

ABSTRACT
Many developmental processes are regulated post-transcriptionally. Such post-transcriptional regulatory mechanisms can now be analyzed by robust single-cell mass spectrometry methods that allow accurate quantification of proteins and their modification in single cells. These methods can enable quantitative exploration of protein synthesis and degradation mechanisms that contribute to developmental cell fate specification. Furthermore, they may support functional analysis of protein conformations and activities in single cells, and thus link protein functions to developmental processes. This Spotlight provides an accessible introduction to single-cell mass spectrometry methods and suggests initial biological questions that are ripe for investigation.
Keywords: Proteomics, Post-transcriptional regulation, Protein degradation, Protein synthesis
Summary: This Spotlight highlights how mass spectrometry analysis of single cells can enable protein quantification and help the identification of their roles in developmental mechanisms.
Introduction
During the early development of many organisms, the genome is not actively transcribed. Thus, early developmental processes are regulated post-transcriptionally based on RNA localization and protein synthesis, transport and degradation. These regulatory processes are essential for normal development, as demonstrated by well-studied mechanisms, such as RNA localization and localized translation (Besse and Ephrussi, 2008; Tomancak et al., 2007). RNA localization and localized transcription, for example, contribute to the formation of the Bicoid protein gradient in fly embryos, and then in turn the Bicoid protein regulates the translation of the caudal mRNA (Dubnau and Struhl, 1996). This example shows how different forms of post-transcriptional regulation (RNA localization, protein synthesis and protein localization) coordinate key events of fly development.
In contrast to this intensively studied example, post-transcriptional regulation involving protein synthesis, localization, modifications and degradation is less well studied in developmental systems because of technological limitations in the comprehensive analysis of proteins in developing embryos. Many embryos are small and highly structured, and this necessitates protein analysis of tiny samples with high spatial and temporal resolution. Traditionally, these analyses have relied on affinity reagents and fluorescent proteins (Levy and Slavov, 2018), which have proven useful for many discoveries, such as characterizing the precision of establishing and reading the Bicoid gradient (Gregor et al., 2007). Yet the limited specificity and throughput of affinity reagents have not allowed comprehensive exploration of post-transcriptional regulation. Such exploration requires sensitive, high-throughput, accurate and specific protein quantification. The aim of this Spotlight is to provide an accessible introduction to the field and highlight opportunities for applying single-cell mass spectrometry (MS) analysis for quantifying post-transcriptional regulatory mechanisms of development.
Accessible single-cell mass-spectrometry
Single-cell protein analysis can be enabled by recent methods for liquid chromatography tandem mass spectrometry (LC-MS/MS) of single cells (Budnik et al., 2018; Schoof et al., 2021; Slavov, 2021a; Zhu et al., 2018). For decades, LC-MS/MS analysis has facilitated quantifying the abundance, and identifying the molecular composition of, intact proteins or, more commonly, peptides produced from protease digestion of the proteins (MacCoss et al., 2023; Zhang et al., 2013). LC-MS/MS involves the separation of the proteins (or peptides) followed by their ionization. The resulting ions are analyzed in MS detectors to precisely measure their mass over charge ratios (m/z). The peptides are then fragmented and the resulting fragment ions are analyzed by tandem MS (denoted as MS2) to measure their m/z. These m/z measurements are then used to determine the amino acid sequence and modifications of the analyzed proteins and peptides (Yates et al., 1995; Zhang et al., 2013). Recent advances in LC-MS/MS have made this method suitable for analyzing proteins from single cells by strategies increasing the efficiency of protein delivery to MS analyzers, improving amino acid sequence identification and increasing throughput (reviewed in Slavov, 2021a). These advances have enabled the quantification of thousands of proteins in individual cells and have motivated the establishment of community guidelines and best practices to facilitate the wider adoption of the technologies (Gatto et al., 2023).
