gel from the expanded to the contracted form. The same polymers will respond to changes in ionic strength brought about by changes in salt concentration. Moreover, replacement of sodium ions in the medium by di- or trivalent cations will cause gel contraction as the multivalent cations cause ionic crosslinks between the polymer chains. This same phenomenon can be induced in the presence of cations that can be reversibly converted from the M+ to the M2+ or M3+ states by electrochemical oxidation or reduction. Use has been made of this effect to construct hydrogel-based devices that will bend or contract when activated electrochemically.43
Figure 7.


Hydrogel volume changes as a function of (A) pH, (B) ion strength, and (C) cation charge for polyphosphazenes with different ratios of OCH2CH2OCH2CH2OCH3 and p-carboxyphenoxy side groups. The side group ratios are: a, 30 mol% p-carboxyphenoxy 70% methoxyethoxyethoxy (MEE); b, 50% p-carboxyphenoxy, 50% MEE; c, 76% p-carboxyphenoxy, 24% MEE; and d, 94% p-carboxyphenoxy, 6% MEE. The polymer labeled MEEP is the bis(methoxyethoxyethoxy) derivative (15).37 The species labeled “ester” is the ethyl ester of polymer 16. Note that the hydrogel volumes from crosslinked polymer 15 and the ethyl ester of 16 are essentially insensitive to pH, ion strength, or different cations, because neither contains an ionic side group.
One of the more unusual phosphazene gel species is the polymer with a polyphosphazene backbone and oxygen-linked tyrosine side groups (Structure 17).44 This polymer has two noteworthy characteristics. First, the size of the tyrosine side groups prevent 100% replacement of chlorine atoms in poly(dichlorophosphazene). This requires that two different substituents must be used – tyrosine and the less-hindered propoxy group. Second, tyrosine is a trifunctional nucleophile in which the amino and carboxylic acid functionalities must be protected before linkage via the phenolic unit to the phosphazene backbone, and then deprotected to allow the gel behavior to become manifest. This polymer shows the unusual behavior evident in Figure 8. Specifically, an expanded gel exists at pH 2 and again near pH 10, but with insolubility evident at pH 6.
Figure 8.

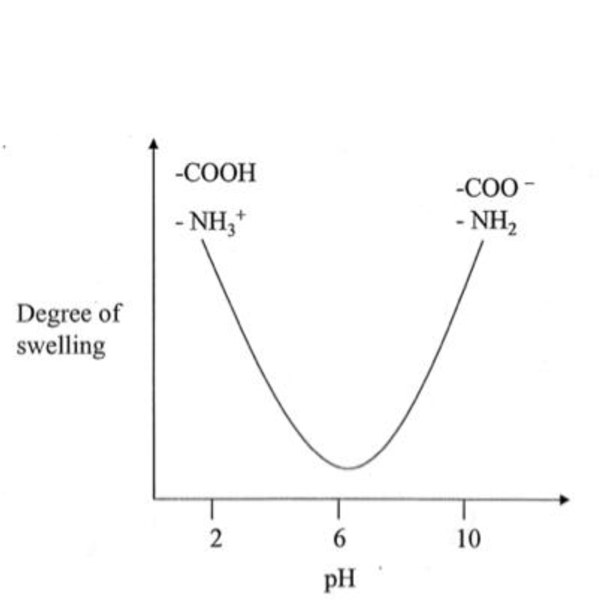
Swelling and de-swelling behavior of a tyrosinyl-propoxy polyphosphazenes (17) as a function of pH.
This is a consequence of the cationic functionality at the low pH and the anionic character at high pH. This behavior does not occur when the cosubstituent group is methoxyethoxyethoxy (highly hydrophilic) or trifluoroethoxy (highly hydrophobic). Thus, the solubility or gel profile of the polymer depends on the presence of the propoxy side groups which provide a balance of hydrophobicity and hydrophilicity. Moreover, reductions in the ratio of propoxy groups compared to tyrosine side groups favors gel formation rather than full solubility at low and high pH even in the absence of radiation crosslinking or divalent cations. At pH 7.4 the water absorption by the gel is more than 400%.
