

Abstract
Background:
Adolescent intermittent ethanol (AIE) exposure causes long-term changes in the brain and behavior of adult male rodents, including persistent induction of innate immune pathways, reductions in hippocampal neurogenic and forebrain cholinergic neuronal markers, and reversal learning deficits. The current study tests the hypothesis that proinflammatory induction mediates AIE-induced (1) loss of adult neurogenesis (i.e., doublecortin (DCX) expressing immature neurons), (2) reductions in forebrain and hippocampal cholinergic markers, and (3) reversal learning deficits.
Methods:
Male and female rats underwent AIE (5.0 g/kg/day ethanol or water, i.g., 2 day-on/2 day-off from postnatal day (PND) 25–54), followed by a 2-week regimen of the anti-inflammatory compound indomethacin (4.0 g/kg/day, PND 56–69) or vehicle, after which one cohort was euthanized for immunohistochemical markers (PND 70) and the second underwent the Morris water maze to assess reversal learning.
Results:
AIE reduced adult (PND 70) DCX+ immunoreactivity (IR) and increased hippocampal expression of the innate immune signal’s high-mobility group box protein 1 (HMGB1 + IR) and cyclooxygenase-2 (COX-2 + IR) in adult male and female rats. AIE also reduced choline acetyltransferase (ChAT+IR) in the basal forebrain and co-labeling of hippocampal vesicular acetylcholine transporter (VAChT+) cholinergic terminals on DCX + IR neurons. Indomethacin treatment after AIE restored molecular endpoints to control levels and rescued AIE-induced reversal learning deficits in the Morris water maze in both sexes. Of note, indomethacin produced several adverse effects selectively in control conditions, highlighting the uniquely beneficial effect of indomethacin in AIE rats.
Conclusions:
These data suggest that in males and females, (1) AIE persistent neuroimmune induction mediates both the loss of adult hippocampal DCX and loss of basal forebrain cholinergic neurons and their innervation to hippocampal targets, and (2) anti-inflammatory indomethacin treatment following AIE that restores these persistent molecular pathologies also restores spatial reversal learning deficits.
Keywords: acetylcholine, alcohol, cognitive flexibility, doublecortin, neuroinflammation
INTRODUCTION
The behavioral and molecular consequences of adolescent intermittent EtOH (AIE) are pernicious, lasting long into adulthood (Crews et al., 2019). Behaviorally, rodents exposed to AIE exhibit deficits in reversal learning (Crews et al., 2019; Gómez-A et al., 2021; Macht, Elchert, & Crews, 2020), which has also been evidenced in binge-drinking college students (Yoo & Kim, 2016), reflecting a loss of cognitive-behavioral flexibility that is frequently associated with neuropsychiatric, neurodegenerative, and substance use disorders in adult humans (Izquierdo et al., 2017). Several neural substrates have been linked to reversal learning, including adult hippocampal neurogenesis (Anacker & Hen, 2017). Newborn neurons are highly excitable, making them an integral cellular mechanism underlying reversal learning in spatial navigation tasks. Interestingly, reversal learning deficits after AIE persist in abstinence as do reductions in adult hippocampal neurogenesis (Broadwater et al., 2014; Coleman et al., 2014; Crews et al., 2006; McClain et al., 2011; Nixon & Crews, 2002). Both can be restored in males with voluntary exercise (Vetreno et al., 2018, 2020). However, the mechanisms underlying both AIE-induced molecular and behavioral deficits and exercise restoration are poorly understood and need to be extended to both sexes.
A potential molecular underpinning of neurogenic deficits after AIE is the induction of proinflammatory innate immune signaling in the hippocampus (Vetreno et al., 2018; Vore et al., 2017, 2021), which may be exacerbated by impaired cholinergic anti-inflammatory feedback. The cholinergic system exerts anti-inflammatory regulation over the innate immune response through nicotinic alpha-7 receptor signaling on microglia and macrophages (Pavlov et al., 2003). Loss of this regulation through decreased cholinergic innervation, reductions in cholinergic release, or cholinergic neuron cell loss is frequently associated with chronic low-grade inflammation and impaired cognition. While often reported in aging and neurodegenerative disorders, disruptions in cholinergic anti-inflammatory control may also underlie the chronic neuroinflammation evidenced after AIE. For example, basal forebrain cholinergic neurons, which project to and innervate the hippocampus, are reduced in adult males after AIE (Vetreno et al., 2020; Vetreno & Crews, 2018), and these reductions are accompanied by blunted behaviorally-evoked acetylcholine efflux in the hippocampus (Reitz et al., 2021). Treatment with the cholinesterase inhibitors donepezil and galantamine after AIE restored the loss of neurogenesis and reversed neuroimmune induction in the hippocampus of adult male rats (Macht et al., 2021; Swartzwelder et al., 2019). Galantamine also reversed AIE induction of the danger-associated molecular pattern high-mobility group box 1 (HMGB1) and the prostaglandin synthesis enzyme cyclooxygenase-2 (Macht et al., 2021) in the granule cell layer of the hippocampus in adult male rats exposed to AIE. These findings suggest that AIE deficits in cholinergic regulation of the hippocampal neuroinflammatory environment may underlie AIE induction of innate immune signaling pathways and loss of adult neurogenesis, contributing to AIE reversal learning deficits. While several studies have suggested that impaired cholinergic function may be linked to neurogenic and behavioral plasticity deficits after AIE, studies have not examined these circuits collectively.
The current study expands upon these observations to test whether proinflammatory inhibition by indomethacin rescues both cholinergic and hippocampal pathology in male and female rats after AIE. Indomethacin is a highly effective nonsteroidal anti-inflammatory drug, which inhibits the prostaglandin synthesis enzymes COX-1 and COX-2, among other mechanisms. Prior studies have demonstrated that EtOH acutely increases COX-2 during EtOH withdrawal (Knapp & Crews, 1999), and AIE persistently increases hippocampal proinflammatory signals in adulthood, including COX-2 (Macht et al., 2021). Therefore, we tested whether indomethacin intervention after AIE (and the established neuropathology was present) could restore neuroinflammatory, neurogenic, and behavioral deficits. We hypothesize that AIE will increase hippocampal HMGB1 and COX-2 proinflammatory signals, decrease cholinergic-hippocampal innervation, reduce newborn neuron marker doublecortin (DCX), and impair reversal learning during a Morris water maze task in adult male and female rats. We further tested whether AIE impacted somatostatin hilar inhibitory interneurons as no studies have examined this cellular population, and loss of inhibitory regulation can be an underlying mechanism of increased neuroinflammation through increased granule cell excitation. Moreover, we hypothesize that indomethacin anti-inflammatory treatment will restore molecular and behavioral endpoints after AIE, emphasizing the importance of neuroimmune signaling and cholinergic loss in AIE pathogenesis.
MATERIALS AND METHODS
Animals
Wistar rats were bred and housed at the University of North Carolina at Chapel Hill in a temperature- (20°C) and humidity-controlled vivarium on a 12/12 h light/dark cycle with lights on at 7:00 a.m. Litters were culled to six males and four females whenever possible on the first postnatal day (PND). On PND 21 (±2), males and females were same-sex pair-housed according to the treatment group using a split-litter design. The experiment consisted of eight groups in a 2 × 2 × 2 design with the factors of sex (male; female), adolescent intermittent exposure (EtOH, AIE; water control, CON), and drug treatment (vehicle, indomethacin). All rats had ad libitum access to food and water throughout the experiment. Studies were conducted in accordance with NIH regulations and with approval of the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill. As described in greater detail below, the experiments consisted of two cohorts. Cohort 1 animals were euthanized for IHC markers on PND 70 (N = 96), and cohort 2 (N = 79) underwent behavioral assessments and were euthanized by PND 105. For more details on n sizes, see Table 1. For an experimental timeline, see Figure 1.
TABLE 1.
n Sizes per group.
| Groups | Cohort | n sizes |
|---|---|---|
|
| ||
| CON-Vehicle | 1 | 13 male; 12 female |
| 2 | 10 male; 10 female | |
| CON-Indo | 1 | 12 male; 13 female |
| 2 | 10 male; 10 female | |
| AI E-Vehicle | 1 | 13 male; 13 female |
| 2 | 10 male; 10 female | |
| AIE-Indo | 1 | 11 male; 9 female |
| 2 | 10 male; 9 female | |
Note: IHC (Cohort 1), Behavior & PCR (Cohort 2).
FIGURE 1.
