

Abstract
Autophagy is a lysosomal degradation system of cytoplasmic components that contributes to cellular homeostasis through the turnover of various biomolecules and organelles, often in a selective manner. Autophagy is closely related to cancer, but its roles in cancer are complicated. It works as either a promoter or suppressor, depending on the stage and type of cancer. In this review, we briefly summarize the basic mechanisms of autophagy and describe the complicated roles of autophagy in cancer. Moreover, we summarize the clinical trials of autophagy inhibitors targeting cancer and the development of more specific autophagy inhibitors for future clinical application.
Keywords: ATG, autophagy, cancer, chloroquine, inhibitor, mTORC1
Autophagy is closely related to cancer, working as either a promoter or suppressor. In this review, we briefly summarize the basic mechanisms of autophagy and describe the complicated roles of autophagy in cancer. Moreover, we summarize the clinical trials of autophagy inhibitors targeting cancer and the development of more specific autophagy inhibitors for future clinical application.
Abbreviations
- AIM
Atg8‐family interacting motif
- ATG
autophagy‐related
- CQ
chloroquine
- ER
endoplasmic reticulum
- FIR
FIP200 interacting region
- HCQ
hydroxychloroquine
- LIR
LC3‐interacting region
- LLPS
liquid–liquid phase separation
- mTORC1
mTOR complex I
- PI3KC3C1
Class III phosphatidylinositol (PI) 3‐kinase complex I
- PI3P
phosphatidylinositol 3‐phosphate
- PPI
protein–protein interaction
- SAR
selective autophagy receptor
1. BASIC MECHANISMS OF AUTOPHAGY
Autophagy is an evolutionarily conserved, intracellular degradation system of cytoplasmic components using lysosomes. Autophagy transports substrates to lysosomes in several ways. The most common type of autophagy is macroautophagy (simply referred to as autophagy), which utilizes de novo‐generated autophagosomes for lysosomal transport. 1 , 2 The most distinguished feature of autophagy is that it degrades almost all components of the cytoplasm, including not only biomolecules, such as proteins, nucleic acids, and lipids, but also all types of organelles and invading microbes. The degradation process of autophagy is often selective, which makes this system a central player in cellular homeostasis. We first briefly summarize the key players in autophagosome formation and selective autophagy.
1.1. Core autophagy machinery
Using budding yeast as a model system, many autophagy‐related (ATG) genes/proteins have been identified as essential for autophagosome formation. They can be divided into six functional groups that are conserved in mammals. 1 The six groups in mammals are as follows: (1) ULK kinase complex consisting of autophagy‐initiating Ser/Thr kinase ULK1 or ULK2, FIP200/RB1CC1, ATG13, and ATG101; (2) Class III phosphatidylinositol 3‐kinase complex I (PI3KC3C1) consisting of VPS34/PIK3C3 (catalytic subunit), PIK3R4, BECN1, ATG14, and NRBF2; (3) lipid scramblase ATG9A; (4) lipid transporting ATG2–WIPI complex; (5) ATG12–ATG5 conjugation system that builds up the ATG12–ATG5–ATG16L1 complex; and (6) the ATG8 (LC3 family and GABARAP family) lipidation system (Figure 1A). Autophagy initiation is mainly regulated by mTOR complex I (mTORC1). In yeast, autophagosome formation proceeds at the pre‐autophagosomal structure, which is organized by liquid–liquid phase separation (LLPS) of the Atg1 complex (yeast counterpart of ULK complex) on the vacuolar membrane. 3 TORC1 inhibits LLPS of the Atg1 complex by directly phosphorylating Atg13 and thereby inhibits autophagy initiation. In mammals, the ULK complex undergoes LLPS to form autophagosome formation sites on the endoplasmic reticulum (ER), which is triggered by calcium transients at the outer surface of the ER membrane. 4 mTORC1 inhibits autophagy by phosphorylating various autophagy‐related factors, including ULK1 and ATG13, 5 but its relationship with the LLPS of the ULK complex remains unknown. 4
FIGURE 1.
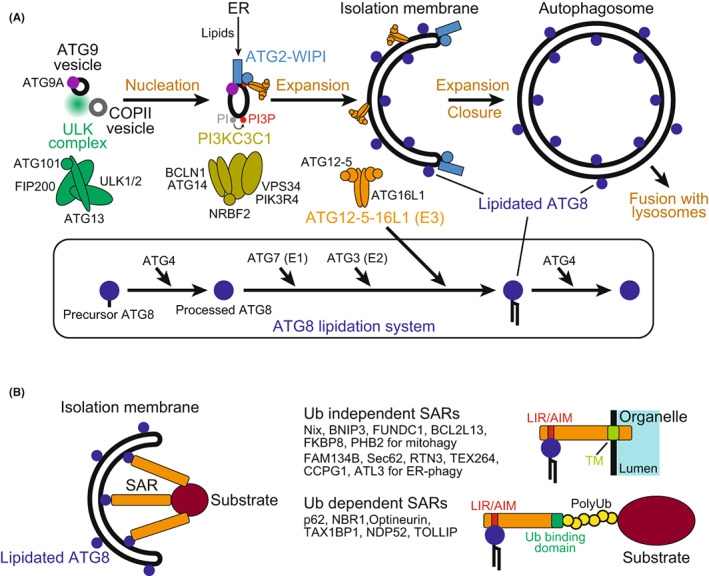
Basic mechanisms of autophagy. (A) Core autophagy machinery mediating autophagosome formation. The ULK complex initiates autophagy by promoting nucleation of isolation membranes. PI3KC3C1 produces PI3P and recruits ATG2–WIPI, which promotes the expansion of isolation membranes by transferring lipids from the endoplasmic reticulum (ER) with the help of ATG9A. ATG12–ATG5–ATG16L1 promotes ATG8 lipidation by functioning as the E3 enzyme, resulting in the decoration of isolation membranes with lipidated ATG8. (B) Basic mechanism of substrate recognition during selective autophagy. Selective substrates are tethered to isolation membranes via lipidated ATG8 and selective autophagy receptors (SARs), promoting their sequestration into autophagosomes. SARs are classified into two types. One is ubiquitin‐independent and directly recognizes substrates (often possessing transmembrane domains in the case of SARs for organelles) and the other is ubiquitin‐dependent and recognizes polyubiquitin chains attached to substrates. Both types possess LC3‐interacting region (LIR)/Atg8‐family interacting motif (AIM) to bind ATG8.
