

Abstract
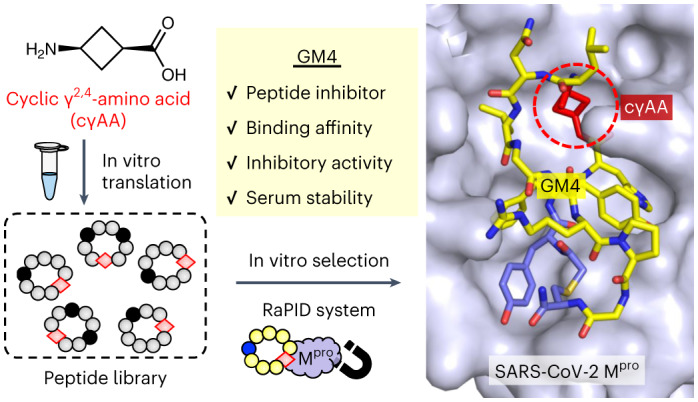
γ-Amino acids can play important roles in the biological activities of natural products; however, the ribosomal incorporation of γ-amino acids into peptides is challenging. Here we report how a selection campaign employing a non-canonical peptide library containing cyclic γ2,4-amino acids resulted in the discovery of very potent inhibitors of the SARS-CoV-2 main protease (Mpro). Two kinds of cyclic γ2,4-amino acids, cis-3-aminocyclobutane carboxylic acid (γ1) and (1R,3S)-3-aminocyclopentane carboxylic acid (γ2), were ribosomally introduced into a library of thioether-macrocyclic peptides. One resultant potent Mpro inhibitor (half-maximal inhibitory concentration = 50 nM), GM4, comprising 13 residues with γ1 at the fourth position, manifests a 5.2 nM dissociation constant. An Mpro:GM4 complex crystal structure reveals the intact inhibitor spans the substrate binding cleft. The γ1 interacts with the S1′ catalytic subsite and contributes to a 12-fold increase in proteolytic stability compared to its alanine-substituted variant. Knowledge of interactions between GM4 and Mpro enabled production of a variant with a 5-fold increase in potency.
Subject terms: Peptides, X-ray crystallography, High-throughput screening, Translation
In vitro screening of a ribosomally synthesized macrocyclic peptide library containing cyclic γ2,4-amino acids (cγAA) afforded the discovery of potent inhibitors of the SARS-CoV-2 main protease (Mpro). A co-crystal structure revealed the contribution of this cγAA to Mpro binding and the proteolytic stability of these macrocycles.
Main
γ-Amino acid residues can induce unique peptide conformations, such as C14-helix, C12/10-helix and C12-turn secondary structures1–10. The γ-amino acids present in natural products play important roles in their biological activities and enable improved proteolytic stability and cell permeability11,12. Pepstatin, isolated from Actinomyces, has two γ-amino acid residues, which contribute to its protease inhibitory activity by enhancing binding to the active sites13–15. Didemnin, an antiviral/antitumor compound, has a γ-amino acid and a macrocyclic scaffold16. Given their remarkable bioactivities and conformational rigidity, macrocyclic peptides containing γ-amino acids are an attractive option for incorporation into novel therapeutic peptides.
Over the past two decades, the ribosomal synthesis and selection-based screening of peptides containing nonproteinogenic amino acids, including N-methyl-l-α-amino, d-amino and β-amino acids, have been accomplished17–26. However, the ribosomal incorporation of γ-amino acids into peptides has been challenging for the following three reasons: (1) efficient self-deacylation of γ-aminoacyl-tRNA via stereoelectronically favoured 5-oxo-trig cyclization, (2) slow accommodation of γ-aminoacyl-tRNAs into the ribosome A site and (3) slow peptidyl transfer of the P-site peptidyl-tRNA onto the γ-amino group of the A-site γ-aminoacyl-tRNA due to poor compatibility with the peptidyl transferase centre of the ribosome27,28. We recently reported that the self-deacylation issue can be circumvented by using structurally constrained cyclic γ2,4-amino acids (cγAAs)29. The cγAAs were charged on an engineered tRNA, referred to as tRNAPro1E2 (Supplementary Fig. 1), the T-stem and D-arm of which were optimized for efficient binding to elongation factor Tu (EF-Tu) and EF-P, thereby accelerating accommodation and peptidyl transfer, respectively30–32. Combination of the flexizyme-assisted33,34 synthesis of cγAA–tRNAPro1E2 and the reconstituted Escherichia coli translation system, referred to as the Flexible In vitro Translation (FIT) system23, that contains EF-Tu and EF-P with their adjusted concentrations enabled the incorporation of cγAAs into nascent peptide chains29,35,36.
Here we report the ribosomal synthesis of a macrocyclic peptide library containing cγAAs and its application to an in vitro display, referred to as the Random nonstandard Peptides Integrated Discovery (RaPID) system23. The library contains two types of cγAAs, cis-3-aminocyclobutane carboxylic acid (γ1) and (1R,3S)-3-aminocyclopentane carboxylic acid (γ2) (Fig. 1a). Owing to the constrained rigid conformations of these cγAAs, they can be exquisite building blocks for preorganization of peptide secondary structures. Therefore, screening of macrocyclic peptide libraries containing cγAAs can lead to molecules with potent binding affinities and peptidase resistance. As a biomedicinally important test target protein, we chose Mpro, which plays an essential role in processing the polyproteins translated from the viral RNA37. Peptidomimetic inhibitors of Mpro have been shown to be therapeutically valuable for the treatment of coronavirus disease 2019 (COVID-19), with one approved for clinical use (nirmatrelvir, PF-07321332)38. Importantly, however, most of these inhibitors contain a reactive warhead (a nitrile in the case of nirmatrelvir) that reacts to form a covalent bond with a Cys residue in the Mpro active site. We took up the challenge of discovering potent ‘non-covalent’ active site binding Mpro inhibitors by applying the RaPID system with cγAA-containing macrocyclic peptide libraries.
Fig. 1. Ribosomal incorporation of cγAAs into a macrocyclic peptide library.

a, Structures of the cγAAs and cβAAs used in this study. b, The reprogrammed codon table that contains ClAcy at the initiator AUG codon, and c, γ1, γ2, β1 and β2 at the elongator AUG, GAG, GUG, GUU and UGU codons, respectively. NNA codons are omitted because they were not used in the mRNA library. c, Construction of the random mRNA library and the corresponding peptide library. Peptides spontaneously macrocyclized between ClAcy and c via a thioether bond. The mRNA and peptide were covalently linked via a puromycin linker.
Results
Selection of macrocyclic peptide binders to SARS-CoV-2 Mpro
To construct a cγAA-containing macrocyclic peptide library, γ1 and γ2 (Fig. 1a) were assigned to GAG and GUG codons using tRNAPro1E2CUC and tRNAPro1E2CAC, respectively (Fig. 1b), employing the FIT system. Two cyclic β2,3-amino acids (cβAAs), (1R,2S)-2-aminocyclopentane carboxylic acid (β1) and (1S,2S)-2-aminocyclopentane carboxylic acid (β2), were introduced at the GUU and UGU codons using tRNAPro1E2GAC and tRNAGluE2GCA, respectively, because we considered that the incorporation not only of cγAA, but also of cβAA, would give more diverse folding possibilities25. For the macrocyclization of the library via thioether bonds, we introduced N-chloroacetyl-d-tyrosine (ClAcy) and d-cysteine (c) at the initiator AUG codon using tRNAfMetCAU and elongator AUG codon using tRNAPro1E2CAU, respectively. The thioether bond spontaneously forms between the N-terminal chloroacetyl group on ClAcy and the thiol group of the c residue (Fig. 1c). These amino acids were precharged onto the respective tRNAs using flexizymes. The peptide library comprised a repeat of 11−14 random residues encoded by GWG and NNU codons (W = A or U; N = A, U, G or C) flanked by the cyclizing ClAcy and c residues. Since multiple cγAA incorporations could not be efficiently achieved, as reported in our previous study29, the library was designed to have only a single γ1 or γ2 appear at the GWG codon. The two cβAAs and the thirteen proteinogenic l-α-amino acids (A, D, F, G, H, I, L, N, P, R, S, T and Y) were assigned at NNU codons. The C-terminal SGGSGG sequence following the c residue was a linker peptide connected to the 3′ end of mRNA via a puromycin linker. To demonstrate the fidelity of translation, model macrocyclic peptides containing any of cγAA and cβAA (γ1, γ2, β1 and β2 assigned at GAG, GUG, GUU and UGU codons, respectively) were synthesized, and their identities were confirmed by matrix-assisted laser desorption/ionization coupled with time of flight mass spectrometry (MALDI-TOF MS; Supplementary Fig. 2).
