

Abstract
Background
Early phase dose-finding (EPDF) trials are crucial for the development of a new intervention and influence whether it should be investigated in further trials. Guidance exists for clinical trial protocols and completed trial reports in the SPIRIT and CONSORT guidelines, respectively. However, both guidelines and their extensions do not adequately address the characteristics of EPDF trials. Building on the SPIRIT and CONSORT checklists, the DEFINE study aims to develop international consensus-driven guidelines for EPDF trial protocols (SPIRIT-DEFINE) and reports (CONSORT-DEFINE).
Methods
The initial generation of candidate items was informed by reviewing published EPDF trial reports. The early draft items were refined further through a review of the published and grey literature, analysis of real-world examples, citation and reference searches, and expert recommendations, followed by a two-round modified Delphi process. Patient and public involvement and engagement (PPIE) was pursued concurrently with the quantitative and thematic analysis of Delphi participants’ feedback.
Results
The Delphi survey included 79 new or modified SPIRIT-DEFINE (n = 36) and CONSORT-DEFINE (n = 43) extension candidate items. In Round One, 206 interdisciplinary stakeholders from 24 countries voted and 151 stakeholders voted in Round Two. Following Round One feedback, one item for CONSORT-DEFINE was added in Round Two. Of the 80 items, 60 met the threshold for inclusion (≥ 70% of respondents voted critical: 26 SPIRIT-DEFINE, 34 CONSORT-DEFINE), with the remaining 20 items to be further discussed at the consensus meeting. The parallel PPIE work resulted in the development of an EPDF lay summary toolkit consisting of a template with guidance notes and an exemplar.
Conclusions
By detailing the development journey of the DEFINE study and the decisions undertaken, we envision that this will enhance understanding and help researchers in the development of future guidelines. The SPIRIT-DEFINE and CONSORT-DEFINE guidelines will allow investigators to effectively address essential items that should be present in EPDF trial protocols and reports, thereby promoting transparency, comprehensiveness, and reproducibility.
Trial registration
SPIRIT-DEFINE and CONSORT-DEFINE are registered with the EQUATOR Network (https://www.equator-network.org/).
Supplementary Information
The online version contains supplementary material available at 10.1186/s12916-023-02937-0.
Keywords: early phase, clinical trials, SPIRIT guideline, CONSORT guideline, dose finding
Background
Early phase dose-finding (EPDF) trials, also typically referred to as phase I, I/II, or dose-escalation trials, are critical in clinical therapy development for a range of interventions that can be given in different doses and be pharmacological (chemical or biological, e.g. drugs, vaccines, cell therapies, gene therapies), non-pharmacological (e.g. radiotherapy, rehabilitation, digital therapies), or a combination thereof. Conducted in healthy volunteers or in participants with a condition or disease, these trials involve interim dosing adaptations (e.g. escalate/de-escalate) and generate data on safety and other information, such as pharmacokinetics, pharmacodynamics, and clinical activity, to enable developers to choose a suitable dosage(s) (dose and schedule) for further clinical testing.
Study protocols can vary greatly in content and quality despite their importance. Incomplete or unclear information in study protocols and final reports hinders the interpretability and replicability of EPDF trials and may impact the overall clinical development timeline as well as lead to erroneous conclusions on safety and efficacy and compromise the safety of trial participants [1, 2].
Trial designs of EPDF trials are evolving. There has been a rise in the use of seamless designs (integrated protocols with several components or phases within a trial) [3], as well as advanced model-assisted or model-based designs (1.6% of published phase I oncology trials in 1991–2006 [4] compared to 8.6% by 2014–2019 [5]). Such trials come with increased complexity and require further transparency in their protocols and trial reports to help readers understand the design, reproduce methods and interpret findings [1, 6, 7].
The Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 [8] statement and the CONsolidated Standards Of Reporting Randomised Trials (CONSORT) 2010 [4] statement have been instrumental in promoting complete and transparent reporting of the minimum essential content in trial protocols and trial reports. Neither the original guidance nor their extensions adequately cover the features of EPDF trials [1, 9]. The DosE-FIndiNg Extensions (DEFINE) study aims to build on the existing SPIRIT 2013 and CONSORT 2010 statements to meet this need and enhance the quality of EPDF trial protocols and their reporting of results across all disease areas [10].
This paper describes the processes, methods and results for the development stages of the DEFINE study. The subsequent consensus meeting and the final checklists will be covered in the forthcoming DEFINE-SPIRIT and DEFINE-CONSORT statement papers.
Overall project aim
The overall aim of this research is to develop evidence-based and consensus-driven guidelines for trial protocols (SPIRIT-DEFINE) and trial reports (CONSORT-DEFINE) for EPDF trials across all disease areas and disseminate them to stakeholders [1].
Methods
Timeline and key parts of the study
To drive delivery of the project, an international Executive Committee (EC) of multidisciplinary experts was formed, and an Independent Expert Panel (IEP) provided oversight and quality control assurances (see Additional file A1). Development of CONSORT-DEFINE commenced in March 2021, followed by SPIRIT-DEFINE in January 2022. The CONSORT-DEFINE protocol was deposited on the EQUATOR network in November 2021, followed by the SPIRIT-DEFINE Protocol in May 2022 [11, 12]. The consensus meeting took place in October 2022.
Literature review and draft checklist generation
The initial generated list of CONSORT-DEFINE candidate items was informed by a methodological review of EPDF trial reports published between 2011 and 2020 to assess their reporting quality [9, 13], based broadly on existing reporting guidelines or recommendations, including CONSORT 2010 [4], SPIRIT 2013 [8], adaptive designs CONSORT extension (ACE) [14], and a proposal for a checklist for phase I dose-finding cancer trials [15], as well as consultation with experts, as described [9, 13].
The draft CONSORT-DEFINE list was further enriched via a review of the published and grey literature, real-world examples analysis, citation tracking, and experts’ recommendations as follows [10]. We conducted two searches on the MEDLINE and EMBASE databases on June 18 and September 7, 2021, respectively, to identify published EPDF literature for CONSORT-DEFINE (see Additional file A2). The following information was extracted from the included articles: (1) potential new or modified candidate items, (2) suggested content for the explanation and elaboration of candidate items, (3) confirmation of already identified items, and (4) general comments.
The resulting draft checklist was externally reviewed by 11 multidisciplinary stakeholders, covering key categories under-represented in the DEFINE Executive Committee (trial management staff, non-oncology clinicians, research ethics committees, journal editors, funders, and patient and public involvement (PPIE) representatives). The trial management staff category was covered via a call for volunteers circulated through the UK Clinical Research Collaboration Trial Managers Network (UKCRC TMN). The reviewers were asked to provide a high-level review of the draft checklist content to include any modifications or suggested additions.
An initial draft of the SPIRIT-DEFINE checklist was prepared, building on SPIRIT 2013 [8] and the draft candidate items identified from the CONSORT-DEFINE development work. Two independent searches were conducted in PubMed for relevant published literature on January 17 and March 17, 2022 (see Additional file 3). We also contacted funding bodies, regulatory agencies, research ethics committee, pharmaceutical companies, contract research organisations (CROs), research institutes/hospitals, Medicines and Healthcare products Regulatory Agency (MHRA)-accredited phase I units, and professional association/consortium to see if they adopted or used a protocol template, guideline, or checklist to write or review EPDF protocols that they were willing to share (see Additional file 3). EPDF trial protocol templates that were freely available on the internet were also examined.
The Delphi process
General principles and scoring system
The draft candidate items for SPIRIT-DEFINE and CONSORT-DEFINE checklists were submitted for consultation and feedback to a wide stakeholder group through a Delphi survey. The Delphi process was conducted according to existing methodological guidance [16–18] and involved inviting participants to score the importance of the candidate items from the draft checklists through two iterative rounds of a web-based survey using DelphiManager, hosted by the University of Liverpool [19]. An importance rating scale of 1 to 9 was used: “not important” (score 1–3), “important but not critical” (score 4–6), “critically important” (score 7–9), and “unable to rate.” The thresholds for dropping items between rounds as well as automatic inclusion in the checklists were pre-specified (Fig. 1).
Fig. 1.

Criteria for dropping items between Delphi survey rounds as well as automatic inclusion in checklists
Prior to the launch of the Delphi survey, pilot rounds were conducted by the Executive Committee to fine-tune the draft checklists, troubleshoot any issues, and confirm the survey platform’s flow and functionality. Additionally, as part of the pilot run, we sent SPIRIT-DEFINE and CONSORT-DEFINE draft checklists to three experienced PPIE representatives to assess if they felt that patient representatives would be able to participate in the rating of the candidate items.
To ensure that the Delphi survey participation reflected the landscape of EPDF trials, key multidisciplinary stakeholder categories, including clinical trial researchers, regulators, ethics committees, journal editors, funders and patient and the public, were defined (see Additional file 4) [10]. Potential participants in EPDF trials were provided with participant information sheets and invited via email to participate in the Delphi survey. They were identified through a wide range of platforms, including targeted mailing lists of professional groups (including UK Experimental Cancer Medicine Centres, UKCRC Registered CTU Network, Adaptive Design Working Group and Statistical Analysis Working Group of MRC-NIHR Trial Methodology Research Partnership, NIHR Statistics Group and International Early Phase Adaptive Trials Workshop participants) and social media (Twitter and LinkedIn). We also invited stakeholders from academia and industry, either found online or recommended by the DEFINE Executive Committee and IEP. Invites were also sent to journal editors and corresponding authors collated from our reviews in Literature review and draft checklist generation section. Moreover, members of the MHRA-accredited phase I units were identified and invited. Individuals recommended by experts, including pharmaceutical company employees and PPI representatives, were also invited. Participants provided informed consent, and demographics, professional characteristics, and country information were collected. Registration rates were monitored continually, with the aim for at least 15 participants in each stakeholder category. Round One took place from March 28 to May 5, 2022, and Round Two from May 30 to June 27, 2022. Registration for Round One was necessary to take part in Round Two.
Analysis
Quantitative analysis
The number of invitation emails sent to potential participants who registered and responded to the survey in each round were presented in a flow diagram. Categorical baseline characteristics were summarised using frequency and percentage and continuous data using median and interquartile range (IQR) for all those who registered and the subsets who responded. The level of agreement between rounds was measured using percentage agreement (the percentage of participants with the same rating between rounds relative to the total responders to all rounds) and weighted Cohen’s kappa coefficient using absolute error weights [20].
Further details of the statistical methods for quantitative analyses are provided in Additional file 5 [20–25].
Qualitative analysis
An inductive thematic analysis using a semantic approach was performed on all free-text comments [26]. Open-ended feedback was extracted from the survey, collated, and uploaded for analysis using NVivo v1.6.2 software; the analysis was conducted by one assessor (SH) and reviewed by the DEFINE research team. After data familiarisation, initial codes were generated for each comment. More than one code could be assigned to a single comment. Initial codes were subsequently grouped into higher-level themes.