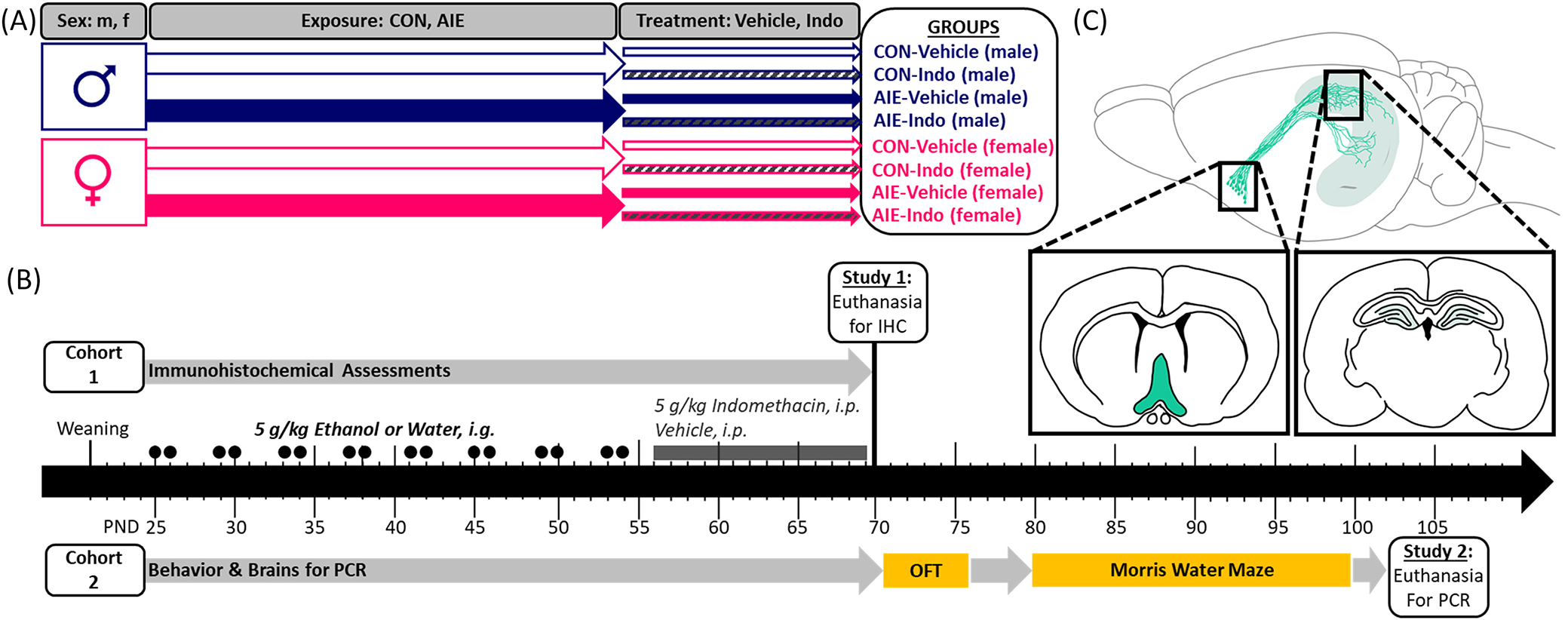
Experimental timeline. (A) The current experiments consisted of eight groups in a 2 × 2 × 2 design with the factors of sex (male, female), exposure (CON, AIE), and treatment (vehicle, Indomethacin). All animals were bred in-house and randomly assigned to treatment groups at weaning (PND 21) in a split-litter design. (B) Two separate cohorts of animals were run for two sets of experiments. The first cohort was utilized for IHC and the second underwent behavior, after which brains were processed for PCR. Starting on PND 25, exposure to either once daily intragastric intubation with water or 5 g/kg EtOH began. Rats were intubated in a 2-day-on/2-day-off cycle throughout adolescence. EtOH/water exposure stopped at PND 54. From PND 56–69, once daily intraperitoneal injections of 4 mg/kg indomethacin or vehicle (0.5% carboxymethylcellulose) began. Rats from Cohort 1 were euthanized on PND 70 for IHC. Cohort 2 continued after this time for behavioral analyses. Rats underwent the open field test to assess locomotion and thigmotaxis and afterwards performed the Morris water maze, which consisted of 6 days of acquisition training followed by 2 days of reversal training. All rats from experiment 2 were euthanized by PND 105 where the hippocampi were dissected for PCR. (C) Histological measures from Cohort 1 were performed on sections from the basal forebrain and dorsal hippocampus. PCR was performed on hippocampal dissections from Cohort 2.
AIE paradigm
In order to model human adolescent binge drinking (4+/5+ drinks in 2 h) (Patrick & Terry-McElrath, 2019) in both exposure pattern and levels of blood EtOH content, a rodent model of adolescent intermittent EtOH (AIE) exposure was developed by the Neurobiology of Adolescent Drinking in Adulthood (NADIA) consortium, described previously (Macht, Elchert, & Crews, 2020). Rats received a single daily intragastric (i.g.) intubation of EtOH (5.0 g/kg) or a comparable volume of water on a 2-day-on/2-day-off schedule (PND 25–54; see Figure 1). This window encompasses peri-adolescent development in rodents and allows the discrete assessment of long-term effects of adolescent EtOH exposure without confounding effects of adult exposure. All rats gained weight across adolescence, discussed in detail in Figure S1.
Blood ethanol content (BEC) was assessed from tail bleeds on PND 38 or PND 54, 1-h post gavage, and then analyzed using a GM7 Analyzer (Analox; London, UK), as described previously (Vetreno & Crews, 2018). There were no differences in BECs between PND 38 or 54, and so, data were collapsed across days. Results were consistent with those reported in previous studies (189 ± 8 mg/dL in males; 144 ± 6 mg/dL in females) (Macht et al., 2021; Macht, Elchert, & Crews, 2020).
Indomethacin treatment
From PND 56–69, rats received daily intraperitoneal (i.p.) injections of vehicle (0.5% carboxymethylcellulose) or 4.0 mg/kg indomethacin, a highly-effective nonsteroidal anti-inflammatory compound. Indomethacin was selected to test the mechanism by which AIE-induced pathology persists in the adult brain. This dosing regimen was selected based on prior studies that suggest that 4.0 mg/kg indomethacin is sufficient to prevent neuroinflammatory damage by EtOH (Pascual et al., 2007; Vetreno et al., 2018; Vetreno & Crews, 2018). The pH of the final solution was verified for tolerability to acidity as indomethacin is known to rapidly ionize in poor buffers with the potential to cause gastric distress.
Rats were weighed daily during this treatment period as indomethacin is not well tolerated under chronic conditions in clinical populations and can induce adverse side effects including weight loss. Three animals were removed from the study due to acute weight loss, attributed to an adverse drug reaction. Indomethacin did not impact bodyweight in any other animals (see Figure S1).
Immunohistochemistry
Cohort 1 rats were euthanized with sodium pentobarbital (100 mg/kg, i.p.) on PND 70 and then transcardially perfused with 4.0% paraformaldehyde/0.1 M phosphate-buffered saline. Brains were removed and postfixed for an additional 24 h, and then sunk in a 30% sucrose/0.1 M PBS solution at 4.0°C over the course of several days. Brains were then frozen and cut in 40 μm serial coronal sections using a MICROM HM450 sliding microtome (Thermo Scientific, Austin, TX). Immunohistochemistry was performed as described previously (Macht et al., 2021). Sections underwent antigen retrieval via a 1-h incubation (70°C) with Citra buffer (BioGenex, #HK080-9K) when necessary, and a 48-h incubation at 4°C in a primary antibody (Table 2). Following, sections were incubated in 1:200 biotinylated anti-goat (Vector Laboratories, #BA-5000), anti-rabbit (BA-1000), or anti-mouse (BA-9200) secondary antibodies for 1 h at room temperature, followed by another 1-h incubation in Vectastain® Elite ABC kit (#PK-6100) for nickel/cobalt-enhanced diaminobenzidine (DAB) visualization. Conversely, Alexa Fluor™ donkey anti-mouse 488 (#A32787) and donkey anti-goat 594 (#A32758) antibodies (1:200) were used for immunofluorescent visualization.
TABLE 2.
Antibody information.
| Antibody | Dilution | Vendor | Catalog number | Application |
|---|---|---|---|---|
|
| ||||
| Mouse anti-doublecortin | 1: 200 | Santa Cruz Biotech | sc-271390 | IHC |
| 1:100 | IF | |||
| Mouse anti-somatostatin | 1:1000 | Santa Cruz Biotech | sc-744556 | IHC |
| Rabbit anti-HMGBl | 1: 1000 | Abcam | ab18256 | IHC |
| Rabbit anti-COX-2 | 1: 2500 | Cell Signaling | 12282S | IHC |
| Goat anti-VAChT | 1:100 | Millipore | ABN100 | IF |
| Goat anti-ChAT | 1: 200 | Millipore | AB144P | IHC |
Histological quantification
Photomicrographs of four representative hippocampal sections from each animal were captured using an Olympus BX50 microscope with a 20× objective and a Sony DCX-390 camera and then quantified using BioQuant Nova Advanced Image Analysis software (R&M Biometric, Nashville, TN). For immunofluorescence analysis, all images were captured using a Nikon DS-RiZ immunofluorescent microscope (Nikon, Inc., Melville, NY) with a 20× objective and then quantified using the Nikon NIS-Elements AR46 software.
The superior and inferior blades of the granule cell layer of the dorsal dentate gyrus were assessed for quantification of VAChT, DCX, HMGB1, and COX-2. Somatostatin interneurons were quantified in the hilar region. Basal forebrain cholinergic neurons include the medial septum, ventral diagonal band, medial forebrain bundle, and the horizontal diagonal band of Broca. Details on region specifics are highlighted in Figure S2 (see Paxinos & Watson, 1986). All future references to the dorsal hippocampus and basal forebrain are selectively referring to these subregions. Sections were analyzed using a modified unbiased stereological technique validated for hippocampal counts as described previously (Macht et al., 2021; Nixon & Crews, 2002).
DCX is a cytoskeletal protein that allows visualization of immature neuroprogenitors and their dendrites, best quantitated by the number of positive immunoreactive (+IR) pixels per total area (mm2). All other immunoreactive markers are expressed in clearly defined cells that were quantified as the sum of IR+ cell numbers per total area. Immunofluorescence results for VAChT were quantified as fluorescent intensity, normalized to the area of the identified region of interest, and then transformed to a percentage of the mean of respective CON-Vehicle rats for each sex to facilitate ease of interpretation. Co-labeling for VAChT with DCX was defined as overlapping immunofluorescent positive labeling, as identified by the Nikon NIS-Elements software platform. As previously reported, these parameters produce highly reproducible results that are nearly identical to those used in traditional unbiased stereology.
Open field test
A 10-min open field test (PND 71–76) was conducted to confirm the equivalent locomotor activity with males being tested across the first 3 days and females being tested across the latter 3 days. The apparatus consisted of an 80 cm × 80 cm black Kydex box with a white gridded floor that was cleaned with Peroxigard between each session. All testing was recorded with EthoVision software. Distance traveled (cm) and latency to enter the inner zone (seconds, s) were assessed as indicators of locomotion and thigmotaxis, respectively.
Morris water maze
The Morris water maze was used to assess spatial and reversal learning. This maze consisted of a 120 cm plastic pool surrounded by spatial cues and pool and filled with room temperature water obscured with nontoxic tempura paint. Following 2-days of single-trial habituation training with a visible platform, the platform was submerged and moved to a new quadrant for six consecutive days of spatial learning trials with three trials per day. For all trials, each rat was gently placed into the water and allowed up to 2 min to swim to the visible platform, after which they were guided to the target, removed from the pool, and dried off. Starting positions changed throughout and were counterbalanced across trials. Following, rats went through 2 days of reversal learning where the platform was moved to the opposite pool quadrant. Noldus EthoVision XT tracking software was used to track each rat, and latency to find the target platform was recorded as an index of learning.
