

Abstract

In this study, a novel series of pyrano[3,2-c]quinoline-1,2,3-triazole hybrids 8a–o were synthesized and evaluated against the α-glucosidase enzyme. All compounds showed significant in vitro inhibitory activity (IC50 values of 1.19 ± 0.05 to 20.01 ± 0.02 μM) compared to the standard drug acarbose (IC50 = 750.0 μM). Among them, 2-amino-4-(3-((1-benzyl-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (compound 8k) demonstrated the best inhibitory effect toward α-glucosidase (IC50 = 1.19 ± 0.05 μM) with a competitive pattern of inhibition. Since compound 8k was synthesized as a racemic mixture, molecular docking and dynamics simulations were performed on R- and S-enantiomers of compound 8k. Based on the molecular docking results, both R- and S-enantiomers of compound 8k displayed significant interactions with key residues including catalytic triad (Asp214, Glu276, and Asp349) in the enzyme active site. However, an in silico study indicated that S- and R-enantiomers were inversely located in the enzyme active site. The R-enantiomer formed a more stable complex with a higher binding affinity to the active site of α-glucosidase than that of the S- enantiomer. The benzyl ring in the most stable complex ((R)-compound 8k) was located in the bottom of the binding site and interacted with the enzyme active site, while the pyrano[3,2-c]quinoline moiety occupied the high solvent accessible entrance of the active site. Thus, the synthesized pyrano[3,2-c]quinoline-1,2,3-triazole hybrids seem to be promising scaffolds for the development of novel α-glucosidase inhibitors.
1. Introduction
Diabetes mellitus (DM) is a chronic disease caused by an inherited or acquired defect in insulin secretion or a decrease in response of organs to secreted insulin.1 The bodies of people with DM do not produce enough insulin, which is responsible for lowering blood sugar levels.2 If diabetics are not treated, their blood glucose levels may increase significantly,3,4 and this may lead to kidney failure, cardiovascular disease, blindness, and impaired wound healing. α-Glucosidase (EC 3.2.1.20) is a carbohydrate-hydrolyzing enzyme, and its catalytic activity, as an intestinal digestive enzyme, results in cleavage of poly- and disaccharides into monosaccharides (glucose), which in turn will raise blood sugar levels.5,6 Therefore, several α-glucosidase inhibitors (AGIs) such as acarbose,7 voglibose,8 miglitol,9 and nojirimycin10 are widely used alone or in combination with insulin injection as treatment of diabetes. However, these drugs are often non-specific agents that may cause certain side effects such as indigestion, bloating, and diarrhoea.11 Although there are common medications to control diabetes, many challenges are encountered in the effective treatment of diabetes, including optimizing common treatments to regulate glucose concentration while reducing side effects. In the recent years, the management and control of diabetes using AGIs have received much attention from pharmacists.12,13
N-heterocyclic compounds especially those with triazole rings such as 1,2,3-triazole have different biological properties such as anti-Alzheimer,14,15 anti-viral,16,17 anti-fungal,18 antibacterial and antitubercular,19 anti-cancer,20 anti-tubulin,21−24 and anti-diabetes25−30 activity. Till date, many scaffolds containing 1,2,3-triaole rings with significant inhibitory activity against α-glucosidase have been reported (some of them are depicted in Figure 1, compounds A, B, and C(25−27)). Besides, quinoline- and quinoline-fused derivatives (Figure 1 compounds D, E, and F) have successfully been applied by medicinal chemists as effective pharmacophores in designing of new α-glucosidase inhibitors.28−30 Based on the abovementioned points and in continuation of our interest in synthesis of novel bioactive agents,31,32 herein, we reported synthesis of a novel series of quinoline-1,2,3-triazole hybrids 8a–o as α-glucosidase inhibitors. These compounds were synthesized via the Cu-catalyzed azide–alkyne cycloaddition method (click chemistry) to produce a 1,2,3-triazole linkage. In vitro evaluation, kinetic, molecular docking, and dynamics studies against α-glucosidase were performed to understand their therapeutic potentials for the treatment of type 2 DM.
Figure 1.
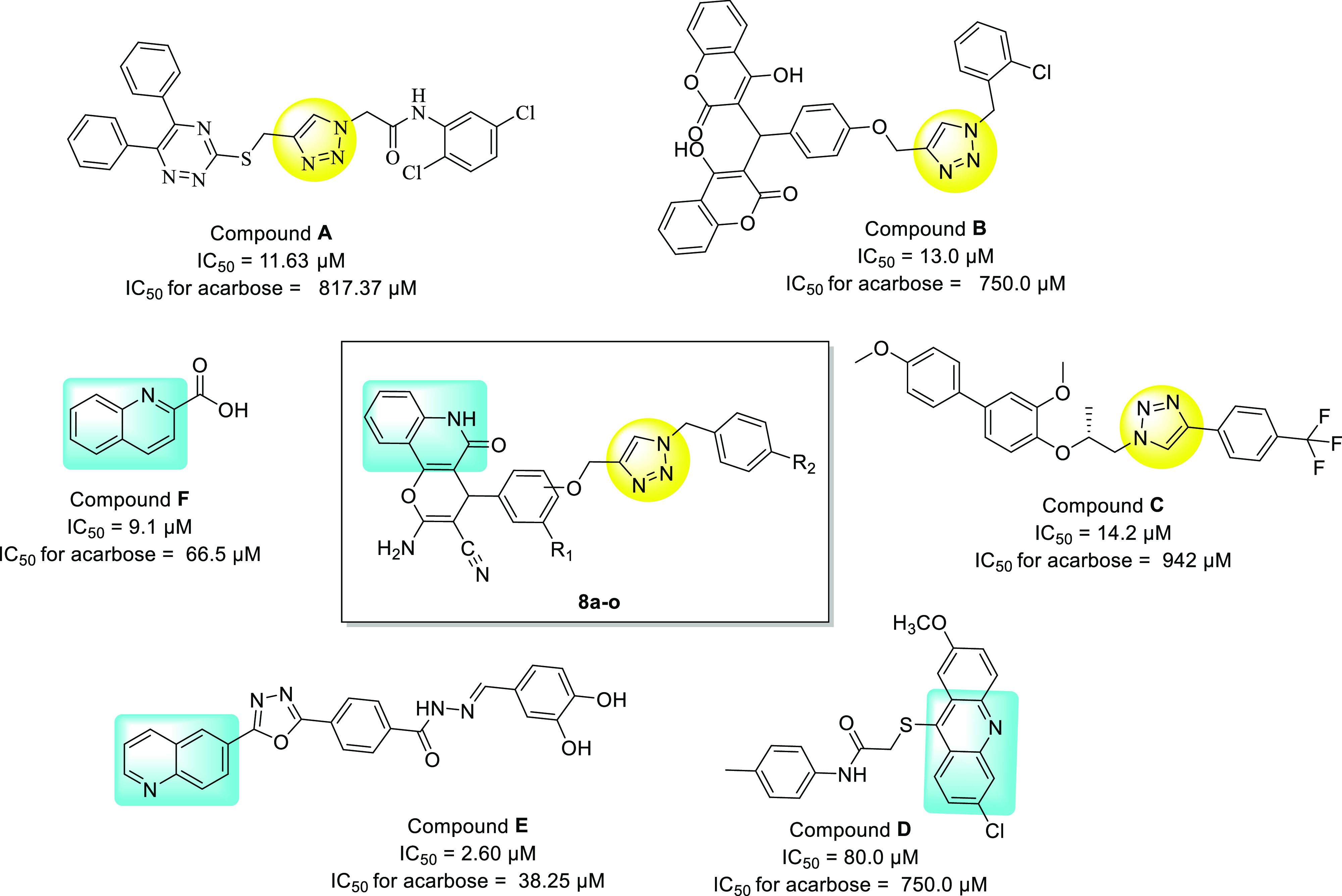
Designed pyrano[3,2-c]quinoline −1,2,3-triazole hybrids based on 1,2,3-triazole and quinoline scaffolds.
2. Results and Discussion
2.1. Chemistry
To synthesize products 8a–o, several steps were performed according to Scheme 1. In the first step, propargylated aldehydes 3a–c were obtained by a reaction between aldehydes 1a–c and propargyl bromide in dimethyl formamide (DMF) at ambient temperature for 24 h.33 In the second step, compounds 6a–c were synthesized by multicomponent reaction between malononitrile, 4-hydroxyquinolin-2(1H)-one, and compounds 3a–c in the presence of a catalytic amount of ammonium acetate in ethanol solvent. In the third step, benzyl bromide derivatives 7a–c were converted to benzyl azide derivatives in DMF and reacted with compounds 6a–c to obtain desired compounds 8a–o in the presence of Et3N, ascorbic acid, and CuSO4·5H2O in DMF/H2O (4:1) at room temperature.14,15 After completion of the reaction, ice was added to the reaction mixture and the resulting precipitates of compounds 8a–o were filtered and washed with water. The structures of all synthesized compounds were confirmed using 1H NMR,13C NMR, mass spectroscopy, and elemental analyses.
Scheme 1. Synthesis of Pyrano[3,2-c]quinoline-1,2,3-triazole Hybrids 8a–o.

2.2. Biological Study
2.2.1. In Vitro α-Glucosidase Inhibitory Activity
The synthesized compounds 8a–o were evaluated for their in vitro inhibitory activity against α-glucosidase and compared to the standard inhibitor acarbose. The obtained results were expressed as mean ± S.E. of three independent experiments in the Table 1. All the synthesized compounds showed significant inhibition against α-glucosidase with an IC50 value of 1.19 ± 0.05 to 23.2 ± 0.8 μM in comparison to acarbose, as a reference drug with an IC50 of 750.0 ± 1.5.
