

Abstract
Platinum-based materials are the most widely used catalysts in propane direct dehydrogenation, which could achieve a balanced activity between both propane conversion and propene formation. One of the core issues of Pt catalysts is how to efficiently activate the strong C–H bond. It has been suggested that adding second metal promoters could greatly solve this problem. In the current work, first-principles calculations combined with machine learning are performed in order to obtain the most promising metal promoters and identify key descriptors for control performance. The combination of three different modes of adding metal promoters and two ratios between promoters and platinum sufficiently describes the system under investigation. The activity of propane activation and the formation of propene are reflected by the increase or decrease of the adsorption energy and C–H bond activation of propane and propene after the addition of promoters. The data of adsorption energy and kinetic barriers from first-principles calculations are streamed into five machine-learning methods including gradient boosting regressor (GBR), K neighbors regressor (KNR), random forest regressor (RFR), and AdaBoost regressor (ABR) together with the sure independence screening and sparsifying operator (SISSO). The metrics (RMSE and R2) from different methods indicated that GBR and SISSO have the most optimal performance. Furthermore, it is found that some descriptors derived from the intrinsic properties of metal promoters can determine their properties. In the end, Pt3Mo is identified as the most active catalyst. The present work not only provides a solid foundation for optimizing Pt catalysts but also provides a clear roadmap to screen metal alloy catalysts.
Introduction
Propene is the cornerstone of the modern chemical industry and the main raw material for the synthesis of several key chemicals such as acrylonitrile, acrylic, plastic, etc.1 Propene is conventionally obtained from fluid catalytic cracking or steam cracking of naphtha and light diesel, which has huge energy consumption and a rather low selectivity.2 In recent years, the production capability of propene could not match the rapidly expanding demands, resulting in the so-called “propene gap”.3 This gap was up to 3.5 million tons in 2020. Therefore, it is urgently needed to develop and optimize effective routes for propene production. On the other hand, the Shale gas revolution not only changes the landscape of worldwide energy supply but also provides an abundant reservoir of light alkanes including methane, ethane, and propane.4 Under this circumstance, the on-purpose catalytic propane dehydrogenation (PDH)5 appeared to be the technology that could revolutionize propene production. Compared to conventional methods, PDH is less energy intensive and can provide a remarkable propene selectivity of up to 90%. Hence, PDH is indeed a promising solution to meet the challenge of the “propene gap”. Due to its merits, PDH has a long history of industrial application, which is first applied in the Pacol process.6 Nowadays, the annual propene obtained from PDH is up to 12 million tons on a global scale. Platinum is one of the most commonly used catalysts in PDH.7 Several industrial PDH processes employed Pt-based materials as catalysts including the Oleflex process,8 Catofin process,9 steam active reforming (STAR) process10 etc. Pt has a superior ability to activate the strong C–H bond in propane11 while keeping the C–C bond intact simultaneously.12 Consequently, it can guarantee high selectivity, which is pivotal to successful industrial application.13
Adding a second metal promoter for Pt catalysts is considered a way to achieve “two birds with one stone”. On the one hand, the second metal will form metal alloys with Pt and then make Pt into small ensembles, which will prevent sintering during reaction and regeneration.14 On the other hand, electronic interactions between Pt and metal promoters induce charge transfer and adjust d-band occupancy, both of which aim to alter the catalytic properties of C–H and C–C bond activation and the binding strength of the reactants and product.15 Therefore, the wisely chosen metal promoter could enhance the stability of Pt and lessen the extent of side reactions.16 Han et al.17 applied a Pt/Al2O3 catalyst modified by Cu in propane dehydrogenation. They concluded that interaction between Pt and Cu increased the energy barrier for C–C bond rupture and weakened propene adsorption; both were beneficial to selectivity and stability. Density functional theory (DFT) calculations carried out by Nykänen et al.18 also revealed the pronounced effect of a Sn promoter for a Pt catalyst in propane dehydrogenation. In fact, Sn is the most studied metal promoter for Pt catalysts and PtSn catalysts; it is also widely applied in the propane dehydrogenation industrial process.19−21 Considering the variety of types and ratios of metal promoters, it seems to be an endless search from an experimental perspective for the best candidate. The above discussions clearly demonstrate that adding second metal is an effective way to enhance selectivity and stability for Pt catalysts in propane dehydrogenation.22 There are various metal promoters reported in the literature that possessed certified activity over pure Pt including Zn,23 Cu,24 Mn,25 V,26 Ga,27 etc. The remaining questions are how to efficiently search for the best metal promoter in a huge configuration space and what is the viable principle behind the selection.
Great efforts have been made in the calculation to search for highly efficient alkane dehydrogenation catalysts. Hook et al.28 defined the difference between the barrier of ethene dehydrogenation and desorption energy as screening criteria to search for effective ethane dehydrogenation catalysts. They concluded that the binding energy of CH3 and CH is an effective descriptor. After screening, they identified that Pt3Pb and PtSb were the most promising candidates with top-layer adding configuration, as shown in Figure 1. Wang et al.22 used a descriptor-based microkinetic simulation to search nonprecious metal alloys for propane dehydrogenation. They identified CH3CHCH2/CH3CH2CH statistically and used it as a descriptor for binding energy. Moreover, they created a decision map from descriptors and found NiMo to be the most promising catalyst for propane dehydrogenation. In a similar way to Wang’s work, Xiao et al.16 also used descriptor-based microkinetic analysis combined with DFT calculation to find PDH catalysts from eight transition metals. The formation energies of CH3CH2* on a terrace and CH* on a stepped surface were two descriptors to characterize the energy aspects of other reaction intermediates and transition states. They found that Fe3Ga1 improved the selectivity and catalytic stability of propene compared with a Pt catalyst, and the theoretical prediction was verified by experiments.
