

Structured Abstract
INTRODUCTION:
Diverse phenotypes, including large brains relative to body size, group living, and vocal learning ability, have evolved multiple times throughout mammalian history. These shared phenotypes may have arisen repeatedly by means of common mechanisms discernible through genome comparisons.
RATIONALE:
Protein-coding sequence differences have failed to fully explain the evolution of multiple mammalian phenotypes. This suggests that these phenotypes have evolved at least in part through changes in gene expression, meaning that their differences across species may be caused by differences in genome sequence at enhancer regions that control gene expression in specific tissues and cell types. Yet the enhancers involved in phenotype evolution are largely unknown. Sequence conservation–based approaches for identifying such enhancers are limited because enhancer activity can be conserved even when the individual nucleotides within the sequence are poorly conserved. This is due to an overwhelming number of cases where nucleotides turn over at a high rate, but a similar combination of transcription factor binding sites and other sequence features can be maintained across millions of years of evolution, allowing the function of the enhancer to be conserved in a particular cell type or tissue. Experimentally measuring the function of orthologous enhancers across dozens of species is currently infeasible, but new machine learning methods make it possible to make reliable sequence-based predictions of enhancer function across species in specific tissues and cell types.
RESULTS:
To overcome the limits of studying individual nucleotides, we developed the Tissue-Aware Conservation Inference Toolkit (TACIT). Rather than measuring the extent to which individual nucleotides are conserved across a region, TACIT uses machine learning to test whether the function of a given part of the genome is likely to be conserved. More specifically, convolutional neural networks learn the tissue- or cell type–specific regulatory code connecting genome sequence to enhancer activity using candidate enhancers identified from only a few species. This approach allows us to accurately associate differences between species in tissue or cell type–specific enhancer activity with genome sequence differences at enhancer orthologs. We then connect these predictions of enhancer function to phenotypes across hundreds of mammals in a way that accounts for species’ phylogenetic relatedness. We applied TACIT to identify candidate enhancers from motor cortex and parvalbumin neuron open chromatin data that are associated with brain size relative to body size, solitary living, and vocal learning across 222 mammals. Our results include the identification of multiple candidate enhancers associated with brain size relative to body size, several of which are located in linear or three-dimensional proximity to genes whose protein-coding mutations have been implicated in microcephaly or macrocephaly in humans. We also identified candidate enhancers associated with the evolution of solitary living near a gene implicated in separation anxiety and other enhancers associated with the evolution of vocal learning ability. We obtained distinct results for bulk motor cortex and parvalbumin neurons, demonstrating the value in applying TACIT to both bulk tissue and specific minority cell type populations. To facilitate future analyses of our results and applications of TACIT, we released predicted enhancer activity of >400,000 candidate enhancers in each of 222 mammals and their associations with the phenotypes we investigated.
CONCLUSION:
TACIT leverages predicted enhancer activity conservation rather than nucleotide-level conservation to connect genetic sequence differences between species to phenotypes across large numbers of mammals. TACIT can be applied to any phenotype with enhancer activity data available from at least a few species in a relevant tissue or cell type and a whole-genome alignment available across dozens of species with substantial phenotypic variation. Although we developed TACIT for transcriptional enhancers, it could also be applied to genomic regions involved in other components of gene regulation, such as promoters and splicing enhancers and silencers. As the number of sequenced genomes grows, machine learning approaches such as TACIT have the potential to help make sense of how conservation of, or changes in, subtle genome patterns can help explain phenotype evolution.

Tissue-Aware Conservation Inference Toolkit (TACIT) associates genetic differences between species with phenotypes. TACIT works by generating open chromatin data from a few species in a tissue related to a phenotype, using the sequences underlying open and closed chromatin regions to train a machine learning model for predicting tissue-specific open chromatin and associating open chromatin predictions across dozens of mammals with the phenotype. [Species silhouettes are from PhyloPic]
Abstract
Protein-coding differences between species often fail to explain phenotypic diversity, suggesting the involvement of genomic elements that regulate gene expression such as enhancers. Identifying associations between enhancers and phenotypes is challenging because enhancer activity can be tissue-dependent and functionally conserved despite low sequence conservation. We developed the Tissue-Aware Conservation Inference Toolkit (TACIT) to associate candidate enhancers with species’ phenotypes using predictions from machine learning models trained on specific tissues. Applying TACIT to associate motor cortex and parvalbumin-positive interneuron enhancers with neurological phenotypes revealed dozens of enhancer–phenotype associations, including brain size–associated enhancers that interact with genes implicated in microcephaly or macrocephaly. TACIT provides a foundation for identifying enhancers associated with the evolution of any convergently evolved phenotype in any large group of species with aligned genomes.
Much of the phenotypic diversity across vertebrates is thought to have arisen from changes in how genes are expressed (1). Variation in phenotypes such as vocal learning (2) and longevity (3) has been linked to patterns of gene expression in relevant brain regions and tissues. Thus, at least some of the genetic differences associated with the evolution of these and other complex phenotypes are likely in enhancers, which we define as distal cis-regulatory genomic elements that are bound by transcription factor (TF) proteins and regulate the expression of associated genes, often through cell type–specific activation (4, 5). For example, limblessness in snakes is associated with sequence divergence and activity loss in a critical enhancer near the Sonic hedgehog gene (6), and mutations in orthologs of this enhancer are associated with polydactyly in humans, mice, and cats (7, 8). Enhancer evolution has been associated with multiple other complex phenotypes, including whisker, penile spine, and brain growth (9).
Recent advances facilitate identifying relationships between enhancer activity and phenotype evolution (10–12). Community genome sequencing efforts such as the Zoonomia Consortium and the Vertebrate Genomes Project have constructed assemblies for hundreds of species from diverse mammalian and vertebrate clades (13, 14). Reference-free multispecies whole-genome alignments that can account for structural rearrangements and tools for extracting orthologs have vastly improved ortholog mapping for noncoding genomic regions (10, 15, 16). In addition, new phylogeny-aware statistical methods have been developed for identifying factors associated with phenotype evolution (17, 18).
Despite these successes, identifying enhancer–phenotype relationships is still a major challenge. Widely used methods to identify conservation and convergent evolution across orthologous genome sequences measure the extent to which the nucleotides within a given region are the same across species (19–21). While these approaches have led to some exciting findings, including the identification of multiple eye enhancers whose functions are lost in blind subterranean mammals (22, 23), such approaches are limited because nucleotide-level sequence conservation is not required for or always sufficient for activity conservation at enhancer orthologs (24). In fact, most enhancer sequences and TF binding sites are under less sequence constraint than promoter and gene sequences (25, 26). For example, a recent study found that the Islet enhancer is conserved in its tissue-specific activation patterns despite low sequence conservation because its TF motifs are in different orders in different species (27, 28). Another study computed average PhastCons scores, which measure the probability that a region is conserved, for house mouse brain enhancers whose rhesus macaque orthologs are not brain enhancers and found a few hundred enhancers that have high sequence conservation (PhastCons scores > 0.5) despite their different activities between species (12, 29). These findings suggest that, even when enhancer sequences are not very conserved at the nucleotide level, they can contain conserved patterns, such as TF motif occurrences, that are predictive of enhancer activity.
Previous studies showed that machine learning models that use DNA sequence to predict enhancer activity in a tissue of interest in one species can accurately predict clade-specific and tissue-specific enhancer activity in species from different mammalian clades (12, 30–32). These findings demonstrate that the sequence patterns associated with enhancer activity in tissues including brain and liver are highly conserved across mammals, even though the patterns’ nucleotide-level conservation is not always high. Leveraging that principle, we recently developed a method for identifying conservation of enhancer activity based on tissue- or cell type–specific regulatory patterns learned by machine learning models rather than conservation of nucleotides (12). Here, we present a framework that builds on this previous work to quantify the association between enhancer activity conservation and specific phenotypes. We apply this framework to open chromatin regions (OCRs), which we use as a proxy for enhancers, to associate open chromatin with brain size and other neural phenotypes and find that many associated candidate enhancers are near relevant genes. This method provides new opportunities to investigate the interplay between DNA sequence and phenotype evolution through gene regulation.
Results
We developed a framework called the Tissue-Aware Conservation Inference Toolkit (TACIT), which identifies candidate enhancers associated with the evolution of phenotypes across multiple clades by integrating machine learning–based predictions of enhancer activity with other comparative genomics advances (13, 17, 18). TACIT uses sequences of candidate enhancers identified experimentally in a small number of species to train machine learning models that predict the probability of enhancer activity of sequences in other genomes at the orthologous regions (13). Models are trained in a specific tissue or cell type that is relevant to a phenotype of interest. TACIT then uses these predictions, treating the probability of enhancer activity as a continuous value, to link candidate enhancers to specific phenotypes while accounting for phylogeny (Fig. 1). In our first application of TACIT, we used OCRs as our candidate enhancers (12, 33–40), convolutional neural networks (CNNs) (41) for our machine learning models, and 222 aligned boreoeutherian mammalian genomes from Zoonomia to identify orthologs (10).
Fig. 1. Overview of TACIT.

We trained a machine learning model using sequences underlying candidate enhancers (indicated in dark red) and non-enhancers (not pictured) to predict enhancer activity in a tissue or cell type of interest. We used the model to predict enhancer activity (darker red arrows indicate higher predicted activity) in that tissue or cell type in hundreds of genomes (13). We associated our predictions with phenotypes using a phylogeny-aware regression and then quantified the significance of the association using an empirical P value. [All silhouettes are from PhyloPic, and the silhouette of Orcinus orca was created by Chris Huh (license: https://creativecommons.org/licenses/by-sa/3.0/) and was not modified (132)]
Nucleotide-level conservation-based metrics do not find brain size–associated genes or regulatory elements
The sequenced genomes and nucleotide alignments of the Zoonomia Project provide the foundation to link differences in genome sequence to differences in complex traits (13). We began by examining brain size, a complex and diverse trait across mammalian species that contributes to human cognitive ability (42). Specifically, we used the brain size residual (deviation of brain mass from the predicted value of brain mass from a regression on body mass) (43, 44) because brain size is highly correlated with overall body size (45, 46) and because we were able to obtain brain size residual annotations for 158 boreoeutherianmammals (43, 44)—primates, lagomorphs, rodents, insectivores, bats, carnivores, pangolins, and ungulates. To explore the sufficiency of existing methods, we applied a previously developed nucleotide conservation–based method called RERconverge (21) to investigate whether there are proteins or motor cortex OCRs whose relative evolutionary rates are associated with the evolution of brain size residual and found no associated proteins and only one associated OCR, which is close in linear but not three-dimensional (3D) space to genes implicated in brain size (47–52).
Convolutional neural networks accurately predict open chromatin status of candidate enhancer OCR orthologs
As an alternative to these approaches, we used our new method, TACIT, which estimates conservation of enhancer activity on the basis of predicted tissue-specific regulatory signatures. We applied TACIT to the motor cortex and liver, both of which have open chromatin data from more than two species, as well as retina and motor cortex parvalbumin-positive (PV+) interneurons, which have open chromatin from only two species; details about the setup for each model are given in the “Model encyclopedia” section of the supplementary text (52). For this first application of TACIT, we used OCRs because accessible regions of the genome are available for TF binding and therefore can serve as a proxy for enhancers. We chose OCRs instead of other metrics of enhancer activity, such as H3K27ac chromatin immunoprecipitation sequencing (ChIP-seq) regions, because open chromatin data are widely available in both tissue and single-cell applications, because OCRs pinpoint functional regulatory sequences with high resolution (16, 52–56), and because several recent studies have suggested that they are more indicative of enhancer activity (52, 57–59).