Comments received per item were grouped based on whether the item was for CONSORT-DEFINE or SPIRIT-DEFINE. Comments were manually summarised by two assessors (DP and OS) according to the number of comments received, the number of participants commenting on the item, the content (the theme) of the comment, and the number of times the theme was repeated for that item. The analysis was conducted independently but was confirmed by all assessors.
Consensus meeting
Following the Delphi survey, a consensus meeting was convened on October 11 and 12, 2022, to finalise the full list of items to be included in the guideline, guided by the information on item importance and level of agreement from the Delphi survey, as well as examples of their use in trial protocols or trial reports. The consensus meeting followed the recommended methodology for such an exercise [27]. International experts in each of the relevant stakeholder categories were invited, ensuring a balance of representation across the categories (see Additional file 4). Results from the consensus meeting and the final checklists will be covered in subsequent DEFINE-SPIRIT and DEFINE-CONSORT statement papers.
Results
Literature review and draft checklist generation
Data were extracted from 476 randomly selected dose-finding trials published between 2011 and 2020 using the bibliographic database MEDLINE (via PubMed), stratified by oncology (n = 238) and non-oncology (n = 238) settings [13]. The findings of the review revealed inconsistent and inadequate reporting of EPDF trials were reported previously. Several items related to EPDF trial aspects were poorly reported, with notable differences between oncology and non-oncology settings. Notably, very few trials provided an accessible protocol (6.3%), statistical analysis plan (3.8%), or lay summary (1.5%). Examples of key items that were poorly reported include specification of planned/maximum sample size, and with justification; definition of the analysis population; and rationale for starting dose [9]. Improvement in trial reporting over time was evident in only a few items, including a marked rise in the use of participant flow diagram (from 15.4% in 2011 to 60.7% in 2020) and a modest increase in sample size justification (from 17.9% in 2011 to 25.0% in 2020).
To identify literature with guidance on writing protocols and reporting of early phase dose-finding trials, we conducted separate searches, as detailed in Additional file 2. Starting with an initial 5291 hits for CONSORT-DEFINE, the articles were screened for duplicates and relevance before being assessed by 11 experts, yielding 47 included articles plus an additional 12 based on the experts’ recommendations. The first search for SPIRIT-DEFINE literature had 265 article hits, and the second search with broader search terms gained 6741 articles, which after screening and assessment yielded 57 included articles, 9 of which were also included in the CONSORT-DEFINE literature search result. Additional file 6 [2–4, 7, 8, 28–124] lists the relevant articles and their allocation to potential candidate items.
When contacted for protocol templates or guidance for EPDF trials, 29 out of 49 (59.2%) organisations responded (see Additional file 3). Amongst those who responded, none of the 8 funding bodies adopted or recommended any EPDF protocol template, while 2 out of 4 pharmaceutical companies and CROs that responded had EPDF templates but could not share them. Thirteen professional organisations, research institutes/hospitals, and MHRA-accredited phase I units responded, with three sharing their protocol template (one EPDF template and two non-EPDF templates). The protocol templates were informative in structuring potential checklist items in an EPDF protocol and in developing some of the wording for Delphi survey items.
For CONSORT-DEFINE, 22 new items were identified as being relevant to the reporting of the EPDF trials (21 for main report and 1 for abstract) and 21 items (20 for main report and 1 for abstract) were modified and expanded to reflect their unique features. Thus, 43 CONSORT-DEFINE candidate items were included in the Delphi survey and sent for a pre-survey review. Six external reviewers provided feedback to help refine the wording and explanation of some candidate items, such as the dose allocation method, which was detailed by specifying the sequence and interval between dosing of participants, e.g. sentinel or staggered dosing; it was also highlighted that there might be some overlap with the CONSORT extension for randomised pilot and feasibility trials [125].
For SPIRIT-DEFINE, 20 new candidate items were identified from all the included literature and 16 of the original SPIRIT items were modified and expanded to encompass the features of EPDF trials. As a result, a total of 36 SPIRIT-DEFINE candidate items were included in the Delphi survey.
Delphi survey
As part of the draft checklists’ pilot testing (General principles and scoring system section), the PPIE representatives advised that the candidate items were too complex and technical for patients or the general public to fully participate in the Delphi survey. To address this, the Executive Committee decided that instead of circulating the DEFINE Delphi survey directly to PPIE platforms, we would instead use a snowballing approach targeting experienced patient partners in clinical trials and asking them to circulate to their experienced PPIE contacts. This feedback also triggered the formation of the protocol-specified PPIE working group to discuss how best to embed the perspectives of patients as key stakeholders to encourage better reporting of EPDF trials and to facilitate the dissemination of the resulting SPIRIT-DEFINE and CONSORT-DEFINE guidelines (further details in Lay summary toolkit secion and Additional file 7) [1, 126–138].
Response rates across rounds
Figure 2 shows the flow of participants who were approached, registered, and responded to the Delphi surveys. During Round One of the Delphi survey, we reviewed the enrolment progress regularly, and we discovered that a large proportion of registered participants were based in Western Europe and North America. A targeted continental search for early phase trials on www.clinicaltrials.gov was conducted to ensure we captured perspectives of stakeholders from wide geographical regions. Additional designated trial contacts located in Eastern Europe, Asia, Africa, and South America were obtained via ClinicalTrials.gov. Out of the 73 additional stakeholders who were invited to participate in the Delphi survey, only 2 registered and 1 participated. A total of 206 participants responded to Round One of the Delphi survey and 151 to Round Two.
Fig. 2.
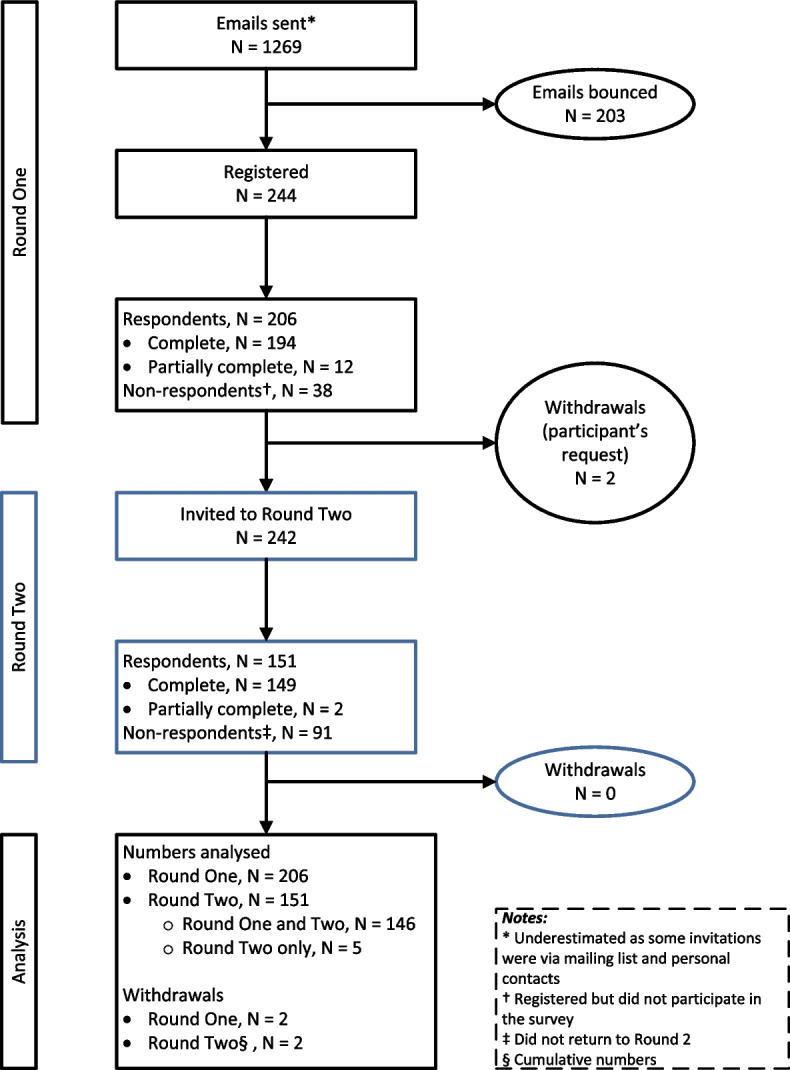
Flow chart of the DEFINE Delphi survey
As part of our pre-planned sensitivity analysis, we carried over the ratings of 59 respondents from Round One to Round Two for the Round One participants who did not take part in Round Two.
Characteristics of registered participants and respondents
The Delphi survey included a wide range of stakeholders, with the majority identifying as statisticians, trial methodologists, data scientists, or quantitative analysts (47.1%) and clinicians or clinical pharmacologists (32.4%) (see Additional file 8, Table A8-1). Most respondents were from academic organisations (77% in Round One). Based on the free-text field provided, participants who responded with “Other” to their experience in early phase trial roles were reclassified as one of the roles/types in the prespecified list (see Additional file 8, Table A8-2). Respondent demographics were relatively consistent across both rounds, with participants from five continents and 24 countries in Round One; most participants were from Europe (62.1%) followed by North America (25.7%) (see Additional file 8, Table A8-1).
Perceptions of proposed candidate items
In Round One, 52 of the 79 candidate items (SPIRIT-DEFINE, n = 25, and CONSORT-DEFINE, n = 27) had at least 70% of respondents rating them as “critically important” (scores 7 to 9). There was no item with 80% or more respondents scoring it as “not important” (scores 1 to 3). Therefore, none was removed after Round One.
Suggestions for new items from participants in Round One were considered by the Executive Committee, and one suggested item, “Access (or link) to code/functions used for simulation studies,” was added to CONSORT-DEFINE. Other suggested items were either already covered by existing items or were not specific to EPDF trials.
In Round Two, 60 of 80 candidate items had at least 70% of respondents rating them as “critically important” (scores 7 to 9) (Fig. 3). All 52 items in Round One that met the inclusion threshold remained highly scored in Round Two.
Fig. 3.

Bar plot of the percentage of respondents scoring each item in Round Two. Items in green text had at least 70% of respondents scoring them as “critically important” (scores 7 to 9)
In our assessment of the stability and consistency of individual ratings of item importance across rounds, we found a reasonably high level of individual agreement. Twenty items had moderate agreement (0.41 to 0.6) and 59 had substantial agreement (0.61 to 0.8). This suggested that respondents had “converged” in their responses (see Additional file 9, Table A9-3).
Visual inspection of the item rating by early phase roles showed that there was no difference for most items except for five candidate items (SPIRIT-DEFINE: planned dosing regimens, additional statistical analyses, location of the protocol, and support for the planned biomarker sub-study; CONSORT-DEFINE: planned dosing regimens, presentation of interim results (see Additional file 9, Figures A9-3 to A9-7)). All these items were also rated as critical by more respondents from Asia than from Europe and North America (see Additional file 9, Figures A9-8 to A9-12).
Detailed analysis of the respondents’ perceptions of the importance of the candidate items and the sensitivity analysis are provided in Additional file 9, Table A9-1.