PCR for the nicotinic cholinergic receptor alpha-7 (nAChR α7)
Male and female rats from cohort 2 were euthanized after PND 100 following a lethal dose of sodium pentobarbital (100 mg/kg, i.p.) and transcardially perfused with sterile 0.1 M phosphate-buffered saline (pH 7.4) as previously described (Vetreno & Crews, 2018). Whole hippocampal sections were dissected on ice, frozen in liquid nitrogen, and then stored at −80°C for later mRNA extraction. Samples were thawed on ice and then homogenized in TRI reagent (Sigma-Aldrich) to facilitate single-step mRNA extraction, after which mRNA was reverse transcribed as previously described (Vetreno & Crews, 2012) and in Figure 1. Beta-actin and GAPDH were used as housekeeping genes to normalize gene expression data for males and females, respectively. Primer sequences (Table 3) are as follows:
TABLE 3.
Primer sequences.
| Primer | (F’) Forward sequence | (R’) Reverse sequence |
|---|---|---|
|
| ||
| Beta-actin | CTACAATGAGCTGCGTGTGGC | CAGGTCCAGACGCAGGATGGC |
| GAPDH | GAGAGAGGCCCAGCTACTCG | CAAATCCGTTCACACCGACC |
| nAChRa7 | TTCTCCTCTATAACAGTGCTGATG | GACCACCCTCCATAGGACCA |
Statistics
All immunohistochemical measurements were analyzed as a 2 (male, female) × 2 (CON, AIE) × 2 (Veh, Indo) analysis of variance (ANOVA) using SPSS Statistics software. For PCR results for nAChR α7, data were run as 2 (male, female) × 2 (CON, AIE) × 2 (Veh, Indo) analysis of variance (ANOVA) using SPSS Statistics software. Because behavioral tests were performed on separate dates for males and females, each sex was analyzed independently. Morris water maze analyses were split between acquisition and reversal learning with the repeated measures factors of test day (acquisition 1–6, reversal 1, 2) and trial number (1, 2, 3). For PCR, all results were normalized to a percentage of the mean for the CON-Vehicle groups for males and females, respectively, and then, data from each sex were run independently. Bonferroni-corrected simple main effects post-hoc analyses were performed when applicable following a significant interaction. Grubb’s outlier analyses were performed on all endpoints and statistical outliers removed when appropriate; n sizes are presented in each graph. A priori simple main effects analyses were performed as indicated below in representative sections when replicating prior results in the field. For all assessments, α = 0.05.
RESULTS
Indomethacin rescues AIE-reduced dentate gyrus DCX immunoreactivity in males and females
Doublecortin is a microtubule-associated protein upregulated in immature developing neurons and lost in mature neurons, providing a commonly used index of neurogenesis. Previous studies have found DCX + IR reduced in males a day after the last EtOH exposure and persists through adulthood (PND 220; Macht et al., 2021; Macht, Crews, & Vetreno, 2020; Vetreno et al., 2018). We now extend these findings to report a loss in DCX + IR after AIE in adult females. Although females exhibited more DCX+ immunoreactivity (IR) in general than males, AIE-reduced DCX + IR in both sexes, p < 0.001. Results further indicate that there was an interaction between EtOH exposure and later indomethacin treatment on adult (PND 70) hippocampal neurogenesis, F(1, 84) = 14.9, p < 0.001, as well as a main effect of sex, F(1, 84) = 7.1, p = 0.01. More specifically, AIE exposure reduced DCX + IR by 33% in males and by 29% in females, suggesting EtOH-induced loss of DCX + IR is similar in magnitude across both sexes. Excitingly, indomethacin rescued this loss of DCX + IR in both sexes, significantly increasing IR by 47% in males and by 36% in females relative to AIE-Vehicle conditions, p’s = 0.02. Collectively, these results suggest that AIE-induced reductions in neurogenesis can be rescued by indomethacin (Figure 2).
FIGURE 2.
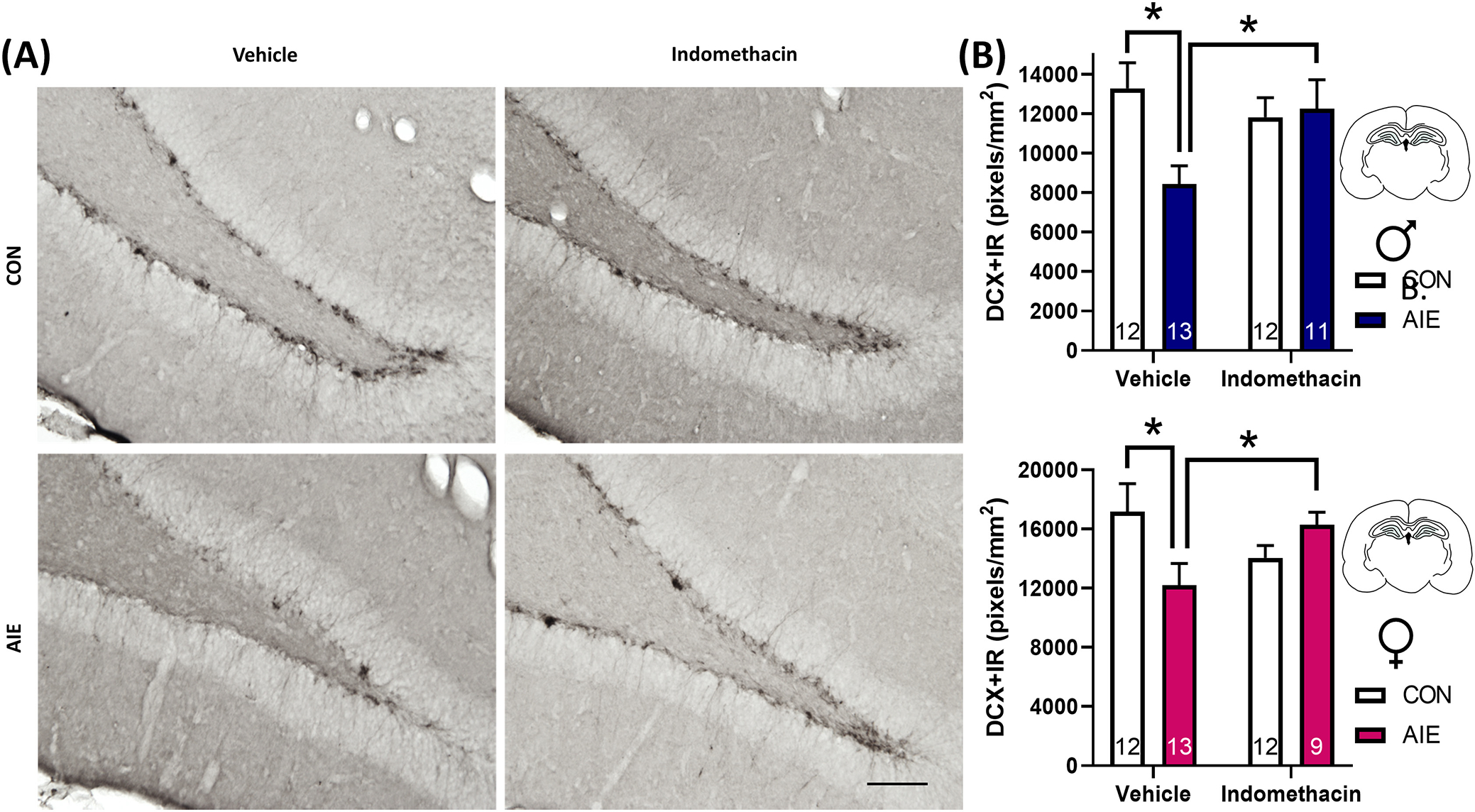
Indomethacin rescues adolescent intermittent EtOH (AIE)-induced loss of the immature neuron marker doublecortin (DCX) in the adult dorsal dentate gyrus in male and female rats. (A) Photomicrographs of DCX+ immunoreactivity (IR). Scale bar indicates 100 μm. (B) Expression of DCX + IR in adulthood is significantly reduced in AIE-treated females by 29% and in AIE-treated males by 33%, p’s < 0.05. Chronic treatment with the nonsteroidal anti-inflammatory compound indomethacin rescues this deficit in both sexes (males: blue, females: pink), suggesting restoring the neuroinflammatory balance in the neurogenic niche is essential to recovering neurogenic deficits after AIE. Sections were quantified as immunoreactive pixels per mm2. Data are expressed as mean ± SEM. *p < 0.05 between indicated groups.
Indomethacin rescues AIE induction of hippocampal innate immune signals in adult male and female rats
Cyclooxygenases, including COX-1 and COX-2, are targets of the nonsteroidal anti-inflammatory drug indomethacin. While COX-1 is constitutively expressed across the body and classically considered a housekeeping enzyme, COX-2 is inducible with robust labeling in the hippocampal formation (Breder et al., 1995). COX-2 can be induced by a variety of insults, including proinflammatory stimulus-driven phosphorylation of nuclear factor kappa B p65 (pNFκB p65) and in response to NMDA receptor-mediated synaptic plasticity (as reviewed by Chen et al., 2002; Yang & Chen, 2008). Results from the current study indicate that AIE and indomethacin treatment interact to influence COX-2 + IR in both males and females, F(1, 85) = 8.6, p = 0.004. There were no sex differences on COX-2 + IR, F(1, 85) = 1.6, p = 0.21. Follow-up analyses indicate that a history of AIE exposure increased COX-2 + IR by 68% in males and by 73% in females, p < 0.001. Indomethacin treatment rescued this neuroinflammatory effect, decreasing COX-2 + IR by 22% in males and by 36% in females relative to AIE-Vehicle conditions, p = 0.04. Indomethacin exhibited some adverse effects in control conditions, increasing COX-2 + IR across both sexes, p = 0.048, suggesting that the beneficial effects of indomethacin on COX-2 are unique to conditions of chronic neuroinflammation. These data indicate that indomethacin treatment mitigates adult hippocampal COX-2 induction after AIE in both sexes (Figure 3).