We limited our focus to OCRs that are likely to function as enhancers, which we defined as nonexonic OCRs that are sufficiently far from the nearest protein-coding transcription start site (TSS) that they would be unlikely to function as promoters and sufficiently short that they would be unlikely to function as super-enhancers (52). We decided to focus on candidate enhancers instead of all OCRs because enhancers and promoters have partially different regulatory codes (60, 61) and because enhancers tend to be better-assembled than promoters owing to their generally lower GC content (62, 63). We chose tissues and cell types that we thought would reveal relationships between open chromatin and complex phenotypes of interest. A logistic regression model trained using TF motif features performed suboptimally (table S1), so we decided to train CNNs, which can automatically learn sequence patterns and pattern combinations that are predictive of open chromatin, enabling them to learn sequences beyond those that match known TF motifs as well as combinations of TF motifs. Since the most-relevant CNN from our previous work (12) and the widely used DeepSEA Beluga model (64), which were trained for tasks related to motor cortex open chromatin prediction (brain and glioblastoma, respectively, open chromatin prediction), had suboptimal motor cortex test set performance (52), we trained models directly for our tasks.
For motor cortex and liver, we trained CNN classifiers to distinguish whether a sequence is an OCR likely to function as an enhancer in one species (positive) or a non-OCR ortholog of a different species’ OCR (negative), as described previously (12). We initially trained CNNs using only house mouse sequences [motor cortex: MouseMotorCortexModel; liver: previously published (12)] to demonstrate that a CNN trained in one species could make accurate predictions in species with different levels of relatedness that were not used in training (fig. S1 and tables S2 and S3) (52). We next trained multispecies CNNs for both motor cortex (MultiSpeciesMotorCortexModel) and liver (MultiSpeciesLiverModel) using data from house mouse (Mus musculus) and Norway rat (Rattus norvegicus) (both in the Glires clade) and from Rhesus macaque (Macaca mulatta) (Euarchonta clade). We also included motor cortex data from Egyptian fruit bat (Rousettus aegyptiacus) and liver data from the domestic cow (Bos taurus) and pig (Sus scrofa) (all Laurasiatheria clade). The models trained on these multispecies datasets achieved overall test set performance area under the receiver operating characteristic curve (AUC) of 0.91 and area under the precision-recall curve (AUPRC) of 0.90 as well as lineage- and tissue-specific OCR accuracy AUC > 0.8 and area under the negative predictive value–specificity curve (AUNPV-Spec.) greater than the fraction of examples in smaller class for all metrics (indicated by white bars in figures) (Fig. 2, A and C; fig. S3A; and tables S4 and S5), far exceeding the performance of the logistic regression (table S1).
Fig. 2. MultiSpeciesMotorCortexModel and MultiSpeciesPVModel performance.

(A and B) Area under the receiver operating characteristic curve (AUC), area under the negative predictive value-specificity curve (AUNPV-Spec.), and area under the precision-recall curve (AUPRC). Results are for the full test set, clade-specific OCRs and non-OCRs, and OCRs shared with another tissue/brain region/cell type (positive) versus tissue/brain region/cell type-specific OCRs in that other tissue/brain region/cell type (negative) [described in the “Detailed description of model performance figures” section of the supplementary materials (52)] for MultiSpeciesMotorCortexModel (A) and MultiSpeciesPVModel (B). Orths., orthologs. The ideal performance is 1, and the horizontal white bar indicates the performance that would be expected from a randomly guessing model, which is the fraction of examples in the minority class for AUNPV-Spec. and AUPRC. (The AUC from random guessing is 0.5.) (C and D) The negative relationship between the average house mouse OCR ortholog MultiSpeciesMotorCortexModel (C) and MultiSpeciesPVModel (D) predictions for Glires species and the time [millions of years ago (MYA)] at which each species diverged from house mouse, where each point corresponds to a different species. The dashed line is the average prediction for the negative test set across all species used to train the model. Prediction standard deviations for MultiSpeciesMotorCortexModel and MultiSpeciesPVModel are given in fig. S2, C and D, respectively. (E and F) Violin plots comparing the first principal component for the embeddings from the first fully connected layer of MultiSpeciesMotorCortexModel (E) and MultiSpeciesPVModel (F) for positives and negatives from each species as well as European rabbit and bottlenose dolphin orthologs of house mouse positives.
We also evaluated the phylogeny-matching correlations, which quantify the relationship between predictions at OCR orthologs and distance from the species in which an OCR was identified, a relationship that we would expect to be negative because open chromatin status is more likely to be different in a species that is more distantly related from the species in which the open chromatin was identified. The phylogeny-matching correlations were Pearson correlation coefficient (r) < −0.95 and Spearman correlation < −0.75 (figs. S2, A, C, and E, and S3, B to F). To determine whether our phylogeny-matching correlation results were likely to be explained by the models learning different sequence embeddings for different species, we computed the first principal component of the outputs of the first fully connected layer of each model and compared the distributions of these for house mouse positives with positives and negatives from each species for which we have open chromatin data, European rabbit (selected because it is the most distantly related Glires species from house mouse in Fig. 2C) orthologs, and bottlenose dolphin (selected because it has a large brain size residual, is a vocal learner, and is not closely related to any species with open chromatin data) orthologs. We found that the first principal component of these embeddings, which explained 34.2 and 34.9% of the variance for MultiSpeciesMotorCortexModel and MultiSpeciesLiverModel, respectively, tended to be more similar between house mouse positives and positives from other species than between house mouse positives and negatives, suggesting that the model is learning a consistent sequence embedding across species (Fig. 2E, fig. S3F, and tables S6 and S7). In addition, the values for the other Glires and bottlenose dolphin orthologs of house mouse positives tended to be distributed in between those of the mouse positives and negatives, with the bottlenose dolphin orthologs tending to have more values closer to those of house mouse negatives, suggesting that the model is learning that OCR orthologs in more distantly related species tend to have sequence compositions more similar to negatives than to positives, matching previously demonstrated trends (Fig. 2E; figs. S2, G, I, and K, and S3F; and tables S6 and S7) (49, 65, 66).
We then used the models to make predictions at house mouse motor cortex OCR orthologs, which we found using the Zoonomia Cactus alignment, as this alignment is reference-free and can account for multiple types of structural rearrangements, including translocations and inversions (10, 67). We obtained orthologs in 222 diverse boreoeutherian Zoonomia mammal genomes, limiting ourselves to the clades for which open chromatin data were available instead of using all 240 mammalian genomes. To further evaluate the reliability of our predictions, we clustered the species hierarchically by comparing the vector of MultiSpeciesMotorCortexModel predictions made on all OCR orthologs in each species and found that the cluster hierarchy was similar to the phylogenetic tree (68), with all but a few species clustering correctly by clade (Fig. 3, fig. S4, and data S1) (52).
Fig. 3. Heatmap of MultiSpeciesMotorCortexModel predictions for a subset of 1000 OCRs, clustered by OCR with predictions as features.

Predictions of OCR ortholog open chromatin are shown for 1000 randomly selected motor cortex OCRs with orthologs in at least 75% of species, with each row corresponding to one OCR and each column corresponding to one species. Predictions are shown on a white (closed) to red (open) scale, with missing (species, OCR) pairs shown in light gray. The OCRs (rows) are ordered according to the results of a hierarchical clustering with Ward’s minimum variance method, where the distance between two OCRs was defined as the cosine similarity of activity predictions in species for which both OCRs have usable orthologs (12). Species are ordered by their position in the phylogenetic tree; the approximate positions of species in selected clades are shown along the bottom, and illustrated species are listed in table S26, with the exception of the bat, which is an Egyptian fruit bat. Species colored black are those with data used in model training, and species colored dark gray are those for which we have only predicted open chromatin.
We then trained CNNs to predict open chromatin in PV+ interneurons and in retina, which required developing a new negative set construction approach owing to having data from only two species (figs. S1, S7, and S9 to S11, and tables S8 to S13) (52). We chose to train models for PV+ interneurons separately from those for bulk motor cortex because, while they are critical in cortical microcircuits and human brain disorders, including schizophrenia (69, 70), they are a minority population, representing 4 to 8% of neurons and 2 to 4% of the total cell population in the mouse cortex (71). Given this sparsity, our bulk motor cortex open chromatin data may not capture OCRs that are specific to PV+ interneurons. In fact, ~30% of mouse PV+ OCRs do not overlap any bulk motor cortex OCRs, including non-reproducible peaks. We began by quantifying the regulatory code conservation of PV+ interneurons and retina by running motif discovery (72) on OCRs from each species for which data were available. For each of PV+ interneurons and retina, we found motifs for many of the same TFs in both species, and some of these TFs have known regulatory roles in PV+ interneurons and retina, respectively (52, 65).
To ensure that CNNs for predicting PV+ interneuron and retina open chromatin could make accurate predictions in species not used for training, we first trained and evaluated CNNs to predict PV+ interneuron (MousePVModel) and retina (MouseRetinaModel) open chromatin using only house mouse sequences (52). We then trained CNNs to predict PV+ interneuron (MultiSpeciesPVModel) and retina (MultiSpeciesRetinaModel) open chromatin using sequences from both house mouse and human. Both MultiSpeciesPVModel and MultiSpeciesRetinaModel achieved AUC > 0.70 and AUPRC and AUNPV-Spec. greater than the fraction of examples in minority class for all criteria as well as phylogeny-matching Pearson r < −0.60 and Spearman correlation < −0.40 (Fig. 2, B, D, and F; figs. S2 and S5, A to F; and tables S14 to S17) (49, 65). Although this performance is not as strong as the performance of MultiSpeciesMotorCortexModel and MultiSpeciesLiverModel, our evaluation sets tended to have lower positive:negative ratios than our evaluation sets for the motor cortex and liver models (tables S8 and S9) owing to the human data being substantially shallower than the datasets for other combinations of tissues and species (37, 40), and the performance is substantially better than would be expected from a randomly guessing model (Fig. 2B and fig. S5A).
We expect models for specific tissues to capture sequence signatures of motifs of TFs involved in those tissues. We evaluated this for our models by comparing the groups of nucleotides the models found to be important to datasets of known TF motifs (figs. S5G and S6 to S8) (52, 73–75). MultiSpeciesMotorCortexModel and MultiSpeciesLiverModel seemed to have learned sequence patterns similar to motifs of TFs involved in motor cortex and liver, respectively, such as MEF2C (myocyte-specific enhancer factor 2C) for motor cortex (76, 77) and HNF4A (hepatocyte nuclear factor 4-alpha) (78, 79) for liver, as well as sequence patterns that do not match any known TF motif (figs. S6 to S8) (52).
Applying TACIT to mammalian phenotypes A framework for associating predicted open chromatin with phenotypes
We applied TACIT to motor cortex and PV+ interneuron OCR orthologs to identify individual OCRs whose predicted open chromatin across species is associated with neurological phenotypes (Fig. 1, table S17, and data S2). We applied the phylolm and phyloglm methods (17) for continuous and binary traits, respectively. These methods are sped-up versions of phylogenetic generalized least squares (80, 81). We used them to test for a relationship between each OCR ortholog’s open chromatin predictions and relevant phenotype annotations across species that cannot be explained by the species phylogeny alone. To minimize false positives, we implemented phylogenetic permulations, which are permutation tests that preserve the general topology of the phenotype tree (18), enabling us to evaluate the significance of each OCR–phenotype relationship against a background distribution of shuffled phenotypes with similar phylogenetic structures (52).
TACIT identifies motor cortex OCRs associated with the evolution of brain size
Applying TACIT with MultiSpeciesMotorCortexModel (figs. S12, A and B, and S13; table S18; and data S3) (52) identified 49 brain size–associated motor cortex OCRs–OCRs associated with brain size residual after Benjamini-Hochberg false discovery rate (FDR) correction (q < 0.15) (82). We note that the 98,912 OCRs we tested with TACIT are the same OCRs that we tested with RERconverge [with the exception of 27 OCRs tested for TACIT that could not be tested for RERconverge with the settings we used (52)] (21), which identified only one association, so these two analyses had approximately the same multiple hypothesis testing burden. Moreover, we found almost no correlation between the TACIT P values and OCR orthologs’ phyloP scores [Pearson r < 0, coefficient of determination (R2) < 0.00129] or distances from the closest TSS (Pearson r < 0, R2 < 0.000286), demonstrating the value in leveraging candidate enhancer activity conservation instead of nucleotide-level conservation and proximity to TSSs in identifying candidate enhancers associated with phenotype evolution (tables S19 and S20) (19, 52, 83).