Although multiple MS approaches have been developed for single-cell proteomics, they fall into a few groups, and the methods within each group are very similar. The methods can be classified on a technical basis as using data-dependent acquisition (DDA), data-independent acquisition (DIA), multiplexed DIA (plexDIA) and tandem mass tags (TMT), and these methods have been reviewed by Gatto et al., (2023) and Sinitcyn et al. (2018). Single-cell proteomics methods can also be classified based on the degree of multiplexing (Fig. 1), which is useful as it helps define throughput expectations. In Fig. 1, one axis for multiplexing corresponds to the number of single cells (barcoded by mass tags) that can be analyzed simultaneously. The other axis for multiplexing corresponds to the number of distinct peptides that can be analyzed simultaneously per MS2 scan. Both types of multiplexing can help increase the throughput of protein analysis, and their combination allows for multiplicative gains, as demonstrated with plexDIA (Derks et al., 2023), a method that multiplexes both single cells and peptides (Fig. 1). These methods can be implemented using accessible hardware (Box 1) and community guidelines for best practices (Gatto et al., 2023). In addition to established methods, multiple opportunities for technological advancements exist and motivate ongoing innovations (Slavov, 2021c).
Fig. 1.
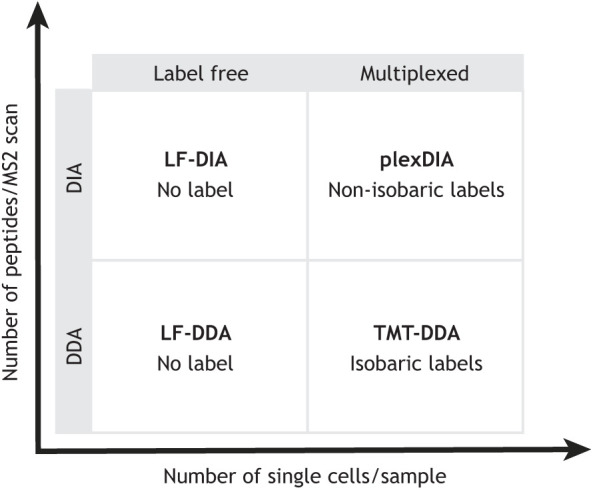
The four major categories of mass spectroscopy methods for protein analysis. All mass spectroscopy (MS) methods fall into one of the four quadrants, defined based on the degree of multiplexing samples (x-axis) and the number of peptides fragmented and analyzed per secondary MS scan (y-axis). Higher degrees of multiplexing favor higher throughput of analysis (Framework for multiplicative scaling of single-cell proteomics, 2023). DDA, data-dependent acquisition (an approach that fragments one peptide sequence at a time); DIA, data-independent acquisition (an approach that fragments multiple peptide sequences at a time); LF, label free; plexDIA, DIA analysis of sample multiplexed with nonisobaric mass tags; TMT, tandem mass tags.
Box 1. Instrumentation required for single-cell mass spectrometry proteomics.
Single-cell protein analysis can be performed using widely available instruments and consumables for both sample preparation and mass spectrometry (MS) analysis. SCoPE2 is an example of a protocol that was optimized to be particularly accessible and to use only common plasticware and MS hardware (Petelski et al., 2021; Specht et al., 2021). SCoPE2 and other methods, such as plexDIA, perform well with widely used and robust chromatography configurations (e.g. commercial chromatographic columns with 75 µm inner diameter) and MS instruments (e.g. timsTOF and orbitrap instruments, even with older instruments, such as Q exactive classic). Although most methods can be implemented with older MS instruments (such as Q exactive classic), newer instruments (such as timsTOF SCP and Exploris 480) generally allow the identification of more peptides and proteins. Other approaches, such as a highly parallel sample preparation by nPOP (Leduc et al., 2022), use more specialized equipment (CellenONE) that is commercially available but may be less accessible due to its cost. Further performance gains can be achieved with custom solutions, such as chromatographic columns with narrow inner diameters that support low flow rates and more efficient peptide delivery (Cong et al., 2020). The existing protocols offer a variety of methods for both sample preparation and MS analysis (Fig. 1), and many of these can be implemented by laboratories experienced in MS analysis of bulk samples.
Quantitative accuracy
The quantitative accuracy of single-cell MS proteomics is fundamentally important for making biological inferences (Franks et al., 2017; Slavov, 2022). For a detailed discussion of accuracy and benchmarks, I refer the reader to Gatto et al. (2023). This section explains how quantification is derived from raw MS data (Fig. 2A) and exemplifies the quantification achieved in a plexDIA experiment (Fig. 2B). As shown in Fig. 2A, the relative levels of a peptide across two single cells can be estimated as the ratios of peptide ions derived from these cells. As multiple estimates can be derived by some methods, such as plexDIA, the consistency of these methods can be used to evaluate the reliability of the quantification. The raw data in Fig. 2A indicate highly consistent estimates and thus support confident quantification. Zooming out from the data for one peptide, Fig. 2B compares relative protein abundance estimates from bulk and single-cell samples. The good correlation between these data suggests that the single-cell protein ratios are consistent with the bulk ratios, and thus supports the feasibility of accurate protein measurements in single human cells.