The mechanism of nucleation of isolation membranes (also known as phagophores) has not been clarified, but ATG9A‐containing vesicles and COPII vesicles are believed to be involved in this process. 1 PI3KC3C1 produces phosphatidylinositol 3‐phosphate (PI3P) at the initial autophagic membranes to recruit the ATG2–WIPI complex. 6 ATG2 then mediates the expansion of isolation membranes by transporting phospholipids from the ER with the help of the lipid scrambling activity of ATG9A. 7 Phospholipid synthesis at the ER has been proposed to contribute to this process. 7 , 8 The ATG12 and ATG8 systems are then recruited to function at the later steps of these initial events. 6 ATG8 is conjugated with phosphatidylethanolamine by consecutive enzymatic reactions similar to those in ubiquitination, which involves ATG4 (cysteine protease that processes ATG8 precursors and deconjugates lipidated ATG8), ATG7 (E1 enzyme), ATG3 (E2 enzyme), and the ATG12–ATG5–ATG16L1 complex (E3 enzyme) (Figure 1A). 1 The ATG12−ATG5−Atg16L1 complex is recruited to the autophagosome formation site by PI3P‐bound WIPI2, which is considered to promote lipidation of ATG8 at isolation membranes. 9 Although the molecular functions of lipidated ATG8 in selective autophagy have been established as described below, those in autophagosome formation are still controversial. Despite the essential role of lipidated ATG8s in isolation membrane expansion in yeast, 1 , 10 they are not essential for isolation membrane expansion in mammals and are proposed to function at the sealing of isolation membranes into autophagosomes, fusion of autophagosomes with lysosomes, and degradation of inner autophagosomal membranes upon fusion with lysosomes. 2
1.2. Selective autophagy
The selectivity of autophagy is generally mediated by lipidated ATG8 and selective autophagy receptors (SARs) (Figure 1B). Lipidated ATG8 is the only known protein that decorates the concave surface of isolation membranes, which can function as a scaffold for recognizing selective autophagy substrates. Although some substrates are directly recognized by lipidated ATG8, substrate recognition is usually mediated by SARs, which interact with lipidated ATG8 using LC3‐interacting region (LIR)/Atg8‐family interacting motif (AIM) and, at the same time, interact with the selective substrates, thereby link them to isolation membranes for selective sequestration into autophagosomes. 11 , 12 SARs recognize the substrates either directly or via polyubiquitin chains attached to the substrates. The former type includes organelle‐anchored transmembrane proteins, such as NIX/BNIP3L, BNIP3, and FUNDC1 at the mitochondria and FAM134B/RETREG1, RTN3, and TEX264 at the ER, which tether each organelle to isolation membranes for selective sequestration. 2 The latter type includes p62/SQSTM1, NBR1, optineurin, TAX1BP1, NDP52/CALCOCO2, and TOLLIP, which mediate selective autophagy of various targets decorated with polyubiquitin chains, including protein aggregates, mitochondria, and invasive microbes. 2 Some SARs of the latter type possess FIP200 interacting region (FIR) and recruit the autophagy‐initiating ULK complex, thereby promoting autophagosome formation in the proximity of selective substrates. Recently, p62 was shown to undergo LLPS to form p62 bodies together with polyubiquitin chains, which recruit the ULK complex to promote autophagosome formation to sequester themselves. 13 , 14
2. AUTOPHAGY AND CANCER
Since autophagy is a critical regulator of cellular metabolism, it is expected to be closely related to cancer. Mutations in ATG genes have been reported in human gastric cancers, liver cancers, and neck squamous cell carcinomas, 15 , 16 , 17 confirming the idea that autophagy is directly involved in human cancer. Here we briefly summarize the current knowledge on the various roles of autophagy in different types and stages of cancers. A recent review has discussed the relationship between autophagy and antitumor immunity. 18
2.1. Various roles of autophagy in cancer
The first report on the relationship between autophagy and cancer mentioned that BECN1 expression and autophagy activity were reduced in breast cancer. 19 Consistently, BECN1 heterozygous disruption in mice resulted in autophagy reduction and a high frequency of tumorigenesis. 20 , 21 Following these findings, tumor formation was reported in systemic ATG5 mosaic knockout mice and liver‐specific ATG7 knockout mice. 22 , 23 The findings of these reports helped establish that autophagy has a role in suppressing tumorigenesis.
While somatic loss of autophagy is associated with tumorigenesis, the loss of autophagy suppresses tumor growth in cells expressing oncogenes, such as KRAS mutants and MMTV‐PyMT. 24 , 25 , 26 , 27 , 28 , 29 , 30 Furthermore, even when ATG is deleted from tumors once formed, tumor growth is suppressed. 31 These observations have established the concept that autophagy plays a promotive role in cancer growth after cells become cancerous, possibly by supplying building blocks such as amino acids and nucleosides. In the metastatic stage, the role of autophagy becomes complicated. A transplantable MMTV‐PyMT tumor model enabling tamoxifen‐inducible deletion of ATGs revealed that autophagy has a suppressive role in breast cancer metastasis. 32 In contrast, autophagy was proposed to promote metastasis by enhancing resistance to anoikis. 33 Thus, autophagy has both inhibitory and promotive roles in cancer metastasis.