We then examined all genes with TSSs within 1 Mb of the 49 brain size–associated OCRs. Of these 49 OCRs, 42 are near genes whose encoded proteins have roles in brain development or brain tumor growth (listed in table S21); 22 of these 42 have orthologs that are physically close to one of those nearby genes in either human or mouse cortices according to chromatin conformation capture data (q < 0.05 for a test of an interaction with the 10-kb bin containing the TSS; 15 of 37 OCR-gene interactions tested in mouse and 13 of 28 OCR-gene interactions tested in human; table S22), potentially reflecting functional enhancer-promoter looping (52, 84). We selected a tolerant FDR threshold of q < 0.15 because we view the reported associations in part as hypotheses for further investigation, and we found potentially relevant gene neighborhoods and chromatin conformation capture data contacts for many OCRs with q values between 0.1 and 0.15 (table S22).
Of the 42 brain size–associated OCRs near brain development and tumor growth genes, 32 are near genes with human mutations implicated in neurological disorders, including 14 OCRs near genes in which mutations have been reported to cause microcephaly or macrocephaly (table S21 and fig. S14, A to N) (52, 85). Furthermore, motor cortex OCRs with human orthologs near [within 1 Mb in Genome Reference Consortium Human Build 38 (hg38) coordinates] genes mutated in microcephaly or macrocephaly tend to have stronger associations with brain size residual than other OCRs. Specifically, OCRs near genes mutated in microcephaly or macrocephaly exhibit a significantly shifted-lower distribution of the number of successful trials out of 10,000 than do other motor cortex OCRs with human orthologs (one-tailed Wilcoxon rank-sum test, P = 0.0127, statistic = −2.23; fig. S12A) (52), where a successful trial is a permulated phenotype that better correlates with the OCR’s predicted activity than the true phenotype. We note that this trend seems to be present but weaker for models with lower test set AUPRC across our evaluation criteria (tables S23 and S24) (52).
One of the brain size–associated OCRs, chr18: 81802310–81802951 (mm10), is ~800 kb downstream from the TSS of the gene Sall3 (spalt-like transcription factor 3). Sall3 is the closest gene upstream and fourth-closest gene overall to this OCR. The three closer genes are Galr1 (galanin receptor 1), Mbp (myelin basic protein), and Zfp236 (zinc finger protein 236), of which Mbp also has a connection to brain development (86). Hi-C from adult human cortex (84) shows that the bin containing the human ortholog of this OCR is close to SALL3 in 3D space (FastHiC q = 1.30 × 10−11; table S22) (87) but does not significantly physically interact with MBP (q = 0.412). This OCR displays a positive association with brain size residual both overall (q = 0.059) and within mammalian clades with especially large variations in brain size residual, including the great apes and cetaceans (Fig. 4A). Sall3 is a member of the conserved spalt-like family of transcription factors, which are important in development in metazoans, and loss of Sall3 in house mice is lethal because it causes a loss in cranial nerve development (88, 89). Although a specific role of Sall3 in the motor cortex has not been described, Sall3 regulates the maturation of neurons in other regions of the mouse brain (89, 90), and Sall3 or SALL3 is expressed in developing house mouse motor neurons (89) and the human cerebral cortex (91).
Fig. 4. Examples of associations between predicted motor cortex OCR ortholog open chromatin and brain size residual.

(A to D) Each point represents an ortholog of the OCR in question in one species; species are grouped along the x axis by clade, as shown by the silhouettes and tree below (C) and (D) (table S26). Points are colored by brain size residual following the scale at the bottom of the figure. The permulations-based Benjamini-Hochberg q-values and the coefficient on the predicted open chromatin returned by phylolm are in the lower right of each panel. The hominoid and cetacean clades are highlighted by gray boxes in each panel, and scatterplots of predicted motor cortex open chromatin versus brain size residual for these clades are in the inset plots in each panel. Note that the lines in the inset plots are not based on the phylogenetic regression we used for TACIT, which we ran across all 222 Boreoeutherian mammals and not in specific clades, are for illustration purposes only. (A) Positive association between predicted motor cortex open chromatin and brain size residual for a motor cortex OCR in the Sall3 locus, chr18:81802310–81802951 (mm10). (B) Positive association between predicted motor cortex open chromatin and brain size residual for a motor cortex OCR in the Lrig1 locus, chr15:40082805–40083380 (mm10). [(C) and (D)] Negative association between predicted motor cortex open chromatin and brain size residual for two motor cortex OCRs in the SATB1 locus, chr17:52351209–52351928 (mm10) and chr2:174466184–174466517 (rheMac8), within Laurasiatheria and Euarchontoglires, respectively. The latter OCR has no orthologs in Lagomorpha, which is omitted from (D). Boreoeutherian mammal-wide panels are shown in fig. S15.
We also identified OCR chr2:75345159–75346046 (rheMac8) as having predicted open chromatin negatively associated with brain size residuals (q = 0.11), with an especially strong negative association in cetaceans and great apes (Fig. 4B). The closest gene to this OCR is LRIG1 (leucine rich repeats and immunoglobulin like domains 1), whose TSSs are ~250 kb upstream of the OCR. LRIG1 slows and delays the differentiation of neural stem cells (92, 93). While this OCR is also near other genes, none of those genes has a known role in brain size. This OCR is in physical proximity to Lrig1 in mouse cortical cells (FitHiC2 q= 0.0100; table S22). It also has strongly significant contact with LRIG1 in the human cortex (FastHiC q = 3.31 × 10−14; table S22), suggesting that this OCR’s 3D connection to the gene it regulates may have been conserved more strongly than its activity in the motor cortex.
We additionally identified two brain size–associated motor cortex OCRs, mm10 chr17: 52351209–52351928 and rheMac8 chr2:174466184–174466517, near SATB1 (SATB homeobox 1)—a gene for which specific mutations can result in either microcephaly or macrocephaly (94) (Fig. 4, C and D, and fig. S14, E and I). For both associations, predicted open chromatin is associated with small brain size residual (q = 0.11 and 0.085, respectively). Their human orthologs are each ~500 kb from the TSS of the gene, where one is upstream and the other is downstream. Satb1/SATB1 is the second-closest gene to each, and the closer genes, Kcnh8 (potassium voltage-gated channel subfamily H member 8) and TBC1D5 (TBC1 domain family member 5), have no known role in brain growth (95, 96). The former OCR does contact Satb1 in mouse cortical cells (FitHiC2 q = 3.49 × 10−3; table S22). The latter OCR does not have an identified mouse ortholog, so we could not evaluate its proximity in mouse; it does not have a significant contact with SATB1 in human cortex (FastHiC q = 0.435; table S22), but, because the human OCR ortholog is predicted to be closed, this does not indicate a lack of relationship between this OCR and SATB1 in small-brained mammals.
The associations seem to be driven in large part by cetaceans (Fig. 4C) and great apes (Fig. 4D), both of which have a large variation in brain size residual (97). In particular, the latter OCR (Fig. 4D) is predicted to be active in all great apes except for humans, the great ape with the largest brain size residual. In humans, most reported cases of SATB1-associated macrocephaly at birth were associated with a mutation that disrupts a large portion of the protein product, whereas microcephaly was usually associated with SATB1 missense mutations (94). This pattern is consistent with the significant negative associations between predicted open chromatin and brain size residual, assuming that the OCRs we identified activate the expression of SATB1. Determining whether an OCR activates or represses gene expression is difficult because many OCRs are bound by both activating and repressive TFs, the motifs of many repressive TFs have never been assayed, and both activation and repression can be done by cofactor proteins that do not directly bind DNA (98–100).
Among the other motor cortex OCRs near genes mutated in macro- and microcephaly is the negatively associated (q = 0.12) OCR chr2:11867277–11867712 (rn6), which is only 69 kb from the Mef2c gene. This OCR has a strong Hi-C contact to MEF2C in human (FastHiC q = 1.16 × 10−23; table S22). In addition to being mutated in a neurodevelopmental disorder that frequently includes microcephaly (76, 101), Mef2c is known to be a critical transcription factor in the brain (76, 102, 103), and its motif was learned by our motor cortex models (figs. S6 and S7).
TACIT identifies PV+ interneuron OCRs associated with the evolution of brain size
We also applied TACIT with MultiSpeciesPVModel to identify PV+ interneuron OCRs whose predicted activities across Euarchontoglires (the clade with primates, rodents, and their closest relatives—we did not have PV+ interneuron open chromatin data from other clades) are associated with brain size residual according to phylolm with phylogenetic permulations (fig. S12C; tables S18 and S25; and data S3). We identified 15 OCRs whose PV+ interneuron predicted open chromatin has an association with species’ brain size residuals after a FDR correction (q < 0.15) (table S25), 12 of which are house mouse OCRs for which predicted open chromatin is associated with having a smaller brain size residual. We identified four PV+ interneuron OCRs that are significantly negatively associated with brain size residual and are within 1 Mb of a gene that is mutated in macrocephaly or microcephaly (fig. S14, O to R, and table S25). Two of those OCRs—chr13:114757413–114757913 (mm10; q = 0.092) and chr13:114793237–114793737 (mm10; q = 0.035)—are, respectively, ~60 kb and ~25 kb from the Mocs2 (molybdenum cofactor synthesis 2) gene, which is the closest gene to both. Both have strong associations with brain size residual within Euarchonta (primates and their closest relatives), especially great apes, and the first also has some association within Glires (rodents and their closest relatives) (Fig. 5 and fig. S14, O and Q). Mocs2 is one of four genes involved in molybdenum cofactor biosynthesis (104). Molybdenum cofactor deficiency in humans is a rare, fatal disease marked by intractable seizures, hypoxia, and microcephaly (105). We also identified an OCR, chr1:95762160–95762660 (mm10; q = 0.041), that is ~100 kb away from the gene St8sia4 (ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 4), which is important for the development and density of interneurons—including PV+ interneurons—in the cortex (106, 107).
Fig. 5. Examples of associations between predicted PV+ interneuron OCR ortholog open chromatin and brain size residual.
