

Abstract
DNA replication stress (RS) causes genomic instability and vulnerability in cancer cells. To counteract RS, cells have evolved various mechanisms involving the ATR kinase signaling pathway, which regulates origin firing, cell cycle checkpoints, and fork stabilization to secure the fidelity of replication. However, ATR signaling also alleviates RS to support cell survival by driving RS tolerance, thereby contributing to therapeutic resistance. Cancer cells harboring genetic mutations and other changes that disrupt normal DNA replication increase the risk of DNA damage and the levels of RS, conferring addiction to ATR activity for sustainable replication and susceptibility to therapeutic approaches using ATR inhibitors (ATRis). Therefore, clinical trials are currently being conducted to evaluate the efficacy of ATRis as monotherapies or in combination with other drugs and biomarkers. In this review, we discuss recent advances in the elucidation of the mechanisms by which ATR functions in the RS response and its therapeutic relevance when utilizing ATRis.
Keywords: ATR inhibitor, ATR kinase, biomarker, cancer therapy, DNA replication stress, replication stress tolerance
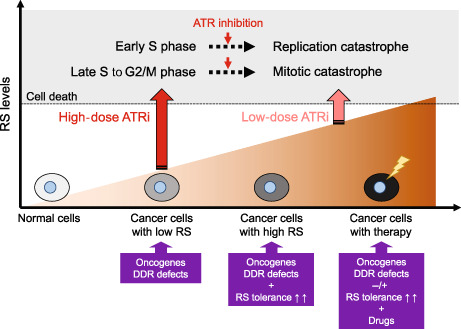
Potential models of action of ataxia telangiectasia and Rad3‐related inhibitors (ATRis) depending on DNA replication stress (RS) levels. Cancer cells often harbor increased RS because of oncogene activation and/or DNA damage response defects and become RS tolerant by acquiring ATR‐mediated RS tolerance mechanisms, thereby conferring susceptibility to therapeutic approaches using ATRis. RS at levels over the threshold, induced by ATRis as monotherapies or in combination with other drugs, can lead to cell death through replication catastrophe and mitotic catastrophe.
Abbreviations
- APOBEC3
apolipoprotein B mRNA editing enzyme catalytic subunit 3
- ARID1A
AT‐rich interactive domain‐containing protein 1A
- ATM
ataxia telangiectasia mutated
- ATRIP
ATR‐interacting protein
- BRCA
breast cancer susceptibility
- CDC25
cell division cycle 25
- CDC45
cell division cycle 45
- CDK
cyclin‐dependent kinase
- CENP
centromere protein
- cGAS
cyclic GMP‐AMP synthase
- Chk1
checkpoint kinase 1
- CI
confidence interval
- CRISPR
clustered regularly interspaced short palindromic repeat
- DDK
DBF4‐dependent kinase
- DSB
double strand break
- ERCC1
excision repair cross‐complementation group 1
- ETAA1
Ewing's tumor‐associated antigen 1
- EXO1
exonuclease 1
- FOXM1
Forkhead box M1
- HGSOC
high‐grade serous ovarian cancer
- LADC
lung adenocarcinoma
- MLL
mixed lineage leukemia
- MRE11
meiotic recombination 11
- PALB2
partner and localizer of BRCA2
- PARP
poly(ADP‐ribose) polymerase
- PCAF
p300/CBP‐associated factor
- PD‐1
programmed death‐1
- PD‐L1
programmed death‐ligand 1
- PFS
progression‐free survival
- PLK
polo‐like kinase
- Polη
DNA polymerase eta
- PrimPol
primase and DNA directed polymerase
- RNF4
ring finger protein 4
- RPA
replication protein A
- SCLC
small cell lung cancer
- SMARCA4
SWI/SNF‐related, matrix‐associated, actin‐dependent regulator of chromatin, subfamily A, member 4
- SMARCAL1
SWI/SNF‐related, matrix‐associated, actin‐dependent regulator of chromatin, subfamily A like 1
- SPOP
speckle‐type POZ protein
- STING
stimulator of interferon genes
- SWI/SNF
switch/sucrose non‐fermentable
- TopBP1
DNA topoisomerase II binding protein 1
- UPF2
up‐frameshift suppressor 2
- XRCC1
X‐ray repair cross complementing protein 1
- XRCC3
X‐ray repair cross complementing protein 3
1. INTRODUCTION
DNA replication is one of the fundamental cellular processes that duplicate genomic DNA during each cell cycle. Although faithful DNA replication is necessary for the preservation of genome integrity, the replication fork is constantly challenged by a wide variety of factors, resulting in altered progression of replication forks, reduced replication fidelity, and DNA breaks. These phenomena during DNA replication are collectively referred to as DNA replication stress (RS), which is a major cause of genome instability. 1 RS is characterized by many different causes, and the definition of RS is constantly evolving. Exogenous factors include DNA lesions that are caused by ultraviolet light, ionizing radiation, and chemical agents such as alkylating agents and cross‐linking agents, while endogenous stress includes reactive oxygen species, metabolic aldehydes, misincorporated ribonucleotide, and abnormal DNA secondary structures. These lesions are generally, but not exclusively, physical obstacles to replication fork progression. 2 Accumulating evidence has revealed that the activation of oncogenes, such as Ras, Myc, and Cyclin E, induces RS by aberrant replication initiation, RNA:DNA hybrids (R‐loops), replication–transcription collisions, and defective nucleotide metabolism, 3 and defects in DNA damage response (DDR) systems lead to DNA synthesis slowdown and/or replication fork stalling because of irreparable DNA lesions. Subsequently, RS caused by these genetic alterations contributes to the further acquisition of genetic mutations and chromosomal aberrations, which promote tumor development. 4 Therefore, RS is considered one of the hallmarks of cancer.
To safeguard the RS, cells have evolved the DDR network. Ataxia telangiectasia and Rad3‐related (ATR) kinase acts as a master regulator of the RS response. RS usually results in stretching ssDNA, which serves as a platform to recruit ATR kinase. Activated ATR stabilizes and restarts the stressed replication fork, suppresses origin firing, activates the cell cycle checkpoint, and facilitates DNA repair. Therefore, ATR has long been considered a tumor suppressor to maintain genome stability in normal cells. However, the mutation in the ATR gene is not common in cancer cells, probably because of its essential role in DNA replication. Importantly, recent studies have shown that ATR signaling contributes to RS tolerance and protects cells from deleterious and chronic RS induced by oncogenes during tumor development. Thus, cancer cells are more highly dependent on ATR signaling compared with normal cells, highlighting the potential of targeting ATR in cancer therapy. When ATR is inhibited, large amounts of ssDNA arise in the genome, resulting in massive fork collapse and cell death, which is termed replication catastrophe. In the absence of a functional checkpoint by ATR inhibition, cells prematurely enter mitosis with increased DNA damage, triggering a mitotic catastrophe. Here, we discuss recent advances in the elucidation of ATR signaling, preclinical data on ATR inhibitors (ATRis) that have led to their entry into clinical trials, and potential biomarkers for predicting ATRi efficacy.