Table 1. Inhibitory Activities of the New Compounds 8a–o Against α-glucosidasea.
| entry | compound | R1 | R2 | IC50 (μM) ± SEa |
|---|---|---|---|---|
| 1 | 8a | H | H | 20.01 ± 0.02 |
| 2 | 8b | H | 4-Me | 9.72 ± 0.18 |
| 3 | 8c | H | 4-F | 12.06 ± 0.03 |
| 4 | 8d | H | 4-Cl | 14.67 ± 0.06 |
| 5 | 8e | H | 4-Br | 4.48 ± 0.03 |
| 6 | 8f | OMe | H | 23.2 ± 0.8 |
| 7 | 8g | OMe | 4-Me | 9.09 ± 0.5 |
| 8 | 8h | OMe | 4-F | 15.9 ± 0.2 |
| 9 | 8i | OMe | 4-Cl | 9.9 ± 0.5 |
| 10 | 8j | OMe | 4-Br | 14.0 ± 0.7 |
| 11 | 8k | H | H | 1.19 ± 0.05 |
| 12 | 8l | H | 4-Me | 14.52 ± 0.05 |
| 13 | 8m | H | 4-F | 8.87 ± 0.03 |
| 14 | 8n | H | 4-Cl | 15.13 ± 0.15 |
| 15 | 8o | H | 4-Br | 9.19 ± 0.17 |
| acarbose | 750 ± 1.5 |
Values are the mean ± standard error of the mean. All experiments were performed at least three times.
Based on the aryloxy phenyl linker between pyrano[3,2-c]quinoline scaffold and benzyl-1,2,3-triazole pendant group, the synthesized compounds 8a–o can be divided into three series: (1) compounds 8a–e with a 4-aryloxy phenyl linker, (2) compounds 8f–j with a 3-methoxy 4-aryloxy phenyl linker, and (3) compounds 8k–o with a 3-aryloxy phenyl linker. Among the synthesized compounds, compound 8k of the third series with un-substituted benzyl moiety showed the best inhibitory activity (IC50 = 1.19 ± 0.05 μM). To evaluate the structure–activity relationships (SARs) for α-glucosidase inhibitory activity, substitution of the hydrogen atom with other chemical groups was carried out at the 4th position of the pendant benzyl moiety.
The IC50 value of the unsubstituted compound 8a was 20.01 ± 0.02 μM. The introduction of methyl, fluoro, chloro, and bromo substituents at the 4th position of the pendant benzyl group led to produce compounds 8b, 8c, 8d, and 8f with IC50 values of 9.72 ± 0.18, 12.06 ± 0.03, 14.67 ± 0.06, and 4.48 ± 0.03 μM, respectively. As presented in Table 1, the replacement of the hydrogen atom with the methyl group, as a hydrophobic electron donating group (EDG), at the 4th position of the benzyl moiety in compound 8b increased its inhibitory activity by twofold. Furthermore, introduction of the electron-withdrawing groups (EWGs) in the benzyl moiety increased inhibitory activity in the following order 4-Br > 4-F > 4-Cl in compounds 8f, 8c, and 8d with IC50 values of 4.48 ± 0.03, 12.06 ± 0.03, and 14.67 ± 0.06 μM, respectively. It seems that the polarizability with the atomic size of substituents on the n-benzyl pendant moiety affected inhibitory activity more than that of the electronic effect. Therefore, bromine with the largest size exhibited better inhibitory activity than fluorine and chlorine.
In the second series, compounds 8f–j had a 3-methoxy-4-aryloxy phenyl linker. Compound 8f with an unsubstituted benzyl moiety exhibited an inhibitory activity (IC50 of 23.2 ± 0.8 μM) similar to compound 8a of the first series (IC50 = 20.01 ± 0.02 μM). Compound 8g with the 4-methyl group at the pendant benzyl moiety exhibited better inhibitory activity (IC50 = 9.9 ± 0.5 μM) compared to compound 8f. Introduction of the EWGs including 4-F, 4-Cl, and 4-Br led to enhanced inhibitory activity in the compounds 8h, 8i, and 8j with IC50 values of 15.9 ± 0.2, 9.0 ± 0.5, and 14.0 ± 0.7 μM, respectively. Also, compound 8g with 4-methyl substitution showed an inhibitory activity (IC50 = 9.09 ± 0.5 μM) similar to compound 8i with 4-chlorine substitution, indicating that both electron-donating (Me) and electron-withdrawing (Cl) effects along with lipophilicity were likely important in the inhibitory activity level. In this series, the inhibitory activities were probably affected by the 3-methoxy group at the linker phenyl ring due to the strict effect and perpendicular conformational to benzyl-1,2,3-triazole and pyrano[3,2-c]quinoline moieties.
In the third series, compounds 8f–j had a 3-aryloxy-phenyl linker. The presence of aryloxy at 3rd position of the phenyl ring could change the conformation of the pendant benzyl-1,2,3-triazole moiety to the linker and another part of the structure. Compound 8k with un-substituted benzyl moiety exhibited the best inhibitory activity (IC50 = 1.19 ± 0.05 μM). Introduction of the methyl group, as the EDG, at the 4th position of the benzyl moiety produced compound 8l and led to decreased inhibitory activity (IC50 = 14.52 ± 0.05) in comparison to compound 8k. Compounds 8m, 8n, and 8o with EDGs including 4-F, 4-Cl, and 4-Br also exhibited decreased inhibitory activity with IC50 values of 8.87, 15.13, and 9.19 μM, respectively. The obtained results revealed that the electron-withdrawing effect of fluorine and the polarizability with the atomic size of bromine in the pendant phenyl group were more critical than lipophilicity.
In general, it is evident from the results that changing the position of the aryloxy-phenyl from 4th-to 3rd position increased the inhibitory activity against α-glucosidase enzyme.
2.2.2. Kinetic Study
The kinetic study of the most active compound 8k against α-glucosidase was performed in order to determine inhibition mode of the synthesized compound. As shown in Figure 2a, the Lineweaver–Burk plot indicated that with increasing concentrations of compound 8k, the Km gradually increased, while Vmax remained unchanged. This finding demonstrated that compound 8k was a competitive inhibitor toward the α-glucosidase enzyme. Furthermore, the plot of the Km versus different concentrations of inhibitor 8k gave an estimate of the inhibition constant (Ki) with the value of 1.3 μM (Figure 2b).
Figure 2.

Kinetic study of compound 8k against the α-glucosidase enzyme. (a) Lineweaver–Burk plot in the absence and presence of different concentrations of compound 8k; (b) secondary plot between Km and various concentrations of compound 8k.
2.2.3. Homology Modeling
Since the crystal structure of the eukaryotic yeast Saccharomyces cerevisiae α-glucosidase is not yet available, homology modeling was used to predict the 3D structure of α-glucosidase. The sequence of S. cerevisiae α-glucosidase is made up of 584 amino acid residues. The NCBI’s BLAST server suggested isomaltase from baker’s yeast as a suitable template for homology modelling. The result of α-glucosidase and isomaltase homology modeling revealed a sequence identity of 72.1%. The quality of the template crystallographic structure was acceptable as 88.9% of residues were located in the favored regions (A, B, and L regions) and 99.8% of residues were placed in the allowed regions (a, b, l, and p regions). The obtained alignment is provided in Figure 3. BLAST results of α-glucosidase and isomaltase showed a score of 909 and an E value of zero (0.0). High-scoring models were selected according to the lowest value of the discrete optimized protein energy (DOPE). The overlay of the homology model of α-glucosidase and isomaltase (3A47) showed a perfect match, especially when considering the backbone atoms (Figure 3). We assessed the quality of the selected models by energetically evaluating the allowed regions of the backbone dihedral angles ψ against φ of amino acid residues in the α-glucosidase structure using the Ramachandran plot (Figure 3). In the final model, 93.2% of residues were located in the favored regions (A, B, and L regions) and 99.8% of residues were placed in the allowed regions (a, b, l, and p regions) as well. The overall quality factor calculated by ERRAT was 86.5%. It should be noted that the ERRAT quality factor, reported for high-resolution structures, is generally around 90% or higher. According to the obtained results, the distribution of distances and angles between the aligned residues in the template and the target protein was comparable.
Figure 3.

Sequence alignment of the α-glucosidase (P53341) and isomaltase (P53051) enzymes. Ramachandran plots of the homology model and the final model obtained from the MD simulation showed minor differences. Overlaid structure of the homology model and isomaltase indicated a perfect match when considering backbone atoms (rmsd = 1.9 Å). rmsd of the backbone during the MD simulation illustrated that the target protein reached a steady state indicating that isomaltase was a good template for homology modeling of the α-glucosidase enzyme.
In this regard, the MODELLER imposed high spatial restraint on the predicted model. To refine the predicted homology model, 50 ns MD simulation was carried out to relax the α-glucosidase structure. Simulation of this enzyme in explicit water revealed minor differences in the backbone dihedral angle values (Figure 3). Backbone rmsd variation of α-glucosidase (0.28 ± 0.017 – rsd <6.03%) (Figure 3) implied that the predicted homology model of α-glucosidase was not affected by the simulation environment dramatically and isomaltase could be a good template for homology modeling of α-glucosidase as well. The dihedral angles ψ against φ of amino acid residues in the α-glucosidase structure were not affected drastically during the MD simulation (99.8 and 99.6% of residues were in the allowed regions before and after the MD simulation). However, the overall 3D quality of the model was modified after the MD simulation and the ERRAT score increased from 86.5%, in the homology model, to 88.9% after the MD simulation.