Qualitative analysis based on open-ended feedback from participants
Following the methodology defined in Analysis section, general comments from Round One were coded and grouped into six higher-level themes: content consideration, feedback on experience of Delphi process, guideline structure, trial participant characteristics, patient and public involvement, and unrelated content (see Additional file 11). The feedback was used to further refine candidate items to improve their applicability and as background information for the consensus meeting discussions.
Inclusion of candidate items in DEFINE checklists
At the end of the Delphi survey, 60 items were automatically recommended for inclusion based on the pre-specified inclusion threshold (Fig. 1). The remaining 20 items (SPIRIT-DEFINE, n = 10, and CONSORT-DEFINE, m = 10) were tabled for discussion at the DEFINE consensus meeting (see Additional file 10).
Expert Focus Group with expertise in non-oncology and healthy volunteers trials
In response to feedback that some candidate items were oncology-focused, after Round Two, we conducted a targeted engagement step via an Expert Focus Group of five international stakeholders with expertise in healthy volunteers and non-oncology EPDF trials to seek their views on the candidate items and identify if refinements to the item description may be required to facilitate their applicability in a non-oncology context. Based on that discussion, some terminologies were changed to ones with broader applicability, such as “dose-limiting toxicity” to “safety measures”; item explanations were enriched to be applicable in all settings (including first-in-human studies), and optionality, i.e. “where applicable,” was added in some cases.
Lay summary toolkit
As described in Delphi survey section, a PPIE working group was formed, consisting of six representatives from both the oncology and non-oncology fields, as well as those with experience in early phase trials. The perspectives of patients and participants on the reporting of EPDF trials were explored. The discussion highlighted the need to develop an easy-to-understand lay summary of the scientific publication of EPDF trials in order to provide increased transparency for patients and the general public. It was also suggested that the provision of a good lay summary exemplar of a published EPDF trial would be helpful to facilitate implementation. This led to the co-development of a lay summary toolkit, which includes a template with guidance notes and an exemplar for the reporting of EPDF trials, taking into consideration the CONSORT-DEFINE candidate items. The drafted template, guidance, and exemplar were then sent for review to three professional experts (a communications media manager, an ethics committee member, and a regulator) to gather feedback and further refinement before a wider consultation with the patient and public involvement and engagement in musculoskeletal research (PIMS) group. The resultant lay summary toolkit provides recommendations of key information that should be reported from the perspective of both patients and the general public (see Additional file 7).
Discussion
The prevalence and importance of early phase trials in the clinical development pipeline, as well as the increasing complexity of the designs used, necessitate thorough, precise, and transparent efforts to ensure that the work can be accurately evaluated and, if necessary, reproduced. The methodological review findings of inconsistent and inadequate reporting of important methodological features in design, conduct, and analysis confirm the need for robust, consensus-driven extensions of SPIRIT 2013 [139] and CONSORT 2010 [9] to provide the research community with the tools needed to accurately report and assess EPDF trials while meeting the standards achieved by the original checklists [4, 8] and existing extensions in the broader clinical trials reporting landscape [140].
SPIRIT-DEFINE and CONSORT-DEFINE were developed using gold-standard methodology to provide robust international evidence and consensus-based guidance. Concurrent development has enabled us to harmonise both guidelines while also achieving increased clarity and a broader perspective on the items required for either or both. This will make it easier for researchers to write the trial protocol and trial report, as well as enable reviewers to assess the final report’s adherence to the protocol as per other recent joint SPIRIT and CONSORT extensions, such as for outcomes reporting [141].
Not many reporting guidelines have provided detailed descriptions of their development processes and results (ACE [14] is one of the exceptions), and we hope that this paper will assist readers in understanding the “DEFINE development journey” as well as enable researchers planning to develop future guidelines or extensions to learn from our experience.
Main strengths
This project’s main strengths were its solid methodological foundations and broad participation. The development of the guidelines was supported by independent oversight and was underpinned by detailed protocols that pre-specified the methods [10].
Strong PPIE engagement was achieved throughout the process, with the co-development of a lay summary toolkit (consisting of a user-friendly template with guidance notes and an exemplar) with our PPIE partners. To the best of our knowledge, this is the first time that a reporting guideline development has embedded within it the production of a toolkit to aid researchers in providing a lay summary of their trial results more effectively to patients and the public. We hope this will encourage future guideline developers to consider this to facilitate implementation. Close engagement will be maintained throughout the dissemination stages.
The comprehensive methodological review included 476 randomly selected trial reports, which were supplemented by subsequent literature reviews.
From the beginning of the process, the DEFINE group sought to engage multiple organisations, both academic and commercial, to ensure that the resulting guidelines would meet the needs of the entire EPDF community. The Delphi survey was successful in involving 206 delegates from 5 continents and 24 countries in a variety of job roles in EPDF trials, exceeding the participation target. It also achieved a high level of consensus across the two rounds, with 60 items meeting the pre-defined threshold.
The dissemination strategy aims to maximise guideline awareness and uptake, including but not limited to dissemination in stakeholder meetings, conferences, peer-reviewed publications, and on the EQUATOR Network and DEFINE project websites.
Main limitations
Despite the above-mentioned key strengths, we encountered several limitations. A combination of approaches was employed to distribute the survey invitations. These included advertising on social media, sending invitations to targeted mailing lists of professional groups, corresponding authors from a random selection of trials, and pre-selected individuals with expertise in the field. For mailing lists of larger groups, as participants were self-selected, the survey results might have been influenced by non-response bias, and we were unable to determine the profile of those who did not sign up to participate. The approach of pre-selecting specific individuals might have introduced certain biases, such as selection bias and sampling bias. Nevertheless, the combination of circulating surveys to larger groups and pre-selected individuals ensured a balanced blend of inclusivity and targeted engagement, maximising the diversity of participants while levering the expertise of key stakeholders, ultimately yielding valuable contributions.
Given that the survey had 80 candidate items and was expected to take around 30 min to complete, 29% of Round One respondents did not return to Round Two, which could be attributed to participant fatigue. To address this, items relevant to both guidelines were presented together, with participants able to save and return later.
Despite the distribution of respondents' characteristics being consistent with the landscape of EPDF clinical trials, the majority of respondents came from academic organisations. Some geographical regions outside of Western Europe and North America were underrepresented, and targeted efforts were made to recruit more participants from these areas. Finally, after Round One, participants were unable to provide feedback on individual items but could provide general comments.
Conclusions
By implementing a robust, comprehensive gold-standard methodological framework for guideline development, SPIRIT-DEFINE and CONSORT-DEFINE will allow investigators to effectively address the essential items that should be included in trial protocols and reporting, thus promoting transparency, completeness, and reproducibility of methods. SPIRIT-DEFINE and CONSORT-DEFINE will also provide a framework for peer review of EPDF trial protocols and reports, including an assessment of the quality of the trial design and methods as well as the risk of bias in the reported outcomes.
By sharing the DEFINE development methods and the decisions undertaken with multi-stakeholder groups, including PPIE partners, we hope it will serve as a model to support future guideline development projects.
Supplementary Information
Additional file 1. Composition of the DEFINE Executive Committee and Independent Expert Panel.
Additional file 2. CONSORT and SPIRIT DEFINE Literature review: Figures A2-1 – A2-2, Tables A2-1. Figure A2-1. PRISMA 2020 flow diagram for the CONSORT-DEFINE literature search and review. Figure A2-2. PRISMA flow diagram for the SPIRIT-DEFINE literature search and review. Table A2-1. Terms used in the PubMed searches and the number of hits from each search terms for SPIRIT-DEFINE.
Additional file 3. Organisations contacted for protocol templates or guidelines and responses.
Additional file 4. Delphi Survey key stakeholder groups and methods of access.
Additional file 5. Further details on the statistical analysis plan.
Additional file 6. SPIRIT-DEFINE and CONSORT-DEFINE items generation for Delphi survey: Table A6-1. Table A6-1. References supporting the proposed candidate items in the DEFINE Delphi survey.
Additional file 7. Toolkit for Lay Summary of EPDF Trial Results.
Additional file 8. Delphi survey participants demographics and harmonisation of roles: Tables A8-1 – A8-2. Table A8-1. Demographics of Delphi survey participants. Table A8-2. Harmonisation of participants’ defined roles.
Additional file 9. Perception of proposed items and sensitivity analysis: Tables A9-1 – A9-3, Figures A9-1 – A9-13. Table A9-1. Summary of categorical and numerical ratings at Round One and 2. Figure A9-1. Percentage of participants changing their numerical scores at Round Two. Figure A9-2. Percentage of participants changing their categorical scores at Round Two. Table A9-2. Number of participants changing their ratings between Round One and Round Two. Table A9-3. Frequency and percentage of perfect agreement. Figure A9-3. Stacked bar plots of candidate item [17 SPIRIT-DEFINE] “Planned dosing regimens presented as a diagram or table, where applicable” by stakeholders. Figure A9-4. Stacked bar plots of candidate item [18 CONSORT-DEFINE] “Planned and delivered dosing regimens presented as a diagram or table, where applicable” by stakeholders. Figure A9-5. Stacked bar plots of candidate item [48 SPIRIT-DEFINE] “Statistical methods for additional analyses” by stakeholders. Figure A9-6. Stacked bar plots of candidate item [69 CONSORT-DEFINE] “Specify if and when results were reported whilst the trial was still ongoing” by stakeholders. Figure A9-7. Stacked bar plots of candidate item [70 SPIRIT-DEFINE] “Where the full trial protocol or the redacted version, with amendments, can be accessed” by stakeholders. Figure A9-8. Stacked bar plots of candidate item [17 SPIRIT-DEFINE] “Planned dosing regimens presented as a diagram or table, where applicable” by Continent. Figure A9-9. Stacked bar plots of candidate item [18 CONSORT-DEFINE] “Planned and delivered dosing regimens presented as a diagram or table, where applicable” by Continent. Figure A9-10. Stacked bar plots of candidate item [48 SPIRIT-DEFINE] “Statistical methods for additional analyses” by Continent. Figure A9-11. Stacked bar plots of candidate item [69 CONSORT-DEFINE] “Specify if and when results were reported whilst the trial was still ongoing” by Continent. Figure A9-12. Stacked bar plots of candidate item [70 SPIRIT-DEFINE] “Where the full trial protocol or the redacted version, with amendments, can be accessed” by Continent. Figure A9-13. Stacked bar plots of candidate item [5 SPIRIT-DEFINE] “Summary of findings from existing correlative biomarker, correlative and associated studies to support planned biomarker sub-study” by Continent.
Additional file 10. List of candidate items to be discussed at the DEFINE Consensus meeting.
Additional file 11. Qualitative analysis. Table A11-1; Figure A11-1. Table A11-1. Themes and codes identified in the general comments received in Delphi survey. Figure A11-1. Tree diagram of the comments received per theme.
Acknowledgements
The authors thank the late Professor Doug Altman for his enthusiasm, inspiration, and significant contribution to the initial conception of this work.