The macrocyclic peptide library was then applied to the RaPID selection against recombinant SARS-CoV-2 Mpro. Translation of the random mRNA library into the peptide library, conjugation of the peptide with the parent mRNA via a puromycin linker and reverse transcription of the mRNA into the cDNA yielded peptide/mRNA/cDNA conjugates (Extended Data Fig. 1a). The library was first subjected to naked magnetic bead treatment to remove bead binders, and then applied to Mpro-immobilized magnetic beads to recover Mpro binders. The cDNA moiety of the recovered peptide/mRNA/cDNA conjugate was amplified by the polymerase chain reaction (PCR) and transcribed into the mRNA library for the next selection round (details in Methods). By repeating this affinity selection, the recovery rate of the Mpro binders was greatly increased at the third round of selection and, even more obviously, in the fourth round, while the recovery of the bead binders did not increase (Extended Data Fig. 1b). Deep sequencing of the cDNA library at the fourth round revealed that the library was enriched with several families of peptides bearing cγAA (Supplementary Table 1 shows the top 100 sequences). Among these, seven macrocyclic peptides containing cγAA were selected for further analysis of their binding affinities, inhibitory activity and proteolytic stability (Table 1, GM1−GM7). GM1, GM2, GM4 and GM6 contain a γ1 residue in their sequences, while GM3, GM5 and GM7 contain γ2. Note that the cβAA-containing peptides were not selected because they were not included in the major peptide families with high read numbers. GM1−GM7 were chemically synthesized on a large scale using the standard solid-phase method without the C-terminal SGGSGG linker, and their purities and identities were confirmed by ultra-performance liquid chromatography (UPLC) and MALDI-TOF MS, respectively (Supplementary Figs. 3 and 4).
Extended Data Fig. 1. RaPID selection against Mpro using a macrocyclic peptide library containing cγAA.

a, Schematic depiction of RaPID display. 1) Puromycin linker ligation to the 3′-end of the mRNA library. 2) Translation of peptides using the reprogrammed genetic code, followed by spontaneous macrocyclization of peptide via a thioether bond. 3) Reverse transcription of mRNA into cDNA. 4) Binding selection of peptides against Mpro immobilized on magnetic beads. 5) Recovery of the bound fraction and amplification of cDNA by PCR. 6) Transcription of cDNA library into mRNA library. 7) Deep sequencing analysis of cDNA library. b, Recovery rate of cDNA after the binding selection at each round. Red and blue bars indicate the recovery rate of Mpro binders and magnetic beads binders, respectively. Bead-binder selection was not performed in the first round.
Table 1.
Binding affinities, inhibitory activities and serum stabilities of the macrocyclic peptides

The thioether-macrocyclic peptides selected by the RaPID system, GM1−GM7, and their mutants are shown. The thioether bond is shown as a blue line where the sulfide atom is omitted. The γ1 and γ2 residues are in red. The table shows sequences, read (%) at the fourth round, kinetic association (ka), kinetic dissociation (kd), KD, IC50 and half-life in human serum (t1/2). Supplementary Fig. 5 shows the sensorgrams of SPR analysis; Fig. 2a and Extended Data Fig. 2 show the results of the Mpro inhibition assay; Fig. 2b and Extended Data Fig. 3 show the results of serum stability assays. −, the kinetic values could not be accurately determined due to low affinity.
Biological activities and stabilities of peptide inhibitors
We first evaluated the binding affinity of GM1−GM7 to Mpro by surface plasmon resonance (SPR). All the peptides exhibited low-to-moderate nanomolar kinetic equilibrium (KD) values (Table 1 and Supplementary Fig. 5). Notably, GM1 and GM4, which contain γ1, and GM5, which contains γ2, have a conserved four residue motif, yFHγX (γX = γ1 or γ2), at their N termini and showed potent affinities, with KD values of 2.3, 5.2 and 5.2 nM, respectively; GM2 has the same motif, but has a higher KD (71 nM). GM6 and GM7, which had relatively low read frequencies, exhibited weak binding compared to GM1−GM5, indicating selection for tight binding peptides. To evaluate contributions of the cγAA residues to potency, we synthesized peptides where the cγAA was substituted with alanine (Table 1, GM1γ14A, GM3γ210A, GM4γ14A and GM5γ24A). In addition to GM1, GM4 and GM5, which share the conserved yFHγX motif, GM3 was selected for alanine substitution because of its high read percentage among peptides without the yFHγX motif. SPR measurements of the alanine mutants revealed that the KD values of GM1γ14A, GM4γ14A and GM5γ24A were comparable or one order higher than those of the original peptides, indicating that the cγAA residue in the conserved yFHγX motif is important for binding. To our surprise, GM3γ210A exhibited an eightfold stronger binding affinity (KD = 7.5 nM), revealing that the contribution of cγAA to the binding affinity is sequence context dependent.
We next evaluated the inhibitory activity of GM1−GM7 against the hydrolytic activity of SARS-CoV-2 Mpro using a reported MS-based method39. GM1−GM7 exhibited inhibition of Mpro; in particular, the yFHγX motif containing GM1, GM4 and GM5, which manifest single-digit nanomolar KD values, showed particularly potent inhibition, with half-maximal inhibitory concentration (IC50) values of 40, 50 and 40 nM, respectively (Table 1, Fig. 2a and Extended Data Fig. 2). The peptides with weaker binding affinity, that is, GM2, GM3, GM6 and GM7, had IC50 values of 1,750–7,930 nM, implying correlation between IC50 and KD values. The inhibitory activities of the alanine mutants were then determined. Notably, despite GM1γ14A having only a fourfold weaker binding affinity compared with GM1, inhibition was ablated, revealing the importance of the cγAA. By contrast, GM3γ210A, GM4γ14A and GM5γ24A retained inhibition activity, showing the context-dependent effects of the cγAA (IC50 = 120, 10 and 50 nM, respectively; Table 1, Fig. 2a and Extended Data Fig. 2).
Fig. 2. Mpro inhibitory activity and serum stability of GM4 and its mutants.

a, Dose response analysis of peptides against Mpro. The Mpro inhibitory activities of peptides were investigated by solid-phase extraction purification coupled to MS analysis using a RapidFire 365 high-throughput sampling robot (Agilent) connected to an iFunnel Agilent 6550 accurate mass quadrupole TOF mass spectrometer39. IC50 values were determined by the mean of three or five independent replicates each performed in technical duplicate. Data are presented as mean values ± standard deviation, s.d. (n = 5 for GM4, GM4γ14A and GM4H3Q; n = 3 for GM4γ14N, GM4H3A and GM4H3E). Extended Data Fig. 2 shows other peptides. b, Serum stability assay of macrocyclic peptides. GM4, GM4γ14A and GM4H3Q were co-incubated with an internal standard peptide in human serum (37 °C). At each time point, the relative intensity of each peptide to the standard peptide was estimated by LC/MS. The relative intensity at 0 h was defined as 100%. Half-lives (t1/2) were determined by analysing the mean of three technical replicates of each sample by nonlinear regression using GraphPad Prism 9. Data are presented as mean values ± s.d. (n = 3). Extended Data Fig. 3 shows results for other peptides.
Extended Data Fig. 2. Representative dose response analyses of inhibition of Mpro by macrocycles.

Conditions: 75 nM Mpro, 4 µM substrate peptide, macrocyclic peptides in 20 mM HEPES, pH 7.5, 50 mM NaCl. See Methods for details. Table 1 gives the recorded IC50 values as the mean of 3 or 5 independent replicates each performed in technical duplicate. Data are presented as mean values ± SD. Number of replicates is indicated above the graph. See Fig. 2 for GM4 and its variants.
