

Abstract
Local hypoxia occurs in most solid tumors and is associated with aggressive disease and therapy resistance. Widespread changes in gene expression play a critical role in the biological response to hypoxia. However, most research has focused on hypoxia‐inducible genes as opposed to those that are decreased in hypoxia. We demonstrate that chromatin accessibility is decreased in hypoxia, predominantly at gene promoters and specific pathways are impacted including DNA repair, splicing, and the R‐loop interactome. One of the genes with decreased chromatin accessibility in hypoxia was DDX5, encoding the RNA helicase, DDX5, which showed reduced expression in various cancer cell lines in hypoxic conditions, tumor xenografts, and in patient samples with hypoxic tumors. Most interestingly, we found that when DDX5 is rescued in hypoxia, replication stress and R‐loop levels accumulate further, demonstrating that hypoxia‐mediated repression of DDX5 restricts R‐loop accumulation. Together these data support the hypothesis that a critical part of the biological response to hypoxia is the repression of multiple R‐loop processing factors; however, as shown for DDX5, their role is specific and distinct.
Keywords: ATACseq, DDX5, hypoxia, R‐loops
Regions of hypoxia occur in solid tumors and are associated with cancer progression and therapy resistance. The repression of specific pathways in response to hypoxia like DNA repair and splicing factors was described previously. Here, we find that genes encoding proteins with roles in the R‐loop interactome are repressed in hypoxia and link this to decreased chromatin accessibility. DDX5 expression was decreased under hypoxia in cell lines (cancer and normal), tumor xenografts and hypoxic tumors from cancer patients leading to reduced R‐loop accumulation and replication stress.
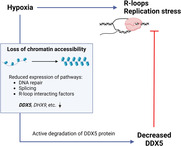
Abbreviations
- ATACseq
assay for transposase‐accessible chromatin with sequencing
- ATCC
American Type Culture Collection
- BNIP3
BCL2 interacting protein 3
- BNIP3L
BCL2 interacting protein 3 like
- CA9
carbonic anhydrase 9
- ChIP‐qPCR
chromatin immunoprecipitation with quantitative real‐time PCR
- CPT
camptothecin
- DAB
3,3′‐diaminobenzidine
- DAPI
4′,6‐diamidino‐2‐phenylindole
- DDX17
DEAD‐box helicase 17
- DDX5
DEAD box protein 5
- DHX9
DExH‐box helicase 9
- DMEM
Dulbecco's Modified Eagle Medium
- DRB
5,6‐dichloro‐1‐β‐d‐ribofuranosylbenzimidazole
- E2F1
E2F transcription factor 1
- EU
5′ethynyluridine
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GO
gene ontology
- H3K27ac
histone 3 lysine 27 acetylation
- H3K27me3
histone 3 lysine 27 trimethylation
- H3K9me3
histone 3 lysine 9 trimethylation
- HIF
hypoxia inducible factor
- IRF3
interferon regulatory factor 3
- IvyGAP
Ivy Glioblastoma Atlas Project
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LDHA
lactate dehydrogenase A
- MCM
minichromosome maintenance complex component
- PHE
public health England
- RPA
replication protein A
- RPMI
Roswell Park Memorial Institute
- SETX
senataxin
- SLC2A1
solute carrier family 2 member 1
- STAT1
signal transducer and activator of transcription 1
- TCGA
The Cancer Genome Atlas
- VEGFA
vascular endothelial growth factor A
1. Introduction
Oxygen is an essential molecule in most biochemical reactions [1]. During the lifetime of a cell, the oxygen concentration can fluctuate and therefore complex mechanisms have evolved to enable the cell to adapt and survive. Hypoxia is associated with pathological conditions such as cancer, stroke, and cardiovascular disease and can lead to cellular damage [2]. In cancer, regions of hypoxia arise when the poorly formed tumor vasculature fails to deliver sufficient oxygen to meet the high metabolic demand of rapidly proliferating cancer cells [3]. The biological response to hypoxia involves a complex transcriptional program including the transactivation of hundreds of genes by the hypoxia inducible factors (HIFs) and the repression of specific processes, for example the DNA repair pathways [4]. Hypoxia switches cellular metabolism from oxidative phosphorylation to aerobic glycolysis, yielding less energy. Therefore, widespread hypoxic‐mediated repression of gene expression has been attributed to reserving cellular energy for vital processes required for cell survival. However, it is also plausible that in specific cases, genes/pathways could be actively repressed as a critical part of the biological response to hypoxia.
R‐loops are three‐stranded nucleic acid structures consisting of an RNA/DNA hybrid and displaced single‐stranded DNA, which originate during transcription [5, 6]. We recently demonstrated that R‐loop levels increase in response to hypoxia (< 0.1% O2) and that the RNA/DNA helicase, Senataxin (SETX), is induced in these conditions [7]. An underlying mechanism for the hypoxia‐mediated accumulation of R‐loops has not been elucidated, and the impact at specific gene loci has not been described. Interestingly, while the SETX helicase was induced in hypoxia, the expression of several other R‐loop‐associated factors (known as the R‐loop interactome) was decreased, suggesting that the loss of these factors could contribute to the overall increase in R‐loop levels [7]. This finding led us to consider that an important biological response to hypoxia is to reduce the expression of R‐loop‐related factors; however, the mechanism behind the global decrease of these factors in hypoxia remains unclear.
We and others have shown that hypoxia leads to widespread epigenetic changes, including a rapid increase in repressive histone methylation marks, for example H3K9me3, which is most likely attributed to impaired function of the oxygen‐dependent histone lysine demethylases [8, 9, 10]. Here, we investigated whether epigenetic changes affecting chromatin accessibility could contribute to the loss of the R‐loop interactome in hypoxic conditions. As both the DNA repair and splicing pathways are repressed in hypoxic conditions and linked to R‐loop formation and resolution, we also assessed whether changes in chromatin accessibility might be responsible for the loss of these pathways [4, 11]. Using the assay for transposase‐accessible chromatin with sequencing (ATAC‐seq), we found a decrease in chromatin accessibility at the promoters of a number of genes encoding RNA processing factors, including R‐loop‐associated helicases. We focused on DDX5, which is an ATP‐dependent DEAD/H‐box RNA helicase, with diverse roles in mRNA splicing, co‐activation of transcription factors, nonsense mediated decay, and resolving R‐loops [12, 13, 14, 15, 16, 17, 18, 19]. We show that restoring DDX5 levels in hypoxia results in further accumulation of replication stress and R‐loops, suggesting that the repression of DDX5 in hypoxia restricts R‐loop accumulation.
2. Materials and methods
2.1. Cell lines and reagents
HCT116, RKO, U87, A549, MCF‐7, MDA‐231, DLD1, H1299, WI‐38, and MRC5 cells (from ATCC, Manassas, VA, USA) were cultured in DMEM with 10% FBS. OE21 and FLO1 cell lines were obtained from PHE culture collections and cultured in RPMI and DMEM, respectively, both supplemented with 10% FBS. RKOHIF‐1α−/− cells were obtained from Dr Denise Chan, UCSF. GL261 mouse glioma cells stably expressing either pEGFP‐N1 or tdTomato (luc+tdT+) were cultured in DMEM with 10% FBS and antibiotics (50 U·mL−1 penicillin, 50 mg·mL−1 streptomycin) [20, 21]. NBE1 cells were cultured in DMEM F12 advanced, glutamax, and 1% FBS while ARPE‐19 cells were cultured in DMEM F12 medium supplemented with 10% FBS (cells from Prof Geoff Higgins, University of Oxford). ReNCell CX, ReNCell VM (from Prof Eric O'Neill, University of Oxford). HCT116 p53+/+ and p53−/− cell lines were a gift from Prof Vogelstein (Johns Hopkins University, Maryland, USA). DharmaFECT 1 transfection reagent (Dharmacon, Lafayette, CO, USA) was used for siRNA transfections according to the manufacturer's instructions; sequences are available in the SI. pSG5‐myc, pSG5‐mycDDX5, and pSG5‐mycDDX5‐NEAD plasmids were a kind gift from Prof Frances Fuller‐Pace (University of Dundee) and DHX9 plasmid from Dr Natalia Gromak (University of Oxford). ppyCAG_RNaseH1_D210N_V5 plasmid was obtained from Addgene, Watertown, MA, USA (#111904). Plasmid transfection was carried out using jetPRIME® (Polyplus, Illkirch, France) transfection reagents according to the manufacturer's protocol.
2.2. Hypoxic conditions and drug treatment
Cells were incubated at < 0.1% O2 in a Bactron chamber (Shel Lab, Cornelius, OR, USA) or at < 0.1–2% O2 in a M35 Hypoxystation (Don Whitley, Shipley, UK) hypoxic chamber in the presence of 5% CO2. The following drugs were used: 25 μg·mL−1 Cycloheximide, 20 μm Emetine, 5 μm MG‐132, 2 μm MLN‐4924, 1 μm Ruxolitinib, 100 μg·mL−1 RNAseA, 1 × 103 U·mL−1 IFNα, 100 μg·mL−1 INFγ, 100 μm 5,6‐dichloro‐1‐β‐d‐ribofuranosylbenzimidazole (DRB), and 10 μm Camptothecin (CPT).
2.3. Western blotting
Cells were washed in PBS and lysed in SDS lysis buffer (10 mm Tris‐Cl, pH 7.5, 0.1 mm EDTA, 0.1 mm EGTA, 0.5% SDS, 0.1 mm β‐mercaptoethanol, protease/phosphatase inhibitors). After blocking in LiCOR blocking buffer, the following primary antibodies were used: DDX5 (Pab204, gift from Prof Frances Fuller‐Pace; clone204, Sigma‐Aldrich, St. Louis, MO, USA), DHX9 (ab26271, Abcam, Cambridge, UK), DDX17 (SQQ‐K14, gift from Prof Frances Fuller‐Pace, Dundee, UK), p53 (DO‐I, Santa Cruz, Dallas, TX, USA), H3K4me3 (07‐473, Millipore, Burlington, MA, USA), H3K9me3 (ab8898, Abcam), H3K27me3 (07‐449, Millipore), H3 total (ab1791, Abcam) β‐actin (AC‐15, Santa Cruz), HIF‐1α (610958, BD Biosciences, San Jose, CA, USA), and GAPDH (6C5, Novus Biologicals, Englewood, CO, USA). IRDye® 680 or IRDye® 800 secondary antibodies were used and the Odyssey infrared system (LI‐COR, Lincoln, NE, USA) to visualize western blots.