Figure 1.
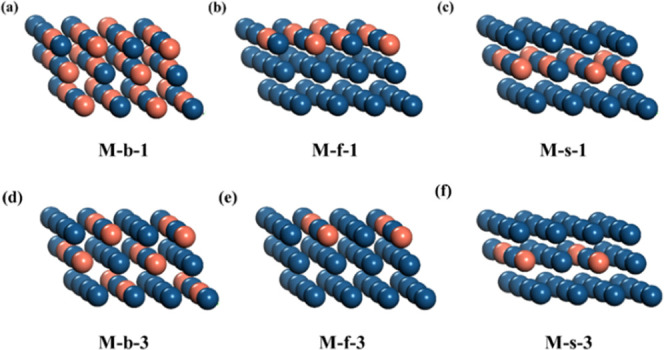
Schematic illustration of the PtM surface, where M represents a metal promoter. Pt is deep blue and M is orange. (a) Uniformly bulk adding Pt/M in a 1:1 ratio (b), top-layer adding Pt/M in a 1:1 ratio (c), sublayer adding Pt/M in a 1:1 ratio, (d) uniformly bulk adding Pt/M in a 3:1 ratio, (e) top-layer adding Pt/M in a 3:1 ratio, and (f) sublayer adding Pt/M in a 3:1 ratio.
Although great progress has been made in screening highly effective PDH catalysts, previous studies are dependent on the chosen descriptor and established linear relation. In some cases, adsorption energy descriptors are not readily available to experimentalists. It is also difficult to relate certain intrinsic properties of metals to changes in their properties. Moreover, the influence of the location of the metal promoter in the catalyst is still unclear. Aiming to resolve these remaining issues, a combined DFT calculation and machine-learning study is performed to screen the metal promoter candidates for Pt catalysts in propane dehydrogenation.29
As shown in Figure 1, three different methods of adding promoters (bulk, surface, subsurface) and two promoter/Pt ratios (1:1 and 1:3) were considered. The adsorption and dehydrogenation capabilities of propane and propene were investigated from first-principles calculations and different machine-learning algorithms. Five different methods including gradient boosting regressor (GBR), random forest regressor (RFR), K neighbors regressor (KNR), AdaBoost regressor (ABR), and SISSO are employed.29−31 Feature engineering is executed to establish the relation between intrinsic properties and activity. The performance of candidates obtained from machine learning is further examined by microkinetic simulation. In the end, a verified machine model is provided, and the design principle of the metal promoter is proposed.
Computational Details
DFT
All of the calculations were carried out by periodic density functional theory with the on-site Coulombic interaction using the Vienna Ab initio Simulation Package (VASP 4.5).30−36 The projected-augmented wave (PAW) method was used to explicate electron–ion interaction.35,37 For valence electrons, a plane-wave basis set was adopted with an energy cutoff of 400 eV. The revised Perdew–Burke–Ernzerhof (RPBE) functional was used as the exchange–correlation functional approximation for chemisorption energetics of molecules on transition-metal surfaces, which is known to yield a good description of adsorption38 and energy barrier39 on transition-metal surfaces. The Brillouin-zone integration was carried out at a 3 × 3 × 1 Monk horst–Pack k-point grid for Pt/M(111). A five-layer slab with a p(2 × 2) supercell is used to model the Pt/M(111) surface, the top three layers are allowed to fully relax in all calculations, and the two bottom layers are fixed at the optimized bulk geometry. The thickness of the vacuum region is 12 Å. The total energy and band structure energy are converged to an accuracy of 1 × 10–5 eV/atom to obtain accurate forces; a force tolerance of 0.03 eV/Å is used in all structure optimizations. Lattice constants of the PtM alloy are obtained by fitting the Birch–Murnaghan equation.40 The calculated lattice constants under different doping elements are listed in Note S2.
The adsorption energy Eads was calculated as
where E(adsorbate/slab) is the total energy of interacting species with the surface slab and Eslab and Eadsorbate are the individual energy of the slab (Pt/M surface) and the adsorbate (isolated C3H8, C3H6 molecules), respectively. The reaction pathways and energy barriers were calculated by using the climbing nudged elastic band (CI-NEB) method.41 The reaction barrier was calculated as the difference between the initial state with the highest image along the pathway.
The EFS and EIS are the total energies of the final state (FS) and the initial state (IS), respectively. The bonding strength is analyzed with COHP42 (crystal orbital Hamilton population analysis).
The microkinetic simulation was performed using the MKMCXX.43 The Gibbs free energy with ZPE correction was obtained from DFT frequency calculations at 800 K. The entropy of gas molecules was calculated by ideal gas approximation, whereas, for surface species, the entropies were calculated in the harmonic approximation. The reaction rates for adsorbed and desorbed species were calculated using Hertz–Knudsen kinetics. The rate constant for the adsorption reaction is calculated as
In the equation, P is the partial pressure of the gaseous molecule, A is the surface area on which the molecule adsorbs, M is the reactant mass, T is the temperature, and kb is Boltzmann’s constant. For the surface reaction, the rate constant for the forward reaction is calculated as
where “X” represents the reacting species, “≠” shows the transition state, “kr” denotes the reaction rate for forward/backward reaction, while “Q” and “h” represent the partition function and Plank’s constant.