Also, we would like to thank all groups and individuals that helped disseminate the survey link, including UKCRC and all other professional groups. The authors thank Xiaoran Lai for his assistance with the revised figure.
The Executive Committee
We would like to thank the DEFINE Executive Committee: Alun Bedding, Andrew Kightley, Christopher Weir, Jeff Evans, Thomas Jaki, Johann De Bono, Munyarazdi Dimairo, Rong Liu, Sally Hopewell, An-Wen Chan, Adrian Mander, and Shing Lee and our collaborators and advisors: Khadija Rantell, Stephen Hahn, Moreno Ursino, and John Kirkpatrick for their significant contribution to this study by providing insight at various stages, reviewing the study data and checklists, and helping to make key decisions for the study.
The Independent Expert Panel
The authors acknowledge the support of Elizabeth Garrett-Mayer, Deborah Ashby, John D. Isaacs, and Melanie Calvert, the members of the Independent Expert Panel, in contributing their expertise and support to this initiative.
The Expert Working Group
We would like to thank the members of our expert working groups, including those involved in patient and participant involvement, non-oncology trials, and healthy volunteer trials: Thomas Jaki, Kathrine Craig, Stephen Senn, Oliver Boix, Richard Peck, Della Ogunleye, Lesley Roberts, Richard Stephens, Emily Lam, Andrew Kightley, and Dawn Richards.
We would also like to thank Elizabeth Abbott, Mark Chegwidden, Annabel Dawson, Rosie Flower, Liz Hammer, Sue Leeder, Cate Middleton, Mike Solomon, and Pamela Spearing from the patient and public involvement and engagement in musculoskeletal research (PIMS) group in Newcastle, as well as Arthur Pratt, Leigh Romaniuk, Julia Bakker, Karen Poole, and Kate Craig for their valuable input in the development of the lay summary toolkit for reporting of early phase dose-finding trial results.
Delphi survey participants
We are thankful to our Delphi survey participants who consented to be named for completing our survey and providing their opinion and feedback: Agnes V Klein, Canada; Aidan Hindley, United Kingdom; Akshay A Shivchhand, India; Alastair Greystoke, United Kingdom; Alessandro Matano, Switzerland; Alexander Ooms, United Kingdom; Alun Bedding, United Kingdom; Andrew Althouse, United States of America; Andrew JT George, United Kingdom; Andy Vail, United Kingdom; Anna Zachariou, United Kingdom; Anne Loeser, United States of America; Annette Kopp-Schneider, Germany; Anthony Joshua, Australia; Aude Espinasse, United Kingdom; Becky M Salisbury, United Kingdom; Benjamin Johnson, United Kingdom; Björn Holzhauer, Switzerland; Carine Bellera, France; Caroline Kelly, United Kingdom; Catey Bunce, United Kingdom; Catherine Fullwood, United Kingdom; Christian Dittrich, Austria; Christian Gluud, Denmark; Christin Henein, United Kingdom; Christophe Le Tourneau, France; Christophe Massard, France; Clare Peckitt, United Kingdom; Conrad Fernandez, Canada; Crescens Tiu, United Kingdom; David Murray, United Kingdom; Dawn Richards, Canada; Deborah Ashby, United Kingdom; Debra J Lett, United Kingdom; Diana R Elbourne, United Kingdom; Elizabeth Garrett-Mayer, United States of America; Elli Bourmpaki, United Kingdom; Elspeth Banks, United Kingdom; Emily Dressler, United States of America; Emily Greenlay, United Kingdom; Eric Frison, France; Fangrong Yan, China; Fiona Thistlethwaite, United Kingdom; Fred Cohen, United States of America; Gaëlle Saint-Hilary, France; Geert Jan Groeneveld, Netherlands; Haitao Pan, United States of America; Helen Brittain, United Kingdom; Hemant Arora, United Kingdom; Herbert H. Loong, China; Ileana Baldi, Italy; Jan Rekowski, United Kingdom; Jean Francois Pittet, United States of America; Jenna Grabey, United Kingdom; Joanna Moschandreas, United Kingdom; Johann de Bono, United Kingdom; John D Isaacs, United Kingdom; Jörg Haier, Germany; Joseph Ross, United States of America; Joshua Savage, United Kingdom; Jost B. Jonas, Germany; Karim M Khan, Canada; Kate Hayward, Australia; Kathrine J Craig, United Kingdom; Katrina Walker, United Kingdom; Kentaro Takeda, United States of America; Kosuke Kashiwabara, Japan; Kyun-Seop Bae, South Korea; Laura Richert, France; Lauren Walker, United Kingdom; Lesley K Seymour, Canada; Lewis L. Hsu, United States of America; Lisa Belin, France; Liz-Anne Lewsley, United Kingdom; Lukas Widmer, Switzerland; Lynley Marshall, United Kingdom; Marcio Augusto Diniz, United States of America; Maria Beatrice Panico, United Kingdom; Marina Pulido, France; Matthew Schipper, United States of America; Matthew Sydes, United Kingdom; Melanie Calvert, United Kingdom; Michael Grayling, United Kingdom; Muhammad Irfan bin Abdul Jalal, Malaysia; Nandi Siegfried, South Africa; Nuria Kotecki, Belgium; Olga Kholmanskikh, Belgium; Olga Solovyeva, United Kingdom; Oliver Boix, Germany; Paul Bycott, United States of America; Pedro A. Torres-Saavedra, United States of America; Peter Dewland, United Kingdom; Peter J Gill, Canada; Philip Drennan, United Kingdom; Rebecca Kristeleit, United Kingdom; Robert M. Golub, United States of America; Roger Dmochowski, United States of America; Rongji Mu, China; Ruitao Lin, United States of America; Sebastian Bate, United Kingdom; Sharon Love, United Kingdom; Siew Wan Hee, United Kingdom; Simon Crabb, United Kingdom; Simon Pacey, United Kingdom; Sonia Fox, United Kingdom; Stefan N. Symeonides, United Kingdom; Stephen Senn, United Kingdom; Susan E. Bates, United States of America; Susan Percy Ivy, United States of America; Susan Smith, United Kingdom; Sze Huey Tan, Singapore; Temsunaro Rongsen Chandola, India; Thomas J. Prior, United States of America; Tom Parke, United Kingdom; Victoria Homer, United Kingdom; Vivianne Shih, Singapore; Yoshiya Tanaka, Japan; Yung-Jue Bang, South Korea Funding.
Abbreviations
- ACE
Adaptive designs CONSORT extension
- CONSORT
Consolidated Standards of Reporting Trials
- CONSORT-DEFINE
CONSORT extension for early phase dose-finding trials
- CRO
Clinical Research Organisation
- DEFINE
Dose-finding extensions
- EC
Executive committee
- EMA
European Medicines Agency
- EPDF
Early phase dose-finding
- EQUATOR
Enhancing the QUAlity and Transparency Of health Research
- IEP
Independent Expert Panel
- IQR
Interquartile range
- MHRA
Medicines and Healthcare products Regulatory Agency
- NCRI
National Cancer Research Institute
- PPIE
Patient and public involvement and engagement
- SPIRIT
Standard Protocol Items: Recommendations for Interventional Trials
- SPIRIT-DEFINE
SPIRIT extension for early phase dose-finding trials
Authors’ contributions
CY and OS had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. CY, AM, AB, AK, TRJE, JdB, SH, SL, TJ, MD, CJW, and AWC were responsible for conception of the DEFINE study. CY, AM, AB, AK, TRJE, JdB, SH, SL, TJ, MD, CJW, AE, OS, and AWC designed the study. AE, DP, AK, RS, LR, DR, and CY conceived and designed the PPIE component. CY, AM, AK, TRJE, JdB, SH, SL, TJ, MD, and CJW obtained the funding for CONSORT-DEFINE. CY, OS, MD, CJW, SWH, AE, MU, DP, AK, TJ, AM, TRJE, SL, SH, KR, AWC, AB, JK, and JdB contributed to the acquisition, analysis, and interpretation of the data. OS, SWH, DP, AE, and CY drafted the manuscript. All authors critically revised and approved the final manuscript.
Funding
The CONSORT-DEFINE part of this study was supported by the UKRI’s MRC-NIHR grant number MR/T044934/1. The SPIRIT-DEFINE part received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. T Jaki received funding from the UK Medical Research Council (MC_UU_00002/14). Christopher J Weir was supported in this work by NHS Lothian via Edinburgh Clinical Trials Unit. Shing Lee was funded through the National Center for Advancing Translational Sciences, National Institutes of Health (Grant Number UL1TR001873). The funders have no involvement in the study design, collection, analysis, interpretation of findings, or reporting. However, research outputs will be published in line with the funders’ publication policy requirements. The SPIRIT-DEFINE part did not receive any external funding. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising from this submission.
Availability of data and materials
Data is shared under a data transfer agreement or a collaboration agreement depending on the nature of the sharing. Requests are via a standard proforma describing the nature of the proposed research and extent of data requirements.
Declarations
Ethics approval and consent to participate
This project was approved by the ICR’s Committee for Clinical Research (reference CCR5460). The Health Research Authority confirmed that Research Ethics Approval is not required. Consent to take part in the Delphi survey was collected from every participant via the web-based survey application. Consent to take part in the consensus meeting was collected ahead of the meeting using a purpose-built registration survey.
Consent for publication
Not applicable.
Competing interests
Professor Johann de Bono has served on advisory boards and received fees from many companies, including Amgen, Astra Zeneca, Astellas, Bayer, Bioxcel Therapeutics, Boehringer Ingelheim, Cellcentric, Daiichi, Eisai, Genentech/Roche, Genmab, GlaxoSmithKline, Harpoon, ImCheck Therapeutics, Janssen, Merck Serono, Merck Sharp & Dohme, Menarini/Silicon Biosystems, Orion, Pfizer, Qiagen, Sanofi Aventis, Sierra Oncology, Taiho, Terumo, and Vertex Pharmaceuticals.
Professor Johann de Bono is an employee of The Institute of Cancer Research, which has received funding or other support for his research work from AZ, Astellas, Bayer, Cellcentric, Daiichi, Genentech, Genmab, GlaxoSmithKline, Janssen, Merck Serono, MSD, Menarini/Silicon Biosystems, Orion, Sanofi Aventis, Sierra Oncology, Taiho, Pfizer, Vertex, and which has a commercial interest in abiraterone, PARP inhibition in DNA repair defective cancers and PI3K/AKT pathway inhibitors (no personal income).
Professor Johann de Bono was named as an inventor with no financial interest in Patent No. 8,822,438 submitted by Janssen that covers the use of abiraterone acetate with corticosteroids. He has been the CI/PI of many industry-sponsored clinical trials.
Professor Johann de Bono is a Senior Investigator at the National Institute for Health Research (NIHR). The views expressed in this article are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Professors Sally Hopewell and An-Wen Chan are members of the SPIRIT-CONSORT Executive Group and leading the current update of the SPIRIT 2013 and CONSORT 2010 reporting guidelines, funded by the UK Medical Research Council National Institute for Health Research Better Methods, Better Research (MR/W020483/1).