FIGURE 3.
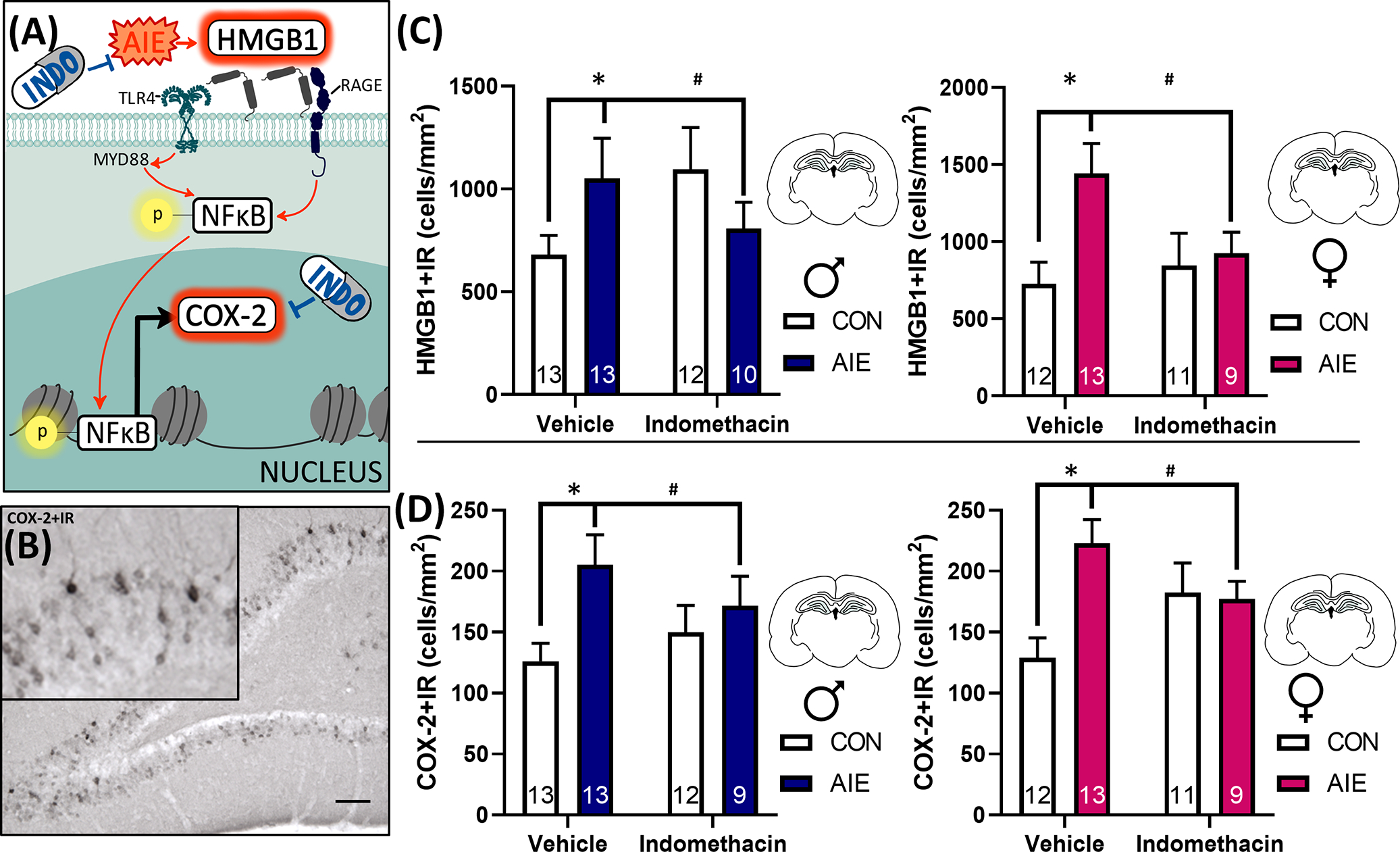
Indomethacin rescues adolescent intermittent EtOH (AIE)-induced HMGB1 and COX-2 proinflammatory signaling in the dorsal dentate gyrus in male and female rats. (A) Extracellular HMGB1 binds to TLR4 and RAGE receptors, which phosphorylate NFκB p65, translocating it to the nucleus where it drives proinflammatory gene transcription, including COX-2. In the current study, we assessed two points on this molecular pathway, HMGB1 and COX-2. AIE increased both HMGB1 and COX-2 immunoreactivity in the hippocampus, and this induction was attenuated by 14 days of indomethacin treatment. (B) Example photomicrograph of COX-2 immunoreactivity (IR). Scale bar indicates 100 μm. (C) Graphs indicate male (blue) and female (pink) changes in HMGB1 + IR in the dorsal dentate gyrus. HMGB1 is persistently increased by AIE in both sexes in adulthood (54% increase in AIE males, 99% increase in AIE females) relative to respective control-vehicle values, and indomethacin rescues this effect, attenuating HMGB1 + IR in both sexes. (D) COX-2 + IR is persistently increased by AIE by 68% in adult males (blue) and by 73% in adult females (pink), and indomethacin attenuates this increase in both sexes. Of note, indomethacin exhibited some adverse effects in control conditions, increasing COX-2 + IR generally, p = 0.048. Collectively, these results suggest that 2 weeks of indomethacin treatment after AIE can rescue neuroimmune induction in both sexes in the dorsal dentate gyrus granular cell layer. Sections were quantified as immunoreactive cells per mm2. Data are expressed as mean ± SEM. *AIE-Vehicle significantly different from CON-Vehicle, p < 0.05; #AIE-Vehicle significantly different from AIE-Indomethacin.
HMGB1 is an endogenously expressed innate immune signaling molecule secreted extracellularly in response to EtOH, cell damage, and other innate immune signals including necrotic cell death. In the current study, AIE interacts with indomethacin treatment to influence HMGB1 + IR in adult male and female rats, F(1, 85) = 7.1, p = 0.009. AIE increased HMGB1 + IR by 54% in males and 99% in females, p = 0.001; this effect was significantly mitigated by indomethacin treatment, reducing HMGB1 + IR by 23% in males and 35% in females relative to AIE-Vehicle conditions, p = 0.03. There was no impact of indomethacin on HMGB1 + IR in control conditions, p = 0.12. Collectively, these results indicate that indomethacin treatment rescues AIE induction of hippocampal HMGB1 and COX2 proinflammatory signaling pathways in both sexes (Figure 3).
Neither AIE nor indomethacin treatment impacted the number of somatostatin neurons in the hilus
Inhibitory control is a critical mechanism underlying the proper maturation and integration of newborn neurons into existing hippocampal circuitry, and loss of inhibitory input can also contribute to neuroinflammatory induction. Somatostatin inhibitory interneurons are highly expressed in the hippocampal hilus where they regulate neuronal activity in the dorsal dentate granule cell layer. However, the impact of AIE on somatostatin+ neurons is unknown. In order to test whether the loss of hilar inhibitory interneurons may contribute to neuropathology after AIE, we examined the impact of AIE and indomethacin treatment on the number of hilar somatostatin interneurons. Although females did exhibit more somatostatin+IR neurons than males, F(1, 84) = 36.8, p < 0.001, there was no effect of AIE or indomethacin on this endpoint, F(1, 84) = 0.46, p = 0.50. These results suggest that AIE and/or indomethacin do not impact somatostatin+ neurons in the hilus and do not contribute to the AIE-induced loss of neurogenesis (Figure 4). Not only does this provide novel information on a never-before examined population of interneurons in the dorsal hippocampus, but it further highlights that AIE does not impact cellular expression ubiquitously, but rather that only specific subpopulations of neurons are sensitive to long-term disruption in cell number by AIE.
FIGURE 4.
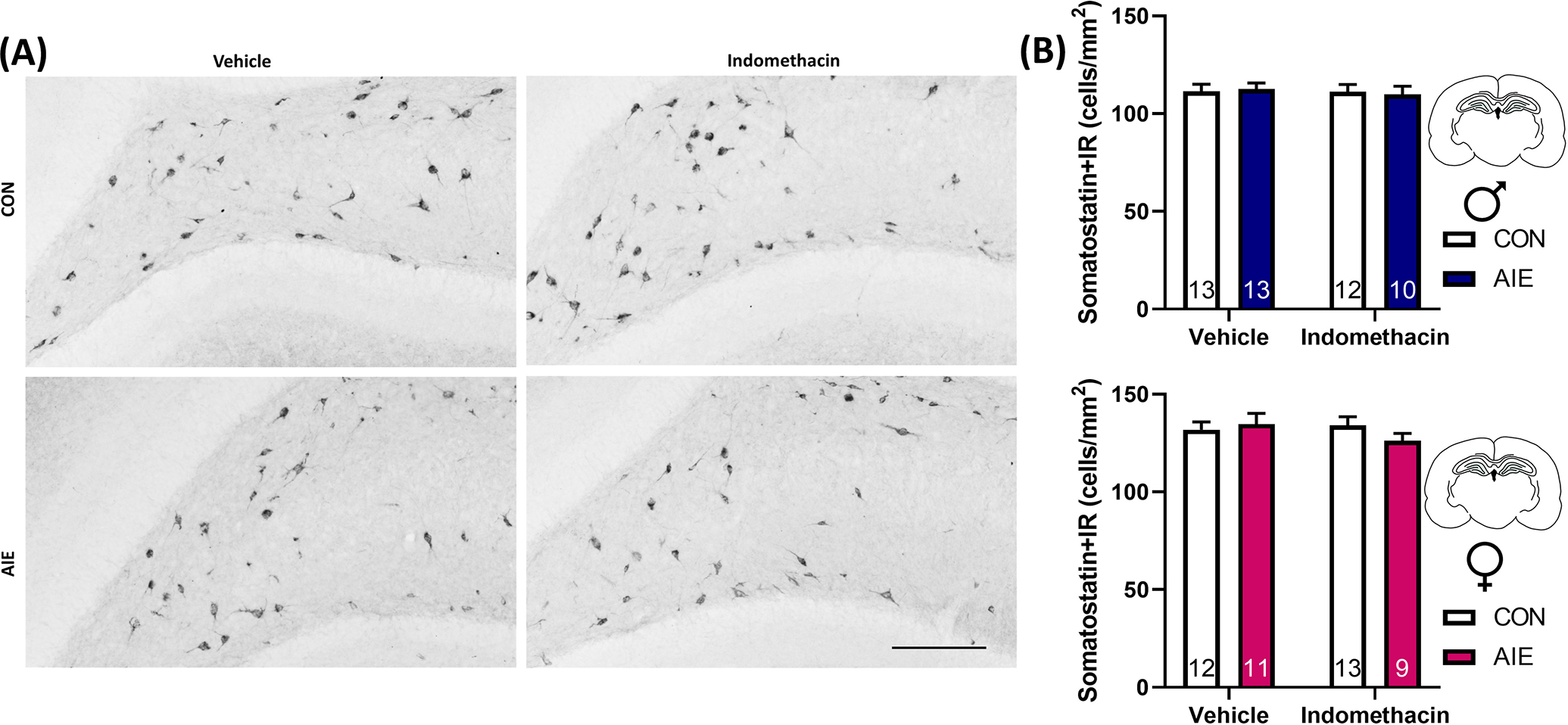
Adolescent intermittent EtOH (AIE) did not impact somatostatin cell number in the hilus. (A) Photomicrographs of somatostatin + immunoreactivity (IR) in the dorsal hippocampus. Scale bar indicates 100 μm. (B) Females (pink) exhibit significantly more hilar somatostatin interneuron expression than males (blue), p < 0.05, but neither AIE nor indomethacin impacted this endpoint. As loss of inhibitory regulation in the hippocampus could lead to excitation-induced neuroinflammation, and somatostatin interneurons provide critical input to maturing newborn neurons, these data suggest somatostatin cell number does not underlie differences in either proinflammatory gene induction or loss of neurogenesis after AIE. Data are expressed as mean ± SEM.
