

Abstract
Osteoclasts (OCs) and regulatory CD4+ T cells (CD4+Tregs) are important components in the tumor microenvironment (TME) of osteosarcoma. In this study, we collected six osteosarcoma samples from our previous study (GSE162454). We also integrated a public database (GSE152048), which included single cell sequencing data of 11 osteosarcoma patients. We obtained 138,192 cells and then successfully identified OCs and CD4+Tregs. Based on the interaction gene set between OCs and CD4+Tregs, patients from GSE21257 were distinguished into two clusters by consensus clustering analysis. Both the tumor immune microenvironment and survival prognosis between the two clusters were significantly different. Subsequently, five model genes were identified by protein–protein interaction network based on differentially upregulated genes of cluster 2. Quantitative RT‐PCR was used to detect their expression in human osteoblast and osteosarcoma cells. A prognostic model was successfully established using these five genes. Kaplan–Meier survival analysis found that patients in the high‐risk group had worse survival (p = 0.029). Therefore, our study first found that cell–cell communication between OCs and CD4+Tregs significantly alters TME and is connected to poor prognosis of OS. The model we constructed can accurately predict prognosis for osteosarcoma patients.
Keywords: CD4+Treg, interaction gene set, osteoclast, osteosarcoma, single‐cell RNA‐seq
This research found that cell–cell communication between osteoclasts and regulatory CD4+ T cells significantly alter the tumor microenvironment and is connected to poor prognosis of osteosarcoma. The model can accurately predict patient prognosis.
Abbreviations
- AUC
area under the ROC curve
- CD4+Treg
regulatory CD4+ T cell
- DEG
differentially expressed gene
- FC
fold change
- FDR
false discovery rate
- GEO
Gene Expression Omnibus
- GSVA
gene set variation analysis
- HR
hazard ratio
- IGS
interaction gene set
- KM
Kaplan–Meier
- LASSO
least absolute shrinkage and selection operator
- MHC
major histocompatibility complex
- NK
natural killer
- OC
osteoclast
- OS
osteosarcoma
- PCA
principal component analysis
- PPI
protein–protein interaction
- qRT‐PCR
quantitative RT‐PCR
- ROC
receiver operating characteristic
- scRNA‐seq
single‐cell RNA‐sequencing
- TME
tumor microenvironment
1. INTRODUCTION
Osteosarcoma, the most prevalent bone‐derived tumor, typically affects adolescents and young children. 1 Worldwide, three to four per million people are diagnosed with OS each year. 2 Since the evolution of systematic chemotherapy, the 5‐year survival rate of OS patients with localized disease has improved to 70%. 3 However, the overall survival rate has not improved significantly in the last 30 years. 4 Tumor microenvironment heterogeneity, chemotherapy resistance, and tumor immune escape are important causes for the poor prognosis. 5 Therefore, it is of great significance to identify patients with high‐risk characteristics of poor survival outcomes.
Tumor microenvironment is a highly specialized, complex, and dynamic environment. The TME is composed of different cellular components and other physicochemical factors. 6 , 7 Frequent cell cross‐talk among them help OS cells remold TME, providing fertile soil for OS proliferation and metastasis. 7 , 8 , 9 Osteoclasts and CD4+Tregs are important components of OS TME. Osteoclasts are the only cells performing the function of osteolysis. Studies have shown that paratumor osteolysis is conducive to the development of OS. 8 In addition, OCs play a key role in immunomodulatory effect. 10 Ibáñez et al. confirmed that OCs had antigen‐presenting functions and could induce CD4+Tregs. 11 , 12 Regulatory CD4+ T cells are immunosuppressive cells that assist tumor cells in immune escape in a variety of cancers. 13 However, studies on whether the interaction between OCs and CD4+Tregs changes the TME and survival prognosis are lacking.
We identified an IGS at the single‐cell level between OCs and CD4+Tregs. Consensus clustering analysis was used to identify clusters with different expression modes of IGS in a GEO cohort. The differences between these two clusters in tumor immunofiltration, biological pathways, and survival prognosis were explored. Subsequently, differential analysis was carried out to obtain differentially upregulated genes of cluster 2. Five model genes were screened by LASSO regression analysis. Then a risk prognosis model was constructed. We found that the model genes were connected to poor prognosis. The prognosis model showed robust predictive performance in KM survival analysis and ROC curve analysis. In addition, we constructed a nomogram to facilitate the use of the risk‐prognosis model.
2. MATERIALS AND METHODS
2.1. Data sources
Figure S1 illustrates the workflow of this study. A part of scRNA‐seq data was obtained from our previous study (GSE162454). 7 In order to obtain more samples, we integrated a public database (GSE152048), which included single‐cell sequencing data from 11 OS patients. 9 The above two data sources of OS scRNA‐seq were utilized to identify the IGS. Bulk RNA sequencing datasets were derived from the GEO database (GSE21257), 14 which was used to construct the risk model and evaluate the prediction accuracy of the risk prognosis model.
2.2. Single‐cell RNA‐seq analysis
We used the “Seurat” package (version 4.0.5) 15 for scRNA‐seq data analysis and the “merge” function to integrate OS data. To obtain high‐quality scRNA‐seq data, we applied the following filtering conditions for each cell: nFeature_RNA >300, nFeature_RNA <4500, and percent.mt <10%.
Next, the “NormalizeData” function of the R package “Seurat” was used to normalize the scRNA‐seq data. The “FindVariableFeatures” function was then used to identify the top 2000 highly variable genes from the standardized scRNA‐seq data. We utilized the “RunPCA” function for PCA, thus achieving the purpose of mapping higher‐dimensional data to lower‐dimensional space and preserving the characteristics of more original data points with fewer data dimensions. We then used the “harmony” package (version 0.1.0) to remove batch effects. 16
In addition, the “FindNeighbors” and “FindClusters” functions in the “Seurat” package were used for cell clustering analysis. The parameters: dim = 1:30, resolution = 0.2. Natural killer/T cells were defined using the marker genes CD2, CD3D, CD3E, CD3G, GNLY, NKG7, KLRD1, and KLRB1; 17 , 18 OCs were defined using ACP5, CTSK, and ATP6V0D2. 19 As we did not study other cells further, we defined them as “other cells”. In addition, the marker genes CD4, TNFRSF18, LAYN, CCR8, FOXP3, BATF, RTKN2, IKZF2, CTLA4, and CTLA4 were used to define CD4+Tregs. 20
Finally, data were visualized using the “DimPlot” function and the “pheatmap” package (version 1.0.12).