Although the concept that autophagy, in general, suppresses tumorigenesis and promotes tumor growth has been established, there exist some conflicting reports. Liver tumors formed by ATG5 deficiency are benign. 23 Furthermore, the frequency of ATG gene mutations is not very high in liver cancer. 34 Further studies are required to determine whether mutations in ATG proteins are actually associated with tumorigenesis in human cancer. Furthermore, the role of autophagy in tumor growth promotion may not hold in some contexts. For example, the deletion of ATGs from KRAS‐induced cancer cell lines did not result in a significant growth defect. 35 Furthermore, autophagy promotes tumor growth in pancreatic ductal adenocarcinoma (PDAC) but suppresses it under PTEN deficiency. 29 , 36 In any case, autophagy might have various roles in different types and stages of cancer; therefore, individual studies are required.
2.2. Cancer‐associated regulation of autophagic activity
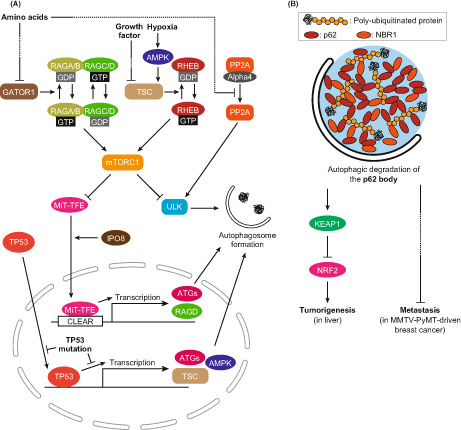
Findings in mouse models suggest that autophagy deficiency is associated with tumorigenesis, while complete deficiency prevents malignant progression. 23 This may reflect the importance of dynamic changes in autophagic activity in actual cancer pathogenesis. In support of this perspective, there are many reports on cancer‐associated regulatory mechanisms for autophagy. The most established regulator of autophagy is mTORC1. mTORC1 directly phosphorylates the ULK complex, which is essential for initiating autophagy (Figure 2A). 5 Besides autophagy, mTORC1 regulates the synthesis of, for instance, proteins, lipids, and nucleotides to promote cell proliferation. 37 mTORC1 is known to be activated in cancer cells; therefore, mTORC1 inhibitors are used as anticancer drugs. mTORC1 is tethered to lysosomes by small GTPases RAGA/B‐RAGC/D and then is allosterically activated by the small GTPase RHEB. 37 GTP‐binding RAGA/B and GDP‐binding RAGC/D form a heterodimer to activate mTORC1. The nucleotide‐binding state of RAGA/B proteins is regulated downstream of amino acid sensors via GATOR1, whereas the nucleotide‐binding state of RHEB is regulated via TSC in the downstream of various cellular environments, such as growth factors and oxygen (Figure 2A). 37 The two groups of GTPases have distinct roles in mTORC1 regulation, and both positive signals are required for mTORC1 activation. mTORC1 plays important roles in the cancer‐associated regulation of autophagic activity.
FIGURE 2.
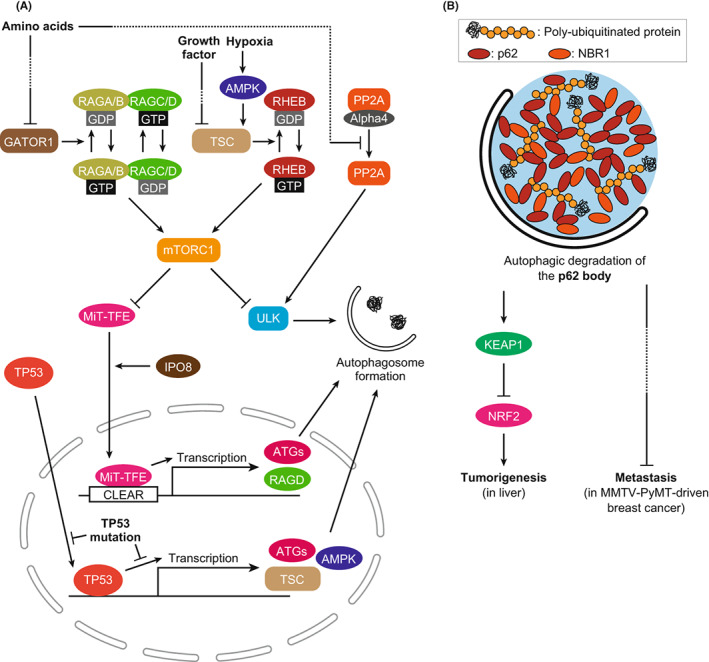
Autophagy and cancer. (A) Cancer‐associated regulation pathways of autophagic activity. The activity of mTORC1 is directly involved in the regulation of autophagy, and its activity is disrupted in many types of cancers. Independent of mTORC1, the PP2A complex regulates autophagy by dephosphorylating the ULK complex. Transcriptional regulation of autophagy by TP53 and MiT‐TFE family proteins is disrupted in cancer cells. (B) Role of autophagic degradation of the p62 body in cancer. The p62 body is an LLPS‐related structure containing polyubiquitinated proteins, p62, and NBR1. In autophagy‐deficient cells, the p62 body accumulates and may trigger cancer pathogenesis. In liver cells, accumulated p62 bodies sequestrate and inactivate KEAP1, leading to tumorigenesis through NRF2 activation. In MMTV‐PyMT–driven breast cancer, accumulated NBR1 in the p62 body induces metastasis via an unknown pathway.
TP53 is one of the best characterized tumor‐suppressor genes, which encodes a transcription factor. Wild‐type TP53 promotes autophagy by regulating transcription of the proteins involving mTORC1 signaling, such as TSC2 and AMPK, as well as autophagy genes such as ULK1, ULK2, and ATG7. 38 Wild‐type TP53 may suppress tumors by activating autophagy in response to stresses received from the environment, such as starvation or DNA damage. However, some cancer‐associated TP53 mutants lose their autophagy‐promoting ability. 38 Since most of the mutations restrict the localization of TP53 in the cytoplasm, autophagy induction by TP53 may be transactivation‐dependent. Consistently, the R175H and R273H mutants have been shown to reduce transactivation of BECN1, ATG12, TSC2, and AMPK. Taken together, tumor‐suppressive autophagy regulation by TP53 is disrupted under cancer‐associated mutations.