We evaluated the half-life of potent cγAA-containing peptides and their mutants in human serum, because in vivo stability is a critical factor in the development of therapeutic peptides. Each peptide and an uncleavable internal standard peptide were co-incubated in human serum at 37 °C, and the relative amount of the remaining sample peptide was estimated by liquid chromatography/MS (LC/MS). The potent inhibitors GM1 and GM4, containing γ1, and GM5, containing γ2, exhibit high peptidase resistance with half-lives (t1/2) of 90 h, 126 h and 32 h, respectively (Table 1, Fig. 2b and Extended Data Fig. 3a). Importantly, by contrast, their alanine mutants show substantially shorter half-lives (t1/2 = 8.3 h, 11 h and 21 h for GM1γ14A, GM4γ14A and GM5γ24A, respectively; Table 1). The enhancement of serum resistance observed for GM4, for instance, was 12-fold. Notably, despite the substitution of γ1 to an Ala residue not diminishing inhibition relative to GM4, the enhancement in serum stability by the γ1 residue is substantial. Moreover, the less potent inhibitors GM2 and GM3 have more than nearly three orders of magnitude shorter half-lives (1.1 h and 2.5 h, respectively) compared to GM4, suggesting that the higher (binding and inhibitory) activity of GM4 is reflected in its higher serum stability, possibly due to its more conformationally constrained fold.
Extended Data Fig. 3. Serum stability assay of macrocyclic peptides.

a, Each of GM1, GM1γ14A, GM2, GM3, GM3γ210A, GM5, and GM5γ24A were co-incubated with an internal standard peptide in human serum at 37 °C. At each time point (0, 0.5, 1, 2, 4, 8, 24, 50, and 100 h), the relative intensity of each peptide with respect to the standard peptide was estimated by LC/MS. The relative intensity at 0 h was defined as 100%. Half-lives (t1/2) were determined by analyzing the mean of 3 technical replicates of each sample by non-linear regression using GraphPad Prism 9. Data are presented as mean values ± SD (n = 3). b,c, Fragments of GM4 (GM4-f1−6) and GM4γ14A (GM4γ14A-f1−3), respectively, after 24 h incubation in human serum as analyzed by LC/MS.
To identify peptidase cleavage sites in serum, the products of GM4 and GM4γ14A after 24 h incubation were analysed by LC/MS. In the case of GM4, six fragments (GM4-f1−GM4-f6, Extended Data Fig. 3b) containing the c-(thioether)-AcyFHγ1L motif were detected, but no fragmentation of the motif itself was apparent. With GM4γ14A, three shorter fragments containing c-(thioether)-AcyF were detected (GM4γ14A-f1−GM4γ14A-f3; Extended Data Fig. 3c), indicating cleavage between F and H of the motif. These results show that the presence of non-canonical residues (one cγAA (γ1), two d-amino acids and a thioether bond) can enable high peptidase resistance by preventing the proteolysis of flanking residues, leading to a remarkable improvement in serum stability.
X-ray crystallographic analysis of the GM4:Mpro complex
To gain structural insights into how GM4, including its γ1 residue, interacts with Mpro, we obtained an X-ray crystal structure of their complex to 1.7 Å resolution (Fig. 3a and Supplementary Table 2). The structure reveals GM4 bound with high occupancy and in the same manner at the active site regions of both monomers in the Mpro dimer. Excellent electron density was observed for all residues in the GM4 macrocycle (Fig. 3c). The overall conformations of Mpro in the GM4 and nirmatrelvir38 (used for comparison) complex structures are very similar (backbone root mean square deviation, 0.34).
Fig. 3. Crystallographic studies reveal the binding mode of the GM4 macrocyclic peptide at the Mpro active site.

a, GM4 binds in the substrate binding cleft of both protomers A and B in the Mpro dimer. b, Structure of GM4; the H3GM4 carbonyl O is indicated with an arrow. c, Polder omit map of GM4 contoured at a level of ±1.5 standard deviation (σ). d, View of GM4 at the active site. The H41 and C145 catalytic dyad is in orange. γ14GM4 (magenta) occupies the S1′ pocket; the side chains of H3GM4, F2GM4 and Acy1GM4 occupy the S1, S2 and S4 pockets, respectively. e,f, Close-ups of the S1 pocket, showing the H3GM4 backbone carbonyl in the oxyanion hole (C145, G143 backbone amides). The backbone NH of H3GM4 is positioned to interact with the H164 backbone CO. The H3GM4 imidazole is positioned to hydrogen bond with the H163 side chain and to interact with the E166 side chain, which interacts with S1 of protomer B, as observed in apo-Mpro structures. Y9GM4 is 3.1 Å and 3.3 Å from the E166 and S1B side chains, respectively. g, Nirmatrelvir with the residues occupying the S1−S4 subsites labelled P1−P4 (the reactive nitrile is indicated with an arrow). h, Superimposition of views from crystal structures of Mpro with GM4 and nirmatrelvir (Protein Data Bank 7VH8 (ref. 60)). GM4 non-covalently interacts at the active site, while the nitrile of nirmatrelvir reacts with C145. Note the similar locations of the nirmatrelvir nitrile-derived N and the H3GM4 carbonyl O.
The four residues in the yFHγ1 motif of GM4 occupy the S4, S2, S1 and S1′ substrate binding subsites, respectively40 (Fig. 3d). H3GM4 is accommodated in the S1 subsite, forming hydrogen bonds with Mpro C145, H164 and H163 (GM4 residues are identified by a subscript; Fig. 3e,f). The F2GM4 residue binds in the S2 subsite, making hydrophobic interactions with M49, M165 and H41. The y1GM4 side chain forms hydrogen bonds with T190 and Q192 in the S4 subsite (Extended Data Fig. 4a).
Extended Data Fig. 4. Views from crystallographic studies.

a, Interaction of the y1GM4 residue at the Mpro active site. The phenolic sidechain of y1GM4 forms polar interactions with T190 and Q192. The backbone NH of y1GM4 is positioned to form H-bonds with the mainchain carbonyl and NH groups of E166, as is typical of P3 residues in peptidomimetic Mpro inhibitors (Ref. 40). b, Intramolecular hydrogen bonds in GM4; H-bonds are formed between γ14GM4 and L7GM4, Acy1GM4 and R10 GM4, and P11GM4 and c13GM4. H-bonds are indicated with dashed black lines; associated distances are in Ångström.
The c13GM4–thioether link, which enables macrocyclization, and the adjacent G12GM4 enable a turn connected to P11GM4, R10GM4 and Y9GM4; none of these residues are positioned to make direct contacts with Mpro in the structure, though these cannot be ruled out in solution. The backbone of GM4 forms three intramolecular hydrogen bonds, that is between γ14GM4 and L7GM4, Acy1GM4 and R10GM4, and P11GM4 and c13GM4 (Extended Data Fig. 4b). The side chains of P11GM4 and Y9GM4 are arranged such that they ‘stack’ with each other, an arrangement positioning the phenolic side chain of Y9GM4 adjacent to the imidazole ring of H3GM4, appearing to lock it into the S1 pocket; the phenolic side chain of Y9GM4 also forms interactions with Ser1, from the other protomer of the Mpro dimer (Fig. 3f).
G8GM4, L7GM4, N6GM4 and L5GM4 form a loop leading to γ14, which is linked to H3GM4. γ14GM4 is positioned to make hydrophobic contact with T24 and T25; the side chains of L5GM4 and N6GM4 are positioned close to the surface of Mpro, with L7GM4 projecting towards H41, C44 and T45. The observation of the unnatural γ14GM4 residue, the cyclobutane ring of which was refined in a near planar conformation, in the P1′ position is striking and implies it contributes to the inhibition or lack of efficient GM4 hydrolysis.
At the active site region, the sulfur of the nucleophilic C145 is positioned close (3.4 Å) to the imidazole ε-nitrogen of H41, which acts as a general base/acid in Mpro catalysis, and to the carbonyl carbon (4.4 Å) of the amide linking H3GM4 and γ14GM4; this carbonyl carbon is positioned in a similar manner to the imine (anion) derived by reaction of the nirmatrelvir nitrile with C145 (Fig. 3g,h).