(E). Micelles, Dendrimers, and Related Structures
Polymer micelles are formed from amphiphilic block copolymers in which one block is hydrophobic and the other is hydrophilic. In phosphazene chemistry both blocks may have a phosphazene backbone but one contains hydrophilic side groups and the other bears hydrophobic substituents. An alternative configuration is where a phosphazene is end-linked to a non-phosphazene polymer with either block being hydrophilic or hydrophobic.45–51 Specific examples are shown in Chart 3.
Chart 3.

Amphiphilic block copolymers that form micelles
Polymer 21 has an unusual behavior.49 Both blocks are hydrophobic, and the polymer is insoluble in water. However, the presence of β-cyclodextrin in the aqueous phase and its ability to encapsulate adamantyl units converts that block to hydrophilic, thus generating an amphiphilic system. In practice this means that micelles that are stable in the presence of the congugate cyclodextrin become unstable as the aqueous phase in diluted and the cyclodextrin is lost. This provides a switching mechanism that could trigger the release of trapped drug molecules.
In block copolymer 23 the hydrophobic block is poly[bis(trifluoroethoxy)phosphazene] whereas the hydrophilic block is poly[(dimethylamino)ethyl methacrylate].46 These molecules were assembled by synthesis of the trifluoroethoxyphosphazene component via the living polymerization/macromolecular substitution route, followed by coupling through the living phosphazene chain end via “click” chemistry and atom transfer radical polymerization of the methacrylate. Micelles formed from these polymers are of narrow size distribution, with diameters that ranged from 100–142 nm depending on the polymer chain length.
In addition we have recently produced a series of densely grafted star- and comb-shaped copolymers with a cyclic or polymeric phosphazene core and polystyrene, poly(tert-butylacrylate), or poly(N-isopropylacrylamide) peripheral structures, molecules that were assembled with the use of ATRP techniques (Figure 6).51 Hydrolysis of the tert-butyl termini of poly(tert-butylacrylate) side chains to carboxylate units yielded negatively charged brush structures with potential access to ion-sensitive behavior in gels (see below). Moreover, the polyphosphazene/poly(N-isopropylacrylamide) brush systems form cylindrical micelles in water with a lower critical solution temperature (LCST) at 33°C, which illustrates the possible utility of these polymers in gel membranes or controlled release applications (see the following section).
Figure 6.

Densely grafted polyphosphazenes produced by ATRP polymerization of styrene, N-isopropylamide, or tert-butyl acrylate.31 Hydrolysis of the tert-butyl acrylate side chains yielded carboxylic acid units.
Related to these block copolymers and micelles is a series of 4-, 6-, and 8-arm dendrimers derived from a diaminobutane core, propylimine branches, and hydrophilic polyphosphazene terminal arms.52 The amphilicity of the dendrimers is generated by the hydrophobic core and the hydrophilic (methoxyethoxyethoxy) side groups that decorate the terminal polyphosphazene chains. Schematic illustrations of these species are shown in Figure 7. In a neutral state these molecules are soluble in water because of the hydrophilicity of the corona. Their utility is their ability to retain hydrophobic model molecules, such as pyrene, in the hydrophobic core, but to release these guests in the presence of aqueous solutions of alkali metal ions which complex with the oligo-ethyleneoxy substituents and swell the corona. This allows water to penetrate into the core and induce a release of the trapped hydrophobic guests. Pyrene is a model for hydrophobic drug molecules. Thus, these dendrimers behave like block copolymer micelles, but with a more stable structure-size relationship.
(F). Organogel Electrolytes
1. Lithium Battery Electrolytes
Polyphosphazenes with linear or branched ethyleneoxy side chains (Chart 2) are gum-like solvents for salts such as lithium triflate. This behavior is attributed to the ability of the oxygen atoms in the side chains to coordinate to lithium cations. Such semi-fluid solutions are ion conductors with room temperature conductivities in the range of 10−4 S/cm. They were originally tested as solvent-free electrolytes for rechargeable lithium batteries, having conductivities at least two orders of magnitude better than solid electrolytes based on the classical system of poly(ethylene oxide) /salt solutions, the traditional standard.53–70 However, conductivities near 10−4 S/cm are roughly an order of magnitude less that the minimum generally required for practical lithium batteries. In current practice, commercial rechargeable lithium batteries use liquid electrolytes based on solutions of lithium salts in volatile flammable solvents such as propylene carbonate or other carbonates. Some much-publicized laptop computer fires (and potential problems with electric vehicle batteries) have been traced in part to the flammability and volatility of these liquid electrolytes.