Indomethacin rescued AIE-induced loss of basal forebrain cholinergic neurons in both males and females
Basal forebrain cholinergic neurons project to the hippocampus where they directly modulate hippocampal neurogenesis through muscarinic receptors and indirectly modulate neurogenesis by providing anti-inflammatory feedback regulation via nicotinic α7 receptors. There were no overarching sex differences in the number of ChAT+IR basal forebrain neurons, F(1, 82) = 0.04, p = 0.84, although there was an interaction between sex and AIE treatment, F(1, 82) = 4.41, p = 0.04, and AIE treatment and indomethacin exposure, F(1, 82) = 10.1, p = 0.002. More specifically, AIE persistently reduced expression of ChAT+ basal forebrain neurons, p = 0.002, and indomethacin rescued this effect, p = 0.005 (Figure 5). However, further follow-up analyses suggest that this effect is largely driven by the significant reduction of ChAT+IR neurons after AIE in vehicle-treated males, p = 0.007, with a trending reduction in vehicle-treated females, p = 0.07. Indomethacin also significantly decreased ChAT+IR neurons in the basal forebrain of control-treated females, p = 0.02, further suggesting that in the absence of prior neuroinflammatory conditions, indomethacin can exhibit adverse effects. Moreover, the loss and restoration of basal forebrain cholinergic neurons after AIE is significantly correlated with the loss and restoration of hippocampal neurogenesis, R = 0.43, p < 0.001. This restoration of the expression of ChAT+ neurons supports a growing body of evidence that AIE causes an epigenetic suppression of the cholinergic neuronal phenotype that can be reversed with anti-inflammatory interventions (Vetreno et al., 2020).
FIGURE 5.
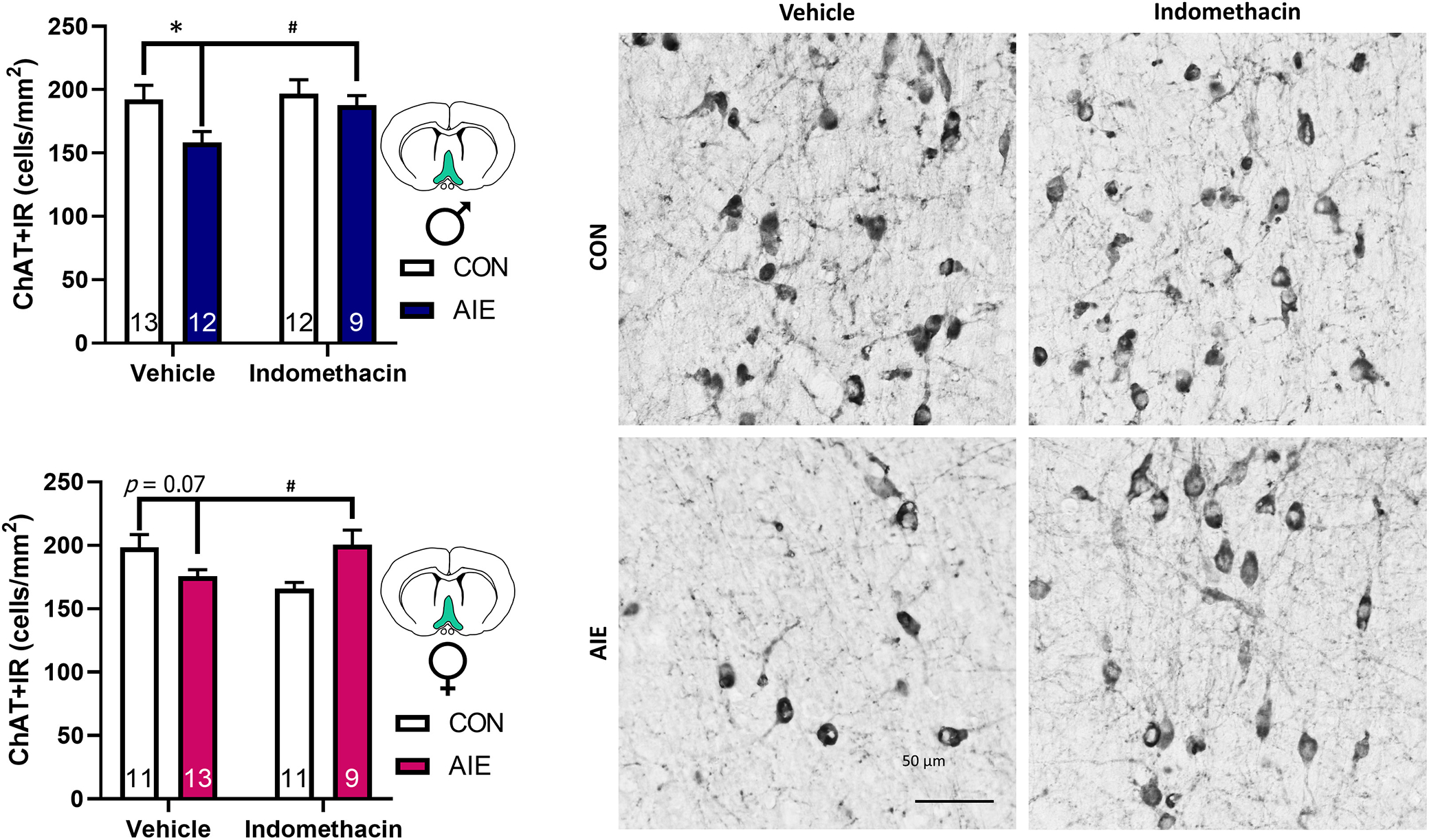
Indomethacin rescues basal forebrain cholinergic neuron number after Adolescent intermittent EtOH (AIE) in both sexes. (A) AIE-reduced ChAT + IR basal forebrain cholinergic neurons by 33% in males (blue) and 11% in females (pink). Indomethacin rescued this effect in both sexes, restoring ChAT + IR cell number to control values in both sexes. Data are expressed as mean ± SEM. (B) Photomicrographs of ChAT labeling of cholinergic neurons in the medial septum of the basal forebrain. For example, photomicrographs were taken at 40× magnification. Fewer ChAT + IR neurons are evidenced in AIE–vehicle-treated rats, but indomethacin restored ChAT + IR cell number after AIE, suggesting that AIE-induced apparent cholinergic cell loss is reversible through anti-inflammatory interventions in adulthood. Scale bar is 50 μm.
Indomethacin rescues AIE-induced loss of dorsal dentate gyrus hippocampal cholinergic innervation in both sexes
Acetylcholine is a well-known anti-inflammatory signal (Tracey, 2002), and our previous studies find cholinesterase inhibition prevents and reverses AIE increases in HMGB1 and other neuroimmune genes (Macht et al., 2022). Here, we used an antibody against the vesicular acetylcholine transporter (VAChT), which identifies cholinergic projections in hippocampal sections, in conjunction with fluorescent labeling of DCX+ cells to determine how the loss of forebrain ChAT+ neurons relate to the expression of cholinergic terminals nearby hippocampal neuroprogenitors. Results indicate that there was a significant interaction between AIE exposure and indomethacin treatment on VAChT co-labeling with DCX + cells, F(1, 85) = 13.57, p < 0.001, as well as an interaction between sex and AIE exposure on VAChT co-labeling with DCX + cells, F(1, 85) = 5.35, p = 0.02. There was also a trend for AIE exposure and indomethacin treatment to interact to impact dorsal hippocampal dentate VAChT levels overall, F(1, 85) = 3.45, p = 0.07. Follow-up analyses indicate that AIE significantly reduced overall VAChT intensity in males, p = 0.03, with a trend toward reduced VAChT levels in females, p = 0.08. Similarly, AIE significantly reduced VAChT levels, which colocalized with DCX-labeled cells in males, p < 0.001, and females, p = 0.04. Indomethacin increased VAChT in control males, p = 0.007, and within AIE-treated males, p < 0.001. While there was no effect of indomethacin in control females, indomethacin increased VAChT labeling in AIE-treated females, p = 0.02. Similarly, indomethacin treatment significantly increased VAChT/DCX co-labeled cells in males, p = 0.03, and females, p = 0.002. Collectively, these results suggest that AIE impairs cholinergic innervation to the dorsal granule cell layer of the hippocampus, with the most robust effects evident on DCX-labeled neurons (Figure 6). For a summary of raw intensity values, see Table 4.
