

Summary
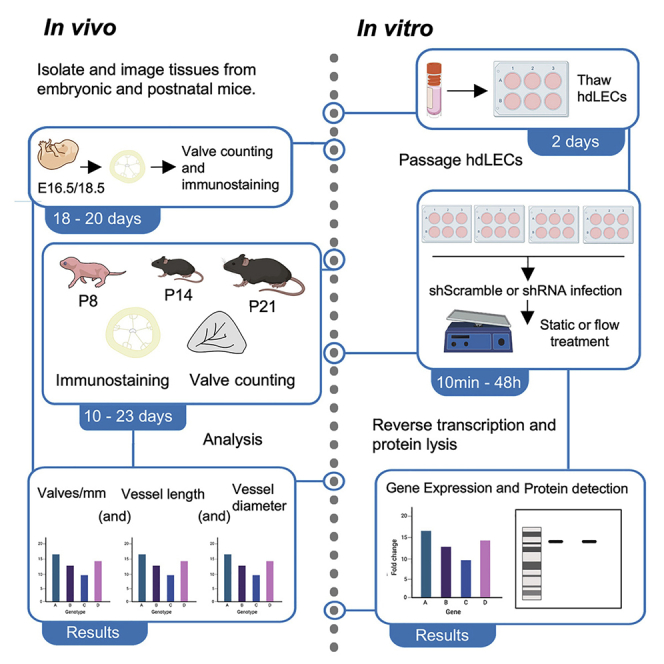
Here we detail a protocol for the isolation and processing of lymphatic enriched tissue of mouse models for the purpose of immunostaining and quantification of lymphatic valves, vessel length, and vessel diameter. Furthermore, we describe an optimized protocol for exposing treated human dermal lymphatic endothelial cells to flow for the purpose of studying lymph shear stress responses via gene expression and protein detection methods. This approach is useful to study lymphatic valve formation driven by oscillatory shear stress.
For complete details on the use and execution of this protocol, please refer to Scallan et al. (2021).1
Subject areas: Cell culture, Developmental biology, Microscopy, Model organisms, Molecular biology, Gene expression
Graphical abstract
Highlights
-
•
Detailed steps for harvesting lymphatic vessel enriched tissues
-
•
Morphological analysis of lymphatic vessels and valves using ex vivo imaging
-
•
Steps for precise quantification of valve number, vessel length, and vessel diameter
-
•
Steps for investigating flow responses in cultured human lymphatic endothelial cells
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Here we detail a protocol for the isolation and processing of lymphatic enriched tissue of mice models for the purpose of immunostaining and quantification of lymphatic valves, vessel length, and vessel diameter. Furthermore, we describe an optimized protocol for exposing treated human dermal lymphatic endothelial cells to flow for the purpose of studying lymph shear stress responses via gene expression and protein detection methods. This approach is useful to study lymphatic valve formation driven by oscillatory shear stress.
Before you begin
The protocols here elucidate the methods and techniques used to image, quantify, and illustrate the research performed for studying the lymphatic network. In vivo and in vitro work have been divided into two sections for the ease of following. These protocols have been used to successfully complete experiments and publish research papers on the many unknowns about the lymphatic system. A few things to remember would be:
-
1.
Although this protocol uses tamoxifen induced gene expression, it can be utilized downstream of various other induced gene expression methods (for example, doxycycline inducible Tet-On/Off system).
-
2.
All in vivo genetic deletions have been performed using a Prox1CreERT2 that targets lymphatic endothelial cells, but not blood endothelial cells.2 However, the protocols listed here can be used for other promoter driven Cre strains.
-
3.
This protocol has been performed using Foxo1flox/flox mice for in vivo experiments, however, it can be used for other floxed mouse strains available.
-
4.Purchase/transfer all mice strains well in advance. The following are time consuming and must be accounted for while beginning experiments.
-
a.Quarantine time of mice must be considered.
-
b.Adequate time for breeding and expanding the colony must be considered.
-
c.Appropriate personnel training must be completed before handling the mice.
-
a.
-
5.
Purchase/transfer human dermal lymphatic endothelial cells and expand the cell line for use in multiple experiments.
-
6.
Purchase drugs, vectors, surgical equipment, and cell culture reagents well in advance.
-
7.
Perform power analysis to know the specific number of animals needed for every experiment to reduce and refine the use of animals.
-
8.
Chart the number of wells of cultured cells that you will need per treatment condition for every in vitro experiment. For example, 2 replicate wells on a 6-well plate are plated for each treatment to extract enough RNA for a qRT-PCR. On the other hand, 3 triplicate wells per treatment are plated to extract enough protein for a western blot.
-
9.
Store coverslips to be used for in vitro immunostaining in 70% EtOH in the Class II laminar flow hood.
Institutional permissions
Mice used in this protocol are housed in the USF Division of Comparative Medicine, which provides an in-house service of pathogen-free animal procurement, husbandry, health surveillance, and quality control. Comparative Medicine operates in accordance with Guide for the Care and Use of Laboratory Animals, the Animal Welfare Regulations, the PHS Policy, the FDA Good Laboratory Practices, and the IACUC Principles and Procedures of Animal Care and Use.
Before you begin this protocol, it is imperative to obtain specific training, permissions, and accreditations to perform in vivo experiments under this protocol. All animal experiments will have to be approved under the institutional body for animal welfare.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical Commercial Assays | ||
| RNeasy® Plus Mini Kit | Qiagen | Cat#169042171 |
| Advantage® RT-for-PCR Kit | Clontech – Takara Bio | Cat#639506 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| UltraPure™ 1 M Tris-HCI Buffer, pH 7.5 | Invitrogen™ | Cat#15567027 |
| Agarose LE | Apex | Cat#20-102GP |
| 16% Paraformaldehyde | Electron Microscopy Sciences | Cat#15710-S |
| Tamoxifen powder 100 mg (store at 4°C) | Sigma-Aldrich | Cat#T5648-1G |
| Safflower seed oil from Carthamus tinctorius seed | Sigma-Aldrich | Cat#S82821 |
| Endothelial cell growth medium MV 2 (store at at 4°C) | PromoCell | Cat#C-22022 |
| SupplementMix Endothelial cell growth medium MV 2 (Store at −20°C) | PromoCell | Cat#C-39226 |
| Human plasma fibronectin (1 mg) | EMD Millipore Corp. | Cat#FC010 |
| 0.05% Trypsin-EDTA (1×) | Gibco | Cat#25300-054 |
| Dulbecco’s PBS (- CaCl, - MgCl) | Gibco | Cat#14190-144 |
| Triton X-100 | Sigma | Cat#T8787 |
| DAPI | MilliporeSigma | Cat#50-874-10001 |
| UltraPure™ Distilled Water (DNase, RNase, Free) | Invitrogen™ | Cat#10977-015 |
| Polybrene | VectorBuilder | Cat#PL0001 |