2.3. Cell–cell communication analysis
We assessed intercellular communication between OCs and CD4+Tregs in TME using CellPhoneDB (version 2.0.0). 21 Interaction gene sets with p < 0.05 were filtered to evaluate the relationship between the different cell clusters.
2.4. Consensus clustering analysis
Based on the expression level of IGS, the “ConsensusClusterPlus” R package (version 1.56.0) was used to identify clusters of the GSE21257 dataset. Principal component analysis was used to show the distribution of the two coagulation clusters, and it was visualized with the “ggplot2” and “scatterplot3d” packages. The overall survival of the two clusters was compared using a KM survival plot, and p = 0.038 indicated statistical significance.
The “ggalluvial” R package (version 0.12.5) was used to create a Sankey diagram to assess the clinical value of clusters. We investigated the relationship between clusters and clinical characteristics, such as age, status, histological subtype, grade, and metastases. Using the “pheatmap” R package (version 1.0.12), we created a heatmap of IGS expression patterns that differ in clusters.
2.5. Gene set variation analysis and specific immune landscape
To understand the distinct biological functions of the various clusters, we undertook GSVA using the “GSVA” R package (version 1.40.1). 22 For the GSVA analysis, the msigdb.v7.2 symbols gene sets (https://www.gsea‐msigdb.org/gsea/msigdb) were utilized. 23 , 24 The Wilcoxon test was applied for statistical analysis, and p < 0.05 was set as the cut‐off criterion.
To explore how the immune landscape differed between the two clusters, we first used the GEO expression data to quantify the infiltration levels of 22 immune cells using the CIBERSORT algorithm for each sample. Then the immune score, stromal score, and the ESTIMATE score were calculated by using the “estimate” package (version 1.0.13). 25 Moreover, we compared the differences in gene expression level between the various clusters for the T cell stimulators and MHCs to confirm the immune characteristics.
The “pRophetic” package (version 0.5) was used to predict chemotherapeutic response by gene expression level in clinical patients. 26 We used an “pRophetic” package to predict the semi‐inhibited concentration of common chemotherapy agents. The effectiveness of these drugs in different clusters was further compared.
2.6. Hub gene selection
Using the “Limma” package (version 3.48.1), DEGs were identified based on OS patients’ expression levels. 27 The DEG selection criteria were as follows: log2FC >0.5, p < 0.05. Using STRING, an online search tool for the PPI networks functional enrichment analysis, 28 a PPI network was constructed with a minimum interaction score of 0.7. Cytoscape software was used for visualizing PPI networks. 29 Two topological algorithms, including Degree and MNC, were used to extract hub genes using a plug‐in called cytoHubba. 30
Subsequently, the LASSO regression model was used to screen out the top 100 core genes in PPI to construct a risk prognosis model: Risk score = Ʃ (Coefi × Expi), 31 where Coefi is the LASSO regression coefficient of the model gene, and Expi is the expression quantity of the corresponding gene. Based on each patient's model gene expression, the risk score for each patient was calculated. The median score was taken as the threshold for separating the high‐risk and low‐risk groups. Furthermore, univariate Cox regression analysis was used to explore the role of model genes in the survival prognosis. At the same time, we used GCSALite, an online Web analysis tool that integrates cancer genome data from The Cancer Genome Atlas and normal tissue data from GTEx, to verify whether the model gene has the same effect in pan‐cancer. 32 Differential analysis of 14 cancers with more than 10 pairs of paired tumor tissue and normal tissue was carried out using the t‐test. The p values were adjusted by FDR; retention significance (FDR <0.05) and FC in genes (FC >2) were retained for graphic visualization. In addition, KM survival plots were plotted using the log‐rank test and retaining p < 0.05 genes in the pan‐cancer survival analyses for 33 cancers.
2.7. Cell culture and qRT‐PCR assay
The human osteoblast (hFOB1.19) and human osteosarcoma cell (HOS) was derived from Fuheng Cell Center (Shanghai Fuheng Cell Center). The HOS were cultured in DMEM (Gibco) presupplemented with 1% penicillin and streptomycin (Gibco) and 10% FBS (Gibco) at 37°C and 5% CO2. hFOB1.19 cells were cultured in hFOB1.19 specific culture medium (Procell) at 33.5°C and 5% CO2.
Total RNA was extracted using the RNA Fast 200 Kit (Feijie Biotechnology). Complementary DNA was produced by reverse transcription of RNA using a cDNA synthesis kit (Takara). On a StepOnePlus Real‐Time PCR System (ABI7500FAST; Applied Biosystems), SYBR Green (Roche) was utilized for qRT‐PCR. After 10 min of PCR at 95°C, 40 cycles of 10 s at 95°C and 1 min at 60°C were carried out. The internal reference gene was GADPH. The 2−ΔΔCt method was used to calculate the relative expression levels of the five model genes in hFOB1.19 and HOS. Table S1 illustrates the qRT‐PCR primer sequences.
2.8. Verification of the model for predictive efficacy
The “pROC” package (version 1.18.0) was used to plot the time‐dependent ROC, and the AUC values for 1, 3, and 5 years were calculated to test the predictive power of the model. 33 We undertook KM survival analyses using log‐rank test for these two groups. In addition, the “rms” package (version 6.3‐0) was used to construct a nomogram for predicting patient outcome based on clinical manifestations. Then the accuracy of the nomogram prediction was evaluated using calibration charts. In addition, the patients’ clinical factors were scored using the “rms” package, visualized by nomogram, and the consistency of the nomogram was evaluated using a calibration plot. Finally, univariable and multivariable Cox regression analyses were used to evaluate whether the risk prognostic model could be used as an independent prognostic factor for OS.