2.2.4. Molecular Docking Study
To identify possible binding modes of the synthesized compounds in the active site of the α-glucosidase enzyme, a molecular docking study was conducted using AutoDock Tools (version 4.2) on the active site of the modeled α-glucosidase enzyme. To obtain a reliable model for the ligand–enzyme complex, ensemble docking was applied to consider the conformational changes of the enzyme. The trajectories of the α-glucosidase enzyme during the 50 ns MD simulation were clustered into 6 conformers using a 2 Å rmsd cut-off. Acarbose and the selected compounds 8e, 8g, and 8k were docked into the active site of 6 conformers of α-glucosidase.
Based on the literature, the catalytic triad in α-glucosidase is composed of Asp214, Glu276, and Asp349 residues, respectively,33 and Asp68, Tyr71, Thr215, Asn241, Phe300, Glu304, Ser308, Pro309, Phe311, His348, Arg439, and His111 residues are also key residues in the enzyme active site.34 Initially, the molecular modeling of the standard inhibitor acarbose was performed to validate the α-glucosidase homology model and docking method. Figure 4 presents the binding mode of acarbose in the active site of the α-glucosidase enzyme. As seen, hydroxyl and amine groups of cyclohexenylamine, as the most important pharmacophoric points of acarbose, formed hydrogen bonds with the catalytic triad (Asp214, Glu276, and Arg439), Tyr71, His348, and Asp68 (Figure 4).31
Figure 4.
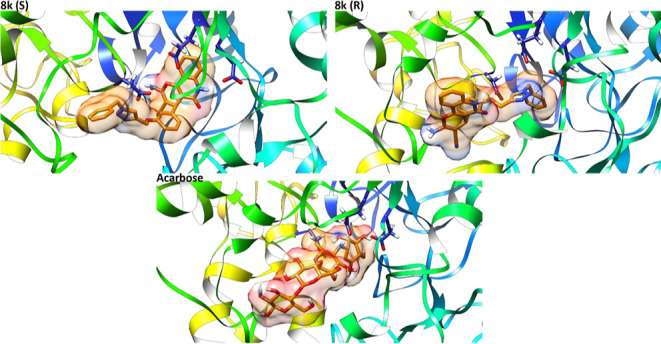
Predicted binding pose of both enantiomers of the compound 8k and acarbose in the active site of the α-glucosidase enzyme. The catalytic triad residues (Asp214, Glu276, and Asp349) are shown by blue sticks. Ligands are represented by orange sticks. As a type I α-glucosidase inhibitor, acarbose formed hydrogen bond via the NH moiety with Glu276. The docking result indicated that the predicted model met the key interactions proposed in the mechanism of action of the enzyme.
The synthesized compounds possessed one chiral center and were produced as a racemic mixture during synthesis. For a comprehensive in silico study, both enantiomers (R and S) were evaluated in a molecular modeling study. The final intermolecular energy (ΔH), torsional free energy (∼−TΔS), estimated free energy of binding (ΔGb), sum of the square of Mulliken charges (∑q2), dipole moment (μ), and quadrupole moment (Q) for acarbose and the three potent compounds 8e, 8g, and 8k are given in Table 2. In the case of acarbose, 95% confidence interval (CI) of predicted ΔGbs in the highest ranked cluster had a wide range after 106 cycles of bootstrapping (3.84 kcal/mol). Even in the limited conformational space of the highest ranked cluster, acarbose experienced a big gap between the minimum (−10.15 kcal/mol) and the maximum (−7.31 kcal/mol) ΔGb. The flexibility of acarbose widened the ΔGb by small conformational changes (rmsd 0–1.98 Å in the highest ranked cluster) that led to less favorable binding mode. Considering 95% CI of the estimated free energy of binding, the difference between affinity of R- and S-enantiomers toward the active site of α-glucosidase was insignificant. So, the docking algorithm could not discriminate between the two enantiomers considering ΔGb. Considering the ΔH and ΔGb values, the docking process was successful in predicting the most potent compound (Table 1). However, the 95% CI showed no significant differences between acarbose and the designed compounds 8e, 8g, and 8k. The charge density (∑q2) could represent the contribution of electrostatic interactions in binding energy. The charge density of the potent compounds was higher than that of the weak ones (acarbose > 8k > 8g > 8e). On the other hand, the same pattern was seen for the quadrupole moments (acarbose > 8g ∼ 8k > 8e). The results indicated that the electrostatic and vdW interactions had a similar importance in the binding process.
Table 2. Results of the Ensemble Docking of Acarbose, 8e, 8g, and 8k into the Active Site of the α-Glucosidase Enzyme in 6 Models Obtained from Clustering of the MD Simulation Trajectoriesa.
| compound | ΔH | –TΔS | ΔGb | 95% CI | ∑q2 | μ | Q | |
|---|---|---|---|---|---|---|---|---|
| acarbose | –13.56 | 1.79 | –11.77 | [−10.53, −6.69] | 2.65 | 6.93 | 27.17 | |
| 8e | R | –13.12 | 1.79 | –11.33 | [−10.49, −10.19] | 1.06 | 5.94 | 8.99 |
| S | –13.38 | 1.79 | –11.59 | [−10.66, −10.28] | 1.06 | 5.95 | 8.99 | |
| 8g | R | –12.47 | 1.79 | –10.68 | [−10.15, −9.82] | 1.18 | 2.82 | 24.37 |
| S | –13.56 | 1.79 | –11.77 | [−10.87, −10.26] | 1.18 | 2.82 | 24.37 | |
| 8k | R | –11.99 | 2.09 | –9.90 | [−10.48, −10.10] | 1.64 | 2.43 | 23.92 |
| S | –13.05 | 2.09 | –10.96 | [−10.57, −10.37] | 1.64 | 2.43 | 23.92 | |
Mulliken charges, dipole moment (μ), and quadrupole moment (Q) were calculated using the semiempirical method AM1. 95% CI of the estimated free energy of binding was calculated by 106 cycles of the bootstrapping method. ΔH: final intermolecular energy, −TΔS: ∼torsional free energy, and ΔGb: estimated free energy of binding (all in kcal/mol)
Figure 4 depicts the binding mode of both enantiomers of compound 8k. Compound (S)-8k occupied the binding site in an extended form. In this orientation, the pyrano[3,2-c]quinoline and benzyl moieties of compound (S)-8k filled the bottom and the entrance of the active site, respectively (Figure 4). (S)-8k established interactions with the key residues including Gly70, Phe158, Gln181, Asp214, Glu276, Glu304, Arg312, His348, and Asp349. The benzyl group of compound (S)-8k formed a π-anion interaction with the carboxylate moiety of Glu304 and a π-alkyl interaction with the side chain of Arg312. The importance of the 1,2,3-triazole ring in proper orientation of these derivatives in the active site of α-glucosidase was determined by forming two hydrogen bonds with key residue Arg312. In addition, the 1,2,3-triazole ring formed a π-anion interaction with residue Asp349. Furthermore, the central phenoxy group made a T-shaped π–π interaction with residue Phe158. In addition, quinolone created a π-anion interaction with Glu276 and π–π stacking interaction with His348. Moreover, the two hydrogen bonds were seen between the carbonyl group and nitrogen atom of the quinolone moiety and the residues Gln181 and Asp214, respectively.
Compound (R)-8k established interactions with the key residues including Phe177, Thr215, His239, Asn241, Glu276, His279, Ser308, Arg312, and Asp348 (Figure 4). The binding mode of the R-enantiomer was inverse to the S-enantiomer. The benzyl group of (R)-8k was placed in the bottom of the active site and formed vdW interactions with Glu276 as one of the catalytic triad residues. The pyrano[3,2-c]quinoline moiety was directed to the entrance of the enzyme active site and interacted with residues His279 and His239 via π–π interactions and the 1,2,3-triazol ring formed vdW and T-shaped π–π interactions with residues Thr215, Asp349, and Phe177, respectively. Furthermore, the CN, NH2, and carbonyl groups of pyrano[3,2-c]quinoline moiety made three hydrogen bonds with residues Arg312, Ser308, and Asn241. Also, the central phenoxy group established a π-cation interaction with Arg312.
The interaction profile of compound 8k showed that most functional groups interacted with the α-glucosidase active site and had an efficient contribution in free energy of binding. Due to the importance of explicit water and the flexibility of the receptor in the binding process, a 100 ns MD simulation was performed on the predicted complexes in the docking approach.
2.2.5. Molecular Dynamics Simulation
100 ns MD simulations were performed in the best ensemble docking models to get more realistic compound 8k (R- and S-enantiomers)-α-glucosidase complexes. The stability of the simulated systems was checked by monitoring total energy and temperature during the 100 ns production run. These thermodynamic parameters converged to an equilibrium state in less than 1 ns from the start of the MD simulation. The average temperature in 100 ns of the MD simulation at 300 K was equal to 300.6 K (±1.3 and 1.2) for S- and R-enantiomers of compound 8g in complex with α-glucosidase. The total energy of the systems was steady during the MD simulation (rsd <0.18%). So, the results indicated that energy conservation was satisfied in the simulations process.
rmsd of the α-glucosidase backbone atoms reached a plateau after 19 ns in both systems (average rmsd of the backbone is 0.30 ± 0.060 nm—rsd <19.6% in the R-enantiomer and 0.25 ± 0.021 nm—rsd <8.3% in the S-enantiomer). The radius of gyration was constant during the whole MD run (rsd <0.23 and 0.59% in R- and S-enantiomers, respectively).