Like the binding modes of substrates (predicted) and inhibitors (as observed by crystallography), the carbonyl of the amide linking H3GM4 and γ14GM4 is positioned in the oxyanion hole, making hydrogen bonds with the C145 NH (2.9 Å) and with the G143 NH (2.9 Å). The ~100° angle between the C145 sulfur and the carbon and oxygen of the amide linking H3 and γ14 is close to that predicted for nucleophilic attack onto an amide carbonyl as observed in studies on serine proteases41. Despite this apparently catalytically productive arrangement, analysis of the electron density map implies a lack of substantial covalent reaction, consistent with solution studies showing a lack of efficient GM4 hydrolysis by Mpro. Lack of reaction appears to be a consequence of multiple interactions made between the peptide macrocycle and Mpro, likely involving P11GM4, Y9GM4 and H3GM4, resulting in tight binding in a catalytically non-productive manner.
SARS-CoV-2 Mpro cleaves after a conserved glutamine residue (P1) in substrates with the following sequence motif: (P2, L/F/V/M (hydrophobic))(P1, Q)↓(P1′, S/A/G/N (small)), where ↓ represents the cleavage site40,42. Interestingly, rather than a glutamine (Q), GM4 has a histidine (H3) at the analogous ‘P1’ position. To investigate the contribution of the H3/P1 residue for activity, we evaluated peptides bearing Q, A or E residues at the analogous position. Among these, GM4H3Q exhibited a sixfold stronger binding affinity (KD = 0.86 nM) and fivefold more potent inhibitory activity (IC50 = 10 nM; Table 1, Fig. 2 and Supplementary Fig. 5). By contrast, GM4H3A manifested no inhibition, and inhibition of GM4H3E was observed only with the highest tested concentration (25 µM). These results are consistent with the known substrate selectivity of Mpro at the P1 position40.
In the case of the P1′ residue, Mpro prefers substrates with small residues, such as G, A and S, although N is also tolerated40. Strikingly, GM4 has γ14 at the P1′ position, likely reflecting the compact size of the γ1 four-membered-ring main-chain residue. In support of this, GM4γ14A (γ1 → A) exhibited a comparable inhibitory activity to that of GM4 (IC50 of GM4γ14A = 10 nM), and GM4γ14N was 20-fold less active (IC50 = 1,000 nM). Despite the almost equivalent Mpro inhibition activities of GM4 and GM4γ14A, the serum stability of GM4 is 12-fold higher (t1/2 = 126 h) than that of GM4γ14A (Fig. 2b). Furthermore, the most potent mutant GM4H3Q bearing γ14 at P1 also exhibited a comparable serum stability (t1/2 = 82 h). These results show how the introduction of a nonstandard amino acid at the P1′ position can prolong the serum stability to non-targeted protease-catalysed hydrolysis while maintaining potency against the targeted protease (Mpro in our case).
Discussion
Our results reveal GM4 and GM4H3Q as highly potent cyclic peptides targeting Mpro. Other peptides and peptidomimetic inhibitors of SARS-CoV-2 Mpro have been reported with varying potencies (Extended Data Fig. 5). Johansen-Leete et al. recently used the RaPID system to screen thioether-macrocyclic peptides containing standard proteinogenic amino acids in the random region between the cyclizing ClAcy and the downstream l-Cys (ref. 43). A macrocyclic peptide, with yLQY residues at the P3−P1′ equivalent positions, exhibiting an inhibition constant (Ki) value of 14 nM against Mpro was identified. The P1 equivalent Q and P1′ equivalent Y residues are on either side of the scissile amide in this peptide, resulting in slow cleavage by Mpro.
Extended Data Fig. 5. Comparison of GM4 and its variants with Mpro substrates and reported Mpro inhibitors.

a, Amino acid sequences around the cleavage site of the Mpro substrate and inhibitors. Superscript numbers indicate references. b, Structures of glutamine analog XQ and peptidomimetics. c, Structures and activities of non-covalent small molecule inhibitors of Mpro.
In the case of the Mpro:GM4 complex crystal structure, well-defined electron density was observed for the complete intact peptide in the active site pocket despite the carbonyl of the amide linking H3GM4 and γ14GM4 being positioned in the oxyanion hole. This observation both highlights the advantage of introducing a cγAA into the library and implies that GM4 and GM4H3Q in part resist hydrolytic cleavage due to the presence of γ1 at the P1′ position. Other groups have also reported a substrate-derived cyclic peptide inhibitor of Mpro (UCI-1 (ref. 44)) and linear peptide inhibitors of Mpro (p13 (ref. 40) and compound 21 (ref. 45)), but the potency is limited to the micromolar range. Peptidomimetic inhibitors, including nirmatrelvir (PF-07321332) and PF-00835231, where the C-terminal residue reacts covalently with the nucleophilic cysteine have been developed38,42,46–49 (Extended Data Fig. 5a,b). XQ, which is present in these inhibitors, is an analogue of the substrate Q/P1 residue wherein the side chain δ-nitrogen is covalently linked to the γ-carbon (Extended Data Fig. 5b, indicated by the blue line) to give a conformationally constrained (S)-γ-lactam, which is well accommodated in the S1 subsite, resulting in improved potency relative to Q in inhibitors46,50. GM4 and GM4H3Q contain a related cγAA ring but are non-covalently binding inhibitors, but notably still exhibit low-nanomolar IC50 values.
Recently, non-covalent small molecule inhibitors against Mpro have also been developed by means of virtual screening and optimization of compounds based on structure–activity relationships51–54. Small molecular inhibitors often have an advantage of better cell membrane permeability than macrocycles and thus are able to inhibit Mpro in cells. In fact, an impressive Mpro non-covalent inhibitor, which is a current clinical candidate, S-217622, exhibited antiviral activity in cells (half-maximal effective concentration (EC50) = 0.37 μM, in Extended Data Fig. 5c)51. Another example of a non-covalent inhibitor, compound 23, is reported to have potent in vitro inhibitory activity (IC50 = 20 nM), but exhibited cellular cytotoxicity (half-maximal cytotoxic concentration (CC50) in Vero E6 cells = 1.15 μM)52. This contrasts with S-217622, for which cell cytotoxicity was not reported, presumably because of substantial medicinal chemistry efforts to optimize in vivo properties, including from a toxicity perspective. However, classical small molecule drugs often suffer from insufficient target specificity and unwanted cell cytotoxicity. By contrast, macrocyclic peptide inhibitors generally have remarkably high target specificities, leading to low cytotoxicity; however, at least for intracellular targets, relatively low membrane permeability can be an issue for peptide macrocycles. Our GM4 and related macrocycles show no cytotoxicity at concentrations as high as 100 µM, but as yet do not manifest obvious antiviral activity in cells, likely due to their poor membrane permeability. Johansen-Leete et al. have reported an Mpro-inhibiting thioether-macrocyclic peptide using the same technology as that used by us, except without elongation reprogramming (compound 1 in Extended Data Fig. 5a)43. The originally discovered peptide showed a potent in vitro efficacy (Ki = 14 nM) but no antiviral activity. However, an increase in cellular uptake, by attaching a cell-penetrating peptide (CPP) at its C-terminal region, resulted in modest but observable antiviral activity. It is known that the addition of CPP to a macrocycle can not only increase cell permeability but also increase cytotoxicity depending on the CPP sequence and cell types used for the study55,56. Since we have witnessed improvements in in vivo potency by reengineering the macrocyclic peptide backbone structures, such as by N-methylation, in our previous works57,58, our ultimate goal is to generate a non-CPP-modified macrocycle with better cell membrane permeability and potent antiviral activity. In the case of the GM4 peptides, our current efforts are directed at reducing molecular weight while maintaining the critical yFHγ1 binding motif59.