3. RESULTS
3.1. Identification of OCs and CD4 +Tregs
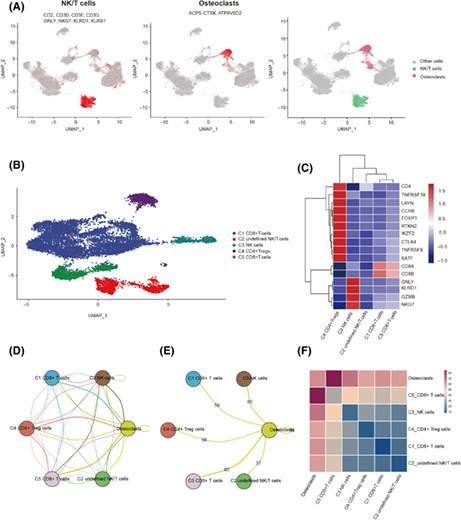
We integrated 17 OS samples to obtain the gene expression profiles of 138,192 cells (Figure S2). We used 2000 genes with the highest relative variability for PCA. Then we removed low‐quality genes, reduced dimensions, and removed batch effects. The OCs and NK/T cells were defined according to the expression of specific marker genes (Figure 1A). Subsequently, NK/T cells were presented by different patients (Figure S3) and a total of five cell clusters including CD4+Tregs were identified (Figure 1B,C). Cell–cell communication analysis was used to obtain IGS between OCs and NK/T cell subsets (Figure 1D). There were 56 ligand receptor pairs between OCs and CD4+Tregs (Figure 1E), which are shown on a heatmap (Figure 1F). We removed the ligand receptor pair that contained the complex and duplicate genes and 25 IGS remained (Table S2).
FIGURE 1.
Cell–cell communication analysis of osteoclasts (OCs) and regulatory CD4+ T cells (CD4+ Tregs). (A) UMAP showing the identification and marker genes of natural killer (NK)/T cells and OCs. Different colors indicate different cells. (B) UMAP showing the NK/T cell subtype. Different colors indicate different cells. (C) Heatmap showing the expression of marker genes in NK/T cell subtypes. The higher the expression, the redder the color. (D) Intercellular communication between OCs and NK/T cell subtypes. The more frequent the cross‐talk between the two cells, the more line segments there are between them. (E) Intercellular communication and number of interaction gene sets between OCs and T cell isoforms. (F) Heatmap showing intercellular communication between OCs and T cell subtypes; the more frequent the cross‐talk between two cells, the redder the color.
3.2. Identification of subtypes in OS
By using consensus clustering analysis, patients in the GSE21257 cohort were divided into two clusters (Figure 2A). Principal component analysis showed that all patients could be roughly divided into two groups, further supporting the presence of two significantly different subtypes (Figure 2B). The KM survival analysis showed that cluster 1 had a better survival prognosis than cluster 2 (log‐rank test, p = 0.038; Figure 2C). We also investigated the connection between clinical characteristics like age, survival status, metastasis, postchemotherapy necrosis rate, and pathological typing. The results showed that patients of cluster 2 accounted for a higher proportion of metastatic patients (χ2‐test, p = 0.0004; Figure 2D). The heatmap showed the expression of IGS in the two clusters (Figure 2E). These results indicated that patients in cluster 2 had higher metastatic outcome and poorer survival prognosis. Therefore, we speculated that OCs and CD4+Tregs might influence tumor development through some underlying mechanism.
FIGURE 2.

Identification of subtypes in osteosarcoma. (A) Two groups of clusters with different expression patterns were obtained. (B) Two clusters were verified by principal component (PC) analysis. Blue represents cluster 1, red represents cluster 2. (C) Kaplan–Meier analysis showing the survival prognosis of two clusters. (D) Sankey diagram showing the connection between clinical characteristics like age, survival status, metastasis, postchemotherapy necrosis rate, and pathological typing and these two clusters. (E) Heatmap showing the expression of interaction gene sets in the two clusters. p < 0.05 was considered statistically significant.
3.3. Tumor microenvironment
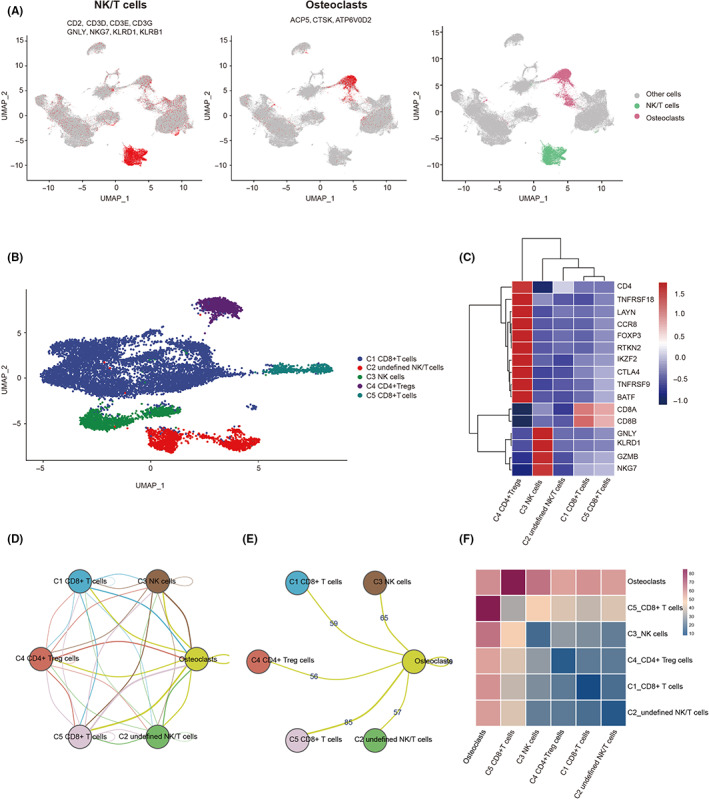
We further investigated the differences in pathway enrichment and tumor immune microenvironment between the two clusters. Results of GSVA showed that cluster 2 was enriched in mediating tumor metastasis, proliferation, angiogenesis, and T cell exhaustion. Cluster 2 enrichment was low in mediating the INF signaling pathway, cell apoptosis, T cell activation response, and immune response to tumor cells (Figure 3A).