The stability of both enantiomers in the active site of α-glucosidase was evaluated using the rmsd matrix (Figure 5). Figure 5 illustrates that compound (R)-8k formed a stable complex in the active site of the enzyme during the 100 ns MD simulation (blue square in the rmsd matrix indicates fluctuation less than 0.15 nm). In the initial steps of the MD simulation, a meta stable state could be tracked (red regions in the rmsd matrix of (R)-8k that finally reach a stable state). In this regard, the ligand passed through a transition state (big blue square) and remained stable in this conformer. These conformational changes caused the (R)-compound 8k enantiomer to drift deeper into the active site of the α-glucosidase enzyme. On the other hand, the (S)-compound 8k enantiomer drifted back out from the active site during the MD simulation time (notice the red and yellow regions in the rmsd matrix and conformational changes during the MD simulation time).
Figure 5.

rmsd matrix and trajectories of compound 8k (R- and S-enantiomers) during the 100 ns MD simulations. The catalytic triad residues Asp214, Glu276, and Asp349 is shown by pink sticks. (S)-8k experienced more conformational variations in the active site of the α-glucosidase enzyme in comparison to the R-enantiomer. (R)-8k seemed to be stable in the whole MD simulation time, but the S-enantiomer started to drift back out of the active site. Notice the distance between the 8k and the triad amino acids.
Interaction profiles of the R- and S-enantiomer of compound 8k revealed that the R-enantiomer was bound to α-glucosidase more effectively. The electrostatic interactions between the two enantiomers and α-glucosidase is inclined toward the R-enantiomer (−60.6 ± 4.3 kcal/mol in comparison to −18.6 ± 5.9 kcal/mol in (S)-8k). Contribution of the vdW interactions was more than that of the electrostatic interactions in binding energy. The vdW interactions of (R)-8k were −53.0 ± 2.8 kcal/mol. The stability and binding energy for both enantiomers indicated that the R-enantiomer (binding energy: −73.6 ± 3.8 kcal/mol) had higher binding affinity to the active site of the enzyme rather than the S-enantiomer (binding energy: −62.4 ± 5.9 kcal/mol). The in silico results were in accordance with the experimental data that related the potency to the substitution on the benzyl ring. The benzyl ring in the most stable complex ((R)-compound 8k) was located in the binding site and interacted with the enzyme active site more than the pyrano[3,2-c]quinoline moiety that occupied high solvent accessible entrance of the active site.
3. Conclusions
The findings of the present study indicated that all of the synthesized pyrano[3,2-c]quinoline-1,2,3-triazole hybrids 8a–o significantly inhibited the α-glucosidase activity of S. cerevisiae. Compound 8k was found to be the most active agent against this enzyme in a competitive manner with an IC50 value of 1.19 ± 0.05. In silico study revealed that both R- and S-enantiomers of compound 8k interacted with critical amino acids in the enzyme active site. However, the R-enantiomer exhibited more stability and binding affinity to the active site of the α-glucosidase enzyme rather than the S-enantiomer. The hydrophobic benzyl moiety of the R-enantiomer showed a tendency to occupy the bottom of the enzyme active site, while the hydrophilic pyrano[3,2-c]quinoline moiety interacted with the active site entrance. Thus, the synthesized pyrano[3,2-c]quinoline-1,2,3-triazole hybrids seem to be promising scaffolds for the development of novel α-glucosidase inhibitors.
4. Experimental Section
Melting points were measured on a Kofler hot stage apparatus and are uncorrected. The NMR (1H and 13C) and IR spectra were obtained by using a Bruker 400-, 300-NMR, and α FT-IR spectrometer on KBr discs, respectively. Elemental analysis was performed on an Elementar Analysen GmbH VarioEL CHNS mode. MS spectra were recorded by an Agilent Technology (HP) mass spectrometer operating at an ionization potential of 70 eV. All chemicals were obtained from Merck and Aldrich.
4.1. Chemistry
4.1.1. General Procedure for the Synthesis of 4-(Prop-2-yn-1-yloxy)benzaldehyde Derivatives (3a–c)
In a 25 mL round-bottomed flask, 1 mmol of benzaldehyde derivatives (1a–c), 1.2 mmol of propargyl bromide 80%, and 1.5 mmol of potassium carbonate in DMF (5 mL) were stirred at room temperature for 24 h. The reaction solution was poured into crushed ice, and then the solid precipitate was filtered and washed with water. The (3a–c) derivatives were used for the next step without further purification.35
4.1.2. General Procedure for the Synthesis of 2-Amino-5-oxo-4-(4-(prop-2-yn-1-yloxy)phenyl)-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (6a–c)
In a 25 mL round-bottomed flask, 1 mmol of derivatives 3a–c, 1.2 mmol of malononitrile, and 4-hydroxyquinoline-2 (1H)-one (1 mmol) in ethanol 98% (5 mL) were stirred for 4 h under reflux conditions. After the reaction was completed, the precipitated product was filtered and washed with ethanol. The 6a–c derivatives were used for the next step without further purification.36
4.1.3. General Procedure for the Synthesis of 2-Amino-4-(4-((1-benzyl-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8a–o)
To generate the benzyl azide derivatives, NaN3 (1.5 mmol) and various benzyl halide derivatives (1.2 mmol) were stirred in DMF (4 mL) at room temperature for 2 h. Intermediates 6a–c, 2 drops of NEt3, ascorbic acid (5% mmol), CuSO4·5H2O (5%mmol), and H2O (1 mL) were added to the previous solution and stirred at room temperature for 24 h to obtain final quinoline-1,2,3-triazole hybrids 8a–o. After completion of the reaction, ice was added to the reaction mixture and the resulting precipitate (8a–o) was filtered and washed with water.17
4.1.3.1. 2-Amino-4-(4-((1-benzyl-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8a)
White solid; yield: 59%, mp 168–171 °C. FT-IR (KBr) νmax (cm–1): 3324, 3188, 2961, 2868, 2197, 1672, 1507, 1381, 1257, 1174, 762; 1H NMR (300 MHz, DMSO-d6): δppm 11.79 (s, 1H, NH), 8.29 (s, 1H, H-triazole), 7.92 (d, J = 7.5 Hz, 1H, H10), 7.60 (t, J = 7.5 Hz, 1H, H9), 7.26–7.40 (m, 9H, NH2, H7, H8, H2″, H3″, H4″, H5″, H6″), 7.13 (d, J = 8.6 Hz, 2H, H2′, H6′), 6.95 (d, J = 8.6 Hz, 2H, H3′, H5′), 5.62 (s, 2H, CH2–O), 5.10 (s, 2H, CH2), 4.46 (s, 1H, H4), 13C NMR (76 MHz, DMSO-d6): δppm 160.98, 159.44, 157.35, 151.45, 138.15, 137.28, 136.44, 133.55, 131.68, 129.23, 128.94, 128.68, 128.63, 128.42, 125.19, 122.28, 120.38, 115.81, 114.98, 112.51, 110.32, 61.53, 58.43, 53.33, 36.33. Anal. Calcd for C29H22N6O3: C, 69.31; H, 4.41; N, 16.72. Found: C, 69.62; H, 4.35; N, 16.50.
4.1.3.2. 2-Amino-4-(4-((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8b)
White solid; yield: 40%, mp 146–148 °C. IR (KBr): FT-IR (KBr) νmax (cm–1): 3457, 3327, 2925, 2863, 2197, 1671, 1648, 1509, 1381, 1257, 1129, 1107, 764; 1H NMR (300 MHz, DMSO-d6): δppm 11.79 (s, 1H, NH), 8.25 (s, 1H, H-triazole), 7.92 (d, J = 8.1 Hz, 1H, H10), 7.59 (t, J = 8.1 Hz, 1H, H9), 7.35 (d, J = 8.1 Hz, 1H, H7), 7.19–7.31 (m, 7H, NH2, H8, H2″, H3″ H5″, H6″), 7.15 (d, J = 8.8 Hz, 2H, H2′, H6′), 6.95 (d, J = 8.8 Hz, 2H, H3′, H5′), 5.55 (s, 2H, CH2–O), 5.09 (s, 2H, CH2), 4.47 (s, 1H, H4), 2.28 (s, 3H, −CH3). 13C NMR (76 MHz, DMSO-d6): δppm 160.96, 159.42, 157.37, 151.43, 138.20, 137.98, 137.29, 133.47, 131.63, 129.76, 128.97, 128.51, 124.92, 122.45, 122.24, 120.42, 115.82, 114.94, 112.52, 110.33, 61.55, 58.39, 53.12, 36.37, 21.17. Anal. Calcd for C30H24N6O3: C, 69.76; H, 4.68; N, 16.27. Found: C, 69.42; H, 4.55; N, 16.10.
4.1.3.3. 2-Amino-4-(4-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8c)
White solid; yield: 61%, mp 192–194 °C. IR (KBr) νmax (cm–1): 3404, 3327, 2925, 2863, 2197, 1671, 1648, 1622, 1509, 1381, 1257, 1129, 1115, 1107, 764; 1H NMR (300 MHz, DMSO-d6): δppm 11.79 (s, 1H, NH), 8.30 (s, 1H, H-triazole), 7.92 (d, J = 8.1 Hz, 1H, H10), 7.59 (t, J = 8.1 Hz, 1H, H9), 7.19–7.44 (m, 8H, NH2, H7, H8, H2″, H3″, H5″, H6″), 7.14 (d, J = 8.5 Hz, 2H, H2′, H6′), 6.95 (d, J = 8.5 Hz, 2H, H3′, H5′), 5.61 (s, 2H, CH2–O), 5.01 (s, 2H, CH2), 4.47 (s, 1H, H4). 13C NMR (76 MHz, DMSO-d6): δppm 163.96, 160.96, 160.73, 159.41, 157.36, 151.43, 138.20, 137.31, 132.76, 132.71, 131.64, 130.87, 130.75, 128.98, 125.03, 122.46, 122.24, 120.42, 116.23, 115.95, 115.81, 114.94, 112.51, 110.32, 61.54, 58.38, 52.50, 36.37. MS: m/z (%) = 520.2 (M+, 1), 361.1 (2), 287.0 (2), 213.0 (3), 133.1 (4), 98 (52), 80 (100). Anal. Calcd for C29H21FN6O3: C, 66.92; H, 4.07; N, 16.15. Found: C, 66.81; H, 4.15; N, 16.20.