Surface Model Construction
The metal (M) promoter candidates include Sn, Zn, In, Ga, Cu, Ag, Ge, Ni, Co, Fe, Au, Nb, Mo, Ru, Ti, Pd, Cr, Hg, V, Tc, and Pb. Different from previous studies15,18,28,44,45 that only considered a single configuration of a PtM catalyst, three different geometric configurations were considered in this work, as shown in Figure 1, to give a more unbiased description. First, M is uniformly mixed with Pt throughout the whole structure, as shown in Figure 1a. Second, M is restrained to be in the first layer of the surface, as shown in Figure 1b. In the third case, M is placed at the sublayer of the surface, as shown in Figure 1c. For each case, two ratios of Pt/M are examined, which are 1:1 and 1:3, respectively. For the latter two cases, only the number of Pt atoms in the layer containing M atoms is considered, regarding calculating the ratio.
ML Methods
All ML algorithms we adopted are conducted by the open-source code Scikit-learn46 in the Python3 environment. Four ML methods were employed to predict the adsorption energy and energy barriers: gradient boosting regressor (GBR),47 K neighbors regressor (KNR),48 random forest regressor (RFR),49 and AdaBoost regressor(ABR).50 Details of four algorithms are listed in Notes S3–S6. To improve the generalization of regression models, the input data collected from DFT computations were randomly shuffled and divided into the training set and test set with an 8:2 ratio. The root-mean-square error (RMSE), the coefficient of determination values (R2 score), and leave-one-out cross-validation were applied to evaluate the performance of the ML models, as described in Notes S7 and S8. In order to avoid overfitting and improve robustness, the parameters were systematically optimized, and the best set of hyperparameters was used in each ML method. The optimal model with hyperparameters that yielded the lowest validation error was further used to predict the adsorption energy and energy barrier in the test set. Details of the parameters of all models can be found in Table S10. Sure independence screening and sparsifying operator (SISSO)51 training was also performed to locate the low-dimensional descriptors to describe the catalytic performance of catalysts. A compressed-sensing-based procedure was used to select one or more most relevant candidate features and construct a linear model of the target property. Next, 10-fold cross-validation (CV10) was employed to test the predictive power of obtained models.
Results and Discussion
Adsorption and Activation of Propene and Propane
The adsorption of propane and propene is an indicator of the activity and selectivity of catalysts. Different adsorption behaviors of propane and propene also reflect the problems associated with coke formation. The typical adsorption configurations of propane and propene are shown in Figure S1. It is revealed that propene has stronger adsorption than propane, as shown in Table S5. As indicated from the ELF analysis shown in Figure S2, the origin of strong adsorption is attributed to the formation of covalent bonds, whereas propane is relatively weak due to physical adsorption. The crystal orbital Hamiltonian population (COHP) analysis also indicates that the adsorption of propene is stronger than that of propane, as shown in Figure S3. The strong adsorption of propene significantly increases the chance of a deep reaction and ultimately reduces the selectivity. The complete adsorption energy values of propane and propene from DFT calculations are shown in Table S5. The metal promoters brought a dramatic change in the adsorption of the reactants/products, as shown in Figure 2a,c. Most metal promoters enhanced the adsorption of propane, which was beneficial to its conversion; concurrently, the adsorption of propene was largely reduced, which facilitated its desorption. Therefore, addition of a metal promoter is indeed an effective way to tune the interaction between the reactant/product and Pt catalysts.
Figure 2.

Comparison between PtM surfaces with pure Pt surfaces regarding propane/propene adsorption and the C–H bond activation barrier. For adsorption energy, negative means the increased binding energy (more exothermic), while positive has the opposite meaning. For barrier, the positive means the increased barrier, while negative has the opposite meaning. More information is listed in Table S8. (a) C3H8 adsorption, (b) barriers of C–H bond activation in propane, (c) C3H6 adsorption, and (d) barriers of C–H bond activation in propene. Each abscissa represents the adsorption energy/barrier on the Pt–M surface minus the adsorption energy/barrier on the pure Pt surface (eV). The central vertical line represents the pure platinum case and is set to 0 eV. The meaning of the catalyst label is indicated in Figure 1.
C–H bond activation in propane and propene is another key descriptor to evaluate the catalytic performance of propane dehydrogenation. First of all, as shown in Table S6, several metal promoters were randomly selected, and the barriers of propane to 2-propyl and propene to 2-propenyl were calculated by the DFT method. Furthermore, it is found that the calculated C–H activation barriers are consistent with the well-established Brønsted–Evans–Polanyi relation,52 as shown in Figure S4, which has been widely applied for metal catalysts in alkane activation.53 Meanwhile, the reaction energy has a linear relationship with the barrier. For the rest of the metal promoters, the BEP relation is also applied to calculate the activation barrier of the C–H bond. The absolute value of the barrier is shown in Table S7, which is obtained from the linear relation shown in Figure S4. Metal promoters have different effects on C–H bond activation compared with pure Pt catalysts, as shown in Figure 2b,d. Ideally, it is expected that the C–H bond activation barriers of propane and propene are decreased or increased after adding a metal promoter, respectively. According to this principle, Mo, Cu, Zn, Sn, Au, Pd, Tc, Ge, and Ti have been identified to be effective promoters.