FIGURE 3.
Physiological characteristics of the two clusters of osteosarcoma subtypes. (A) Heatmap showing the differences in enrichment in different gene sets between the two clusters. The deeper the red, the higher the enrichment. (B) Violin diagram showing the infiltration of 22 immune cells in two clusters. (C) Scatter plots shows immune score, stromal score, and ESTIMATE score of two clusters. (D) Boxplot showing the expression of human leukocyte antigen genes in two clusters. (E) Boxplot showing the expression of T‐cell stimulating factor genes in two clusters. (F) Heatmap showing 40 statistically significant drugs between cluster 1 and cluster 2. (G) Boxplot showing IC50 values of two clusters against classic osteosarcoma chemotherapy drugs. *p < 0.05, **p < 0.01, ***p < 0.001.
CIBERSORT showed that cluster 1 was characterized by high infiltration of CD8+ T cells, whereas cluster 2 was characterized by high infiltration of M0 type macrophages and neutrophils (Wilcoxon test, p < 0.05; Figure 3B). Furthermore, the ESTIMATE algorithm showed that immune score, stromal score, and ESTIMATE score of cluster 1 were substantially higher than cluster 2 (Wilcoxon test, p < 0.05; Figure 3C). Genes related to T cell stimulators and MHC were highly expressed in cluster 1 (Wilcoxon test, p < 0.05; Figure 3D,E). The above results indicated that TME in cluster 2 had worse immune infiltration. In addition, we used the “pRRophetic” R package to assess the response of each patient to 138 kinds of chemotherapy agents for GSE21257. The IC50 differences between cluster 1 and cluster 2 were compared by Wilcoxon test, and 40 statistically significant drugs were finally obtained (Figure 3F). Among the classic chemotherapy drugs for OS, the IC50 of doxorubicin in cluster 2 was lower (Wilcoxon test, p = 0.009), suggesting that cluster 2 has a better therapeutic effect on doxorubicin. Methotrexate tended to be lower in cluster 1, and cisplatin tended to be more sensitive in cluster 2 pairs (Figure 3G).
3.4. Construction of a risk‐based prognostic model
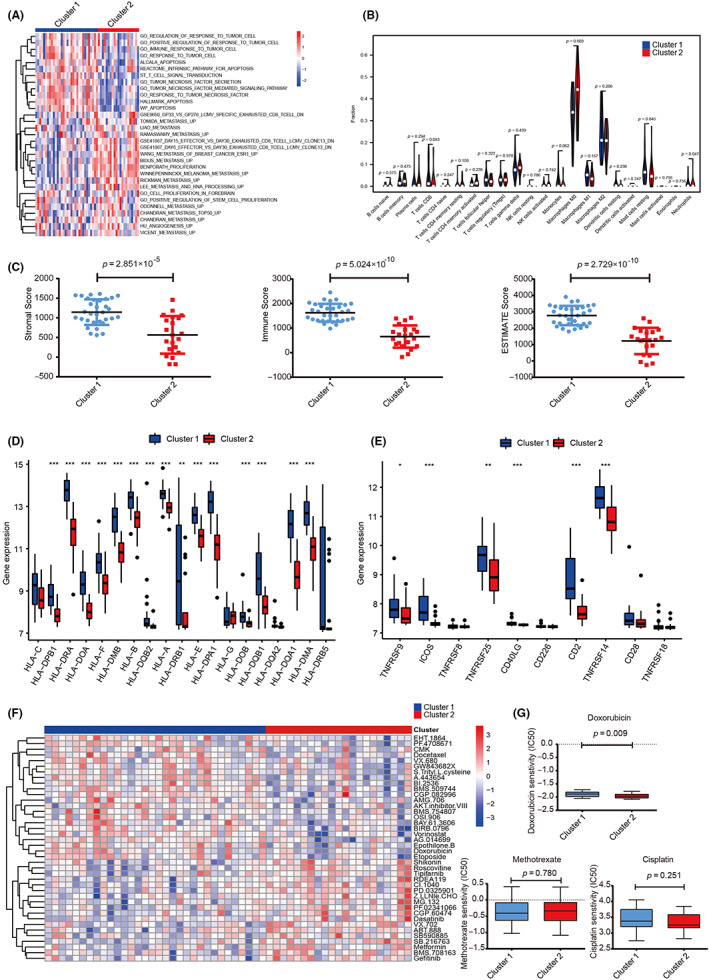
Because of the poor prognosis in cluster 2, it is particularly significant to identify differential genes in cluster 2 and construct accurate prognostic models. Therefore, 330 differential upregulated genes in cluster 2 were selected with p < 0.05 and log2 FC >0.5 (Figure 4A). The PPI network based on cluster 2 differentially upregulated genes was obtained using the STRING database (Figure 4B). We unexpectedly found that the genes obtained by the MNC and DEGREE algorithms were consistent. Thus, these genes can be considered as hub genes (Figure 4C). Next, we used LASSO regression analysis to select the optimal prognostic biomarkers from 100 hub genes. The “glmnet” package was used for 1000 times cross‐validation, and five genes corresponding to the minimum lambda (DDX27, CCT6A, PNN, DTL, and RPS15) were selected to establish the subsequent model (Figure 4D,E).
FIGURE 4.