An alternative to both the unplasticized gum electrolytes and organic solvent electrolytes are organogel electrolytes based on a polyphosphazene either dissolved in an organic solvent or plasticized by small amounts of a solvent such as propylene carbonate. Flammability experiments have shown that such systems can be fire-resistant due to the phosphorus-nitrogen polymer backbone, and they have ionic conductivities above the 10−3 limit.71,72 For example, the addition of 25 wt% of the high polymer 15 to a propylene carbonate based electrolyte forms a viscous gel with the flammability reduced by 90% compared to the polymer-free electrolyte while maintaining an ionic conductivity of 2.5 × 10−3 Scm-1. Even as little as 10% of an organic solvent dissolved in polymer 15 or its analogues raises the ionic conductivity close to the 10−3 Scm−1 target. Covalent crosslinking of the polymer chains by gamma irradiation (before solvent absorption) has little effect on the ionic conductivity. Clearly the reorientational freedom of the side groups and the backbone is retained after light crosslinking, since these factors are crucial to ion transport.
The mechanism of ion conduction in these systems is still under investigation.68–70,73 It is known that increases in the salt concentration beyond a certain point raise the Tg of 15 due to reversible cation-coordinative crosslinking between oxygen atoms on different side chains and this leads to reduced ion transport. An earlier idea was that ionic conduction occurs mainly via lithium ion transport, in which cation hopping occurs from one etheric side group to another. However, recent studies73 suggest that a significant fraction of the charge transport in these electrolytes is due to anion migration. The mechanism of anion transport is still under investigation. Magnesium salts can be employed in place of lithium in this electrolyte and this may be significant for the future development of magnesium batteries.69 Related polyphosphazene gel systems have also been studied as possible electrolytes for primary seawater batteries.74
2. Gel Electrolytes for Dye-Based Solar Cells
Polymeric or oligomeric gel electrolytes are also of considerable interest in the field of dye-based photovoltaic cells.75 These cells consist of a porous titanium dioxide semiconductor electrode separated from a counter electrode by an electrolyte. The TiO2 electrode must be highly porous to provide a large surface area for the attachment of ruthenium bipyridine-based and related dye molecules. Traditionally, the high surface area has been provided by the use of TiO2 nanospheres, but recent studies have also used nanotubes and nanorods.76,77 Photons passing through the porous titanium dioxide electrode activate dye molecules which inject an electron into that electrode for transfer into an external circuit and thence to the counter electrode. However, the resultant oxidized dye molecules are sensitive to decomposition unless reduced rapidly to the neutral state. This is usually accomplished by an iodide/triiodide couple in an organic solvent. Electrons injected from the counter electrode reduce the oxidized salt, which is then available to reduce more dye molecules. The normal solvent used in the electrolyte is a volatile organic liquid such as acetonitrile. This is a weak link in the process since loss of the solvent at elevated temperatures, even from a well-sealed cell, will permanently damage the device. Thus, a need exists for non-volatile electrolytes, with polymers being good candidates because of their high molecular weights.
Both high molecular weight polyphosphazenes and short chain or cyclic phosphazene oligomers have been studied for this role.76,77 High molecular weight polyphosphazenes such as 15 cannot penetrate into the interstices of nanosphere electrodes and are thus inefficient electrolytes. Solvent-free liquid phase small molecule cyclic phosphazenes are too viscous. However, short-chain phosphazene oligomers with oligo-ethyleneoxy side chains have a lower cross section, and are currently under investigation. In all these cases, conversion of the polymer or oligomeric electrolyte to an organogel by the addition of small amounts of an organic solvent enhances the electrolyte behavior while lowering the volatility. Incidentally, organophosphates with oligo-ethyleneoxy substituents are also effective as solvents and electrolytes, presumably because of their ability to access dye molecules buried deep in the electrode structure.78
3. Fuel Cell Membranes
Finally, some of the materials employed in polymer membrane fuel cells also fall into the category of soft matter in the sense that they are amphiphilic ionic polymers swollen by water. These are important for uses in hydrogen-driven or direct methanol fuel cells. The traditional membranes in such devices are typically hydrophobic polymers with sulfonic acid or sulfonimide units forming part of the polymer side group structure. Such materials self assemble into hydrophobic and hydrophilic domains, with proton conduction occurring through the water-filled hydrophilic channels. A typical fuel cell membrane is Nafion - a sulfonated fluorocarbon polymer.