Feature Engineering
Generally, features should be values that can be easily queried in a database or easily computed. Also, it might be helpful to link it to the intrinsic properties of the metal promoter itself. In order to reduce redundancy, it is better that there is no linear relationship between parameter features. On the basis of these criteria, 21 physical and chemical properties are initially selected as the features of ML models. As shown in Table S9, these descriptors consist of four different categories: atomic structures, physical properties of metal, description of the electronic structure, and representation of the alloy surface. Although the above features cover a wide range of properties of metal promoters, they still seem a little cumbersome; moreover, some of them might be closely correlated. Therefore, these features are further screened. The Pearson correlation coefficient matrix is used to identify the correlation between the random feature pairs, as shown in Figure 3. Features that have a high correlation (|p|) with others are removed. For example, |p| between P and M is 0.96, and that between Hf and Mp is 0.93, both of which belong to a high positive correlation. These features will cause large interference. After preliminary screening, the number of primary features is reduced to 15, with a reasonable cutoff (|p| < 0.854). The features such as χ, Im, and ρ have a low linear correlation with all other features, indicating that they are independent.
Figure 3.

Pearson correlation analysis of the initial features. The meaning of the feature symbol is explained in Table S9.
Performance of the ML Model
The original data obtained from DFT calculations were normalized and preprocessed. To reduce the risk of overfitting, the test data set was reproduced by adding uniformly distributed random noises on a scale of −2 to 2%, and the training procedure was executed 500 times for each ML model. Then, the training set of the preprocessed data was analyzed by four ML regression algorithms including GBR, KNR, RFR, and ABR (detailed information about parameters can be found in Table S10).
Using the aforementioned optimized features (Table S11), the regression of adsorption energy and C–H bond activation barriers obtained from each model are summarized in Figure 4. The root-mean-square error (RMSE) and the coefficient of determination values (R2 score) are determined accordingly. From regression, RMSE and R2 scores clearly indicated that the GBR model had a similar performance for both the training and test data sets, with the former being less than 0.04 and the latter being above 0.8, respectively. Other models have much inferior metrics of the training set and the test set, as shown in Table S13. Therefore, GBR is identified as the best model among examined methods. Moreover, leave-one-out cross-validation was used in order to further verify the reliability of the GBR model, as shown in Figure S5, and there was no significant change in the RMSE of the training set and the test set.
Figure 4.

Comparison between DFT calculations and machine learning with metrics of RMSE and R2 of the test set. The first row shows propane adsorption energy, the second row shows propene adsorption energy, and the third row shows the C–H bond activation barrier. (a) RFR, (b) KNR, (c) ABR, and (d) GBR.
From the feature importance analysis shown in Figure S6, the most relevant features are revealed. For propane adsorption, the foremost two factors are lattice constants (a) and doping configuration (ST), while the most important factors affecting the adsorption of propylene are the atomic radius of the metal promoter (rd) and ST. For C–H bond activation, they are transferred charges (qm) and rd. It is assuring that the most decisive factors are related to the intrinsic properties of metals, which are easily acquired. This relation is not established in previous work.16,22,28
The predictions of adsorption energy and C–H activation barriers from the GBR model are shown in Table S12. In order to screen out highly effective second metal promoters, it should meet the following requirements: propane adsorption energy is larger than −0.51 eV, propene adsorption energy is less than −1.79 eV, the energy barrier of propane to 2-propyl is less than 0.76 eV, and propene to 2-propenyl is bigger than 1.02. The threshold values are taken from pure Pt surfaces. Under these criteria, it can guarantee suitable conversion of reactants and favorable selectivity, resulting in excellent yields. It was found that the best candidate is Mo from screening. In addition to Mo metal, several candidates with good performance were selected by the same screening method, such as Sc, Mn, etc.
The C–H bond activation pattern is further explored by using SISSO. A pool of candidate features is first constructed iteratively by combining 14 low-cost primary features listed in Table S14. The performance of the SISSO method is shown in Figure 5. RMSE, the averaged RMSE, and the distribution of errors of the activation barriers of propane (Eb8) and propene (Eb6) with 10-fold cross-validation (CV10) are collected as an evaluation index of the descriptor dimension. As shown in Figure 5a, CV10 starts to increase when the dimension is 4, and its optimal dimension can be determined to be four combined with the RMSE value. We employ the identified 4D SISSO model formula, as shown in Table 1, for the high-throughput screening of promoters. The outcome is shown in Table S15. With the same screening conditions as mentioned before, Zn, V, Mo, and Ti were identified as the most promising candidates. In particular, Mo was found to have the best performance, which is consistent with GBR model prediction.
Figure 5.

SISSO errors and their distribution for the C–H activation barrier of propane (Eb8) and propene (Eb6). (a) RMSE and the averaged RMSE of the 10-fold cross-validation. Distribution of errors for the best models vs DFT results for (b) Eb8 and (c) Eb6.