Hub gene selection and prognostic model construction. (A) Volcano map showing differentially expressed genes in cluster 2, with red dots representing differentially upregulated genes. (B) Protein–protein interaction (PPI) network of cluster 2's differentially upregulated genes. (C) Top 100 core genes in PPI obtained by Degree and MNC topological algorithms. (D, E) LASSO regression analysis selected the optimal prognostic biomarkers from 100 hub genes.
To explore the prediction potential of the model for OS survival, we undertook univariate Cox regression analysis in the GSE21257 dataset. The results showed that genes DDX27 (HR = 2.026, p = 0.008), CCT6A (HR = 2.198, p = 0.007), PNN (HR = 1.744, p = 0.028), and PRS15 (HR = 3.898, p < 0.001) were all injury factors (Figure 5A). We undertook KM survival analysis and found that patients in the high‐risk group had worse survival rate in the GSE21257 dataset (log‐rank test, p = 0.029; Figure 5B). Furthermore, each patient was given a risk score based on the expression level of the model gene. Patients were divided into high‐ and low‐risk groups based on the median risk score. We found that there were more deaths in the high‐risk group (Figure 5C). The results of qRT‐PCR showed that the five model genes were expressed in hFOB1.19 and HOS. DDX27, CCT6A, PNN, and RPS15 were significantly highly expressed in HOS (Figure 5D). We then used the GCSALite online database to analyze the role of model genes in pan‐cancer. Interestingly, the results showed that the model genes were highly expressed in 14 cancer types (t‐test, FC >2, p < 0.05; Figure 5E). In the pan‐cancer analysis, the model genes had a statistically significant effect on prognosis in 16 types of cancer (Figure 5F). These results indicated that the prognostic model based these five genes could effectively predict prognosis.
FIGURE 5.
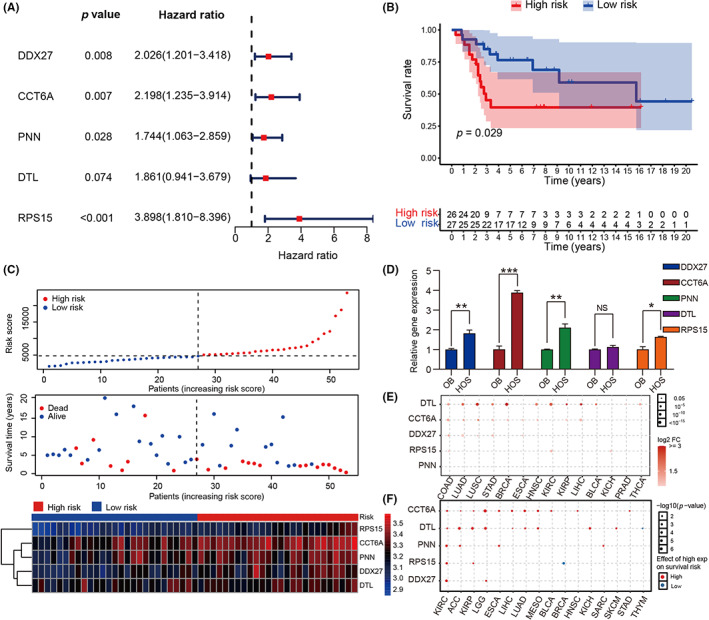
Clinical implications of model genes and prognostic model. (A) Effects of five model genes on survival of patients with osteosarcoma were analyzed by univariate Cox regression risk analysis. (B) Survival analysis between cluster 1 and cluster 2. (C) Top panel, distribution of risk scores. Middle panel, each patient's survival status. Bottom panel, heatmap showing the expression of the five model genes. (D) Quantitative RT‐PCR detected the expression level of the five model genes in hFOB1.19 (OB) and HOS. (E) Expression of the five model genes in pan‐cancer. The deeper the red, the higher the expression in pan‐cancer; the larger the dot, the smaller the p value. (F) Survival analysis of the five model genes in pan‐cancer showing that higher expression level (red) was associated with worse survival. The larger the point, the smaller the p value. *p < 0.05, **p < 0.01, ***p < 0.001. FC, fold change; NS, no statistically significant.
3.5. Validation of model
In order to verify the predictive accuracy of the model, ROC was plotted, and the predicted AUC values were 0.673, 0.810, and 0.762 in 1, 3, and 5 years, respectively (Figure 6A). We constructed a nomogram to quantify clinical factors to predict 1‐, 3‐, and 5‐year survival rates (Figure 6B). We then plotted a calibration plot to evaluate the predictive performance of the nomogram, and the results showed that the working curve predicted by the nomogram was close to the standard curve (Figure 6C). Univariate and multivariate Cox regression analyses showed that the grade and risk prognosis model could be used as independent prognostic factors (Figure 6D,E). The above results indicate that the predictive efficacy of the prognosis model we constructed was robust and powerful, which could accurately stratify patients and predict the prognosis.
FIGURE 6.