Several groups have explored the possibility that the electrochemical stability of the polyphosphazene backbone and the ease of introduction of different side groups may offer a means for tailoring fuel cell membranes in ways that are difficult or impossible with other polymers. An additional reason for this interest is because the inorganic backbone is resistant to decomposition by the free radicals generated at the oxygen reduction electrode. Mixed substituent sulfonated, phosphonated, or sulfonimidated aryloxy groups79–85 linked to a phosphazene backbone have shown the most promise. The non-functionalized hydrophobic cosubstituent aryl rings provide some protection for the backbone against the strongly acidic media found in the membrane. Moreover, many of these membranes are not swelled by or soluble in methanol, an advantage over Nafion-type structures for direct methanol applications. Research on these membranes is continuing.
(G). Summary
The variety of different soft materials accessible through the phosphazene platform opens up many avenues for future research and applications. Three factors are responsible for the synthetic and use-oriented utility of the polyphosphazene system. The first is the flexibility of the inorganic backbone, a property that can be traced to the low torsional barrier of the covalent phosphorus-nitrogen bonds, which in turn can be rationalized in terms of 3d-orbital involvement at the phosphorus atoms or to zwitterionic structures. Any explanation of the skeletal bonding must be tempered by the knowledge that these polymers are electrical insulators rather than semiconductors. The skeleton is also transparent throughout the visible spectrum and to the 220 nm region in the ultraviolet. Thus, classical long-range electron delocalization phenomena are not found, and some kind of limited P-N-P delocalization, insulated from the flanking units, may be responsible. The second factor is the stability of the inorganic skeleton to oxidation, reduction, and photochemical, or thermal bond cleavage, provided appropriate side groups are attached to phosphorus. The third, and most crucial factor is the access to an extremely broad portfolio of nucleophilic reagents for chlorine replacement reactions with poly(dichlorophosphazene) that alone or in different combinations can yield diverse physical and chemical properties.
Although it is clear that the structure-property permutations in the phosphazene system are extremely broad, it is useful to identify a few of the most promising systems at this time in each category of soft matter. In the area of elastomers, the mixed-substituent fluoroalkoxyphosphazene polymers have proved to be the most useful, although the other emerging candidates may prove to be equally interesting in the coming years. The block copolymers could become significant in the future, but their utility is currently restricted by the small-scale employed for synthesis of the monomer for the living cationic polymerization process. Among the hydrogels, the polymers with alkyleneoxy and ionic co-substituent side chains have been studied in greatest detail and are most appropriate for responsive membrane applications. The organogels utilized as lithium or magnesium battery or solar cell electrolytes appear to have a promising future, but more scale-up and development work is needed before this becomes a practical reality.
Many aspects of polyphosphazene chemistry illustrate how long-range fundamental research can move far ahead of the existing technology, but is still dependent on the pace at which engineers can utilize new materials and incorporate them into devices.
Figure 9.

Diagrammatic illustration of the structure of three dendrimers. The inner cores consist of hydrophobic tetramethylene and trimethylene chains, while the corona is made up of phosphazene chains with hydrophilic methoxyethoxyethoxy side groups. Linker units between the segments are not shown for clarity.
Acknowledgements
The information summarized in this review was obtained through the work of numerous coworkers who are mentioned in the reference list. Different aspects of the research were sponsored by the U.S. Department of Energy, the National Institutes of Health, and the Department of Defense.