2. ATR SIGNALING PATHWAY IN RESPONSE TO RS
2.1. Mechanisms of ATR activation
A broad spectrum of genomic insults that activate ATR, including replication interference, DSBs, and other types of DNA lesions, commonly expose ssDNA, which is immediately bound and protected by RPA. 5 The RPA‐coated ssDNA is directly recognized by ATRIP, a regulatory partner protein of ATR, which recruits the ATR–ATRIP complex to ssDNA. 6 The recruitment of multiple ATR–ATRIP complexes to RPA–ssDNA promotes the autophosphorylation of ATR at T1989, one of the first markers of ATR activation. 7 In addition, two ATR activator proteins, TopBP1 and ETAA1, are also recruited to ssDNA and stimulate ATR kinase activity by directly interacting with the ATR‐activating domain. TopBP1 is recruited at ssDNA–dsDNA junctions where the Rad17 complex and Rad9–RAD1–HUS1 (9–1–1) complexes form the scaffold for TopBP1 recruitment. 8 , 9 Unlike TopBP1, ETAA1 directly accumulates at the RPA–ssDNA via its two RPA‐binding motifs. 10 , 11 Alternatively, a fraction of ATR that is recruited to the RPA–ssDNA distal to ssDNA–dsDNA junctions is activated through Nbs1 in a TopBP1‐dependent manner at replication‐associated DSBs. 12 The detailed mechanisms of ATR activation have recently been reviewed elsewhere. 13 , 14 These multiple modes of ATR activation can allow ATR to phosphorylate different substrates, thereby carrying out diverse functions in the DDR, as discussed below (Figure 1).
FIGURE 1.
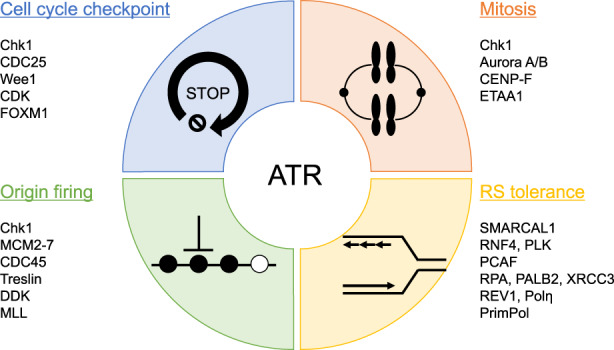
Ataxia telangiectasia and Rad3‐related (ATR) signaling network in the DNA damage response (DDR). ATR kinase plays a critical role in regulating DDR, particularly during DNA replication. ATR activates the cell cycle checkpoint pathway that halts the cell cycle in response to DNA damage or replication stress (RS). ATR regulates DNA replication by inhibiting the replicative helicase, preventing further origin firing. When a replication fork stalls, ATR phosphorylates some proteins that are involved in stabilizing the fork, preventing its collapse, promoting repair, and restarting forks to promote RS tolerance mechanisms. ATR also functions in mitosis to ensure chromosome segregation. These functions are controlled by the indicated representative factors directly or indirectly regulated by ATR or Chk1. By coordinating these processes, ATR helps to maintain genomic integrity.
2.2. Cell cycle checkpoint
One crucial function of ATR signaling is to regulate the G2/M cell cycle checkpoint following DNA damage. In response to DNA damage, Chk1 is phosphorylated by ATR at multiple sites, stimulating the kinase activity of Chk1. Activated Chk1 phosphorylates and inactivates CDC25 phosphatases (known as CDC25A, CDC25B, and CDC25C), which positively regulate CDK activity by removing its inhibitory phosphorylation. 15 Chk1 also phosphorylates and activates Wee1 kinase, which negatively regulates CDK1 activity by adding inhibitory phosphorylation. 16 Interestingly, a recent study showed that, until the S phase ends, ETAA1‐mediated ATR activation restricts CDK1‐dependent FOXM1 phosphorylation and prevents mitotic gene expression by enforcing the S/G2 checkpoint. 17 These signaling pathways delay cell cycle progression for recovery from DNA damage and stalled replication forks.
2.3. Origin firing
Inhibition of CDK activity by ATR–Chk1 signaling not only regulates the cell cycle checkpoint but also limits origin firing. The origin recognition complex initially loads MCM2–7 complexes as inactive double hexamers onto DNA. 18 Helicase activation requires CDC45 binding, which occurs following the CDK‐dependent phosphorylation of Treslin and DDK‐mediated phosphorylation of the MCM2–7 complex. 19 Following DNA damage, ATR–Chk1 signaling downregulates the kinase activities of CDK and DDK, and thereby prevents CDC45 loading and helicase activation to limit origin firing. 14 In another mechanism, ATR phosphorylates and stabilizes MLL on chromatin, where it methylates histone H3 lysine 4 at the late replication origin and inhibits CDC45 loading. 20 Based on these mechanisms, ATR inhibition triggers unscheduled origin firing, generates excessive ssDNA that exhausts the nuclear pool of RPA, and increases fork breakage, resulting in replication catastrophe. 21
2.4. RS tolerance
Acute and severe RS often give up continuous DNA synthesis, resulting in senescence and cell death. However, cells have evolved RS tolerance (also known as DNA damage tolerance) mechanisms aimed at allowing sustainable DNA replication to overcome lesions, which support cell survival. RS tolerance mechanisms mainly include fork reversal/template switching, translesion DNA synthesis (TLS), and repriming. These pathways are regulated by fine‐tuned mechanisms based on the genetic background and the extent of DNA damage (for more detail, see these reviews 22 , 23 ). ATR also plays a key role in the regulation of RS tolerance and contributes to fork stability.
Fork reversal can be conceptually divided into the following two steps: first, the formation of four‐way junction structures by coordinately annealing the two newly synthesized daughter strands, and second, fork restart through the removal of DNA lesions or template switching. ATR phosphorylates the DNA translocase SMARCAL1, thereby limiting its fork regression activity and preventing aberrant fork processing through SLX4‐associated nucleases. 24 In ATR‐deficient cells, suppression of RNF4 and/or PLK reduces SLX4‐mediated DSB formation. 25 PCAF‐mediated histone H4 acetylation at stalled forks promotes fork degradation by MRE11 and EXO1 nucleases in BRCA‐deficient cells. 26 ATR phosphorylates PCAF to limit its association and excessive fork degradation. These findings suggest that ATR functions in replication fork protection to prevent aberrant remodeling of stalled forks, thereby avoiding excessive nucleolytic processing of the replicating genome.
Another aspect of RS tolerance regulated by ATR involves the restart of replicated forks. ATR phosphorylates two of the TLS polymerases, REV1 and Pol η, suggesting that ATR may facilitate lesion bypass through continuous DNA synthesis without repairing the lesion. 27 , 28 , 29 ATR also phosphorylates several proteins that promote RAD51‐dependent replication restart pathways including template switching, fork reversal and repair, and homologous recombination. At the stressed fork, ATR phosphorylates RPA facilitating recruitment of PALB2 and BRCA2. 30 When DSBs occur, ATR also phosphorylates PALB2 enhancing interaction with BRCA1, promoting RAD51 filament formation. 31 In addition, ATR‐dependent XRCC3 phosphorylation is required for chromatin loading of RAD51 and homologous recombination (HR)‐mediated recovery of collapsed replication forks. 32 These findings suggest that ATR contributes to fork remodeling and restart by manipulating HR factors. Other processes that promote fork restart involve PrimPol‐mediated repriming, which reinitiates DNA synthesis beyond a DNA lesion, leaving unreplicated ssDNA gaps to be filled post‐replicatively through either TLS or template switching. A recent report showed that the expression level and repriming activity of PrimPol are controlled in an ATR‐dependent manner. 33 In addition, chemically induced RS induces PrimPol phosphorylation dependent on ATR and Chk1, indicating that ATR–Chk1 signaling‐dependent phosphorylation of PrimPol is a critical switch to turn on its repriming activity. 34 Importantly, PrimPol phosphorylation reduces undamaged cell fitness but recovers damaged cell fitness, suggesting that PrimPol needs to be tightly regulated during DNA replication to prevent aberrant repriming, fork speeding, and chromosomal breakage, which can increase the risk of genomic instability. Collectively, ATR signaling not only protects the stalled fork from nuclease‐mediated degradation but also seems to promote PrimPol‐dependent repriming to overcome RS.