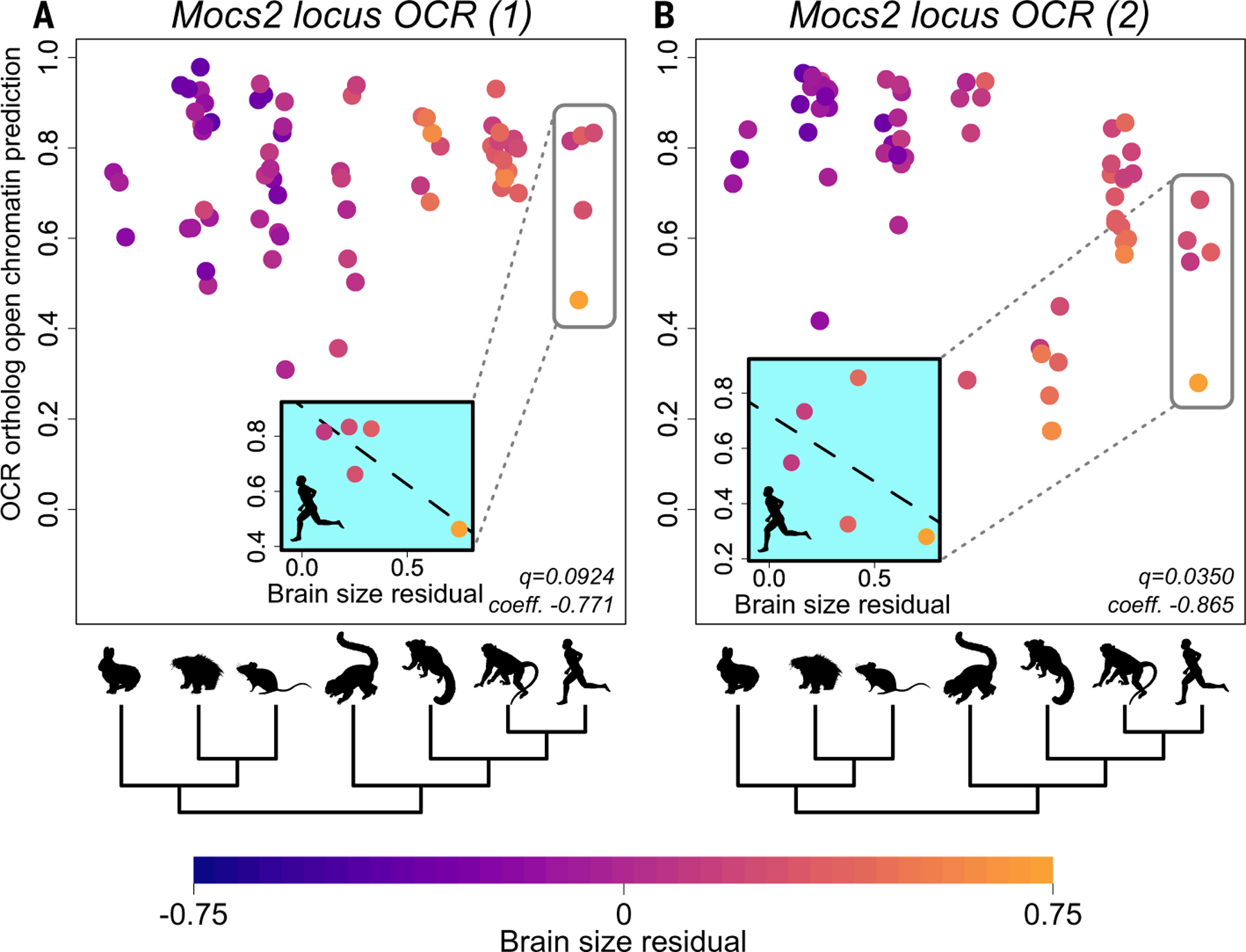
(A and B) Each point represents an ortholog of the OCR in question in one species; species are grouped along the x axis by clade, as shown by the silhouettes and tree below (table S26). Points are colored by brain size residual following the scale at the bottom of the figure. The permulations-based Benjamini-Hochberg q-values the coefficient and the predicted open chromatin returned by phylolm are in the lower right of each panel. Negative association within Euarchontoglires between predicted PV+ interneuron open chromatin and brain size residual of two PV+ interneuron OCRs in the Mocs2 locus, chr13:114757413–114757913 (mm10) (A) and chr13:114793237–114793737 (mm10) (B), respectively. The hominoid clade is highlighted by a gray box in each panel, and scatterplots of predicted PV+ interneuron open chromatin versus brain size residual in Hominoidea are in the inset plots. Note that the lines in the inset plots are for illustration purposes only and are not based on the phylogenetic regression we used for TACIT; we ran the phylogenetic regression across all Euarchontoglires and not in specific clades.
Notably, there is no overlap between the bulk motor cortex OCRs and PV+ interneuron OCRs with predicted activity that are significantly associated with brain size residual. In fact, no house mouse OCR ortholog from either set is within 3 Mb of a house mouse OCR ortholog from the other set, suggesting that the OCRs are involved in regulating different genes. We also used MultiSpeciesLiverModel to identify liver OCRs associated with brain size residual (q < 0.15) and found that none of those OCRs overlapped the associated motor cortex OCRs (tables S18 and S27 and data S3) (52); only one liver OCR is within 1 Mb of a motor cortex or a PV+ interneuron OCR with an association. This highlights the complementary information provided by using TACIT OCRs from different tissues as well as from using both bulk and specific cell type data.
TACIT identifies PV+ interneuron and motor cortex OCRs in loci associated with the evolution of solitary and group living
Next, we used TACIT with a targeted approach to examine relationships between predicted PV+ interneuron open chromatin from MultiSpeciesPVModel and social organization including solitary living, which we define as spending little time with nonprogeny members of the same species outside of mating, as well as heterogeneous group-living lifestyles (108). PV+ interneurons are implicated in regulating social behaviors and in neuropsychiatric disorders with social components such as autism spectrum disorder (ASD) and schizophrenia in humans (109). Molecular evidence for PV+ interneuron involvement suggests associated transcriptional changes. For example, PVALB was the most strongly down-regulated transcript in ASD brain tissue compared with healthy controls and in animal models of monogenetic neurodevelopmental syndromic disorders (110, 111), and single-nucleus RNA sequencing performed on brain tissue of humans with schizophrenia revealed substantially affected gene expression in PV+ interneurons (112, 113). Manipulation of psychiatric genes in PV+ interneurons induced social deficits in mice, whereas similar manipulations in other neuronal cell types had different effects (114). Given the impact of PV+ interneuron gene expression on social behaviors, we hypothesized that selection on PV+ interneuron open chromatin may be associated with social structure transitions in mammals.
Before investigating our results, we evaluated the presence of a biologically plausible signal within TACIT results for PV+ interneurons and solitary living using the MultiSpeciesPVModel enhancer activity predictions genome-wide with 10,000 trials (table S18 and data S3). To define a set of candidate enhancers likely to be enriched for neuronal function and potentially social function, we divided PV+ OCRs into two groups: those that overlapped a schizophrenia-associated genetic variant (115) and those that did not. Despite a small foreground size, the set of PV+ interneuron OCRs with schizophrenia-associated variants had a somewhat shifted-lower distribution of number of successful trials out of 10,000 for association with solitary living compared with the distribution for other PV+ interneuron OCRs (one-tailed Wilcoxon rank-sum, P = 0.078, statistic = −1.42; fig. S12D) (52). That is, OCRs overlapping schizophrenia-associated single-nucleotide polymorphisms were, overall, more likely to have a stronger association with solitary living than with a null phenotype with a similar tree topology compared with other OCRs, lending support to the candidate enhancer-phenotype prediction outputs from TACIT.
One challenge of using TACIT is that tens to hundreds of thousands of OCRs are tested, so substantial multiple hypothesis correction is necessary. The number of tested OCRs can be limited ifa small number of genomic loci have been hypothesized to be involved in a trait. For solitary living and group living, we chose to focus on the 1,661,222-bp Williams-Beuren Syndrome (WBS) deletion region (Fig. 6A), where haploinsufficiency causes increased sociability, intellectual disability, and enhanced verbal fluency in human patients and deletion causes a decrease in nose-to-nose sniffing in mice (116). This region has also been proposed to be associated with sociability differences between dogs and wolves (117), but this is not functionally resolved owing to fully confounded phylogenetic relationships and social traits in canines. TACIT provides an opportunity to assess social living strategy-enhancer associations within the WBS locus across many mammals while accounting for phylogenetic relationships.
Fig. 6. Associations between predicted PV+ interneuron and motor cortex OCR ortholog open chromatin and solitary living.

(A) Human WBS deletion region. The locations of the PV+ interneuron and motor cortex OCRs [(B) and (C)] near the gene GTF2IRD1 are in yellow and green, respectively. (B) Marginal negative association between predicted PV+ interneuron open chromatin and solitary living of a PV+ interneuron OCR near GTF2IRD1 and GTF2I, chr5:134485808–134486308 (mm10). (C) Negative association between predicted motor cortex open chromatin and solitary living of a motor cortex OCR near GTF2IRD1 and GTF2I, chr3:42408296–42408946 (rheMac8). In (B) and (C), each point represents an ortholog in one species; points are grouped along the x axis by the clade of the species represented, as shown by the silhouettes and tree below (C) (table S26). Points are colored to indicate solitary versus nonsolitary living following the key at the lower right. The permulations-based Benjamini-Hochberg q-value and the coefficient for the predicted open chromatin returned by phyloglm are shown in the lower right of (B) and (C).
When applying TACIT to only the WBS locus, we identified a house mouse PV+ interneuron OCR (out of two OCRs in this locus) 29 kb upstream of Gtf2ird1 (general transcription factor II I repeat domain–containing 1) and ~168 kb upstream of Gtf2i (general transcription factor II I) that was positively associated with group living (q = 0.043) and negatively associated with solitary living (q = 0.14) (Fig. 6B, table S18, and data S3). To evaluate whether this association was limited to PV+ interneurons, we also evaluated the relationship between predicted bulk motor cortex open chromatin from MultiSpeciesMotorCortexModel and solitary as well as group living (table S18 and data S3). We found one OCR with both a significant negative association with solitary living (q = 8.5 × 10−3) (Fig. 6C) and a significant positive association with group living (q = 0.016). This OCR’s human ortholog (OCR was originally found in macaque) is in an intron of GTF2IRD1 that is ~27 kb from its nearest TSS and ~177 kb from the TSS for GTF2I but does not overlap the OCR identified for PV+ interneurons. We also found a second OCR with some negative association (q = 0.094) with group living. Of the 27 protein-coding genes in the WBS locus, Gtf2i is the only gene with a duplication associated with separation anxiety and a heterozygous deletion associated with increased nose-to-nose contact in mice (118, 119). We additionally evaluated the relationship between predicted liver open chromatin and solitary as well as group living using MultiSpeciesLiverModel but did not obtain any statistically significant relationships after multiple hypothesis correction.
TACIT identifies OCRs associated with the evolution of vocal learning
We applied TACIT to vocal learning, the ability to modify vocal output as a result of social experience, which has convergently evolved across mammals and been associated with convergent patterns of gene expression in the motor cortex (2, 120, 121). We identified dozens of OCRs displaying convergent patterns of predicted open chromatin after FDR correction (q < 0.15) for motor cortex tissue (MultiSpeciesMotorCortexModel) and for PV+ interneurons (MultiSpeciesPVModel), which are described in more depth in our other manuscript (35). One of the motor cortex OCRs lies 88 kb from Vip (vasoactive intestinal peptide), whose expression in the motor cortex has been associated with vocal learning (2). Another OCR is 715 kb from TSHZ3 (teashirt zinc finger homeobox 3) (35). TSHZ3 is involved in the formation of corticostriatal circuits, which play a central role in vocal learning behavior in mammals and birds, and its disruption in the human population is associated with a form of autism that includes delayed or disrupted speech acquisition (121, 122).
Discussion
We sought to use the hundreds of aligned genomes of the Zoonomia project to discover genetic variation across placental mammals associated with the evolution of complex neural phenotypes. We first applied RERconverge (21, 22, 123) to identify brain size residual–associated accelerated or constrained nucleotide-level conservation across genes and candidate enhancers for 158 species. Despite the large number of genomes and reliable phenotype annotations, we found only one significantly associated locus, although we cannot rule out that alternative methods for detecting convergent evolution in aligned genes or enhancers could still find associated regions. While RERconverge and other nucleotide-level conservation-based approaches have identified enhancers associated with phenotypes that overlap some of the most conserved noncoding regions of the genome (22, 124), we realized that such methods’ utility is limited in regions with high functional conservation but low to moderate nucleotide-level conservation.
To overcome the limitations in using the alignment of individual nucleotides as a proxy for conservation, we present TACIT, a method for associating genotypes to phenotypes using machine learning predictions of tissue- or cell type–specific open chromatin. TACIT accounts for the conservation of enhancer activity in the presence of low sequence conservation and can capture the tissue- and cell type–specificity of enhancer activity (12) through machine learning models that learn the conserved regulatory code underlying enhancer activity in a tissue or cell type of interest. We provide a community resource of annotated predicted open chromatin for more than 400,000 OCRs from four tissues and cell types across 222 mammalian species by making it available on the University of California, Santa Cruz (UCSC) Genome Browser (https://genome.ucsc.edu/cgi-bin/hgGateway?genome=Homo_sapiens&hubUrl=https://cgl.gi.ucsc.edu/data/cactus/241=mammalian-2020v2-hub/hub.txt) (125).