In PDAC, MiT‐TFE family transcription factors may regulate autophagy. MiT‐TFE family proteins activate the transcription of genes with CLEAR sequences in their promoters, including ATG and lysosomal genes (Figure 2A). 39 MiT‐TFE family proteins are phosphorylated by mTORC1 and remain in the cytoplasm, and when mTORC1 is inactivated, they translocate to the nucleus. One of the transcription targets of MiT‐TFE family proteins is RAGD, a regulator of mTORC1. 37 MiT‐TFE protein levels are elevated in renal cell carcinoma, melanoma, and PDAC cells, and those cells exhibit RAGD‐mediated mTORC1 activation. 40 Furthermore, in PDAC, MiT‐TFE family proteins accumulate and translocate to the nucleus independent of mTORC1 activity. 41 For the mechanism, it is known that IPO8 induces nuclear translocation of MiT‐TFE family proteins in an mTORC1‐independent manner (Figure 2A). 41 These reports suggest that autophagy is promoted in PDAC by the MiT‐TFE family despite the activation of mTORC1.
Protein phosphatase PP2A/PPP2CA, a tumor suppressor, is also involved in mTORC1‐independent autophagy induction in PDAC. 42 PP2A is responsible for the dephosphorylation of the ULK complex, which is required for autophagy initiation. In PDAC cell lines, the PP2A complex has higher phosphatase activity for the ULK complex. Phosphatase activity of the PP2A complex is suppressed by binding to the inhibitory protein Alpha4/IGBP1. Importantly, upon starvation, the PP2A complex is dissociated from Alpha4 and is activated independently of mTORC1 (Figure 2A). Correlatively, starvation conditions induce autophagy at higher levels than pharmacological mTORC1 inhibition. These observations support the idea that PP2A activates autophagy in an mTORC1‐independent manner.
2.3. Mechanisms for autophagy‐related cancer pathogenesis
We have discussed the various roles and regulations of autophagy in the stages of cancer. More detailed mechanisms for cancer pathogenesis and autophagy have also been extensively studied. p62‐mediated signaling is the best characterized mechanism for autophagy‐related tumorigenesis. 43 p62 forms LLPS‐related structures in the cell through its self‐interactions and binding to polyubiquitin chains, which are called p62 bodies. 44 In autophagy‐deficient cells, p62 is accumulated and phosphorylated. 45 Phosphorylated p62 at S349 binds to the cytoplasmic E3 ubiquitin ligase KEAP1, which is responsible for ubiquitination of the transcription factor NRF2 for proteasomal degradation (Figure 2B). Phosphorylated p62 sequestrates KEAP1 into the p62 body and inactivates it, resulting in the activation of NRF2 by preventing its proteasomal degradation. 22 Activated NRF2 contributes to cell survival and proliferation by promoting the transcription of genes involved in the oxidative stress response and in glucose and glutamine metabolism, leading to tumorigenesis. 46 p62 accumulation is an essential step of this pathway because intracellular ubiquitin‐positive aggregates and the sizes of liver tumors in autophagy‐deficient mice are reduced by the simultaneous deletion of p62. 23 Therefore, autophagy suppresses tumorigenesis via constitutive degradation of p62 in the liver.
Selective autophagy is also thought to be involved in the metastasis phase. NBR1, a homolog of p62, interacts with p62 and polyubiquitin chains and is included in the p62 body. 47 , 48 In a transplantable MMTV‐PyMT‐induced breast cancer model, autophagy deficiency caused NBR1 accumulation that promoted metastasis (Figure 2B). 32 , 49 Consistently, the metastasis promoted by autophagy deficiency was significantly suppressed by the simultaneous deletion of NBR1. 32 However, the mechanism is largely unknown and requires further assessment. Accordingly, degradation of the p62 body by selective autophagy plays an important role in tumorigenesis and metastasis.
As described above, autophagy has a promotive role in tumor growth. One of the reasons is that autophagy provides cells with degradation products, such as amino acids, as nutrients. In general, cancer cells show increased bioenergetic and biosynthetic demand. Furthermore, developed cancer shows hypoxia that is caused by an imbalance between oxygen delivery and oxygen consumption. Autophagy may supply nutrients to cancers by releasing degradation products to the cytoplasm. 50 Consistently, autophagic activity is increased by mTORC1 inhibition during hypoxia (Figure 2A). 51 Furthermore, in autophagy‐deficient, KRAS‐transformed tumor cells, levels of TCA metabolites and mitochondrial respiration are not maintained during starvation. 28 Taken together, cancer cells seem to rely on autophagy for nutrients during the proliferation step. This condition is often referred to as “autophagy addiction.” 28 Notably, it has been reported that autophagy is dispensable for KRAS‐driven tumor growth, suggesting that it depends on the type and stage of cancer. 35
In summary, autophagy changes its role as the cancer progresses. Therefore, pharmacological modulation of autophagic activity at the right time could be a new therapeutic strategy against cancer. Currently, drug development to achieve this is underway at the clinical level, with information provided below.