Most importantly, our strategy for the construction of a cγAA-containing macrocyclic peptide library and its application for the RaPID system has demonstrated that unprecedented protease inhibitors containing cγAAs can be generated. It is striking that crystallography reveals that the γ1 residue of GM4 sits deeply in the Mpro active site, and that the compactness of γ1 plays a critical role in organizing a unique turn conformation, resulting in resistance to proteolytic cleavage of GM4. The general ability of cγAAs in the preorganization of turn conformations thus appears to be advantageous in obtaining potent, compact and stable macrocycles with drug-like properties.
Methods
Preparation of flexizymes and tRNAs
Flexizymes (dFx and eFx) and tRNAs for charging cγAAs, cβAAs and d-amino acids were transcribed in vitro using the T7 RNA polymerase from the corresponding template DNAs and prepared by extension and PCR (Supplementary Table 3 for primer sequences). PCR products were purified by phenol/chloroform extraction and ethanol precipitation. The transcription reaction was then carried out using the following mixture: 40 mM Tris-HCl buffer (pH 8.0), 22.5 mM MgCl2, 10 mM dithiothreitol, 1 mM spermidine, 0.01% Triton X-100, 3.75 mM NTP mix, 0.04 U μl–1 RNasin RNase inhibitor (Promega, N2615) and 120 nM T7 RNA polymerase (37 °C, 16 h). For transcription of tRNAs, 5 mM GMP was added to the above solution to introduce a monophosphate at the 5′ end of tRNAs. The resulting RNA transcripts were then treated with RQ1 DNase (Promega, M6101) at 37 °C for 30 min and purified by 12% (flexizymes) or 8% (tRNAs) denaturing polyacrylamide gel electrophoresis (PAGE) containing 6 M urea.
Preparation of aminoacyl-tRNAs
Preparation of aminoacyl-tRNA employed the flexizyme method25,33,29. The cγAAs, cβAAs and d-cysteine (c) were pre-activated as their 3,5-dinitrobenzyl esters; N-chloroacetyl-d-tyrosine (ClAcy) was activated as its cyanomethyl ester. The activated amino acids were charged onto the respective tRNAs using flexizymes (dFx for 3,5-dinitrobenzyl ester or eFx for cyanomethyl ester). Aminoacylation was carried out at 4 °C for 16 h for cγAA and cβAA, 6 h for c or 2 h for ClAcy in the following mixture: 600 mM MgCl2, 20% DMSO solvent, 25 μM dFx or eFx, 25 μM tRNA and 5 mM activated amino acid. The reaction pH was adjusted by bicine-KOH (pH 8.7) for cγAAs and cβAAs, or HEPES-KOH buffer (pH 7.5) for c and ClAcy. The reaction was stopped by addition of ×4 volume of 0.3 M sodium acetate (pH 5.2) and ×10 volume of ethanol. The resulting aminoacyl-tRNAs were purified by ethanol precipitation.
Translation of peptides
Ribosomal synthesis of the model peptides and the peptide library was carried out using the modified FIT system (37 °C, 40 min)32,34, which employs 3 μM initiation factor 2 (IF2), 20 μM EF-Tu, 5 μM EF-P and 0.1 μM elongation factor G (EF-G), and the following components: 50 mM HEPES-KOH (pH 7.6), 100 mM KOAc, 12.3 mM Mg(OAc)2, 2 mM ATP, 2 mM GTP, 1 mM CTP, 1 mM UTP, 20 mM creatine phosphate, 2 mM spermidine, 1 mM dithiothreitol, 1.5 mg ml–1 E. coli total tRNA, 1.2 μM E. coli ribosome, 2.7 μM IF1, 1.5 μM IF3, 0.25 μM release factor 2 (RF2), 0.17 μM RF3, 0.5 μM ribosome recycling factor (RRF), 4 μg ml–1 creatine kinase, 0.1 μM T7 RNA polymerase, 3 μg ml–1 myokinase, 0.1 μM inorganic pyrophosphatase, 0.1 μM nucleotide diphosphate kinase, l-α-amino acids (500 μM each G/H/N/P/R/T/Y, 250 μM each D/F/I/L/S/K and 125 μM A), 0.73 μM AlaRS, 0.03 μM ArgRS, 0.38 μM AsnRS, 0.13 μM AspRS, 0.09 μM GlyRS, 0.02 μM HisRS, 0.4 μM IleRS, 0.04 μM LeuRS, 0.11 μM LysRS, 0.68 μM PheRS, 0.16 μM ProRS, 0.04 μM SerRS, 0.09 μM ThrRS, 0.02 μM TyrRS, 50 μM γ1-tRNAPro1E2CUC, 100 μM γ2-tRNAPro1E2CAC, 20 μM β1-tRNAPro1E2GAC, 20 μM β2-tRNAGluE2GCA, 20 μM ClAcy-tRNAfMetCAU and 20 μM c-tRNAPro1E2CAU. For translation of model peptides, a 0.04 μM DNA template was added to the above solution for transcription/translation coupled reactions. The DNA templates were prepared by extension and PCR (Supplementary Table 4 for the primer sequences). For the translation of the peptide library, a 1.5 μM mRNA library conjugated to puromycin linker was used.
MALDI-TOF MS of translated model peptides
The model peptides with a C-terminal FLAG-tag (DYKDDDDK) were translated and diluted with an equal volume of ×2 TBS buffer (100 mM Tris-HCl (pH 7.6), 300 mM NaCl), and then incubated with 10 μl of ANTI-FLAG M2 affinity gel (Sigma, A2220) at 25 °C for 30 min. The gel beads were washed with 50 μl TBS buffer (50 mM Tris-HCl (pH 7.6), 150 mM NaCl); the peptides were eluted from the beads by adding 20 μl of 0.2% (v/v) trifluoroacetic acid (TFA). The peptides were desalted with SPE C-tip (Nikkyo Technos) and eluted with 1.2 μl of 80% (v/v) acetonitrile and 0.5% (v/v) acetic acid solution containing 50%-saturated α-cyano-4-hydroxycinnamic acid. MALDI-TOF MS was carried out using an ultrafleXtreme instrument (Bruker Daltonics) in reflector/positive mode. Peptide calibration standard II (Bruker Daltonics, 8222570) was used for the external mass calibration.
Production of recombinant SARS-CoV-2 Mpro
Mpro was prepared as reported, and assays with it were performed exclusively using freshly purified recombinant Mpro solution39,61. Refrozen Mpro samples exhibited reduced activity and were not used.
RaPID selection of peptides against SARS-CoV-2 Mpro
The random mRNA library was ligated with a puromycin linker at the 3′ end, and then added to the above described translation mixture (Extended Data Fig. 1a, step 1). The peptide library was translated at 37 °C for 40 min in 150 μl (for the first round of selection) or 10 μl (for the second to fourth rounds) of the FIT system and incubated at 25 °C for 5 min to conjugate the translated peptide with the corresponding mRNA–puromycin (step 2). A ×0.04 volume of 500 mM ethylenediaminetetraacetic acid (EDTA; pH 8.0) was then added and incubated (37 °C, 10 min) to dissociate ribosomes from the mRNA–peptide conjugates. Reverse transcription (42 °C, 30 min) used the NNUGWGAUG.R38 primer (5′-TTTCCGCCCCCCGTCCTAACCTCCGCTACCTCCACTCA-3′) and M-MLV reverse transcriptase lacking RNase H activity (Promega, M3682; step 3). The resulting cDNA/mRNA/peptide conjugates were subjected to naked Dynabead streptavidin (Thermo Fisher, DB11206) treatment (4 °C, 15 min) three times to remove bead-binding peptides; the supernatant was then applied to Mpro-immobilized Dynabeads (4 °C, 15 min; step 4). The beads were washed with 100 μl ice-cold TBS-T buffer (50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 0.05% (v/v) Tween 20) three times. Note that the removal of bead-binding peptides was not performed for the first selection round. Then, 100 μl of ×1 PCR buffer (10 mM Tris-HCl (pH 9.0), 50 mM KCl, 0.1% (v/v) Triton X-100, 0.25 mM dNTP, 2.5 mM MgCl2, 0.25 μM T7.F53 primer (5′-GGCGTAATACGACTCACTATAGGGTTGAACTTTAAGTAGGAGATATATCCATG-3′) and 0.25 μM NNUGWGAUG.R38 primer) was added to the beads; the cDNAs were eluted at 95 °C for 5 min and PCR amplified to make a cDNA library (step 5). To estimate the recovery rate of cDNA, 1 μl of the elute was mixed with 19 μl of ×1 PCR buffer containing SYBR Green I (Lonza, 50513) and Taq DNA polymerase; amounts of cDNA were quantified by real-time PCR.