2.4. Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation method was adapted from Cook et al. [22], with some modifications. 2 × 106 HCT116 cells were used per each immunoprecipitation (IP) reaction, and 15 units of micrococcal nuclease (MNase, Worthington, Lakewood, NJ, USA) was used to digest the chromatin for the ChIP reaction. Four microgram of antibody was used for each IP reaction: H3 (ab1791, Abcam), H3K27ac (#8173), and normal rabbit IgG (#2729S, Cell Signaling Technology, Danvers, MA, USA) together with 20 μL of prewashed Protein A/G Dynabeads Chromatin digestion was assessed using TapeStation electrophoresis system (Agilent Technologies, Santa Clara, CA, USA). Desired DNA digestion yielded a mono‐ and polynucleosome pattern with a range of DNA fragments 100–1000 bp. Purified input and ChIP samples were subjected to qPCR with Applied Biosystems™ Fast SYBR™ Green Master Mix (4385612, Applied Biosystem, Waltham, MA, USA) according to the manufacturer's instructions using quantstudio (Waltham, MA, USA) 12 K Flex Real‐Time PCR System. Primers were used for DDX5 promoter locus (forward primer: ATGTTCCTTCGTCTGCCTCGA, reverse primer: CTTGCTTTTTGTGTGGGGATT) and DDX5 gene body locus (forward primer: TGAAAACACTGCCTGCATTTT, reverse primer: AATTGCAGAAATGACTGCAGT). Percent of input was determined for total H3, H3K27ac, and IgG control IP samples (2−(sample Ct − adjusted input Ct)). Fold enrichment for H3K27ac and IgG was determined relative to H3. Mean and standard deviation from three biological replicates was plotted.
2.5. 5'EU staining
Cells were incubated with 5'ethynyluridine (EU; 0.5 mm), then fixed in 4% paraformaldehyde for 10 min, and permeabilized in 0.1% Triton‐X 100 for 10 min. Click‐iT Alexa Fluor 647 labeling kit (ThermoFisher, Waltham, MA, USA) was used to measure nascent transcription in the entire nucleus. Images were captured using a LSM710 confocal microscope (Carl Zeiss, Oberkochen, Germany). Image processing and analysis were performed using the image j software (Bethesda, MA, USA). The 5'EU mean nuclear intensity signal was determined using the image j plugin/algorithm, measured as previously [7]. A minimum of 100 cells were included per condition.
2.6. RPA foci staining
Cells transfected with DDX5WT were fixed in 4% paraformaldehyde for 10 min, permeabilized in 0.1% Triton‐X 100 for 10 min, and blocked with 2% BSA in 0.1% Tween‐20 in PBS for 1 h. Cells were then incubated with RPA32 antibody (2208, Cell Signaling) for 2 h at 37 °C followed by Alexa fluor rat 594 secondary antibody (A‐11007, Invitrogen, Waltham, MA, USA) for 1 h at room temperature. Coverslips were mounted using mounting medium Prolong® Gold with DAPI (P36962, Invitrogen). Images were captured using a LSM710 confocal microscope (Carl Zeiss). Image processing and analysis were performed using the image j software.
2.7. R‐loop quantification
Cells transfected with RNase H1D21ON V5 were extracted in ice‐cold PBS/0.2% Triton‐X 100 for 2 min, fixed in 4% paraformaldehyde for 10 min, permeabilized in 0.1% Triton‐X 100 for 10 min, and blocked with 1% BSA /FBS in PBS for 1 h. Cells were then incubated with V5 antibody (R96025, ThermoFisher Scientific) followed by Alexa fluor mouse 488 secondary antibody (A11029, Invitrogen) for 1 h at room temperature. Coverslips were mounted using mounting medium Prolong® Gold with DAPI (P36962, Invitrogen). Images were captured using a LSM710 confocal microscope (Carl Zeiss). Image processing and analysis were performed using the image j software. The V5 mean nuclear intensity signal was determined using the image j plugin/algorithm, measured as previously [7]. A minimum of 100 cells were included per condition.
2.8. Immunohistochemistry and immunofluorescence on xenograft sections
Animal procedures involving HCT116 and OE21 xenografts were performed in accordance with current UK legislation and were approved by the University of Oxford Biomedical Services Ethical Review Committee, Oxford, UK (project license 30/2922). Mouse were housed in individually ventilated cages in groups of five and given food and water ad libitum. Animals were handled in accordance with the Animals (Scientific Procedures) Act 1986 and the University of Oxford ethical guidelines. HCT116 and OE21 cells were grown as xenograft tumors as previously described [23]. Briefly, 6–8‐week‐old female athymic nude mice (BALB/c nude; Charles River, Wilmington, MA, UK) were injected subcutaneously into the flank with 5 × 106 HCT116 cells in serum‐free DMEM. In addition, 6–8‐week‐old female CD‐1 nude mice (Charles River) were injected subcutaneously into the flank with 5 × 106 OE21 cells in 50% (v/v) matrigel and serum‐free RPMI. Two hours before the tumor was harvested, mice were injected intraperitoneally with 60 mg·kg−1 of pimonidazole. Tumors were harvested before reaching 500 mm3 (according to the formula V = W × D × L × π/6). Hypoxic regions were visualized by pimonidazole staining with hypoxyprobe 1 antibody (clone 4.3.11.3, Hypoxyprobe) after dewaxing and antigen retrieval with 10 mm sodium citrate buffer (pH 6.0). In addition, serial sections were stained for DDX5 (pAb204, Millipore), or no primary antibody (negative control), followed by HRP‐conjugated secondary antibody incubation. Staining was developed with 3,3′‐Diaminobenzidine (DAB, Vector Labs, Newark, CA, USA), and nuclei were counterstained with hematoxylin. Images were obtained using an Aperio Scanner (Leica Biosystems, Wetzlar, Germany).
Orthotopic mouse GL261 glioma growth was conducted under the protocol approved by the Local Ethics Committee for Animal Experimentation at the Nencki Institute of Experimental Biology in Warsaw (project lincense 1019/2020). Animals were housed in individually ventilated cages in groups of five and given food and water ad libitum. Male C57BL/6 mice (12 weeks) were anesthetized with 4% isofluorane and maintained at 1.5% isofluorane in oxygen during the tumor implantation procedure. A total of 8 × 104 luc+tdT+ GL261 gLioma cells in 1 μL of DMEM were inoculated into a right striatum at the rate of 0.25 μL per minute using a stereotactic apparatus. The coordinates for injection were +1 mm anterior–posterior, −2 mm lateral, and −3 mm dorsal‐ventral in relation to bregma. After 25 days of postimplantation, animals were intraperitoneally injected with 60 mg·kg−1 of pimonidazole and 2 h later were anesthetized, sacrificed, and perfused with PBS and subsequently with 4% paraformaldehyde (PFA) in PBS. Brains with tumors were then removed and fixed additionally with 4% PFA in PBS for 24 h, followed by immersion in 30% sucrose for 48 h and mounting in OCT freezing medium on dry ice. Coronal sections 10 μm in size were collected using a cryostat. Sections were subjected to an antigen retrieval with 10 mm sodium citrate buffer (pH 6.0) and costained with hypoxyprobe 1 and DDX5 (ab126730, Abcam) antibodies was carried out. Nuclei were counterstained with DAPI. Images were acquired using a Zeiss LSM800 confocal microscope.
2.9. qPCR
RNA samples were prepared using TRIzol (Invitrogen/Life Technologies). cDNA was synthesized from total RNA using the Verso Kit (Thermo Scientific). qPCR was performed with the SYBR Green PCR Master Mix Kit (Applied Biosystems) in a 7500 fast real‐time pcr thermocycler with the v2.0.5 software (Applied Biosystems). Primer sequences are included in the Appendix S1. Data were normalized to 18S housekeeping gene. mRNA fold change was calculated using the 2−ΔΔCt method. The qPCR graphs show the mean ± SEM of three biological replicates, and statistical significance was analyzed with two‐tailed Student's t‐tests for each gene.
2.10. Gene expression correlations in TCGA datasets
RNA‐sequencing data (RNA Seq V2 RSEM) for 382 colorectal adenocarcinoma, 166 glioblastoma, 408 bladder urothelial carcinoma, 1100 breast invasive carcinoma, 501 lung squamous cell carcinomas, and 517 lung adenocarcinomas patient sample datasets were extracted from the TCGA project, which can be accessed through cbioportal (http://www.cbioportal.org/). To examine the correlation of DDX5 against the hypoxia signature, raw data for each sequenced gene were rescaled to set the median equal to 1, and the hypoxia signature was determined by quantifying the median expression of genes from the hypoxic signature [24]. Log10 conversion of the hypoxia signature was plotted against Log10 conversion of raw data for DDX5 (also rescaled to set the median equal to 1).
Correlations and statistical significance were determined by calculating Spearman's rho rank correlation coefficients and two‐tailed P‐values using hmisc package in r studio (Boston, MA, USA).
2.11. IvyGAP human glioblastoma data
Expression of hypoxia‐inducible genes (VEGFA, SLC2A1, CA9, BNIP3, BNIP3L, and LDHA) and candidate genes belonging to the GO Splicing pathway (GO:0008380), whose promoters had decreased ATACseq signal in hypoxic conditions in our GL261 dataset, has been tested in the transcriptional atlas of human glioblastoma, where samples for the RNAseq analysis had been collected via microdissection from anatomic structures within glioblastoma (Ivy Glioblastoma Atlas Project; http://glioblastoma.alleninstitute.org/) [25]. Microvascular Proliferation and Pseudopalisading Cells Around Necrosis regions were taken into account, which should clearly distinguish between oxygenated and hypoxic environments. The presented heatmap based on the expression z‐scores was generated with an online selection tool at http://glioblastoma.alleninstitute.org/.