4.1.3.4. 2-Amino-4-(4-((1-(4-chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8d)
White solid; yield: 57%, mp 140–142 °C. 1H NMR (300 MHz, DMSO-d6): δppm 11.78 (s, 1H, NH), 8.30 (s, 1H, H-triazole), 7.92 (d, J = 7.2 Hz, 1H, H10), 7.59 (t, J = 7.2 Hz, 1H, H9), 7.26–7.47 (m, 8H, NH2, H7, H8, H2″, H3″, H5″, H6″), 7.14 (d, J = 8.3 Hz, 2H, H2′, H6′), 6.95 (d, J = 8.3 Hz, 2H, H3′, H5′), 5.62 (s, 2H, CH2–O), 5.11 (s, 2H, CH2), 4.47 (s, 1H, H4). 13C NMR (76 MHz, DMSO-d6): δppm 160.95, 159.42, 157.36, 151.43, 138.20, 137.32, 135.47, 133.36, 131.64, 130.42, 129.25, 128.98, 125.19, 122.45, 122.23, 115.82, 114.95, 112.52, 110.33, 61.55, 58.39, 52.50, 36.37. Anal. Calcd for C29H21ClN6O3: C, 64.87; H, 3.94; N, 15.65. Found: C, 65.71; H, 4.10; N: 15.41.
4.1.3.5. 2-Amino-4-(4-((1-(4-bromobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8e)
White solid; yield:51%, mp 203–205 °C. 1H NMR (300 MHz, DMSO-d6): δppm 11.79 (s, 1H, NH), 8.30 (s, 1H, H-triazole), 7.92 (d, J = 8.1 Hz, 1H, H10), 7.57–7.61 (m, 3H, H9, H5″, H3″), 7.27–7.36 (m, 6H, NH2, H7, H8, H2″, H6″), 7.14 (d, J = 8.7 Hz, 2H, H2′, H6′), 6.96 (d, J = 8.7 Hz, 2H, H3′, H5′), 5.61 (s, 2H, CH2–O), 5.11 (s, 2H, CH2), 4.47 (s, 1H, H4). 13C NMR (76 MHz, DMSO-d6): δppm 160.95, 159.41, 157.36, 151.43, 138.20, 137.32, 135.89, 132.18, 131.64, 130.73, 128.98, 122.45, 122.23, 121.92, 115.82, 114.94, , 112.51, 110.32, 61.54, 58.38, 52.56, 36.37. MS: m/z (%) = 583.1 (M + 2+, 1), 581.1.1 (M+, 1), 279.0 (2), 238.0 (3), 213 (4), 149 (7), 133.1 (4), 98 (52), 80 (100). Anal. Calcd for C29H21BrN6O3: C, 59.91; H, 3.64; N, 14.45. Found: C, 60.11; H, 3.59; N: 14.48.
4.1.3.6. 2-Amino-4-(4-((1-benzyl-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxyphenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8f)
White solid; yield: 83%, mp 185–186 °C. IR (KBr) νmax (cm–1): 3325, 3179, 3003, 2867, 2194, 1673, 1629, 1559, 1378, 1029, 755; 1H NMR (400 MHz, DMSO-d6): δppm 11.78 (s, 1H, NH), 8.26 (s, 1H, H-triazole), 7.91 (d, J = 6.5 Hz, 1H, H10), 7.58 (t, J = 6.5 Hz, 1H, H9), 7.28–7.38 (m, 7 H, H7, H8, H2″, H3″, H4″, H5″, H6″), 7.25 (s, 2H–NH2), 7.03 (d, J = 8.3 Hz, 1H, H5′), 6.87 (s, 1H, H2′), 6.56 (d, J = 8.3 Hz, 1H, H6′), 5.60 (s, 2H, CH2–O), 5.06 (s, 2H, CH2), 4.47 (s, 1H, H4), 3.69 (s, 3H, −OMe), 13C NMR (100 MHz, DMSO-d6): δppm 160.51, 159.03, 151.01, 148.68, 146.31, 137.70, 137.50, 135.98, 131.14, 131.09, 128.72, 128.10, 127.92, 124.74, 121.94, 121.74, 119.02, 115.31, 113.74, 112.02, 111.73, 109.72, 61.76, 57.71, 55.41, 52.78, 36.13. MS: m/z (%) = 532.2 (M+, 1), 279.0 (2), 212.9 (4), 133.1 (4), 98 (50), 79.9 (100). Anal. Calcd for C30H24N6O4: C, 67.66; H, 4.54; N, 15.78. Found: C, 67.79; H, 4.49; N, 15.82.
4.1.3.7. 2-Amino-4-(3-methoxy-4-((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8g)
White solid; yield: 85%, mp 164–168 °C. IR (KBr) νmax (cm–1): 3438, 2924, 2866, 1674, 1628, 1588, 1512, 1379, 1269, 1136, 1031, 759; 1H NMR (300 MHz, DMSO-d6): δppm 11.80 (s, 1H, NH), 8.24 (s, 1H, H-triazole), 7.92 (d, J = 7.7 Hz, 1H, H10), 7.59 (t, J = 7.7 Hz, 1H, H9), 7.17–7.37 (m, 8H, NH2, H7, H8, H2″, H3″ H5″, H6″), 7.03 (d, J = 8.2 Hz, 1H, H5′), 6.88 (s, 1H, s, 1H, H2′), 6.67 (d, J = 8.2 Hz, 1H, H6′), 5.55 (s, 2H, CH2–O), 5.06 (s, 2H, CH2), 4.49 (s, 1H, H4), 3.89 (s, 3H, −OMe), 2.28 (s, 3H, −Me), 13C NMR (76 MHz, DMSO-d6): δppm 161.03, 159.53, 151.52, 149.16, 146.83, 138.21, 137.99, 133.45, 131.66, 129.80, 129.76, 128.59, 128.53, 122.45, 122.25, 119.54, 115.82, 114.17, 112.53, 112.19, 110.21, 62.26, 58.21, 55.90, 53.12, 36.64, 21.18. MS: m/z (%) = 546.2 (M+, 1), 279.0 (2), 167.1 (25), 125.1 (100), 89 (12). Anal. Calcd for C31H26N6O4: C, 68.12; H, 4.79; N, 15.38. Found: C, 68.19; H, 4.69; N, 15.58.
4.1.3.8. 2-Amino-4-(4-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxyphenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8h)
White solid; yield: 88%, mp 146–148 °C. 1H NMR (400 MHz, DMSO-d6): δppm 11.86 (s, 1H, NH), 8.33 (s, 1H, H-triazole), 7.98 (d, J = 7.8 Hz, 1H, H10), 7.66 (t, J = 7.8, 1H, H9), 7.28–7.48 (m, 8H, NH2, H7, H8, H2″, H3″, H5″, H6″), 7.11 (d, J = 8.0 Hz, 1H, H5′), 6.95 (s, 1H, s, 1H, H2′), 7.43 (d, J = 8.0 Hz, 1H, H6′), 5.67 (s, 2H, CH2–O), 5.13 (s, 2H, CH2), 4.55 (s, 1H, H4), 3.76 (s, 3H, −OMe), 13C NMR (100 MHz, DMSO-d6): δppm 163.06, 160.61, 160.52, 159.00, 151.01, 148.63, 146.30, 137.69, 137.50, 132.22, 132.20, 131.15, 130.36, 130.28, 124.62, 121.95, 121.74, 119.03, 115.68, 115.47, 115.30, 113.63, 112.01, 111.65, 109.68, 61.71, 57.69, 55.38, 51.97, 36.13. MS: m/z (%) = 550.2 (M+, 1), 336.1 (2), 279.1 (7), 149.1 (45), 80 (13), 73 (100). Anal. Calcd for C30H23FN6O4: C, 65.5; H, 4.21; N, 15.27. Found: C, 65.33; H, 4.25; N, 15.46.
4.1.3.9. 2-Amino-4-(4-((1-(4-chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxyphenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8i)
White solid; yield: 69%, mp174–176 °C. IR (KBr): 3333, 3179, 2929, 2854, 2193, 1674, 1628, 1597, 1511, 1378, 1269, 1137, 1016, 763; 1H NMR (300 MHz, DMSO-d6): δppm 11.80 (s, 1H, NH), 8.30 (s, 1H, H-triazole), 7.92 (d, J = 7.6 Hz, 1H, H10), 7.59 (t, J = 7.6 Hz, 1H, H9), 7.27–7.50 (m, 8H, NH2, H7, H8, H2″, H3″, H5″, H6″), 7.04 (d, J = 8.2, 1H, H5′), 6.89 (s, 1H, H2′), 6.67 (d, J = 8.2 Hz, 1H, H6′), 5.62 (s, 2H, CH2–O), 5.07 (s, 2H, CH2), 4.49 (s, 1H, H4), 3.70 (s, 3H, −OMe), 13C NMR (76 MHz, DMSO-d6): δppm 161.03, 159.54, 151.54, 149.19, 146.83, 138.21, 138.06, 135.42, 133.38, 131.65, 130.78, 130.50, 130.45, 129.25, 129.18, 122.45, 122.26, 119.57, 115.83, 114.24, 112.53, 112.24, 110.20, 62.28, 58.22, 55.92, 52.53, 36.65. Anal. Calcd for C30H23ClN6O4: C, 63.55; H, 4.09; N, 14.82. Found: C, 63.43; H, 4.15; N: 14.96.