Solid-phase peptide synthesis
Macrocyclic peptides were synthesized by standard Fmoc solid-phase peptide synthesis using a Syro I automated peptide synthesizer (Biotage). Fmoc-protected amino acids and coupling reagents were from Merck, Watanabe Chemical Industries or Enamine. NovaPEG Rink Amide Resin (54 mg, 25 μmol) was incubated with N,N-dimethylformamide (DMF; room temperature, 1 h). Each Fmoc-protected amino acid was coupled at 30 °C for 40 min on the resin in a DMF solution containing 0.2 M Fmoc-protected amino acid (6 equiv.), 0.2 M 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (5 equiv.), 0.2 M 1-hydroxybenzotriazole (5 equiv.) and 0.1 M N,N-diisopropylethylamine (12 equiv.). After washing the resin five times with 600 μl DMF, the Fmoc group was deprotected with 600 μl of 40% (v/v) piperidine in DMF at 30 °C for 12 min. Coupling of the Fmoc-protected amino acid and Fmoc deprotection were repeated accordingly. After automated peptide synthesis, 0.2 M chloroacetyl N-hydroxysuccinimide ester (8 equiv.) in N-methylpyrrolidone was added to the resin; the mixture was incubated at room temperature for 1 h with rotation. After washing the resin subsequently with DMF five times and with dichloromethane five times, the resin-bound peptides were treated with 2 ml of a solution of 92.5% (v/v) TFA, 2.5% (v/v) water, 2.5% triisopropylsilane and 2.5% 3,6-dioxa-1,8-octanedithiol at room temperature for 3 h with rotation to deprotect and cleave off from the resin. The resulting linear peptides were precipitated with diethyl ether and dissolved in 10 ml of 80% (v/v) DMSO and 0.1% TFA in water. Following the addition of 200 μl of 0.5 M tris(2-carboxyethyl) phosphine and triethylamine to adjust the pH to 8, the peptide mixture was incubated with rotation at room temperature for 10 h to form a thioether bond between the N-terminal chloroacetamide and thiol group of the downstream cysteine. Macrocyclization of the peptides was confirmed by MALDI-TOF MS; the crude peptides were purified by reverse-phase high-performance LC (Shimadzu). The purities of the peptides were evaluated by UPLC (Shimadzu) using a reverse-phase column (ACQUITY UPLC BEH C18, 1.7 μm, 2.1 × 150 mm; Waters) with a linear gradient from 10% buffer B to 70% buffer B. Buffer A was water with 0.1% (v/v) TFA; buffer B was acetonitrile with 0.1% (v/v) TFA.
Binding kinetics analysis of peptides by SPR
The binding affinities of selected peptides and Mpro were analysed by SPR using a Biacore T200 instrument (Cytiva) at 25 °C. The composition of the running buffer was 10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.05% (v/v) Tween 20 and 0.1% (v/v) DMSO. Biotin-tagged Mpro was immobilized on a Biacore sensor chip CAP (Cytiva) to a surface density of 1,500–2,000 response units following the immobilization protocols of the Biotin CAPture Kit (Cytiva). The kinetic constants were determined by a single-cycle kinetics method by the injection of five different concentrations (twofold dilution series) of each peptide at a flow rate of 30 μl min–1. Binding sensorgrams were fitted to the standard 1:1 interaction model and analysed using Biacore evaluation software.
Solid-phase extraction coupled to MS inhibition assays
Inhibition of Mpro was measured by solid-phase extraction purification coupled to MS analysis using a RapidFire 365 high-throughput sampling robot (Agilent) connected to an iFunnel Agilent 6550 accurate mass quadrupole TOF spectrometer as reported39. In brief, the cyclic peptides were dispensed in an 11-point, threefold dilution series across 384-well plates using an acoustic Echo Dispenser machine (LabCyte). Formic acid and DMSO were used as positive and negative controls, respectively. The assay was adapted for a reaction volume of 20 μl with a final condition of 75 nM Mpro and 4 µM 37-mer substrate (ALNDFSNSGSDVLYQPPQTSITSAVLQ/SGFRKMAFPS-NH2). Reactions were incubated (15 min) and then quenched by the addition of 10% (v/v) aqueous formic acid (5 μl per well). C-terminal product peptide-derived SGFRKMAF-NH2 (10 μl per well) was employed as the internal standard; data were extracted and processed as reported.
Serum stability assays
A synthetic macrocyclic peptide (10 μM) and an internal standard peptide (10 μM) (Supplementary Fig. 6a for its structure) were mixed and incubated in human serum (Cosmo Bio, 12181201) at 37 °C for up to 100 h. We have previously shown that the standard peptide is peptidase resistant, with no degradation occurring under these experimental conditions25. At each time point (0, 0.5, 1, 2, 4, 8, 24, 50 and 100 h), 4 μl of the mixture was removed and quenched by adding 12 μl methanol. Following centrifugation (15,000g, 25 °C, 3 min), 10 μl of the supernatant was mixed with 40 μl of 1% (v/v) TFA. Following centrifugtion (15,000g, 25 °C, 3 min), the supernatant was collected for LC/MS analysis, which used a reverse-phase column (ACQUITY UPLC BEH C18, 1.7 μm, 2.1 × 150 mm; Waters) and a Xevo G2-XS QTof system (Waters) with a linear gradient from 1% B to 60% B. Buffer A was water with 0.1% (v/v) formic acid; buffer B was acetonitrile with 0.1% (v/v) formic acid. The percentages of remaining peptides were determined by the peak area integration of the chromatograms.
Crystallization, data collection and structure solution
Mpro was thawed and diluted to 6 mg ml–1 using 20 mM HEPES (pH 7.5) and 50 mM NaCl. GM4 was diluted into the protein solution to a final concentration of 10 mM, which was incubated for two hours at room temperature prior to dispensing plates. The drop composition was 0.15 μl protein ligand solution, 0.3 μl 11% (v/v) polyethylene glycol (PEG) 4K, 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES) pH 6.5 and 0.05 μl Mpro crystal seed stock. The Mpro crystal seed stock was prepared by crushing Mpro crystals with a pipette tip; suspending them in 30% PEG 4K, 5% (v/v) DMSO and 0.1 M MES pH 6.5; and vortexing for 60 s with ~10 glass beads (1.0 mm diameter, BioSpec Products). The reservoir solution was 11% (v/v) PEG 4K, 5% (v/v) DMSO and 0.1 M MES pH 6.5. Crystals were grown by the sitting drop vapour diffusion method (20 °C) and appeared within 24 h, reaching full size within 36 h; they were harvested after one week.
Data collection and structure determination
All diffraction data were collected at 100 K with a wavelength of 0.9762 Å on beamline I03 at the Diamond Light Source. Data were processed using Dials62 via Xia263 and Aimless64 within CCP4i2 (ref. 65). The datasets were phased using Molrep66 using the Mpro apo structure (Protein Data Bank 6YB7). Ligand restraints were generated using AceDRG67. Crystal structures were manually rebuilt in Coot and refined using Refmac68 and PDBredo69.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41557-023-01205-1.
Supplementary information
Supplementary Figs. 1–6.
Top 100 peptide sequences of Mpro binders after the fourth round of selection.
Acknowledgements
This work was supported by a Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for JSPS Fellows (JP22J12466) to T.M.; a Grant-in-Aid for Scientific Research (B) (JP18H02080), a Grant-in-Aid for Scientific Research (A) (22H00439), and a Grant-in-Aid for Challenging Research (Pioneering) (JP21K18233) to T.K.; and a Grant-in-Aid for Specially Promoted Research (JP20H05618) to H.S. The Oxford researchers thank the COVID-19 Research Response Fund and King Abdulaziz University, Saudi Arabia, for funding. This research was funded in part by the Wellcome Trust (106244/Z/14/Z) and the European Research Council under the European Union’s Horizon 2020 Research and Innovation Programme (101003111). For the purpose of open access, the authors have applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. We thank Diamond for beamtime through the COVID-19 dedicated call (proposal ID MX27088) and the Diamond MX group for their support and expertise. T.R.M. was supported by the Biotechnology and Biological Sciences Research Council (BB/M011224/1).