| Sodium azide (NaN3) | Sigma-Aldrich | Cat#S2002 |
| Sodium hydroxide (NaOH) | Sigma-Aldrich | Cat#S5881 |
| TaqMan™ Fast Univ. PCR Master mix 2× | ThermoFisher Scientific | Cat#01238930 |
| Bacterial and Virus Strains | ||
| pLV[shRNA]-EGFP/Puro-U6>Scramble_shRNA | VectorBuilder | Cat#LVL(VB151023-10034)-C |
| pLV[shRNA]-EGFP/Puro-U6>hFOXO1[shRNA#2] | VectorBuilder | Made to order |
| Experimental Models: Cell Lines | ||
| hdLEC (juvenile foreskin) | Promocell® | Cat#C12216 |
| Experimental Models: Organisms/Strains | ||
| Mouse: STOCK Foxo1tm1Rdp/J, Strain #:024756, adult, both female and male. | Jackson Laboratory | RRID: IMSR_JAX:024756 |
| Mouse: Prox1-GFP, adult, both female and male. | Choi et al.3 | N/A |
| Mouse: Prox1CreERT2, adult, both female and male. | Bazigou et al.2 | N/A |
| Software and Algorithms | ||
| Zen microscopy software | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html. |
| ImageLab Software | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z. |
| Biotek Gen5 microplate reader and imager software | Agilent | https://www.biotek.com/products/software-robotics-software/gen5-microplate-reader-and-imager-software/. |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/. |
| NIH – Fiji (ImageJ) | Biowulf | https://hpc.nih.gov/apps/Fiji.html. |
| BioRender | BioRender | https://biorender.com/ |
| Other | ||
| Ketaset® ketamine hydroschloride injection | Zoetis manufacturing and research | Cat#537921 |
| ProLong™ Diamond Antifade Mountant | Invitrogen™ | Cat#P36961 |
| Balance XSR104 | Mettler-Toledo GmbH | Cat#30355485 |
| Revolver™ Tube Mixer | Labnet Inc. | Cat#H5600 |
| FS Drybath Stdrd heating block | Fisherbrand | Cat#88860023 |
| Centrifuge 5430R | Eppendorf | Cat#5428IG027636 |
| Biometra TRIO 48 | Analytik jena | Cat#8462070723 |
| ChemiDoc™ XRS+ system | Bio-Rad | Cat#1708265 |
| Vari-mix™ test tube rocker | ThermoScientific | Cat#12-815-3Q |
| Stereomicroscope Stemi 508 | Carl Zeiss | Cat#3963001136 |
| Axiocam 208 color | Carl Zeiss | Cat#4265709000000 |
| AxioZoom.V16 | Carl Zeiss | https://www.zeiss.com/microscopy/en/products/light-microscopes/stereo-and-zoom-microscopes/axio-zoom-v16-for-biology.html#downloads. |
| Xcite Xylis light source | Excelitas | Cat#XT720S |
| Axiocam 506 mono | Carl Zeiss | https://www.zeiss.com/microscopy/en/products/cameras/axiocam-506-mono.html. |
| Laser confocal miscroscope TCS SP8 X | Leica Microsystems | Cat#1593102204 |
| QuantStudio™ 6 Flex Real-Time PCR System | Applied Biosystems™ | Cat#4485697 |
Step-by-step method details
In vivo experiments
Tamoxifen preparation
Timing: 18 h
Protocol to prepare tamoxifen.
-
1.The following steps are to prepare tamoxifen in safflower oil.
-
a.In a 50 mL tube, add 0.5 mL of 100% EtOH.
-
b.Measure 200 mg tamoxifen powder on tare paper using a METTLER TOLEDO scale for accurate measurements.
-
c.Dissolve 200 mg tamoxifen powder in the 0.5 mL 100% EtOH.
-
d.Mix 9.5 mL safflower oil into the tube and tape it to a test tube rocker for ∼16 h mixing.
-
e.Seal the cap with Parafilm and cover the entire 50 mL tube with aluminum foil to protect tamoxifen from light exposure.
 Pause point: mix for ∼16 h.
Pause point: mix for ∼16 h. -
f.Next day, make sure the powder is all dissolved before aliquoting desired amount of tamoxifen into each tube.Note: The amount of each aliquot depends on the volume of tamoxifen needed for every experiment. The leftover tamoxifen after every experiment is discarded to avoid freeze-thaw cycles.
-
g.Dissolved tamoxifen can be stored at −20°C for long term.
 CRITICAL: All tamoxifen material should be handled while wearing laboratory gloves and mask.
CRITICAL: All tamoxifen material should be handled while wearing laboratory gloves and mask.
-
a.
To identify plugged dams and calculate the date for the collection of embryos and postnatal pups
The following protocol is for the identification of a plugged dam.
-
2.For best results, mate one male mouse and 3 female mice in one cage.
-
a.Check plugs early in the morning every day.
-
b.The morning of seeing the plug is Embryonic day (E) 0.5.
-
c.Inject tamoxifen (TM) intraperitoneally for pregnant dams and subcutaneously for postnatal pups on required dates. Troubleshooting problem 5.
-
d.Calculate the date of analysis based on the plug observation.
-
a.
Isolating tissues from embryos and pups for immunostaining
This protocol breaks down the steps involved in isolating and processing mouse embryonic and postnatal tissues for whole-mount immunostaining. (Figure 1).
-
3.For embryonic tissue collection, tamoxifen (TM) injected pregnant dams are euthanized by CO2 asphyxiation followed by cervical dislocation. Embryonic tissues are harvested in the following manner:
-
a.Place the euthanized pregnant dam face up on a flat surface.
-
b.Wet her abdomen with 70% EtOH and create a superficial incision in the skin using dissecting scissors without cutting through the peritoneal sac.
-
c.Cut across the skin longitudinally to reveal the peritoneal sac.
-
d.Create an incision in the peritoneal membrane and cut across longitudinally to expose the contents of the peritoneum.
-
e.Bare the uterine horn to unveil the number of embryos.Note: At this point, label and keep ready 1.5 mL Eppendorf tubes for embryonic tail collection for genotyping.
-
d.Excise the uterine horn from the body and place in ice cold 1× PBS.
-
e.Cut through the uterine membrane and uncover individual embryos.
-
f.Peel the amniotic sac enveloping individual embryos and separate the embryos from the placenta.Note: Keep a 10 cm dish with ice cold 1× PBS ready.
-
g.Transfer the embryos to the 10 cm dish with ice cold 1X PBS.
-
h.Clip off about 2 mm of the tail and place the tails at the bottom of a 1.5 mL Eppendorf tube for genotyping.CRITICAL: To prevent cross contamination of DNA during genotyping, 1) replace with fresh PBS if the solution is bloody; 2) keep embryos away from each other in the dish; and 3) clean the scissors used for tailing with 70% EtOH between each embryo.
