

Abstract
Small extracellular vesicles (sEV) contain various microRNAs (miRNAs) and play crucial roles in the tumor metastatic process. Although miR‐29b levels in peritoneal exosomes were markedly reduced in patients with peritoneal metastases (PM), their role has not been fully clarified. In this study, we asked whether the replacement of miR‐29b can affect the development of PM in a murine model. UE6E7T‐12, human bone marrow‐derived mesenchymal stem cells (BMSCs), were transfected with miR‐29b‐integrating recombinant lentiviral vector and sEV were isolated from culture supernatants using ultracentrifugation. The sEV contained markedly increased amounts of miR‐29b compared with negative controls. Treatment with transforming growth factor‐β1 decreased the expression of E‐cadherin and calretinin with increased expression of vimentin and fibronectin on human omental tissue‐derived mesothelial cells (HPMCs). However, the effects were totally abrogated by adding miR‐29b‐rich sEV. The sEV inhibited proliferation and migration of HPMCs by 15% (p < 0.005, n = 6) and 70% (p < 0.005, n = 6), respectively, and inhibited adhesion of NUGC‐4 and MKN45 to HPMCs by 90% (p < 0.0001, n = 5) and 77% (p < 0.0001, n = 5), respectively. MicroRNA‐29b‐rich murine sEV were similarly obtained using mouse BMSCs and examined for in vivo effects with a syngeneic murine model using YTN16P, a highly metastatic clone of gastric cancer cell. Intraperitoneal (IP) transfer of the sEV every 3 days markedly reduced the number of PM from YTN16P in the mesentery (p < 0.05, n = 6) and the omentum (p < 0.05, n = 6). Bone marrow mesenchymal stem cell‐derived sEV are a useful carrier for IP administration of miR‐29b, which can suppress the development of PM of gastric cancer.
Keywords: exosome, gastric cancer, mesothelial cell, miR‐29b, peritoneal metastases
miR‐29b incorporated in mesenchymal stem cells (MSCs) derived exsosomes strongly inhibits mesothelial mesenchymal transition in vitro. Intraperitoneal transfer of the miR‐29b‐rich exosomes suppresses the development of peritoneal metastasis from gastric cancer in murine model.
Abbreviations
- BMSC
bone marrow‐derived mesenchymal stem cell
- EMT
epithelial–mesenchymal transition
- EV
extracellular vesicle
- FN
fibronectin
- HPF
high power field
- HPMC
human omental tissue‐derived mesothelial cell
- IP
intraperitoneal
- MC
mesothelial cell
- miR
microRNA
- miRNA
microRNA
- MMT
mesothelial mesenchymal transition
- MSC
mesenchymal stromal cell
- NC
negative control
- PM
peritoneal metastases
- PMC
peritoneal mesothelial cell
- RFP
red fluorescent protein
- sEV
small extracellular vesicle
- TGF‐β1
transforming growth factor‐β1
1. INTRODUCTION
Peritoneal metastases frequently occur in patients with gastric cancer, which is associated with an extremely poor prognosis. 1 , 2 , 3 Although PM are likely to develop from free IP tumor cells exfoliated from the serosal surface of a primary tumor, 4 , 5 the detailed mechanisms leading to the formation of PM have not been fully elucidated. Emerging evidence suggests that tumor‐secreted EV contains various functional molecules such as proteins, DNA, and various types of RNAs, 6 , 7 , 8 conveying potential biological information to target organs and creating a premetastatic niche to establish organotypic metastases. 9 , 10 In particular, miRNAs in sEV circulating in the blood have been shown to be dysregulated in the circulating blood of patients with cancer 11 and are used as noninvasive biomarkers for early diagnosis and evidence of tumor progression in various types of malignancies. 12 , 13 , 14
In comparison to exosomes in serum, the molecular composition and functions of sEV in the peritoneal cavity are less understood, presumably due to the difficulty of obtaining samples from patients. However, previous studies have shown that sEV derived from gastric 15 , 16 , 17 , 18 or ovarian 19 cancer cells as well as from malignant ascites from patients 20 are efficiently incorporated in MCs and induces MMT, which facilitates the adhesion of tumor cells to MC and enhances development and growth of PM from gastric cancer. In addition, inhibition of sEV secretion by Rab27b knockdown in tumor cells suppressed PM formation in a murine model. 21 Based on these results, it is supposed that sEV in the peritoneal cavity also play key roles in the formation of metastases on peritoneal surfaces. 22 , 23
In previous studies, we undertook a comprehensive analysis of miRNA in sEV contained in the IP fluid (ascites/peritoneal lavage fluid) from patients with gastric cancer and found that the miR‐29 family, especially miR‐29b, is markedly reduced in peritoneal sEV in patients with PM. 24 Among the patients with advanced gastric cancer with serosal exposure (T4) who underwent curative gastrectomy, miR‐29b was markedly downregulated in patients who developed peritoneal recurrence as compared with those without recurrence. 25 As miR‐29b is well known to have suppressive effects on tumor progression, 26 , 27 , 28 we hypothesized that exogenous replacement of miR‐29b in sEV might suppress PM. In a previous study, we found that IP transfer of miR‐29b mimics together with atelocollagen can partially suppress PM partly though the effects of MC. 29 In the present study, therefore, we sought to determine how the antitumor effects of miR‐29b are modified if administered in the form of encapsulation in sEV. As a source of EVs, we used BMSCs, because MSCs are known to be well‐suited for mass production of sEV for drug delivery. 30 , 31
2. MATERIALS AND METHODS
2.1. Reagents
Rabbit mAbs to E‐cadherin, calretinin, and FN, were purchased from Abcam and Cell Signaling Technology. Recombinant TGF‐β1 was purchased from R&D Systems. Rabbit anti‐vimentin mAb and anti‐rabbit IgG conjugated with Alexa Fluor 488 or Alexa Fluor 595, anti‐mouse IgG conjugated with Alexa Fluor 647 were obtained from Invitrogen. We obtained DAPI from Dojindo. Anti‐mouse CD31, CD34, CD44, CD49d, CD73, and CD90 were obtained from BioLegend. Anti‐human CD9 and CD63 were obtained from BD Biosciences. The lentivirus plasmid of miR‐29b precursor and negative control miRNA as well as the pLV‐miRNA Expression Vector System were purchased from Biosettica. The sequence of the oligonucleotides used for miR‐29b precursor (hsa‐mir‐29b‐1) is as follows:
CUUCAGGAAGCUGGUUUCAUAUGGUGGUUUAGAUUUAAAUAGUGAUUGUCUAGCACCAUUUGAAAUCAGUGUUCUUGGGGG.
2.2. Cell culture
NUGC‐4 human gastric cancer cells were obtained from Riken, and a BMSC line, UE6E7T‐12, was obtained from JCRB cell bank. The cells were cultured in DMEM supplemented with 10% FBS (Sigma), 100 U/mL penicillin, and 100 mg/mL streptomycin (Life Technologies) at 37°C in a 5% CO2 cell culture incubator.