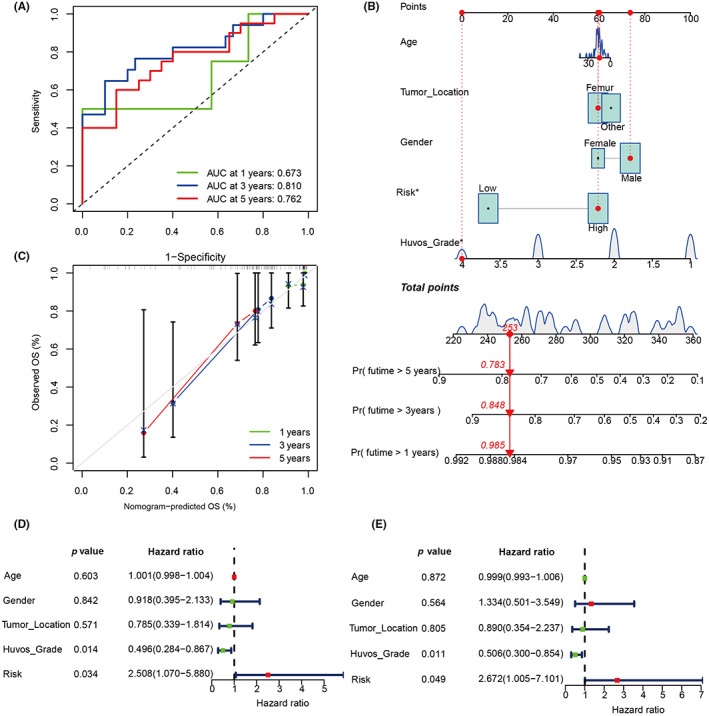
Validation of model to predict the prognosis of patients with osteosarcoma. (A) Area under the receiver operating characteristic curve (AUC) analysis of the prognostic model. (B) Nomogram to predict patient prognosis based on prognostic model and clinical manifestations. (C) A calibration plot evaluating the consistency of the nomogram. OS, overall survival. (D, E) Univariate and multivariate Cox regression analyses of the prognostic model and clinical features.
4. DISCUSSION
Recent research has gradually revealed the important role of TME in the development, metastasis, drug resistance, and other biological behaviors of OS. 7 , 8 , 9 , 10 , 34 However, the effect of intercellular interaction between OCs and CD4+Tregs in the TME on the survival of OS patients remains unknown. Therefore, based on our previous study, this research comprehensively analyzed the potential value of IGS between OCs and CD4+Tregs in predicting prognosis.
The scRNA‐seq technique is the most advanced method to decipher the heterogeneity and complexity of TME. 35 , 36 Bulk RNA‐seq technology has advantages in the measurement of gene expression pattern, subtype expression, and prognosis assessment. 37 Fewer scholars have combined the scRNA‐seq technology and bulk RNA‐seq technology to identify the IGS of intracellular interactions and established clinical prognostic models. 38 In the present study, scRNA‐seq was used to identify OCs and CD4+Tregs, and 56 IGS were obtained. We then used bulk RNA sequencing data to obtain two clusters with different IGS expression patterns based on the GSE21257 queue. Five model genes (DDX27, CCT6A, PNN, DTL, RPS15) related to prognosis were finally obtained by LASSO regression analysis and a risk prognosis model was successfully constructed. ROC analysis results showed that the AUC values in the 1‐, 3‐ and 5‐years were 0.673, 0.810 and 0.762, respectively, suggesting that our model had a good performance in predicting the survival and prognosis. In addition, the calibration plot showed that the working curves of Nomograph predicting the survival rate of 1, 3 and 5 years are all close to the ideal standard curve. In brief, our study constructed a robust model that can accurately predict the outcomes for patients, and is expected to be an effective tool in clinical practice.
In addition, we found that these five model genes were harmful factors in the prognosis by univariate COX regression analysis. The results of qRT‐PCR showed that the five model genes were expressed in hFOB1.19 and HOS. DDX27, CCT6A, PNN, and RPS15 were significantly highly expressed in HOS. Research has shown that overexpression of PNN can enhance the vitality of OS cells, improve their ability of proliferation, adhesion, and invasion, and inhibit apoptosis. 39 Jiang et al. 40 found that CCT6A in Ewing sarcoma is closely related to prognosis. In addition, CCT6A has been shown to have cancer‐promoting effects in a variety of cancers. 41 , 42 DDX27 is a member of the DEAD‐box helicase family. Tang et al. 43 confirmed in vivo and in vitro experiments that DDX27 can promote tumor proliferation, invasion, and metastasis, thus leading to poor prognosis of patients. Research has also shown that DTL can degrade programmed cell death 4 (PDCD4) and promote the proliferation and migration of cancer cells. 44 It has been confirmed that downregulated DTL can inhibit tumor progression. 45 RPS15 is the encoding gene of 40S ribosomal protein S15, which plays an important role in the progression of cancer. 46 The RPS15 mutation makes chronic lymphocytic leukemia more aggressive. 47 In summary, our five model genes are associated with occurrence, development, metastasis, and poor prognosis of cancer.
Osteoclasts are differentiated from mononuclear macrophages and can perform the function of antigen presentation independently. 11 Osteoclasts attract CD8+ T cells and CD4+ T cells around them by secreting T cell chemokines. 11 , 48 , 49 Osteoclasts can process soluble antigens, express MHC, and secrete T cell stimulators, which then induce CD4+ Treg proliferation and activation. 11 , 48 , 50 In addition, OCs form a local immunosuppressive TME by expressing interleukin‐10 in cells involved in inflammation and immunosuppression. 11 CD4+ Tregs are the main immunosuppressive cells in TME and play a key role in tumor immune escape by expressing cosuppressive molecules or secreting immunosuppressive cytokines. 13 , 51 This study found that that CD8+ T cells were highly enriched in cluster 1 patients, and M0 macrophages were highly enriched in cluster 2 patients. CD8+ T cells play a crucial role in directly killing tumor cells and are an important cellular component of the immune system for immune surveillance. 52 In addition, the immune score, stromal score, and ESTIMATE score of cluster 2 were considerably lower than those of cluster 1, indicating a lower degree of immune infiltration in the TME of cluster 2 patients. The MHC is located in the short arm of chromosome 6 and involved in the body's immune surveillance. It is related to adaptive T cell immunity and is an important component of innate immunity. 53 , 54 Our results show that cluster 2 patients had lower expression levels of MHC genes and T‐cell stimulating factor genes. Thus, it is reasonable to speculate that the function and infiltration of immune cells was inhibited by cell interaction between OCs and CD4+ Tregs, which leads to poor prognosis.
Despite the promising findings of our study, there are some limitations. Our findings need to be further validated in cell and animal studies. Additionally, the sample size of this study is insufficient, which could bring bias to the results.