Fig. 2.

Quantitative accuracy of single-cell proteomics. (A) Raw mass spectrometry (MS) data from Derks et al. (2023) for the quantification of the peptide SSQPLASK from the protein high mobility group AT-hook 1 (HMGA1). The precursor peptide ions from two single cells, one pancreatic ductal adenocarcinoma (PDAC) cell and one U-937 (pro-monocytic model cell line) cell (orange panel), and their corresponding fragment ions (Σ fragments; green panel) are shown. Both the peptide (MS1-level) and the fragment (MS2-level) ions are measured in multiple scans (labeled 1, 2 and 3), and each of these measurements can be used to estimate the abundance of the peptide. Adapted, with permission, from Gatto et al. (2023). (B) Comparison of relative protein levels measured by plexDIA in single cells (y-axis) and in bulk samples composed of 100 cells (x-axis) (Derks et al., 2023). XIC, extracted ion chromatograms.
These examples illustrate that single-cell MS can provide accurate quantification when following established protocols and guidelines (Gatto et al., 2023; Petelski et al., 2021). I recommend that experiments include benchmarks for evaluating the degree to which the potential for quantitative accuracy of single-cell MS has been achieved (Gatto et al., 2023; Petelski et al., 2021).
MS proteomics of developmental systems
Proteins in oocytes are maternally deposited, and thus the proteomes of oocytes, zygotes and early embryos are likely to be mostly independent from the corresponding transcriptomes. This expectation is strongly supported by MS analysis of human oocytes, which indicates little correspondence between the abundances of protein and transcript products from the same gene (Guo et al., 2022; Virant-Klun et al., 2016). Similarly, throughout development and homeostasis, the correlation between the abundance of transcript and protein products of a gene remains weak for many genes (van den Berg et al., 2023; Franks et al., 2017). These observations underscore the importance of analyzing protein dynamics during development, especially as key regulatory processes are likely to involve protein transport, synthesis and degradation.
Without reviewing the field comprehensively, the few examples in this paragraph are chosen to illustrate biological discoveries enabled by protein analysis. For example, X. laevis embryos are a model of development that have been frequently analyzed by MS proteomics and metabolomics, partly because of the large size of oocytes and blastomeres during early development stages. This analysis has involved capillary microsampling of developing cells, and has identified cellular heterogeneity at the 16-cell stage (Lombard-Banek et al., 2016) and small molecules that may influence cell fates (Onjiko et al., 2015). Another recent discovery in X. laevis includes the observation that maternally deposited nuclear proteins, such as transcription factors and RNA polymerases, enter the nucleus sequentially. This timing of entry corresponds to the timing of downstream transcription (Nguyen et al., 2022). Post-transcriptional regulation continues to play major roles at later developmental stages after the zygotic genome activation, as indicated by the low correlations between RNA and protein dynamics in mouse embryos developing from the two-cell stage to the morula stage (Gao et al., 2017). Similarly, protein and RNA dynamics exhibit substantial differences during in vitro stem cell differentiation (van den Berg et al., 2023). Some of these differences are due to the time delay introduced by protein synthesis and degradation, whereas other differences reflect active regulatory processes (van den Berg et al., 2023).
Perspectives
With the recent establishment of robust methods and community standards (Gatto et al., 2023), the stage is set for applications driven by biological questions. Below, I outline a few questions that might be addressed by MS analysis.
Biological questions
What regulatory mechanisms control protein synthesis?
First, MS allows the direct measurement of protein synthesis rates based on pulsing amino acids labeled with stable isotopes and chasing their incorporation in newly synthesized proteins (Doherty et al., 2009). These protein synthesis rates can then be associated with the levels of possible regulators (such as RNA-binding proteins) and, subsequently, these associations can be tested by perturbation experiments. Second, direct measurements of proteins may provide evidence for changes in the stoichiometry of protein complexes, such as the ribosomes. Such changes have been observed in population average measurements of embryonic stem cells (Slavov et al., 2015) and in differentiating single cells (Budnik et al., 2018). Further analysis is needed to test whether these observations reflect causal regulatory mechanisms; MS offers powerful tools for such investigations (Emmott et al., 2019; Petelski and Slavov, 2020).
Which proteins contribute to cell fate specification?