2.3. Isolation and culture of HPMCs
Human greater omentum (3–5 cm3) was obtained from patients who underwent sleeve gastrectomy with written informed consent. Human omental tissue‐derived mesothelial cells were isolated as described elsewhere. 32 In brief, cells were collected from omental samples, placed in a half TrypLE Express (Thermo Fisher Scientific) with pure PBS, and incubated at 37°C for 2 h. The supernatants were collected after filtration through 100 μm‐pore nylon mesh, then centrifuged at 420 g at 4°C for 5 min. Explants were cultured in DMEM with 20% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin into collagen‐coated 10 cm2 tissue culture dishes. All procedures were carried out in accordance with guidelines and regulations of the Declaration of Helsinki and all experimental protocols were approved by the Institutional Review Boards of Jichi Medical University (approval number: RIN‐A‐21‐048). Written informed consent was obtained from all patients who provided omental samples for this study.
2.4. Isolation and identification of mouse BMSCs
Bone marrow obtained from the femurs of 6 weeks old C57BL/6 mice was used to isolate the primary MSCs. The BMSCs were cultured using a MesenCult Expansion Kit (Veritas) according to the manufacturer's instructions. The BMSCs at passage 3 were incubated with 10 μL of Fc‐blocker for 10 min and immunostained with mAbs for 1 h at room temperature. After washing and labeling of dead cells with 7‐AAD (Invitrogen), cells were analyzed using a FACSCalibur and CellQuest Pro Software (Becton Dickinson).
2.5. Lentiviral transfection of human/mouse BMSCs and isolation of sEV
The lentivirus incorporating miR‐29b precursor and RFP was created using the pLV‐miRNA Expression Vector System according to the manufacturer's instructions. 33 UE6E7T‐12 and mouse BMSCs were harvested at a density of 1 × 105 cells/well in 6‐well plates and then incubated at 37°C in DMEM and 5% CO2. After 12 h, cells were transfected with recombinant lentiviral vector integrated with miR‐29b or miR‐NC vector with 10 μg/mL polyacrylamide (Sigma‐Aldrich) to assist the internalization of virus particles. The MOI was 10. After 20 h, the medium was replaced with fresh culture medium. Three days after transfection, more than 90% of BMSCs were identified to be positive for RFP. The BMSCs were additionally cultured for 48 h, and EVs were extracted from culture supernatants by ultracentrifugation. In brief, supernatants were centrifuged at 2000 g for 10 min to remove floating cells and filtered through a 200 nm filter (Millipore) to remove cell debris. Then the supernatants were ultracentrifuged at 150,000 g for 70 min at 4°C. The presence of isolated EVs was confirmed using an HT‐7700 transmission electron microscope (Hitachi High‐Technologies). Size distribution and number of EVs were determined using NanoSight LM10 (Malvern). The FACSCaliber analysis was applied to detect CD9 or CD63 on the surface of EVs. Purified EVs were incubated with 4 μm diameter aldehyde/sulfate latex beads (Invitrogen) in PBS for 15 min at room temperature under gentle agitation. After washing with PBS, samples were resuspended in PBS and incubated for 1 h with Abs to CD9 and CD69 and then with Alexa Fluor 647‐conjugated secondary Ab and analyzed using a FACSCalibur flow cytometer and CellQuest Pro Software.
2.6. Sample collection, miRNA purification, and digital PCR
Cell line miRNAs were isolated from cell pellets using the miRNeasy kit (Qiagen) according to the manufacturer's instructions. Experiments were carried out in singleplex using the QuantStudio 3D‐Digital PCR System platform using the QuantStudio 3D‐Digital PCR Master Mix V2 (Thermo Fisher Scientific), according to the manufacturer's instructions. A negative control was included in each analysis. The target gene miR‐29b was analyzed in triplicate for both samples and controls. We prepared 15 μL of reaction mix containing 7.5 μL of 2× QuantStudio 3D Digital PCR Master Mix (Life Technologies), 0.75 μL of 20× TaqMan‐MGB‐FAM‐probe assay, 5.0 μL diluted cDNA (50 ng/μL), and 1.75 μL nuclease‐free water (Qiagen). Loaded chips underwent thermocycling following specific amplification conditions: 96°C for 8 min, 40 cycles at 60°C for 2 min, and 98°C for 30 s, followed by a final extension step at 60°C for 2 min. The expression levels of miR‐29b were determined using a quantification analysis module on Thermo Fisher Cloud (Thermo Fisher Scientific).
2.7. Immunofluorescence observation
The BMSC‐derived sEV were stained with PKH26 using Cell Linker Kits for General Cell Membrane Labeling (Sigma‐Aldrich) as described previously. 34 The HPMCs or gastric cancer cells (1 × 104) were plated in 24‐well plates at 70%–80% confluence and incubated with labeled sEV for 24 h.
To examine the effects on MMT, the HPMCs (5 × 104) were incubated with 10 ng/mL TGF‐β1 in 24‐well collagen‐coated plates and transfected with miR‐29b or NC‐containing sEV for 48 h. Cells were washed with PBS, fixed in 4% paraformaldehyde for 10 min at 37°C, and permeabilized with 0.5% Tween‐20 in PBS for 20 min. Subsequently, cells were blocked for 1 h with 3% BSA in PBS and incubated for 1 h at room temperature with mAbs to E‐cadherin (1:200), calretinin (1:500), vimentin (1:1000), and FN (1:150). Cells were then washed three times with PBS and incubated for 30 min with secondary anti‐rabbit Abs conjugated with Alexa Fluor 488 or Alexa Fluor 595 (1:2000). Finally, the nucleus was counterstained with DAPI (1:1000) for 5 min. Glass coverslips were placed on slides and the preparations were visualized under a fluorescence microscope (Keyense).
2.8. Cell proliferation assay
Human omental tissue‐derived mesothelial cells (1.0 × 104 cells) transfected with miR‐29b or NC contained in sEV were cultured with or without TGF‐β1 in 96 well culture plates for 24 h and incubated with The 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) (Dojindo) diluted in normal culture media at 37°C for an additional 3 h. Proliferation rates were quantified with a microtiter plate reader (Spectra Rainbow; Tecan).
2.9. Cell migration assay
Human omental tissue‐derived mesothelial cells (5.0 × 105 cells) and gastric cancer cells transfected with miR‐29b or NC containing sEV were cultured with or without TGF‐β1 for 48 h. Cells were resuspended in DMEM and seeded on an 8 μm‐pore membrane in culture inserts of a 24‐well plate and 10% FBS added to the lower chamber as a chemoattractant. After 24 h, nonmigrated cells were gently removed with a cotton swab. Migrated cells on the lower surface of the culture insets were stained with Dif‐Quick (Sysmex) and counted under the microscope.
2.10. Cell adhesion assay
Human omental tissue‐derived mesothelial cells (5 × 104 cells) were transfected with miR‐29b or NC containing sEV and cultured with or without TGF‐β1 in 24‐well culture plates. Fluorescein labeled NUGC‐4 or MKN‐45 (1 × 104 cells) were added and incubated for 15 min for all cells to attach to the HPMC monolayer. After gentle washing with warmed media three times, the number of NUGC‐4 cells remaining attached on the HPMCs was counted under a fluorescent microscope.