2.5. Mitosis
While ATR inhibition during DNA replication increases genomic instability in the S phase, cells upon uncontrolled replication prematurely enter mitosis and overlap DNA synthesis with chromosome condensation, resulting in mitosis defects. 35 The mitosis defects in ATR‐absent cells are partially rescued by CDK1 inhibition, suggesting that ATR activates the cell cycle checkpoint to minimize the level of unreplicated DNA prior to mitotic onset. Surprisingly, a recent study revealed that ATR localizes to centromeres through Aurora A‐regulated association with CENP‐F, allowing ATR to engage RPA‐coated centromeric R‐loops, and activated ATR at centromeres stimulates Aurora B through Chk1, preventing chromosome instability. 36 In addition, TopBP1‐ and ETAA1‐dependent phosphoproteomics revealed TopBP1 to be a primary ATR activator for RS, while ETAA1 regulates mitotic ATR signaling. 37 Although these findings clearly indicate a mitosis‐specific ATR role, the functions of ATR in mitosis remain largely unknown. Therefore, further understanding the action of ATR inhibition in mitosis will provide a rationale for ATRi therapy.
3. CANCER THERAPEUTICS WITH ATRis
In 2011, the first potent and selective ATRi, VE‐821, was discovered by Vertex Pharmaceuticals. VE‐821 induces cancer cell killing and reversibly inhibits cell cycle progression in normal cells, suggesting that ATR is a promising target for cancer therapy. 38 Subsequently, bioavailable berzosertib (M6620, VE‐822, VX‐970, recently licensed to Merck KGaA), an improved analog of VE‐821, entered clinical trials. In 2013, AstraZeneca also developed a preclinical ATRi, AZ20, and its improved analog, ceralasertib (AZD6738), which can be orally administered. Other orally administered ATRis, gartisertib (M4344, VX‐803) and tuvusertib (M1774) by Merck, elimusertib (BAY1895344) by Bayer, camonsertib (RP‐3500, recently licensed to Roche) by Repare, ART0380 by Artios, IMP9064 by Impact, ATG‐018 by Antengene, and ATRN‐119 by Aprea were discovered. These ATRis are currently being tested preclinically and have progressed to phase I/II clinical trials as monotherapies and in combination with DNA‐damaging agents or molecular targeted drugs (Table 1).
TABLE 1.
Selected ongoing and completed clinical trials with ATRis.
| ATR inhibitor | Interventions | Cancers | Phase | Study start | Status | NCT number | Result reported |
|---|---|---|---|---|---|---|---|
| Monotherapy | |||||||
| Berzosertib | Alone | Advanced solid tumor, leiomyosarcoma, osteosarcoma | II | 2019 | Completed | NCT03718091 | |
| Ceralasertib | Alone | Relapsed/refractory CLL, PLL or B‐cell lymphoma | I | 2013 | Completed | NCT01955668 | |
| Alone | Head and neck squamous cell carcinoma | I | 2017 | Completed | NCT03022409 | ||
| Alone | Metastatic TNBC | II | 2018 | Recruiting | NCT03801369 | ||
| Alone | Neoadjuvant chemotherapy‐resistant residual TNBC | II | 2019 | Recruiting | NCT03740893 | ||
| Alone | Progressive MDS or CMML | I | 2019 | Recruiting | NCT03770429 | ||
| Alone | Advanced solid tumor | II | 2020 | Recruiting | NCT04564027 | ||
| Elimusertib | Alone | Advanced solid tumor and lymphomas | I | 2017 | Recruiting | NCT03188965 | [40] |
| Alone | Relapsed or refractory solid tumor | I/II | 2021 | Recruiting | NCT05071209 | ||
| IMP9064 | Alone | Advanced solid tumor | I | 2022 | Recruiting | NCT05269316 | |
| ATG‐018 | Alone | Advanced solid tumor and hematological malignancy | I | 2022 | Recruiting | NCT05338346 | |
| ATRN‐119 | Alone | Advanced solid tumor | I/II | 2023 | Recruiting | NCT04905914 | |
| ART0380 | Alone | Advanced solid tumor | II | 2023 | Not yet recruiting | NCT05798611 | |
| Chemotherapy combinations | |||||||
| Berzosertib | Irinotecan, Gemcitabine, Cisplatin, Cisplatin + Gemcitabine or Etoposide or Carboplatin | Advanced solid tumor | I | 2012 | Completed | NCT02157792 | [39,43, 44, 45, 46] |
| Topotecan | Small‐cell cancer | I/II | 2015 | Active, not recruiting | NCT02487095 | [41, 42] | |
| Cisplatin + Gemcitabine | Metastatic urothelial cancer | II | 2016 | Active, not recruiting | NCT02567409 | ||
| Cisplatin + Radiotherapy | Advanced head and neck cancer | I | 2016 | Active, not recruiting | NCT02567422 | ||
| Gemcitabine | Platinum‐resistant recurrent ovarian or primary peritoneal fallopian tube cancer | II | 2016 | Active, not recruiting | NCT02595892 | [49] | |
| Irinotecan | Advanced solid tumor | I | 2016 | Active, not recruiting | NCT02595931 | ||
| Carboplatin + Gemcitabine | Recurrent platinum‐sensitive epithelial ovarian, peritoneal, and fallopian tube cancer | I | 2016 | Active, not recruiting | NCT02627443 | ||
| Cisplatin + Capecitabine + Radiotherapy | Esophageal cancer and other solid tumors | I | 2018 | Completed | NCT03641547 | ||
| Alone, Carboplatin + Paclitaxel | Advanced solid tumor | I | 2018 | Active, not recruiting | NCT03309150 | ||
| Carboplatin | Metastatic castration‐resistant prostate cancer | II | 2018 | Active, not recruiting | NCT03517969 | ||
| Topotecan | Relapsed small‐cell lung cancer | II | 2019 | Active, not recruiting | NCT03896503 | ||
| Irinotecan | Progressive TP53 mutant gastric and gastroesophageal junction cancer | II | 2020 | Active, not recruiting | NCT03641313 | ||
| Topotecan | Relapsed platinum‐resistant SCLC | II | 2021 | Active, not recruiting | NCT04768296 | ||
| Lurbinectedin | Small‐cell cancer and high‐grade neuroendocrine cancer | I/II | 2021 | Recruiting | NCT04802174 | ||
| Sacituzumab govitecan | SCLC, EP‐SCNC and HR‐deficient cancer resistant to PARP Inhibitors | I/II | 2021 | Recruiting | NCT04826341 | ||
| Topotecan | Advanced solid tumor | I | 2022 | Recruiting | NCT05246111 | ||