Initial hypotheses related to this question may be generated based on observing differential protein abundance across blastomeres. Subsequently, these differentially abundant proteins can be associated with developmental fates. Such associations can then be tested computationally, based on conditioning on confounders (Slavov, 2022), and experimentally, based on degrading the associated proteins and observing the effects on developmental fates.
Which proteins determine the functional viability of oocytes?
Single-cell proteomics can also help with more-clinical questions, such as identifying, in oocytes, protein markers that are more likely to support successful in vitro fertilization. Such marker identification demands single-cell analysis to globally investigate protein variation associated with fertilization outcomes.
The above questions exemplify feasible types of analysis that can benefit from direct protein quantification in single cells. However, they are not exhaustive. When the awareness of the technical capabilities of single-cell MS technologies increases, developmental biologists will undoubtedly pose many additional questions that may be resolved by these technologies.
Functional measurements
As discussed above, many questions may be addressed based on measuring protein abundances in the single cells of developing embryos. Yet many regulatory processes depend on protein activities, which may not be reflected in the abundance. Such examples include conformational changes, binding interactions and post-translational modifications (PTMs). These additional levels of protein functionality may also be analyzed by single-cell MS, albeit such analysis is more challenging. The easiest approach is to perform a variable search for PTMs (Yates et al., 1995), although such a search is likely to identify few modified peptides in single cells (Orsburn et al., 2022). To increase the probability of quantifying PTMs of interest, one may prioritize their analysis by real-time MS instrument control. Such prioritization was recently demonstrated with the quantification of proteolytically cleaved peptides in primary macrophages (Huffman et al., 2023) and the approach can be expanded to a wide range of PTMs, including phosphorylation and ubiquitylation. This can enable their consistent PTM analysis across single cells while simultaneously increasing the depth and dynamic range of proteome coverage (Huffman et al., 2023).
Another example of functional measurements includes the quantification of protein conformation by footprinting methods, such as covalent protein painting (Bamberger et al., 2021). This approach labels lysine residues exposed on the protein surfaces in living cells. As the labeling is highly efficient, it may be extended to achieve single-cell sensitivity (Slavov, 2021b). Although this has not yet been demonstrated, it exemplifies the type of functional protein measurements that may be achieved by MS analysis of single cells from developing embryos.
Outlook
Single-cell proteomics is still a young field bustling with excitement and method development that promises continued technological improvements. Yet the field also offers robust and accessible methods (Gatto et al., 2023; Petelski et al., 2021) that can be implemented on widely used MS instruments. Such methods are readily deployed to begin answering long-standing questions in developmental biology.
Acknowledgements
I thank Jason Derks for help with preparing Fig. 2B.
Footnotes
Funding
This work was supported by an Allen Distinguished Investigator award through the Paul G. Allen Frontiers Group, by a Seed Networks Award from the Chan Zuckerberg Initiative (CZF2019-002424) and by a R01 award from the National Institute of General Medical Sciences from the National Institutes of Health to N.S. (R01GM144967). Deposited in PMC for release after 12 months.
References
- Bamberger, C., Pankow, S., Martínez-Bartolomé, S., Ma, M., Diedrich, J., Rissman, R. A. and Yates, J. R. (2021). Protein footprinting via covalent protein painting reveals structural changes of the proteome in Alzheimer's disease. J. Proteome Res. 20, 2762-2771. 10.1021/acs.jproteome.0c00912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse, F. and Ephrussi, A. (2008). Translational control of localized mRNAs: restricting protein synthesis in space and time. Nat. Rev. Mol. Cell Biol. 9, 971-980. 10.1038/nrm2548 [DOI] [PubMed] [Google Scholar]