2.11. Syngeneic peritoneal metastasis model of gastric cancer
C57BL/6N mice were purchased from CLEA and allowed to adapt to their new surroundings for 7 days. YTN16P (1 × 105 cells, 500 μL), a highly metastatic clone of the murine gastric cancer cell line YTN16, 35 was IP injected into C57BL/6N mice. Mice were then IP injected with PBS (500 μL) or sEV (40 μg/500 μL) isolated from BMSCs transfected with lentivirus integrating miRNA‐29b‐ (Exo‐miR‐29b) or miR‐NC‐ (Exo miR‐NC), every 3 days, seven times from day 0 to day 18. Mice were killed on day 21 and peritoneal metastases were evaluated based on the number of macroscopic nodules on the mesentery and the omental surface. All procedures were approved by the Animal Care Committee of Jichi Medical University (No. 20043‐01) and carried out in accordance with the Guiding Principles in the Care and Use of Animals approved by the Council of the American Physiological Society.
2.12. Statistical analysis
Data are represented as mean ± SD. The significance of the differences between groups was assessed with one‐way ANOVA using GraphPad Prism 8. Differences were considered significant when the p value was less than 0.05.
3. RESULTS
3.1. MicroRNA‐29b‐containing sEV derived from human BMSCs suppress MMT of HPMCs
UE6E7T‐12 was transfected with lentiviral vector integrated with miR‐29b or miR‐NC, and the EV were purified from the supernatant by ultracentrifugation. A scanning electron microscope and nanoparticle tracking analysis showed that most of the vesicles were 100–200 nm in diameter (Figure 1A,B). Flow cytometry revealed that EVs highly expressed CD9 and CD63 (Figure 1C). These data suggest the purified EVs are categorized as sEV. The expression of miR‐29b in BMSCs and their sEV was strongly increased in the sEV transfected miR‐29b with 3D‐PCR (Figure 1D). The sEV labeled with PKH26 were incorporated in HPMCs more clearly than MKN‐45 or NUGC‐4 cells for 24 h (Figure 1E). The HPMCs cocultured with miR‐29b‐containing EVs for 48 h showed markedly increased expression of miR‐29b (Figure S1).
FIGURE 1.
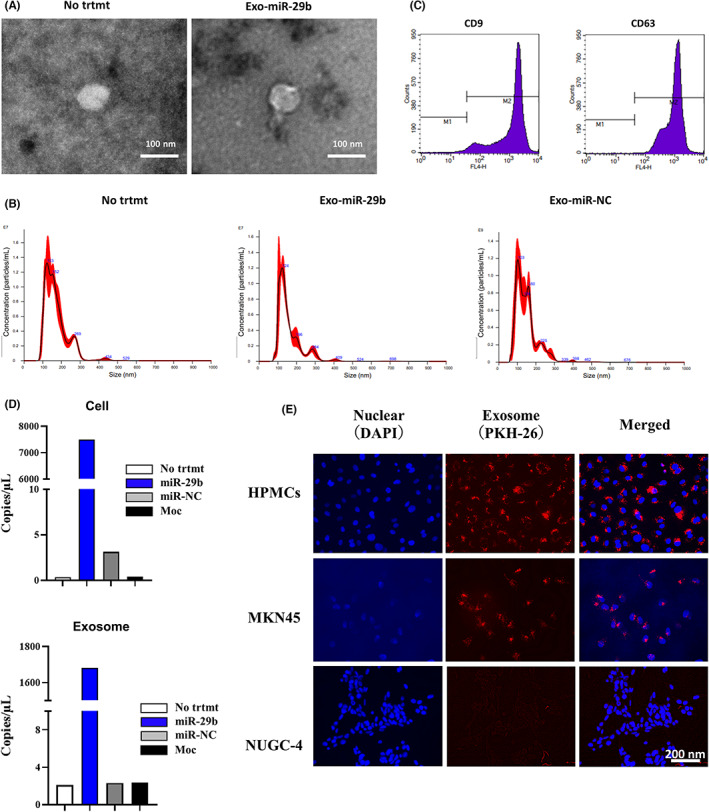
Identification of extracellular vesicles (EVs) derived from human bone marrow‐derived mesenchymal stem cells (BMSCs). (A) EVs isolated from supernatant of human BMSCs (UE6E7T‐12) (No treatment [trtmt]), BMSCs transfected with microRNA (miR)‐29b (Exo‐miR‐29b), or negative control (Exo‐miR‐NC) were dropped onto a copper grid, dyed with uranyl acetate, and observed using an HT‐7700 transmission electron microscope. Magnification, 40,000×. (B) Identification of EVs by nanoparticle tracking analysis. EV samples were observed for size distribution and number of extracellular vesicles by NanoSight LM10. (C) Detection of EV surface antigens by flow cytometry. Latex beads were incubated with EV and EV‐coated beads were analyzed for the presence of mAbs to CD9 or CD63 by a flow cytometer. (D) miR‐29b expression in human BMSCs (UE6E7T‐12) and corresponding EVs were quantified with 3D‐digital PCR. UE6E7T‐12 was transfected with recombinant lentiviral vector containing miR‐29b or miR‐negative control (miR‐NC) vector or vector alone (Moc) with 10 μg/mL polyacrylamide. After 20 h, the medium was replaced with fresh culture medium. Three days after transfection, EVs were extracted from the UE6E7T‐12 supernatant by ultracentrifugation. miRNAs were isolated from cell pellets and EVs using the miRNeasy kit. Experiments were performed in singleplex by the QuantStudio 3D Digital PCR System platform. (E) EVs derived from UE6E7T‐12 were stained with PKH26 and cocultured with HPMCs, MKN‐45, or NUGC‐4 cells for 24 h and observed with fluorescein microscopy. Magnification, 400×.
After 48 h of culture with 10 ng/mL TGF‐β1, HPMCs changed their morphology from round to spindle shape. Stimulation with TGF‐β1 significantly downregulated the expression of E‐cadherin and calretinin, but markedly enhanced vimentin expression (Figure 2). However, addition of miR‐29b‐containing sEV (Exo‐miR‐29b), but not NC (Exo‐miR‐NC), significantly suppressed the morphological changes with reduced expression of vimentin and restored the expression of E‐cadherin and calretinin induced by TGF‐β1 (Figure 2).
FIGURE 2.
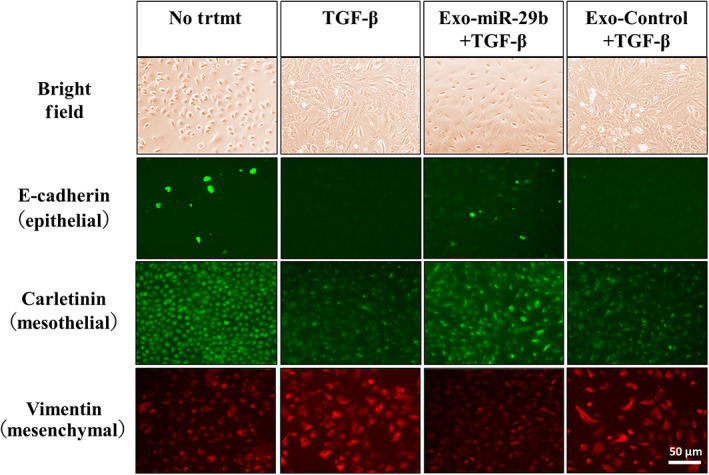
MicroRNA (miR)‐29b‐containing small extracellular vesicles (sEV) suppresses phenotypic changes of human omentum tissue‐derived mesothelial cells (HPMCs) stimulated with transforming growth factor‐β1 (TGF‐β1). HPMCs were cultured with 10 ng/mL TGF‐β1 for 48 h. In some wells, HPMCs were transfected with miR‐29b in sEV (Exo‐miR‐29b) or negative control miR (Exo‐Control) and cultured with TGF‐β1. HPMCs were immunostained with mAbs to E‐cadherin, calretinin, and vimentin and their expressions were observed with fluorescein microscopy. No trtmt, without stimulation with TGF‐β1. Magnification, 400×.