-
i.Now the embryos are ready for tissue collection under the dissecting microscope.
-
i.Euthanize the embryos by decapitation.
-
ii.To collect E16.5 and E18.5 axillary and back skin, pin the embryos onto a sylgard 184 pad and create an incision on the ventral lower abdominal skin using spring scissors and cut across the abdomen longitudinally to expose the peritoneal sac (Figure 1).
-
iii.Now separate the layer of skin from the peritoneal sac gently using Iris forceps and from mid abdomen to back left of the fetus, cutting connections whenever necessary.
-
iv.Repeat on the right side.
-
v.You will now be able to pin out the skin, vascular side up, on a sylgard 184 pad.
-
vi.To collect the mesentery, create an incision in the peritoneal membrane on the ventral lower abdomen using spring scissors and cut across longitudinally towards the neck exposing the organs of the peritoneal cavity (Figure 2).
-
vii.Push the intestines and stomach towards the left using the blunt end of the iris forceps, snipping the connections between the mesentery and other organs wherever necessary.
-
viii.Isolate the mesentery from the duodenum to the ileum from the body by excising the cecum and the stomach.
-
ix.Pin out the mesentery on a sylgard 184 pad starting from the duodenum in a clockwise manner into a ring.Note: Create multiple rings of the mesentery if necessary.Note: Perform every step with caution as the embryonic skin and mesentery are extremely fragile and will rip. You will need intact lymphatic vessels stretching from the mesenteric lymph node to the gut for immunostaining.
-
i.
-
j.Discard the embryos along with the dam in a red biohazard bag.
-
k.To fix embryonic tissues, use 1% paraformaldehyde (PFA).
-
i.Fix tissues in 1% PFA in PBS (1×) + NaN3 (0.05%) for ∼16 h at 4°C. Troubleshooting problem 4.Pause point: Overnight tissue fixation for ∼16 hours.
-
ii.Next day, wash the tissues 3 times, 10 min each with PBS (1×) + NaN3 (0.05%) on an orbital shaker at 4°C.
-
i.
-
a.
-
4.For postnatal tissue collection of pre-weaning and weanling pups, TM injected pups are euthanized with an intraperitoneal ketamine overdose followed by cervical dislocation.
-
a.For mesentery collection from P8, P14, and P21 mice, place the pup ventrally on a flat surface and wet the ventral skin with 70% EtOH.
-
i.Create an incision on the lower abdominal skin and cut longitudinally across the skin using dissecting scissors to reveal the peritoneal sac (Figure 2, white segmented line).
-
ii.Create an incision in the peritoneal membrane and cut across longitudinally to expose the contents of the peritoneum (Figure 2, black segmented line).
-
iii.Use the iris forceps to move the intestines along with the stomach to the left of the body, cutting connections between the intestines and other organs wherever necessary.
-
iv.Collect the mesentery from the duodenum to ileum by excising the stomach and the cecum and pin the mesentery as a ring (or multiple rings) on a sylgard 184 pad under the dissecting microscope. Troubleshoot problem 1.
-
v.Fix the mesenteries with 2% PFA for ∼16 h on an orbital shaker at 4°C. Troubleshooting problem 4.
-
vi.The next day, wash the tissue 3 times, 10 min each with PBS (1×) + NaN3 (0.05%) on an orbital shaker at 4°C.Note: Make sure not to rip the lymphatic vessels as you will need structurally intact lymphatic vessels for immunostaining.
-
i.
-
b.For ear collection from P21 pups, excise the ears from the head (Figure 8):
-
i.Place the ear with the inner side facing up under the dissecting microscope on a flat surface.
-
ii.Use a feather scalpel to slice the edge of the bottom corners of the ear to create a gap between the inner side and the outer side of the ear with the cartilage layer between these two sides.
-
iii.Use precision tweezers to remove the inner side skin together with the cartilage layer from the outer side skin to reveal the underlying vascular dermis on the outer side skin.Note: Take care to not break the cartilage layer or to leave any cartilage on the vascular dermis of the outer side skin. It will be difficult to image the ear lymphatics with a cartilage layer on top.
-
iv.Pin down the outer side skin of the ear, vascular side up on a sylgard 184 pad along the edge.
-
v.Fix ear tissue in 2% PFA in PBS (1×) + NaN3 (0.05%) for ∼16 h on an orbital shaker at 4°C. Troubleshooting problem 4.Pause point: Overnight tissue fixation for ∼16 hours.
-
vi.The next day, wash the tissue 3 times, 10 min each with PBS (1×) + NaN3 (0.05%) on an orbital shaker at 4°C.
-
i.
-
c.To collect diaphragm from P21 pups, cut through the sternum to split the ribcage and reveal organs of the pleural cavity. The diaphragm will be at the end of the ribcage and above the liver (Figure 2).
-
i.Hold the costal muscles with the iris forceps and snip at the muscle to release the diaphragm from the lower ribcage.
-
ii.Snip all around the costal muscles to release the diaphragm from the ribcage and liver.
-
iii.Finally separate the diaphragm from the vertebral column.
-
iv.Pin the diaphragm on a sylgard 184 pad with the central tendon domain side up.Note: Clean the central tendon domain off any connective tissue because the connective tissue can hamper imaging (Figure 3).
-
v.Fix the diaphragm in 2% PFA in PBS (1×) + NaN3 (0.05%) for ∼16 h on an orbital shaker at 4°C. Troubleshooting problem 4.Pause point: Overnight tissue fixation for ∼16 hours.
-
vi.The next day, wash the tissue 3 times, 10 min each time on an orbital shaker at 4°C with PBS (1×) + NaN3 (0.05%).
-
i.
-
a.
-
5.
After fixation, all tissues can be stored at 4°C in PBS (1×) + NaN3 (0.05%) within one week for further use (Figure 4).
Figure 1.