| Gartisertib | Alone, Carboplatin | Advanced solid tumor | I | 2015 | Completed | NCT02278250 | |
| Tuvusertib | Temozolomide | Advanced solid tumor, hematopoietic and lymphoid tumor | I/II | 2023 | Not yet recruiting | NCT05691491 | |
| Ceralasertib | Paclitaxel | Refractory cancer | I | 2015 | Completed | NCT02630199 | [48] |
| Gemcitabine | Advanced solid tumor | I | 2019 | Recruiting | NCT03669601 | ||
| Trastuzumab deruxtecan | HER2‐positive advanced solid tumor | I | 2021 | Recruiting | NCT04704661 | ||
| Elimusertib | Cisplatin, Cisplatin + Gemcitabine | Advanced solid tumor | I | 2021 | Recruiting | NCT04491942 | |
| Irinotecan, Topotecan | Advanced solid tumor | I | 2021 | Recruiting | NCT04514497 | ||
| FOLFIRI | Metastatic colorectal and gastric/gastroesophageal cancer | I | 2021 | Recruiting | NCT04535401 | ||
| Gemcitabine | Advanced solid tumor | I | 2021 | Recruiting | NCT04616534 | ||
| ART0380 | Alone, Gemcitabine, Irinotecan | Advanced or metastatic solid tumor | I/II | 2021 | Recruiting | NCT04657068 | |
| PARPi combinations | |||||||
| Berzosertib | Veliparib + Cisplatin | Refractory solid tumor | I | 2017 | Completed | NCT02723864 | |
| Gartisertib | Niraparib | PARPi‐resistant recurrent ovarian cancer | I | 2023 | Not yet recruiting | NCT04149145 | |
| Tuvusertib | Alone, Niraparib | Advanced solid tumor | I | 2019 | Recruiting | NCT04170153 | |
| Ceralasertib | Olaparib | Advanced solid tumor | II | 2015 | Terminated | NCT02576444 | [61] |
| Olaparib | Platinum‐refractory extensive‐stage SCLC | II | 2016 | Active, not recruiting | NCT02937818 | ||
| Olaparib | Advanced breast cancer | II | 2016 | Recruiting | NCT03182634 | ||
| Olaparib | Advanced TNBC | II | 2018 | Active, not recruiting | NCT03330847 | ||
| Olaparib | Recurrent ovarian cancer | II | 2018 | Recruiting | NCT03462342 | [60] | |
| Olaparib | Recurrent ovarian, primary peritoneal, or fallopian tube cancer | II | 2018 | Recruiting | NCT03579316 | ||
| Olaparib | Relapsed SCLC | II | 2018 | Completed | NCT03428607 | ||
| Olaparib | Prostate cancer | II | 2019 | Active, not recruiting | NCT03787680 | ||
| Olaparib | IDH1 and IDH2 mutant tumor | II | 2019 | Recruiting | NCT03878095 | ||
| Alone, Olaparib | Gynecological cancer with ARID1A loss or no loss | II | 2019 | Recruiting | NCT04065269 | ||
| Olaparib | Advanced or metastatic germline BRCA mutated breast cancer | II | 2020 | Recruiting | NCT04090567 | ||
| Olaparib | Recurrent osteosarcoma | II | 2020 | Recruiting | NCT04417062 | ||
| Elimusertib | Niraparib | Advanced solid tumor | I | 2020 | Recruiting | NCT04267939 | |
| Camonsertib | Alone, Talazoparib, Gemcitabine | Advanced solid tumor | I/II | 2020 | Recruiting | NCT04497116 | |
| Niraparib, Olaparib | Advanced solid tumor | I/II | 2021 | Recruiting | NCT04972110 | ||
| Olaparib | DDR‐deficient relapsed/refractory CLL | I/II | 2022 | Recruiting | NCT05405309 | ||
| Immunotherapy combinations | |||||||
| Berzosertib | Avelumab + Carboplatin | PARPi‐resistant recurrent ovarian, primary peritoneal, or fallopian tube cancer | I | 2019 | Completed | NCT03704467 | |
| Carboplatin + Gemcitabine + Pembrolizumab | Advanced NSCLC | I/II | 2020 | Recruiting | NCT04216316 | ||
| Avelumab | DDR‐deficient metastatic or unresectable solid tumor | I/II | 2020 | Recruiting | NCT04266912 | ||
| Tuvusertib | M4076, Avelumab | Advanced solid tumor | I | 2022 | Recruiting | NCT05396833 | |
| Ceralasertib | Alone, Carboplatin, Olaparib, AZD5305, Durvalumab | Advanced solid tumor | I/II | 2014 | Recruiting | NCT02264678 | [47] |
| Durvalumab | NSCLC | II | 2015 | Active, not recruiting | NCT02664935 | ||
| Alone, Durvalumab | NSCLC | II | 2017 | Recruiting | NCT03334617 | ||
| Durvalumab | Advanced solid tumor | I | 2018 | Recruiting | NCT05514132 | ||
| Alone, Olaparib, Durvalumab | Advanced solid tumor | II | 2019 | Recruiting | NCT03682289 | ||
| Durvalumab | Gastric cancer and melanoma | II | 2019 | Active, not recruiting | NCT03780608 | [67, 68] | |
| Durvalumab | NSCLC | II | 2019 | Recruiting | NCT03833440 | ||
| Durvalumab | Refractory biliary tract cancer | II | 2020 | Recruiting | NCT04298008 | ||
| Durvalumab, Olaparib | Advanced biliary tract cancer | II | 2020 | Recruiting | NCT04298021 | ||
| Durvalumab | Relapsed SCLC | II | 2020 | Active, not recruiting | NCT04361825 | ||
| Cisplatin or Carboplatin + Etoposide + Durvalumab | Extensive‐stage SCLC | II | 2021 | Recruiting | NCT04699838 | ||
| Durvalumab + Nab‐paclitaxel | Advanced TNBC | II | 2022 | Recruiting | NCT05582538 | ||
| Elimusertib | Pembrolizumab | Advanced solid tumor | I | 2019 | Active, not recruiting | NCT04095273 | |
| Pembrolizumab + Radiotherapy | Recurrent head and neck cancer | I | 2021 | Recruiting | NCT04576091 | ||
| Other combinations | |||||||
| Berzosertib | Whole‐brain radiotherapy | Brain metastases from lung cancer | I | 2016 | Active, not recruiting | NCT02589522 | |
| Radiotherapy | Chemotherapy‐resistant triple‐negative and ER/PR‐positive, HER2‐negative breast cancer | I | 2019 | Recruiting | NCT04052555 | ||
| Tuvusertib | Peposertib | Advanced solid tumor | I | 2023 | Not yet recruiting | NCT05687136 | |
| Ceralasertib | Acalabrutinib | Relapsed/refractory aggressive non‐Hodgkin's lymphoma | I | 2018 | Completed | NCT03527147 | |
| Alone, Acalabrutinib | Relapsed or refractory CLL | I | 2020 | Active, not recruiting | NCT03328273 | ||
| Camonsertib | RP‐6306 | Advanced solid tumor | I | 2021 | Recruiting | NCT04855656 | |
| Radiotherapy | Metastatic cancer with ATM mutation | I/II | 2022 | Recruiting | NCT05566574 | ||
Note: These clinical trial data are from ClinicalTrials.gov (April 2023). Ongoing clinical trials include the status active, not recruiting, recruiting, and not yet recruiting.