The proliferation of HPMCs was not significantly altered by TGF‐β. However, if pretreated with Exo‐miR‐29b, their proliferation was slightly decreased (n = 5, p < 0.05; Figure 3A). The chemotactic migration of HPMCs was significantly increased after stimulation with TGF‐β1 (48.3 ± 14.5 vs. 140.9 ± 16.1 counts/HPF, n = 6, p < 0.005). However, when pretreated with Exo‐miR‐29b, the number of migrated cells was greatly reduced compared with Exo‐miR‐NC (36.7 ± 5.4 vs. 124.9 ± 14.8 counts/HPF, n = 6, p < 0.005; Figure 3B,C).
FIGURE 3.
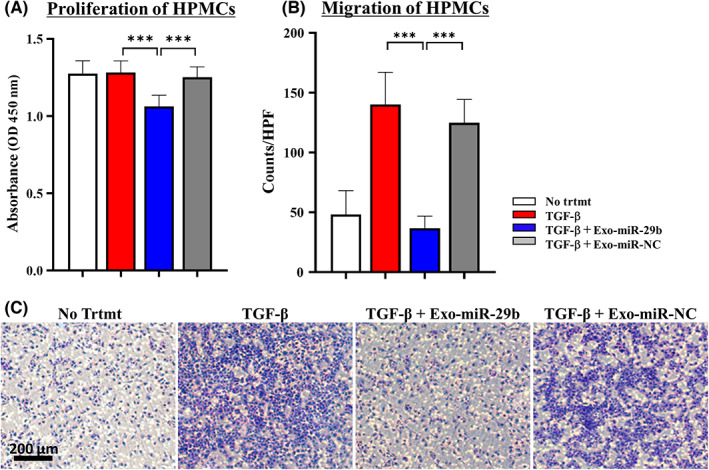
MicroRNA (miR)‐29b‐containing small extracellular vesicles (Exo‐miR‐29b) suppresses the proliferation and migration of human omentum tissue‐derived mesothelial cells (HPMCs) stimulated with transforming growth factor‐β1 (TGF‐β1). (a) Proliferation of HPMCs was determined by MTT assay. Data show mean ± SD in one of five different experiments. (b, c) Chemotactic migration of HPMCs to 10% FBS was evaluated with the number of cells that migrated to the lower surface of culture insets for 24 h. Data show mean ± SD in one of three different experiments. ***p < 0.005. HPF, high power field; NC, negative control; No trtmt, without stimulation with TGF‐β1; OD, optical density.
In adhesion experiments, a few NUGC‐4 and MKN‐45 cells attached to the HPMC monolayer without stimulation. However, after stimulation of HPMCs with TGF‐β1, the number of NUGC‐4 and MKN‐45 cells attached to the HPMCs markedly increased (NUGC‐4: 4.7 ± 1.6 vs. 31.1 ± 10.8 counts/HPF, n = 5, p < 0.001; MKN‐45: 16.3 ± 4.2 vs. 390.9 ± 30.4 counts/HPF, n = 5, p < 0.0001; Figure 4A,B). When HPMCs were treated with Exo‐miR‐29b, the enhancing effects of TGF‐β1 on the adhesion of NUGC‐4 and MKN‐45 cells were totally abrogated (NUGC‐4: 4.5 ± 1.1 vs. 47.6 ± 26.5 counts/HPF, n = 5, p < 0.0001; MKN‐45: 67.7 ± 8.9 vs. 290.2 ± 45.1 counts/HPF, n = 5, p < 0.0001; Figure 4A,B).
FIGURE 4.
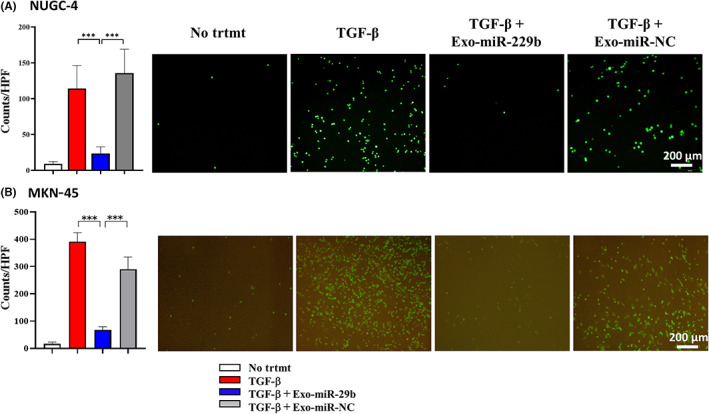
MicroRNA (miR)‐29b‐containing small extracellular vesicles (Exo‐miR‐29b) suppresses the attachment of gastric cancer cells to human omentum tissue‐derived mesothelial cells (HPMCs) stimulated with transforming growth factor‐β1 (TGF‐β1). HPMCs were cultured then fluorescein‐labeled NUGC‐4 (A) and MKN‐45 (B) cells were added and incubated for 15 min on the mesothelial cell monolayer. After gentle washing three times with warmed media, the number of NUGC‐4 cells remaining attached were counted under a fluorescence microscope. Data show mean ± SD in one of five different experiments ***p < 0.005. HPF, high power field; NC, negative control; No trtmt, without stimulation with TGF‐β1.
As FN was reported to be an important molecule mediating the adhesion between tumor cells and mesothelial cells, the effect on the expression of FN on HPMCs was next examined. Although FN was faintly detected on the surface of resting HPMCs, the expression was markedly enhanced after stimulation with TGF‐β1 (Figure 5). However, when Exo‐miR‐29b was present with TGF‐β1 stimulation, the expression of FN was not increased significantly (Figure 5).
FIGURE 5.
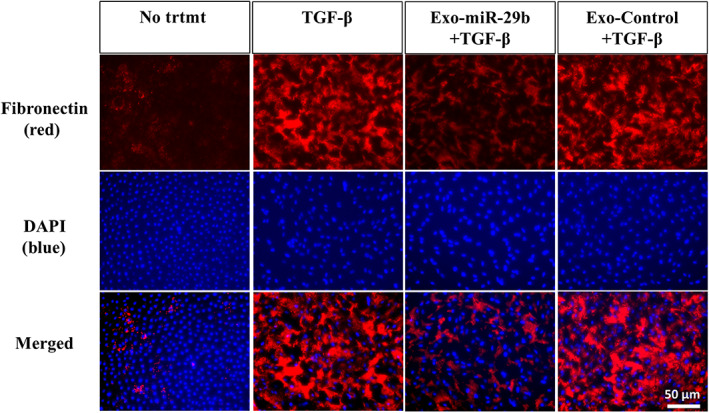
MicroRNA (miR)‐29b‐containing small extracellular vesicles (Exo‐miR‐29b) suppresses the expression of fibronectin 1 (FN) on human omentum tissue‐derived mesothelial cells (HPMCs) stimulated with transforming growth factor‐β1 (TGF‐β1). HPMCs were immunostained with anti‐ FN mAb (red) followed by Alexa Fluor 594‐conjugated secondary Ab or DAPI (blue) and observed with fluorescein microscopy under corresponding wavelengths. The two photographs were then superimposed. Magnification, 400×.