3. CANCER TREATMENT BY DEVELOPING AUTOPHAGY INHIBITORS
3.1. Current status of the application of autophagy inhibitors in clinical trials
The development of autophagy inhibitors has been an important step in obtaining powerful tools for the mechanistic study of autophagy and for the advancement of novel therapeutic agents targeting autophagy. The discoveries of representative autophagy inhibitors, such as 3‐methyladenine 52 and Wortmannin, 53 a PI3K inhibitors, and Bafilomycin A1, 54 a vacuolar H + ‐ATPases (V‐ATPases) inhibitor, have accumulated a large body of knowledge on the relationship between autophagy and cancer. However, these inhibitors have significant side effects that cannot be ignored, making their application in clinical trials difficult. The only exception is chloroquine (CQ) and its analog hydroxychloroquine (HCQ), which have been used in most current clinical trials (Table 1), although it was reported that the anti‐tumor growth effect of chloroquine might be unrelated to its inhibitory effect on autophagy. 35
TABLE 1.
Phase II clinical trials of autophagy inhibition for cancer therapy
| Conditions | Interventions | Number enrolled | Comment | NCT number | Reference |
|---|---|---|---|---|---|
| Hepatocellular cancer | Sorafenib HCQ | 68 | Response rate increased from 2% to 25%. | NCT03037437 | 62 |
| Non‐small cell lung cancer advanced non‐small cell lung cancer recurrent non‐small cell lung cancer | Paclitaxel Carboplatin HCQ Bevacizumab | 32 | The objective response rate (ORR) and total clinical benefit rate were 47% and 68%, respectively. | NCT01649947 | 61 |
| Colorectal cancer | Vorinostat Regorafenib HCQ | 44 | Showed good tolerability and improved anti‐tumor immunity | NCT02316340 | 58 |
| Rectal cancer colon cancer metastasis adenocarcinoma | HCQ Oxaliplatin Leucovorin 5‐fluorouracil Bevacizumab | 50 | The ORR was 68% with an 11% complete response (CR) rate. | NCT01206530 | 63 |
| Pancreatic cancer | Gemcitabine Abraxane HCQ | 104 | Evans grade histopathologic response was statistically improved (p = 0.00016). | NCT01978184 | 59 |
| Metastatic clear cell renal cell carcinoma | HCQ RAD001 | 40 | Disease control [stable disease + partial response (PR)] occurred in 22 of 33 (67%) evaluable patients. | NCT01510119 | 60 |
| Prostate cancer | HCQ | 64 | Minimal toxicity and some activity in prostate‐specific antigen (PSA) progression after local treatment | NCT00726596 | 64 |
| Carcinoma, intraductal, noninfiltrating DCIS ductal carcinoma in situ | CQ Standard Dose (500 mg/week) CQ Low dose (250 mg/week) Procedure: Breast Biopsy | 12 | PCNA proliferation index of DCIS lesions was reduced compared to untreated controls (p = 0.001). | NCT01023477 | 65 |
| Advanced BRAF mutant melanoma | HCQ Trametinib 2 mg daily dabrafenib 150 mg orally twice a day | 50 | Showed high tolerability and response rate (RR), but failed to meet success criteria for 1‐year PFS rate. | NCT02257424 | 66 |
| Advanced adenocarcinoma metastatic adenocarcinoma | HCQ Abraxane Gemcitabine | 119 | Overall survival at 12 months, the primary endpoint, was not improved. | NCT01506973 | 67 |
| Brain and central nervous system tumors | HCQ TMZ RT | 92 | RT and TMZ combined with HCQ (600 mg/d, dose‐limiting toxicity) did not consistently achieve autophagy inhibition, nor was significant improvement in overall survival observed. | NCT00486603 | 68 |
Abbreviations: CQ, chloroquine; HCQ, hydroxychloroquine; RAD001, Everolimus; RT, Radiation; TMZ, temozolomide.
In the cytoplasm, CQ exists in a nonprotonated state that allows it to diffuse freely across the plasma membrane and organelle membrane, while in acidic compartments, such as lysosomes, it is protonated, significantly inhibiting its diffusion into the cytoplasm. The intralysosomal enrichment of CQ due to its lysosomotropic properties is thought to cause osmotic pressure and membrane damage in lysosomes, which can, in turn, inhibit the degradation of autophagic substrates in lysosomes. 55 , 56 CQ is already being widely used as an antimalarial agent, and its tolerability is known to be high, making its application in clinical trials feasible. 57 CQ and HCQ have shown efficacy against cancers of various tissues in phase II clinical trials (Table 1). Several phase II clinical trials have shown that CQ/HCQ, in combination with conventional chemotherapy, such as sorafenib, FOLFOX, and bevacizumab (Avastin), amplifies anticancer activity. 58 , 59 , 60 , 61 , 62 , 63 In some tumor tissues, such as prostate cancer and ductal carcinoma, CQ/HCQ alone has improved symptoms, indicating the importance of antiautophagy therapy in cancer treatment. 64 , 65 However, for highly malignant tumors, such as advanced BRAF‐mutant melanoma and advanced adenocarcinoma, the enhancement of anticancer activity by HCQ has been reported to be partial. 66 , 67 Furthermore, for brain and central nervous system tumors, autophagy inhibition requires high doses of HCQ, and 600 mg/day of HCQ with no dose‐limiting toxicity likely does not provide sufficient autophagy inhibition. No significant improvement in overall survival has been observed. 68 This raises the challenge of developing molecules that inhibit autophagy more potently and with less toxicity than HCQ in some tumor tissues.
3.2. Development of inhibitors targeting autophagy‐related proteins
The results of clinical trials involving CQ/HCQ have demonstrated the importance of antiautophagy therapy. Since mid‐2010s, several molecules that inhibit autophagy directly with high specificity have been developed, and they target autophagy‐related enzymes such as ULK1/ULK2, VPS34, ATG7, and ATG4A/ATG4B (Table 2). 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 , 83 , 84 In the 2020s, molecules that inhibit the protein–protein interaction (PPI) of autophagy‐related proteins have been developed as highly specific autophagy inhibitors, including SUPR4B1W that targets ATG4B‐LC3 PPI, which was identified by scanning unnatural protease‐resistant mRNA display 85 ; staple peptides that target ATG5‐ATG16L1 PPI, which was designed from the crystal structure of the ATG5–ATG16L1 complex 86 ; and a compound that inhibits BECN1–ATG14 PPI without affecting the Beclin1–UVRAG interaction. 87 The development of these more specific autophagy inhibitors is important for developing more effective and less adverse antitumor drugs. However, they are not yet undergoing clinical trials.