3.1. Monotherapy and chemotherapy combinations
Berzosertib is the first‐in‐class ATR inhibitor and has been tested as a monotherapy. Berzosertib monotherapy was well tolerated, with no dose‐limiting toxicities observed and a durable complete response was observed in a patient with advanced‐stage colorectal cancer harboring ATM loss and ARID1A mutation. 39 Elimusertib monotherapy was well tolerated, with antitumor activity including partial responses against cancers with ATM loss. 40
Early clinical trials with ATRis were often conducted using them in combination with chemotherapy‐inducing RS. Antimetabolites (e.g., gemcitabine) inhibit the elongation of newly synthesized DNA strands by blocking nucleotide synthesis and by acting as nucleotide analogs that lead to chain termination. DNA crosslinkers (e.g., cisplatin and carboplatin) and topoisomerase inhibitors (e.g., topotecan and irinotecan) generate obstacles to fork progression. Consequently, these chemotherapeutic agents increase RS and synergize with ATRis. The first reported phase I study of ATRi demonstrated that combined berzosertib with topotecan was partially effective in platinum‐refractory SCLC. 41 , 42 Other phase I studies of berzosertib in combination with chemotherapy, including gemcitabine and cisplatin, also showed preliminary efficacy signs. 43 , 44 , 45 , 46 In phase I studies of ceralasertib in combination with carboplatin or paclitaxel, patients with low ATM or mutated ATM had complete or partial responses. 47 , 48 Notably, the first randomized phase II study of ATRi was evaluated based on its use in combination with gemcitabine versus gemcitabine alone in platinum‐resistant HGSOC. 49 The median progression‐free survival in all patients was 22.9 weeks (90% CI: 17.9–72.0) for the combination group versus 14.7 weeks (90% CI: 9.7–36.7) for the monotherapy group (p = 0.044). Interestingly, this benefit of the combination group was seen in patients with a platinum‐free interval of 3 months or less compared with those with more than 3 months to less than 6 months. While these preliminary data from early phase studies for ATRis in combination with DNA‐damaging chemotherapy showed antitumor activity, the chemotherapy combinations were associated with higher rates of adverse events. 39 Further late‐stage clinical evaluation of the chemotherapy combinations is warranted and the identification of predictive biomarkers of response is the critical next step.
3.2. PARPi combinations
In the past decade, PARP inhibitors (PARPis) have been clinically used to treat tumors with defects in HR, such as BRCA1/2. 50 PARPis generate DNA lesions through the following two general actions: catalytic inhibition of PARP1 and trapping PARP1 on damaged DNA; these actions render synthetic lethality with HR deficiency. Recently, it has also been proposed that the accumulation of ssDNA gaps behind replication forks is the primary genotoxic lesion enhancing PARPi sensitivity. PARPi induces ssDNA gaps on the leading strand behind replication forks via PrimPol‐mediated repriming 33 , 51 , 52 and on the lagging strand via defects in Okazaki fragment processing. 53 PARPi‐induced ssDNA gaps generated in the first S phase persist in the second S phase, where BRCA1/2‐deficient cells fail to activate ATR and suppress origin firing, resulting in increased fork collapse. In addition, ATRi combined with PARPi abrogated the PARPi‐induced G2/M checkpoint and synergistically increased chromosome aberrations. 54 , 55 In fact, several preclinical studies have described the synergistic therapeutic efficacy of ATRis with PARPis in tumor cells harboring BRCA1/2, ATM, and RNASEH2 deficiency and alternative lengthening of the telomere system. 54 , 55 , 56 , 57 Furthermore, ATRi combined with PARPi overcomes PARPi‐ or platinum‐resistant ovarian cancer and PARPi‐resistant BRCA‐deficient cancer models. 55 , 58 , 59 Therefore, these preclinical data support the clinical development of ATRis in combination with PARPis. In reported phase II studies of ceralasertib in combination with olaparib, one study in 12 patients with platinum‐resistant high‐grade ovarian cancer observed no objective response. 60 In another study, two out of five patients with tumors harboring the ATM mutation achieved complete response or stable disease. Of seven patients with PARP inhibitor‐resistant HGSOC, one achieved a partial response and five had stable disease. 61 Further testing of larger populations and randomized trials are warranted.
3.3. Immunotherapy combinations
Other proposed combination drugs with ATRis are immune checkpoint inhibitors (ICIs), such as monoclonal antibodies targeting PD‐1/PD‐L1. Accumulating evidence indicates that, upon RS, DNA fragments might be released from the nucleus into the cytosol, causing cGAS–STING pathway activation, 62 leading to T‐cell priming and recruitment and boosting the efficacy of immunotherapies. 63 Moreover, ATRi downregulates PD‐L1 expression by activating the CDK1–SPOP pathway and sensitizes tumor cells to T‐cell‐mediated killing. 64 , 65 These results provide a rationale for combination therapy with ATRis and ICIs. Preclinical studies showed beneficial results by combination therapy with ATRi and ICI and by triple therapy with ATRi, ICI, and radiation. 65 , 66 In phase II studies with ceralasertib in combination with durvalumab, 9 of 30 patients with gastric cancer and 7 of 31 patients with melanoma achieved partial responses. 67 , 68
Overall, ATRis exhibit preliminary antitumor activities in these clinical studies when RS is increased by genetic alterations or by drug combinations, but many ongoing clinical studies are still awaiting results. Whereas ATM loss seems to be a useful biomarker for predicting vulnerability to ATRi monotherapy and combination therapy, the identification of further predictive biomarkers is critical for the success of therapeutic approaches using ATRis.
4. BIOMARKERS
To optimize ATRi therapy, in vitro and in vivo assays have identified several potential biomarkers associated with the response to ATRis. These biomarkers may be categorized into four groups (Table 2). The first group is associated with cell signaling pathways activated in response to RS. As fundamental models, various types of RS inducers generally increase the phosphorylated forms of ATR, Chk1, and RPA32 as markers of ATR signaling activation and γH2A.X levels as markers of DNA damage. 13 These phosphorylated forms reflect the cellular response to low or high levels of RS, indicating the utility of immunohistochemistry assays based on specific antibodies. The second group includes genetic alterations leading to RS. Activation of oncogenes such as Ras, Myc, and Cyclin E can elicit RS through different mechanisms and increase susceptibility to ATRis. 69 , 70 , 71 In addition, APOBEC3A/B activities impose a unique type of RS by generating abasic sites at replication forks and rendering cancer cells susceptible to ATRis. 72 The third group includes loss of function in DDR and DNA repair. Cells harboring defects in DDR and DNA repair are highly dependent on alternative DDR pathways, resulting in synthetic lethality with additional DDR inhibition because of unbearable DNA damage. Consistently, ATRis are toxic in ATM‐, p53‐, or HR‐deficient tumor cells. 38 , 59 , 73 Deficiencies in XRCC1, ERCC1, ARID1A, and RNASEH2, which are involved in DNA repair, were also identified as predictive biomarkers of ATRi susceptibility. 74 , 75 , 76 BRCA1/2 deficiency, especially in the context of PARPi‐ or Cisplatin‐resistant tumors, confers ATRi alone or ATRi and PARPi combination sensitivity. 55 , 59 The fourth group is biomarkers resistant to ATRis. Cells with low levels of CDC25A and UPF2 were resistant to ATRi, 77 , 78 suggesting the possibility of previously unknown mechanisms underlying resistance to ATRis. The clinical utility of these biomarkers is currently under active investigation (Table 1), and further validation is clearly needed to stratify patients who will respond to ATRi therapy.