3.2. Isolation of miR‐29b‐containing sEV derived from murine BMSCs
After three passages of primary cultures of murine bone marrow, a large of number of spindle‐shaped cells was obtained (Figure 6A) Most of the cells were positive for CD44, CD73, and CD90 but negative for CD31, CD34, and CD49d (Figure 6B). As these phenotypes were consistent with BMSCs, we undertook transfection of miR‐29b‐ or miR‐NC‐integrated lentiviral vector, and isolated sEV extracted from their supernatants by ultracentrifugation. A scanning electron microscope and nanoparticle tracking analysis showed that the peak size of the vesicles was approximately 130 nm with positive expression of CD9 and CD63 (Figure 7A–C). The sEV derived from miR‐29b‐transfected BMSCs contained markedly higher levels of miR‐29b than in the NC (Figure 8A). As in the human system, PKH26‐stained sEV derived from mouse BMSCs were incorporated in mouse peritoneal mesothelial cells more efficiently than YTN16P (Figure 8B).
FIGURE 6.
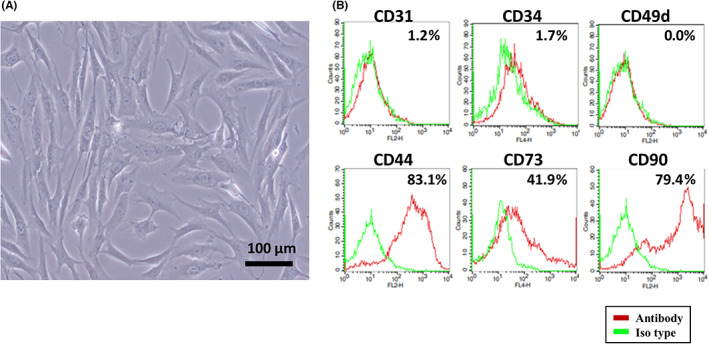
Culture of murine bone marrow derived mesenchymal stem cells (BMSCs). Primary BMSCs were isolated from the femurs of 6‐week‐old C57BL/6N mice and cultured using MesenCult Expansion Kit. (A) Morphological observation of mouse BMSCs. (B) Detection of mouse BMSC surface antigens by flow cytometry. BMSCs at passage 3 were stained with mAbs to CD31, CD34, CD44, CD49d, CD73, and CD90 and their expressions were examined using FACSCalibur.
FIGURE 7.
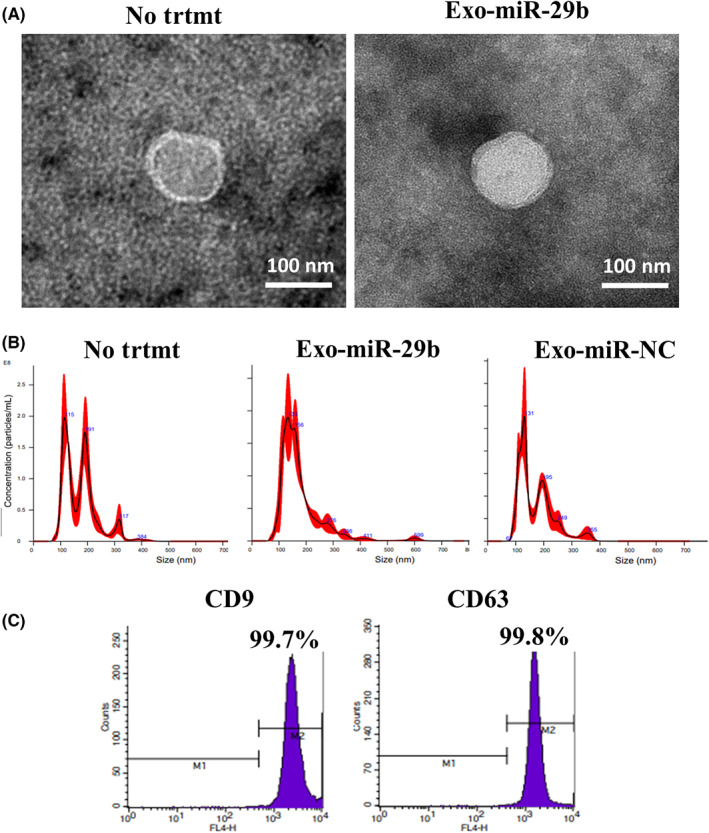
Identification of extracellular vesicles (EVs) derived from murine bone marrow‐derived mesenchymal stem cells (BMSCs). EVs were isolated from supernatant of mouse BMSCs or microRNA (miR)‐29b transfected BMSCs (Exo‐miR‐29b) and their size (A,B) and antigen expression (C) were examined. (A) Magnification, 40,000×. NC, negative control; No trtmt, without treatment.
FIGURE 8.
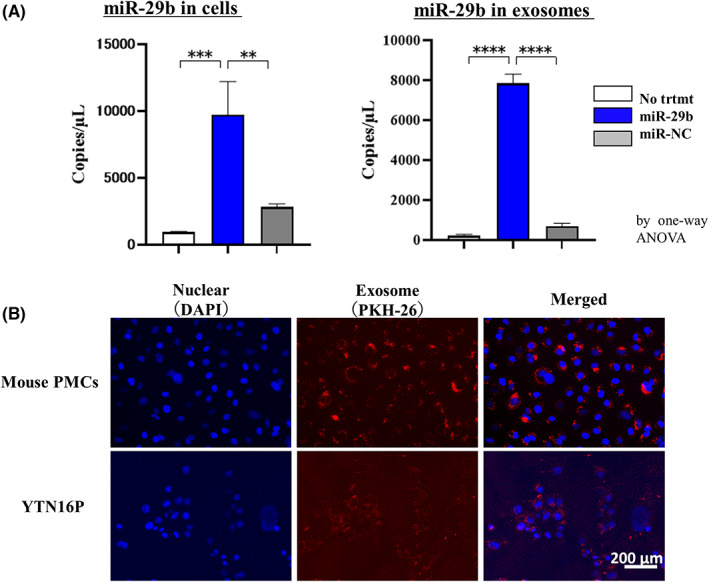
MicroRNA (miR)‐29b‐containing small extracellular vesicles (sEV) derived from murine bone marrow‐derived mesenchymal stem cells (BMSCs). (A) miR‐29b expression in mouse BMSCs and corresponding sEV was quantified with 3D‐digital PCR. **p < 0.01; ***p < 0.005; **** p < 0.001. Data show mean ± SD in one of three different experiments. (B) Mouse omentum‐derived mesothelial cells (mouse PMCs) and YTN16P were cultured with PKH26‐stained sEV derived from mouse BMSCs for 24 h and observed with fluorescein microscopy. Magnification, 400×.