Extended data
Source data
Raw data of inhibitory activity assays and serum stability assays.
Author contributions
T.M., T.R.M., C.D.O., L.B., T.K., M.A.W., C.J.S. and H.S. wrote the manuscript. T.M., T.R.M., A.T., C.D.O., L.B., M.A.M., E.S., P.L., C.S.-D. and H.M. carried out the experiments and analysed the data. N.T., T.K., M.A.W., A.K., C.J.S. and H.S. supervised the programme.
Peer review
Peer review information
Nature Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Data availability
Coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under accession code 7Z4S. Other data supporting this study are available in the Supplementary Information. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
is available for this paper at 10.1038/s41557-023-01205-1.
Supplementary information
The online version contains supplementary material available at 10.1038/s41557-023-01205-1.
References
- 1.Hintermann T, Gademann K, Jaun B, Seebach D. γ-Peptides forming more stable secondary structures than α-peptides: synthesis and helical NMR-solution structure of the γ-hexapeptide analog of H-(Val-Ala-Leu)2-OH. Helv. Chim. Acta. 1998;81:983–1002. doi: 10.1002/hlca.19980810514. [DOI] [Google Scholar]
- 2.Seebach D, Brenner M, Rueping M, Jaun B. γ2-, γ3-, and γ2,3,4-amino acids, coupling to γ-hexapeptides: CD spectra, NMR solution and X-ray crystal structures of γ-peptides. Chem. Eur. J. 2002;8:573–584. doi: 10.1002/1521-3765(20020201)8:3<573::AID-CHEM573>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 3.Chatterjee S, et al. Expanding the peptide β-turn in αγ hybrid sequences: 12 atom hydrogen bonded helical and hairpin turns. J. Am. Chem. Soc. 2009;131:5956–5965. doi: 10.1021/ja900618h. [DOI] [PubMed] [Google Scholar]
- 4.Vasudev PG, Chatterjee S, Shamala N, Balaram P. Gabapentin: a stereochemically constrained γ amino acid residue in hybrid peptide design. Acc. Chem. Res. 2009;42:1628–1639. doi: 10.1021/ar9001153. [DOI] [PubMed] [Google Scholar]
- 5.Basuroy K, et al. Unconstrained homooligomeric γ-peptides show high propensity for C14 helix formation. Org. Lett. 2013;15:4866–4869. doi: 10.1021/ol402248s. [DOI] [PubMed] [Google Scholar]
- 6.Giuliano MW, et al. Evaluation of a cyclopentane-based γ-amino acid for the ability to promote α/γ-peptide secondary structure. J. Org. Chem. 2013;78:12351–12361. doi: 10.1021/jo401501g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giuliano MW, et al. A γ-amino acid that favors 12/10-helical secondary structure in α/γ-peptides. J. Am. Chem. Soc. 2014;136:15046–15053. doi: 10.1021/ja5076585. [DOI] [PubMed] [Google Scholar]
- 8.Konda M, Kauffmann B, Rasale DB, Das AK. Structural and morphological diversity of self-assembled synthetic γ-amino acid containing peptides. Org. Biomol. Chem. 2016;14:4089–4102. doi: 10.1039/C6OB00380J. [DOI] [PubMed] [Google Scholar]
- 9.Misra R, et al. Structural dimorphism of achiral α,γ-hybrid peptide foldamers: coexistence of 12- and 15/17-helices. Chem. Eur. J. 2017;23:3764–3772. doi: 10.1002/chem.201605753. [DOI] [PubMed] [Google Scholar]
- 10.Debnath S, Ghosh S, Pandit G, Satpati P, Chatterjee S. Effect of differential geminal substitution of γ amino acid residues at the (i + 2) position of αγ turn segments on the conformation of template β-hairpin peptides. J. Org. Chem. 2021;86:11310–11323. doi: 10.1021/acs.joc.1c00351. [DOI] [PubMed] [Google Scholar]
- 11.Frackenpohl J, Arvidsson PI, Schreiber JV, Seebach D. The outstanding biological stability of β- and γ-peptides toward proteolytic enzymes: an in vitro investigation with fifteen peptidases. ChemBioChem. 2001;2:445–455. doi: 10.1002/1439-7633(20010601)2:6<445::AID-CBIC445>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 12.Bockus AT, et al. Probing the physicochemical boundaries of cell permeability and oral bioavailability in lipophilic macrocycles inspired by natural products. J. Med. Chem. 2015;58:4581–4589. doi: 10.1021/acs.jmedchem.5b00128. [DOI] [PubMed] [Google Scholar]
- 13.Umezawa H, Aoyagi T, Morishima H, Matsuzaki M, Hamada M. Pepstatin, a new pepsin inhibitor produced by actinomycetes. J. Antibiot. Tokyo. 1970;23:259–262. doi: 10.7164/antibiotics.23.259. [DOI] [PubMed] [Google Scholar]
- 14.Marks N, Grynbaum A, Lajtha A. Pentapeptide (pepstatin) inhibition of brain acid proteinase. Science. 1973;181:949–951. doi: 10.1126/science.181.4103.949. [DOI] [PubMed] [Google Scholar]
- 15.Katoh I, Yasunaga T, Ikawa Y, Yoshinaka Y. Inhibition of retroviral protease activity by an aspartyl proteinase inhibitor. Nature. 1987;329:654–656. doi: 10.1038/329654a0. [DOI] [PubMed] [Google Scholar]
- 16.Rinehart KL, Jr., et al. Didemnins: antiviral and antitumor depsipeptides from a Caribbean tunicate. Science. 1981;212:933–935. doi: 10.1126/science.7233187. [DOI] [PubMed] [Google Scholar]
- 17.Wang L, Xie J, Schultz PG. Expanding the genetic code. Annu. Rev. Biophys. Biomol. Struct. 2006;35:225–249. doi: 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- 18.Dedkova LM, Fahmi NE, Golovine SY, Hecht SM. Enhanced d-amino acid incorporation into protein by modified ribosomes. J. Am. Chem. Soc. 2003;125:6616–6617. doi: 10.1021/ja035141q. [DOI] [PubMed] [Google Scholar]
- 19.Fujino T, Goto Y, Suga H, Murakami H. Reevaluation of the d-amino acid compatibility with the elongation event in translation. J. Am. Chem. Soc. 2013;135:1830–1837. doi: 10.1021/ja309570x. [DOI] [PubMed] [Google Scholar]
- 20.Fujino T, Goto Y, Suga H, Murakami H. Ribosomal synthesis of peptides with multiple β-amino acids. J. Am. Chem. Soc. 2016;138:1962–1969. doi: 10.1021/jacs.5b12482. [DOI] [PubMed] [Google Scholar]
- 21.Guillen Schlippe YV, Hartman MCT, Josephson K, Szostak JW. In vitro selection of highly modified cyclic peptides that act as tight binding inhibitors. J. Am. Chem. Soc. 2012;134:10469–10477. doi: 10.1021/ja301017y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Passioura T, Katoh T, Goto Y, Suga H. Selection-based discovery of druglike macrocyclic peptides. Annu. Rev. Biochem. 2014;83:727–752. doi: 10.1146/annurev-biochem-060713-035456. [DOI] [PubMed] [Google Scholar]
- 23.Yamagishi Y, et al. Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol. 2011;18:1562–1570. doi: 10.1016/j.chembiol.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 24.Imanishi S, et al. In vitro selection of macrocyclic d/l-hybrid peptides against human EGFR. J. Am. Chem. Soc. 2021;143:5680–5684. doi: 10.1021/jacs.1c02593. [DOI] [PubMed] [Google Scholar]
- 25.Katoh T, Sengoku T, Hirata K, Ogata K, Suga H. Ribosomal synthesis and de novo discovery of bioactive foldamer peptides containing cyclic β-amino acids. Nat. Chem. 2020;12:1081–1088. doi: 10.1038/s41557-020-0525-1. [DOI] [PubMed] [Google Scholar]
- 26.Katoh T, Suga H. In vitro selection of foldamer-like macrocyclic peptides containing 2-aminobenzoic acid and 3-aminothiophene-2-carboxylic acid. J. Am. Chem. Soc. 2022;144:2069–2072. doi: 10.1021/jacs.1c12133. [DOI] [PubMed] [Google Scholar]
- 27.Trobro S, Åqvist J. Mechanism of peptide bond synthesis on the ribosome. Proc. Natl. Acad. Sci. USA. 2005;102:12395–12400. doi: 10.1073/pnas.0504043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohshiro Y, et al. Ribosomal synthesis of backbone-macrocyclic peptides containing γ-amino acids. ChemBioChem. 2011;12:1183–1187. doi: 10.1002/cbic.201100104. [DOI] [PubMed] [Google Scholar]