Pipeline of embryonic tissue collection and processing
Figure 2.

Representative figure of mouse anatomy and dissection technique
(A) Animal in supine position with incision line (segmented white line).
(B) Animal with the peritoneal membrane exposed with incision line (segmented black line).
(C) The red circles highlight the external ears; the blue oblong highlights the diaphragm; and the white square highlights the mesentery.
Figure 8.

Diagrammatic representation of obtaining P21 ear skin dermis for valve analysis
Figure 3.
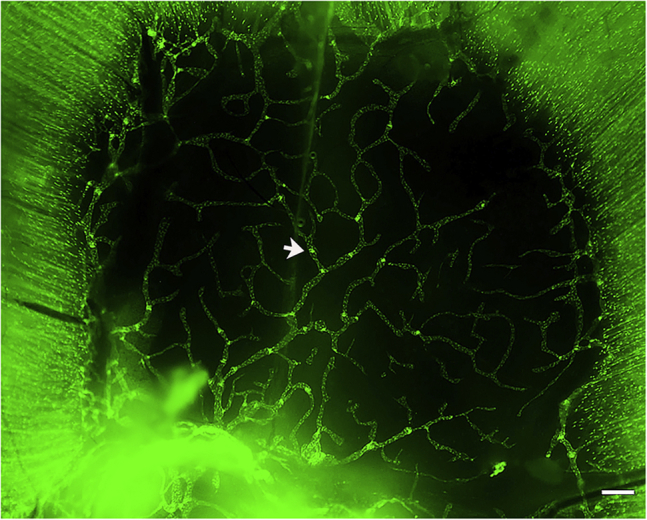
Representative GFP positive 25× image of a P21 diaphragm
Arrow points towards a GFPhigh valve. Scale bar is 250 μm.
Figure 4.

Pipeline of postnatal tissue collection and processing
To process and genotype tail clips
This protocol lists the steps in processing DNA from embryonic and postnatal mouse tail clips for genotyping.
-
6.About 2 mm tail is snipped from embryos or postnatal pups and placed in a 1.5 mL boil safe tube for processing.Note: Set a heating block to 95°C and keep it ready.
-
i.For E16.5 tails: submerge the tail in 100 μL 50 mM NaOH.For E18.5 and postnatal tails: submerge the tail in 200 μL 50 mM NaOH.
-
ii.Place the tubes in 95°C heating block for 25 min.
-
iii.Let the samples cool down.
-
iv.Neutralize the samples with 1 M Tris-HCl pH 7.5.For E16.5 tails: 20 μL 1 M Tris-HCl pH 7.5.For E18.5 and postnatal tails: 40 μL 1 M Tris-HCl pH 7.5.
-
v.Vortex samples to mix reagents.
-
vi.Centrifuge the samples at 13,000 g (10,730 rpm at 20°C–22°C) for 3 min.
-
vii.Use immediately or store at −20°C.
-
i.
-
7.
Perform a standard PCR on a Biometra TRIO thermal cycler (analytik jena) to genotype the tail samples.
-
8.
Cast an agarose gel and run the PCR product according to standard protocol to separate genomic bands according to size.
-
9.
Image the gel using a BioRad ChemiDoc XRS+ system.
To perform immunostaining on tissues collected from embryos and postnatal mice
Outlined here are the steps to perform immunostaining on embryonic and postnatal tissues collected from mice.
The tissues are pinned on sylgard 184 pads on 35 mm petri dishes. All steps are performed at 4°C on an orbital shaker, unless stated otherwise.
-
10.Day 1:
-
a.Permeabilize the fixed tissues using 4 mL of PBS (1×) + NaN3 (0.05%) + 0.3% Triton (PBST – permeabilization buffer) per dish for at least 1 h.Optional: The samples can be in PBST over an hour.
-
b.Block the samples in 3% donkey serum in 2 mL PBST per dish for at least 2 h.Optional: The blocking step can go over 2 hours.Note: Prepare your primary antibodies in 1.5 mL PBST per dish. Triple staining can be performed on the samples. But make sure not to stain two antibodies raised in the same species.
-
c.Incubate the samples with the prepared primary antibodies and mix for ∼16 h at 4°C.Pause point: Incubation of samples in primary antibodies for ∼16 h.
-
a.
-
11.Day 2:
-
a.Wash the samples with 4 mL PBST, 5 times, and 10 min each time.Note: Prepare secondary antibodies in 1.5 mL PBST in a dark bodied tube.
-
b.Incubate the samples in secondary antibodies for 1.5 h on an orbital shaker at 20°C–22°C in a black-out box to prevent light exposure.
-
c.Wash the samples with 4 mL PBST, 5 times, 10 min each.Optional: Conjugated antibodies can be incubated together with the secondary antibodies.Note: Prepare DAPI in PBS (1×) + NaN3 (0.05%) in a dark bodied tube (2 mL per dish).
-
d.Incubate samples in DAPI (1 μg/mL) for 5 min.
-
e.Wash samples with 4 mL PBS (1×) + NaN3 (0.05%), once for 5 min.
-
f.Mount the tissues on the slides with mounting media.
-
g.Carefully place a cover slip on top of the tissues without any bubbles.CRITICAL: If you plan to mount on another day, make sure to store the tissues in mounting media at 4°C. Do not store the samples in PBS (1×) + NaN3 (0.05%).
-
h.Image the immunostained slides using:
-
i.A Zeiss V16 fluorescence microscope. Troubleshooting problem 4.
-
ii.A Leica SP8 confocal fluorescence microscope.
-
i.
-
a.
To image tissues for lymphatic valve counting, measuring lymphatic vessel length, and diameter.
This protocol lists the steps involved in ex vivo imaging of mouse tissues for lymphatic valve counting.
-
12.A GFP reporter mouse line was bred with the experimental mice for visualization of lymphatic vessels and valves.
-
a.A Zeiss V16 fluorescence microscope was used to capture images of GFP positive mesenteric lymphatic vessels.
-
a.
-
13.
To count lymphatic valves in embryonic mesentery, image the whole ring of pinned out mesenteric lymphatic vessels under a Zeiss V16 fluorescence microscope with the Zen Pro software (Figures 5 and 6).
-
14.To count lymphatic valves in postnatal mesentery, images of each mesenteric collecting lymphatic vessel and its branches were captured under a Zeiss V16 microscope (Figures 5 and 7).
-
a.Overdose and euthanize the TM injected postnatal pup with ketamine followed by cervical dislocation.
-
b.Place the euthanized pup abdomen side up on a flat surface.
-
c.Wet the abdominal skin using 70% EtOH and create an incision on the skin.
-
d.Cut longitudinally towards the neck to reveal the peritoneal sac.
-
e.Create an incision on the peritoneal membrane and cut longitudinally towards the neck to expose the organs of the peritoneum and pleural cavity.
-
f.Make lateral cuts on the skin all the way to the back to expose the ribcage and the organs inferior to it (Figure 5).
-
g.Place the animal on its left side, so the right side is facing up, on a flat surface.
-
h.Place a sylgard 184 semicircular pad next to the belly of the animal to serve as a stage to pin the mesentery for easy imaging (Figure 5).Note: Use 1× PBS to hydrate the mesentery at all times of dissection and imaging. It is critical to not let the organ dry.
-
i.To identify the first lymphatic vessel at the duodenum arising from the mesenteric lymph node, use the iris forceps and carefully expose the part of the mesentery inferior to the pancreas.
-
j.Place pins wherever necessary to expose the full length of the vessel and capture an image of the GFP positive mesentery under the V16 microscope using the Zen Pro software.
-
k.Acquire images in the GFP channel under low and high magnification of each collecting lymphatic vessel by exposing the mesenteric lymphatic vessels of the duodenum, jejunum, and ileum.
- l.
-
a.
-
15.To count lymphatic valves in P21 ears, excise and pin the ear, inner vascular side up, on a sylgard 184 pad as explained in step 2b of "Isolating tissues from embryos and pups for immunostaining" (Figure 8).
-
a.Image the ear tissue under a fluorescence microscope (Figure 9).
-
b.Higher magnification images, for example 32×, will be useful for valve quantification.
-
a.
Note: The collecting lymphatic vessels are abundant in valves, and they are located towards the bottom of ear. Hence, it will be useful to capture higher magnification images of the lower half of the ear (Figure 9).
Figure 5.