TABLE 2.
Inhibitors targeting autophagy‐related factors.
| Targeted ATG | Compound | IC50 | Reference |
|---|---|---|---|
| ULK1/ULK2 | SBI‐0206965 | 108 nM/711 nM | 69 |
| MRT67307 | 45 nM/38 nM | 70 | |
| MRT68921 | 2.9 nM/1.1 nM | ||
| Compound 6 | 8 nM/− | 71 | |
| ULK‐100 | 1.6 nM/2.6 nM | 72 | |
| ULK‐101 | 8.3 nM/30 nM | ||
| VPS34 | SAR405 | 1.2 nM | 73 |
| VPS34‐IN1 | 25 nM | 74 | |
| PIK‐III | 18 nM | 75 | |
| Compound 31 | 2 nM | 76 | |
| SB02024 | 14 nM | 77 | |
| ATG7 | Compound 18 | 48 nM | 78 |
| Compound 19 | 52 nM | ||
| Compound 37 | 62 nM | ||
| Atg4A/Atg4B | NSC185058 | −/50 μM | 79 |
| Tioconazol | 1.3 μM/1.8 μM | 80 | |
| LV320 | 35.5 μM/24.5 μM | 81 | |
| S130 | 7.11 μM/3.24 μM | 82 | |
| FMK‐9a | −/80 nM | 83 | |
| UAMC‐2526 | −/− | 84 | |
| ATG4B‐LC3 PPI | SUPR4B1W | 120 nM (KD) | 85 |
| ATG5‐ATG16L1 PPI | Peptide 10 | 12 nM (KD) | 86 |
| Beclin1‐ATG14L PPI | Cmpound 19 | 33.9 uM | 87 |
Note: (−): not applicable, not available.
FUNDING INFORMATION
This work was supported in part by JSPS KAKENHI Grant Numbers JP19H05707, JP21H02072 (to N.N.N.), JP22J01742 (to Y.H.), and JP21K06173 (to Y.O.), CREST, Japan Science and Technology Agency Grant Number JPMJCR20E3 (to N.N.N.), and grants from the Takeda Science Foundation (to N.N.N.).
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to declare.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Reviewer Board: N/A.
Informed consent: N/A.
Registry and the registration no. of the study/trial: N/A.
Animal studies: N/A.
ACKNOWLEDGEMENTS
We would like to thank all Noda's laboratory members for helpful discussions.
Hama Y, Ogasawara Y, Noda NN. Autophagy and cancer: Basic mechanisms and inhibitor development. Cancer Sci. 2023;114:2699‐2708. doi: 10.1111/cas.15803
REFERENCES
- 1. Nakatogawa H. Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol. 2020;21:439‐458. [DOI] [PubMed] [Google Scholar]
- 2. Morishita H, Mizushima N. Diverse cellular roles of autophagy. Annu Rev Cell Dev Biol. 2019;35:453‐475. [DOI] [PubMed] [Google Scholar]
- 3. Fujioka Y, Alam JM, Noshiro D, et al. Phase separation organizes the site of autophagosome formation. Nature. 2020;578:301‐305. [DOI] [PubMed] [Google Scholar]
- 4. Zheng Q, Chen Y, Chen D, et al. Calcium transients on the ER surface trigger liquid‐liquid phase separation of FIP200 to specify autophagosome initiation sites. Cell. 2022;185:4082‐4098 e22. [DOI] [PubMed] [Google Scholar]
- 5. Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132‐139. [DOI] [PubMed] [Google Scholar]
- 6. Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Noda NN. Atg2 and Atg9: intermembrane and interleaflet lipid transporters driving autophagy. Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866:158956. [DOI] [PubMed] [Google Scholar]
- 8. Ogasawara Y, Cheng J, Tatematsu T, et al. Long‐term autophagy is sustained by activation of CCTbeta3 on lipid droplets. Nat Commun. 2020;11:4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12‐5‐16L1. Mol Cell. 2014;55:238‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maruyama T, Alam JM, Fukuda T, et al. Membrane perturbation by lipidated Atg8 underlies autophagosome biogenesis. Nat Struct Mol Biol. 2021;28:583‐593. [DOI] [PubMed] [Google Scholar]
- 11. Noda NN, Ohsumi Y, Inagaki F. Atg8‐family interacting motif crucial for selective autophagy. FEBS Lett. 2010;584:1379‐1385. [DOI] [PubMed] [Google Scholar]
- 12. Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol. 2020;432:80‐103. [DOI] [PubMed] [Google Scholar]
- 13. Turco E, Witt M, Abert C, et al. FIP200 claw domain binding to p62 promotes autophagosome formation at ubiquitin condensates. Mol Cell. 2019;74:330‐346 e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kageyama S, Gudmundsson SR, Sou YS, et al. p62/SQSTM1‐droplet serves as a platform for autophagosome formation and anti‐oxidative stress response. Nat Commun. 2021;12:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kang MR, Kim MS, Oh JE, et al. Frameshift mutations of autophagy‐related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol. 2009;217:702‐706. [DOI] [PubMed] [Google Scholar]
- 16. An CH, Kim MS, Yoo NJ, Park SW, Lee SH. Mutational and expressional analyses of ATG5, an autophagy‐related gene, in gastrointestinal cancers. Pathol Res Pract. 2011;207:433‐437. [DOI] [PubMed] [Google Scholar]
- 17. Keulers TG, Koch A, van Gisbergen MW, et al. ATG12 deficiency results in intracellular glutamine depletion, abrogation of tumor hypoxia and a favorable prognosis in cancer. Autophagy. 2022;18:1898‐1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xia H, Green DR, Zou W. Autophagy in tumour immunity and therapy. Nat Rev Cancer. 2021;21:281‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672‐676. [DOI] [PubMed] [Google Scholar]
- 20. Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809‐1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077‐15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Inami Y, Waguri S, Sakamoto A, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takamura A, Komatsu M, Hara T, et al. Autophagy‐deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25:1510‐1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rosenfeldt MT, O'Prey J, Morton JP, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296‐300. [DOI] [PubMed] [Google Scholar]
- 26. Rao S, Tortola L, Perlot T, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. [DOI] [PubMed] [Google Scholar]