We applied TACIT to identify tissue- and cell type–specific OCRs whose predicted open chromatin status across species is associated with brain size residual, solitary living, group living, and vocal learning, including OCRs near genes that were previously implicated in these phenotypes, providing potential mechanisms for how these genes are regulated. Specifically, we identified motor cortex and PV+ interneuron OCRs associated with brain size residual that are near genes whose mutations are associated with microcephaly and macrocephaly in humans. While many of these genes are known for roles in brain development that may influence brain size, the OCRs that regulate them may continue to be open in the adult brain. We also found motor cortex OCRs with a strong brain size residual association in cetaceans, providing candidate mechanisms for the evolution of brain size beyond human-specific deletions identified in earlier work (9). In addition, OCRs within the WBS deletion region that are associated with solitary living reside near a critical gene for WBS presentation and a gene associated with social behavior in mice (118, 119). Genome wide, the associations of PV+ interneuron OCRs with solitary living are correlated with whether the OCR overlaps a genome-wide association study (GWAS) hit for schizophrenia, which suggests that OCRs involved in the evolution of phenotypes may also be involved in related disorders. To be confident that the OCRs we identified have enhancer activity that differs between species, we would need to use reporter assays to test the OCR orthologs’ enhancer activity in multiple species. Unfortunately, current technology limits largescale reporter assays to cell lines, and there is no cell line that captures the transcriptional regulatory program of motor cortex and PV+ interneurons or protocol for differentiating these specific cell types from induced pluripotent stem cells. In addition, to thoroughly demonstrate that these OCRs regulate the nearby genes associated with the phenotypes, we would need to do experiments such as CRISPR followed by RNA quantitative polymerase chain reaction to knock out the OCR and show that the knockout causes a change in the expression of the nearby gene, but doing such experiments for more than one OCR at a time is currently feasible in only cell lines. Furthermore, considering genes with TSSs within 1 Mb may limit our ability to identify real gene–OCR relationships (126), and data measuring 3D genome interactions is not currently available from motor cortex in species other than human and house mouse or from PV+ interneurons in any species. As such data become available at higher resolution and in additional species, tissues, and cell types, our ability to link candidate enhancers associated with phenotypes to the genes they likely regulate will improve.
While we previously used data from at least three species for model training (12), in this study, we developed a strategy for negative set construction that allowed us to train accurate models using data from only two species. This enabled us to train models that accurately predict whether sequence differences across species in PV+ interneuron OCR orthologs are associated with PV+ interneuron open chromatin changes, demonstrating that the regulatory code is conserved across Euarchontoglires not only at the bulk tissue level but also in a specific neuronal cell type. We have found that, when the relevant data were available, including data from more clades enabled us to accurately predict OCRs in more distantly related species (12). With our confident predictions in diverse clades, we identified OCRs associated with phenotypes in a variety of clades, such as the OCR near Lrig1 associated with the evolution of brain size residual in the Cetacea infraorder within Laurasiatheria (the clade that includes bats, carnivorans, ungulates, and their close relatives). Predictions in more species also provide us with the power to identify OCRs exhibiting weaker associations with a phenotype across multiple lineages, such as the OCR near SALL3 associated with the evolution of brain size residual in both Euarchonta and Laurasiatheria.
Unlike phyloP or PhastCons scores, the broad application of TACIT is limited by the availability of high-quality enhancer activity data from the same tissue or cell type in multiple species. TACIT requires enhancer activity data from at least two species for evaluating the corresponding machine learning models, and different datasets may need to be filtered differently depending on data quality and genome size. Biases due to data quality and filtering need to be evaluated before model evaluations are done on held-out test sets. Additionally, predictions are currently limited to identifiable orthologs of experimentally identified candidate enhancers, meaning that we are not able to capture enhancers that are not active in the experimentally assayed species, cell types, developmental stages, or conditions or use enhancers that cannot be aligned with existing alignment methods, which are more common when applying TACIT to more distantly related species. Furthermore, our approach assumes that the evolution of a phenotype is controlled by the same candidate enhancer across species. There are likely many phenotypes controlled by genes that are not activated by the same enhancer in every species, as previous studies have shown that many enhancers are deleted or inserted via transposable elements in some species despite the expression of the genes they regulate being conserved (127, 128). We also treat missing or unusable OCR orthologs as missing data, but some of these may have been lost during evolution, making them negatives. Moreover, neither our models nor our phenotype annotations are perfect, which could cause incorrect association results, and our lack of known positive and negative open chromatin–phenotype associations often makes evaluating the amount of noise that TACIT can tolerate infeasible. Finally, our approach assumes that the regulatory code in our tissue or cell type of interest is conserved across the species in which we are making predictions, an assumption that may be violated in some tissues and cell types. For example, this may explain the suboptimal performance of MouseRetinaModel in predicting Euarchonta-specific open and closed chromatin (129, 130).
Exciting extensions to our approach include training models to predict whether sequence differences cause changes in candidate enhancer activity genome-wide, jointly modeling cross-species predicted activity of enhancers near the same gene, using genome quality and the predicted open chromatin of OCRs in closely related species to determine when a lack of a usable OCR ortholog should be treated as a non-OCR, and evaluating more-lenient definitions of an enhancer for smaller genomes. TACIT could also be extended to identify promoters or noncoding RNAs associated with phenotype evolution by training models to predict the promoter or noncoding RNA activity at these elements’ orthologs.
With the Zoonomia Cactus alignment of >200 mammalian genomes (10) and the wealth of publicly available enhancer activity data from matching tissues and cell types in human, house mouse, and other species, TACIT can currently be applied to identify candidate enhancers associated with the evolution of many mammalian phenotypes. Because TACIT requires enhancer activity data from tissues or cell types of interest in only a few species, it can be used to associate losses of enhancer activity with changes in a phenotype even in challenging-to-study species for which we have genomes but cannot collect tissue samples. In addition, although we trained our models for TACIT using open chromatin and CNNs, TACIT can also be applied using other assays of enhancer activity, such as H3K27ac and EP300 ChIP-seq, and using other machine learning modeling methods, such as support vector machines (30). Candidate enhancers associated with the evolution of phenotypes near genes with mutations or expression differences involved in diseases related to those phenotypes may provide mechanistic insights. We anticipate that, as more genomes and regulatory genomics data become available, TACIT will allow us to discover regulatory mechanisms governing a wide range of phenotypes.
Methods summary
We obtained open chromatin data from motor cortex, liver, PV+ interneurons, and retina from multiple species, mapped and filtered the reads, called peaks, and obtained reproducible peaks. We used the sequences underlying the reproducible peaks to train a machine learning model for predicting open chromatin in each tissue and cell type. We identified orthologs of the reproducible peaks from each tissue and cell type in 222 boreoeutherian mammals and used the corresponding machine learning models to predict open chromatin in that tissue or cell type in each species. We associated the predictions with phenotype annotations for brain size, solitary and group living, and vocal learning using phylolm for continuous and phyloglm for binary traits, computed empirical P values using phylogenetic permulations, and corrected P values using the Benjamini-Hochberg procedure (17, 18, 82).
Supplementary Material
ACKNOWLEDGMENTS
We thank C. Ellington, D. Levesque, K. Lord, and the members of the Pfenning lab for useful discussions and suggestions. We thank P. Sullivan for curating the brain size residual annotations; A. Hindle for providing annotations of which mammals spend time underground; and M. Chikina, A. Kowalczyk, and E. Saputra for consulting with us about phylogenetic permulations. We also thank the reviewers for fantastic feedback that substantially improved this manuscript. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), through the Pittsburgh Supercomputing Center Bridges and Bridges-2 Compute Clusters, which was supported by National Science Foundation grants TG-BIO200055 and ACI-1548562 (131). Portions of this research were conducted on Lehigh University’s Research Computing infrastructure, which is partially supported by NSF award 2019035.
Funding:
Funding was provided by a Carnegie Mellon University Computational Biology Department Lane Fellowship (I.M.K.); NIH NIDA DP1DA046585 grant (D.E.S., M.E.W., X.Z., A.R.B., and A.R.P.); NSF grant 2046550 (I.M.K. and A.R.P.); an Alfred P. Sloan Foundation Research Fellowship (I.M.K., M.E.W., and A.R.P.); the Carnegie Mellon University Computational Biology Department (C.S.); NSF Graduate Research Fellowship Program grant DGE1252522 (A.J.L.); NSF Graduate Research Fellowship Program grant DGE1745016 (A.J.L.); a Carnegie Mellon University Summer Undergraduate Research Fellowship (D.E.S.); NIH NIDA Fellowship grant F30DA053020 (B.N.P.); NIH UG3-MH-120094 (K.P.); NSF grant 2022046 (D.P.G.); NIH NHGRI R01HG008742 grant (E.K.K.); and a Swedish Research Council Distinguished Professor Award (K.L.-T.).