3.3. Intraperitoneal injection of miR‐29b‐contaning murine sEV suppresses peritoneal metastasis in a syngeneic murine model
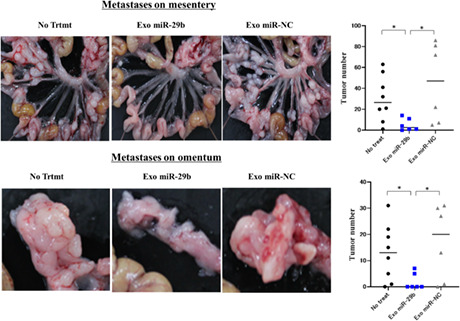
When YTN16P (1.0 × 105 cells) was IP injected into syngeneic C57BL/6N mice, many metastatic nodules developed on the omentum as well as mesentery at day 21. However, when Exo‐miR‐29b derived from mouse BMSCs was repeatedly injected at a concentration of 40 μg/500 μL every 3 days from day 0 to day 18, the number of metastatic nodules on the mesentery markedly decreased (30.3 ± 22.0 vs. 5.2 ± 5.9, n = 6, p < 0.05), which was not observed using Exo‐miR‐NC (45.5 ± 38.2, n = 6; Figure 9). Similarly, the number of omental metastases was significantly reduced with Exo‐miR‐29b, but not Exo‐miR‐NC (13.0 ± 10.9 vs. 2.0 ± 3.2, n = 6, 17.0 ± 14.3, n = 6, p < 0.05; Figure 9).
FIGURE 9.
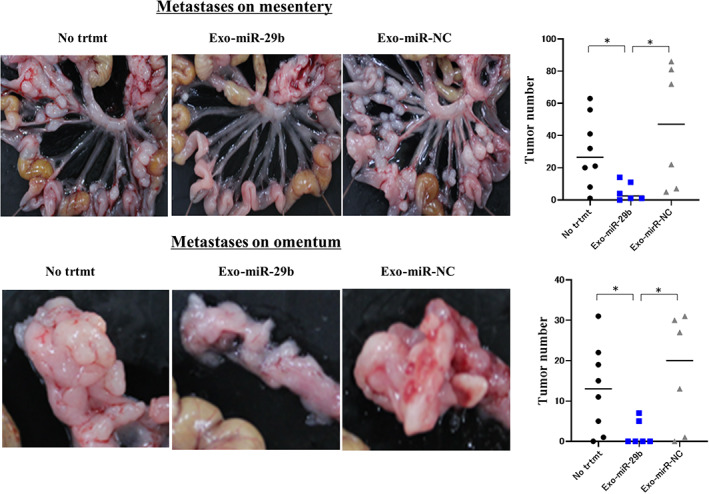
Intraperitoneal (IP) transfer of microRNA (miR)‐29b‐containing small extracellular vesicles (sEV) suppresses peritoneal metastasis in a mouse syngeneic model. C57BL6/N mice were IP injected with YTN16P (1.0 × 105 cells, 500 μL) and then IP injected with PBS (500 μL) or sEV (40 μg/500 μL) isolated from murine bone marrow‐derived mesenchymal stem cells transfected with lentivirus integrating miRNA‐29b (Exo‐miR‐29b) or negative control (Exo miR‐NC) every 3 days from day 0 to day 18. Mice were killed on day 21 and peritoneal metastases were evaluated based on the number of macroscopic nodules on the mesentery and the omental surface. Data show mean ± SD in one of the two different experiments. *p < 0.05. No trtmt, without treatment.
4. DISCUSSION
The peritoneum is composed of a single layer of flat peritoneal MCs; it functions as the first barrier against bacterial invasion and tumor attachment under physiological conditions. 36 , 37 In the process of PM formation, however, these MCs lose their intercellular junctions, increase their migratory capacity, and produce substantial amounts of ECM components as well as inflammatory and angiogenic factors. The phenotypic and functional change of MCs is referred to as MMT and considered to be a crucial component in the development of a premetastatic niche for disseminated tumor cells to form PM. 5 , 38 , 39 , 40 Recent studies have suggested that specific miRNAs contained in exosomes are critically involved in the induction of MMT and PM formation from gastric cancer. 17 , 18
In the present study, we focused on miR‐29b based on the results of previous translational studies from this laboratory 24 , 25 and examined whether local replacement of miR‐29b‐contaning sEV in the peritoneal cavity would suppress MMT and PM. The BMSC‐derived sEV were used as carriers of miR‐29b, as sEV are excellent for delivery of functional cargo to targeted cells. 41 , 42 MicroRNA‐29b was then introduced into mouse BMSCs as well as a human BMSC cell line, UE6E7T‐12, using a lentivirus system and succeeded in the isolation of miR‐29‐rich sEV from the culture supernatant. The sEV contained markedly high amounts of miR‐29b and were efficiently incorporated in human and mouse omentum‐derived peritoneal mesothelial cells, rendering them attractive as delivery vehicles for miRNA.
The miR‐29b‐rich sEV (Exo‐miR‐29b) strongly inhibit MMT of HPMCs and attachment of tumor cells, presumably partly through the downregulation of FN on HPMC. The characteristic changes of MCs are exactly the same as in the results of our previous study in which miR‐29b was introduced to HPMCs using lipofection. 29 However, in vivo results showed marked differences. In the previous study, atelocollagen‐mixed miR‐29b was IP injected in a murine model, which reduced the number of PM on the greater omentum but not on the mesentery or parietal peritoneum. Although the reason is unclear, it is speculated that miR‐29b transferred with atelocollagen was degraded before suppressing PM in the whole abdominal cavity, as miRNAs are generally degraded rapidly by RNA‐degrading enzymes that are abundant in the body. 43 In this study, however, IP transfer of Exo‐miR‐29b clearly suppressed the development of PM not only in the omentum but also on the mesentery. These results indicate that sEV are more suitable than atelocollagen as the carrier of miR‐29b for delivery to the peritoneal space.
In recent years, sEV have attracted a great deal of attention not only for diagnostic but also for therapeutic purposes. 44 , 45 For example, exosome‐mediated delivery of inhibitors for specific miRNAs could possibly antagonize the effect on growth, migration, and chemoresistance of gastric cancer cells to retrieve drugs. 46 , 47 In particular, MSCs are known to be ideal candidates as producers of exosomes for drug delivery. 30 There is an increasing body of clinical evidence demonstrating safe transplantation of MSCs, which suggests that transplanting MSC‐derived sEV would be unlikely to lead to serious adverse effects. 48 Mesenchymal stromal cells have the ability to exert immunosuppressive effects that would enhance the longevity of allogeneic MSC‐derived sEV as drug delivery vehicles and bioavailability of their drug cargo. 49 , 50 Recently, Naseri et al have reported that administration of miR‐142‐3p encapsulated in BMSC‐derived sEV suppresses tumor formation in breast cancer. 51
The results of the present study clearly suggest that IP administration of miR‐29b when encapsulated in sEV potently inhibits the development of PM partly though the inhibition of MMT induced by TGF‐β1. In particular, miR‐29b reduced the expression of FN1 on mesothelial cells and inhibited the tumor cell attachment to mesothelial cells, which is the initial and critical step in the process of PM. This is consistent with the results of previous studies showing that blocking the TGF‐β signal inhibits MMT, leading to the inhibition of PM from gastric cancer. 52 , 53
The precise molecular mechanisms of miR‐29b to suppress the process of PM remain unclear, which is the limitation of this study. However, numerous studies have already shown that miR‐29b suppresses the proliferation and migration of tumor cells through the inhibition of CKD, CDC42, PI3K/AKT signal, and the p53‐mediated apoptotic pathway. 28 , 54 MicroRNA‐29b is also shown to suppress EMT though the direct inhibition of a number of EMT‐related genes such as β‐catenin (CTNNB1), integrin β1 (ITGB1), lysyl oxidase like 2 (LOXL2), and mucin‐1 (MUC1) as well as many ECM proteins (e.g., COL1A1, COL3A1, and LAMA2). 28 , 54 We confirmed that Exo‐miR‐29b also suppressed the migration and tended to reduce the proliferation of human (NUGC‐4) and murine (YTN16P) gastric cancer cells, possibly though the inhibition of these molecules (Figure S2). Given that these EMT‐related genes are largely shared with mesothelial cells, these molecular mechanisms are considered to be involved in MMT. In addition, it is well known that miR‐29 can suppress fibrosis, angiogenesis, and immune responses. 28 , 55 Taken together, miR‐29b is supposed to suppress the PM through the effects both on tumor and host cells through multiple molecular mechanisms.