Professor Jeffry Evans declares the following conflicts of interest:
Honoraria for Consultancies (payable to the employing institution) Ascelia, Bayer, Bristol Myers Squibb, Celgene, Clovis, Eisai, Genentech, Immunova, Jennerex/Transgene, Nucana, Karus Therapeutics, MSD, Otsuka, Roche, Seagen, Medivir, Bicycle Therapeutics.
Honoraria for speaker’s fees (payable to employing institution) Astra Zeneca, Ascelia, Bayer, Bristol Myers Squibb, Celgene, Eisai, GlaxoSmithKline, Nucana, MSD, Roche, Medivir, United Medical.
Support of costs of commercial clinical trials (payable to employing institution) Astra Zeneca, Basilea, Bayer, Celgene, MiNa Therapeutics, Roche, Pfizer, Sierra, Lilly, Eisai, Glaxo Smith Kline, Novartis, Bicycle Therapeutics, Halozyme, Johnson & Johnson, CytomX, Vertex, Plexxikon, Boehringer, Athinex, Adaptimmune, Bristol-Myers Squibb, MSD, Medivir, Versatem, Nucana, Immnuocore, Berg, Beigene, Iovance, Modulate, BiolinerX, Merck Serono, Nurix Therapeutics, T3P, Janssen Clovis, Sanofi-Aventis, Starpharma, UCB, Sapience, Seagen, Avacta, Codiak.
Support to attend international conferences (personal) Bayer, Celgene, Roche, Bristol-Myers Squibb, PIerre-Fabre, MSD.
Dr Sarah Hughes receives funding from the National Institute of Health and Care (NIHR) Oxford-Birmingham Blood and Transplant Research Unit, the NIHR Applied Research Collaboration (ARC) West Midlands, and UK SPINE. Dr Hughes declares personal fees from Aparito Limited, Astra Zeneca, CIS Oncology, Cochlear Limited, and ICON plc. The views expressed in this article are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Dr Moreno Ursino acted as consultant for eXYSTAT, Saryga, PTC Therapeutics International Ltd., ImCheck Therapeutics. He also has projects, ongoing and ended, with Sanofi.
Dr Shing Lee served as a consultant for PTC Therapeutics.
Dr Mander is currently employed by GSK.
All other authors declare no conflicts of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Yap C, Bedding A, de Bono J, Dimairo M, Espinasse A, Evans J, Hopewell S, Jaki T, Kightley A, Lee S, et al. The need for reporting guidelines for early phase dose-finding trials: dose-finding CONSORT extension. Nat Med. 2022;28(1):6–7. doi: 10.1038/s41591-021-01594-1. [DOI] [PubMed] [Google Scholar]
- 2.Mariani L, Marubini E. Content and quality of currently published phase II cancer trials. J Clin Oncol. 2000;18(2):429–436. doi: 10.1200/JCO.2000.18.2.429. [DOI] [PubMed] [Google Scholar]
- 3.Hobbs BP, Barata PC, Kanjanapan Y, Paller CJ, Perlmutter J, Pond GR, Prowell TM, Rubin EH, Seymour LK, Wages NA, et al. Seamless designs: current practice and considerations for early-phase drug development in oncology. J Natl Cancer Inst. 2019;111(2):118–128. doi: 10.1093/jnci/djy196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulz KF, Altman DG, Moher D, Group C CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMC Med. 2010;8:18. doi: 10.1186/1741-7015-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Araujo DV, Oliva M, Li K, Fazelzad R, Liu ZA, Siu LL. Contemporary dose-escalation methods for early phase studies in the immunotherapeutics era. Eur J Cancer. 2021;158:85–98. doi: 10.1016/j.ejca.2021.09.016. [DOI] [PubMed] [Google Scholar]
- 6.EMA. Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products. 2017. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-strategies-identify-mitigate-risks-first-human-early-clinical-trials-investigational_en.pdf. [DOI] [PMC free article] [PubMed]
- 7.Lorch U, O'Kane M, Taubel J. Three steps to writing adaptive study protocols in the early phase clinical development of new medicines. BMC Med Res Methodol. 2014;14:84. doi: 10.1186/1471-2288-14-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan AW, Tetzlaff JM, Altman DG, Laupacis A, Gotzsche PC, Krleza-Jeric K, Hrobjartsson A, Mann H, Dickersin K, Berlin JA, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200. doi: 10.7326/0003-4819-158-3-201302050-00583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yap C, Solovyeva O, Yin Z, Martin J, Manickavasagar T, Weir C, Lee S, Dimairo M, Liu R, Kightley A, et al. Assessing the reporting quality of early phase dose-finding trials. Ann Oncol. 2022;33:S24–S24. doi: 10.1016/j.annonc.2022.01.018. [DOI] [Google Scholar]
- 10.Espinasse A, Solovyeva O, Dimairo M, Weir C, Jaki T, Mander A, Kightley A, Evans J, Lee S, Bedding A, et al. SPIRIT and CONSORT extensions for early phase dose-finding clinical trials: the DEFINE (DosE-FIndiNg Extensions) study protocol. BMJ Open. 2023;13(3):e068173. doi: 10.1136/bmjopen-2022-068173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DEFINE study Protocol writing group. CONSORT-DEFINE (dose finding extensions): development of a CONSORT extension for early-phase dose-finding trials (CONSORT-DEFINE) [https://www.equator-network.org/wp-content/uploads/2022/05/DF-CONSORT-protocol-v1.2_FINAL.pdf].
- 12.DEFINE study Protocol writing group: SPIRIT–DEFINE (dose finding): development of a SPIRIT extension for early-phase dose finding trials. [https://www.equator-network.org/wp-content/uploads/2022/05/SPIRIT-protocol-v1.0-06052022_FINAL1.pdf]
- 13.Solovyeva O, Weir CJ, Lee S, Dimairo M, Espinasse A, Martin JWB, Manickavasagar T. Liu, R. Kightley A, De Bono J, Yap C. Reporting quality of early phase dose-finding clinical trials: a rapid methodological review protocol [https://osf.io/7pyds/].
- 14.Dimairo M, Pallmann P, Wason J, Todd S, Jaki T, Julious SA, Mander AP, Weir CJ, Koenig F, Walton MK, et al. The adaptive designs CONSORT Extension (ACE) statement: a checklist with explanation and elaboration guideline for reporting randomised trials that use an adaptive design. BMJ. 2020;369:m115. doi: 10.1136/bmj.m115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zohar S, Lian Q, Levy V, Cheung K, Ivanova A, Chevret S. Quality assessment of phase I dose-finding cancer trials: proposal of a checklist. Clin Trials. 2008;5(5):478–485. doi: 10.1177/1740774508096653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diamond IR, Grant RC, Feldman BM, Pencharz PB, Ling SC, Moore AM, Wales PW. Defining consensus: a systematic review recommends methodologic criteria for reporting of Delphi studies. J Clin Epidemiol. 2014;67(4):401–409. doi: 10.1016/j.jclinepi.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Hasson F, Keeney S, McKenna H. Research guidelines for the Delphi survey technique. J Adv Nurs. 2000;32(4):1008–1015. [PubMed] [Google Scholar]
- 18.von der Gracht HA. Consensus measurement in Delphi studies review and implications for future quality assurance. Technol Forecast Soc. 2012;79(8):1525–1536. doi: 10.1016/j.techfore.2012.04.013. [DOI] [Google Scholar]
- 19.COMET-Initiative: DelphiManager [https://www.comet-initiative.org/delphimanager/].
- 20.Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33(1):159–174. doi: 10.2307/2529310. [DOI] [PubMed] [Google Scholar]
- 21.Dorai-Raj S. binom: binomial confidence intervals for several parameterizations. 2022. [Google Scholar]
- 22.Canty A, Ripley BD. boot: Bootstrap R (S-Plus) Functions. 2021. [Google Scholar]
- 23.Efron B. Bootstrap methods: another look at the jackknife. Annals of Statistics. 1979;7(1):1–26, 26. doi: 10.1214/aos/1176344552. [DOI] [Google Scholar]
- 24.Gamer M, Lemon J, Fellows I, Singh P. irr: various coefficients of interrater reliability and agreement. 2019. [Google Scholar]
- 25.Revelle W. psych: procedures for personality and psychological research. 2022. [Google Scholar]
- 26.Clarke V, Braun V. Thematic analysis: a practical guide. Los Angeles: SAGE; 2022. [Google Scholar]
- 27.Moher D, Schulz KF, Simera I, Altman DG. Guidance for developers of health research reporting guidelines. PLoS Med. 2010;7(2):e1000217. doi: 10.1371/journal.pmed.1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The European Parliament and the Council of the European Union. Annex I of Regulation (EU) No 536/2014 on Clinical Trials on Medicinal Products for Human Use and repealing Directive 2001/20/EC. In. Edited by Union TEPatCotE: Official Journal of the European Union 54–64. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32014R0536.
- 29.Malik L, Mejia A, Parsons H, Ehler B, Mahalingam D, Brenner A, Sarantopoulos J, Weitman S. Predicting success in regulatory approval from Phase I results. Cancer Chemother Pharmacol. 2014;74(5):1099–1103. doi: 10.1007/s00280-014-2596-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arbuck SG. Workshop on phase I study design. Ninth NCI/EORTC New Drug Development Symposium, Amsterdam, March 12, 1996. Ann Oncol. 1996;7(6):567–573. doi: 10.1093/oxfordjournals.annonc.a010672. [DOI] [PubMed] [Google Scholar]
- 31.Beck L, Witt R, Nesper-Brock M, Milde T, Hettmer S, Frühwald MC, Rössig C, Fischer M, Reinhardt D, Taylor LA, et al. A study of regulatory challenges of pediatric oncology phase I/II trial submissions and guidance on protocol development. Clin Pharmacol Ther. 2021;110(4):1025–1037. doi: 10.1002/cpt.2319. [DOI] [PubMed] [Google Scholar]
- 32.Bird SM, Bailey RA, Grieve AP, Senn S. Statistical issues in first-in-human studies on BIA 10–2474: neglected comparison of protocol against practice. Pharm Stat. 2017;16(2):100–106. doi: 10.1002/pst.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du Y, Yin J, Sargent DJ, Mandrekar SJ. An adaptive multi-stage phase I dose-finding design incorporating continuous efficacy and toxicity data from multiple treatment cycles. J Biopharm Stat. 2019;29(2):271–286. doi: 10.1080/10543406.2018.1535497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.EMA. ICH Topic E3 Structure and Content of Clinical Study Reports In. 1996. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-e-3-structure-content-clinical-study-reports-step-5_en.pdf.
- 35.EMA. Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products. 2017. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-strategies-identify-mitigate-risks-first-human-early-clinical-trials-investigational_en.pdf. [DOI] [PMC free article] [PubMed]
- 36.EMA. ICH Guideline E8 (R1) on General Considerations for Clinical Studies. 2022. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-e-8-general-considerations-clinical-trials-step-5_en.pdf.