2.12. ATACseq sequencing and data processing
ATACseq was performed as previously described with some modifications [26]. While still in hypoxic conditions, GL261 cells were fixed with 1% formaldehyde (Thermo Scientific) for 10 min and then quenched with 0.125 m glycine for 5 min at room temperature. After returning to normal conditions (21% O2), 25 000 cells were washed and subsequently lysed in cold lysis buffer (10 mm Tris–HCl, pH 7.4, 10 mm NaCl, 3 mm MgCl2 and 0.1% IGEPAL CA‐630) for 5 min on ice. Cells were then centrifuged at 500 × g for 8 min and cell pellets resuspended in transposition reaction according to the standard ATACseq protocol using 2.5 μL of Tn5 enzyme per 25 000 of cells [27]. After transposition, cells were centrifuged and reverse crosslinked (50 mm Tris‐Cl, 1 mm EDTA, 1% SDS, 0.2 m NaCl, 200 μg·mL−1 proteinase K) at 65 °C with a gentle overnight shaking. Zymo DNA Clean & Concentrator‐5 columns (ZymoResearch, Irvine, CA, USA) were used to purify DNA, and the sequencing libraries were prepared according to the original ATACseq protocol [27]. The ATACseq libraries were assessed for appropriate quality using a Bioanalyzer 2100 and subjected to paired‐end sequencing (2 × 76 bp) using NovaSeq 6000 (Illumina, San Diego, CA, USA) at the Laboratory of Sequencing (Nencki Institute). The quality of raw fastq data was assessed using the fastqc software (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were trimmed using Trimmomatic to remove adapter and transposase sequence with option ‘sliding window 5 : 20’ and with a cutoff from 5′ and 3′ end set to 3 and 15, respectively [28]. Reads with a length of less than 50 bp were discarded. Then, paired‐end reads were aligned to mm10 using bowtie2 aligner v2.2.5 with parameters set to ‐very sensitive and ‐X2000 [29]. Duplicate reads were subsequently removed with picard (http://picard.sourceforge.net), and reads mapping to the mitochondrial genome were also removed. Only properly paired reads with mapping quality score (MAPQ) > 30 were kept for downstream analysis using samtools view with option ‐q 30 [30]. Peaks were called using macs2 v.2.1.1.2 with parameters set to ‐f BAPME ‐q 0.001 –nomodel –shift 0 [30]. The obtained peaks were then filtered for peaks overlapping mm10 ENCODE blacklisted genomic regions. To visualize the overlap of open genomic regions (peaks) from different conditions, the makeVennDiagram function from the chippeakanno package was used with minoverlap parameter set to 200 [31]. All sequencing track Fig.s were generated by IGV using a normalized bigWig file created with deepTools (bamCoverage) and normalized to genome coverage – RPGC [32, 33]. ATACseq data files are available at https://www.ncbi.nlm.nih.gov/geo under accession number GSE200757.
2.13. Differential analysis of ATACseq peaks
Changes in chromatin accessibility were assessed using deseq2 [34]. To create a set of consensuses ATAC peaks for the read count matrix, the following steps were performed. First, peaks for all replicates in each condition were intersected and only peaks with overlap > 200 bp were included. Then, obtained peaks from all conditions were merged with reduction of overlapping regions using the ‘reduce’ function from the genomicranges package to generate consensus peak lists [35]. Peaks with FDR < 0.05 and |log2 fold change| ≥ 0.6 were classified as significantly different. Alternative ATACseq differential accessibility analysis was performed with csaw r package using aforementioned ATAC peak sets with either a trimmed mean of M values (TMM) or nonlinear loss‐based normalization method [36, 37].
2.14. Annotation for differential ATACseq peaks to genomic features
The peaks were annotated to genomic features using ChIPseeker with promoter region defined as ±2000 bp around the transcription start site (TSS) [38]. Each peak was annotated to only one genomic feature according to the default annotation priority in ChIPseeker. To obtain all the genes which had peaks were assigned around their promoter region, a custom‐written script in r was used based on biomart and genomicranges packages [35, 39]. Functional enrichment analysis of differentially accessible regions nearby genes was performed using Gene Ontology, (GO) KEGG and Reactome databases [40, 41, 42].
2.15. Statistical analysis and data processing
Statistical analyses for qPCRs, ChIP‐qPCR, immunofluorescent analysis of DDX5 expression, 5'EU incorporation, RPA foci measurement, and R‐loop quantification were performed using the graphpad prism software (GraphPad Software Inc., San Diego, CA, USA). As indicated in the fig. legends, statistical tests included two‐tailed Student's t‐tests ns, nonsignificant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
3. Results
To investigate the potential role of epigenetic changes in the regulation of the R‐loop interactome in hypoxia, we employed a murine glioblastoma model, GL261, which is known to develop hypoxic areas when grown orthotopically (Fig. S1A) [43]. We assessed the changes in chromatin marks associated with a repressive heterochromatin state in hypoxia. GL261 cells were exposed to either moderate (1% O2) or severe (< 0.1% O2) hypoxia, and changes in chromatin marks were determined. As expected, we observed a marked increase in H3K9 and H3K27 trimethylation [8, 9, 44]. However, we also observed an increase in H3K4 trimethylation, an activating histone modification. In all cases, increased methylation was most pronounced in severe hypoxia (< 0.1% O2) and was reduced to near normoxic levels as quickly as 1 h after reoxygenation, further emphasizing oxygen‐dependent changes in chromatin (Fig. 1A and Fig. S1B,C). In addition, hypoxia (< 0.1% O2) led to the loss of histone acetylation mark H3K27ac, which labels active promoters and enhancers (Fig. S1B,C).
Fig. 1.
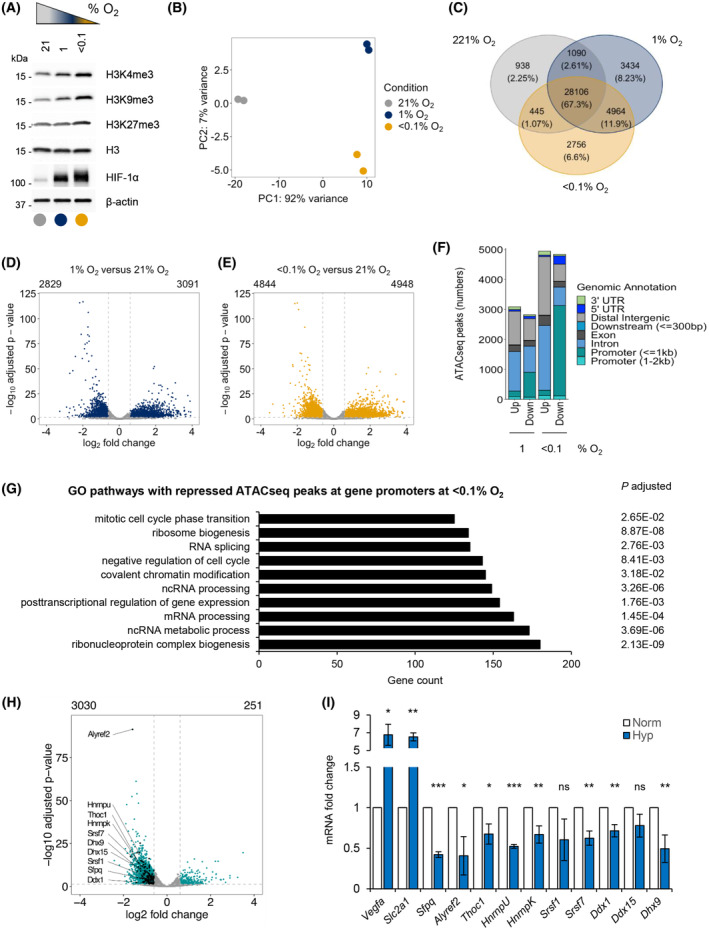
Oxygen‐dependent chromatin alterations lead to a loss of promoter accessibility of numerous pathways, including RNA processing factors. (A) GL261 cell line was exposed to 21%, 1% or < 0.1% O2 for 16 h and subjected to western blotting for the histone modifications indicated. HIF‐1α was used as a hypoxia control. A representative western blot of three biological replicates is shown. (B) GL261 cells were exposed to 16 h of normoxia (21% O2) or two conditions of hypoxia (1% and < 0.1% O2). Samples were fixed and ATACseq carried out. Principal component analysis of ATACseq peaks for the three oxygen tensions is shown. (C) Venn diagram showing hypoxia specific or common ATACseq peaks detected in all oxygen conditions from (B). The percentage of peaks in the total amount of peaks is shown for each condition. (D) A volcano plot showing differentially altered ATACseq peaks in 1% O2 versus 21% O2. Statistically significant peaks with FDR < 0.05 and |log2 fold change| ≥ 0.6 are marked in blue. (E) A volcano plot showing differentially altered ATACseq peaks in < 0.1% O2 versus 21% O2. Statistically significant peaks with FDR < 0.05 and |log2 fold change| ≥ 0.6 are marked in yellow. (F) Differentially regulated ATACseq peaks from parts (D and E) annotated to distinct genomic regions at 1% or < 0.1% O2 in relation to normoxic control. ‘Up’ or ‘Down’ marks significantly increased or decreased peaks, respectively, for each hypoxic condition in relation to normoxia. (G) Top 10 Gene Ontology (GO) pathways (based on the highest gene count) associated with genes that had decreased ATACseq peaks at their promoters. Adjusted P value indicates statistical significance for each pathway shown. (H) A volcano plot showing differentially regulated ATACseq peaks at the gene promoters in cells treated with < 0.1% O2, with significantly up‐regulated 251 peaks and 3030 downregulated peaks. Statistically significant peak changes were annotated with green dots (FDR < 0.05 and |log2 fold change| ≥ 0.6). Black dots mark all significantly downregulated peaks for gene promoters of genes from the GO pathway ‘RNA splicing’ (GO:0008380) and some names of these genes are indicated (full list in Table S4). (I) qPCR validation of hypoxia‐dependent repression of splicing factors indicated in (H) in GL261 cell line treated for 16 h at < 0.1% O2. Mean ± standard deviation expression is shown from three biological repeats. Vegfa and Slc2a1 were used as positive hypoxia‐inducible controls. Statistical significance was calculated with two‐tailed Student's t‐test for each gene expression tested in hypoxia compared to normoxia (*P < 0.05, **P < 0.01, ***P < 0.001, ns, nonsignificant).