FIGURE 6.
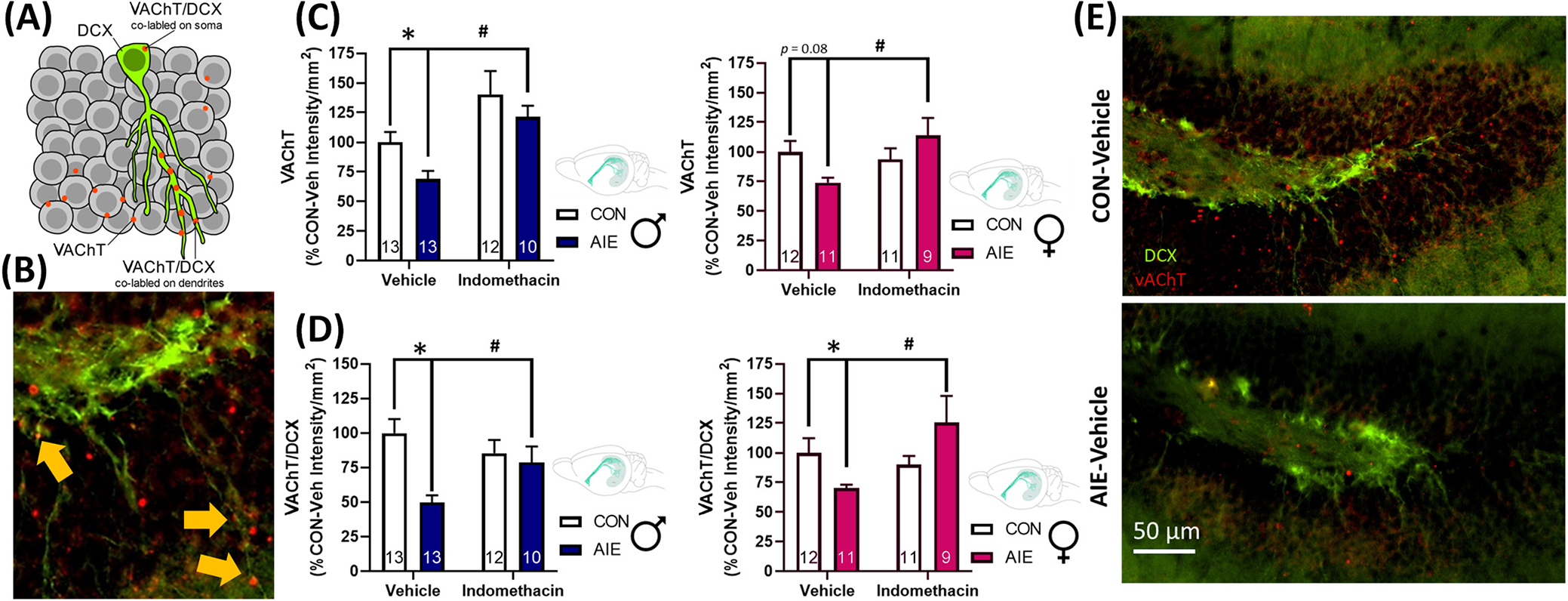
Indomethacin restores the loss of VAChT expression on DCX-labeled neurons in the dorsal hippocampus. (A) Diagram of co-labeling of VAChT on DCX neurons. VAChT labeling appears as puncta (red) on DCX (green) dendrites and soma. (B) Example of co-labeling of VAChT on DCX + cells, identified by yellow arrows. (C) AIE significantly reduced expression of the cholinergic terminal marker VAChT in males (blue) with a trend to reduce overall VAChT expression in females (pink), p = 0.08. Within AIE-treated rats, indomethacin significantly increased VAChT expression in both sexes. All data are expressed as a percentage of respective CON-Vehicle cohorts. This indicates that indomethacin increases cholinergic innervation of the dorsal dentate gyrus. (D) While VAChT is robustly expressed throughout the granule cell layer, it is particularly robust at the connection between the granule cell layer and the molecular layer, where the dendritic arbors of newborn granular neurons reach. When we assessed the colocalization of VAChT on DCX cell bodies and their respective dendritic arbors, we found that AIE produced a marked reduction of VAChT/DCX colocalization relative to controls in both sexes. Excitingly, indomethacin rescued this deficit, significantly increasing VAChT expression in males and females within AIE-treated rats. (E) Photomicrographs of DCX and VAChT labeling of AIE-Vehicle and CON-Vehicle rats. Scale bar is 50 μm. VAChT is expressed in red, and DCX is expressed in green. High levels of VAChT puncta are evidenced throughout the granule cell layer of CON-Vehicle rat with a substantial reduction evidenced in the AIE-Vehicle rat. Notably, the remaining VAChT puncta in the AIE-treated rat are not expressed near DCX-labeled cells. Data are expressed as mean ± SEM. *AIE-Vehicle significantly different from CON-Vehicle; #AIE-Indomethacin significantly different from AIE-Vehicle.
TABLE 4.
Raw intensity values by the group of VAChT and VAChT/DCX co-expression.
| Sex | Exposure | Treatment | VAChT/DCX | VAChT |
|---|---|---|---|---|
|
| ||||
| Male | CON | Vehicle | 13,398,199 ±1,373,461 | 88,496,891 ±7,577,810 |
| Indomethacin | 11,449,966 ±1,215,481 | 124,341,639 ±16,743,818 | ||
| AIE | Vehicle | 6,656,608 ±692,326 | 61,038,105 ±6,068,309 | |
| Indomethacin | 10,541,181 ±1,334,030 | 107,221,265 ±7,365,337 | ||
| Female | CON | Vehicle | 9,644,772 ±1,138,240 | 83,948,418 ±7,385,835 |
| Indomethacin | 8,673,847 ±661,810 | 78,622,995 ±7,165,448 | ||
| AIE | Vehicle | 6,758,999 ±275,748 | 62,142,386 ±3,239,119 | |
| Indomethacin | 12,087,339 ±1,815,482 | 95,612,863 ±10,199,976 | ||
Note: Data expressed as Mean ± SEM, bolded values indicate significant differences from CON-Veh cohorts.
Hippocampal nAChR α7 mRNA expression is reduced after AIE
The nAChR α7 is a mediator through which the cholinergic system provides regulatory anti-inflammatory feedback (Conejero-Goldberg et al., 2008, p. 7). Here, we show that AIE significantly reduced adult hippocampal mRNA expression of nAChR α7 by 21% in males and by 38% in females, F(1, 26) = 7.8, p = 0.01 and F(1, 35) = 14.6, p < 0.001, respectively. Indomethacin increased hippocampal nAChR α7 mRNA expression by 10% in AIE-treated male rats, resulting in nAChR α7 mRNA expression, which was insignificantly different from control values (p = 0.34). These results suggest that reductions in nAChR α7 expression accompany retraction of cholinergic innervation of the hippocampus, such that loss of cholinergic nAChR α7 could contribute to the persistent neuroimmune induction and neurogenic reduction evidenced in the adult hippocampus after AIE in both sexes (see Table 5).
TABLE 5.
Adolescent intermittent EtOH (AIE) reduces hippocampal nAChRa7 mRNA in both sexes.
| Sex | Exposure | Treatment | nAChRa7 mRNA |
|---|---|---|---|
|
| |||
| Male | CON | Vehicle | 100 ±6 |
| Indomethacin | 96 ±7 | ||
| AIE | Vehicle | 79 ±3 | |
| Indomethacin | 89 ±4 | ||
| Female | CON | Vehicle | 100 ±11 |
| Indomethacin | 91 ±8 | ||
| AIE | Vehicle | 62 ±7 | |
| Indomethacin | 61 ±7 | ||
Note: mRNA data expressed as a percent of CON-Vehicle for males and females, respectively; Mean ± SEM, bolded values indicate significant differences from CON-Veh cohorts.
Locomotion is not impacted by AIE or indomethacin
Locomotor behavior was assessed in an open field test. There was no difference between any groups on total distance traveled or latency to enter the center zone in males, F(1, 36) = 0.930 and F(1, 36) = 1.12, p > 0.05. Similarly, there was no difference between any groups on total distance traveled or latency to enter the inner zone in females, F(1, 35) = 1.14, p > 0.05 and F(1, 35) = 0.34, p > 0.05. These results suggest that neither AIE nor indomethacin impacted locomotor ability or thigmotaxis in either sex (Figure 7).
FIGURE 7.