Diagrammatic representation of performing ex-vivo fluorescence microscopy to analyze lymphatic valves
Figure 6.

Representative GFP positive images of E18.5 mesentery for valve counting
(A) Low magnification image of a GFP positive E18.5 mesentery showing the entire mesenteric ring.
(B) Higher magnification image of a GFP positive E18.5 mesentery showing a single mesenteric lymphatic vessel.
White arrows indicate GFPhigh valves. Scale bars are 500 μm.
Figure 7.

Representative GFP positive P14 mesentery imaged with Zen Pro software
(A) A 12.5× image of P14 collecting lymphatic vessels.
(B) A 32× image of the white box area from image A.
Scale bars are 500 μm. White arrows indicate GFPhigh valves.
Figure 9.

GFP positive P21 ear skin
(A) A 12.5× image of a GFP positive P21 ear skin.
(B) A 32× image of the white squared area in A. Arrows point towards GFPhigh valves.
Scale bars are 500 μm.
In vitro experiments
This section highlights the steps involved in the culturing and passaging of human dermal lymphatic endothelial cells (hdLECs) before performing experiments.
Culturing human dermal lymphatic endothelial cells
The protocol below enlists the steps in thawing, culturing, and passaging previously frozen hdLECs.
-
16.All steps are performed in a Class II laminar flow hood unless mentioned otherwise. While thawing cells:
-
a.Coat required number of wells of 6-well plates with fibronectin – 2 μg/cm2 fibronectin in 1 mL DPBS per well.Note: The number of wells is calculated based on seeding 1 × 105 cells per well.Note: Fibronectin coating supports hdLECs to have flow responses.
-
b.Incubate at 37°C, 5% CO2 for 30 min in a humidified cell culture incubator.Note: hdLECs are stored in liquid N2 for long-term storage.
-
c.Prepare complete endothelial cell growth medium MV 2 by adding supplement mix to the endothelial cell basal medium MV2 according to manufacturer’s instructions.Note: Keep ready a 15 mL tube with 9 mL of complete Endothelial Cell Growth Medium MV 2 in a Class II laminar flow hood.
-
d.Thaw frozen hdLECs cryovial in a 37°C water bath until very little frozen solution remains.
-
e.Quickly and generously spray the vial with 70% EtOH, wipe the tube.
-
f.Transfer the contents into the 15 mL tube containing Endothelial Cell Growth Medium MV 2.
-
g.Centrifuge the cellular suspension for 5 min at 400 g at 20°C–22°C.
-
h.Quickly remove the supernatant media and resuspend the pellet with 1 mL of Endothelial Cell Growth Medium MV 2.
-
i.Pipette up and down to create a homogenous cell resuspension.
-
j.Add the remaining Endothelial Cell Growth Medium MV 2 to make the full volume cell resuspension (1.5 mL media in each well).
-
k.Remove the fibronectin using a vacuum aspiration system (e.g., VACTRAP™) inside of the hood and add 1.5 mL of cell resuspension to each well.
-
l.Rock the plate back and forth and side to side to ensure equal distribution of cells.
-
m.Incubate the cells at 37°C, 5% CO2 in a cell culture incubator.
-
a.
-
17.While passaging cultured cells:Note: Coat required wells of 6-well plates with 2 μg/cm2 fibronectin per well and incubate in a humidified cell culture incubator for 30 min.Note: Seed 1 × 105 cells per well of a 6-well plate.
-
a.Remove Endothelial Cell Growth Medium MV 2 from the cultured wells and wash the cells with 1 mL of DPBS.
-
b.Incubate the wells with 1 mL of 0.5% Trypsin-EDTA for 3 min at 37°C, 5% CO2 in a cell culture incubator.Note: After 3 min, check if cells are floating in the well. If not, incubate for another minute and check again. Repeat if necessary.
-
c.Once cells have detached from the plate, add 1 mL of Endothelial Cell Growth Medium MV 2 to stop trypsinization.
-
d.Collect the cell suspension in a 15 mL tube and centrifuge for 5 min at 400 g at 20°C–22°C to obtain a pellet and supernatant.
-
e.Remove the supernatant and resuspend the cell pellet with 1 mL of Endothelial Cell Growth Medium MV 2.
-
f.Pipette up and down to create a homogenous cell resuspension.
-
g.For counting cell number using LUNA-II automated cell counter:
-
i.Mix 10 μL of cell resuspension with 10 μL of trypan blue dye.
-
ii.Load 10 μL of cell-dye mixture onto the commercial slides and read the concentration and the viability of the cells on the cell counter.
-
iii.The viability of the cells should be > 90% and around 1.2 × 106 per mL can be obtained from 3 confluent wells of a 6-well plate.
-
iv.Add the required amount of Endothelial Cell Growth Medium MV 2 to make up required volume.
-
i.
-
h.Pipette 1.5 mL of cell resuspension into each fibronectin coated well of 6-well plates and incubate at 37°C, 5% CO2 in a cell culture incubator.
-
a.
-
18.To prepare for immunostaining on cultured cells:
-
a.Using forceps, place a 70% EtOH soaked 22 × 22 (mm2) coverslip standing up on each well of a 6-well plate.
-
b.Air-dry the coverslip in the Class II laminar flow hood for 30 mins (Figure 10).
-
c.Drop the coverslip into the plate making sure it is lying flat.
-
d.Coat the coverslip containing wells with fibronectin (2 μg/cm2) and incubate in the humidified cell culture incubator for 30 min.
-
e.Remove the fibronectin from the wells using the vacuum aspiration system (e.g., VACTRAP™).
-
f.Passage hdLECs onto the coverslip containing wells.
-
a.
Figure 10.