The results of this study strongly suggest that miR‐29b might be clinically useful to prevent PM in patients with advanced gastric cancer. The results also suggest that sEV are a hopeful tool for drug delivery of miRNA drugs. There are still few clinical application examples of nanomedicine therapeutic drugs using sEV, especially targeting cancer. 56 However, modification of the surface structure of sEV could further enhance the targeting efficiency to specific cell types. 57 , 58 Therefore, induction of MC‐specific molecules on sEV might further increase the inhibition on PM. Further studies are warranted to develop stable production and preservation methods to maintain the biological quality of sEV.
AUTHOR CONTRIBUTIONS
Y.Kim, H.O., H.M., and J.K. conceived and designed the experiments and performed the experiments. Y.Kim, Y.Kan, T.K., H.O., and R.T. performed experiments. K.K., S.S., H.Y., and Y.S. provided the omental samples. Y.Kim, A.K.L, N.S, and J.K. wrote the main manuscript.
FUNDING INFORMATION
This work was supported by the Japan Society for the Promotion of Science (20K07704) and subsidized by JKA through its promotion funds from KEIRIN RACE.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Review Board: The protocol was approved by the Institutional Review Boards of Jichi Medical University (approval number: RIN‐A‐21‐048).
Informed consent: Written informed consent was obtained from all patients who provided omental samples for this study.
Registry and the registration no. of the study/trial: N/A.
Animal study: The protocol of the animal study was approved by Institutional Review Board of the Animal Care Committee of Jichi Medical University (No. 20043‐01).
Supporting information
Figure S1.
Figure S2.
ACKNOWLEDGMENTS
We thank N. Nishiaki, J. Shinohara, H. Hatakeyama, and I. Nieda for technical and clerical work.
Kimura Y, Ohzawa H, Miyato H, et al. Intraperitoneal transfer of microRNA‐29b‐containing small extracellular vesicles can suppress peritoneal metastases of gastric cancer. Cancer Sci. 2023;114:2939‐2950. doi: 10.1111/cas.15793
REFERENCES
- 1. Soucisse ML, Liauw W, Hicks G, Morris DL. Early postoperative intraperitoneal chemotherapy for lower gastrointestinal neoplasms with peritoneal metastasis: a systematic review and critical analysis. Pleura Peritoneum. 2019;4:20190007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thomassen I, van Gestel YR, van Ramshorst B, et al. Peritoneal carcinomatosis of gastric origin: a population‐based study on incidence, survival and risk factors. Int J Cancer. 2014;134:622‐628. [DOI] [PubMed] [Google Scholar]
- 3. Yonemura Y, Ishibashi H, Mizumoto A, et al. The development of peritoneal metastasis from gastric cancer and rationale of treatment according to the mechanism. J Clin Med. 2022;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sodek KL, Murphy KJ, Brown TJ, Ringuette MJ. Cell‐cell and cell‐matrix dynamics in intraperitoneal cancer metastasis. Cancer Metastasis Rev. 2012;31:397‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun F, Feng M, Guan W. Mechanisms of peritoneal dissemination in gastric cancer. Oncol Lett. 2017;14:6991‐6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ruivo CF, Adem B, Silva M, Melo SA. The biology of cancer exosomes: insights and new perspectives. Cancer Res. 2017;77:6480‐6488. [DOI] [PubMed] [Google Scholar]
- 7. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213‐228. [DOI] [PubMed] [Google Scholar]
- 8. Pegtel DM, Gould SJ. Exosomes. Annu Rev Biochem. 2019;88:487‐514. [DOI] [PubMed] [Google Scholar]
- 9. Hoshino A, Costa‐Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell‐to‐cell mediators of metastasis. Cancer Cell. 2016;30:836‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matsuzaki J, Ochiya T. Circulating microRNAs and extracellular vesicles as potential cancer biomarkers: a systematic review. Int J Clin Oncol. 2017;22:413‐420. [DOI] [PubMed] [Google Scholar]
- 13. Nedaeinia R, Manian M, Jazayeri MH, et al. Circulating exosomes and exosomal microRNAs as biomarkers in gastrointestinal cancer. Cancer Gene Ther. 2017;24:48‐56. [DOI] [PubMed] [Google Scholar]
- 14. Sindhu KJ, Venkatesan N, Karunagaran D. MicroRNA interactome multiomics characterization for cancer research and personalized medicine: an expert review. Omics. 2021;25:545‐566. [DOI] [PubMed] [Google Scholar]
- 15. Arita T, Ichikawa D, Konishi H, et al. Tumor exosome‐mediated promotion of adhesion to mesothelial cells in gastric cancer cells. Oncotarget. 2016;7:56855‐56863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deng G, Qu J, Zhang Y, et al. Gastric cancer‐derived exosomes promote peritoneal metastasis by destroying the mesothelial barrier. FEBS Lett. 2017;591:2167‐2179. [DOI] [PubMed] [Google Scholar]
- 17. Li Q, Li B, Li Q, et al. Exosomal miR‐21‐5p derived from gastric cancer promotes peritoneal metastasis via mesothelial‐to‐mesenchymal transition. Cell Death Dis. 2018;9:854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu M, Zhang N, He S, Lu X. Exosomal miR‐106a derived from gastric cancer promotes peritoneal metastasis via direct regulation of Smad7. Cell Cycle. 2020;19:1200‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakamura K, Sawada K, Kinose Y, et al. Exosomes promote ovarian cancer cell invasion through transfer of CD44 to peritoneal mesothelial cells. Mol Cancer Res. 2017;15:78‐92. [DOI] [PubMed] [Google Scholar]
- 20. Wei M, Yang T, Chen X, et al. Malignant ascites‐derived exosomes promote proliferation and induce carcinoma‐associated fibroblasts transition in peritoneal mesothelial cells. Oncotarget. 2017;8:42262‐42271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nambara S, Masuda T, Hirose K, et al. Rab27b, a regulator of exosome secretion, is associated with peritoneal metastases in gastric cancer. Cancer Genomics Proteomics. 2023;20:30‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li K, Chen Y, Li A, Tan C, Liu X. Exosomes play roles in sequential processes of tumor metastasis. Int J Cancer. 2019;144:1486‐1495. [DOI] [PubMed] [Google Scholar]
- 23. Chen X, Wang H, Huang Y, et al. Comprehensive roles and future perspectives of exosomes in peritoneal metastasis of gastric cancer. Front Oncol. 2021;11:684871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ohzawa H, Kumagai Y, Yamaguchi H, et al. Exosomal microRNA in peritoneal fluid as a biomarker of peritoneal metastases from gastric cancer. Ann Gastroenterol Surg. 2020;4:84‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ohzawa H, Saito A, Kumagai Y, et al. Reduced expression of exosomal miR29s in peritoneal fluid is a useful predictor of peritoneal recurrence after curative resection of gastric cancer with serosal involvement. Oncol Rep. 2020;43:1081‐1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chou J, Lin JH, Brenot A, Kim JW, Provot S, Werb Z. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA‐29b expression. Nat Cell Biol. 2013;15:201‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alizadeh M, Safarzadeh A, Beyranvand F, et al. The potential role of miR‐29 in health and cancer diagnosis, prognosis, and therapy. J Cell Physiol. 2019;234:19280‐19297. [DOI] [PubMed] [Google Scholar]