Since hypoxia‐mediated increases in methylation marks could have a repressive or activating influence on gene expression, we investigated chromatin accessibility. We carried out ATACseq analysis on GL261 cells exposed to hypoxic (1 or < 0.1% O2) or normoxic (21% O2) conditions. To prevent any reoxygenation‐dependent changes in the chromatin that could occur during sample processing, we employed a modification of ATACseq that allows fixation of cells prior to lysis [26]. Principal component analysis (PCA) showed a clear separation of ATACseq profiles between normoxic and the two hypoxic conditions (Fig. 1B). Overall, we detected 41 733 ATACseq peaks, of which 67.3% were detected in all conditions, 2.25% were specific to normoxia, 8.23% were specific to 1% O2, and 6.6% were specific to < 0.1% O2 conditions (Fig. 1C). We then carried out a differential peak analysis between hypoxic and normoxic conditions and identified a total of 5920 peaks with differential chromatin accessibility at 1% O2 and 9792 peaks with differential chromatin accessibility at < 0.1% O2, in relation to the normoxic control (Fig. 1D,E). Of these, 3091 peaks were significantly increased, and 2829 peaks were significantly decreased at 1% O2, while 4948 peaks were significantly increased, and 4844 peaks were significantly decreased at < 0.1% O2 (Fig. 1D,E and Table S1). Overall, severe hypoxia led to a greater number of significant ATACseq peak changes, which reflects the oxygen‐dependent changes in histone methylation that are known to contribute to the chromatin accessibility (Fig. 1A and Fig. S1B,C) [45, 46]. Next, we carried out genomic annotation of the differentially regulated ATACseq peaks in hypoxia (1 and < 0.1% O2; Fig. 1F and Fig. S1D). One of the most notable differences between the response to moderate (1% O2) and severe (< 0.1% O2) hypoxia was the number of ATACseq peaks lost at promoter regions, with nearly 1000 peaks lost in 1% O2 and over 3000 peaks lost in < 0.1% O2. The oxygen dependency was further emphasized when a ratio between a percentage of decreased peaks at specific genomic annotations in severe versus moderate hypoxia was evaluated (Fig. S1E,F), where again the highest percentage of decreased ATACseq peaks at promoter regions was in response to severe hypoxia compared with moderate hypoxia (Fig. S1F and Table S2). Moreover, the majority of repressed promoter peaks identified in moderate hypoxia (789 of 852) were included in the peaks repressed in severe hypoxia (Fig. S1G). However, severe hypoxia (< 0.1% O2) repressed additional promoters in comparison with moderate hypoxia (1% O2), demonstrating the oxygen dependency of this process. Since severe hypoxia induced the greatest loss of accessible promoters, we performed a functional enrichment analysis of the genes with significantly decreased ATACseq peaks at promoter regions in severe hypoxia using three databases: Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Reactome [40, 41, 42]. We identified several signaling pathways associated with the peaks decreased in severe hypoxia, many of which were related to RNA metabolism and processing, including ribonucleotide complex biogenesis, ncRNA metabolic process, or mRNA processing and splicing. The top 10 significantly downregulated pathways from GO analysis based on the highest gene count associated with each pathway are shown (Fig. 1G, full list of significantly associated pathways Table S3). In order to ensure robust ATACseq results, we employed two additional data normalization approaches, as it was recently shown that choice of analysis can impact differential chromatin accessibility results [37]. Using two alternative normalization pipelines, we found that the impact of hypoxia on the chromatin accessibility remained qualitatively similar as determined with the initial deseq2 analysis. We observed changes in the number of significantly upregulated or downregulated ATACseq peaks between every method used; however, the effect on the repression of accessible promoters in conditions of severe hypoxia, including genes affecting RNA processing and splicing was observed with all methods (Fig. S2).
To validate the results of our ATACseq analysis, we focused on genes involved in splicing, as components of this pathway have previously been shown to be transcriptionally repressed in hypoxia [11]. From the list of the spliceosome genes, we identified a number of splicing factors as having decreased ATACseq peaks in hypoxia (Table S4) and confirmed that 10 of them were repressed at the mRNA level (Fig. 1H,I). This was in contrast to the mRNA of known HIF targets, Slc2a1 or Vegfa, which are induced in response to hypoxia [47]. Moreover, the functional analyses of decreased ATACseq peaks in gene promoter regions led to the identification of other pathways previously reported as decreased in hypoxia, including the DNA repair pathways; base excision repair, homology‐directed repair, double strand break repair, and nonhomologous end‐joining (Reactome analysis, Table S3) [4]. Overall, our data identify a global mechanism of hypoxia‐induced repression of multiple pathways linking previously reported mRNA repression with decreased chromatin accessibility.
Loss of chromatin accessibility at the promoters of genes coding for RNA‐processing factors supports our hypothesis that the repression of genes involved in R‐loops could be mediated by epigenetic changes, as many of these genes have been identified in the R‐loop interactome. The RNA and RNA/DNA helicases Ddx1, Ddx3x, Ddx5, Ddx18, Ddx21, Ddx27, Ddx39b, Dhx9, Dhx15, Srsf1, Xrn2, and Prkdc are all components of the R‐loop interactome and were all found to have reduced chromatin accessibility in their promoters in hypoxia (Fig. S3A and Table S5) [48]. Of these helicases, we have shown previously that DHX9 mRNA expression is repressed in hypoxia (< 0.1% O2) therefore supporting a link between these ATACseq data and gene expression in hypoxia (Fig. S3B) [7]. In contrast, chromatin accessibility did not significantly change at the SETX gene promoter (Fig. S3C), consistent with the recently identified increase in SETX expression in response to hypoxia (< 0.1% O2) [7]. We then asked whether the chromatin accessibility changes in hypoxia detected with ATACseq were mirrored by expression of identified R‐loop and spliceosome interacting genes. We investigated this hypothesis using the transcriptional atlas of human glioblastoma, where RNAseq analysis was performed on samples micro‐dissected from specific regions within the glioblastoma, including well‐oxygenated ‘Microvascular Proliferation’ regions, as well as ‘Pseudopalisading Cells Around Necrosis,’ which are likely to be hypoxic (Ivy Glioblastoma Atlas Project; http://glioblastoma.alleninstitute.org/) [25]. A heatmap generated from the z‐scores of this data showed a high expression of known hypoxia‐inducible genes (VEGFA, SLC2A1, CA9, BNIP3, BNIP3L, and LDHA) in Pseudopalisading Cells Around Necrosis. In contrast, the expression of numerous genes from the GO Splicing pathway (GO:0008380) and the R‐loop interactome, whose promoters had decreased ATACseq signal in hypoxic conditions in our dataset, were downregulated at the mRNA level in human glioblastoma samples, emphasizing the clinical relevance of our ATACseq findings (Fig. 2A). One of the novel hypoxia‐repressed targets identified here in ATACseq analysis and confirmed with the IvyGAP dataset was DDX5, a RNA helicase that was recently reported as a crucial factor in resolving R‐loops (Fig. 2C and Fig. S3D) [15, 16, 17, 18, 19].
Fig. 2.
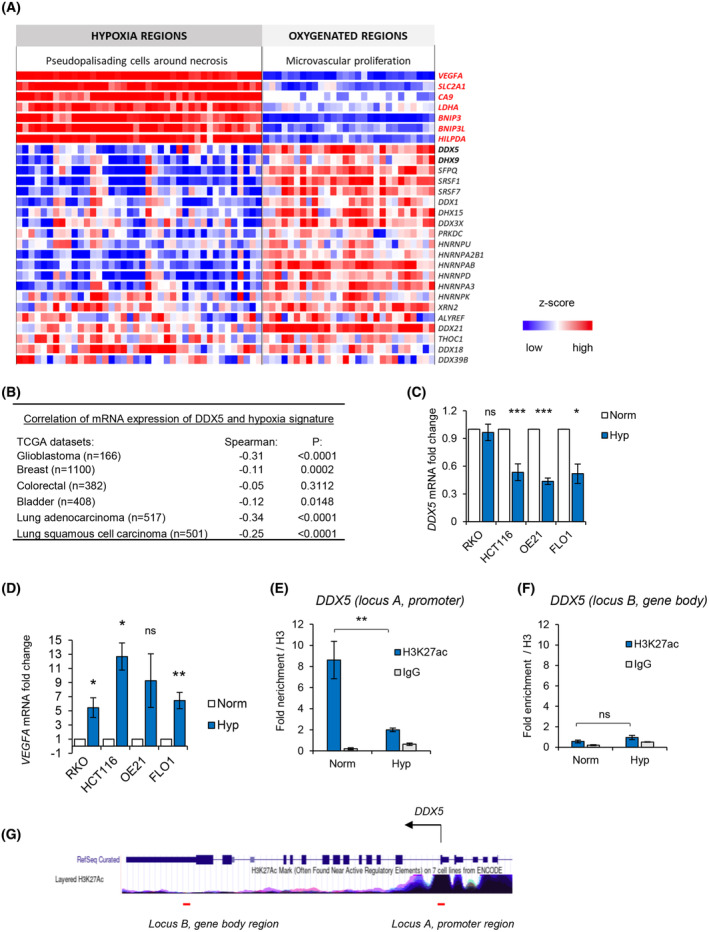
Loss of chromatin accessibility in hypoxia is mirrored by gene expression changes, including the loss of mRNA expression of DDX5. (A) A heatmap showing a z‐score expression of hypoxia markers as well as RNA processing factors in two anatomical regions of glioblastoma, including ‘microvascular proliferation’ region and ‘pseudopalisading cells around necrosis.’ Data was accessed via IvyGAP Glioblastoma project at http://glioblastoma.alleninstitute.org/ and selected genes are shown. Each column represents a separate sample for a given region and rows show expression of particular genes in these samples. (B) Expression of DDX5 (mRNA) was correlated with the hypoxia metagene signature in the indicated TCGA cancer patient cohorts: breast invasive carcinoma, colorectal adenocarcinoma, glioblastoma, bladder urothelial carcinoma, lung adenocarcinoma, and lung squamous cell carcinoma. The numbers of patient samples are shown in brackets. Spearman's rank correlation coefficient and P values are shown for the Log10 median expression of DDX5 and hypoxic signature. (C, D) The indicated cell lines were exposed to hypoxia (< 0.1% O2) for 18 h. Expression of DDX5 (C) and VEGFA (D) was determined by qPCR and normalized to 18S. Statistical significance was calculated with two‐tailed Student's t‐test for each cell line based on three biological replicates (*P < 0.05, **P < 0.01, ***P < 0.001, ns, nonsignificant). (E, F) HCT116 cells were exposed to 16 h of hypoxia (< 0.1% O2) followed by chromatin immunoprecipitation with H3, H3K27ac and IgG antibodies and qPCR analysis. Fold enrichment for H3K27ac and IgG normalized to total H3 enrichment is shown as mean ± standard deviation from three independent experiments. Statistical significance was calculated with two‐tailed Student's t‐test (**P < 0.01, ns, nonsignificant). (G) The DDX5 locus taken from the UCSC genome browser (https://genome.ucsc.edu/) at the human GRCh37/hg19 genome assembly is shown with a track underneath showing the layered H3K27ac profile across the DDX5 gene. Red lines underneath the track indicate the binding site of ChIP‐qPCR primers amplifying the promoter region (locus A) and gene body region (locus B) used in ChIP‐qPCR analysis in (E and F).