Table 1. Identified Equation from SISSO Models for Activation of Propane and Propene.
| equation for the energy barrier of C–H bond activation predicted based on SISSO analysis | |
|---|---|
| propane | 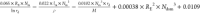 |
| propene | 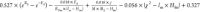 |
To further verify the predictions from ML regression, the TOF of predicted PtM catalysts is calculated from microkinetic simulation, as shown in Figure 6a. Indeed, Mo, Sc, Mn, Zn, Cu, and Ni promoters obtained from machine learning do have improved performance compared with pure Pt catalysts. Among them, Mo has the highest TOF. Therefore, Pt3Mo, with the configuration shown in Figure 1b and a ratio of 3:1, is identified to be the best catalyst candidate for propane dehydrogenation. Besides, an increased TOF of propene formation and the reduced apparent activation energy of Pt3Mo, as shown in Figure 6b,c, further validated its excellent activity and selectivity over conventional Pt catalysts through the investigated temperature range.
Figure 6.

(a) Calculated TOF of propane conversion of PtM (M = Mo, Sc, Mn, Zn, Cu, Ni, Ga). The dashed line represents the case of pure Pt. (b) TOF of C3H6 formation of Pt and Pt3Mo. (c) Apparent activation energy of Pt and Pt3Mo.
On the other hand, experimental reports of PtMo alloy dehydrogenation catalysts are rare. However, a few available experimental studies indicated the promising potential of PtMo catalysts. Kondarides et al.55 applied a PtMo6 cluster supported on MgO in butane, isobutane, and propane. They found that PtMo catalysts demonstrated higher yields and better stability than the monometallic counterparts of either Pt or Mo catalysts. It is also noted that the selectivity of PtMo to alkene is over 97%. Boufaden et al.56 synthesized a series of PtMo catalysts by varying the Pt/Mo ratio and applied in dehydrogenation of methylcyclohexane to toluene. This study uncovered that PtMo bimetallic catalysts had an improved activity over monometallic catalysts with the highest reaction rate at around 9 × 10–2 mole h–1g–1. Other predicted metal promoters such as Zn,57 Ni,58 and Cu59 are also reported to have excellent performance from experimental studies. The predictions from the current work will give new impetus to the experimental exploration of a PtMo bimetallic catalyst in a dehydrogenation reaction.
Conclusions
In summary, a combined first-principles calculations and machine learning study is performed to identify the effective metal promoter for Pt catalysts in propane dehydrogenation. The adsorption strength of propane/propene was calculated and compared, and the origin of deep reaction tendency was revealed. ELF and COHP analyses showed that the bonding properties of propane and propene were physical adsorption and covalent chemisorption, respectively. The addition of a metal promoter brought a significant change in adsorption. It is found that the adsorption energy of propane is increased while the counterpart of propene is decreased. In particular, the latter change will lessen the probability of a deep reaction and enhance the selectivity. The calculated dehydrogenation barriers of propane and propene are consistent with the well-established BEP relation. The features are mainly selected from the intrinsic properties of the metal, which are easy to access. Pearson correlation analysis refined the initial features and reduced redundant parameters. Based on DFT calculated adsorption energy and kinetic barriers, four methods including GBR, KNR, RFR, and ABR are performed for regression. GBR is verified to be the optimal method considering RMSE and R2 metrics. Moreover, a compressed-sensing method, SISSO, is also applied, which draws a good agreement with the outcome from GBR. On the other hand, properties such as lattice constants, metal radius, and adding methods are identified as the most relevant descriptors. TOF and apparent activation energy are calculated to verify the performance of the chosen candidate from machine learning. In the end, Mo with a surface ratio of 1:3 with Pt was identified as the most effective promoter, which outperformed pure Pt catalysts. Besides, Sc, Mn, Zn, and Cu also exceeded the performance of Pt. Overall, the current work supplied several concrete metal promoter candidates and lay out an effective model for screening metal alloy catalysts for propane dehydrogenation.
Acknowledgments
This work was supported by the Natural Science Foundation of Liao Ning Province (2021-MS-004), the Shenyang Normal University (BS202208), and the Program for Excellent Talents at Shenyang Normal University. It was also supported by the Fundamental Research Funds for the Central Universities, China.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c02675.