- 37.Gaydos B, Anderson KM, Berry D, Burnham N, Chuang-Stein C, Dudinak J, Fardipour P, Gallo P, Givens S, Lewis R, et al. Good practices for adaptive clinical trials in pharmaceutical product development. Drug Inf J. 2009;43(5):539–556. doi: 10.1177/009286150904300503. [DOI] [Google Scholar]
- 38.Hoffmann TC, Glasziou PP, Boutron I, Milne R, Perera R, Moher D, Altman DG, Barbour V, Macdonald H, Johnston M, et al. Better reporting of interventions: template for intervention description and replication (TIDieR) checklist and guide. BMJ. 2014;348:g1687. doi: 10.1136/bmj.g1687. [DOI] [PubMed] [Google Scholar]
- 39.Petroni GR, Wages NA, Paux G, Dubois F. Implementation of adaptive methods in early-phase clinical trials. Stat Med. 2017;36(2):215–224. doi: 10.1002/sim.6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piantadosi S. Clinical Trials as Experimental Designs. In: Piantadosi S, editor. Clinical Trials: A Methodologic Perspective. 2nd ed. Wiley; 2005.
- 41.Pijls-Johannesma M, van Mastrigt G, Hahn SM, De Ruysscher D, Baumert BG, Lammering G, Buijsen J, Bentzen SM, Lievens Y, Kramar A, et al. A systematic methodology review of phase I radiation dose escalation trials. Radiother Oncol. 2010;95(2):135–141. doi: 10.1016/j.radonc.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 42.PMDA . Guidance for establishing safety in first-in-human studies during drug development. 2018. [Google Scholar]
- 43.van der Laan JW, Benson CT, Janssens W, Bos J, Stahl E, Brady JT, Wändel-Liminga U, Corriol-Rohou S, Forster R, Hartmann A, et al. Shared Learnings on the New EMA First-in-Human and Early Clinical Trial Guideline: Proceedings From a DIAlogue Session at DIA Europe 2018. Ther Innov Regul Sci. 2020;54(2):462–467. doi: 10.1007/s43441-019-00077-3. [DOI] [PubMed] [Google Scholar]
- 44.Wages NA, Horton BJ, Conaway MR, Petroni GR. Operating characteristics are needed to properly evaluate the scientific validity of phase I protocols. Contemp Clin Trials. 2021;108:106517. doi: 10.1016/j.cct.2021.106517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wheeler GM, Mander AP, Bedding A, Brock K, Cornelius V, Grieve AP, Jaki T, Love SB, Odondi L, Weir CJ, et al. How to design a dose-finding study using the continual reassessment method. BMC Med Res Methodol. 2019;19(1):18. doi: 10.1186/s12874-018-0638-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang F, Zhu J, Mo M, Zhou CM, Jia HX, Xie L, Zheng Y, Zhang S. Role of industry funders in oncology RCTs published in high-impact journals and its association with trial conclusions and time to publication. Ann Oncol. 2018;29(10):2129–2134. doi: 10.1093/annonc/mdy305. [DOI] [PubMed] [Google Scholar]
- 47.Rivoirard R, Langrand-Escure J, Oriol M, Tinquaut F, Chauvin F, Rancoule C, Magne N, Bourmaud A. Evaluation of the quality of the reporting of phase II clinical trials in oncology: a systematic review. Crit Rev Oncol Hematol. 2018;125:78–83. doi: 10.1016/j.critrevonc.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 48.Shepshelovich D, Goldvaser H, Wang L, Abdul Razak AR, Bedard PL. Comparison of reporting phase I trial results in ClinicalTrials.gov and matched publications. Invest New Drugs. 2017;35(6):827–833. doi: 10.1007/s10637-017-0510-8. [DOI] [PubMed] [Google Scholar]
- 49.Shen J, Swift B, Mamelok R, Pine S, Sinclair J, Attar M. Design and conduct considerations for first-in-human trials. Clin Transl Sci. 2019;12(1):6–19. doi: 10.1111/cts.12582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pallmann P, Bedding AW, Choodari-Oskooei B, Dimairo M, Flight L, Hampson LV, Holmes J, Mander AP, Odondi L, Sydes MR, et al. Adaptive designs in clinical trials: why use them, and how to run and report them. BMC Med. 2018;16(1):29. doi: 10.1186/s12916-018-1017-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wikler D. Must research benefit human subjects if it is to be permissible? J Med Ethics. 2017;43(2):114–117. doi: 10.1136/medethics-2015-103123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pasqualetti G, Gori G, Blandizzi C, Del Tacca M. Healthy volunteers and early phases of clinical experimentation. Eur J Clin Pharmacol. 2010;66(7):647–653. doi: 10.1007/s00228-010-0827-0. [DOI] [PubMed] [Google Scholar]
- 53.Malik L, Mejia A. Informed consent for phase I oncology trials: form, substance and signature. J Clin Med Res. 2014;6(3):205–208. doi: 10.14740/jocmr1803w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Habets MG, van Delden JJ, Bredenoord AL. The social value of clinical research. BMC Med Ethics. 2014;15:66. doi: 10.1186/1472-6939-15-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gore L, Ivy SP, Balis FM, Rubin E, Thornton K, Donoghue M, Roberts S, Bruinooge S, Ersek J, Goodman N, et al. Modernizing Clinical Trial Eligibility: Recommendations of the American Society of Clinical Oncology-Friends of Cancer Research Minimum Age Working Group. J Clin Oncol. 2017;35(33):3781–3787. doi: 10.1200/JCO.2017.74.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang SM, Lamborn KR, Kuhn JG, Yung WK, Gilbert MR, Wen PY, Fine HA, Mehta MP, DeAngelis LM, Lieberman FS, et al. Neurooncology clinical trial design for targeted therapies: lessons learned from the North American Brain Tumor Consortium. Neuro Oncol. 2008;10(4):631–642. doi: 10.1215/15228517-2008-021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Helen Diller Family Comprehensive Cancer Centre (HDFCCC). Phase I protocol template [https://cancer.ucsf.edu/sites/cancer.ucsf.edu/files/itr/sm_files/HDFCCC_Phase1_Protocol_Template_current.docx].
- 58.Cancer Therapy Evaluation Program (CTEP). Generic Protocol Template for Cancer Treatment Trial [https://ctep.cancer.gov/protocolDevelopment/docs/CTEP_Generic_Protocol_Template_for_Cancer_Treatment_Trial.docx].
- 59.Belorgey C, Pletan Y, Goehrs JM. Round Table No GXIX: Adaptation of the clinical trials directive: recommendations on the contents of a dossier for the request for authorisation of the first trials in human subjects. Therapie. 2004;59(3):329–347. doi: 10.2515/therapie:2004062. [DOI] [PubMed] [Google Scholar]
- 60.Dalton EJ, Churilov L, Lannin NA, Corbett D, Campbell BCV, Hayward KS. Multidimensional phase I dose ranging trials for stroke recovery interventions: key challenges and how to address them. Neurorehabil Neural Repair. 2021;35(8):663–679. doi: 10.1177/15459683211019362. [DOI] [PubMed] [Google Scholar]
- 61.FDA. Considerations for the design of early-phase clinical trials of cellular and gene therapy products: guidance for industry. 2015. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-design-early-phase-clinical-trials-cellular-and-gene-therapy-products.
- 62.Khan T, Stewart M, Blackman S, Rousseau R, Donoghue M, Cohen K, Seibel N, Fleury M, Benettaib B, Malik R, et al. Accelerating pediatric cancer drug development: challenges and opportunities for pediatric master protocols. Ther Innov Regul Sci. 2019;53(2):270–278. doi: 10.1177/2168479018774533. [DOI] [PubMed] [Google Scholar]
- 63.Kimmelman J, London AJ, Ravina B, Ramsay T, Bernstein M, Fine A, Stahnisch FW, Emborg ME. Launching invasive, first-in-human trials against Parkinson's disease: ethical considerations. Mov Disord. 2009;24(13):1893–1901. doi: 10.1002/mds.22712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Margolin K, Synold T, Longmate J, Doroshow JH. Methodologic guidelines for the design of high-dose chemotherapy regimens. Biol Blood Marrow Transplant. 2001;7(8):414–432. doi: 10.1016/S1083-8791(01)80009-4. [DOI] [PubMed] [Google Scholar]
- 65.Paller CJ, Bradbury PA, Ivy SP, Seymour L, LoRusso PM, Baker L, Rubinstein L, Huang E, Collyar D, Groshen S, et al. Design of phase I combination trials: recommendations of the Clinical Trial Design Task Force of the NCI Investigational Drug Steering Committee. Clin Cancer Res. 2014;20(16):4210–4217. doi: 10.1158/1078-0432.CCR-14-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Staunton C. Informed consent for HIV cure research in South Africa: issues to consider. BMC Med Ethics. 2015;16:3. doi: 10.1186/1472-6939-16-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Gerven J, Bonelli M. Commentary on the EMA Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products. Br J Clin Pharmacol. 2018;84(7):1401–1409. doi: 10.1111/bcp.13550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Korn EL, Arbuck SG, Pluda JM, Simon R, Kaplan RS, Christian MC. Clinical trial designs for cytostatic agents: are new approaches needed? J Clin Oncol. 2001;19(1):265–272. doi: 10.1200/JCO.2001.19.1.265. [DOI] [PubMed] [Google Scholar]
- 69.Dahlberg SE, Gray RJ. Pragmatic approaches to address expansion cohort design. Cancer. 2018;124(16):3290–3292. doi: 10.1002/cncr.31574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hamberg P, Ratain MJ, Lesaffre E, Verweij J. Dose-escalation models for combination phase I trials in oncology. Eur J Cancer. 2010;46(16):2870–2878. doi: 10.1016/j.ejca.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 71.Homer V, Yap C, Bond S, Holmes J, Stocken D, Walker K, Robinson EJ, Wheeler G, Brown S, Hinsley S, et al. Early phase clinical trials extension to guidelines for the content of statistical analysis plans. BMJ. 2022;376:e068177. doi: 10.1136/bmj-2021-068177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garnett C, Bonate PL, Dang Q, Ferber G, Huang D, Liu J, Mehrotra D, Riley S, Sager P, Tornoe C, et al. Scientific white paper on concentration-QTc modeling. J Pharmacokinet Pharmacodyn. 2018;45(3):383–397. doi: 10.1007/s10928-017-9558-5. [DOI] [PubMed] [Google Scholar]
- 73.Smoragiewicz M, Bogaerts J, Calvo E, Marabelle A, Perrone A, Seymour L, Shalabi A, Siu LL, Tabernero J, Giaccone G, et al. Design and conduct of early clinical studies of immunotherapy agent combinations: recommendations from the task force on Methodology for the Development of Innovative Cancer Therapies. Ann Oncol. 2018;29(11):2175–2182. doi: 10.1093/annonc/mdy398. [DOI] [PubMed] [Google Scholar]
- 74.Pledger G. Proof of efficacy trials: choosing the dose. Epilepsy Res. 2001;45(1–3):23–28. doi: 10.1016/S0920-1211(01)00208-X. [DOI] [PubMed] [Google Scholar]