Preparing coverslips for in vitro immunostaining
To expose cultured human dermal lymphatic endothelial cells to oscillatory flow
Lymph flow is oscillatory; hence, lymphatic vessels experience oscillatory shear stress (OSS), which is crucial for the expression of valve genes.4 To perform in vitro experiments on hdLECs exposed to flow conditions, an optimized rocker technique that simulates the OSS experienced by lymphatic endothelial cells (LECs) on hdLECs in 6-well plates was applied to study flow responses. The following are detailed steps for an experiment that requires hdLEC exposure to flow.
-
19.Seed 1 × 105 cells per well to start the culture.
-
a.Remove Endothelial Cell Growth Medium MV 2 media from 90%–95% confluent cells.
- b.
-
c.Incubate the plates in a cell culture incubator at 37°C, 5% CO2.
-
d.After designated treatment time, replace with fresh complete endothelial cell growth medium and dispose the media with lentiviruses in freshly made 10% bleach.
-
e.Add 1.5 mL Endothelial Cell Growth Medium MV 2 media for static conditions and 1 mL Endothelial Cell Growth Medium MV 2 media for flow conditions.Note: Prepare the rocker in the incubator. Set the rocker to 18 rpm at a 48° which is optimized rocking for lymphatic in vitro experiments.5
-
f.Place the plate on the rocker in a humidified cell culture incubator for required time.
-
i.To quantify the expression levels of valve genes using quantitative real-time polymerase chain reaction (qRT-PCR), western blot, and/or immunostaining, cells are exposed to flow conditions for 48 h (Figure 11).
-
ii.To quantify phosphorylated proteins using western blot, and/or immunostaining, cells are exposed to flow conditions for a shorter period of time (10/30/60 min) (Figure 11).Note: Some protocols might require 10/30/60 min of flow exposure. hdLECs once taken off flow must be quickly processed for respective experiments.Note: Infection of lentiviral shRNA is not required for flow treatment. hdLECs without viral infection or with different types of infection or transfection (e.g., overexpression or plasmid transfection) are suitable for flow treatment as long as cells are fully confluent before flow starts.
-
i.
-
a.
Figure 11.

Pipeline of in vitro experiments performed on hdLECs
Quantitative real time polymerase chain reaction
This protocol describes the steps to perform RNA extraction, reverse transcription, and cDNA synthesis for a qRT-PCR using cultured hdLECs.
-
20.To collect RNA from cultured hdLECs:
-
a.Follow manufacturer’s protocol to extract total RNA from hdLECs using Qiagen RNeasy® Plus Mini Kit.CRITICAL: Use RNase free tubes for RNA extraction.Note: Use 350 μL buffer RLT plus 3.5 μL β-mercaptoethanol (BME) to collect RNA from two wells of a 6-well plate with fully confluent cells.
-
b.After adding 15–25 μL RNase free water to the spin column membrane, wait 1 min to allow the water to soak into the membrane before centrifuging.
-
c.After extracting RNA, pipette 2 μL RNase free water control onto two microspots of the Take3 microvolume plate and pipette 2 μL of samples onto the microspot.
-
d.Read the plate using an absorbance reader for RNA concentration.
-
i.Use a nucleic acid quantification protocol without lid to read the plate.
-
ii.The A260/A280 ratio should be between 1.9 and 2.2 for the purest RNA extraction. Troubleshooting problem 3.
-
iii.Store the RNA samples at −80°C for long-term storage.
-
i.
-
a.
-
21.For reverse transcription and cDNA synthesis:
-
a.Thaw RNA samples on ice.
-
b.Perform reverse transcription using 1,000 ng (1 μg) RNA sample (Figure 12).
- c.
-
d.Post the PCR, dilute cDNA with Ultrapure distilled water (PCR water) at 1:16 for qRT-PCR.
-
e.RNA and cDNA can be stored at −80°C for long term use.
-
a.
-
22.Perform a qRT-PCR to quantify gene expression.Note: For every gene to be detected, each cDNA sample requires 3 wells as triplicates. The TaqMan probes are stored at −20°C, so thaw them on ice well in advance.
-
a.Prepare the PCR master mix by pipetting the TaqMan fast mix and the Taqman probe into an Eppendorf tube.Note: Multiply the number of wells based on the number of genes to be detected. For example, if the expression levels of 5 genes are to be analyzed, the total number of wells for preparing the master mix will be 3 × 5 = 15 wells. To compensate for the pipetting error, prepare the master mix for an extra well. So, it will be 16 wells (Table 1).
Table 1.
qRT-PCR mix for each geneReagent Per well (μL) Total 3 wells (μL) TaqMan 2× 5 5∗3 = 15 Probe 20× 0.5 0.5∗3 = 1.5 cDNA template (1:16) 4.5 Not in the master mix and will be pipetted onto each triplicate well on the plate Total 10 -
b.Pipette 5.5 μL PCR master mix for a probe into each well of a 384 well plate.
-
c.Add the 4.5 μL cDNA template after.Note: Use GAPDH as control.
-
d.Seal the qPCR plate with a transparent optical adhesive cover.
-
e.Run a qRT-PCR on a QuantStudio 6 real-time PCR system.
-
i.Run program for a fast standard curve.
-
ii.The program should take about 32 min.Note: Choose the program for qRT-PCR according to the type of TaqMan PCR master mix.
-
i.
-
a.
Figure 12.