To explore potential links between DDX5 and hypoxia in cancer, we asked whether DDX5 expression and a validated hypoxia signature correlated in TCGA cancer patient cohorts (glioblastoma, breast, colorectal, bladder, lung adenocarcinoma, and lung squamous cell carcinoma). We found a significant inverse correlation between DDX5 mRNA and the hypoxic signature in five of the six tested datasets (Fig. 2B and Fig. S4). These findings suggest that DDX5 mRNA expression is repressed in hypoxic tumors. This association was further verified in cancer cell lines using qPCR. In three of the tested cell lines (HCT116, OE21, and FLO1), we observed significant repression of DDX5 mRNA (~ 50% of the normoxic control) in contrast to the HIF‐1 target VEGFA which was induced in hypoxia (Fig. 2C,D). To test whether DDX5 mRNA repression could be directly linked with chromatin accessibility changes at the DDX5 promoter, we carried out ChIP‐qPCR for H3K27ac. H3K27ac, which labels active gene promoters and enhancers, was significantly enriched at the promoter region of DDX5 in normoxic conditions. However, this enrichment was lost in response to hypoxia (Fig. 2E,G). In contrast, H3K27ac was not enriched in the DDX5 gene body distal to the promoter, in normoxic or hypoxic conditions (Fig. 2F,G). Overall, these data support our ATACseq suggesting that DDX5 expression might be repressed at the transcriptional level, via restriction of the accessible chromatin at the promoter of DDX5 gene and loss of active chromatin marks, including H3K27ac.
Next, we asked whether changes in chromatin accessibility and mRNA expression of DDX5 correlated with changes in protein expression. DDX5 protein was significantly decreased in cells exposed to hypoxia (< 0.1% O2) and was quickly reversed when oxygen was restored to normoxic levels reflecting the changes to the histone modification marks during hypoxia and reoxygenation (Fig. 3A and Fig. S1C). DHX9 was also included and found to decrease in response to hypoxia (Fig. 3A). As the HIF‐1 transcription factor is known to regulate chromatin in hypoxic conditions, we further investigated the oxygen‐dependency of DDX5 repression and possible HIF involvement, using a range of hypoxic conditions (< 0.1 or 2% O2) as well as the hypoxia mimetics CoCl2 and DFO [49]. Exposure to 2% O2, CoCl2 or DFO, had little or no effect on DDX5 expression, indicating that the repression of DDX5 is unlikely to be dependent on HIF‐mediated signaling (Fig. S5A,B). To confirm this hypothesis, RKO cells lacking HIF‐1α were exposed to hypoxia and the levels of DDX5 determined compared with the control cell line. As expected, DDX5 levels decreased in both cell lines demonstrating that this occurs independently of HIF‐1α stabilization (Fig. 3B,C). As DDX5 expression has not previously been linked to the response to hypoxia, we investigated its expression in a panel of human cell lines. In all the tested cancer cell lines, including lung, esophageal, colorectal, bladder, bone, and glioma cells, DDX5 protein was repressed in response to hypoxia (< 0.1% O2; Fig. 3D–I and Fig. S5C–E). Notably, a similar change occurred in noncancer MRC‐5 cells, indicating that the hypoxia‐mediated repression of DDX5 is not restricted to cancer cells (Fig. S5F).
Fig. 3.
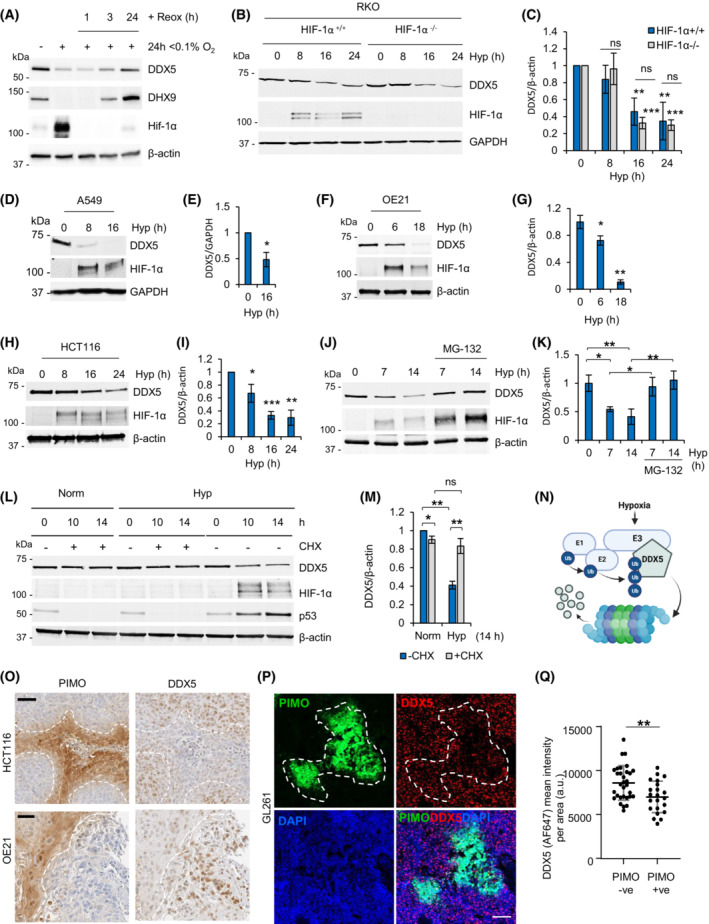
Hypoxia leads to the active repression of DDX5 protein. (A) GL261 cells were exposed to hypoxia (Hyp, 0.1% O2) for 24 h followed by reoxygenation (to 21% O2) for the indicated times following hypoxia. Western blotting was carried out for the antibodies shown (representative of three independent experiments). (B, C) RKOHIF‐1α+/+ and RKOHIF‐1α−/− were exposed to hypoxia (< 0.1% O2) for the times indicated and subjected to western blotting with the antibodies shown (representative of at least three biological replicates). Densitometry is shown in part (C). (D–I) A549, OE21, and HCT116 cell lines were exposed to hypoxia (Hyp, 0.1% O2) for the times indicated followed by western blotting for DDX5 as well as HIF‐1α (hypoxic marker) and β‐actin (loading control). Representative blots from at least three independent experiments are shown. Densitometry for the biological replicates showing hypoxia‐dependent repression of DDX5 is shown. (J, K) HCT116 cells were exposed to hypoxia (Hyp, < 0.1% O2) and MG‐132 (5 μm) as indicated and western blotting was carried out with the antibodies shown. Densitometry for three biological replicates is shown in part (K). (L, M) HCT116 cells were treated with 25 μg·mL−1 cycloheximide (CHX) in hypoxic (Hyp, < 0.1% O2) or normoxic (Norm, 21% O2) conditions for the times indicated and analyzed with western blotting. Densitometry for three biological replicates is shown in part (M). (N) A scheme showing hypothesis of hypoxia‐inducible degradation of DDX5 via proteasome. The scheme was drawn with BioRender.com. (O) Sequential sections from HCT116 and OE21 tumor xenografts using the cell lines indicated (HCT116 or OE21) were stained for hypoxia with antipimonidazole hyoxyprobe‐1 (PIMO) and DDX5 antibodies. Nuclei were counterstained with hematoxylin Scale bar = 50 μm. The outline of PIMO‐positive (brown) staining is shown by the dashed white line. (P) PFA‐fixed GL261 tumor xenografts were sectioned and subjected to immunofluorescent staining for hypoxia (PIMO) and DDX5. Nuclei were counterstained with DAPI. Scale bar = 100 μm. The outline of PIMO‐positive (FITC) staining is shown by the dashed white line. (Q) Mean fluorescence intensity ± standard deviation of DDX5 signal (labeled with Alexa Fluor‐647) in GL261 tumors from part (P) was measured in hypoxic (PIMO +ve) and normoxic (PIMO −ve) image areas using zen2 software (Zeiss). A total of 22 hypoxic areas and 31 normoxic areas were analyzed and two‐tailed nonparametric Mann–Whitney test shows statistical significance (P‐value 0.007, **).
DDX5 is known to interact with its paralog DDX17 and depletion of DDX5 has been shown to upregulate DDX17 [14]. Therefore, we asked whether loss of DDX5 expression in hypoxia would lead to increased expression of DDX17. When we depleted DDX5 using siRNA, the levels of DDX17 increased significantly in normoxic conditions confirming the compensatory relationship between DDX17 and DDX5 (Fig. S5G,H). However, in hypoxic conditions, where DDX5 is repressed, the expression of DDX17 did not change, indicating that this compensatory relationship does not occur when DDX5 is repressed by hypoxia. Together, these data suggest that DDX5 is repressed in an oxygen‐dependent manner, and this event does not result in increased expression of its paralog, DDX17.
Next, we tested the involvement of the proteasome pathway in the downregulation of DDX5 and confirmed that DDX5 protein was degraded via the proteasome in hypoxia by cotreating cells with a proteasome inhibitor, MG‐132 (Fig. 3J,K). We then went on to investigate the half‐life of the DDX5 protein in hypoxia and in normoxia and surprisingly, and found that blocking global protein translation with cycloheximide rescued DDX5 expression in hypoxia (Fig. 3L,M). Using emetine, as an alternative agent to block translation, we confirmed that hypoxia‐mediated downregulation of DDX5 could be reversed by blocking translation (Fig. S5I,J). These data suggest that active translation is required in hypoxic conditions to decrease the levels of DDX5 and led to the hypothesis that an E3 ligase induced in hypoxia could target DDX5 for proteasomal degradation (Fig. 3N). In support of this hypothesis, we observed a partial, but consistent, rescue of DDX5 repression in hypoxia in the presence of a neddylation inhibitor, MLN4924, indicating the potential involvement of an E3 ubiquitin ligase/s, from the Cullin‐RING type family (Fig. S5K).