- 75.Paoletti X, Postel-Vinay S. Phase I-II trial designs: how early should efficacy guide the dose recommendation process? Ann Oncol. 2018;29(3):540–541. doi: 10.1093/annonc/mdy044. [DOI] [PubMed] [Google Scholar]
- 76.LoRusso PM, Boerner SA, Seymour L. An overview of the optimal planning, design, and conduct of phase I studies of new therapeutics. Clin Cancer Res. 2010;16(6):1710–1718. doi: 10.1158/1078-0432.CCR-09-1993. [DOI] [PubMed] [Google Scholar]
- 77.Iasonos A, Gönen M, Bosl GJ. Scientific review of phase i protocols with novel dose-escalation designs: how much information is needed? J Clin Oncol. 2015;33(19):2221–2225. doi: 10.1200/JCO.2014.59.8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Booth CM, Calvert AH, Giaccone G, Lobbezoo MW, Seymour LK, Eisenhauer EA. Endpoints and other considerations in phase I studies of targeted anticancer therapy: recommendations from the task force on Methodology for the Development of Innovative Cancer Therapies (MDICT) Eur J Cancer. 2008;44(1):19–24. doi: 10.1016/j.ejca.2007.07.034. [DOI] [PubMed] [Google Scholar]
- 79.Casali PG, Bruzzi P, Bogaerts J, Blay JY. Rare Cancers Europe Consensus P: Rare Cancers Europe (RCE) methodological recommendations for clinical studies in rare cancers: a European consensus position paper. Ann Oncol. 2015;26(2):300–306. doi: 10.1093/annonc/mdu459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kurzrock R, Lin CC, Wu TC, Hobbs BP, Pestana RM, Hong DS. Moving beyond 3+3: the future of clinical trial design. Am Soc Clin Oncol Educ Book. 2021;41:e133–e144. doi: 10.1200/EDBK_319783. [DOI] [PubMed] [Google Scholar]
- 81.Mi G, Bian Y, Wang X, Zhang W. SPA: single patient acceleration in oncology dose-escalation trials. Contemp Clin Trials. 2021;105:106378. doi: 10.1016/j.cct.2021.106378. [DOI] [PubMed] [Google Scholar]
- 82.Horton BJ, O'Quigley J, Conaway MR. Consequences of performing parallel dose finding trials in heterogeneous groups of patients. JNCI Cancer Spectr. 2019;3(2):pkz013. doi: 10.1093/jncics/pkz013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tighiouart M, Piantadosi S, Rogatko A. Dose finding with drug combinations in cancer phase I clinical trials using conditional escalation with overdose control. Stat Med. 2014;33(22):3815–3829. doi: 10.1002/sim.6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reijntjes S, Albayaty M, Bush J, Cheriyan J, Cromie A, Koch A, Hammond M, Mair S, Scholes P, Lorch U, et al. The Association for Human Pharmacology in the Pharmaceutical Industry London Meeting 2018: Brexit and other challenges in early phase drug development. Front Pharmacol. 2018;9:1301. doi: 10.3389/fphar.2018.01301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Senn S, Amin D, Bailey RA, Bird SM, Bogacka B, Colman P, Garrett A, Grieve A, Lachmann P, First-in WPSI. Statistical issues in first-in-man studies. J R Stat Soc a Stat. 2007;170:517–579. doi: 10.1111/j.1467-985X.2007.00481.x. [DOI] [Google Scholar]
- 86.Ponzano S, Blake K, Bonelli M, Enzmann H. European Medicines Agency Committee for Human Medicinal Products "First-in-Human Guideline Drafting G: Promoting safe early clinical research of novel drug candidates: a European Union Regulatory Perspective. Clin Pharmacol Ther. 2018;103(4):564–566. doi: 10.1002/cpt.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Senderowicz AM. Information needed to conduct first-in-human oncology trials in the United States: a view from a former FDA medical reviewer. Clin Cancer Res. 2010;16(6):1719–1725. doi: 10.1158/1078-0432.CCR-09-2766. [DOI] [PubMed] [Google Scholar]
- 88.Milton MN, Horvath CJ. The EMEA guideline on first-in-human clinical trials and its impact on pharmaceutical development. Toxicol Pathol. 2009;37(3):363–371. doi: 10.1177/0192623309332997. [DOI] [PubMed] [Google Scholar]
- 89.Suh HY, Peck CC, Yu KS, Lee H. Determination of the starting dose in the first-in-human clinical trials with monoclonal antibodies: a systematic review of papers published between 1990 and 2013. Drug Des Devel Ther. 2016;10:4005–4016. doi: 10.2147/DDDT.S121520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mishra A, Sarangi SC, Reeta K. First-in-human dose: current status review for better future perspectives. Eur J Clin Pharmacol. 2020;76(9):1237–1243. doi: 10.1007/s00228-020-02924-x. [DOI] [PubMed] [Google Scholar]
- 91.Derhaschnig U, Gilbert J, Jäger U, Böhmig G, Stingl G, Jilma B. Combined integrated protocol/basket trial design for a first-in-human trial. Orphanet J Rare Dis. 2016;11(1):134. doi: 10.1186/s13023-016-0494-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Francillon A, Pickering G, Belorgey C. Exploratory clinical trials: implementation modes & guidelines, scope and regulatory framework. Therapie. 2009;64(3):149–159. doi: 10.2515/therapie/2009018. [DOI] [PubMed] [Google Scholar]
- 93.Harrington JA, Wheeler GM, Sweeting MJ, Mander AP, Jodrell DI. Adaptive designs for dual-agent phase I dose-escalation studies. Nat Rev Clin Oncol. 2013;10(5):277–288. doi: 10.1038/nrclinonc.2013.35. [DOI] [PubMed] [Google Scholar]
- 94.Marti GE, Bauer S, Puri RK, Noguchi PD. Regulatory review of cellular and gene therapies: an overview of the process. Transfus Sci. 1994;15(4):323–329. doi: 10.1016/0955-3886(94)90163-5. [DOI] [PubMed] [Google Scholar]
- 95.Gunhan BK, Weber S, Seroutou A, Friede T. Phase I dose-escalation oncology trials with sequential multiple schedules. BMC Med Res Methodol. 2021;21(1):69. doi: 10.1186/s12874-021-01218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wheeler GM, Sweeting MJ, Mander AP. Toxicity-dependent feasibility bounds for the escalation with overdose control approach in phase I cancer trials. Stat Med. 2017;36(16):2499–2513. doi: 10.1002/sim.7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chow SC, Corey R. Benefits, challenges and obstacles of adaptive clinical trial designs. Orphanet J Rare Dis. 2011;6:79. doi: 10.1186/1750-1172-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zohar S, O'Quigley J. Identifying the most successful dose (MSD) in dose-finding studies in cancer. Pharm Stat. 2006;5(3):187–199. doi: 10.1002/pst.209. [DOI] [PubMed] [Google Scholar]
- 99.Coates S, Taubel J, Lorch U. Practical risk management in early phase clinical trials. Eur J Clin Pharmacol. 2019;75(4):483–496. doi: 10.1007/s00228-018-02607-8. [DOI] [PubMed] [Google Scholar]
- 100.MHRA. Phase I Accreditation Scheme Requirements. Version 4.1. August 12, 2022. London: Medicines and Healthcare products Regulatory Agency (UK); 2022. Available from: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1102629/1._MHRA_Phase_I_Accreditation_Scheme_Requirements_v4.1_12_August_2022.pdf.
- 101.Prowell TM, Theoret MR, Pazdur R. Seamless oncology-drug development. N Engl J Med. 2016;374(21):2001–2003. doi: 10.1056/NEJMp1603747. [DOI] [PubMed] [Google Scholar]
- 102.Kurzrock R, Stewart DJ. Compliance in early-phase cancer clinical trials research. Oncologist. 2013;18(3):308–313. doi: 10.1634/theoncologist.2012-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boissel JP, Durieu I, Girard P, Nony P, Chauvin F, Haugh M. Dose-ranging trials: guidelines for data collection and standardized descriptions. Control Clin Trials. 1995;16(5):319–330. doi: 10.1016/0197-2456(95)00002-X. [DOI] [PubMed] [Google Scholar]
- 104.Breithaupt-Groegler K, Coch C, Coenen M, Donath F, Erb-Zohar K, Francke K, Goehler K, Iovino M, Kammerer KP, Mikus G, et al. Who is a 'healthy subject'?-consensus results on pivotal eligibility criteria for clinical trials. Eur J Clin Pharmacol. 2017;73(4):409–416. doi: 10.1007/s00228-016-2189-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Caldwell S, Sima C, Jameson G, Fleck S, Weiss GJ. Factors influencing time to determination of the recommended phase 2 dose in phase 1 clinical trials. Am J Clin Oncol. 2013;36(2):146–150. doi: 10.1097/COC.0b013e31824370a3. [DOI] [PubMed] [Google Scholar]
- 106.Mehrotra DV. A note on the draft International Council for Harmonisation guidance on estimands and sensitivity analysis. Clin Trials. 2019;16(4):339–344. doi: 10.1177/1740774519844259. [DOI] [PubMed] [Google Scholar]
- 107.Potter DM. Phase I studies of chemotherapeutic agents in cancer patients: a review of the designs. J Biopharm Stat. 2006;16(5):579–604. doi: 10.1080/10543400600860295. [DOI] [PubMed] [Google Scholar]
- 108.van den Bogert CA, Cohen AF, Leufkens HGM, van Gerven JMA. Pharmacological vs. classical approaches in the design of first in man clinical drug trials. Br J Clin Pharmacol. 2017;83(12):2807–2812. doi: 10.1111/bcp.13422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen Z, Yuan Y, Li Z, Kutner M, Owonikoko T, Curran WJ, Khuri F, Kowalski J. Dose escalation with over-dose and under-dose controls in phase I/II clinical trials. Contemp Clin Trials. 2015;43:133–141. doi: 10.1016/j.cct.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bonnett LJ, Davies GR. Quality of outcome reporting in phase II studies in pulmonary tuberculosis. Trials. 2015;16:518. doi: 10.1186/s13063-015-1050-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.FDA . ive designs for clinical trials of drugs and biologics: guidance for industry. 2019. [Google Scholar]
- 112.O'Neill RT, Temple R. The prevention and treatment of missing data in clinical trials: an FDA perspective on the importance of dealing with it. Clin Pharmacol Ther. 2012;91(3):550–554. doi: 10.1038/clpt.2011.340. [DOI] [PubMed] [Google Scholar]
- 113.Parasrampuria DA, Benet LZ. Inclusion of placebos and blinding for ascending dose first-in-human studies and other underpowered phase 1 studies has not been justified and on balance is not useful. Basic Clin Pharmacol Toxicol. 2015;117(1):44–51. doi: 10.1111/bcpt.12352. [DOI] [PubMed] [Google Scholar]
- 114.Senn S. Ethical and Practical Issues in Phase 1 Trials in Healthy Volunteers. In: Gould AL, editor. Statistical Methods for Evaluating Safety in Medical Product Development. 1st ed. John Wiley & Sons; 2015.