References
- 1.Allcock HR, Chemistry and Applications of Polyphosphazenes, Wiley Interscience, 2003. (725 pages). [Google Scholar]
- 2.Allcock HR and Kugel RL, J. Am. Chem. Soc, 1965, 87, 4216–4217. [Google Scholar]
- 3.Allcock HR, Kugel RL and Valan KJ, Inorg. Chem, 1966, 5, 1709–1715. [Google Scholar]
- 4.Allcock HR and Kugel RL, Inorg. Chem, 1966, 5, 1716–1718. [Google Scholar]
- 5.Honeyman CH, Manners I, Morrissey CT and Allcock HR, J. Am. Chem. Soc, 1995, 117, 7035–7036. [Google Scholar]
- 6.Allcock HR, Crane CA, Morrissey CT, Nelson JM, Reeves SD, Honeyman CH and I Manners, Macromolecules, 1996, 29, 7740–7747. [Google Scholar]
- 7.Allcock HR, Nelson JM, Reeves SD, Honeyman CH and I. Manners, Macromolecules, 1997, 30, 50–56. [Google Scholar]
- 8.Nelson JM and Allcock HR, R. H, Macromolecules, 1997, 30, 1854–1856. [Google Scholar]
- 9.Allcock HR, Reeves SD, Nelson JM, Crane CA and Manners I, Macromoleculs, 1997, 30, 2213–2215. [Google Scholar]
- 10.De Jaeger R and Potin Ph., Ch. 2 in Synthesis and Characterization of Poly(organophosphazenes) (Gleria M; De Jaeger R Eds.) Nova Science Publishers, New York: 2004. [Google Scholar]
- 11.D’Halluin G, De Jaeger R, Chambrette JP and Potin Ph., Macromolecules 1992, 25, 1254. [Google Scholar]
- 12.Allcock HR and Morozowich NL, Polymer Chemistry, 2012, 3, 578–590. [Google Scholar]
- 13.Allcock HR, Kugel R. l. and Stroh EG, Macromolecules, 1972, 5, 231–232. [Google Scholar]
- 14.Allcock HR, McDonnell GS and Desorcie JL, Macromolecules,1990, 23, 3873–3877.5 [Google Scholar]
- 15.Flindt ER and Rose H, Z. Anorg. Allgem. Chem, 1977, 428, 204. [Google Scholar]
- 16.Wisian-Neilson P and Neilson RH, J. Am. Chem. Soc, 1980, 102, 2848 [Google Scholar]
- 17.Neilson RH and Wisian-Neilson P, Chem. Rev, 1988, 88, 541. [Google Scholar]
- 18.Wisian-Neilson P, Jung J–H and Potluri SK, ACS Symposium Series 2006, 917, 335–346. [Google Scholar]
- 19.Montague R,A and Matyjaszewski K, J. Am. Chem. Soc. 1990, 112, 6721. [Google Scholar]
- 20.Matyjaszewski K, Moore MM and White ML, Macromolecules 1993, 26, 6741 [Google Scholar]
- 21.Kojima M and Magill JH, Polymer, 1985, 26, 1971–1978. [Google Scholar]
- 22.Singh A, Seely L and Allcock HR, Langmuir, 2005, 21, 11604–11607. [DOI] [PubMed] [Google Scholar]
- 23.Allcock HR, Steely L, Singh A and Hindenlang M, Chapter in Superhydrophobic Surfaces (Carre A and Mittal K. l., eds.), VSP Publ., Leiden, The Netherlands, 2009, 127–138. [Google Scholar]
- 24.Richter GM, Stampfle U, Stampfl S, Rehnitz C, Holler S, Schnabel P and Grunze M, Investigative Radiology, 2005, 40, 210–218. [DOI] [PubMed] [Google Scholar]
- 25.Rose SH, J. Polym. Sci. Ser. B, 1968, 6, 837–839 [Google Scholar]
- 26.Tate DP, J. Polym. Sci. Polymer Symposia, 1974, 48, 33–45. [Google Scholar]
- 27.Singler RE, Schnieder NS, Hagnauer GL, Polym. Sci. Eng, 1975, 15, 321–338. [Google Scholar]
- 28.Kolich CH, Klobucar WD and Books JT, U.S. Patent, 1990, 4,945,139. [Google Scholar]
- 29.Gettleman L, Farris CL, LeBouef RJ, U.S. Patent, 1984, 4,432,730. [Google Scholar]