Zoonomia Consortium
Gregory Andrews1, Joel C. Armstrong2, Matteo Bianchi3, Bruce W. Birren4, Kevin R. Bredemeyer5, Ana M. Breit6, Matthew J. Christmas3, Hiram Clawson2, Joana Damas7, Federica Di Palma8,9, Mark Diekhans2, Michael X. Dong3, Eduardo Eizirik10, Kaili Fan1, Cornelia Fanter11, Nicole M. Foley5, Karin Forsberg-Nilsson12,13, Carlos J. Garcia14, John Gatesy15, Steven Gazal16, Diane P. Genereux4, Linda Goodman17, Jenna Grimshaw14, Michaela K. Halsey14, Andrew J. Harris5, Glenn Hickey18, Michael Hiller19,20,21, Allyson G. Hindle11, Robert M. Hubley22, Graham M. Hughes23, Jeremy Johnson4, David Juan24, Irene M. Kaplow25,26, Elinor K. Karlsson1,4,27, Kathleen C. Keough17,28,29, Bogdan Kirilenko19,20,21, Klaus-Peter Koepfli30,31,32, Jennifer M. Korstian14, Amanda Kowalczyk25,26, Sergey V. Kozyrev3, Alyssa J. Lawler4,26,33, Colleen Lawless23, Thomas Lehmann34, Danielle L. Levesque6, Harris A. Lewin7,35,36, Xue Li1,4,37, Abigail Lind28,29, Kerstin Lindblad-Toh3,4, Ava Mackay-Smith38, Voichita D. Marinescu3, Tomas Marques-Bonet39,40,41,42, Victor C. Mason43, Jennifer R. S. Meadows3, Wynn K. Meyer44, Jill E. Moore1, Lucas R. Moreira1,4, Diana D. Moreno-Santillan14, Kathleen M. Morrill1,4,37, Gerard Muntané24, William J. Murphy5, Arcadi Navarro39,41,45,46, Martin Nweeia47,48,49,50, Sylvia Ortmann51, Austin Osmanski14, Benedict Paten2, Nicole S. Paulat14, Andreas R. Pfenning25,26, BaDoi N. Phan25,26,52, Katherine S. Pollard28,29,53, Henry E. Pratt1, David A. Ray14, Steven K. Reilly38, Jeb R. Rosen22, Irina Ruf54, Louise Ryan23, Oliver A. Ryder55,56, Pardis C. Sabeti4,57,58, Daniel E. Schäffer25, Aitor Serres24, Beth Shapiro59,60, Arian F. A. Smit22, Mark Springer61, Chaitanya Srinivasan25, Cynthia Steiner55, Jessica M. Storer22, Kevin A. M. Sullivan14, Patrick F. Sullivan62,63, Elisabeth Sundström3, Megan A. Supple59, Ross Swofford4, Joy-El Talbot64, Emma Teeling23, Jason Turner-Maier4, Alejandro Valenzuela24, Franziska Wagner65, Ola Wallerman3, Chao Wang3, Juehan Wang16, Zhiping Weng1, Aryn P. Wilder55, Morgan E. Wirthlin25,26,66, James R. Xue4,57, Xiaomeng Zhang4,25,26
1Program in Bioinformatics and Integrative Biology, UMass Chan Medical School, Worcester, MA 01605, USA. 2Genomics Institute, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 3Department of Medical Biochemistry and Microbiology, Science for Life Laboratory, Uppsala University, Uppsala 751 32, Sweden. 4Broad Institute of MIT and Harvard, Cambridge, MA 02139, USA. 5Veterinary Integrative Biosciences, Texas A&M University, College Station, TX 77843, USA. 6School of Biology and Ecology, University of Maine, Orono, ME 04469, USA. 7The Genome Center, University of California Davis, Davis, CA 95616, USA. 8Genome British Columbia, Vancouver, BC, Canada. 9School of Biological Sciences, University of East Anglia, Norwich, UK. 10School of Health and Life Sciences, Pontifical Catholic University of Rio Grande do Sul, Porto Alegre 90619–900, Brazil. 11School of Life Sciences, University of Nevada Las Vegas, Las Vegas, NV 89154, USA. 12Biodiscovery Institute, University of Nottingham, Nottingham, UK. 13Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala 751 85, Sweden. 14Department of Biological Sciences, Texas Tech University, Lubbock, TX 79409, USA. 15Division of Vertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA. 16Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA. 17Fauna Bio Incorporated, Emeryville, CA 94608, USA. 18Baskin School of Engineering, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 19Faculty of Biosciences, Goethe-University, 60438 Frankfurt, Germany. 20LOEWE Centre for Translational Biodiversity Genomics, 60325 Frankfurt, Germany. 21Senckenberg Research Institute, 60325 Frankfurt, Germany. 22Institute for Systems Biology, Seattle, WA 98109, USA. 23School of Biology and Environmental Science, University College Dublin, Belfield, Dublin 4, Ireland. 24Department of Experimental and Health Sciences, Institute of Evolutionary Biology (UPF-CSIC), Universitat Pompeu Fabra, Barcelona 08003, Spain. 25Department of Computational Biology, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 26Neuroscience Institute, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 27Program in Molecular Medicine, UMass Chan Medical School, Worcester, MA 01605, USA. 28Department of Epidemiology & Biostatistics, University of California San Francisco, San Francisco, CA 94158, USA. 29Gladstone Institutes, San Francisco, CA 94158, USA. 30Center for Species Survival, Smithsonian’s National Zoo and Conservation Biology Institute, Washington, DC 20008, USA. 31Computer Technologies Laboratory, ITMO University, St. Petersburg 197101, Russia. 32Smithsonian-Mason School of Conservation, George Mason University, Front Royal, VA 22630, USA. 33Department of Biological Sciences, Mellon College of Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 34Senckenberg Research Institute and Natural History Museum Frankfurt, 60325 Frankfurt am Main, Germany. 35Department of Evolution and Ecology, University of California Davis, Davis, CA 95616, USA. 36John Muir Institute for the Environment, University of California Davis, Davis, CA 95616, USA. 37Morningside Graduate School of Biomedical Sciences, UMass Chan Medical School, Worcester, MA 01605, USA. 38Department of Genetics, Yale School of Medicine, New Haven, CT 06510, USA. 39Catalan Institution of Research and Advanced Studies (ICREA), Barcelona 08010, Spain. 40CNAG-CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona 08036, Spain. 41Department of Medicine and Life Sciences, Institute of Evolutionary Biology (UPF-CSIC), Universitat Pompeu Fabra, Barcelona 08003, Spain. 42Institut Català de Paleontologia Miquel Crusafont, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Barcelona, Spain. 43Institute of Cell Biology, University of Bern, 3012 Bern, Switzerland. 44Department of Biological Sciences, Lehigh University, Bethlehem, PA 18015, USA. 45BarcelonaBeta Brain Research Center, Pasqual Maragall Foundation, Barcelona 08005, Spain. 46CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona 08003, Spain. 47Department of Comprehensive Care, School of Dental Medicine, Case Western Reserve University, Cleveland, OH 44106, USA. 48Department of Vertebrate Zoology, Canadian Museum of Nature, Ottawa, ON K2P 2R1, Canada. 49Department of Vertebrate Zoology, Smithsonian Institution, Washington, DC 20002, USA. 50Narwhal Genome Initiative, Department of Restorative Dentistry and Biomaterials Sciences, Harvard School of Dental Medicine, Boston, MA 02115, USA. 51Department of Evolutionary Ecology, Leibniz Institute for Zoo and Wildlife Research, 10315 Berlin, Germany. 52Medical Scientist Training Program, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261, USA. 53Chan Zuckerberg Biohub, San Francisco, CA 94158, USA. 54Division of Messel Research and Mammalogy, Senckenberg Research Institute and Natural History Museum Frankfurt, 60325 Frankfurt am Main, Germany. 55Conservation Genetics, San Diego Zoo Wildlife Alliance, Escondido, CA 92027, USA. 56Department of Evolution, Behavior and Ecology, School of Biological Sciences, University of California San Diego, La Jolla, CA 92039, USA. 57Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA 02138, USA. 58Howard Hughes Medical Institute, Harvard University, Cambridge, MA 02138, USA. 59Department of Ecology and Evolutionary Biology, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 60Howard Hughes Medical Institute, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 61Department of Evolution, Ecology and Organismal Biology, University of California Riverside, Riverside, CA 92521, USA. 62Department of Genetics, University of North Carolina Medical School, Chapel Hill, NC 27599, USA. 63Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden. 64Iris Data Solutions, LLC, Orono, ME 04473, USA. 65Museum of Zoology, Senckenberg Natural History Collections Dresden, 01109 Dresden, Germany. 66Allen Institute for Brain Science, Seattle, WA 98109, USA.
Footnotes
Competing interests: E.K.K. is on the advisory board of Fauna Bio. All other authors declare that they have no competing interests.
Diversity and inclusion: One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Data and materials availability: Publicly available ATAC-seq data were obtained from Gene Expression Omnibus accessions GSE161374, GSE146897, GSE137311, and GSE159815; China National GeneBank accession CNP0000198; and ArrayExpress accession E-MTAB-2633. Unpublished ATAC-seq data generated by the Pfenning lab can be found under accession GSE187366. The tree used for the phenotype association pipeline can be obtained in (68). Publicly available genomes and annotations were downloaded from NCBI Assembly and the UCSC Genome Browser. Publicly available human Hi-C data were accessed at http://hugin2.genetics.unc.edu/Project/hugin/. Mouse cortex Dip-C data were downloaded from Gene Expression Omnibus (accession GSE146397). Motif discovery results and machine learning models can be found at http://daphne.compbio.cs.cmu.edu/files/ikaplow/TACITSupplement/(65). Machine learning model predictions can be obtained from the UCSC Genome Browser (https://genome.ucsc.edu/cgi-bin/hgGateway?genome=Homo_sapiens&hubUrl=https://cgl.gi.ucsc.edu/data/cactus/241-mammalian-2020v2-hub/hub.txt). New code for this work can be found in Zenodo (133).
License information: Copyright © 2023 the authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original US government works. https://www.science.org/about/science-licenses-journal-article-reuse
SUPPLEMENTARY MATERIALS
science.org/doi/10.1126/science.abm7993
Materials and Methods
Supplementary Text
Figs. S1 to S15
Tables S1 to S27
References (134–360)
REFERENCES AND NOTES
- 1.King MC, Wilson AC, Evolution at two levels in humans and chimpanzees. Science 188, 107–116 (1975). doi: 10.1126/science.1090005; pmid: 1090005 [DOI] [PubMed] [Google Scholar]
- 2.Pfenning AR et al. , Convergent transcriptional specializations in the brains of humans and song-learning birds. Science 346, 1256846 (2014). doi: 10.1126/science.1256846; pmid: 25504733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fushan AA et al. , Gene expression defines natural changes in mammalian lifespan. Aging Cell 14, 352–365 (2015).doi: 10.1111/acel.12283; pmid: 25677554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wray GA, The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet 8, 206–216 (2007). doi: 10.1038/nrg2063; pmid: 17304246 [DOI] [PubMed] [Google Scholar]
- 5.Villar D, Flicek P, Odom DT, Evolution of transcription factor binding in metazoans—mechanisms and functional implications. Nat. Rev. Genet 15, 221–233 (2014). doi: 10.1038/nrg3481; pmid: 24590227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kvon EZ et al. , Progressive loss of function in a limb enhancer during snake evolution. Cell 167, 633–642.e11 (2016). doi: 10.1016/j.cell.2016.09.028; pmid: 27768887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lettice LA, Hill AE, Devenney PS, Hill RE, Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum. Mol. Genet 17, 978–985 (2008). doi: 10.1093/hmg/ddm370; pmid: 18156157 [DOI] [PubMed] [Google Scholar]
- 8.Furniss D et al. , A variant in the sonic hedgehog regulatory sequence (ZRS) is associated with triphalangeal thumb and deregulates expression in the developing limb. Hum. Mol. Genet 17, 2417–2423 (2008). doi: 10.1093/hmg/ddn141; pmid: 18463159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLean CY et al. , Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 471, 216–219 (2011). doi: 10.1038/nature09774; pmid: 21390129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armstrong J et al. , Progressive Cactus is a multiple-genome aligner for the thousand-genome era. Nature 587, 246–251 (2020). doi: 10.1038/s41586-020-2871-y; pmid: 33177663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stefen C et al. , Phenotyping in the era of genomics: MaTrics – a digital character matrix to document mammalian phenotypic traits coded numerically. bioRxiv 2021.01.17.426960 [Preprint] (2021). 10.1101/2021.01.17.426960. [DOI]
- 12.Kaplow IM et al. , Inferring mammalian tissue-specific regulatory conservation by predicting tissue-specific differences in open chromatin. BMC Genomics 23, 291 (2022). doi: 10.1186/s12864-022-08450-7; pmid: 35410163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoonomia Consortium A comparative genomics multitool for scientific discovery and conservation. Nature 587, 240–245 (2020). doi: 10.1038/s41586-020-2876-6; pmid: 33177664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rhie A et al. , Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746 (2021). doi: 10.1038/s41586-021-03451-0; pmid: 33911273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hickey G, Paten B, Earl D, Zerbino D, Haussler D, HAL: A hierarchical format for storing and analyzing multiple genome alignments. Bioinformatics 29, 1341–1342 (2013). doi: 10.1093/bioinformatics/btt128; pmid: 23505295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Kaplow IM, Wirthlin M, Park TY, Pfenning AR, HALPER facilitates the identification of regulatory element orthologs across species. Bioinformatics 36, 4339–4340 (2020). doi: 10.1093/bioinformatics/btaa493; pmid: 32407523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho L. s. T., Ané C, A linear-time algorithm for Gaussian and non-Gaussian trait evolution models. Syst. Biol 63, 397–408 (2014). doi: 10.1093/sysbio/syu005; pmid: 24500037 [DOI] [PubMed] [Google Scholar]
- 18.Saputra E, Kowalczyk A, Cusick L, Clark N, Chikina M, Phylogenetic permulations: A statistically rigorous approach to measure confidence in associations in a phylogenetic context. Mol. Biol. Evol 38, 3004–3021 (2021). doi: 10.1093/molbev/msab068; pmid: 33739420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A, Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20, 110–121 (2010). doi: 10.1101/gr.097857.109; pmid: 19858363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Z, PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol 24, 1586–1591 (2007). doi: 10.1093/molbev/msm088; pmid: 17483113 [DOI] [PubMed] [Google Scholar]