TABLE 2.
Categories of biomarkers for predicting ATRi efficacy.
| Biomarkers associated with cell signaling pathways activated in response to replication stress | p‐ATR |
| p‐Chk1 | |
| p‐RPA32 | |
| γH2A.X | |
| Biomarkers of genetic alterations leading to replication stress | MYC overexpression |
| RAS mutation | |
| Cyclin E overexpression | |
| APOBEC3A/B overexpression | |
| EWS translocation | |
| MLL translocation | |
| Biomarkers of loss of function in DNA repair and DDR | ARID1A deficiency |
| RNASEH2 deficiency | |
| ATM deficiency | |
| p53 deficiency | |
| ERCC1 deficiency | |
| XRCC1 deficiency | |
| PARPi‐ or platinum‐resistant BRCA1/2 deficiency | |
| RAD51 deficiency | |
| SLFN11 deficiency | |
| SMARCA4 deficiency | |
| Biomarkers resistant to ATRis | CDC25A deficiency |
| UPF2 deficiency |
As mentioned above, genome‐wide CRISPR or si/shRNA screens enable a comprehensive search for predictive biomarkers of ATRis and have been used to identify some genetic biomarker candidates. One of the concerns is that these screening models have evaluated the response to ATRis under acute RS after knockout or knockdown but lack a process to become RS tolerant. Alternatively, recent studies have clarified that RS tolerance can overcome acute RS and promote cancer survival. As cells with oncogenic alterations seem to acquire RS tolerance mechanisms during the long process of tumorigenesis, cancer cells may no longer be highly dependent on the ATR response against such RS induced by acute loss of biomarkers. Therefore, a strategy to examine biomarkers after the chronic loss of candidate factors would allow for the identification of biomarkers that can account for acute RS and RS tolerance.
We previously identified a deficiency in SMARCA4, a core component of the SWI/SNF chromatin remodeling complex, as a predictive biomarker of ATRi efficacy using LADC cell line‐based screening. 79 As a chronic response, SMARCA4‐deficient cells increase heterochromatin formation and thereby elevate RS, rendering them dependent on ATR‐mediated RS tolerance for survival. However, in the absence of SMARCA4, ATRi‐induced acute RS causes severe ssDNA exposure on nascent DNA near the reversed forks around heterochromatin in a Mre11‐dependent manner, leading to replication catastrophe. Thus, SMARCA4 loss synergistically confers ATRi susceptibility by increasing heterochromatin‐associated RS and by allowing Mre11 to destabilize reversed forks. In agreement with our results, Gupta and colleagues also reported that SMARCA4 loss led to clinically relevant gene expression changes related to RS and prereplication functions in LADC patients and in a mouse model; these changes activated ATR signaling. 80 The dependence on ATR under replication defects provides a possible explanation for how lung cancer cells tolerate SMARCA4 loss and confer ATRi susceptibility. Although what kind of RS tolerance mechanisms are at play in SMARCA4‐deficient cancer cells awaits further investigation, ATRis may target not only acute RS but also RS tolerance in SMARCA4‐deficient cells. Thus, SMARCA4 deficiency can be a beneficial predictive biomarker of ATRi efficacy.
5. PERSPECTIVE
ATR kinase is a master regulator in response to RS, contributing to both genomic integrity in normal cells and RS tolerance in cancer cells. Importantly, preclinical evidence has suggested that targeting ATR might be a selective strategy for cancer cells, but not normal cells, making ATR an attractive target; furthermore, the causes and levels of RS in cancer cells might be critical determinants of ATRi efficacy. Here, we provide potential models of ATRi action depending on RS levels in cells (Figure 2). Cancer cells with low RS due to factors such as activation of oncogenes or defects in DDR might require high doses of ATRis to reach the RS threshold to induce cell death. When cancer cells can adapt to RS by acquiring ATR‐mediated RS tolerance mechanisms, they become more addicted to ATR signaling for survival. Therefore, RS tolerance dependency might be a new type of predictive biomarker of ATRi efficacy. Finally, cancer cells treated with chemotherapeutic agents such as cisplatin and PARPi exhibit very high RS, which may be sufficient by itself to reach the RS threshold or to synergize with low doses of ATRis. As RS causes cell cycle arrest at S and/or G2 phase in an ATR‐dependent manner, ATR inhibition can induce replication catastrophe in early S phase cells, promote early mitotic entry and predominantly induce mitotic catastrophe in cells in late S phase or G2 phase. However, RS levels are determined in a highly complex manner by multiple factors and are associated with ATRi susceptibility. Therefore, the development of a method to accurately measure RS in cancer cells is expected to open further possibilities for ATRi therapy. Hopefully, this knowledge will be integrated into clinical development, and ATRis will become the new drugs of choice in the fight against cancer in the future.
FIGURE 2.
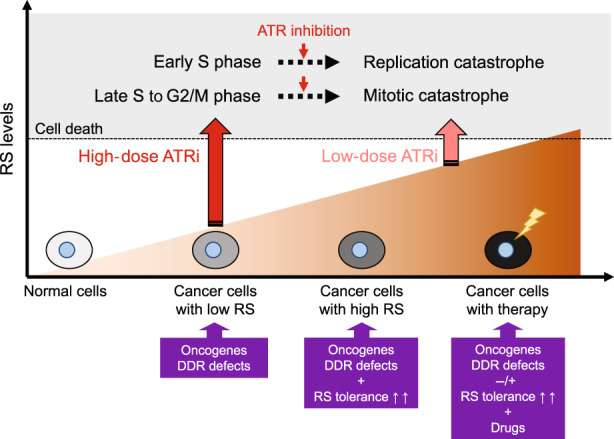
Potential models of action of ataxia telangiectasia and Rad3‐related inhibitors (ATRis) depending on DNA replication stress (RS) levels. Because their RS stems from a high rate of DNA replication and/or genomic instability, cancer cells often have an increased reliance on the ATR signaling pathway compared with normal cells, conferring susceptibility to therapeutic approaches using ATRis. Cancer cells harboring oncogene activation and/or DNA damage response defects survive with low levels of RS and might require high doses of ATRis to reach the RS threshold that induces cell death. When cancer cells acquire ATR‐mediated RS tolerance, they are more dependent on ATR signaling for survival. In addition, cancer cells treated with RS‐inducing drugs exhibit high levels of RS that synergize with low doses of ATRis to induce cell death. Finally, RS at levels over the threshold, induced by ATRis, can lead to cell death through replication catastrophe in early S cells and mitotic catastrophe in late S to G2/M phase cells.