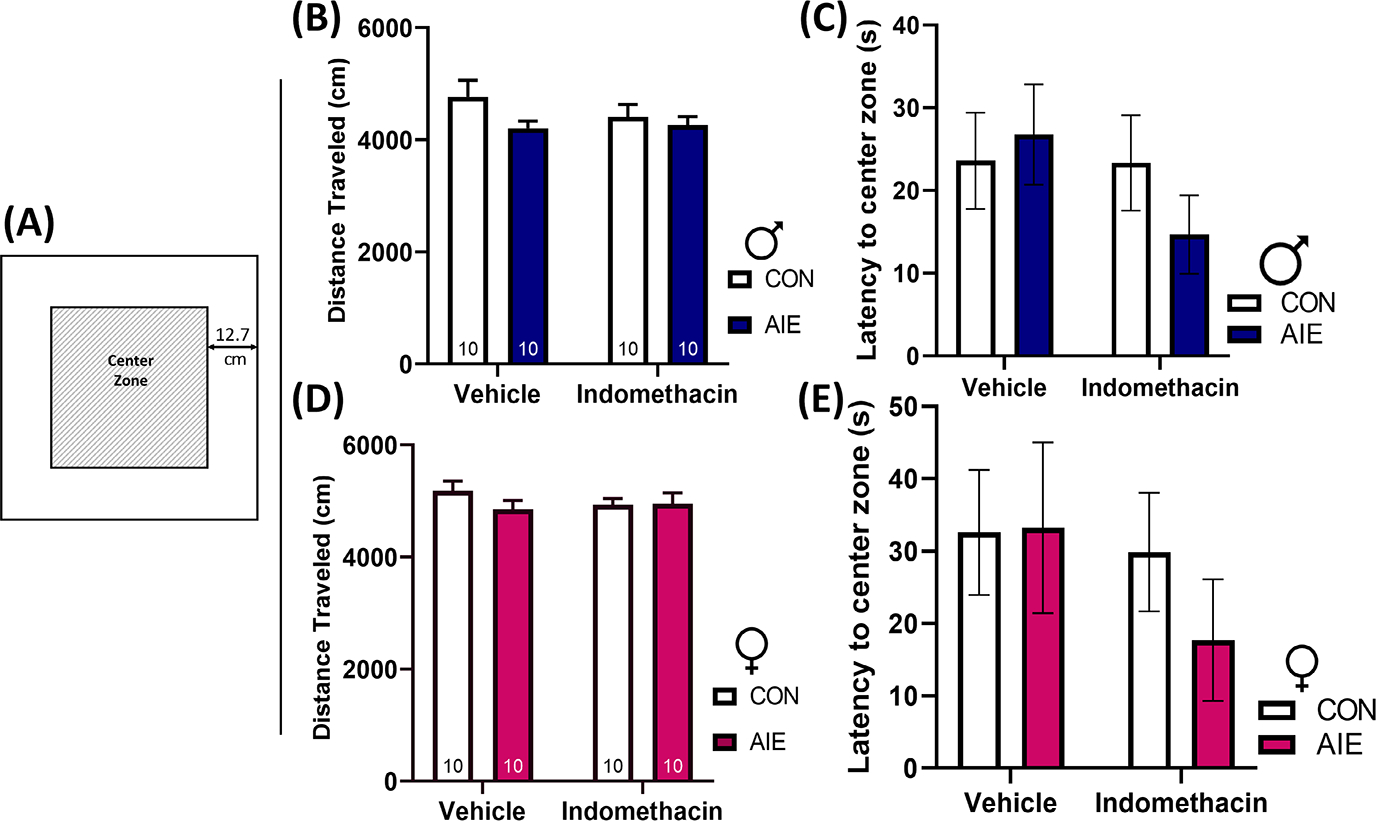
Adolescent intermittent EtOH (AIE) did not impact distance traveled or thigmotaxis behaviors in male or female rats. (A) An open field test was performed as a locomotor assessment prior to the Morris water maze. Example of the open field apparatus. The center zone was identified as 12.7 cm from the perimeter wall. Increased latency to enter the center zone can be indicative of anxiety-type behavior. All testing was performed under low light (<40 lux). The test consisted of a total of 10 min of free exploratory time. Distance traveled was calculated as centimeters (cm). (B) Neither AIE nor indomethacin treatment impacted distance traveled in males. (C) Neither AIE nor indomethacin treatment impacted latency to enter the center zone in males. (D) Neither AIE nor indomethacin treatment impacted distance traveled in females. (E) Neither AIE nor indomethacin treatment impacted latency to enter the center zone in females. Collectively, these results suggest that neither AIE nor indomethacin treatment impacted locomotor behavior, thus eliminating a potential confounder of test results from the Morris water maze. Data are expressed as mean ± SEM.
Morris water maze: AIE reversal learning impairments in males are reversed by indomethacin
The Morris water maze was used to assess both spatial learning and reversal learning, a component of cognitive flexibility that has previously been reported to be reduced in males following AIE exposure (Coleman et al., 2011; Macht, Elchert, & Crews, 2020; Sircar & Sircar, 2005; Vetreno & Crews, 2018). Results from the current study indicate that the time (latency) that male rats from every group took to find the platform decreased significantly across trial and day, indicating that all rats demonstrated learning and improved within and across testing days, F(2, 70) = 7.2, p = 0.001, and F(5, 175) = 35.3, p < 0.001, respectively. There was no effect of AIE exposure or indomethacin treatment on the acquisition of the initial target platform location in males, F(10, 350) = 1.0, p = 0.44. When the target platform location was reversed, there was a significant interaction between AIE exposure and indomethacin treatment on latency to find the platform, F(1, 36) = 5.1, p = 0.03. In addition, there was a significant interaction between test day and trial number, indicating that animals significantly improved across trials over the two reversal days, F(2, 72) = 9.5, p < 0.001. Follow-up analyses indicate that AIE-exposed rats took significantly longer to find the new target platform on the first trial of the second day of reversal learning, p = 0.004. Indomethacin reversed this effect, significantly improving performance in AIE–indomethacin-treated rats relative to AIE-Vehicle counterparts, p = 0.02. These data suggest that indomethacin rescues reversal learning deficits in an allocentric spatial learning task in AIE males.
Morris water maze: AIE impairments in spatial navigation in females are reversed by indomethacin
The time (latency) that female rats from every group took to find the platform decreased significantly across trial and day, indicating that all rats demonstrated learning and improved within and across testing days, F(2, 70) = 18.3, p < 0.001, and F(5, 175) = 71.0, p < 0.001, respectively. However, unlike in males, AIE exposure and indomethacin treatment did interact across time in female rats to affect spatial acquisition learning, F(5, 175) = 3.0, p = 0.01. More specifically, female rats exposed to EtOH took significantly longer to find the target platform on days 3 and 5, p’s = 0.01. Indomethacin rescued this effect such that AIE-exposed females treated with indomethacin performed no differently than control counterparts, p’s > 0.05. This effect was most robust on the third test day, where indomethacin treatment significantly improved the acquisition of the target platform location on day 3 in AIE-exposed females, p = 0.01. Interestingly, indomethacin slightly impaired spatial learning acquisition in controls, prolonging swim times on days 2 and 6, p’s = 0.01 and 0.03. These data indicate that AIE selectively impairs spatial learning in females and that this deficit can be rescued by indomethacin (Figure 8).
FIGURE 8.
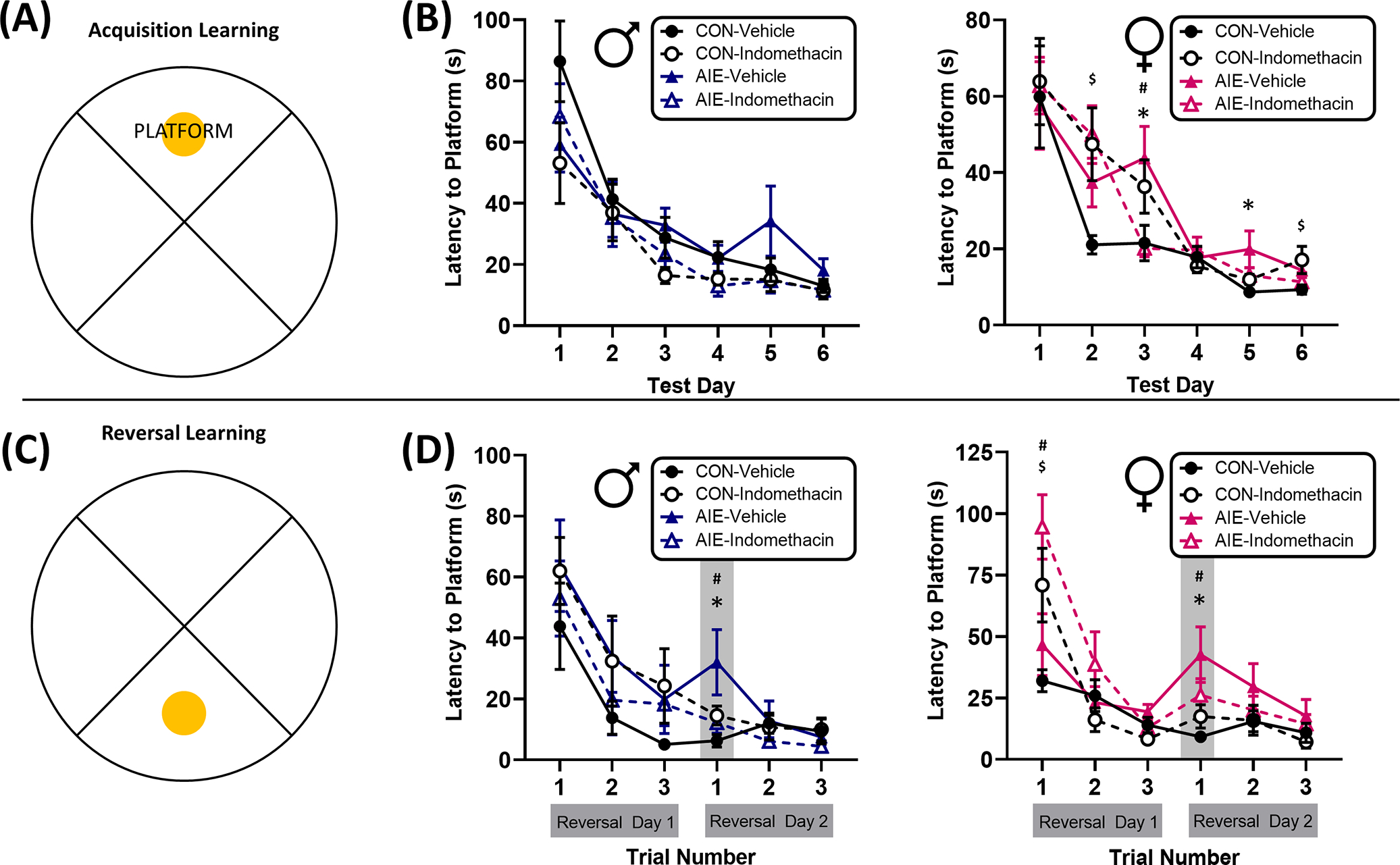
Indomethacin rescued reversal learning deficits in both sexes during a Morris water maze task. (A) Diagram of quadrants and platform location during acquisition training sessions. Rats were given three training trials per day with start locations assigned randomly and in a counterbalanced fashion to one of the opposing three quadrants. (B) Acquisition training for males (blue) and females (pink) consisted of three training sessions per day over a course of six consecutive days. Latency to find the platform was recording, with increased latency indicative of reduced spatial memory of the platform location. Consistent with previous studies, adolescent intermittent EtOH (AIE)-exposed male rats did not exhibit any deficits in learning during the spatial task. All groups significantly improved latency to find the platform over the course of the six test days. By contrast, AIE-exposed females did exhibit transient learning deficits on days 3 and 5. Indomethacin rescued this spatial learning deficit most robustly on day 3 in AIE-treated rats. (C) Diagram of the reversed platform location during the 2 days of reversal training. Rats were given three training trials per day, consistent with acquisition training, and with start locations assigned randomly and in a counterbalanced fashion to one of the opposing three quadrants. (D) AIE-exposed male and female rats took significantly longer to find the new platform location, particularly on the first trial of the second day of training. Indomethacin rescued this reversal learning deficit in both sexes. Surprisingly, indomethacin selectively impaired reversal learning in both control and AIE-treated females on the first trial of the first day of reversal learning, but this effect was transient and all indomethacin-treated rats did equally well to control counterparts by the second trial of the first day. These data suggest that indomethacin restores reversal learning capacity in both males and females after AIE. *AIE-Vehicle significantly different from CON-Vehicle; #AIE-Indomethacin significantly different from AIE-Vehicle; $CON-Indomethacin significantly different from CON-Vehicle.