- 21.Kowalczyk A et al. , RERconverge: An R package for associating evolutionary rates with convergent traits. Bioinformatics 35, 4815–4817 (2019). doi: 10.1093/bioinformatics/btz468; pmid: 31192356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Partha R et al. , Subterranean mammals show convergent regression in ocular genes and enhancers, along with adaptation to tunneling. eLife 6, e25884 (2017). doi: 10.7554/eLife.25884; pmid: 29035697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roscito JG et al. , Phenotype loss is associated with widespread divergence of the gene regulatory landscape in evolution. Nat. Commun 9, 4737 (2018). doi: 10.1038/s41467-018-07122-z; pmid: 30413698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang S et al. , Functionally conserved enhancers with divergent sequences in distant vertebrates. BMC Genomics 16, 882 (2015). doi: 10.1186/s12864-015-2070-7; pmid: 26519295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Villar D et al. , Enhancer evolution across 20 mammalian species. Cell 160, 554–566 (2015). doi: 10.1016/j.cell.2015.01.006; pmid: 25635462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindblad-Toh K et al. , A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011). doi: 10.1038/nature10530; pmid: 21993624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snetkova V et al. , Ultraconserved enhancer function does not require perfect sequence conservation. Nat. Genet 53, 521–528 (2021). doi: 10.1038/s41588-021-00812-3; pmid: 33782603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong ES et al. , Deep conservation of the enhancer regulatory code in animals. Science 370, eaax8137 (2020). doi: 10.1126/science.aax8137; pmid: 33154111 [DOI] [PubMed] [Google Scholar]
- 29.Siepel A et al. , Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15, 1034–1050 (2005). doi: 10.1101/gr.3715005; pmid: 16024819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L, Fish AE, Capra JA, Prediction of gene regulatory enhancers across species reveals evolutionarily conserved sequence properties. PLOS Comput. Biol 14, e1006484 (2018). doi: 10.1371/journal.pcbi.1006484; pmid: 30286077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelley DR, Cross-species regulatory sequence activity prediction. PLOS Comput. Biol 16, e1008050 (2020). doi: 10.1371/journal.pcbi.1008050; pmid: 32687525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minnoye L et al. , Cross-species analysis of enhancer logic using deep learning. Genome Res 30, 1815–1834 (2020). doi: 10.1101/gr.260844.120; pmid: 32732264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srinivasan C et al. , Addiction-associated genetic variants implicate brain cell type- and region-specific cis-regulatory elements in addiction neurobiology. J. Neurosci 41, 9008–9030 (2021). doi: 10.1523/JNEUROSCI.2534-20.2021; pmid: 34462306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wirthlin M et al. , The regulatory evolution of the primate fine-motor system. bioRxiv 2020.10.27.356733 [Preprint] (2020). 10.1101/2020.10.27.356733. [DOI]
- 35.Wirthlin ME et al. , Vocal learning-associated convergent evolution in mammalian proteins and regulatory elements. bioRxiv 2022.12.17.520895 [Preprint] (2022). 10.1101/2022.12.17.520895. [DOI] [PMC free article] [PubMed]
- 36.Halstead MM et al. , A comparative analysis of chromatin accessibility in cattle, pig, and mouse tissues. BMC Genomics 21, 698 (2020). doi: 10.1186/s12864-020-07078-9; pmid: 33028202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bakken TE et al. , Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature 598, 111–119 (2021). doi: 10.1038/s41586-021-03465-8; pmid: 34616062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li YE et al. , An atlas of gene regulatory elements in adult mouse cerebrum. Nature 598, 129–136 (2021). doi: 10.1038/s41586-021-03604-1; pmid: 34616068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miesfeld JB et al. , The Atoh7 remote enhancer provides transcriptional robustness during retinal ganglion cell development. Proc. Natl. Acad. Sci. U.S.A 117, 21690–21700 (2020). doi: 10.1073/pnas.2006888117; pmid: 32817515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cherry TJ et al. , Mapping the cis-regulatory architecture of the human retina reveals noncoding genetic variation in disease. Proc. Natl. Acad. Sci. U.S.A 117, 9001–9012 (2020). doi: 10.1073/pnas.1922501117; pmid: 32265282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le Cun Y et al. , Handwritten digit recognition: Applications of neural network chips and automatic learning. IEEE Commun. Mag 27, 41–46 (1989). doi: 10.1109/35.41400 [DOI] [Google Scholar]
- 42.Mitchell C, Silver DL, Enhancing our brains: Genomic mechanisms underlying cortical evolution. Semin. Cell Dev. Biol 76, 23–32 (2018). doi: 10.1016/j.semcdb.2017.08.045; pmid: 28864345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burger JR, George MA Jr., C. Leadbetter, F. Shaikh, The allometry of brain size in mammals. J. Mammal 100, 276–283 (2019). doi: 10.1093/jmammal/gyz043 [DOI] [Google Scholar]
- 44.Herculano-Houzel S, The remarkable, yet not extraordinary, human brain as a scaled-up primate brain and its associated cost. Proc. Natl. Acad. Sci. U.S.A 109 (suppl. 1), 10661–10668 (2012). doi: 10.1073/pnas.1201895109; pmid: 22723358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Montgomery SH et al. , The evolutionary history of cetacean brain and body size. Evolution 67, 3339–3353 (2013). doi: 10.1111/evo.12197; pmid: 24152011 [DOI] [PubMed] [Google Scholar]
- 46.Tsuboi M et al. , Breakdown of brain-body allometry and the encephalization of birds and mammals. Nat. Ecol. Evol 2, 1492–1500 (2018). doi: 10.1038/s41559-018-0632-1; pmid: 30104752 [DOI] [PubMed] [Google Scholar]
- 47.Sullivan PF et al. , Leveraging base pair mammalian constraint to understand genetic variation and human disease. Science 380, eabn2937 (2023). doi: 10.1123/science.abn2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kundaje A et al. , Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). doi: 10.1038/nature14248; pmid: 25693563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ji L, Kim N-H, Huh S-O, Rhee HJ, Depletion of inositol polyphosphate 4-phosphatase II suppresses callosal axon formation in the developing mice. Mol. Cells 39, 501–507 (2016). doi: 10.14348/molcells.2016.0058; pmid: 27109423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li D et al. , Pathogenic variants in SMARCA5, a chromatin remodeler, cause a range of syndromic neurodevelopmental features. Sci. Adv 7, eabf2066 (2021). doi: 10.1126/sciadv.abf2066; pmid: 33980485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou L, Talebian A, Meakin SO, The signaling adapter, FRS2, facilitates neuronal branching in primary cortical neurons via both Grb2- and Shp2-dependent mechanisms. J. Mol. Neurosci 55, 663–677 (2015). doi: 10.1007/s12031-014-0406-4; pmid: 25159185 [DOI] [PubMed] [Google Scholar]
- 52.Materials and methods are available as supplementary materials.
- 53.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). doi: 10.1038/nmeth.2688; pmid: 24097267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buenrostro JD et al. , Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). doi: 10.1038/nature14590; pmid: 26083756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma S, Zhang Y, Profiling chromatin regulatory landscape: Insights into the development of ChIP-seq and ATAC-seq. Mol. Biomed 1, 9 (2020). doi: 10.1186/s43556-020-00009-w; pmid: 34765994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y et al. , Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). doi: 10.1186/gb-2008-9-9-r137; pmid: 18798982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang T, Zhang Z, Dong Q, Xiong J, Zhu B, Histone H3K27 acetylation is dispensable for enhancer activity in mouse embryonic stem cells. Genome Biol 21, 45 (2020). doi: 10.1186/s13059-020-01957-w; pmid: 32085783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rickels R et al. , Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat. Genet 49, 1647–1653 (2017). doi: 10.1038/ng.3965; pmid: 28967912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fu S et al. , Differential analysis of chromatin accessibility and histone modifications for predicting mouse developmental enhancers. Nucleic Acids Res 46, 11184–11201 (2018). doi: 10.1093/nar/gky753; pmid: 30137428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andersson R, Sandelin A, Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet 21, 71–87 (2020). doi: 10.1038/s41576-019-0173-8; pmid: 31605096 [DOI] [PubMed] [Google Scholar]
- 61.Nguyen TA et al. , High-throughput functional comparison of promoter and enhancer activities. Genome Res 26, 1023–1033 (2016). doi: 10.1101/gr.204834.116; pmid: 27311442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoeppner MP et al. , An improved canine genome and a comprehensive catalogue of coding genes and non-coding transcripts. PLOS ONE 9, e91172 (2014). doi: 10.1371/journal.pone.0091172; pmid: 24625832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao T, Duan Z, Genchev GZ, Lu H, Closing human reference genome gaps: identifying and characterizing gap-closing sequences. G3 10, 2801–2809 (2020). doi: 10.1534/g3.120.401280; pmid: 32532800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou J et al. , Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet 50, 1171–1179 (2018). doi: 10.1038/s41588-018-0160-6; pmid: 30013180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaplow IM, TACITSupplement; http://daphne.compbio.cs.cmu.edu/files/ikaplow/TACITSupplement/.
- 66.Roller M et al. , LINE retrotransposons characterize mammalian tissue-specific and evolutionarily dynamic regulatory regions. Genome Biol 22, 62 (2021). doi: 10.1186/s13059-021-02260-y; pmid: 33602314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paten B et al. , Cactus: Algorithms for genome multiple sequence alignment. Genome Res 21, 1512–1528 (2011). doi: 10.1101/gr.123356.111; pmid: 21665927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Foley NM et al. , A genomic time scale for placental mammal evolution. Science 380, eabl8189 (2023). doi: 10.1123/science.abl8189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McColgan P, Joubert J, Tabrizi SJ, Rees G, The human motor cortex microcircuit: Insights for neurodegenerative disease. Nat. Rev. Neurosci 21, 401–415 (2020). doi: 10.1038/s41583-020-0315-1; pmid: 32555340 [DOI] [PubMed] [Google Scholar]
- 70.Gonzalez-Burgos G, Cho RY, Lewis DA, Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol. Psychiatry 77, 1031–1040 (2015). doi: 10.1016/j.biopsych.2015.03.010; pmid: 25863358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rudy B, Fishell G, Lee S, Hjerling-Leffler J, Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol 71, 45–61 (2011). doi: 10.1002/dneu.20853; pmid: 21154909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Machanick P, Bailey TL, MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011). doi: 10.1093/bioinformatics/btr189; pmid: 21486936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shrikumar A, Greenside P, Kundaje A, Learning important features through propagating activation differences. Proc. Mach. Learn. Res 70, 3145–3153 (2017). [Google Scholar]
- 74.Lundberg SM, Lee S-I, A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst 31, 4768–4777 (2017). [Google Scholar]
- 75.Weirauch MT et al. , Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014). doi: 10.1016/j.cell.2014.08.009; pmid: 25215497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harrington AJ et al. , MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. eLife 5, e20059 (2016). doi: 10.7554/eLife.20059; pmid: 27779093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen YC et al. , Foxp2 controls synaptic wiring of corticostriatal circuits and vocal communication by opposing Mef2c. Nat. Neurosci 19, 1513–1522 (2016). doi: 10.1038/nn.4380; pmid: 27595386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Babeu JP, Boudreau F, Hepatocyte nuclear factor 4-alpha involvement in liver and intestinal inflammatory networks. World J. Gastroenterol 20, 22–30 (2014). doi: 10.3748/wjg.v20.i1.22; pmid: 24415854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alpern D et al. , TAF4, a subunit of transcription factor II D, directs promoter occupancy of nuclear receptor HNF4A during post-natal hepatocyte differentiation. eLife 3, e03613 (2014). doi: 10.7554/eLife.03613; pmid: 25209997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grafen A, The phylogenetic regression. Philos. Trans. R. Soc. London Ser. B 326, 119–157 (1989). doi: 10.1098/rstb.1989.0106; pmid: 2575770 [DOI] [PubMed] [Google Scholar]
- 81.Ives AR, Garland T Jr., Phylogenetic logistic regression for binary dependent variables. Syst. Biol 59, 9–26 (2010). doi: 10.1093/sysbio/syp074; pmid: 20525617 [DOI] [PubMed] [Google Scholar]
- 82.Benjamini Y, Hochberg Y, Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc 57, 289–300 (1995). [Google Scholar]
- 83.Frankish A et al. , GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, D766–D773 (2019). doi: 10.1093/nar/gky955; pmid: 30357393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Giusti-Rodríguez P et al. , Using three-dimensional regulatory chromatin interactions from adult and fetal cortex to interpret genetic results for psychiatric disorders and cognitive traits. bioRxiv 406330 [Preprint] (2019). 10.1101/406330. [DOI]
- 85.McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, OMIM: An Online Catalog of Human Genes and Genetic Disorders; https://omim.org/.