4.1.3.10. 2-Amino-4-(4-((1-(4-bromobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxyphenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8j)
White solid; yield: 64%, mp 180–183 °C. 1H NMR (400 MHz, DMSO-d6): δppm 11.78 (s, 1H, NH), 8.27 (s, 1H, H-triazole), 7.90 (d, J = 8.0 Hz, 1H, H10), 7.57–7.60 (m, 3H, H9, H3″, H5″), 7.25–7.35 (m, 6 H, NH2, H7, H8, H2″, H6″), 7.03 (d, J = 8.2 Hz, 1H, H5′), 6.87 (s, 1H, s, 1H, H2′), 6.66 (d, J = 8.2 Hz, 1H, H6′), 5.59 (s, 2H, CH2–O), 5.06 (s, 2H, CH2), 4.47 (s, 1H, H4), 3.69 (s, 3H, −OMe), 13C NMR (100 MHz, DMSO-d6): δppm 160.51, 159.01, 151.01, 148.66, 146.32, 137.69, 137.52., 135.37, 131.66, 131.15, 130.23, 124.82, 121.94, 121.75, 121.41, 119.05, 115.31, 113.68, 112.01, 111.69, 109.69, 61.75, 57.70, 55.40, 52.03, 36.13. Anal. Calcd for C30H23BrN6O4: C, 58.93; H, 3.79; N, 13.74. Found: C, 59.13; H, 3.85; N, 13.89.
4.1.3.11. 2-Amino-4-(3-((1-benzyl-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8k)
White solid; yield: 63%, mp 203–233 °C. IR (KBr): 3191, 3151, 2923, 2861, 2189, 1671, 1550, 1384, 1258, 1106, 1026, 763; 1H NMR (400 MHz, DMSO-d6): δppm 11.88 (s, 1H, NH), 8.39 (s, 1H, H-triazole), 8.00 (d, J = 8.2 Hz, 1H, H10), 7.67 (t, J = 8.2 Hz,1H, H9), 7.37–7.46 (m, 9 H, NH2, H7, H8, H1″, H2″, H3″, H5″, H6″), 7.31 (t, J = 8.0 Hz, 1H, H5′), 7.01 (d, J = 8.0 Hz, 1H, H4′), 6.92 (s, 1H, H2′), 6.88 (d, J = 8.0 Hz, 1H, H6′), 5.68 (s, 2H, CH2–O), 5.19 (s, 2H, CH2), 4.59 (s, 1H, H4), 13C NMR (100 MHz, DMSO-d6): δppm 160.45, 159.00, 157.94, 151.25, 146.01, 137.75, 135.95, 131.20, 129.54, 128.72, 128.09, 127.93, 124.78, 121.95, 121.76, 119.88, 119.81, 115.34, 114.44, 112.19, 111.98, 109.39, 60.99, 57.57, 52.81, 36.56. MS: m/z (%) = 502.2 (M+, 1), 336.1 (2), 288.1 (5), 91.1 (30), 64 (100). Anal. Calcd for C29H22N6O3: C, 69.31; H, 4.41; N, 16.72. Found: C, 69.42; H, 4.57; N, 16.66.
4.1.3.12. 2-Amino-4-(3-((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8l)
White solid; yield: 40%, mp 222–226 °C. IR (KBr): 3371, 3176, 2921, 2862, 2196, 1679, 1591, 1490, 1415, 1376, 1257, 1162, 1009, 768; 1H NMR (300 MHz, DMSO-d6): δppm 11.79 (s, 1H, NH), 8.27 (s, 1H, H-triazole), 7.92 (d, J = 7.0 Hz, 1H, H10), 7.60 (t, J = 7.0 Hz, 1H, H9), 7.19–7.37 (m, 9 H, 2H–NH2, H7, H10, H5́, H2″, H3″ H5″ H6″), 6.93 (d, J = 8.4 Hz, 1H, H4′), 6.79–6.84 (m, 2H, H2′, H6′), 5.54 (s, 2H, CH2–O), 5.10 (s, 2H, CH2), 4.50 (s, 1H, H4), 2.29 (s, 3H, −Me). 13C NMR (76 MHz, DMSO-d6): δppm 160.96, 159.52, 158.45, 151.75, 146.51, 143.36, 138.27, 137.95, 133.46, 131.71, 130.05, 129.76, 128.48, 125.06, 122.47, 122.27, 120.37, 115.85, 114.95, 112.72, 112.49, 109.91, 61.49, 58.08, 53.12, 37.07, 21.17. Anal. Calcd for C30H24N6O3: C, 69.76; H, 4.68; N, 16.27. Found: C, 69.62; H, 4.57; N, 16.16.
4.1.3.13. 2-Amino-4-(3-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8m)
White solid; yield: 55%, mp 228–231 °C. IR (KBr): 3325, 3190, 2926, 2862, 2191, 1673, 1602, 1552, 1511, 1411, 1382, 1268, 1167, 1021, 762; 1H NMR (400 MHz, DMSO-d6): δppm 11.87 (s, 1H, NH), 8.39 (s, 1H, H-triazole), 8.00 (d, J = 8.2 Hz, 1H, H10), 7.67 (t, J = 8.2 Hz, 1H, H9), 7.27–7.50 (m, 9 H, H8, H7, NH2, H5′, H2″, H3″, H5″, H6″), 7.01 (d, J = 8.0 Hz, 1H, H4′), 6.92 (s, 1H, H2′), 6.89 (d, J = 8.0 Hz, 1H, H6′), 5.68 (s, 2H, CH2–O), 5.18 (s, 2H, CH2), 4.58 (s, 1H, H4). 13C NMR (100 MHz, DMSO-d6): δppm 163.06, 160.45, 159.00, 157.92, 151.24, 146.01, 142.93, 137.75, 132.21, 132.19, 131.20, 130.31, 130.23, 129.54, 129.44, 124.63, 121.95, 121.76, 119.88, 119.80, 115.67, 115.45, 115.34, 114.45, 112.19, 111.97, 109.38, 60.98, 57.56, 51.99, 36.55. Anal. Calcd for C29H21FN6O3: C, 66.92; H, 4.07; N, 16.15. Found: C, 66.51; H, 4.17; N, 16.13.
4.1.3.14. 2-Amino-4-(3-((1-(4-chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8n)
White solid; yield: 56%, mp 213–216 °C. 1H NMR (300 MHz, DMSO-d6): δppm 11.82 (s, 1H, NH), 8.33 (s, 1H, H-triazole), 7.94 (d, J = 7.9 Hz, 1H, H10), 7.59 (t, J = 7.9 Hz 1H, H9), 7.25–7.45 (m, 9 H, H7, H8, NH2, H5′, H2″, H3″, H5″, H6″), 6.94 (d, J = 8.0 Hz, 1H, H4′), 6.88 (s, 1H, H2′), 6.84 (d, J = 8.0 Hz, 1H, H6′), 5.62 (s, 2H, CH2–O), 5.13 (s, 2H, CH2), 4.54 (s, 1H, H4). 13C NMR (76 MHz, DMSO-d6): δppm 161.01, 159.53, 158.45, 151.79, 146.55, 143.49, 138.27, 135.40, 133.37, 131.69, 130.36, 130.07, 129.22, 125.28, 122.46, 122.28, 120.44, 120.37, 115.86, 115.01, 112.72, 112.51, 109.90, 61.50, 58.15, 52.53, 37.10. Anal. Calcd for C29H21ClN6O3: C, 64.87; H, 3.94; N, 15.65. Found: C, 64.73; H, 4.05; N, 15.54.
4.1.3.15. 2-Amino-4-(3-((1-(4-bromobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile (8o)
White solid; yield: 43%, mp 153–155 °C. 1H NMR (300 MHz, DMSO-d6): δppm 11.81 (s, 1H, NH), 8.33 (s, 1H, H-triazole), 7.93 (d, J = 9.0 Hz, 1H, H10), 7.56–7.61 (m, 3H, H9, H2″, H6″), 7.25–7.35 (m, 7 H, 2H–NH2, H7, H8, H5′, H3″, H5″), 6.94 (d, J = 8.0 Hz, 1H, H4′), 6.86 (s, 1H, H2′) −6.87 (d, J = 8.0 Hz, 1H, H6′), 5.60 (s, 2H, CH2–O), 5.12 (s, 2H, CH2), 4.53 (s, 1H, H4). 13C NMR (76 MHz, DMSO-d6): δppm 160.99, 159.52, 158.44, 151.78, 146.54, 143.48, 138.27, 135.84, 132.16, 131.70, 130.67, 130.06, 125.30, 122.46, 122.28, 121.91, 120.42, 120.36, 119.90, 115.86, 115.00, 112.71, 112.50, 109.90, 61.49, 58.11, 52.58, 37.09. MS: m/z (%) = 583.2 (M+2+, 1), 581.2 (M+, 1), 369.1 (2), 279.1 (5), 238.1 (7), 149.0 (60), 80 (100). Anal. Calcd for C29H21BrN6O3: C, 59.91; H, 3.64; N, 14.42. Found: C, 59.98; H, 3.81; N, 14.38.