Detailed process of ML-DFT; details of lattice constants; computational methods of GBR, KNR, RFR, ABR, and adsorption configurations of propane and propene; Bader charge and COHP analysis; BEP relation; leave-one-out cross-validation average RMSE; importance of features, error distributions of SISSO, adsorption energy, and energy barriers obtained from DFT calculations; list of features for ML models, parameters of ML models, adsorption energy and energy barriers obtained from ML prediction; RMSE and R2 score of six models; and identified equation from SISSO models (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Qi W.; Yan P.; Su D. S. Oxidative Dehydrogenation on Nanocarbon: Insights into the Reaction Mechanism and Kinetics Via in Situ Experimental Methods. Acc. Chem. Res. 2018, 51, 640–648. 10.1021/acs.accounts.7b00475. [DOI] [PubMed] [Google Scholar]
- Solowey D. P.; Mane M. V.; Kurogi T.; Carroll P. J.; Manor B. C.; Baik M.-H.; Mindiola D. J. A New and Selective Cycle for Dehydrogenation of Linear and Cyclic Alkanes under Mild Conditions Using a Base Metal. Nat. Chem. 2017, 9, 1126–1132. 10.1038/nchem.2795. [DOI] [PubMed] [Google Scholar]
- Cavani F.; Ballarini N.; Cericola A. Oxidative Dehydrogenation of Ethane and Propane: How Far from Commercial Implementation?. Catal. Today 2007, 127, 113–131. 10.1016/j.cattod.2007.05.009. [DOI] [Google Scholar]
- Li X. Y.; Pei C. L.; Gong J. L. Shale Gas Revolution: Catalytic Conversion of C-1-C-3 Light Alkanes to Value-Added Chemicals. Chem 2021, 7, 1755–1801. 10.1016/j.chempr.2021.02.002. [DOI] [Google Scholar]
- Bricker J. C. Advanced Catalytic Dehydrogenation Technologies for Production of Olefins. Top. Catal. 2012, 55, 1309–1314. 10.1007/s11244-012-9912-1. [DOI] [Google Scholar]
- Bhasin M. M.; McCain J. H.; Vora B. V.; Imai T.; Pujado P. R. Dehydrogenation and Oxydehydrogenation of Paraffins to Olefins. Appl. Catal., A 2001, 221, 397–419. 10.1016/S0926-860X(01)00816-X. [DOI] [Google Scholar]
- Yang M.-L.; Zhu Y.-A.; Fan C.; Sui Z.-J.; Chen D.; Zhou X.-G. Density Functional Study of the Chemisorption of C1, C2 and C3 Intermediates in Propane Dissociation on Pt(111). J. Mol. Catal. A: Chem. 2010, 321, 42–49. 10.1016/j.molcata.2010.01.017. [DOI] [Google Scholar]
- Jacob T.; Goddard W. A. Chemisorption of (Chx and C2hy) Hydrocarbons on Pt(111) Clusters and Surfaces from Dft Studies. J. Phys. Chem. B 2005, 109, 297–311. 10.1021/jp0463868. [DOI] [PubMed] [Google Scholar]
- Won W.; Lee K. S.; Lee S.; Jung C. Repetitive Control and Online Optimization of Catofin Propane Process. Comput. Chem. Eng. 2010, 34, 508–517. 10.1016/j.compchemeng.2009.12.011. [DOI] [Google Scholar]
- Buker K.; Donnermeyer S.; Gehrke H.; Heinritz-Adrian M. Star Process (R) for the on-Purpose Production of Propylene. Oil Gas Eur. Mag. 2010, 36, 40–44. [Google Scholar]
- Wang Y. L.; Hu P.; Yang J.; Zhu Y. A.; Chen C. C-H Bond Activation in Light Alkanes: A Theoretical Perspective. Chem. Soc. Rev. 2021, 50, 4299–4358. 10.1039/D0CS01262A. [DOI] [PubMed] [Google Scholar]
- Ju T. Y.; Yang H. Q.; Li F. M.; Li X. Y.; Hu C. W. Activation of Propane C-H and C-C Bonds by a Diplatinum Cluster: Potential Energy Surfaces and Reaction Mechanisms. Struct. Chem. 2014, 25, 471–481. 10.1007/s11224-013-0311-4. [DOI] [Google Scholar]
- Zhang J.; Liu X.; Blume R.; Zhang A.; Schloegl R.; Su D. S. Surface-Modified Carbon Nanotubes Catalyze Oxidative Dehydrogenation of N-Butane. Science 2008, 322, 73–77. 10.1126/science.1161916. [DOI] [PubMed] [Google Scholar]
- Jiang F.; Zeng L.; Li S. R.; Liu G.; Wang S. P.; Gong J. L. Propane Dehydrogenation over Pt/Tio2-Al2o3 Catalysts. ACS Catal. 2015, 5, 438–447. 10.1021/cs501279v. [DOI] [Google Scholar]
- Zha S.; Sun G.; Wu T.; Zhao J.; Zhao Z. J.; Gong J. Identification of Pt-Based Catalysts for Propane Dehydrogenation Via a Probability Analysis. Chem. Sci. 2018, 9, 3925–3931. 10.1039/C8SC00802G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L.; Hu P.; Sui Z. J.; Chen D.; Zhou X. G.; Yuan W. K.; Zhu Y. A. Rational Design of Intermetallic Compound Catalysts for Propane Dehydrogenation from a Descriptor-Based Microkinetic Analysis. J. Catal. 2021, 404, 32–45. 10.1016/j.jcat.2021.09.008. [DOI] [Google Scholar]