- Budnik, B., Levy, E., Harmange, G. and Slavov, N. (2018). SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 19, 161. 10.1186/s13059-018-1547-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, Y., Liang, Y., Motamedchaboki, K., Huguet, R., Truong, T., Zhao, R., Shen, Y., Lopez-Ferrer, D., Zhu, Y. and Kelly, R. T. (2020). Improved Single-Cell Proteome Coverage Using Narrow-Bore Packed NanoLC Columns and Ultrasensitive Mass Spectrometry. Anal. Chem. 92, 2665-2671. 10.1021/acs.analchem.9b04631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derks, J., Leduc, A., Wallmann, G., Huffman, R. G., Willetts, M., Khan, S., Specht, H., Ralser, M., Demichev, V. and Slavov, N. (2023). Increasing the throughput of sensitive proteomics by plexDIA. Nat. Biotechnol. 41, 50-59. 10.1038/s41587-022-01389-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty, M. K., Hammond, D. E., Clague, M. J., Gaskell, S. J. and Beynon, R. J. (2009). Turnover of the human proteome: determination of protein intracellular stability by dynamic SILAC. J. Proteome Res. 8, 104-112. 10.1021/pr800641v [DOI] [PubMed] [Google Scholar]
- Dubnau, J. and Struhl, G. (1996). RNA recognition and translational regulation by a homeodomain protein. Nature 379, 694-699. 10.1038/379694a0 [DOI] [PubMed] [Google Scholar]
- Emmott, E., Jovanovic, M. and Slavov, N. (2019). Ribosome stoichiometry: from form to function. Trends Biochem. Sci. 44, 95-109. 10.1016/j.tibs.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Framework for multiplicative scaling of single-cell proteomics. (2023). Nat. Biotechnol. 41, 23-24. 10.1038/s41587-022-01411-1 [DOI] [PubMed] [Google Scholar]
- Franks, A., Airoldi, E. and Slavov, N. (2017). Post-transcriptional regulation across human tissues. PLoS Comput. Biol. 13, e1005535. 10.1371/journal.pcbi.1005535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, Y., Liu, X., Tang, B., Li, C., Kou, Z., Li, L., Liu, W., Wu, Y., Kou, X., Li, J.et al. (2017). Protein expression landscape of mouse embryos during pre-implantation development. Cell Rep. 21, 3957-3969. 10.1016/j.celrep.2017.11.111 [DOI] [PubMed] [Google Scholar]
- Gatto, L., Aebersold, R., Cox, J., Demichev, V., Derks, J., Emmott, E., Franks, A. M., Ivanov, A. R., Kelly, R. T., Khoury, L.et al. (2023). Initial recommendations for performing, benchmarking and reporting single-cell proteomics experiments. Nat. Methods 20, 375-386. 10.1038/s41592-023-01785-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor, T., Tank, D. W., Wieschaus, E. F. and Bialek, W. (2007). Probing the limits to positional information. Cell 130, 153-164. 10.1016/j.cell.2007.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y., Cai, L., Liu, X., Ma, L., Zhang, H., Wang, B., Qi, Y., Liu, J., Diao, F., Sha, J.et al. (2022). Single-cell quantitative proteomic analysis of human oocyte maturation revealed high heterogeneity in in vitro-matured oocytes. Mol. Cell. Proteomics 21, 100267. 10.1016/j.mcpro.2022.100267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman, R. G., Leduc, A., Wichmann, C., Di Gioia, M., Borriello, F., Specht, H., Derks, J., Khan, S., Khoury, L., Emmott, E.et al. (2023). Prioritized mass spectrometry increases the depth, sensitivity and data completeness of single-cell proteomics. Nat. Methods 20, 714-722. 10.1038/s41592-023-01830-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc, A., Huffman, R. G., Cantlon, J., Khan, S. and Slavov, N. (2022). Exploring functional protein covariation across single cells using nPOP. Genome Biol. 23, 261. 10.1186/s13059-022-02817-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, E. and Slavov, N. (2018). Single cell protein analysis for systems biology. Essays Biochem. 62, 595-605. 10.1042/EBC20180014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard-Banek, C., Moody, S. A. and Nemes, P. (2016). Single–cell mass spectrometry for discovery proteomics: quantifying translational cell heterogeneity in the 16–cell frog (Xenopus) embryo. Angew. Chem. Weinheim Bergstr. Ger. 128, 2500-2504. 10.1002/ange.201510411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccoss, M. J., Alfaro, J. A., Faivre, D. A., Wu, C. C., Wanunu, M. and Slavov, N. (2023). Sampling the proteome by emerging single-molecule and mass spectrometry methods. Nat. Methods 20, 339-346. 10.1038/s41592-023-01802-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, T., Costa, E. J., Deibert, T., Reyes, J., Keber, F. C., Tomschik, M., Stadlmeier, M., Gupta, M., Kumar, C. K., Cruz, E. R.et al. (2022). Differential nuclear import sets the timing of protein access to the embryonic genome. Nat. Commun. 