4.2. In Vitro α-Glucosidase Inhibition Assay
The α-Glucosidase enzyme (from S. cerevisiae, EC3.2.1.20, 20 U/mg) and the substrate (p-nitrophenyl glucopyranoside) were purchased from Sigma-Aldrich. An appropriate concentration of the enzyme was prepared in potassium phosphate buffer (pH 6.8, 50 mM), and 4,5-diarylimidazole-1,2,3-triazole hybrids 8a–o were dissolved in DMSO (final concentration 10%). The enzyme (20 μL), different concentrations of the synthesized compounds (20 μL), and potassium phosphate buffer (135 μL) were added to a 96-well plate and incubated at 37 °C for 10 min. Then, the substrate (25 μL, 4 mM) was added to each well of the plate and incubated at 37 °C for 20 min. Finally, the change in the absorbance was measured at 405 nm by using a Gen5 spectrophotometer (Power wave xs2, BioTek, America). DMSO, as a solvent control, and acarbose, as the standard inhibitor drug, were used. The percentage of inhibition for the synthesized compounds 8a–o, the control, and acarbose was calculated by using the following formula
The IC50 values of the tested agents were determined from the nonlinear regression curve using the logit method.37
4.3. Kinetic Study
The α-glucosidase solution (1 U/mL, 20 μL) was incubated with different concentrations of compound 8k (0, 0.5, 1.25, and 2.5 μM) at 30 °C for 15 min. The enzymatic reaction was initiated by adding various concentrations of p-nitrophenyl glucopyranoside as the substrate substance (1–10 mM). Then, change in the absorbance was measured for 20 min at 405 nm by using a spectrophotometer (Gen5, Power wave xs2, BioTek, America).37
4.4. Homology Modeling
The sequence of the eukaryotic yeast S. cerevisiae α-glucosidase was retrieved from the NCBI database (accession number P53341). The BLAST module in the NCBI was utilized to search the best template with the known structures in the Protein Data Bank.34 According to the obtained results, the isomaltase enzyme from the baker’s yeast (accession number P53051, PDB ID: 3A4A) at 1.60 Å resolution was selected as a template for the homology modeling study. A pairwise sequence alignment was generated using CLUSTALW based on the default parameters.38 Following the sequence alignment, the structure of the isomaltase enzyme was used as a template for building an α-glucosidase model with the Modeller 9v24 software.39 A number of 106 conformations of α-glucosidase was generated. The best model was selected with the lowest value of the DOPE. The general stereochemical quality of the final selected model was evaluated using the Ramachandran plot provided by PROCHECK.40
4.5. Docking Study
The 3D structures of acarbose and the selected compounds 8e, 8g, and 8k were optimized by ORCA using the PBE0/def2-TZVP method.41 AM1BCC charges and torsional degree of freedom were assigned for each ligand. Polar hydrogen atoms and Koullman charges were added to the enzyme structure using AutoDockTools-1.5.4. AutoGrid maps for each atom type in the ligands, were calculated with 0.375 Å spacing between grid points. The center of the grid box was placed at x = 85.87, y = 69.585, and z = 40.072. The dimensions of the active site box were set at 60 × 60 × 60 Å. Flexible ligand dockings were accomplished for the selected compounds. 200 GA runs, 27,000 maximum generations, a gene mutation rate of 0.02, and a crossover rate of 0.8 were set for the LGA method. The lowest energy conformation of the highest populated cluster was selected for the analysis. Graphic visualizations were done by Chimera software.
4.6. MD Simulation
The MD simulation was performed using the GROMACS-2019 package with a standard AMBER99SB-ILDN force field. Ligand atom types were assigned by using ANTECHAMBER. Partial atomic charges were calculated using the AM1BCC method. The predicted complex of the α-glucosidase enzyme and the selected compounds was centered in a dodecahedron box and solvated in water molecules represented by the TIP3P model. Sixteen water molecules were replaced with nine Na+ atoms to get electro-neutrality. Linear constraint solver was used to constrain all bonds to their initial length. The particle mesh Ewald method with a cutoff length of 14 Å and cubic interpolation was used for both long range electrostatic and van der Waals interactions. Energy minimization was carried out using 10,000 cycles of steepest descent. The system was heated to 300 K in 100 ps and was equilibrated in NVT and NPT ensembles in each 100 ps. For production run, all of the constraints were removed and the 100 ns MD simulation was carried out in the NPT ensemble.
Acknowledgments
This work was supported by Vice-chancellor for Research and Technology of Hamadan University of Medical Sciences with project no. 140106084786.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c00133.
Spectral data for all compounds (FT-IR, NMR, and mass spectra) (PDF)
Author Contributions
S.E. synthesized and analyzed all of the compounds. A.E. and H.G. performed the molecular dynamics study. M.F. and S.M. performed in vitro α-glucosidase study. A.K. and M.M. provided some starting materials. Z.N. designed and edited the manuscript. All authors contributed to the subsequent revision of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Deng Y.; Li N.; Wu Y.; Wang M.; Yang S.; Zheng Y.; Deng X.; Xiang D.; Zhu Y.; Xu P.; Zhai Z.; Zhang D.; Dai Z.; Gao J. Global, Regional, and National Burden of Diabetes-Related Chronic Kidney Disease From 1990 to 2019. Front. Endocrinol. 2021, 12, 672350. 10.3389/fendo.2021.672350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- León-Jiménez D.; Miramontes-González J. P.; Márquez-López L.; Astudillo-Martín F.; Beltrán-Romero L. M.; Moreno-Obregón F.; Escalada-San Martín J. Basal insulin analogues in people with diabetes and chronic kidney disease. Diabetic Med. 2022, 39, e14679 10.1111/dme.14679. [DOI] [PubMed] [Google Scholar]
- Andreadi A.; Bellia A.; Di Daniele N.; Meloni M.; Lauro R.; Della-Morte D.; Lauro D. The molecular link between oxidative stress, insulin resistance, and type 2 diabetes: A target for new therapies against cardiovascular diseases. Curr. Opin. Pharmacol. 2022, 62, 85–96. 10.1016/j.coph.2021.11.010. [DOI] [PubMed] [Google Scholar]
- Bradley P. Hypothesis: Enhanced glucose availability and insulin resistance enhances an activated immune system and accounts for the obesity paradox. Clin. Obes. 2022, 12, e12521 10.1111/cob.12521. [DOI] [PubMed] [Google Scholar]
- Fadem S. Z.Staying Healthy with Kidney Disease; Springer, 2022. [Google Scholar]
- Lin C.-F.; Liu H.-C.; Lin S.-Y. Kidney Function and Risk of Physical and Cognitive Impairment in Older Persons with Type 2 Diabetes at an Outpatient Clinic with Geriatric Assessment Implementation. Diabetes, Metab. Syndr. Obes.: Targets Ther. 2022, 15, 79–91. 10.2147/dmso.s341935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escandón-Rivera S.; González-Andrade M.; Bye R.; Linares E.; Navarrete A. s.; Mata R. α-Glucosidase Inhibitors from Brickellia cavanillesii. J. Nat. Prod. 2012, 75, 968–974. 10.1021/np300204p. [DOI] [PubMed] [Google Scholar]
- Shen X.; Saburi W.; Gai Z.; Kato K.; Ojima-Kato T.; Yu J.; Komoda K.; Kido Y.; Matsui H.; Mori H.; et al. Structural analysis of the α-glucosidase HaG provides new insights into substrate specificity and catalytic mechanism. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015, 71, 1382–1391. 10.1107/s139900471500721x. [DOI] [PubMed] [Google Scholar]
- Hossain U.; Das A. K.; Ghosh S.; Sil P. C. An overview on the role of bioactive α-glucosidase inhibitors in ameliorating diabetic complications. Food Chem. Toxicol. 2020, 145, 111738. 10.1016/j.fct.2020.111738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt W.; Hanefeld M.; Fischer S.; Schulze J. Efficacy of α-glucosidase inhibitors on lipids in NIDDM subjects with moderate hyperlipidaemia. Eur. J. Clin. Invest. 1994, 24, 45–49. 10.1111/j.1365-2362.1994.tb02256.x. [DOI] [PubMed] [Google Scholar]
- Chapel C.; Garcia C.; Roingeard P.; Zitzmann N.; Dubuisson J.; Dwek R. A.; Trepo C.; Zoulim F.; Durantel D. Antiviral effect of α-glucosidase inhibitors on viral morphogenesis and binding properties of hepatitis C virus-like particles. J. Gen. Virol. 2006, 87, 861–871. 10.1099/vir.0.81503-0. [DOI] [PubMed] [Google Scholar]
- Tundis R.; Loizzo M.; Menichini F. Natural products as α-amylase and α-glucosidase inhibitors and their hypoglycaemic potential in the treatment of diabetes: an update. Mini-Rev. Med. Chem. 2010, 10, 315–331. 10.2174/138955710791331007. [DOI] [PubMed] [Google Scholar]