- Han Z. P.; Li S. R.; Jiang F.; Wang T.; Ma X. B.; Gong J. L. Propane Dehydrogenation over Pt-Cu Bimetallic Catalysts: The Nature of Coke Deposition and the Role of Copper. Nanoscale 2014, 6, 10000–10008. 10.1039/C4NR02143F. [DOI] [PubMed] [Google Scholar]
- Nykänen L.; Honkala K. Density Functional Theory Study on Propane and Propene Adsorption on Pt(111) and Ptsn Alloy Surfaces. J. Phys. Chem. C 2011, 115, 9578–9586. 10.1021/jp1121799. [DOI] [Google Scholar]
- Ma Z. H.; Jiang A. J.; Li S.; Li J.; Sun L. Y.; An C. H. Synthesis of Pt-Sn Nanoalloy Catalysts with Enhanced Performance in the Dehydrogenation of Propane. Micro Nano Lett. 2019, 14, 794–798. 10.1049/mnl.2018.5024. [DOI] [Google Scholar]
- Nykänen L.; Honkala K. Selectivity in Propene Dehydrogenation on Pt and Pt3sn Surfaces from First Principles. ACS Catal. 2013, 3, 3026–3030. 10.1021/cs400566y. [DOI] [Google Scholar]
- Hauser A. W.; Gomes J.; Bajdich M.; Head-Gordon M.; Bell A. T. Subnanometer-Sized Pt/Sn Alloy Cluster Catalysts for the Dehydrogenation of Linear Alkanes. Phys. Chem. Chem. Phys. 2013, 15, 20727–20734. 10.1039/c3cp53796j. [DOI] [PubMed] [Google Scholar]
- Wang T.; Cui X. J.; Winther K. T.; Abild-Pedersen F.; Bligaard T.; Norskov J. K. Theory-Aided Discovery of Metallic Catalysts for Selective Propane Dehydrogenation to Propylene. ACS Catal. 2021, 11, 6290–6297. 10.1021/acscatal.0c05711. [DOI] [Google Scholar]
- Ingale P.; Knemeyer K.; Preikschas P.; Ye M. Y.; Geske M.; D’Alnoncourt R. N.; Thomas A.; Rosowski F. Design of Ptzn Nanoalloy Catalysts for Propane Dehydrogenation through Interface Tailoring Via Atomic Layer Deposition. Catal. Sci. Technol. 2021, 11, 484–493. 10.1039/D0CY01528H. [DOI] [Google Scholar]
- Sun S. J.; Sun G. D.; Pei C. L.; Zhao Z. J.; Gong J. L. Origin of Performances of Pt/Cu Single-Atom Alloy Catalysts for Propane Dehydrogenation. J. Phys. Chem. C 2021, 125, 18708–18716. 10.1021/acs.jpcc.1c04295. [DOI] [Google Scholar]
- Wu Z. W.; Bukowski B. C.; Li Z.; Milligan C.; Zhou L.; Ma T.; Wu Y.; Ren Y.; Ribeiro F. H.; Delgass W. N.; et al. Changes in Catalytic and Adsorptive Properties of 2 Nm Pt3mn Nanoparticles by Subsurface Atoms. J. Am. Chem. Soc. 2018, 140, 14870–14877. 10.1021/jacs.8b08162. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Zhou R. J.; Chang Q. Y.; Sui Z. J.; Zhou X. G.; Chen D.; Zhu Y. A. Tailoring Catalytic Properties of V2o3 to Propane Dehydrogenation through Single-Atom Doping: A Dft Study. Catal. Today 2021, 368, 46–57. 10.1016/j.cattod.2020.02.023. [DOI] [Google Scholar]
- Chang Q. Y.; Wang K. Q.; Sui Z. J.; Zhou X. G.; Chen D.; Yuan W. K.; Zhu Y. A. Rational Design of Single-Atom-Doped Ga2o3 Catalysts for Propane Dehydrogenation: Breaking through Volcano Plot by Lewis Acid-Base Interactions. ACS Catal. 2021, 11, 5135–5147. 10.1021/acscatal.0c05454. [DOI] [Google Scholar]
- Hook A.; Celik F. E. Predicting Selectivity for Ethane Dehydrogenation and Coke Formation Pathways over Model Pt-M Surface Alloys with Ab Initio and Scaling Methods. J. Phys. Chem. C 2017, 121, 17882–17892. 10.1021/acs.jpcc.7b03789. [DOI] [Google Scholar]
- Jones R. O. Density Functional Theory: Its Origins, Rise to Prominence, and Future. Rev. Mod. Phys. 2015, 87, 897–923. 10.1103/RevModPhys.87.897. [DOI] [Google Scholar]
- Kresse G.; Furthmüller J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. 10.1016/0927-0256(96)00008-0. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Abinitio Molecular-Dynamics for Liquid-Metals. Phys. Rev. B 1993, 47, 558–561. 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Ab-Initio Molecular-Dynamics Simulation of the Liquid-Metal Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. 10.1103/PhysRevB.49.14251. [DOI] [PubMed] [Google Scholar]
- Kresse G. Ab-Initio Molecular-Dynamics for Liquid-Metals. J. Non-Cryst. Solids 1995, 192-193, 222–229. 10.1016/0022-3093(95)00355-X. [DOI] [Google Scholar]
- Kresse G.; Joubert D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Kresse G.; Hafner J. Norm-Conserving and Ultrasoft Pseudopotentials for First-Row and Transition-Elements. J. Phys.: Condens. Matter 1994, 6, 8245–8257. 10.1088/0953-8984/6/40/015. [DOI] [Google Scholar]
- Blöchl P. E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- Hammer B.; Hansen L. B.; Norskov J. K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59, 7413–7421. 10.1103/PhysRevB.59.7413. [DOI] [Google Scholar]