13, 5887. 10.1038/s41467-022-33429-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onjiko, R. M., Moody, S. A. and Nemes, P. (2015). Single-cell mass spectrometry reveals small molecules that affect cell fates in the 16-cell embryo. Proc. Natl. Acad. Sci. USA 112, 6545-6550. 10.1073/pnas.1423682112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsburn, B. C., Yuan, Y. and Bumpus, N. N. (2022). Insights into protein post-translational modification landscapes of individual human cells by trapped ion mobility time-of-flight mass spectrometry. Nat. Commun. 13, 7246. 10.1038/s41467-022-34919-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petelski, A. A. and Slavov, N. (2020). Analyzing ribosome remodeling in health and disease. Proteomics 20, e2000039. 10.1002/pmic.202000039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petelski, A. A., Emmott, E., Leduc, A., Huffman, R. G., Specht, H., Perlman, D. H. and Slavov, N. (2021). Multiplexed single-cell proteomics using SCoPE2. Nat. Protoc. 16, 5398-5425. 10.1038/s41596-021-00616-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoof, E. M., Furtwängler, B., Üresin, N., Rapin, N., Savickas, S., Gentil, C., Lechman, E., Keller, U. A. D., Dick, J. E. and Porse, B. T. (2021). Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 12, 3341. 10.1038/s41467-021-23667-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinitcyn, P., Rudolph, J. D. and Cox, J. (2018). Computational methods for understanding mass spectrometry–based shotgun proteomics data. Annu. Rev. Biomed. Data Sci. 1, 207-234. 10.1146/annurev-biodatasci-080917-013516 [DOI] [Google Scholar]
- Slavov, N. (2021a). Single-cell protein analysis by mass spectrometry. Curr. Opin. Chem. Biol. 60, 1-9. 10.1016/j.cbpa.2020.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov, N. (2021b). Measuring protein shapes in living cells. J. Proteome Res. 20, 3017. 10.1021/acs.jproteome.1c00376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov, N. (2021c). Driving single cell proteomics forward with innovation. J. Proteome Res. 20, 4915-4918. 10.1021/acs.jproteome.1c00639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov, N. (2022). Learning from natural variation across the proteomes of single cells. PLoS Biol. 20, e3001512. 10.1371/journal.pbio.3001512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov, N., Semrau, S., Airoldi, E., Budnik, B. and Van Oudenaarden, A. (2015). Differential stoichiometry among core ribosomal proteins. Cell Rep. 13, 865-873. 10.1016/j.celrep.2015.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht, H., Emmott, E., Petelski, A. A., Huffman, R. G., Perlman, D. H., Serra, M., Kharchenko, P., Koller, A. and Slavov, N. (2021). Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 22, 50. 10.1186/s13059-021-02267-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomancak, P., Berman, B. P., Beaton, A., Weiszmann, R., Kwan, E., Hartenstein, V., Celniker, S. E. and Rubin, G. M. (2007). Global analysis of patterns of gene expression during Drosophila embryogenesis. Genome Biol. 8, R145. 10.1186/gb-2007-8-7-r145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Berg, P. R., Bérenger-Currias, N. M. L. P., Budnik, B., Slavov, N. and Semrau, S. (2023). Integration of a multi-omics stem cell differentiation dataset using a dynamical model. PLoS Genet. 19, e1010744. 10.1371/journal.pgen.1010744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virant-Klun, I., Leicht, S., Hughes, C. and Krijgsveld, J. (2016). Identification of Maturation-Specific Proteins by Single-Cell Proteomics of Human Oocytes. Mol. Cell. Proteomics 15, 2616-2627. 10.1074/mcp.M115.056887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates, J. R., 3rd, Eng, J. K., Mccormack, A. L. and Schieltz, D. (1995). Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67, 1426-1436. 10.1021/ac00104a020 [DOI] [PubMed] [Google Scholar]
- Zhang, Y., Fonslow, B. R., Shan, B., Baek, M.-C. and Yates, J. R.3rd (2013). Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 113, 2343-2394. 10.1021/cr3003533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y., Clair, G., Chrisler, W. B., Shen, Y., Zhao, R., Shukla, A. K., Moore, R. J., Misra, R. S., Pryhuber, G. S., Smith, R. D.et al. (2018). Proteomic analysis of single mammalian cells enabled by microfluidic nanodroplet sample preparation and ultrasensitive NanoLC-MS. Angew. Chem. Int. Ed Engl. 57, 12370-12374. 10.1002/anie.201802843 [DOI] [PMC free article] [PubMed] [Google Scholar]