- 27. Santanam U, Banach‐Petrosky W, Abate‐Shen C, Shen MM, White E, DiPaola RS. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016;30:399‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo JY, Xia B, White E. Autophagy‐mediated tumor promotion. Cell. 2013;155:1216‐1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo JY, Chen HY, Mathew R, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karsli‐Uzunbas G, Guo JY, Price S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4:914‐927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marsh T, Kenific CM, Suresh D, et al. Autophagic degradation of NBR1 restricts metastatic outgrowth during mammary tumor progression. Dev Cell. 2020;52:591‐604 e596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peng YF, Shi YH, Ding ZB, et al. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy. 2013;9:2056‐2068. [DOI] [PubMed] [Google Scholar]
- 34. Cancer Genome Atlas Research Network . Electronic address wbe, cancer genome atlas research N. comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327‐1341 e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eng CH, Wang Z, Tkach D, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci U S A. 2016;113:182‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rosenfeldt MT, O'Prey J, Flossbach L, et al. PTEN deficiency permits the formation of pancreatic cancer in the absence of autophagy. Cell Death Differ. 2017;24:1303‐1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi Y, Norberg E, Vakifahmetoglu‐Norberg H. Mutant p53 as a regulator and target of autophagy. Front Oncol. 2021;10:607149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. La Spina M, Contreras PS, Rissone A, Meena NK, Jeong E, Martina JA. MiT/TFE family of transcription factors: An evolutionary perspective. Front Cell Dev Biol. 2020;8:609683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Di Malta C, Siciliano D, Calcagni A, et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. 2017;356:1188‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perera RM, Stoykova S, Nicolay BN, et al. Transcriptional control of autophagy‐lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wong PM, Feng Y, Wang J, Shi R, Jiang X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun. 2015;6:8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Katsuragi Y, Ichimura Y, Komatsu M. Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Current Opinion in Toxicology. 2016;1:54‐61. [Google Scholar]
- 44. Noda NN, Wang Z, Zhang H. Liquid‐liquid phase separation in autophagy. J Cell Biol. 2020;219:e202004062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ichimura Y, Waguri S, Sou YS, et al. Phosphorylation of p62 activates the Keap1‐Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618‐631. [DOI] [PubMed] [Google Scholar]
- 46. Robledinos‐Anton N, Fernandez‐Gines R, Manda G, Cuadrado A. Activators and inhibitors of NRF2: a review of their potential for clinical development. Oxid Med Cell Longev. 2019;2019:9372182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kirkin V, Lamark T, Sou YS, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505‐516. [DOI] [PubMed] [Google Scholar]
- 48. Sanchez‐Martin P, Sou YS, Kageyama S, Koike M, Waguri S, Komatsu M. NBR1‐mediated p62‐liquid droplets enhance the Keap1‐Nrf2 system. EMBO Rep. 2020;21:e48902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marsh T, Tolani B, Debnath J. The pleiotropic functions of autophagy in metastasis. J Cell Sci. 2021;134:jcs247056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tang T, Yang ZY, Wang D, et al. The role of lysosomes in cancer development and progression. Cell Biosci. 2020;10:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Daskalaki I, Gkikas I, Tavernarakis N. Hypoxia and selective autophagy in cancer development and therapy. Front Cell Dev Biol. 2018;6:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Seglen PO, Gordon PB. 3‐Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889‐1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Blommaart EF, Krause U, Schellens JP, Vreeling‐Sindelarova H, Meijer AJ. The phosphatidylinositol 3‐kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240‐246. [DOI] [PubMed] [Google Scholar]
- 54. Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H‐4‐II‐E cells. Cell Struct Funct. 1998;23:33‐42. [DOI] [PubMed] [Google Scholar]
- 55. Homewood CA, Warhurst DC, Peters W, Baggaley VC. Lysosomes, pH and the anti‐malarial action of chloroquine. Nature. 1972;235:50‐52. [DOI] [PubMed] [Google Scholar]
- 56. Villamil Giraldo AM, Appelqvist H, Ederth T, Ollinger K. Lysosomotropic agents: impact on lysosomal membrane permeabilization and cell death. Biochem Soc Trans. 2014;42:1460‐1464. [DOI] [PubMed] [Google Scholar]
- 57. Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol. 2009;625:220‐233. [DOI] [PubMed] [Google Scholar]
- 58. Arora SP, Tenner L, Sarantopoulos J, et al. Modulation of autophagy: a phase II study of vorinostat plus hydroxychloroquine versus regorafenib in chemotherapy‐refractory metastatic colorectal cancer (mCRC). Br J Cancer. 2022;127:1153‐1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zeh HJ, Bahary N, Boone BA, et al. A randomized phase II preoperative study of autophagy inhibition with high‐dose Hydroxychloroquine and gemcitabine/nab‐paclitaxel in pancreatic cancer patients. Clin Cancer Res. 2020;26:3126‐3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Haas NB, Appleman LJ, Stein M, et al. Autophagy inhibition to augment mTOR inhibition: a phase I/II trial of Everolimus and Hydroxychloroquine in patients with previously treated renal cell carcinoma. Clin Cancer Res. 2019;25:2080‐2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Malhotra J, Jabbour S, Orlick M, Riedlinger G, Joshi S, Guo JY. Modulation of autophagy with hydroxychloroquine in patients with advanced non‐small cell lung cancer (NSCLC): a phase Ib study. J Clin Oncol. 2018;36:e21138. [Google Scholar]