- Hammer B. Adsorption, Diffusion, and Dissociation of No, N and O on Flat and Stepped Ru(0001). Surf. Sci. 2000, 459, 323–348. 10.1016/S0039-6028(00)00467-2. [DOI] [Google Scholar]
- Murnaghan F. D. The Compressibility of Media under Extreme Pressures. Proc. Natl. Acad. Sci. U.S.A. 1944, 30, 244–247. 10.1073/pnas.30.9.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkelman G.; Uberuaga B. P.; Jonsson H. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. 10.1063/1.1329672. [DOI] [Google Scholar]
- Dronskowski R.; Blochl P. E. Crystal Orbital Hamilton Populations (Cohp) - Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations. J. Phys. Chem. A 1993, 97, 8617–8624. 10.1021/j100135a014. [DOI] [Google Scholar]
- Filot I. A. W.; van Santen R. A.; Hensen E. J. M. The Optimally Performing Fischer-Tropsch Catalyst. Angew. Chem., Int. Ed. 2014, 53, 12746–12750. 10.1002/anie.201406521. [DOI] [PubMed] [Google Scholar]
- Yang M.-L.; Zhu Y.-A.; Zhou X.-G.; Sui Z.-J.; Chen D. First-Principles Calculations of Propane Dehydrogenation over Ptsn Catalysts. ACS Catal. 2012, 2, 1247–1258. 10.1021/cs300031d. [DOI] [Google Scholar]
- Liu X.; Lang W.-Z.; Long L.-L.; Hu C.-L.; Chu L.-F.; Guo Y.-J. Improved Catalytic Performance in Propane Dehydrogenation of Ptsn/Γ-Al2o3 Catalysts by Doping Indium. Chem. Eng. J. 2014, 247, 183–192. 10.1016/j.cej.2014.02.084. [DOI] [Google Scholar]
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Friedman J. H. Greedy Function Approximation: A Gradient Boosting Machine. Ann. Stat. 2001, 29, 1189–1232. 10.1214/aos/1013203451. [DOI] [Google Scholar]
- Devroye L.; Gyorfi L.; Krzyzak A.; Lugosi G. On the Strong Universal Consistency of Nearest-Neighbor Regression Function Estimates. Ann. Stat. 1994, 22, 1371–1385. 10.1214/aos/1176325633. [DOI] [Google Scholar]
- Breiman L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar]
- Tavallali P.; Yazdi M.; Khosravi M. R. Robust Cascaded Skin Detector Based on Adaboost. Multimedia Tools Appl. 2019, 78, 2599–2620. 10.1007/s11042-018-6385-7. [DOI] [Google Scholar]
- Wei A. R.; Ye H.; Guo Z. L.; Xiong J. Sisso-Assisted Prediction and Design of Mechanical Properties of Porous Graphene with a Uniform Nanopore Array. Nanoscale Adv. 2022, 4, 1455–1463. 10.1039/D1NA00457C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logadottir A.; Rod T. H.; Norskov J. K.; Hammer B.; Dahl S.; Jacobsen C. J. H. The Bronsted-Evans-Polanyi Relation and the Volcano Plot for Ammonia Synthesis over Transition Metal Catalysts. J. Catal. 2001, 197, 229–231. 10.1006/jcat.2000.3087. [DOI] [Google Scholar]
- Latimer A. A.; Kulkarni A. R.; Aljama H.; Montoya J. H.; Yoo J. S.; Tsai C.; Abild-Pedersen F.; Studt F.; Norskov J. K. Understanding Trends in C-H Bond Activation in Heterogeneous Catalysis. Nat. Mater. 2017, 16, 225–229. 10.1038/nmat4760. [DOI] [PubMed] [Google Scholar]
- Overholser B. R.; Sowinski K. M. Biostatistics Primer: Part 2. Nutr. Clin. Pract. 2008, 23, 76–84. 10.1177/011542650802300176. [DOI] [PubMed] [Google Scholar]
- Kondarides D. I.; Tomishige K.; Nagasawa Y.; Lee U.; Iwasawa Y. Characterization and Performance of a Ptmo6 /Mgo Catalyst for Alkane-to-Alkene Conversion. J. Mol. Catal. A: Chem. 1996, 111, 145–165. 10.1016/1381-1169(96)00117-3. [DOI] [Google Scholar]
- Boufaden N.; Akkari R.; Pawelec B.; Fierro J. L. G.; Zina M. S.; Ghorbel A. Dehydrogenation of Methylcyclohexane to Toluene over Partially Reduced Silica-Supported Pt-Mo Catalysts. J. Mol. Catal. A: Chem. 2016, 420, 96–106. 10.1016/j.molcata.2016.04.011. [DOI] [Google Scholar]
- Chen S.; Zhao Z. J.; Mu R. T.; Chang X.; Luo J.; Purdy S. C.; Kropf A. J.; Sun G. D.; Pei C. L.; Miller J. T.; et al. Propane Dehydrogenation on Single-Site Ptzn4 Intermetallic Catalysts. Chem 2021, 7, 387–405. 10.1016/j.chempr.2020.10.008. [DOI] [Google Scholar]
- Liu Y. T.; Li Y. Y.; Ge M.; Chen X. Y.; Guo M. Q.; Zhang L. H. Perovskite-Derived Pt-Ni/Zn(Ni)Tio3/Sio2 Catalyst for Propane Dehydrogenation to Propene. Catal. Lett. 2019, 149, 2552–2562. 10.1007/s10562-019-02857-w. [DOI] [Google Scholar]
- Lee H.; Kim W. I.; Jung K. D.; Koh H. L. Effect of Cu Promoter and Alumina Phases on Pt/Al2o3 for Propane Dehydrogenation. Korean J. Chem. Eng. 2017, 34, 1337–1345. 10.1007/s11814-017-0020-6. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.