- 30.Allcock HR and Coggio WD, Macromolecules, 1993, 26, 764–771. [Google Scholar]
- 31.Chen C, Allcock HR, 2012, Unpublished studies. [Google Scholar]
- 32.Weikel AL, Lee DK, Krogman NR, and Allcock HR, Polymer Eng. and Sci, 2011, 51, 1693–1700. [Google Scholar]
- 33.Prange R, and Allcock HR, Macromolecules 1999, 32, 6390–6392. [Google Scholar]
- 34.Nelson JM, Primrose AP, Hartle TJ, Allcock HR and I. Manners, Macromolecules 1998, 31, 5206–5214. [Google Scholar]
- 35.Allcock HR, Kwon S, Riding GH, Fitzpatrick RJ, and Bennett JL, Biomaterials, 1988, 9, 509–513. [DOI] [PubMed] [Google Scholar]
- 36.Allcock HR, Pucher SR, Turner ML and Fitzpatrick RJ, Macromolecules, 1992, 25, 5573–5577. [Google Scholar]
- 37.Allcock HR, Pucher S,R and Visscher KB, Biomaterials, 1994, 15, 502–506. [DOI] [PubMed] [Google Scholar]
- 38.Allcock HR, Fitzpatrick RJ and Visscher KB, Chemistry of Materials, 1992, 4, 775–780. [Google Scholar]
- 39.Allcock HR and Dudley GK, Macromolecules, 1996, 29, 1313. [Google Scholar]
- 40.Allcock HR and Ambrosio AMA, Biomaterials, 1996, 17, 2295–2302. [DOI] [PubMed] [Google Scholar]
- 41.Kim J, Chun C, Kim B, Hong JM, Cho J-K, Lee SH and Song S-C, Biomaterials, 2012, 33, 218–224. [DOI] [PubMed] [Google Scholar]
- 42.Park M-R, Chun C, Hahn S-W, Ki M-H., Cho C-S and Song S-C, J. Controlled Release, 2010, 147, 359–367. [DOI] [PubMed] [Google Scholar]
- 43.Fei S-T, Phelps MVB, Wang Y, Barrett E, Gandhi F and Allcock HR, Soft Matter, 2006, 2, 397–401. [DOI] [PubMed] [Google Scholar]
- 44.Allcock HR, Singh A, A.Ambrosio AM and Laredo WR, Biomacromolecules, 2003, 4. 1646–1653. [DOI] [PubMed] [Google Scholar]
- 45.Chang Y, Lee SC, Kim KT, Kim C, Reeves SD and Allcock HR, Macromolecules, 2001. 34, 269–274 [Google Scholar]
- 46.Liu X, Tian Z, Chen C and Allcock HR, Macromolecules, 2012, 45, 1417–1426. [Google Scholar]
- 47.Chang Y, Powell ES and Allcock HR, J. Polymer Sci. Part A, 2005, 43, 2912–2920. [Google Scholar]
- 48.Allcock HR, Powell ES, Chang Y, and Kim C, Macromolecules, 2004, 37, 7163–7167. [Google Scholar]
- 49.Cho SY and Allcock HR, Macromolecules, 2009, 42, 4484–4490. [Google Scholar]
- 50.Allcock HR, Cho SY and Steely LB, Macromolecules, 2006, 39, 8334–8338. [Google Scholar]
- 51.Tian Z, Liu X, Chen C and Allcock HR, Macromolecules, 2012, 45, 2502–2508. [Google Scholar]
- 52.Cho SY, and Allcock HR, Macromolecules, 2007, 40, 3115–3121. [Google Scholar]
- 53.Blonsky PM, Shriver DF, Austin P and Allcock HR, J. Am. Chem. Soc. 1984, 106, 6854–6855. [Google Scholar]
- 54.Allcock HR, Austin PE, Neenan TX, Sisko JT, Blonsky PM, Shriver DF, Macromolecules 1986, 19, 1508–1512. [Google Scholar]
- 55.Blonsky PM, Shriver DF, Austin P, and Allcock HR, Polym. Mater. Sci. Eng. 1985, 53, 118–122. [Google Scholar]
- 56.Blonsky PM, Shriver DF, Austin P and Allcock HR, Solid State Ionics 1986, 19, 258–264. [Google Scholar]
- 57.Bennett JL, Dembek AA, Allcock HR, Heyen BJ, and Shriver DF, Chem. Mater. 1989, 1, 14–16. [Google Scholar]
- 58.Allcock HR, Pucher SR, Turner ML and Fitzpatrick RJ, Macromolecules 1992, 25, 5573–5577. [Google Scholar]