- 86.Deber CM, Reynolds SJ, Central nervous system myelin: Structure, function, and pathology. Clin. Biochem 24, 113–134 (1991). doi: 10.1016/0009-9120(91)90421-A; pmid: 1710177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu Z, Zhang G, Wu C, Li Y, Hu M, FastHiC: A fast and accurate algorithm to detect long-range chromosomal interactions from Hi-C data. Bioinformatics 32, 2692–2695 (2016). doi: 10.1093/bioinformatics/btw240; pmid: 27153668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Celis JF, Barrio R, Regulation and function of Spalt proteins during animal development. Int. J. Dev. Biol 53, 1385–1398 (2009). doi: 10.1387/ijdb.072408jd; pmid: 19247946 [DOI] [PubMed] [Google Scholar]
- 89.Parrish M et al. , Loss of the Sall3 gene leads to palate deficiency, abnormalities in cranial nerves, and perinatal lethality. Mol. Cell. Biol 24, 7102–7112 (2004). doi: 10.1128/MCB.24.16.7102-7112.2004; pmid: 15282310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Harrison SJ, Parrish M, Monaghan AP, Sall3 is required for the terminal maturation of olfactory glomerular interneurons. J. Comp. Neurol 507, 1780–1794 (2008). doi: 10.1002/cne.21650; pmid: 18260139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Uhlén M et al. , Tissue-based map of the human proteome. Science 347, 1260419 (2015). doi: 10.1126/science.1260419; pmid: 25613900 [DOI] [PubMed] [Google Scholar]
- 92.Jeong D et al. , LRIG1-mediated inhibition of EGF receptor signaling regulates neural precursor cell proliferation in the neocortex. Cell Rep 33, 108257 (2020). doi: 10.1016/j.celrep.2020.108257; pmid: 33053360 [DOI] [PubMed] [Google Scholar]
- 93.Marqués-Torrejón MÁ et al. , LRIG1 is a gatekeeper to exit from quiescence in adult neural stem cells. Nat. Commun 12, 2594 (2021). doi: 10.1038/s41467-021-22813-w; pmid: 33972529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.den Hoed J et al. , Mutation-specific pathophysiological mechanisms define different neurodevelopmental disorders associated with SATB1 dysfunction. Am. J. Hum. Genet 108, 346–356 (2021). doi: 10.1016/j.ajhg.2021.01.007; pmid: 33513338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bauer CK, Schwarz JR, Ether-à-go-go K+ channels: Effective modulators of neuronal excitability. J. Physiol 596, 769–783 (2018). doi: 10.1113/JP275477; pmid: 29333676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Borg Distefano M et al. , TBC1D5 controls the GTPase cycle of Rab7b. J. Cell Sci 131, jcs216630 (2018). doi: 10.1242/jcs.216630; pmid: 30111580 [DOI] [PubMed] [Google Scholar]
- 97.Ridgway SH, Brownson RH, Van Alstyne KR, Hauser RA, Higher neuron densities in the cerebral cortex and larger cerebellums may limit dive times of delphinids compared to deep-diving toothed whales. PLOS ONE 14, e0226206 (2019). doi: 10.1371/journal.pone.0226206; pmid: 31841529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gaston K, Jayaraman PS, Transcriptional repression in eukaryotes: Repressors and repression mechanisms. Cell. Mol. Life Sci 60, 721–741 (2003). doi: 10.1007/s00018-003-2260-3; pmid: 12785719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Adachi M, Monteggia LM, Decoding transcriptional repressor complexes in the adult central nervous system. Neuropharmacology 80, 45–52 (2014). doi: 10.1016/j.neuropharm.2013.12.024; pmid: 24418103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lupo A et al. , KRAB-zinc finger proteins: a repressor family displaying multiple biological functions. Curr. Genomics 14, 268–278 (2013). doi: 10.2174/13892029113149990002; pmid: 24294107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cooley Coleman JA et al. , Comprehensive investigation of the phenotype of MEF2C-related disorders in human patients: A systematic review. Am. J. Med. Genet. A 185, 3884–3894 (2021). doi: 10.1002/ajmg.a.62412; pmid: 34184825 [DOI] [PubMed] [Google Scholar]
- 102.Pai EL-L et al. , Maf and Mafb control mouse pallial interneuron fate and maturation through neuropsychiatric disease gene regulation. eLife 9, e54903 (2020). doi: 10.7554/eLife.54903; pmid: 32452758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brown AR et al. , An in vivo massively parallel platform for deciphering tissue-specific regulatory function. bioRxiv 2022.11.23.517755 [Preprint] (2022). 10.1101/2022.11.23.517755. [DOI]
- 104.Leimkühler S, Freuer A, Araujo JAS, Rajagopalan KV, Mendel RR, Mechanistic studies of human molybdopterin synthase reaction and characterization of mutants identified in group B patients of molybdenum cofactor deficiency. J. Biol. Chem 278, 26127–26134 (2003). doi: 10.1074/jbc.M303092200; pmid: 12732628 [DOI] [PubMed] [Google Scholar]
- 105.Bayram E et al. , Molybdenum cofactor deficiency: Review of 12 cases (MoCD and review). Eur. J. Paediatr. Neurol 17, 1–6 (2013). doi: 10.1016/j.ejpn.2012.10.003; pmid: 23122324 [DOI] [PubMed] [Google Scholar]
- 106.Kröcher T et al. , A crucial role for polysialic acid in developmental interneuron migration and the establishment of interneuron densities in the mouse prefrontal cortex. Development 141, 3022–3032 (2014). doi: 10.1242/dev.111773; pmid: 24993945 [DOI] [PubMed] [Google Scholar]
- 107.Curto Y, Alcaide J, Röckle I, Hildebrandt H, Nacher J, Effects of the genetic depletion of polysialyltransferases on the structure and connectivity of interneurons in the adult prefrontal cortex. Front. Neuroanat 13, 6 (2019). doi: 10.3389/fnana.2019.00006; pmid: 30787870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lukas D, Clutton-Brock TH, The evolution of social monogamy in mammals. Science 341, 526–530 (2013). doi: 10.1126/science.1238677; pmid: 23896459 [DOI] [PubMed] [Google Scholar]
- 109.Ferguson BR, Gao W-J, PV interneurons: Critical regulators of E/I Balance for prefrontal cortex-dependent behavior and psychiatric disorders. Front. Neural Circuits 12, 37 (2018). doi: 10.3389/fncir.2018.00037; pmid: 29867371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schwede M et al. , Strong correlation of downregulated genes related to synaptic transmission and mitochondria in post-mortem autism cerebral cortex. J. Neurodev. Disord 10, 18 (2018). doi: 10.1186/s11689-018-9237-x; pmid: 29859039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Phan BN et al. , A myelin-related transcriptomic profile is shared by Pitt-Hopkins syndrome models and human autism spectrum disorder. Nat. Neurosci 23, 375–385 (2020). doi: 10.1038/s41593-019-0578-x; pmid: 32015540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reiner BC et al. , Single-nuclei transcriptomics of schizophrenia prefrontal cortex primarily implicates neuronal subtypes. bioRxiv 2020.07.29.227355 [Preprint] (2021). 10.1101/2020.07.29.227355. [DOI]
- 113.Ruzicka WB et al. , Single-cell dissection of schizophrenia reveals neurodevelopmental-synaptic axis and transcriptional resilience. medRxiv 2020.11.06.20225342 [Preprint] (2020). 10.1101/2020.11.06.20225342. [DOI]
- 114.Smith AL, Jung E-M, Jeon BT, Kim W-Y, Arid1b haploinsufficiency in parvalbumin- or somatostatin-expressing interneurons leads to distinct ASD-like and ID-like behavior. Sci. Rep 10, 7834 (2020). doi: 10.1038/s41598-020-64066-5; pmid: 32398858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Trubetskoy V et al. , Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022). doi: 10.1038/s41586-022-04434-5; pmid: 35396580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kopp N, McCullough K, Maloney SE, Dougherty JD, Gtf2i and Gtf2ird1 mutation do not account for the full phenotypic effect of the Williams syndrome critical region in mouse models. Hum. Mol. Genet 28, 3443–3465 (2019). doi: 10.1093/hmg/ddz176; pmid: 31418010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.vonHoldt BM et al. , Structural variants in genes associated with human Williams-Beuren syndrome underlie stereotypical hypersociability in domestic dogs. Sci. Adv 3, e1700398 (2017). doi: 10.1126/sciadv.1700398; pmid: 28776031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mervis CB et al. , Duplication of GTF2I results in separation anxiety in mice and humans. Am. J. Hum. Genet 90, 1064–1070 (2012). doi: 10.1016/j.ajhg.2012.04.012; pmid: 22578324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martin LA, Iceberg E, Allaf G, Consistent hypersocial behavior in mice carrying a deletion of Gtf2i but no evidence of hyposocial behavior with Gtf2i duplication: Implications for Williams-Beuren syndrome and autism spectrum disorder. Brain Behav 8, e00895 (2017). doi: 10.1002/brb3.895; pmid: 29568691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wirthlin M et al. , A modular approach to vocal learning: Disentangling the diversity of a complex behavioral trait. Neuron 104, 87–99 (2019). doi: 10.1016/j.neuron.2019.09.036; pmid: 31600518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jarvis ED, Learned birdsong and the neurobiology of human language. Ann. N. Y. Acad. Sci 1016, 749–777 (2004). doi: 10.1196/annals.1298.038; pmid: 15313804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chabbert D et al. , Postnatal Tshz3 deletion drives altered corticostriatal function and autism spectrum disorder–like behavior. Biol. Psychiatry 86, 274–285 (2019). doi: 10.1016/j.biopsych.2019.03.974; pmid: 31060802 [DOI] [PubMed] [Google Scholar]
- 123.Partha R, Kowalczyk A, Clark NL, Chikina M, Robust method for detecting convergent shifts in evolutionary rates. Mol. Biol. Evol 36, 1817–1830 (2019). doi: 10.1093/molbev/msz107; pmid: 31077321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Langer BE, Roscito JG, Hiller M, REforge associates transcription factor binding site divergence in regulatory elements with phenotypic differences between species. Mol. Biol. Evol 35, 3027–3040 (2018). doi: 10.1093/molbev/msy187; pmid: 30256993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kent WJ et al. , The human genome browser at UCSC. Genome Res 12, 996–1006 (2002). doi: 10.1101/gr.229102; pmid: 12045153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rao SSP et al. , A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). doi: 10.1016/j.cell.2014.11.021; pmid: 25497547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Berthelot C, Villar D, Horvath JE, Odom DT, Flicek P, Complexity and conservation of regulatory landscapes underlie evolutionary resilience of mammalian gene expression. Nat. Ecol. Evol 2, 152–163 (2018). doi: 10.1038/s41559-017-0377-2; pmid: 29180706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dukler N, Huang Y-F, Siepel A, Phylogenetic modeling of regulatory element turnover based on epigenomic data. Mol. Biol. Evol 37, 2137–2152 (2020). doi: 10.1093/molbev/msaa073; pmid: 32176292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Volland S, Esteve-Rudd J, Hoo J, Yee C, Williams DS, A comparison of some organizational characteristics of the mouse central retina and the human macula. PLOS ONE 10, e0125631 (2015). doi: 10.1371/journal.pone.0125631; pmid: 25923208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wu Y, Wang H, Wang H, Hadly EA, Rethinking the origin of primates by reconstructing their diel activity patterns using genetics and morphology. Sci. Rep 7, 11837 (2017). doi: 10.1038/s41598-017-12090-3; pmid: 28928374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Towns J et al. , XSEDE: accelerating scientific discovery. Comput. Sci. Eng 16, 62–74 (2014). doi: 10.1109/MCSE.2014.80 [DOI] [Google Scholar]
- 132.Huh C, Orcinus orca, PhyloPic; http://phylopic.org/image/880129b5-b78b-40a9-88ad-55f7d1dc823f/.
- 133.Kaplow IM, Schäffer DE, Srinivasan C, Lawler AJ, Sestili HH, pfenninglab/TACIT: TACIT_conditionalpValuesUpdated, version 0.1.4, Zenodo (2023); 10.5281/zenodo.7829847. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.