- 28. Kwon JJ, Factora TD, Dey S, Kota J. A systematic review of miR‐29 in cancer. Mol Ther Oncolytics. 2019;12:173‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kimura Y, Ohzawa H, Miyato H, et al. MiR‐29b may suppresses peritoneal metastases through inhibition of the mesothelial‐mesenchymal transition (MMT) of human peritoneal mesothelial cells. Sci Rep. 2022;12:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yeo RW, Lai RC, Zhang B, et al. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv Drug Deliv Rev. 2013;65:336‐341. [DOI] [PubMed] [Google Scholar]
- 31. Ahmadi M, Mahmoodi M, Shoaran M, Nazari‐Khanamiri F, Rezaie J. Harnessing normal and engineered mesenchymal stem cells derived exosomes for cancer therapy: opportunity and challenges. Int J Mol Sci. 2022;23:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Riera M, McCulloch P, Pazmany L, Jagoe T. Optimal method for isolation of human peritoneal mesothelial cells from clinical samples of omentum. J Tissue Viability. 2006;16:22‐24. [DOI] [PubMed] [Google Scholar]
- 33. Voorhoeve PM, le Sage C, Schrier M, et al. A genetic screen implicates miRNA‐372 and miRNA‐373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169‐1181. [DOI] [PubMed] [Google Scholar]
- 34. Puzar Dominkus P, Stenovec M, Sitar S, et al. PKH26 labeling of extracellular vesicles: characterization and cellular internalization of contaminating PKH26 nanoparticles. Biochim Biophys Acta Biomembr. 2018;1860:1350‐1361. [DOI] [PubMed] [Google Scholar]
- 35. Kumagai Y, Futoh Y, Miyato H, et al. Effect of systemic or intraperitoneal Administration of Anti‐PD‐1 antibody for peritoneal metastases from gastric cancer. In Vivo. 2022;36:1126‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kiyasu Y, Kaneshima S, Koga S. Morphogenesis of peritoneal metastasis in human gastric cancer. Cancer Res. 1981;41:1236‐1239. [PubMed] [Google Scholar]
- 37. Mutsaers SE, Wilkosz S. Structure and function of mesothelial cells. Cancer Treat Res. 2007;134:1‐19. [DOI] [PubMed] [Google Scholar]
- 38. Sandoval P, Jimenez‐Heffernan JA, Guerra‐Azcona G, et al. Mesothelial‐to‐mesenchymal transition in the pathogenesis of post‐surgical peritoneal adhesions. J Pathol. 2016;239:48‐59. [DOI] [PubMed] [Google Scholar]
- 39. Gordillo CH, Sandoval P, Munoz‐Hernandez P, Pascual‐Anton L, Lopez‐Cabrera M, Jimenez‐Heffernan JA. Mesothelial‐to‐mesenchymal transition contributes to the generation of carcinoma‐associated fibroblasts in locally advanced primary colorectal carcinomas. Cancers (Basel). 2020;12:499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saito H, Fushida S, Harada S, et al. Importance of human peritoneal mesothelial cells in the progression, fibrosis, and control of gastric cancer: inhibition of growth and fibrosis by tranilast. Gastric Cancer. 2018;21:55‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome‐mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654‐659. [DOI] [PubMed] [Google Scholar]
- 42. Simpson RJ, Jensen SS, Lim JW. Proteomic profiling of exosomes: current perspectives. Proteomics. 2008;8:4083‐4099. [DOI] [PubMed] [Google Scholar]
- 43. de Abreu RC, Ramos CV, Becher C, et al. Exogenous loading of miRNAs into small extracellular vesicles. J Extracell Vesicles. 2021;10:e12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rajput A, Varshney A, Bajaj R, Pokharkar V. Exosomes as new generation vehicles for drug delivery: biomedical applications and future perspectives. Molecules. 2022;27:7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Allegra A, Petrarca C, Di Gioacchino M, Casciaro M, Musolino C, Gangemi S. Exosome‐mediated therapeutic strategies for Management of Solid and Hematological Malignancies. Cell. 2022;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang X, Zhang H, Bai M, et al. Exosomes serve as nanoparticles to deliver anti‐miR‐214 to reverse chemoresistance to cisplatin in gastric cancer. Mol Ther. 2018;26:774‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ji R, Zhang X, Gu H, et al. miR‐374a‐5p: a new target for diagnosis and drug resistance therapy in gastric cancer. Mol Ther Nucleic Acids. 2019;18:320‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Peterson MF, Otoc N, Sethi JK, Gupta A, Antes TJ. Integrated systems for exosome investigation. Methods. 2015;87:31‐45. [DOI] [PubMed] [Google Scholar]
- 49. Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC. Suppression of allogeneic T‐cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation. 2003;75:389‐397. [DOI] [PubMed] [Google Scholar]
- 50. Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815‐1822. [DOI] [PubMed] [Google Scholar]
- 51. Naseri Z, Oskuee RK, Forouzandeh‐Moghadam M, Jaafari MR. Delivery of LNA‐antimiR‐142‐3p by mesenchymal stem cells‐derived exosomes to breast cancer stem cells reduces tumorigenicity. Stem Cell Rev Rep. 2020;16:541‐556. [DOI] [PubMed] [Google Scholar]
- 52. Miao ZF, Zhao TT, Wang ZN, et al. Transforming growth factor‐beta1 signaling blockade attenuates gastric cancer cell‐induced peritoneal mesothelial cell fibrosis and alleviates peritoneal dissemination both in vitro and in vivo. Tumour Biol. 2014;35:3575‐3583. [DOI] [PubMed] [Google Scholar]
- 53. Lv ZD, Zhao WJ, Jin LY, et al. Blocking TGF‐beta1 by P17 peptides attenuates gastric cancer cell induced peritoneal fibrosis and prevents peritoneal dissemination in vitro and in vivo. Biomed Pharmacother. 2017;88:27‐33. [DOI] [PubMed] [Google Scholar]
- 54. Jiang H, Zhang G, Wu JH, Jiang CP. Diverse roles of miR‐29 in cancer (review). Oncol Rep. 2014;31:1509‐1516. [DOI] [PubMed] [Google Scholar]
- 55. Horita M, Farquharson C, Stephen LA. The role of miR‐29 family in disease. J Cell Biochem. 2021;122:696‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Weng Z, Zhang B, Wu C, et al. Therapeutic roles of mesenchymal stem cell‐derived extracellular vesicles in cancer. J Hematol Oncol. 2021;14:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ohno S, Takanashi M, Sudo K, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21:185‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Alvarez‐Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341‐345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.