FUNDING INFORMATION
Japan Society for the Promotion of Science (JSPS), Grant/Award Number: 18KK0235 and 22H03745 to B.S., 22K18033 to K.Y.
ETHICS STATEMENTS
Approval of the research protocol by an Institutional Reviewer Board: N/A.
Informed Consent: N/A.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: N.A.
CONFLICT OF INTEREST STATEMENT
B.S. reports receiving research grants from Merck KGaA and AstraZeneca. The other author has no potential conflicts of interest.
ACKNOWLEDGMENTS
We apologize to the investigators whose relevant work we could not cite for space reasons.
Yano K, Shiotani B. Emerging strategies for cancer therapy by ATR inhibitors. Cancer Sci. 2023;114:2709‐2721. doi: 10.1111/cas.15845
REFERENCES
- 1. Saxena S, Zou L. Hallmarks of DNA replication stress. Mol Cell. 2022;82:2298‐2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kotsantis P, Petermann E, Boulton SJ. Mechanisms of oncogene‐induced replication stress: jigsaw falling into place. Cancer Discov. 2018;8:537‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annu Rev Pathol. 2015;10:425‐448. [DOI] [PubMed] [Google Scholar]
- 5. Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA‐ssDNA complexes. Science. 2003;300:1542‐1548. [DOI] [PubMed] [Google Scholar]
- 7. Liu S, Shiotani B, Lahiri M, et al. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell. 2011;43:192‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9‐Hus1‐Rad1 (9‐1‐1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007;21:1472‐1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR‐ATRIP complex. Cell. 2006;124:943‐955. [DOI] [PubMed] [Google Scholar]
- 10. Haahr P, Hoffmann S, Tollenaere MA, et al. Activation of the ATR kinase by the RPA‐binding protein ETAA1. Nat Cell Biol. 2016;18:1196‐1207. [DOI] [PubMed] [Google Scholar]
- 11. Bass TE, Luzwick JW, Kavanaugh G, et al. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol. 2016;18:1185‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shiotani B, Nguyen HD, Hakansson P, et al. Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep. 2013;3:1651‐1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yazinski SA, Zou L. Functions, regulation, and therapeutic implications of the ATR checkpoint pathway. Annu Rev Genet. 2016;50:155‐173. doi: 10.1146/annurev-genet-121415-121658 [DOI] [PubMed] [Google Scholar]
- 14. Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 2017;18:622‐636. doi: 10.1038/nrm.2017.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karlsson‐Rosenthal C, Millar JB. Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006;16:285‐292. [DOI] [PubMed] [Google Scholar]
- 16. O'Connell MJ, Raleigh JM, Verkade HM, Nurse P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 1997;16:545‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saldivar JC, Hamperl S, Bocek MJ, et al. An intrinsic S/G(2) checkpoint enforced by ATR. Science. 2018;361:806‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF. Concerted loading of Mcm2‐7 double hexamers around DNA during DNA replication origin licensing. Cell. 2009;139:719‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yekezare M, Gomez‐Gonzalez B, Diffley JF. Controlling DNA replication origins in response to DNA damage—inhibit globally, activate locally. J Cell Sci. 2013;126:1297‐1306. [DOI] [PubMed] [Google Scholar]
- 20. Liu H, Takeda S, Kumar R, et al. Phosphorylation of MLL by ATR is required for execution of mammalian S‐phase checkpoint. Nature. 2010;467:343‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Toledo LI, Altmeyer M, Rask MB, et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155:1088‐1103. [DOI] [PubMed] [Google Scholar]
- 22. Quinet A, Tirman S, Cybulla E, Meroni A, Vindigni A. To skip or not to skip: choosing repriming to tolerate DNA damage. Mol Cell. 2021;81:649‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cybulla E, Vindigni A. Leveraging the replication stress response to optimize cancer therapy. Nat Rev Cancer. 2022;23:6‐24. doi: 10.1038/s41568-022-00518-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Couch FB, Bansbach CE, Driscoll R, et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013;27:1610‐1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ragland RL, Patel S, Rivard RS, et al. RNF4 and PLK1 are required for replication fork collapse in ATR‐deficient cells. Genes Dev. 2013;27:2259‐2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim JJ, Lee SY, Choi JH, Woo HG, Xhemalce B, Miller KM. PCAF‐mediated histone acetylation promotes replication fork degradation by MRE11 and EXO1 in BRCA‐deficient cells. Mol Cell. 2020;80:327‐344 e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sabbioneda S, Bortolomai I, Giannattasio M, Plevani P, Muzi‐Falconi M. Yeast Rev1 is cell cycle regulated, phosphorylated in response to DNA damage and its binding to chromosomes is dependent upon MEC1. DNA Repair (Amst). 2007;6:121‐127. [DOI] [PubMed] [Google Scholar]
- 28. Pages V, Santa Maria SR, Prakash L, Prakash S. Role of DNA damage‐induced replication checkpoint in promoting lesion bypass by translesion synthesis in yeast. Genes Dev. 2009;23:1438‐1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gohler T, Sabbioneda S, Green CM, Lehmann AR. ATR‐mediated phosphorylation of DNA polymerase eta is needed for efficient recovery from UV damage. J Cell Biol. 2011;192:219‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murphy AK, Fitzgerald M, Ro T, et al. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol. 2014;206:493‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Buisson R, Niraj J, Rodrigue A, et al. Coupling of homologous recombination and the checkpoint by ATR. Mol Cell. 2017;65:336‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Somyajit K, Basavaraju S, Scully R, Nagaraju G. ATM‐ and ATR‐mediated phosphorylation of XRCC3 regulates DNA double‐strand break‐induced checkpoint activation and repair. Mol Cell Biol. 2013;33:1830‐1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quinet A, Tirman S, Jackson J, et al. PRIMPOL‐mediated adaptive response suppresses replication fork reversal in BRCA‐deficient cells. Mol Cell. 2020;77:461‐474 e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mehta KPM, Thada V, Zhao R, et al. CHK1 phosphorylates PRIMPOL to promote replication stress tolerance. Sci Adv. 2022;8:eabm0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eykelenboom JK, Harte EC, Canavan L, et al. ATR activates the S‐M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep. 2013;5:1095‐1107. [DOI] [PubMed] [Google Scholar]
- 36. Kabeche L, Nguyen HD, Buisson R, Zou L. A mitosis‐specific and R loop‐driven ATR pathway promotes faithful chromosome segregation. Science. 2018;359:108‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bass TE, Cortez D. Quantitative phosphoproteomics reveals mitotic function of the ATR activator ETAA1. J Cell Biol. 2019;218:1235‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reaper PM, Griffiths MR, Long JM, et al. Selective killing of ATM‐ or p53‐deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7:428‐430. [DOI] [PubMed] [Google Scholar]
- 39. Yap TA, O'Carrigan B, Penney MS, et al. Phase I trial of first‐in‐class ATR inhibitor M6620 (VX‐970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol. 2020;38:3195‐3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yap TA, Tan DSP, Terbuch A, et al. First‐in‐human trial of the Oral ataxia telangiectasia and RAD3‐related (ATR) inhibitor BAY 1895344 in patients with advanced solid tumors. Cancer Discov. 2021;11:80‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thomas A, Redon CE, Sciuto L, et al. Phase I study of ATR inhibitor M6620 in combination with Topotecan in patients with advanced solid tumors. J Clin Oncol. 2018;36:1594‐1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thomas A, Takahashi N, Rajapakse VN, et al. Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell. 2021;39:566‐579 e567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Middleton MR, Dean E, Evans TRJ, et al. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX‐970) combined with gemcitabine +/− cisplatin in patients with advanced solid tumours. Br J Cancer. 2021;125:510‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shapiro GI, Wesolowski R, Devoe C, et al. Phase 1 study of the ATR inhibitor berzosertib in combination with cisplatin in patients with advanced solid tumours. Br J Cancer. 2021;125:520‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Plummer R, Dean E, Arkenau HT, et al. A phase 1b study evaluating the safety and preliminary efficacy of berzosertib in combination with gemcitabine in patients with advanced non‐small cell lung cancer. Lung Cancer. 2022;163:19‐26. [DOI] [PubMed] [Google Scholar]