In conclusion, we established an efficient and accurate prognostic model to help clinicians evaluate the prognosis of patients with OS. In addition, our study revealed the effect of cell interactions between OCs and CD4+ Tregs on immune infiltration of osteosarcoma TME and related signaling pathways, which could be beneficial for related experimental studies.
AUTHOR CONTRIBUTIONS
LFC, THJ, FWY, and LY managed and designed the research. LXT, LXD, and LK gathered data. LSY and LJM analyzed a related article and compiled background information. LFC, LSJ, and ZCY analyzed data and wrote articles. ZXL and WQJ organized and assembled pictures.
FUNDING INFORMATION
This work was supported by: the Natural Science Foundation of Guangxi Province (grant number: 2020GXNSFAA259088); Youth Science Foundation of Guangxi Medical University (grant number: GXMUYSF202129 and GXMUYSF202313); Youth Science and Technology Project of the First Affiliated Hospital of Guangxi Medical University (grant number: 201903038); the National Natural Science Foundation of China (grant number: 81960768 and 82260814); and Self‐raised project of Guangxi Zhuang Autonomous Region Health Committee (grant number: Z20200737).
CONFLICT OF INTEREST STATEMENT
The author declares no conflict of interest.
ETHICS STATEMENTS
Approval of the research protocol by an institutional review board: The acquisition of OS samples was approved by the Ethics Committee of the First Affiliated Hospital of Guangxi Medical University (approval number: KY‐E‐097).
Informed consent: The patients/participants were properly informed and provided their written informed consent to participate in this study. Registry and registration no. of the study/trial: N/A.
Animal studies: N/A.
Supporting information
Figures S1–S3
Tables S1–S2
ACKNOWLEDGMENTS
The authors are indebted to those who supported this study's implementation.
Li F, Tang H, Luo X, et al. Interaction gene set between osteoclasts and regulatory CD4+ T cells can accurately predict the prognosis of patients with osteosarcoma. Cancer Sci. 2023;114:3014‐3026. doi: 10.1111/cas.15821
Feicui Li and Haijun Tang contributed equally to this work.
Contributor Information
Wenyu Feng, Email: fengwenyu7381@126.com.
Yun Liu, Email: liuyun200450250@sina.com.
DATA AVAILABILITY STATEMENT
The OS scRNA‐seq data of prior treatment has been uploaded to the GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162454). The OS scRNA‐seq data of posttreatment and bulk RNA sequencing data can be accessed from the GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152048; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE21257).
REFERENCES
- 1. Meltzer PS, Helman LJ. New horizons in the treatment of osteosarcoma. N Engl J Med. 2021;385:2066‐2076. [DOI] [PubMed] [Google Scholar]
- 2. Sheng G, Gao Y, Yang Y, Wu H. Osteosarcoma and metastasis. Front Oncol. 2021;11:780264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kager L, Tamamyan G, Bielack S. Novel insights and therapeutic interventions for pediatric osteosarcoma. Future Oncol. 2017;13:357‐368. [DOI] [PubMed] [Google Scholar]
- 4. Gill J, Gorlick R. Advancing therapy for osteosarcoma. Nat Rev Clin Oncol. 2021;18:609‐624. [DOI] [PubMed] [Google Scholar]
- 5. Chen C, Xie L, Ren T, Huang Y, Xu J, Guo W. Immunotherapy for osteosarcoma: fundamental mechanism, rationale, and recent breakthroughs. Cancer Lett. 2021;500:1‐10. [DOI] [PubMed] [Google Scholar]
- 6. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79:4557‐4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu Y, Feng W, Dai Y, et al. Single‐cell Transcriptomics reveals the complexity of the tumor microenvironment of treatment‐naive osteosarcoma. Front Oncol. 2021;11:709210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corre I, Verrecchia F, Crenn V, Redini F, Trichet V. The osteosarcoma microenvironment: a complex but targetable ecosystem. Cells. 2020;9:1‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou Y, Yang D, Yang Q, et al. Single‐cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat Commun. 2020;11:6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heymann MF, Lézot F, Heymann D. The contribution of immune infiltrates and the local microenvironment in the pathogenesis of osteosarcoma. Cell Immunol. 2019;343:103711. [DOI] [PubMed] [Google Scholar]
- 11. Ibáñez L, Abou‐Ezzi G, Ciucci T, et al. Inflammatory osteoclasts prime TNFα‐producing CD4(+) T cells and express CX(3) CR1. J Bone Mineral Res. 2016;31:1899‐1908. [DOI] [PubMed] [Google Scholar]
- 12. Madel MB, Ibáñez L, Wakkach A, et al. Immune function and diversity of osteoclasts in Normal and pathological conditions. Front Immunol. 2019;10:1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xie M, Wei J, Xu J. Inducers, attractors and modulators of CD4(+) Treg cells in non‐small‐cell lung cancer. Front Immunol. 2020;11:676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buddingh EP, Kuijjer ML, Duim RA, et al. Tumor‐infiltrating macrophages are associated with metastasis suppression in high‐grade osteosarcoma: a rationale for treatment with macrophage activating agents. Clin Cancer Res. 2011;17:2110‐2119. [DOI] [PubMed] [Google Scholar]
- 15. Hao Y, Hao S, Andersen‐Nissen E, et al. Integrated analysis of multimodal single‐cell data. Cell. 2021;184:3573‐3587.e3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single‐cell data with harmony. Nat Methods. 2019;16:1289‐1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim N, Kim HK, Lee K, et al. Single‐cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun. 2020;11:2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Azizi E, Carr AJ, Plitas G, et al. Single‐cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174:1293‐1308.e1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aliprantis AO, Ueki Y, Sulyanto R, et al. NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest. 2008;118:3775‐3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang X, Ge R, Chen H, et al. Follicular helper CD4(+) T cells, follicular regulatory CD4(+) T cells, and inducible Costimulator and their roles in multiple sclerosis and experimental autoimmune encephalomyelitis. Mediators Inflamm. 2021;2021:2058964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Efremova M, Vento‐Tormo M, Teichmann SA, Vento‐Tormo R. CellPhoneDB: inferring cell‐cell communication from combined expression of multi‐subunit ligand‐receptor complexes. Nat Protoc. 2020;15:1484‐1506. [DOI] [PubMed] [Google Scholar]