- 29.Katoh T, Suga H. Ribosomal elongation of cyclic γ-amino acids using a reprogrammed genetic code. J. Am. Chem. Soc. 2020;142:4965–4969. doi: 10.1021/jacs.9b12280. [DOI] [PubMed] [Google Scholar]
- 30.Dale T, Sanderson LE, Uhlenbeck OC. The affinity of elongation factor Tu for an aminoacyl-tRNA is modulated by the esterified amino acid. Biochemistry. 2004;43:6159–6166. doi: 10.1021/bi036290o. [DOI] [PubMed] [Google Scholar]
- 31.Katoh T, Wohlgemuth I, Nagano M, Rodnina MV, Suga H. Essential structural elements in tRNApro for EF-P-mediated alleviation of translation stalling. Nat. Commun. 2016;7:11657. doi: 10.1038/ncomms11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katoh T, Iwane Y, Suga H. Logical engineering of D-arm and T-stem of tRNA that enhances d-amino acid incorporation. Nucleic Acids Res. 2017;45:12601–12610. doi: 10.1093/nar/gkx1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murakami H, Ohta A, Ashigai H, Suga H. A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nat. Methods. 2006;3:357–359. doi: 10.1038/nmeth877. [DOI] [PubMed] [Google Scholar]
- 34.Goto Y, Katoh T, Suga H. Flexizymes for genetic code reprogramming. Nat. Protoc. 2011;6:779–790. doi: 10.1038/nprot.2011.331. [DOI] [PubMed] [Google Scholar]
- 35.Lee J, Schwarz KJ, Kim DS, Moore JS, Jewett MC. Ribosome-mediated polymerization of long chain carbon and cyclic amino acids into peptides in vitro. Nat. Commun. 2020;11:4304. doi: 10.1038/s41467-020-18001-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adaligil E, Song A, Cunningham CN, Fairbrother WJ. Ribosomal synthesis of macrocyclic peptides with linear γ4- and β-hydroxy-γ4-amino acids. ACS Chem. Biol. 2021;16:1325–1331. doi: 10.1021/acschembio.1c00292. [DOI] [PubMed] [Google Scholar]
- 37.V’kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021;19:155–170. doi: 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Owen DR, et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 39.Malla TR, et al. Mass spectrometry reveals potential of β-lactams as SARS-CoV-2 Mpro inhibitors. Chem. Commun. 2021;57:1430–1433. doi: 10.1039/D0CC06870E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan HTH, et al. Discovery of SARS-CoV-2 Mpro peptide inhibitors from modelling substrate and ligand binding. Chem. Sci. 2021;12:13686–13703. doi: 10.1039/D1SC03628A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilmouth RC, et al. Structure of a specific acyl-enzyme complex formed between β-casomorphin-7 and porcine pancreatic elastase. Nat. Struct. Biol. 1997;4:456–462. doi: 10.1038/nsb0697-456. [DOI] [PubMed] [Google Scholar]
- 42.Zhang L, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johansen-Leete J, et al. Antiviral cyclic peptides targeting the main protease of SARS-CoV-2. Chem. Sci. 2022;13:3826–3836. doi: 10.1039/D1SC06750H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kreutzer AG, et al. A cyclic peptide inhibitor of the SARS-CoV-2 main protease. Eur. J. Med. Chem. 2021;221:113530. doi: 10.1016/j.ejmech.2021.113530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ullrich S, et al. Challenges of short substrate analogues as SARS-CoV-2 main protease inhibitors. Bioorg. Med. Chem. Lett. 2021;50:128333. doi: 10.1016/j.bmcl.2021.128333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoffman RL, et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Citarella A, Scala A, Piperno A, Micale N. SARS-CoV-2 Mpro: a potential target for peptidomimetics and small-molecule inhibitors. Biomolecules. 2021;11:607. doi: 10.3390/biom11040607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin Z, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 49.Dai W, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dragovich PS, et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as l-glutamine replacements. J. Med. Chem. 1999;42:1213–1224. doi: 10.1021/jm9805384. [DOI] [PubMed] [Google Scholar]
- 51.Unoh Y, et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 2022;65:6499–6512. doi: 10.1021/acs.jmedchem.2c00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang CH, et al. Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent. Sci. 2021;7:467–475. doi: 10.1021/acscentsci.1c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rossetti GG, et al. Non-covalent SARS-CoV-2 Mpro inhibitors developed from in silico screen hits. Sci. Rep. 2022;12:2505. doi: 10.1038/s41598-022-06306-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Glaser J, et al. Hit expansion of a noncovalent SARS-CoV-2 main protease inhibitor. ACS Pharmacol. Transl. Sci. 2022;5:255–265. doi: 10.1021/acsptsci.2c00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cardozo AK, et al. Cell-permeable peptides induce dose- and length-dependent cytotoxic effects. Biochim. Biophys. Acta Biomembr. 2007;1768:2222–2234. doi: 10.1016/j.bbamem.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 56.El-Andaloussi S, Järver P, Johansson, Henrik J, Langel Ü. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: a comparative study. Biochem. J. 2007;407:285–292. doi: 10.1042/BJ20070507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawamura A, et al. Highly selective inhibition of histone demethylases by de novo macrocyclic peptides. Nat. Commun. 2017;8:14773. doi: 10.1038/ncomms14773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rogers JM, et al. In vivo modulation of ubiquitin chains by N-methylated non-proteinogenic cyclic peptides. RSC Chem. Biol. 2021;2:513–522. doi: 10.1039/D0CB00179A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vinogradov AA, Yin Y, Suga H. Macrocyclic peptides as drug candidates: recent progress and remaining challenges. J. Am. Chem. Soc. 2019;141:4167–4181. doi: 10.1021/jacs.8b13178. [DOI] [PubMed] [Google Scholar]
- 60.Zhao Y, et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell. 2022;13:689–693. doi: 10.1007/s13238-021-00883-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Douangamath A, et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020;11:5047. doi: 10.1038/s41467-020-18709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Winter G, et al. DIALS as a toolkit. Protein Sci. 2022;31:232–250. doi: 10.1002/pro.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Winter G, Lobley CMC, Prince SM. Decision making in xia2. Acta Crystallogr. D. 2013;69:1260–1273. doi: 10.1107/S0907444913015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr. D. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Potterton L, et al. CCP4i2: the new graphical user interface to the CCP4 program suite. Acta Crystallogr. D. 2018;74:68–84. doi: 10.1107/S2059798317016035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr. D. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 67.Long F, et al. AceDRG: a stereochemical description generator for ligands. Acta Crystallogr. D. 2017;73:112–122. doi: 10.1107/S2059798317000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 69.Joosten RP, Joosten K, Murshudov GN, Perrakis A. PDB_REDO: constructive validation, more than just looking for errors. Acta Crystallogr. D. 2012;68:484–496. doi: 10.1107/S0907444911054515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figs. 1–6.
Top 100 peptide sequences of Mpro binders after the fourth round of selection.
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under accession code 7Z4S. Other data supporting this study are available in the Supplementary Information. Source data are provided with this paper.