- 46. Telli ML, Tolaney SM, Shapiro GI, et al. Phase 1b study of berzosertib and cisplatin in patients with advanced triple‐negative breast cancer. NPJ Breast Cancer. 2022;8:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yap TA, Krebs MG, Postel‐Vinay S, et al. Ceralasertib (AZD6738), an Oral ATR kinase inhibitor, in combination with carboplatin in patients with advanced solid tumors: a phase I study. Clin Cancer Res. 2021;27:5213‐5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim ST, Smith SA, Mortimer P, et al. Phase I study of Ceralasertib (AZD6738), a novel DNA damage repair agent, in combination with weekly paclitaxel in refractory cancer. Clin Cancer Res. 2021;27:4700‐4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Konstantinopoulos PA, Cheng SC, Wahner Hendrickson AE, et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum‐resistant high‐grade serous ovarian cancer: a multicentre, open‐label, randomised, phase 2 trial. Lancet Oncol. 2020;21:957‐968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mateo J, Lord CJ, Serra V, et al. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol. 2019;30:1437‐1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Berti M, Ray Chaudhuri A, Thangavel S, et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol. 2013;20:347‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Simoneau A, Xiong R, Zou L. The trans cell cycle effects of PARP inhibitors underlie their selectivity toward BRCA1/2‐deficient cells. Genes Dev. 2021;35:1271‐1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cong K, Peng M, Kousholt AN, et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol Cell. 2021;81:3128‐3144 e3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lloyd RL, Wijnhoven PWG, Ramos‐Montoya A, et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM‐deficient cancer cells. Oncogene. 2020;39:4869‐4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim H, Xu H, George E, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun. 2020;11:3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zimmermann M, Bernier C, Kaiser B, et al. Guiding ATR and PARP inhibitor combinationswith chemogenomic screens. Cell Rep. 2022;40:111081. [DOI] [PubMed] [Google Scholar]
- 57. Roulston A, Zimmermann M, Papp R, et al. RP‐3500: a novel, potent, and selective ATR inhibitor that is effective in preclinical models as a monotherapy and in combination with PARP inhibitors. Mol Cancer Ther. 2022;21:245‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Murai J, Feng Y, Yu GYK, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. 2016;7:76534‐76550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yazinski SA, Comaills V, Buisson R, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor‐resistant BRCA‐deficient cancer cells. Genes Dev. 2017;31:318‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shah PD, Wethington SL, Pagan C, et al. Combination ATR and PARP inhibitor (CAPRI): a phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum‐resistant epithelial ovarian cancer. Gynecol Oncol. 2021;163:246‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mahdi H, Hafez N, Doroshow D, et al. Ceralasertib‐mediated ATR inhibition combined with Olaparib in advanced cancers harboring DNA damage response and repair alterations (Olaparib combinations). JCO Precis Oncol. 2021;5:1432‐1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Técher H, Pasero P. The replication stress response on a narrow path between genomic instability and inflammation. Front Cell Dev Biol. 2021;9:702584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ragu S, Matos‐Rodrigues G, Lopez BS. Replication stress, DNA damage, inflammatory cytokines and innate immune response. Genes (Basel). 2020;11:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sun LL, Yang RY, Li CW, et al. Inhibition of ATR downregulates PD‐L1 and sensitizes tumor cells to T cell‐mediated killing. Am J Cancer Res. 2018;8:1307‐1316. [PMC free article] [PubMed] [Google Scholar]
- 65. Tang Z, Pilie PG, Geng C, et al. ATR inhibition induces CDK1‐SPOP signaling and enhances anti‐PD‐L1 cytotoxicity in prostate cancer. Clin Cancer Res. 2021;27:4898‐4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sheng H, Huang Y, Xiao Y, et al. ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J Immunother Cancer. 2020;8:e000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kim R, Kwon M, An M, et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti‐PD‐1 therapy. Ann Oncol. 2022;33:193‐203. [DOI] [PubMed] [Google Scholar]
- 68. Kwon M, Kim G, Kim R, et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced gastric cancer. J Immunother Cancer. 2022;10:e005041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gilad O, Nabet BY, Ragland RL, et al. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage‐dependent manner. Cancer Res. 2010;70:9693‐9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jones RM, Mortusewicz O, Afzal I, et al. Increased replication initiation and conflicts with transcription underlie cyclin E‐induced replication stress. Oncogene. 2013;32:3744‐3753. [DOI] [PubMed] [Google Scholar]
- 71. Cottini F, Hideshima T, Suzuki R, et al. Synthetic lethal approaches exploiting DNA damage in aggressive myeloma. Cancer Discov. 2015;5:972‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Buisson R, Lawrence MS, Benes CH, Zou L. APOBEC3A and APOBEC3B activities render cancer cells susceptible to ATR inhibition. Cancer Res. 2017;77:4567‐4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Krajewska M, Fehrmann RS, Schoonen PM, et al. ATR inhibition preferentially targets homologous recombination‐deficient tumor cells. Oncogene. 2015;34:3474‐3481. [DOI] [PubMed] [Google Scholar]
- 74. Mohni KN, Kavanaugh GM, Cortez D. ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res. 2014;74:2835‐2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Williamson CT, Miller R, Pemberton HN, et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun. 2016;7:13837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang C, Wang G, Feng X, et al. Genome‐wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene. 2019;38:2451‐2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ruiz S, Mayor‐Ruiz C, Lafarga V, et al. A genome‐wide CRISPR screen identifies CDC25A as a determinant of sensitivity to ATR inhibitors. Mol Cell. 2016;62:307‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. O'Leary PC, Chen H, Doruk YU, et al. Resistance to ATR inhibitors is mediated by loss of the nonsense‐mediated decay factor UPF2. Cancer Res. 2022;82:3950‐3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kurashima K, Kashiwagi H, Shimomura I, et al. SMARCA4 deficiency‐associated heterochromatin induces intrinsic DNA replication stress and susceptibility to ATR inhibition in lung adenocarcinoma. NAR Cancer. 2020;2:zcaa005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gupta M, Concepcion CP, Fahey CG, et al. BRG1 loss predisposes lung cancers to replicative stress and ATR dependency. Cancer Res. 2020;80:3841‐3854. [DOI] [PMC free article] [PubMed] [Google Scholar]