- 115.Aarons L, Karlsson MO, Mentre F, Rombout F, Steimer JL, van Peer A, Experts CB. Role of modelling and simulation in phase I drug development. Eur J Pharm Sci. 2001;13(2):115–122. doi: 10.1016/S0928-0987(01)00096-3. [DOI] [PubMed] [Google Scholar]
- 116.EMA . Guidelines on Data Monitoring Committees In. 2005. [Google Scholar]
- 117.PMDA . Guideline on Data Monitoring Committee. 2013. [Google Scholar]
- 118.Coulman KD, Nicholson A, Shaw A, Daykin A, Selman LE, Macefield R, Shorter GW, Cramer H, Sydes MR, Gamble C, et al. Understanding and optimising patient and public involvement in trial oversight: an ethnographic study of eight clinical trials. Trials. 2020;21(1):543. doi: 10.1186/s13063-020-04495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Genske A, Engel-Glatter S. Rethinking risk assessment for emerging technology first-in-human trials. Med Health Care Philos. 2016;19(1):125–139. doi: 10.1007/s11019-015-9660-7. [DOI] [PubMed] [Google Scholar]
- 120.Rengelshausen J, Breithaupt-Groegler K, Donath F, Erb-Zohar K, Hardman T, Mikus G, Plassmann S, Wensing G, Sourgens H. How to interpret an investigator's brochure for meaningful risk assessment: results of an AGAH discussion forum. Ther Innov Regul Sci. 2021;55(3):612–618. doi: 10.1007/s43441-021-00257-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yap C, Billingham LJ, Cheung YK, Craddock C, O'Quigley J. Dose transition pathways: the missing link between complex dose-finding designs and simple decision-making. Clin Cancer Res. 2017;23(24):7440–7447. doi: 10.1158/1078-0432.CCR-17-0582. [DOI] [PubMed] [Google Scholar]
- 122.Foster M, Fergusson DA, Hawrysh T, Presseau J, Kekre N, Schwartz S, Castillo G, Asad S, Fox G, Atkins H, et al. Partnering with patients to get better outcomes with chimeric antigen receptor T-cell therapy: towards engagement of patients in early phase trials. Res Involv Engagem. 2020;6:61. doi: 10.1186/s40900-020-00230-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Collaborative initiative IRAS. Integrated Research Application System (IRAS) form Question A6–1. [https://www.myresearchproject.org.uk/help/hlpcollatedqsg-iras.aspx#2297].
- 124.World Health Organisation. WHO Trial Registration Data Set (version 1.3.1). https://www.who.int/clinical-trials-registry-platform/network/who-daWHOta-set.
- 125.Eldridge SM, Chan CL, Campbell MJ, Bond CM, Hopewell S, Thabane L, Lancaster GA. group Pc: CONSORT 2010 statement: extension to randomised pilot and feasibility trials. BMJ. 2016;355:i5239. doi: 10.1136/bmj.i5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Clinical Trials Ontario. Child and Adolescent Migraine Prevention (CHAMP) brief summary report [https://www.ctontario.ca/cms/wp-content/uploads/2022/05/Populated-CommuniKIDS-Template-May-2022-Final-002.pdf].
- 127.Pfizer. Study to evaluate the safety of combinations of avelumab, binimetinib and talazoparib in patients with advanced cancer [https://www.pfizer.com/science/clinical-trials/plain-language-study-results-summaries/study-evaluate-safety-combinations].
- 128.Pfizer. Study to evaluate safety of axitinib in combination with MK-3475 in patients with advanced renal cell cancer [https://www.pfizer.com/science/clinical-trials/plain-language-study-results-summaries/study-evaluate-safety-axitinib].
- 129.Servier. Clinical Trial Lay summary: study that tested the combination of 2 medicines for people with glioblastoma, a kind of brain cancer: a new drug called S49076 with a marketed drug called bevacizumab. [https://clinicaltrials.servier.com/wp-content/uploads/CL1-49076-002-lay-summary.pdf].
- 130.UCB. Clinical study results: a study to learn if certolizumab pegol works in Chinese participants with active rheumatoid arthritis [https://www.ucb.com/_up/ucb_com_clinical_studies/documents/ra0044-lay_summary_approved_v2_0.pdf].
- 131.UCB. Study Summary: To look at the transfer of certolizumab pegol across the placenta (CRIB). [https://www.ucb.com/_up/ucb_com_rd/documents/up0017-lay-summary-eu.pdf].
- 132.HRA. Writing a plain language (lay) summary of your research findings. [https://www.hra.nhs.uk/planning-and-improving-research/best-practice/writing-plain-language-lay-summary-your-research-findings/].
- 133.NIHR. Plain English Summaries [https://www.nihr.ac.uk/documents/plain-english-summaries/27363].
- 134.Barnes A, Patrick S. Lay summaries of clinical study results: an overview. Pharmaceut Med. 2019;33(4):261–268. doi: 10.1007/s40290-019-00285-0. [DOI] [PubMed] [Google Scholar]
- 135.Brown CM, Leithold LH, Sroka-Saidi K, Schindler TM. Lay summaries for phase I trials in healthy volunteers. Med Writing. 2020;29(4):24. [Google Scholar]
- 136.European Commission. Good lay summary practice guidance. [https://health.ec.europa.eu/latest-updates/good-lay-summary-practice-guidance-2021-10-04_en].
- 137.Clinical Trials Ontario. Plain language result summaries. [https://www.ctontario.ca/patients-public/resources-for-engaging-patients/toolkit-to-improve-clinical-trial-participants-experiences/plain-language-summaries/#tab12].
- 138.ICON PLC. Plain language summaries keeping the end in mind: key considerations for creating plain language summaries. [https://www.iconplc.com/insights/regulatory/keeping-the-end-in-mind-key-considerations-for-creating-plain-language-summaries/].
- 139.Villacampa G, Patel D, Zheng H, McAleese J, Rekowski J, Solovyeva O, Yin Z, Yap C. Assessing the reporting quality of early phase dose-finding trial protocols: a methodological review. eClinicalMedicine. 2023;60:102020. doi: 10.1016/j.eclinm.2023.102020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Turner L, Shamseer L, Altman DG, Weeks L, Peters J, Kober T, Dias S, Schulz KF, Plint AC, Moher D. Consolidated standards of reporting trials (CONSORT) and the completeness of reporting of randomised controlled trials (RCTs) published in medical journals. Cochrane Database Syst Rev. 2012;11:MR000030. doi: 10.1002/14651858.MR000030.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Butcher NJ, Monsour A, Mew EJ, Chan AW, Moher D, Mayo-Wilson E, Terwee CB, Chee ATA, Baba A, Gavin F, et al. Guidelines for reporting outcomes in trial reports: The CONSORT-Outcomes 2022 Extension. JAMA. 2022;328(22):2252–2264. doi: 10.1001/jama.2022.21022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Composition of the DEFINE Executive Committee and Independent Expert Panel.
Additional file 2. CONSORT and SPIRIT DEFINE Literature review: Figures A2-1 – A2-2, Tables A2-1. Figure A2-1. PRISMA 2020 flow diagram for the CONSORT-DEFINE literature search and review. Figure A2-2. PRISMA flow diagram for the SPIRIT-DEFINE literature search and review. Table A2-1. Terms used in the PubMed searches and the number of hits from each search terms for SPIRIT-DEFINE.
Additional file 3. Organisations contacted for protocol templates or guidelines and responses.
Additional file 4. Delphi Survey key stakeholder groups and methods of access.
Additional file 5. Further details on the statistical analysis plan.
Additional file 6. SPIRIT-DEFINE and CONSORT-DEFINE items generation for Delphi survey: Table A6-1. Table A6-1. References supporting the proposed candidate items in the DEFINE Delphi survey.
Additional file 7. Toolkit for Lay Summary of EPDF Trial Results.
Additional file 8. Delphi survey participants demographics and harmonisation of roles: Tables A8-1 – A8-2. Table A8-1. Demographics of Delphi survey participants. Table A8-2. Harmonisation of participants’ defined roles.
Additional file 9. Perception of proposed items and sensitivity analysis: Tables A9-1 – A9-3, Figures A9-1 – A9-13. Table A9-1. Summary of categorical and numerical ratings at Round One and 2. Figure A9-1. Percentage of participants changing their numerical scores at Round Two. Figure A9-2. Percentage of participants changing their categorical scores at Round Two. Table A9-2. Number of participants changing their ratings between Round One and Round Two. Table A9-3. Frequency and percentage of perfect agreement. Figure A9-3. Stacked bar plots of candidate item [17 SPIRIT-DEFINE] “Planned dosing regimens presented as a diagram or table, where applicable” by stakeholders. Figure A9-4. Stacked bar plots of candidate item [18 CONSORT-DEFINE] “Planned and delivered dosing regimens presented as a diagram or table, where applicable” by stakeholders. Figure A9-5. Stacked bar plots of candidate item [48 SPIRIT-DEFINE] “Statistical methods for additional analyses” by stakeholders. Figure A9-6. Stacked bar plots of candidate item [69 CONSORT-DEFINE] “Specify if and when results were reported whilst the trial was still ongoing” by stakeholders. Figure A9-7. Stacked bar plots of candidate item [70 SPIRIT-DEFINE] “Where the full trial protocol or the redacted version, with amendments, can be accessed” by stakeholders. Figure A9-8. Stacked bar plots of candidate item [17 SPIRIT-DEFINE] “Planned dosing regimens presented as a diagram or table, where applicable” by Continent. Figure A9-9. Stacked bar plots of candidate item [18 CONSORT-DEFINE] “Planned and delivered dosing regimens presented as a diagram or table, where applicable” by Continent. Figure A9-10. Stacked bar plots of candidate item [48 SPIRIT-DEFINE] “Statistical methods for additional analyses” by Continent. Figure A9-11. Stacked bar plots of candidate item [69 CONSORT-DEFINE] “Specify if and when results were reported whilst the trial was still ongoing” by Continent. Figure A9-12. Stacked bar plots of candidate item [70 SPIRIT-DEFINE] “Where the full trial protocol or the redacted version, with amendments, can be accessed” by Continent. Figure A9-13. Stacked bar plots of candidate item [5 SPIRIT-DEFINE] “Summary of findings from existing correlative biomarker, correlative and associated studies to support planned biomarker sub-study” by Continent.
Additional file 10. List of candidate items to be discussed at the DEFINE Consensus meeting.
Additional file 11. Qualitative analysis. Table A11-1; Figure A11-1. Table A11-1. Themes and codes identified in the general comments received in Delphi survey. Figure A11-1. Tree diagram of the comments received per theme.
Data Availability Statement
Data is shared under a data transfer agreement or a collaboration agreement depending on the nature of the sharing. Requests are via a standard proforma describing the nature of the proposed research and extent of data requirements.