Finally, we analyzed tumor xenografts (HCT116, OE‐21 and GL261) for both hypoxia (using pimonidazole) and DDX5 expression. In all three tumor types, an inverse correlation was observed between hypoxia (pimonidazole positive) and DDX5 expression, confirming that hypoxia represses DDX5 protein expression in vivo (Fig. 3O–Q). Together, these data suggest that expression of DDX5 is regulated at multiple levels, including the decreased accessibility of the chromatin at the promoter of DDX5, decreased DDX5 mRNA, and enhanced protein degradation in hypoxia.
We hypothesized that the widespread repression of R‐loop‐related factors in hypoxia could in part explain the increase in R‐loops in hypoxic conditions [7]. We investigated whether rescuing DDX5 in hypoxia could reduce the accumulation of R‐loops in hypoxia with the obvious caveat that rescuing the expression of a single component of the R‐loop interactome could have little impact. However, as R‐loops are transcription dependent and DDX5 is a transcription cofactor, we first determined whether rescuing DDX5 in hypoxia increased this activity. To begin, we measured changes in global transcription using 5'EU incorporation in cells exposed to hypoxia (< 0.1% O2) with and without DDX5 overexpression. Rescuing DDX5 expression in hypoxia in both A549 and HCT116 cells had no impact on 5'EU incorporation (Fig. 4A,B and Fig. S5A–C). Furthermore, as DDX5 is a known cofactor of E2F1 and we have previously shown that some E2F target genes are repressed in hypoxia [50], we also overexpressed DDX5 in hypoxia and tested expression of known E2F target genes: MCM2, MCM4, MCM5, MCM6, MCM7, and CDC6. As expected, we observed a clear decrease in protein expression of these E2F target genes in hypoxia; however, levels were not rescued when DDX5 was restored in hypoxia (Fig. S5D). We then investigated the effect of restoring DDX5 expression in hypoxia on R‐loop accumulation. DDX5 was expressed in cells and exposed to hypoxia (< 0.1% O2), and used the catalytic‐dead mutant RNase H1D210N to visualize R‐loops [51, 52]. In addition, we included a DDX5 helicase mutant (DDX5NEAD) and DHX9 as controls. Surprisingly, we found that rescuing DDX5 in hypoxia led to a significant accumulation of R‐loops and importantly that this was dependent on DDX5 helicase activity (Fig. 4C–E). In contrast, when we restored the expression of DHX9, R‐loop levels were significantly reduced both in normoxia and in hypoxia. To confirm this surprising finding, we verified the role of DDX5 in R‐loop accumulation in hypoxia in a second cell line, HCT116. Again, we found that when DDX5 expression was restored in cells exposed to hypoxia, the levels of R‐loops increased further (Fig. S5E–G). These data demonstrate that in hypoxic conditions, the restoration of DDX5‐mediated helicase activity increases R‐loop levels and that this is specific to the DDX5 helicase, as overexpression of DHX9 abrogated R‐loops. Increasing R‐loops in hypoxia would be predicted to lead to a further accumulation of replication stress [7]. To investigate this hypothesis, we determined the impact of rescuing DDX5 expression in hypoxia on the levels of RPA foci as a marker of replication stress. A significant increase in the number of RPA foci per cell was seen in hypoxic cells with overexpressed DDX5 (Fig. 4F,G). Overall, these data suggest that hypoxia‐mediated loss of DDX5 expression acts to restrict replication stress through decreased R‐loop accumulation.
Fig. 4.
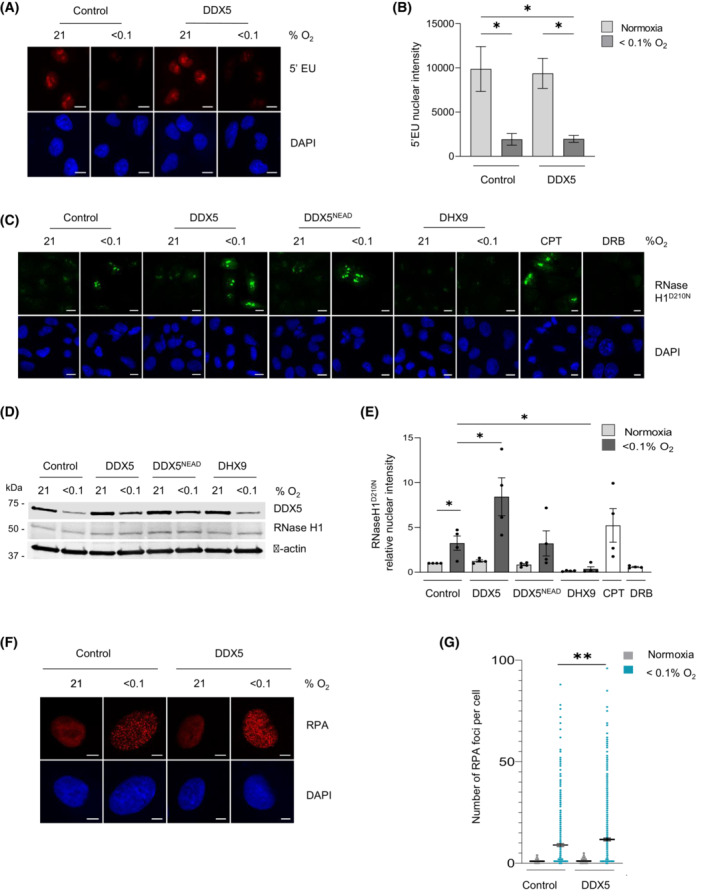
Rescuing DDX5 expression in hypoxia leads to accumulation of R‐loops. (A) A549 cells were transfected with myc‐tagged DDX5 or control vector and exposed to hypoxia (8 h) with 5'EU (0.5 mm) added for the final hour. Staining for 5'EU was then carried out. Representative images are shown (scale bar = 10 μm). 5'EU staining in red, DAPI (blue) shows the nucleus. (B) Nuclear Intensity of 5'EU staining from A was determined. Data represents the mean expression and SEM from three independent experiments. Statistical significance was calculated with an unpaired Student's t‐test for each indicated condition (*P < 0.05). (C) A549 cells were co‐transfected with RNase H1D210N together with myc‐tagged DDX5, myc‐tagged DDX5NEAD, or with DHX9 and exposed to hypoxia for 18 h. CPT (10 μm, 20 min) or DRB (100 μm, 1 h) were used as a positive and negative controls for R‐loops, respectively. Representative images for R‐loop visualization with V5‐RNase H1D210N fluorescence are shown (scale bar = 10 μm). V5 fluorescence is shown in green, DAPI (blue) shows the nucleus. (D) Western blot analysis confirms expression of DDX5 and RNase H1D210N across conditions shown in part (C). (E) Quantification of fluorescent intensity from part (C). A minimum of 100 cells were included per condition. Data represent the mean expression and SEM from four independent experiments. Statistical significance was determined with an unpaired Student's t‐test for each indicated condition (*P < 0.05). (F) A549 cells were transfected as indicated and staining for RPA foci was then carried out. Representative images are shown (scale bar = 10 μm). RPA staining in red, DAPI (blue) shows the nucleus. (G) Quantification of the number of foci per cell in part (F) was determined. Data represents the mean expression and SEM from three independent experiments, each dot represents a cell and a minimum of 100 cells were imaged per treatment. Statistical significance was calculated with an unpaired Student's t‐test for each indicated condition (**P < 0.01).
4. Discussion
We demonstrate that many genes involved in R‐loop formation and resolution (the R‐loop interactome) are repressed in hypoxic conditions, with the notable exception of SETX. Our ATACseq analysis suggests that in most cases, the repression of the R‐loop interactome correlates with decreased chromatin accessibility, likely leading to reduced transcription. Changes in chromatin accessibility in hypoxia have been previously characterized in hypoxia‐induced cardiac damage, adaptation to high‐altitude, or cancer cell lines, and both chromatin condensation and increased open chromatin at various loci, including hypoxia‐responsive genes, were reported [49, 53, 54, 55]. While changes to chromatin in hypoxia have been largely attributed to changes in chromatin modifying enzymes, it is also likely that changes in transcription factor activity in these conditions may impact chromatin and ultimately accessibility/gene expression [56]. Recently, ATACseq was also used to demonstrate that the type I interferon pathway is downregulated in hypoxia and that this is dependent on decreased chromatin accessibility at genes with motifs for STAT1 and IRF3 [57]. Here, we found further pathways downregulated through this mechanism in hypoxia, including DNA repair, splicing, and the R‐loop interactome. It is plausible that the reduced expression of genes in all of these pathways contributes to R‐loop levels in hypoxia.
Evidence to support the hypothesis that the repression of these genes is part of the biological response to hypoxia as opposed to an energy saving measure comes from our finding that in the case of DDX5, multiple mechanisms exist in hypoxia to reduce expression. Our data suggest increased protein degradation in hypoxia via a hypoxia‐inducible E3 ubiquitin ligase, potentially as a means of quickly reducing DDX5 levels in addition to the loss of chromatin accessibility. In addition, the induction of miR‐462 and ‐731 in hypoxia in a HIF‐dependent manner has been shown to repress DDX5 in zebrafish, further supporting our finding that multiple mechanisms exist to reduce DDX5 in hypoxia [58]. Surprisingly, whilst we predicted that loss of the R‐loop interactome contributed to R‐loop levels in hypoxia, we found that this was not a general response as rescuing DDX5 increased R‐loops whilst the opposite effect was detected when DHX9 was restored. Notably, our studies have focused on the global levels of R‐loops and do not address changes at specific genomic locations.
Why restoring DDX5 expression in hypoxia leads to further R‐loop accumulation above the level observed in hypoxia is unclear. Previous reports have all shown that expression of DDX5 leads to R‐loop resolution, that is, a decrease in R‐loop levels [15, 16, 17, 18, 19]. To our knowledge, expression of DDX5 in hypoxia is the first example of a context where DDX5 has been linked to R‐loop accumulation. One hypothesis is that this is related to the changes in splicing also observed in these conditions, as recruitment of the spliceosome onto nascent mRNA attenuates R‐loop formation [59]. In support of this, overexpression of DHX9 in the absence of key splicing factors has been shown to increase R‐loop accumulation [60]. However, when we restored DHX9 in hypoxia, we found an almost complete abrogation of R‐loops, although notably this also occurred in the normoxic samples, making the impact of DHX9 in hypoxia difficult to interpret.