- 59.Allcock HR, Napierala ME, Cameron CG and O’Connor SJM, Macromolecules 1996, 29, 1951–1956. [Google Scholar]
- 60.Allcock HR, Kuharcik SE, Reed CS and Napierala ME, Macromolecules 1996, 29, 3384–3389. [Google Scholar]
- 61.Allcock HR, O’Connor SMJ, Olmeijer DL, Napierala ME and Cameron CG, Macromolecules 1996, 29, 7544–7552. [Google Scholar]
- 62.Allcock HR, Napierala ME, Olmeijer DL, Cameron CG, Kuharcik SE, Reed CS and O’Connor SJM, Proc. Int. Conf on Solid Polymeric Electrolytes (Thomas J ed.) Uppsala, Sweden, Electrochimica Acta, 1997, 1–6. [Google Scholar]
- 63.Allcock HR, Ravikiran R and O’Connor SJM, Macromolecules 1997, 30, 3184–3190. [Google Scholar]
- 64.Allcock HR, Napierala ME, Best SA and Merz KM, Macromolecules, 1999, 32, 732–741. [Google Scholar]
- 65.Allcock HR, Olmeijer DL and O’Connor SJM, Macromolecules 1998, 31, 753–759. [Google Scholar]
- 66.Allcock HR, Sunderland NJ, Ravikiran R, and Nelson JM, Macromolecules 1998, 31, 8026–8035. [Google Scholar]
- 67.Allcock HR; Olmeijer DL, Macromolecules 1998, 31, 8036–8046. [Google Scholar]
- 68.York S, Kellam EC, Allcock HR and Frech R, Electrochim. Acta 2001, 46, 1553–1557. [Google Scholar]
- 69.Allcock HR and Kellam EC, Solid State Ionics, 2003, 156, 401–414. [Google Scholar]
- 70.Frech R, York S, Allcock HR and Kellam EC, C. E; Macromolecules 2004, 23, 8699–8702. [Google Scholar]
- 71.Fei S-T, and Allcock HR, Mater. Res. Soc. Symp. 2009,1127-T01–05. [Google Scholar]
- 72.Fei S-T, and Allcock HR, Power Sources, 2010, 195 (7), 2082–2088. [Google Scholar]
- 73.Lee D and Allcock HR, Solid State Ionics, 2010, 181, 1721–1726. [Google Scholar]
- 74.Welna DT, Stone DA and Allcock HR, R. H, Chemistry of Materials, 2006, 18, 4486–4492. [Google Scholar]
- 75.Gratzel MJJ, Photochemistry and Photobiology C, Photochemistry Reviews, 2003, 4, 145–153. [Google Scholar]
- 76.Lee S-HA, Jackson AMS, Hess A, Fei S-T, Pursel SM, Basham J, Grimes CA, Horn MW, Allcock HR and Mallouk TE, J. Phys. Chem. 2010, 114, 15234–15242 [Google Scholar]
- 77.S.-T.; Fei S-H Lee A, Pursel SM, Bashem J, Hess A, Grimes CA, Horn MW, Mallouk TE and Allcock HR, Power Sources 2011, 196, 5223–5230. [Google Scholar]
- 78.Hess A and Allcock HR, Unpublished studies. [Google Scholar]
- 79.Guo Q, Pintauro PN, Tang H and O’Connor S, J. Membrane Sci. 1999, 154, 175–181. [Google Scholar]
- 80.Wycisk R and Pintauro PN, J. Membrane Sci. 1996, 119, 155–60. [Google Scholar]
- 81.Tang H, and Pintauro PN, J. Appl. Polym. Sci. 2001, 79, 49–59. [Google Scholar]
- 82.Fedkin MV, Zhou X, Hofmann MA, Chalkova E, Weston JA, Allcock HR and Lvov SN, Mater. Lett. 2002, 52, 192–196. [Google Scholar]
- 83.Allcock HR, Hofmann MA, Wood RM, Macromolecules 2001, 34, 6915–6921. [Google Scholar]
- 84.H. R., M. A., Ambler CM, Lvo SN, Zho X, Chalkova E and Weston J, J. Membrane Sci., 2002, in press. [Google Scholar]
- 85.Lee D, Saito T, Benesi A, Hickner MT, and H MT. Allcock R, J Phys.Chem. 2011, 115(5), 776–783. [DOI] [PubMed] [Google Scholar]