Preparing RNA samples for reverse transcription
Immunostaining of hdLECs
The following protocol lists out the steps involved in in vitro staining of cultured hdLECs.
-
23.Day 1:
-
a.Culture the cells on the glass coverslips.
-
b.When the cells are ready, remove the media and wash the cells with PBS once.
-
c.Fix them with 4% PFA for 12 min at 20°C–22°C.
-
d.Wash three times with PBS (1X)+NaN3 (0.05%), 5 min each time.Note: You can store the cells in 4 degrees at this step. But make sure that the cells don’t dry, and bacteria contamination does not occur during storage.
-
e.Permeabilize the cells with 0.1% triton+PBS (1X)+NaN3 (0.05%) for 20 min at 20°C–22°C.
-
f.Block the cells with 3% donkey serum in PBS (1X)+NaN3 (0.05%) (blocking buffer) for 30 min to 1 h at 20°C–22°C.
-
g.Dilute the antibodies in the blocking buffer according to the concentration.
-
h.Add primary antibodies.Note: For square coverslips, 200 μL blocking buffer is sufficient.
-
i.Incubate in the wet box for ∼16 h at 4°C.
-
a.
-
24.Day 2:
-
a.Wash three times with PBS (1X)+NaN3 (0.05%), 5 min each time.
-
b.Dilute the secondary antibodies in the PBS (1X)+NaN3 (0.05%) according to the concentration.
-
c.Add secondary antibodies.
-
d.Incubate in the wet box for 1 h at 20°C–22°C.
-
e.Wash three times with PBS (1X)+NaN3 (0.05%), 5 min each time.
-
f.Mount with mounting media and image.
-
a.
Expected outcomes
Typical hdLECs RNA yield from two wells of a 6-well plate is ∼6,000 ng (Figure 13). Pay attention to the A260/A280 ratio. It should be between 1.9 to 2.2 for the purest RNA extraction.
Figure 13.

Obtained RNA concentrations from treated hdLECs exposed to static or flow conditions
Quantification and statistical analysis
-
1.To count valves per vessel length in embryonic and postnatal mice.
-
a.The protocol is common to embryonic and postnatal mesenteric collecting lymphatic vessels.
-
i.Use the segmented line tool in Fiji to outline the individual vessels of the mesentery (Figure 14).
-
ii.Save the lines on an ROI tool and measure the length of each line by clicking the measure button on the ROI display (Figure 15).Note: Select Analyze < Set measurement < Min and max gray value.
-
iii.Use the multipoint tool in Fiji to identify GFPhigh valves on the collecting lymphatic vessels (Figures 16 and 17).
-
iv.Sum the length of the vessels and the number of valves.
-
v.The number of valves per mm can be calculated by:
-
vii.For example, the expected valves per mm of E18.5 Foxo1flox/flox mesentery is 1.7 valves/mm.1
-
v.Run the power analysis to determine the number of animals for each experiment. Male and female embryos are utilized. Significance is achieved at p < 0.05 and calculated using GraphPad Prism.
-
i.
-
a.
-
2.To quantify lymphatic valves of P21 ear skin, both males and females were used. Significance was achieved at p < 0.05 and calculated using GraphPad Prism.
-
a.Use the 32× GFP images of the ear skin for the quantification. Select one where you can easily differentiate the collecting lymphatic vessels and the GFPhigh valves are easily identifiable (Figure 14).
-
b.Using segmented line tool on Fiji, outline the length of the collecting lymphatic vessel and save it in an ROI. Repeat the same on at least 3 collecting lymphatic vessels on one ear skin (Figure 18).
-
c.Using the multi-point tool in Fiji, number the GFPhigh valves (Figures 16 and 18).
-
d.Use the valves per mm formula to calculate valves per mm on a P21 ear skin.
-
a.
-
3.To quantify lymphatic vessel diameter, at least 3 lymphatic collecting vessels from each ear were used. Both male and female pups are used for this analysis. Significance was achieved at p < 0.05 and calculated using Prism.
-
4.To quantify qRT-PCR data. The original file will be in “.eds” format. Click on the file and open using the design and analysis software on ThermoFisher connect ™.
-
a.Name samples and targets and assign samples and targets to selected wells.
-
b.Click analyze. A standard curve will be generated for each gene.
-
c.On the “actions” menu, select “Export” and an excel file will get saved to your dashboard.
-
d.Save this excel file with all sheets combined from your dashboard.
-
e.Use the results sheet of the excel document to calculate fold change.
-
f.All sample Cq values will be normalized to GAPDH.
-
g.The following table will summarize the calculation of fold change (Table 2).
-
h.If you want to calculate the relative fold change between X and Y, then the ddCq must be calculated taking one of the samples to be a control sample, hence, you would be calculating the fold change of X compared to Y. The control sample is always normalized to itself, thereby harboring a ddCq value of 0 and a fold change of 1.
-
i.In this example, X changes 8-fold comparted to Y.
-
a.
Figure 14.

Segmented line tool on NIH-Fiji
Figure 15.

Measuring vessel length using Fiji
(A) Outlining vessel length using segmented line tool in Fiji.
(B) The saved ROI.
(C) Measurements of the ROI.
Figure 16.

Multi-point tool on NIH-Fiji
The red box marks the multi-point tool.
Figure 17.

Counting valves using multi-point tool in Fiji
(A) 12.5× image of the mesenteric collecting lymphatic vessel.
(B) A 32× magnification of the white box in A showing valve counting using multi-point tool in Fiji.
Scale bars are 500 μm.
Figure 18.

Quantification of collecting lymphatic valves of P21 ear skin
(A) Outlining vessel length.
(B) The saved ROI (Upper panel). Results showing length (lower panel). Click the “measure” tool to obtain length. Add the lengths to obtain total length.
(C) Numbering GFPhigh valves.
Figure 19.