A caveat of having chosen DDX5 as an exemplar for the R‐loop interactome is that DDX5 has numerous functions which are independent of R‐loops [12, 13, 14, 61, 62, 63]. For example, it is plausible that repression of DDX5 could be a part of the biological adaption to reduced oxygen levels as shRNA‐mediated depletion of DDX5 in lung cancer cells leads to reduced oxygen consumption through changes in mitochondrial respiration, oxidative phosphorylation, and reduced intracellular succinate [64]. Therefore, reduction of DDX5 expression in hypoxia may alter numerous alternative pathways, which in turn may also indirectly affect R‐loop levels, making the direct effect of DDX5 on R‐loop levels difficult to determine.
One of the aspects of DDX5 that intrigued us was the finding that is overexpressed or amplified in a wide range of cancers [65]. Initially, our finding that hypoxia leads to loss of DDX5 seemed contradictory to the finding that it is overexpressed in cancers. However, when considered together, this demonstrates that whilst hypoxic tumors express less DDX5, the levels are increased compared with normal tissues. This in turn suggests that the DDX5 gene amplification in cancers could occur to compensate for the repressive effect of hypoxia on DDX5. Together, our data highlight the significant energy‐consuming processes a hypoxic cell employs to turn off the expression of specific genes such as DDX5 and, not surprisingly, that cancers circumvent this biological response.
5. Conclusions
In summary, the repressive impact of hypoxia on gene expression has been described and in particular with reference to genes involved in the DNA repair and splicing pathways. Here, we demonstrate that numerous genes included in the R‐loop interaction are also repressed in hypoxia and that this is likely due to increased chromatin compaction. We have focused on the RNA helicase, DDX5 and demonstrated that multiple mechanisms exist to reduce expression in hypoxia, the impact of which is decreased replication stress and R‐loop accumulation.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
KBL coconceived the project; conceived, designed, performed or supervised, interpreted the majority of the experiments; and wrote the manuscript. MD performed ATACseq experiments, analyses and validation; and contributed to the writing of the manuscript. HE performed R‐loop assays. J(utta)M performed qPCR analyses. EB contributed to the writing of the manuscript. AJG provided feedback and supervision. J(akub)M assisted with the ATACseq analyses and advised on the manuscript. BK advised on the manuscript and facilitated ATACseq experiments. EMH coconceived the project; conceived, designed, and interpreted the majority of the experiments; and wrote the manuscript. All authors commented on the manuscript.
Supporting information
Fig. S1. Oxygen‐dependent changes in the chromatin accessibility.
Fig. S2. ATACseq data analysis using alternative normalization methods.
Fig. S3. Hypoxia‐dependent chromatin accessibility changes at R‐loop interacting factors.
Fig. S4. Correlation between DDX5 expression and hypoxia metagene in TCGA cancer patient samples.
Fig. S5. Hypoxia‐dependent DDX5 repression in multiple cell lines.
Fig. S6. Effects of rescue of DDX5 expression in hypoxia on transcription and R‐loops in HCT116 cells.
Table S1. ATACseq differential peak analysis.
Table S2. ATACseq differential peak analysis of promoter regions.
Table S3. ATACseq KEGG, GO, and REACTOME pathway analysis at promoters repressed under severe hypoxia.
Table S4. ATACseq – spliceosome regulation in hypoxia.
Table S5. ATACseq – R‐loop‐interactome.
Appendix S1. Primer sequences.
Acknowledgements
The authors would like to thank Drs Monica Olcina and Tiffany Ma (University of Oxford) for their insightful feedback on our manuscript. KBL and EB were supported by a CRUK grant C5255/A23755 (awarded to EMH). KBL was also supported by CRUKDF 0715‐KL grant (awarded to KBL and EMH). MD and KBL were supported by National Science Centre (Poland) grant 2019/33/B/NZ1/01556 (awarded to KBL). HE was supported by Medical Research Council‐UKRI grant MC_UU_00001/8 (awarded to AJG). We acknowledge Ms Beata Kaza for preparing GL261 cryo‐sections. We acknowledge Mr Paweł Segit, Mr Karol Jacek and Mr Adria‐Jaume Roura Canalda for guidance on ATACseq data analysis. We acknowledge Ms Paulina Szadkowska, Dr Bartlomiej Gielniewski and Dr Bartosz Wojtas for sequencing the ATACseq samples.
Contributor Information
Katarzyna B. Leszczynska, Email: k.leszczynska@nencki.edu.pl.
Ester M. Hammond, Email: ester.hammond@oncology.ox.ac.uk.
Data availability statement
The data that support the findings of this study (Figs [Link], [Link] and Tables [Link], [Link]) are available in the Supporting Information of this article. The ATACseq data that support the findings of this study are openly available in Gene Expression Omnibus repository at https://www.ncbi.nlm.nih.gov/geo, reference number GSE200757.
References
- 1. Raymond J, Segrè D. The effect of oxygen on biochemical networks and the evolution of complex life. Science. 2006;311:1764–7. 10.1126/science.1118439 [DOI] [PubMed] [Google Scholar]
- 2. Liao C, Zhang Q. Understanding the oxygen‐sensing pathway and its therapeutic implications in diseases. Am J Pathol. 2020;190:1584–95. 10.1016/j.ajpath.2020.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hammond EM, Asselin MC, Forster D, O'Connor JPB, Senra JM, Williams KJ. The meaning, measurement and modification of hypoxia in the laboratory and the clinic. Clin Oncol. 2014;26:277–88. 10.1016/j.clon.2014.02.002 [DOI] [PubMed] [Google Scholar]
- 4. Chan N, Pires IM, Bencokova Z, Coackley C, Luoto KR, Bhogal N, et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010;70:8045–54. 10.1158/0008-5472.CAN-10-2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Santos‐Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nat Rev Genet. 2015;16:583–97. 10.1038/nrg3961 [DOI] [PubMed] [Google Scholar]
- 6. Crossley MP, Bocek M, Cimprich KA. R‐loops as cellular regulators and genomic threats. Mol Cell. 2019;73:398–411. 10.1016/j.molcel.2019.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ramachandran S, Ma TS, Griffin J, Ng N, Foskolou IP, Hwang M‐S, et al. Hypoxia‐induced SETX links replication stress with the unfolded protein response. Nat Commun. 2021;12:3686. 10.1038/s41467-021-24066-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olcina MM, Foskolou IP, Anbalagan S, Senra JM, Pires IM, Jiang Y, et al. Replication stress and chromatin context link ATM activation to a role in DNA replication. Mol Cell. 2013;52:758–66. 10.1016/j.molcel.2013.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Batie M, Frost J, Frost M, Wilson JW, Schofield P, Rocha S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science. 2019;363:1222–6. 10.1126/science.aau5870 [DOI] [PubMed] [Google Scholar]
- 10. Olcina MM, Leszczynska KB, Senra JM, Isa NF, Harada H, Hammond EM. H3K9me3 facilitates hypoxia‐induced p53‐dependent apoptosis through repression of APAK. Oncogene. 2016;35:793–9. 10.1038/onc.2015.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brady LK, Wang H, Radens CM, Bi Y, Radovich M, Maity A, et al. Transcriptome analysis of hypoxic cancer cells uncovers intron retention in EIF2B5 as a mechanism to inhibit translation. PLoS Biol. 2017;15:e2002623. 10.1371/journal.pbio.2002623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Giraud G, Terrone S, Bourgeois CF. Functions of DEAD box RNA helicases DDX5 and DDX17 in chromatin organization and transcriptional regulation. BMB Rep. 2018;51:613–22. 10.5483/BMBRep.2018.51.12.234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dardenne E, PolayEspinoza M, Fattet L, Germann S, Lambert MP, Neil H, et al. RNA helicases DDX5 and DDX17 dynamically orchestrate transcription, miRNA, and splicing programs in cell differentiation. Cell Rep. 2014;7:1900–13. 10.1016/j.celrep.2014.05.010 [DOI] [PubMed] [Google Scholar]
- 14. Geißler V, Altmeyer S, Stein B, Uhlmann‐Schiffler H, Stahl H. The RNA helicase Ddx5/p68 binds to hUpf3 and enhances NMD of Ddx17/p72 and Smg5 mRNA. Nucleic Acids Res. 2013;41:7875–88. 10.1093/nar/gkt538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Villarreal OD, Mersaoui SY, Yu Z, Masson JY, Richard S. Genome‐wide R‐loop analysis defines unique roles for DDX5, XRN2, and PRMT5 in DNA/RNA hybrid resolution. Life Sci Alliance. 2020;3:e202000762. 10.26508/LSA.202000762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kang HJ, Eom H j, Kim H, Myung K, Kwon HM, Choi JH. Thrap3 promotes R‐loop resolution via interaction with methylated DDX5. Exp Mol Med. 2021;53:1602–11. 10.1038/s12276-021-00689-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu Z, Mersaoui SY, Guitton‐Sert L, Coulombe Y, Song J, Masson J‐Y, et al. DDX5 resolves R‐loops at DNA double‐strand breaks to promote DNA repair and avoid chromosomal deletions. NAR Cancer. 2020;2:zcaa028. 10.1093/narcan/zcaa028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mersaoui SY, Yu Z, Coulombe Y, Karam M, Busatto FF, Masson J, et al. Arginine methylation of the DDX 5 helicase RGG/RG motif by PRMT 5 regulates resolution of RNA:DNA hybrids. EMBO J. 2019;38:e100986. 10.15252/embj.2018100986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sessa G, Gómez‐González B, Silva S, Pérez‐Calero C, Beaurepere R, Barroso S, et al. BRCA2 promotes DNA‐RNA hybrid resolution by DDX5 helicase at DNA breaks to facilitate their repair‡. EMBO J. 2021;40:e106018. 10.15252/embj.2020106018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sliwa M, Markovic D, Gabrusiewicz K, Synowitz M, Glass R, Zawadzka M, et al. The invasion promoting effect of microglia on glioblastoma cells is inhibited by cyclosporin A. Brain. 2007;130:476–89. 10.1093/brain/awl263 [DOI] [PubMed] [Google Scholar]