- 62. Arora SP, Moseley JL, Tenner LL, et al. Phase II study of modulation of sorafenib (SOR)‐induced autophagy using hydroxychloroquine (HCQ) in advanced hepatocellular cancer (HCC): planned interim efficacy and safety analysis. J Clin Oncol. 2021;39:305. [Google Scholar]
- 63. O'Hara MH, Karasic TB, Vasilevskaya I, et al. Phase II trial of the autophagy inhibitor hydroxychloroquine with FOLFOX and bevacizumab in front line treatment of metastatic colorectal cancer. J Clin Oncol. 2017;35:3545. [Google Scholar]
- 64. George MA, Mayer TM, Moore D, Chen CX, White E, DiPaola RS. Autophagic cell death with hydroxychloroquine in patients with hormone‐dependent prostate‐specific antigen progression after local therapy for prostate cancer. J Clin Oncol. 2017;35:102. [Google Scholar]
- 65. Espina VA, Liotta L, Rassulova S, et al. PINC trial: preventing invasive breast neoplasia with chloroquine. Cancer Res. 2017;77:CT140. [Google Scholar]
- 66. Mehnert JM, Mitchell TC, Huang AC, et al. BAMM (BRAF autophagy and MEK inhibition in melanoma): a phase I/II trial of Dabrafenib, Trametinib, and Hydroxychloroquine in advanced BRAFV600‐mutant melanoma. Clin Cancer Res. 2022;28:1098‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Karasic TB, O'Hara MH, Loaiza‐Bonilla A, et al. Effect of gemcitabine and nab‐paclitaxel with or without Hydroxychloroquine on patients with advanced pancreatic cancer: a phase 2 randomized clinical trial. JAMA Oncol. 2019;5:993‐998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rosenfeld MR, Ye XB, Supko JG, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10:1359‐1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Egan DF, Chun MGH, Vamos M, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015;59:285‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Petherick KJ, Conway OJL, Mpamhanga C, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)‐dependent autophagy. J Biol Chem. 2015;290:11376‐11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lazarus MB, Novotny CJ, Shokat KM. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem Biol. 2015;10:257‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Martin KR, Celano SL, Solitro AR, et al. A potent and selective ULK1 inhibitor suppresses autophagy and sensitizes cancer cells to nutrient stress. Iscience. 2018;8:74‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ronan B, Flamand O, Vescovi L, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10:1013‐1019. [DOI] [PubMed] [Google Scholar]
- 74. Bago R, Malik N, Munson MJ, et al. Characterization of IIPS34‐IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3‐phosphate‐binding SGK3 protein kinase is a downstream target of class ill phosphoinositide 3‐kinase. Biochem J. 2014;463:413‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dowdle WE, Nyfeler B, Nagel J, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16:1069‐1079. [DOI] [PubMed] [Google Scholar]
- 76. Pasquier B, El‐Ahmad Y, Filoche‐Romme B, et al. Discovery of (2S)‐8[(3R)‐3‐Methylmorpholin‐4‐yl]‐1‐(3‐methyl‐2‐oxobutyl)‐2‐(trifluoromethyl)‐3,4‐dihydro‐2H‐pyrimido[1,2‐a]pyrimidin‐6‐one: a novel potent and selective inhibitor of Vps34 for the treatment of solid tumors. J Med Chem. 2015;58:376‐400. [DOI] [PubMed] [Google Scholar]
- 77. Noman MZ, Parpal S, Van Moer K, et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti‐PD‐1/PD‐L1 immunotherapy. Sci Adv. 2020;6:eaax7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Huang SC, Adhikari S, Brownell JE, et al. Discovery and optimization of pyrazolopyrimidine sulfamates as ATG7 inhibitors. Bioorgan . Med Chem. 2020;28:115681. [DOI] [PubMed] [Google Scholar]
- 79. Akin D, Wang SK, Habibzadegah‐Tari P, et al. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy. 2014;10:2021‐2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu PF, Tsai KL, Hsu CJ, et al. Drug repurposing screening identifies Tioconazole as an ATG4 inhibitor that suppresses autophagy and sensitizes cancer cells to chemotherapy. Theranostics. 2018;8:830‐845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bosc D, Vezenkov L, Bortnik S, et al. A new quinoline‐based chemical probe inhibits the autophagy‐related cysteine protease ATG4B. Sci Rep‐Uk. 2018;8:11653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fu YY, Hong L, Xu JC, et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy. 2019;15:295‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Qiu ZX, Kuhn B, Aebi J, et al. Discovery of Fluoromethylketone‐based Peptidomimetics as covalent ATG4B (Autophagin‐1) inhibitors. ACS Med Chem Lett. 2016;7:802‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kurdi A, Cleenewerck M, Vangestel C, et al. ATG4B inhibitors with a benzotropolone core structure block autophagy and augment efficiency of chemotherapy in mice. Biochem Pharmacol. 2017;138:150‐162. [DOI] [PubMed] [Google Scholar]
- 85. Gray JP, Uddin MN, Chaudhari R, et al. Directed evolution of cyclic peptides for inhibition of autophagy. Chem Sci. 2021;12:3526‐3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cui J, Ogasawara Y, Kurata I, et al. Targeting the ATG5‐ATG16L1 protein‐protein interaction with a hydrocarbon‐stapled peptide derived from ATG16L1 for autophagy inhibition. J Am Chem Soc. 2022;144:17671‐17679. [DOI] [PubMed] [Google Scholar]
- 87. Pavlinov I, Salkovski M, Aldrich LN. Beclin 1‐ATG14L protein‐protein interaction inhibitor selectively inhibits autophagy through disruption of VPS34 complex I. J Am Chem Soc. 2020;142:8174‐8182. [DOI] [PubMed] [Google Scholar]