Measuring collecting lymphatic vessel diameter using NIH-Fiji
(A) A 32× image of ear skin displaying outlined width of collecting lymphatic vessel using straight line tool in Fiji.
(B) Zoomed in view of the white box area in A.
(C) ROI for A. By clicking the measure tool on ROI manager, obtain the value of vessel diameter.
(D) Calculate the average value of vessel diameter using the sum of lengths marked in the red box.
Table 2.
Fold change calculation
| Sample | Target | Cq | Av Cq | Av GAPDH Cq | dCq | ddCq | Fold change (Rq) |
| X1 | PROX1 | 25 | =Av (X1,X2,X3) = 24 | 17 | =Av Cq - Av GAPDH Cq = 7 | = Av dCq(X) – AvdCq(Y) = (-3) | =2(-ddCq) = 2(-(-3)) = 8 |
| X2 | PROX1 | 24 | |||||
| X3 | PROX1 | 26 | |||||
| Y1 | PROX1 | 28 | =Av (Y1,Y2,Y3) = 27 | 17 | =Av Cq - Av GAPDH Cq = 10 | = Av dCq(Y) – AvdCq(Y) = 0 | = 2(-(-0)) = 1 |
| Y2 | PROX1 | 27 | |||||
| Y3 | PROX1 | 29 |
Limitations
No major limitations are expected in the execution of the above protocols. The protocols have been tested and performed multiple times by trained and experienced personnel and have led to many published research articles.1,6,7 Having said that, some aspects of the protocol demand attention. It is imperative to know working with animals requires patience and planning. Identifying a plugged dam does not guarantee conception and it requires routine daily checks to identify a plugged dam. In vivo experiments require extended waiting periods and being resourceful with animals should be our top priority. Although the in vivo oscillatory shear stress that collecting lymphatic vessels experience is 1–12 dyn/cm2, the maximum shear stress than can be generated is 0.3 dyn/cm2 on the test tube rocker under 18 rpm with an incline of 24° on each side.5 An alternative shear stress generating system, the Ibidi pump system, was used in the lab, however, the ThermoFisher Scientific digital test tube rocker flow method generates more cell lysate material to work with in the in vitro experiments with a proper flow response.
Troubleshooting
Problem 1
GFP expression is lost during tissue collection.
Potential solution
Collecting and imaging tissues take time. It is highly possible that the GFP expression might be lost due to cell death caused by tissue dehydration. For example, mesentery, ear skin and diaphragm are sensitive organs that lose GFP expression if dehydrated (related to steps 3 and 4). To avoid such circumstances, periodically hydrate the remaining organs with ice cold 1× PBS.
Problem 2
Lentivirus infection causes cell death.
Potential solution
To increase cell survival, the duration of viral treatment and the amount of viruses should be optimized based on the response of cultured hdLECs. For example, if 48 h lentiviral infection with MOI 120 causes severe cell death, a shorter treatment (e.g., 24 h) and/or a lower MOI should be tested (relevant to step 19). However, it is required to confirm that any changes of the viral infection should still result in significant knockdown of the gene of interest.
Problem 3
RNA concentration too low for reverse transcription.
Potential solution
Expand the number of wells to increase the total amount of RNA. The protocol calls for 1,000 ng RNA sample for reverse transcription, however, you can use anywhere from 500 ng to 1,000 ng. If the A260/A280 ratio less than 1.9, it is advisable to not use the sample as it might contain genomic DNA (relevant to step 20).
Problem 4
Some immunostaining antibodies are sensitive to the concentration of PFA.
Potential solution
The concentration of PFA used to fix tissues should be optimized according to the antibodies for immunostaining. For example, 2% PFA is generally used for fixing postnatal tissues, but some antibodies work better after 1% PFA fixation for postnatal tissues (relevant to steps, 3, 4, 10, and 11).
Problem 5
Deletion efficiency is low in vivo.
Potential solution
The amount and the duration of administered tamoxifen is dependent on the CreERT2 and the flox strains. The deletion efficiency can be assessed by using the RosamT/mG reporter line and/or by immunostaining the protein to be deleted (relevant to step 2).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ying Yang (yingyang@usf.edu).
Materials availability
This study did not generate new unique reagents. Any information regarding this protocol is available from the lead contact.
Acknowledgments
This work was performed under funding from National Heart, Lung, and Blood Institute (NHLBI) grants R01 HL145397 to Y.Y. We thank Dr. Taija Makinen of Uppsala University for sharing the Prox1CreERT2 mice. We thank Dr. Young Kwon Hong at the Keck School of Medicine of USC for sharing the Prox1-GFP mice. Figures 1, 2, 4, 5, 8, 10, and 11 are created with BioRender.com.
Author contributions
Conceptualization, Supervision, and Funding Acquisition, Y.Y.; Methodology, Formal Analysis, and Writing – Original Draft, Y.Y., R.B.; Validation, Investigation, and Writing – Review & Editing, Y.Y., L.A.K., R.B. All authors contributed to and approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Data and code availability
This study did not analyze/generate any new unique data and codes.
References
- 1.Scallan J.P., Knauer L.A., Hou H., Castorena-Gonzalez J.A., Davis M.J., Yang Y. Foxo1 deletion promotes the growth of new lymphatic valves. J. Clin. Invest. 2021;131:e142341. doi: 10.1172/JCI142341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bazigou E., Lyons O.T., Smith A., Venn G.E., Cope C., Brown N.A., Makinen T. Genes regulating lymphangiogenesis control venous valve formation and maintenance in mice. J. Clin. Invest. 2011;121:2984–2992. doi: 10.1172/JCI58050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi I., Chung H.K., Ramu S., Lee H.N., Kim K.E., Lee S., Yoo J., Choi D., Lee Y.S., Aguilar B., Hong Y.K. Visualization of lymphatic vessels by Prox1-promoter directed GFP reporter in a bacterial artificial chromosome-based transgenic mouse. Blood. 2011;117:362–365. doi: 10.1182/blood-2010-07-298562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sweet D.T., Jiménez J.M., Chang J., Hess P.R., Mericko-Ishizuka P., Fu J., Xia L., Davies P.F., Kahn M.L. Lymph flow regulates collecting lymphatic vessel maturation in vivo. J. Clin. Invest. 2015;125:2995–3007. doi: 10.1172/JCI79386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cha B., Geng X., Mahamud M.R., Fu J., Mukherjee A., Kim Y., Jho E.H., Kim T.H., Kahn M.L., Xia L., et al. Mechanotransduction activates canonical Wnt/beta-catenin signaling to promote lymphatic vascular patterning and the development of lymphatic and lymphovenous valves. Genes Dev. 2016;30:1454–1469. doi: 10.1101/gad.282400.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y., Cha B., Motawe Z.Y., Srinivasan R.S., Scallan J.P. VE-cadherin is required for lymphatic valve formation and maintenance. Cell Rep. 2019;28:2397–2412.e4. doi: 10.1016/j.celrep.2019.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogunsina O., Banerjee R., Knauer L.A., Yang Y. Pharmacological inhibition of FOXO1 promotes lymphatic valve growth in a congenital lymphedema mouse model. Front. Cell Dev. Biol. 2022;10:1024628. doi: 10.3389/fcell.2022.1024628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not analyze/generate any new unique data and codes.