- 22. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinform. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739‐1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Becht E, Giraldo NA, Lacroix L, et al. Estimating the population abundance of tissue‐infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Geeleher P, Cox N, Huang RS. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PloS One. 2014;9:e107468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szklarczyk D, Gable AL, Nastou KC, et al. The STRING database in 2021: customizable protein‐protein networks, and functional characterization of user‐uploaded gene/measurement sets. Nucleic Acids Res. 2021;49:D605‐D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498‐2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub‐networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tibshirani R. The lasso method for variable selection in the Cox model. Stat Med. 1997;16:385‐395. [DOI] [PubMed] [Google Scholar]
- 32. Liu CJ, Hu FF, Xia MX, Han L, Zhang Q, Guo AY. GSCALite: a web server for gene set cancer analysis. Bioinformatics. 2018;34:3771‐3772. [DOI] [PubMed] [Google Scholar]
- 33. Robin X, Turck N, Hainard A, et al. pROC: an open‐source package for R and S+ to analyze and compare ROC curves. BMC Bioinformat. 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang C, Tian Y, Zhao F, et al. Bone microenvironment and osteosarcoma metastasis. Int J Mol Sci. 2020;21:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jovic D, Liang X, Zeng H, Lin L, Xu F, Luo Y. Single‐cell RNA sequencing technologies and applications: a brief overview. Clin Transl Med. 2022;12:e694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Suvà ML, Tirosh I. Single‐cell RNA sequencing in cancer: lessons learned and emerging challenges. Mol Cell. 2019;75:7‐12. [DOI] [PubMed] [Google Scholar]
- 37. Thind AS, Monga I, Thakur PK, et al. Demystifying emerging bulk RNA‐Seq applications: the application and utility of bioinformatic methodology. Brief Bioinform. 2021;22:1‐16. [DOI] [PubMed] [Google Scholar]
- 38. Chen Z, Yang X, Bi G, et al. Ligand‐receptor interaction atlas within and between tumor cells and T cells in lung adenocarcinoma. Int J Biol Sci. 2020;16:2205‐2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Qin G, Wu X. Hsa_circ_0032463 acts as the tumor promoter in osteosarcoma by regulating the miR‐330‐3p/PNN axis. Int J Mol Med. 2021;47:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jiang J, Liu C, Xu G, et al. CCT6A, a novel prognostic biomarker for Ewing sarcoma. Medicine. 2021;100:e24484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ying Z, Tian H, Li Y, et al. CCT6A suppresses SMAD2 and promotes prometastatic TGF‐β signaling. J Clin Invest. 2017;127:1725‐1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zeng G, Wang J, Huang Y, et al. Overexpressing CCT6A contributes to cancer cell growth by affecting the G1‐to‐S phase transition and predicts a negative prognosis in hepatocellular carcinoma. Onco Targets Ther. 2019;12:10427‐10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang J, Chen H, Wong CC, et al. DEAD‐box helicase 27 promotes colorectal cancer growth and metastasis and predicts poor survival in CRC patients. Oncogene. 2018;37:3006‐3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chang X, Jian L. LncRNA ZFPM2‐AS1 drives the progression of nasopharyngeal carcinoma via modulating the downstream miR‐3612/DTL signaling. Anticancer Drugs. 2022;33:523‐533. [DOI] [PubMed] [Google Scholar]
- 45. Chen YC, Chen IS, Huang GJ, et al. Targeting DTL induces cell cycle arrest and senescence and suppresses cell growth and colony formation through TPX2 inhibition in human hepatocellular carcinoma cells. Onco Targets Ther. 2018;11:1601‐1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Landau DA, Tausch E, Taylor‐Weiner AN, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ntoufa S, Gerousi M, Laidou S, et al. RPS15 mutations rewire RNA translation in chronic lymphocytic leukemia. Blood Adv. 2021;5:2788‐2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kiesel JR, Buchwald ZS, Aurora R. Cross‐presentation by osteoclasts induces FoxP3 in CD8+ T cells. J Immunol. 2009;182:5477‐5487. [DOI] [PubMed] [Google Scholar]
- 49. Grassi F, Manferdini C, Cattini L, et al. T cell suppression by osteoclasts in vitro. J Cell Physiol. 2011;226:982‐990. [DOI] [PubMed] [Google Scholar]
- 50. Li H, Hong S, Qian J, Zheng Y, Yang J, Yi Q. Cross talk between the bone and immune systems: osteoclasts function as antigen‐presenting cells and activate CD4+ and CD8+ T cells. Blood. 2010;116:210‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Granito A, Muratori L, Lalanne C, et al. Hepatocellular carcinoma in viral and autoimmune liver diseases: role of CD4+ CD25+ Foxp3+ regulatory T cells in the immune microenvironment. World J Gastroenterol. 2021;27:2994‐3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. St Paul M, Ohashi PS. The roles of CD8(+) T cell subsets in antitumor immunity. Trends Cell Biol. 2020;30:695‐704. [DOI] [PubMed] [Google Scholar]
- 53. Mellet J, Tshabalala M, Agbedare O, Meyer P, Gray CM, Pepper MS. Human leukocyte antigen (HLA) diversity and clinical applications in South Africa. S Afr Med J. 2019;109:29‐34. [DOI] [PubMed] [Google Scholar]
- 54. Madden K, Chabot‐Richards D. HLA testing in the molecular diagnostic laboratory. Virchows Archiv. 2019;474:139‐147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S3
Tables S1–S2
Data Availability Statement
The OS scRNA‐seq data of prior treatment has been uploaded to the GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162454). The OS scRNA‐seq data of posttreatment and bulk RNA sequencing data can be accessed from the GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152048; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE21257).