- 21. Ochocka N, Segit P, Walentynowicz KA, Wojnicki K, Cyranowski S, Swatler J, et al. Single‐cell RNA sequencing reveals functional heterogeneity of glioma‐associated brain macrophages. Nat Commun. 2021;12:1151. 10.1038/s41467-021-21407-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cook A, Mieczkowski J, Tolstorukov MY. Single‐assay profiling of nucleosome occupancy and chromatin accessibility. Curr Protoc Mol Biol. 2017;2017:21.34.1–21.34.18. 10.1002/cpmb.45 [DOI] [PubMed] [Google Scholar]
- 23. Leszczynska KB, Foskolou IP, Abraham AG, Anbalagan S, Tellier C, Haider S, et al. Hypoxia‐induced p53 modulates both apoptosis and radiosensitivity via AKT. J Clin Invest. 2015;125:2385–98. 10.1172/JCI80402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Buffa FM, Harris AL, West CM, Miller CJ. Large meta‐analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br J Cancer. 2010;102:428–35. 10.1038/sj.bjc.6605450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Puchalski RB, Shah N, Miller J, Dalley R, Nomura SR, Yoon JG, et al. An anatomic transcriptional atlas of human glioblastoma. Science. 2018;360:660–3. 10.1126/science.aaf2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen X, Shen Y, Draper W, Buenrostro JD, Litzenburger U, Cho SW, et al. ATAC‐see reveals the accessible genome by transposase‐mediated imaging and sequencing. Nat Methods. 2016;13:1013–20. 10.1038/nmeth.4031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC‐seq: a method for assaying chromatin accessibility genome‐wide. Curr Protoc Mol Biol. 2015;2015:21.29.1–9. 10.1002/0471142727.mb2129s109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Langmead B, Salzberg SL. Fast gapped‐read alignment with bowtie 2. Nat Methods. 2012;9:357–9. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10:giab008. 10.1093/gigascience/giab008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, et al. ChIPpeakAnno: a Bioconductor package to annotate ChIP‐seq and ChIP‐chip data. BMC Bioinformatics. 2010;11:237. 10.1186/1471-2105-11-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep‐sequencing data analysis. Nucleic Acids Res. 2016;44:W160–5. 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9:e1003118. 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lun ATL, Smyth GK. Csaw: a Bioconductor package for differential binding analysis of ChIP‐seq data using sliding windows. Nucleic Acids Res. 2015;44:e45. 10.1093/nar/gkv1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reske JJ, Wilson MR, Chandler RL. ATAC‐seq normalization method can significantly affect differential accessibility analysis and interpretation. Epigenetics Chromatin. 2020;13:22. 10.1186/s13072-020-00342-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu G, Wang LG, He QY. ChIP seeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–3. 10.1093/bioinformatics/btv145 [DOI] [PubMed] [Google Scholar]
- 39. Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4:1184–91. 10.1038/nprot.2009.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carbon S, Dietze H, Lewis SE, Mungall CJ, Munoz‐Torres MC, Basu S, et al. Expansion of the gene ontology knowledgebase and resources: the gene ontology consortium. Nucleic Acids Res. 2017;45:D331–8. 10.1093/nar/gkw1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–80. 10.1093/nar/gkh063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48:D498–503. 10.1093/nar/gkz1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pombo Antunes AR, Scheyltjens I, Lodi F, Messiaen J, Antoranz A, Duerinck J, et al. Single‐cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat Neurosci. 2021;24:595–610. 10.1038/s41593-020-00789-y [DOI] [PubMed] [Google Scholar]
- 44. Dobrynin G, McAllister TE, Leszczynska KB, Ramachandran S, Krieg AJ, Kawamura A, et al. KDM4A regulates HIF‐1 levels through H3K9me3. Sci Rep. 2017;7:11094. 10.1038/s41598-017-11658-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Klemm SL, Shipony Z, Greenleaf WJ. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet. 2019;20:207–20. 10.1038/s41576-018-0089-8 [DOI] [PubMed] [Google Scholar]
- 46. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. 10.1016/j.cell.2007.02.005 [DOI] [PubMed] [Google Scholar]
- 47. Choudhry H, Harris AL. Advances in hypoxia‐inducible factor biology. Cell Metab. 2018;27:281–98. 10.1016/j.cmet.2017.10.005 [DOI] [PubMed] [Google Scholar]
- 48. Cristini A, Groh M, Kristiansen MS, Gromak N. RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R‐loop‐associated DNA damage. Cell Rep. 2018;23:1891–905. 10.1016/j.celrep.2018.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Batie M, Frost J, Shakir D, Rocha S. Regulation of chromatin accessibility by hypoxia and HIF. Biochem J. 2022;479:767–86. 10.1042/bcj20220008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pires IM, Bencokova Z, Milani M, Folkes LK, Li JA, Stratford MR, et al. Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 2010;70:925–35. 10.1158/0008-5472.CAN-09-2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen L, Chen JY, Zhang X, Gu Y, Xiao R, Shao C, et al. R‐ChIP using inactive RNase H reveals dynamic coupling of R‐loops with transcriptional pausing at gene promoters. Mol Cell. 2017;68:745–57.e5. 10.1016/j.molcel.2017.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Teloni F, Michelena J, Lezaja A, Kilic S, Ambrosi C, Menon S, et al. Efficient pre‐mRNA cleavage prevents replication‐stress‐associated genome instability. Mol Cell. 2019;73:670–83.e12. 10.1016/j.molcel.2018.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ward MC, Banovich NE, Sarkar A, Stephens M, Gilad Y. Dynamic effects of genetic variation on gene expression revealed following hypoxic stress in cardiomyocytes. Elife. 2021;10:e57345. 10.7554/eLife.57345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li Y, Gruber JJ, Litzenburger UM, Zhou Y, Miao YR, LaGory EL, et al. Acetate supplementation restores chromatin accessibility and promotes tumor cell differentiation under hypoxia. Cell Death Dis. 2020;11:102. 10.1038/s41419-020-2303-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xin J, Zhang H, He Y, Duren Z, Bai C, Chen L, et al. Chromatin accessibility landscape and regulatory network of high‐altitude hypoxia adaptation. Nat Commun. 2020;11:4928. 10.1038/s41467-020-18638-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lara‐Astiaso D, Weiner A, Lorenzo‐Vivas E, Zaretsky I, Jaitin DA, David E, et al. Immunogenetics. Chromatin state dynamics during blood formation. Science. 2014;345:943–9. 10.1126/science.1256271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miar A, Arnaiz E, Bridges E, Beedie S, Cribbs AP, Downes DJ, et al. Hypoxia induces transcriptional and translational downregulation of the type i IFN pathway in multiple cancer cell types. Cancer Res. 2020;80:5245–56. 10.1158/0008-5472.CAN-19-2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huang CX, Chen N, Wu XJ, Huang CH, He Y, Tang R, et al. The zebrafish miR‐462/miR‐731 cluster is induced under hypoxic stress via hypoxia‐inducible factor 1α and functions in cellular adaptations. FASEB J. 2015;29:4901–13. 10.1096/fj.14-267104 [DOI] [PubMed] [Google Scholar]
- 59. Bonnet A, Grosso AR, Elkaoutari A, Coleno E, Presle A, Sridhara SC, et al. Introns protect eukaryotic genomes from transcription‐associated genetic instability. Mol Cell. 2017;67:608–21.e6. 10.1016/j.molcel.2017.07.002 [DOI] [PubMed] [Google Scholar]
- 60. Chakraborty P, Huang JTJ, Hiom K. DHX9 helicase promotes R‐loop formation in cells with impaired RNA splicing. Nat Commun. 2018;9:4346. 10.1038/s41467-018-06677-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mazurek A, Luo W, Krasnitz A, Hicks J, Scott Powers R, Stillman B. DDX5 regulates DNA replication and is required for cell proliferation in a subset of breast cancer cells. Cancer Discov. 2012;2:812–25. 10.1158/2159-8290.CD-12-0116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nicol SM, Bray SE, Derek Black H, Lorimore SA, Wright EG, Lane DP, et al. The RNA helicase p68 (DDX5) is selectively required for the induction of p53‐dependent p21 expression and cell‐cycle arrest after DNA damage. Oncogene. 2013;32:3461–9. 10.1038/onc.2012.426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nyamao RM, Wu J, Yu L, Xiao X, Zhang FM. Roles of DDX5 in the tumorigenesis, proliferation, differentiation, metastasis and pathway regulation of human malignancies. Biochim Biophys Acta Rev Cancer. 2019;1871:85–98. 10.1016/j.bbcan.2018.11.003 [DOI] [PubMed] [Google Scholar]
- 64. Xing Z, Russon MP, Utturkar SM, Tran EJ. The RNA helicase DDX5 supports mitochondrial function in small cell lung cancer. J Biol Chem. 2020;295:8988–98. 10.1074/jbc.ra120.012600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dai TY, Cao L, Yang ZC, Li YS, Tan L, Ran XZ, et al. P68 RNA helicase as a molecular target for cancer therapy. J Exp Clin Cancer Res. 2014;33:64. 10.1186/s13046-014-0064-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Oxygen‐dependent changes in the chromatin accessibility.
Fig. S2. ATACseq data analysis using alternative normalization methods.
Fig. S3. Hypoxia‐dependent chromatin accessibility changes at R‐loop interacting factors.
Fig. S4. Correlation between DDX5 expression and hypoxia metagene in TCGA cancer patient samples.
Fig. S5. Hypoxia‐dependent DDX5 repression in multiple cell lines.
Fig. S6. Effects of rescue of DDX5 expression in hypoxia on transcription and R‐loops in HCT116 cells.
Table S1. ATACseq differential peak analysis.
Table S2. ATACseq differential peak analysis of promoter regions.
Table S3. ATACseq KEGG, GO, and REACTOME pathway analysis at promoters repressed under severe hypoxia.
Table S4. ATACseq – spliceosome regulation in hypoxia.
Table S5. ATACseq – R‐loop‐interactome.
Appendix S1. Primer sequences.
Data Availability Statement
The data that support the findings of this study (Figs [Link], [Link] and Tables [Link], [Link]) are available in the Supporting Information of this article. The ATACseq data that support the findings of this study are openly available in Gene Expression Omnibus repository at https://www.ncbi.nlm.nih.gov/geo, reference number GSE200757.