- Mahmood N. A review of α-amylase inhibitors on weight loss and glycemic control in pathological state such as obesity and diabetes. Comp. Clin. Pathol. 2016, 25, 1253–1264. 10.1007/s00580-014-1967-x. [DOI] [Google Scholar]
- Najafi Z.; Mahdavi M.; Saeedi M.; Karimpour-Razkenari E.; Asatouri R.; Vafadarnejad F.; Moghadam F. H.; Khanavi M.; Sharifzadeh M.; Akbarzadeh T. Novel tacrine-1, 2, 3-triazole hybrids: In vitro, in vivo biological evaluation and docking study of cholinesterase inhibitors. Eur. J. Med. Chem. 2017, 125, 1200–1212. 10.1016/j.ejmech.2016.11.008. [DOI] [PubMed] [Google Scholar]
- Najafi Z.; Mahdavi M.; Saeedi M.; Karimpour-Razkenari E.; Edraki N.; Sharifzadeh M.; Khanavi M.; Akbarzadeh T. Novel tacrine-coumarin hybrids linked to 1, 2, 3-triazole as anti-Alzheimer’s compounds: In vitro and in vivo biological evaluation and docking study. Bioorg. Chem. 2019, 83, 303–316. 10.1016/j.bioorg.2018.10.056. [DOI] [PubMed] [Google Scholar]
- Karypidou K.; Ribone S. R.; Quevedo M. A.; Persoons L.; Pannecouque C.; Helsen C.; Claessens F.; Dehaen W. Synthesis, biological evaluation and molecular modeling of a novel series of fused 1,2,3-triazoles as potential anti-coronavirus agents. Bioorg. Med. Chem. Lett. 2018, 28, 3472–3476. 10.1016/j.bmcl.2018.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y.; Liu Z.; Liu J.; Huang B.; Kang D.; Zhang H.; De Clercq E.; Daelemans D.; Pannecouque C.; Lee K.-H.; et al. Targeting the entrance channel of NNIBP: Discovery of diarylnicotinamide 1, 4-disubstituted 1, 2, 3-triazoles as novel HIV-1 NNRTIs with high potency against wild-type and E138K mutant virus. Eur. J. Med. Chem. 2018, 151, 339–350. 10.1016/j.ejmech.2018.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son Hai D.; Thi Thu Ha N.; Tien Tung D.; Thi Kim Giang N.; Thi Thu Huong N.; Huu Anh H.; Thi Kim Van H.; Ngoc Toan V.; Toan D. N.; Thanh N. D. Synthesis, biological evaluation and induced fit docking simulation study of d-glucose-conjugated 1 H-1, 2, 3-triazoles having 4 H-pyrano [2, 3-d] pyrimidine ring as potential agents against bacteria and fungi. New J. Chem. 2022, 46, 8303–8323. 10.1039/d1nj05330b. [DOI] [Google Scholar]
- Bangalore P. K.; Vagolu S. K.; Bollikanda R. K.; Veeragoni D. K.; Choudante P. C.; Misra S.; Sriram D.; Sridhar B.; Kantevari S. Usnic acid enaminone-coupled 1, 2, 3-triazoles as antibacterial and antitubercular agents. J. Nat. Prod. 2019, 83, 26–35. 10.1021/acs.jnatprod.9b00475. [DOI] [PubMed] [Google Scholar]
- Srinivas Reddy M.; Swamy Thirukovela N.; Narsimha S.; Ravinder M.; Kumar Nukala S. Synthesis of fused 1, 2, 3-triazoles of Clioquinol via sequential CuAAC and CH arylation; in vitro anticancer activity, in silico DNA topoisomerase II inhibitory activity and ADMET. J. Mol. Struct. 2022, 1250, 131747. 10.1016/j.molstruc.2021.131747. [DOI] [Google Scholar]
- Singh H.; Singh J. V.; Gupta M. K.; Saxena A. K.; Sharma S.; Nepali K.; Bedi P. M. S. Triazole tethered isatin-coumarin based molecular hybrids as novel antitubulin agents: Design, synthesis, biological investigation and docking studies. Bioorg. Med. Chem. Lett. 2017, 27, 3974–3979. 10.1016/j.bmcl.2017.07.069. [DOI] [PubMed] [Google Scholar]
- Singh H.; Kumar M.; Nepali K.; Gupta M. K.; Saxena A. K.; Sharma S.; Bedi P. M. S. Triazole tethered C5-curcuminoid-coumarin based molecular hybrids as novel antitubulin agents: Design, synthesis, biological investigation and docking studies. Eur. J. Med. Chem. 2016, 116, 102–115. 10.1016/j.ejmech.2016.03.050. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Gupta M. K.; Saxena A. K.; Bedi P. M. S. Triazole linked mono carbonyl curcumin-isatin bifunctional hybrids as novel anti tubulin agents: Design, synthesis, biological evaluation and molecular modeling studies. Bioorg. Med. Chem. 2015, 23, 7165–7180. 10.1016/j.bmc.2015.10.013. [DOI] [PubMed] [Google Scholar]
- Singh J.; Sharma S.; Saxena A. K.; Nepali K.; Bedi P. M. S. Synthesis of 1, 2, 3-triazole tethered bifunctional hybrids by click chemistry and their cytotoxic studies. Med. Chem. Res. 2013, 22, 3160–3169. 10.1007/s00044-012-0312-7. [DOI] [Google Scholar]
- Wang G.; Peng Z.; Wang J.; Li X.; Li J. Synthesis, in vitro evaluation and molecular docking studies of novel triazine-triazole derivatives as potential α-glucosidase inhibitors. Eur. J. Med. Chem. 2017, 125, 423–429. 10.1016/j.ejmech.2016.09.067. [DOI] [PubMed] [Google Scholar]
- Avula S. K.; Khan A.; Rehman N. U.; Anwar M. U.; Al-Abri Z.; Wadood A.; Riaz M.; Csuk R.; Al-Harrasi A. Synthesis of 1H-1,2,3-triazole derivatives as new α-glucosidase inhibitors and their molecular docking studies. Bioorg. Chem. 2018, 81, 98–106. 10.1016/j.bioorg.2018.08.008. [DOI] [PubMed] [Google Scholar]
- Asgari M. S.; Mohammadi-Khanaposhtani M.; Kiani M.; Ranjbar P. R.; Zabihi E.; Pourbagher R.; Rahimi R.; Faramarzi M. A.; Biglar M.; Larijani B.; et al. Biscoumarin-1,2,3-triazole hybrids as novel anti-diabetic agents: Design, synthesis, in vitro α-glucosidase inhibition, kinetic, and docking studies. Bioorg. Chem. 2019, 92, 103206. 10.1016/j.bioorg.2019.103206. [DOI] [PubMed] [Google Scholar]
- Mohammadi-Khanaposhtani M.; Rezaei S.; Khalifeh R.; Imanparast S.; Faramarzi M. A.; Bahadorikhalili S.; Safavi M.; Bandarian F.; Nasli Esfahani E.; Mahdavi M.; et al. Design, synthesis, docking study, α-glucosidase inhibition, and cytotoxic activities of acridine linked to thioacetamides as novel agents in treatment of type 2 diabetes. Bioorg. Chem. 2018, 80, 288–295. 10.1016/j.bioorg.2018.06.035. [DOI] [PubMed] [Google Scholar]
- Taha M.; Ismail N. H.; Imran S.; Wadood A.; Rahim F.; Ali M.; Rehman A. U. Novel quinoline derivatives as potent in vitro α-glucosidase inhibitors: in silico studies and SAR predictions. MedChemComm 2015, 6, 1826–1836. 10.1039/c5md00280j. [DOI] [Google Scholar]
- Lee H.-W.; Yang J.-Y.; Lee H.-S. Quinoline-2-carboxylic acid isolated from Ephedra pachyclada and its structural derivatives show inhibitory effects against α-glucosidase and α-amylase. J. Korean Soc. Appl. Biol. Chem. 2014, 57, 441–444. 10.1007/s13765-014-4156-3. [DOI] [Google Scholar]
- Chehardoli G.; Gholamhoseini P.; Ebadi A.; Ziaei M.; Akbarzadeh T.; Saeedi M.; Mahdavi M.; Khoshneviszadeh M.; Najafi Z. 6-Methoxy-1-tetralone Derivatives Bearing an N-Arylpyridinium Moiety as Cholinesterase Inhibitors: Design, Synthesis, Biological Evaluation, and Molecular Docking Study. ChemistrySelect 2022, 7, e202201977 10.1002/slct.202201977. [DOI] [Google Scholar]
- Sadafi Kohnehshahri M.; Chehardoli G.; Bahiraei M.; Akbarzadeh T.; Ranjbar A.; Rastegari A.; Najafi Z. Novel tacrine-based acetylcholinesterase inhibitors as potential agents for the treatment of Alzheimer’s disease: Quinolotacrine hybrids. Mol. Diversity 2022, 26, 489–503. 10.1007/s11030-021-10307-2. [DOI] [PubMed] [Google Scholar]
- Liu M.; Zhang W.; Qiu L.; Lin X. Synthesis of butyl-isobutyl-phthalate and its interaction with -glucosidase in vitro. J. Biochem. 2011, 149, 27–33. 10.1093/jb/mvq110. [DOI] [PubMed] [Google Scholar]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. 10.1016/s0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Somakala K.; Amir M.; Sharma V.; Wakode S. Synthesis and pharmacological evaluation of pyrazole derivatives containing sulfonamide moiety. Monatsh. fur Chem. 2016, 147, 2017–2029. 10.1007/s00706-016-1694-x. [DOI] [Google Scholar]
- Lei M.; Ma L.; Hu L. A green, efficient, and rapid procedure for the synthesis of 2-amino-3-cyano-1, 4, 5, 6-tetrahydropyrano [3, 2-c] quinolin-5-one derivatives catalyzed by ammonium acetate. Tetrahedron Lett. 2011, 52, 2597–2600. 10.1016/j.tetlet.2011.03.061. [DOI] [Google Scholar]
- Adib M.; Peytam F.; Rahmanian-Jazi M.; Mahernia S.; Bijanzadeh H. R.; Jahani M.; Mohammadi-Khanaposhtani M.; Imanparast S.; Faramarzi M. A.; Mahdavi M.; et al. New 6-amino-pyrido[2,3-d]pyrimidine-2,4-diones as novel agents to treat type 2 diabetes: A simple and efficient synthesis, α-glucosidase inhibition, molecular modeling and kinetic study. Eur. J. Med. Chem. 2018, 155, 353–363. 10.1016/j.ejmech.2018.05.046. [DOI] [PubMed] [Google Scholar]
- Thompson J. D.; Higgins D. G.; Gibson T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šali A.; Blundell T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. 10.1107/s0021889892009944. [DOI] [Google Scholar]
- Neese F.; Wennmohs F.; Becker U.; Riplinger C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. 10.1063/5.0004608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.