When the target platform location was reversed, there were significant interactions across days and trials by indomethacin exposure, F(2, 70) = 6.6, p = 0.002, and a trend for interaction between indomethacin treatment and AIE exposure across reversal test days, F(1, 35) = 3.3, p = 0.08. Follow-up analyses indicate that as with males, AIE exposure selectively impaired reversal learning on the first trial of the second day of reversal learning, p = 0.002. Excitingly, indomethacin rescued this deficit, improving retention of the reversed platform location in AIE-treated rats on the first trial of reversal day 2, p = 0.008. There was a surprising drug effect of indomethacin in control and AIE-exposed females to increase latency to locate the new target platform location, p’s = 0.03 and 0.008, respectively. However, this effect was transient and there was no effect of indomethacin on subsequent trials, p’s > 0.05. Excitingly, these data replicate AIE male reversal deficits and now extend to females where AIE impairs both spatial learning and reversal learning in females. In both sexes, indomethacin treatment rescues these deficits (Figure 8).
DISCUSSION
We report here for the first time in male and female rats that indomethacin treatment reverses AIE-induced loss of ChAT+ basal forebrain cholinergic projection neurons, hippocampal VChAT+IR, and loss of hippocampal DCX+ progenitors and inhibits increases in neuroimmune signals (HMGB1, COX-2). These findings are consistent with AIE-induced atrophy of the septohippocampal cholinergic circuit and reduced hippocampal mRNA expression of the anti-inflammatory cholinergic receptor nAChRα7. Newborn hippocampal neurons and basal forebrain cholinergic neurons appear to be uniquely susceptible to long-term deficits after AIE, as hippocampal somatostatin interneuron numbers were unchanged. This further suggests that AIE selectively disrupts subpopulations of neurons even within a brain region. Surprisingly, there was a general lack of sex differences in the persistent neurogenic, cholinergic, and neuroimmune consequences of AIE, suggesting that the effects of AIE on these molecular markers are not dependent on sex hormones. AIE also impaired reversal learning in both sexes, and while AIE females exhibited a modest spatial learning deficit not evidenced in males, indomethacin treatment was able to restore cognitive-behavioral deficits across both sexes. Indomethacin also restored the hippocampal inflammatory environmental milieu after AIE, indicating that increases in brain innate immune signals are likely mechanistic mediators of adult reversal learning and neurogenic deficits in both sexes after AIE, driving adult brain and behavioral pathology. However, indomethacin treatment also caused some adverse consequences in control-treated rats, emphasizing that this anti-inflammatory intervention is uniquely beneficial in conditions of chronic underlying neuroinflammation.
Previous studies indicate that AIE decreases adult neurogenesis, evidenced by the loss of DCX immunoreactivity in male rats through PND 220 (Vetreno & Crews, 2015). These EtOH-mediated effects are unique to adolescence as adult intermittent EtOH exposure does not result in long-term reductions in adult DCX immunoreactivity (Broadwater et al., 2014). The current study expands upon these findings, indicating that AIE persistently reduces DCX immunoreactivity in both adult males and females to a similar extent. Interestingly, females exhibit slightly higher raw counts of DCX+ immunostaining in adulthood in both control and AIE groups, which is consistent with prior work that females do not exhibit as significant of an age-related decline in pre-to postpubertal DCX labeling as is evidenced in males (Siddiqui & Romeo, 2019).
More excitingly, the current study establishes that indomethacin direct anti-inflammatory intervention can reverse AIE induction of hippocampal HMGB1 and COX-2 and restore the loss of DCX immunoreactivity in both sexes. This finding expands upon previous studies in males indicating that indirect anti-inflammatory interventions via either voluntary exercise (Vetreno et al., 2018) or increased cholinergic anti-inflammatory feedback effects by the cholinesterase inhibitors galantamine and donepezil (Pavlov et al., 2003; Reardon, 2016; Tracey, 2002) restore hippocampal and basal forebrain pathology after AIE (Macht et al., 2021; Swartzwelder et al., 2019). Thus, while exercise is a far more viable clinical intervention, indomethacin’s efficacy suggests persistent AIE-induced neuroinflammation is a critical mechanism underlying the loss of hippocampal neurogenesis.
Previous studies report indomethacin treatment during AIE blocks the development of hippocampal pathology in males (Vetreno et al., 2018), now extended in the current study to indomethacin reversal of pathology in male and female rats. Given that some studies find that adolescent females exhibit greater sickness behaviors than their male counterparts following an acute lipopolysaccharide innate immune challenge (Sharma et al., 2018), one might expect greater neuroimmune susceptibility to AIE in adolescent females. However, the current study finds that males and females exhibit similar molecular induction of proinflammatory indicators HMGB1 and COX-2 in the dentate granule cell layer, suggesting that the long-term effects of adolescent intermittent binge EtOH exposure on hippocampal HMGB1 and COX-2 are not sexually divergent. This finding is in line with other studies, which suggest that brain COX-2 expression exhibits estrogen-mediated sexual dimorphism in preoptic hypothalamic areas but not the hippocampus (Amateau & McCarthy, 2004). Of note, while indomethacin is known to have inhibitory effects on COX-1 and COX-2, the current study did not examine the impact of AIE and indomethacin on COX-1 expression in the hippocampus. This remains an interesting future area of investigation as some more recent review articles have encouraged a reassessment of the historical roles of COX-1 as housekeeping and COX-2 as inflammatory (López & Ballaz, 2020).
In parallel to our findings following AIE, hippocampal HMGB1 also does not follow sexually divergent expression following adolescent stress: male and female adolescent mice exposed to a cold physiological stressor exhibited equivalent induction of poststress hippocampal HMGB1 (Xu et al., 2019). Therefore, while future studies should examine whether other brain regions, such as the preoptic area, exhibit sex-mediated neuroimmune responses to AIE, the current study indicates that sex does not mediate these AIE neuroimmune signaling effects in the hippocampus.
Adolescent EtOH exposure also produces persistent decreases in basal forebrain cholinergic neuron markers, such as ChAT (Coleman et al., 2011; Vetreno & Crews, 2018). The current study expands these observations, indicating that both male and female rats exhibit reductions in cholinergic cell bodies in the basal forebrain in adulthood after AIE, although AIE males exhibited a reduction slightly greater in magnitude than AIE females relative to controls (33% reduction in AIE males and 11% in females). Basal forebrain cholinergic neurons project to the cortex and the hippocampus along the septohippocampal pathway. Here, we report for the first time that AIE also reduces adult hippocampal expression of VAChT in both sexes, particularly when colocalized with the terminals of immature neurons, labeled by DCX. VAChT is responsible for the vesicular packing of acetylcholine for release, and therefore it effectively outlines cholinergic terminals. Interestingly, although VAChT labeling is expressed throughout the hippocampus, the punctate marker is densest at distal edges of the granular cell layer, where the dendritic arbors of newborn neurons grow toward and extend into the molecular layer. This could suggest that loss of VAChT terminals on DCX neurons after AIE may impair neurite outgrowth and synapse development of a role, which has been supported by in vitro studies, contributing to the loss of adult newborn neuron survivability (Sternfeld et al., 1998). In support of this, several studies have also demonstrated loss of the basal forebrain cholinergic system directly reduces newborn neuron proliferation, differentiation, and survival (Cooper-Kuhn et al., 2004; Mohapel et al., 2005), consistent with a loss of trophic factor drive. In fact, other studies have found AIE reduces hippocampal BDNF in parallel with loss of neurogenesis (Sakharkar et al., 2016). Taken together with the current findings, these data suggest that AIE-induced loss of hippocampal cholinergic synapses may be linked to loss of cholinergic-mediated trophic support and loss of DCX+ progenitors in both sexes.
In addition to resulting in a loss of trophic support, loss of cholinergic innervation to the hippocampus may contribute to the increases in the proinflammatory drive after AIE through failed cholinergic inhibitory feedback mechanisms of the neuroinflammatory response (Pavlov et al., 2003, 2009). Acetylcholine provides anti-inflammatory regulatory control by activating nAChR α7 to reduce microglial proinflammatory cytokine production and release (for review see Mizrachi et al., 2021), and we report here a decrease in hippocampal nAChR α7 mRNA in males and females after AIE. Interestingly, AIE-induced loss and anti-inflammatory treatment-induced restoration of basal forebrain cholinergic neuron phenotypic expression and hippocampal neurogenesis often occur in tandem, suggesting these molecular changes are strongly linked. For example, the cholinesterase inhibitors galantamine and donepezil restore both basal forebrain cholinergic neuron expression and hippocampal neurogenesis after AIE (Crews et al., 2021; Macht et al., 2021; Swartzwelder et al., 2019). The current study expands upon these previous findings to show that anti-inflammatory treatment with indomethacin restores not only hippocampal DCX expression after AIE but also basal forebrain cholinergic neuron expression and cholinergic innervation in the dorsal granule cell layer of the hippocampus in both sexes, suggesting that restoring the proinflammatory balance within the hippocampal environmental milieu includes restoration of this septohippocampal circuit.
Disruptions in neuroimmune signaling (Vetreno et al., 2018; Vetreno & Crews, 2018), hippocampal neurogenesis (Anacker & Hen, 2017; Clelland et al., 2009; Kempermann, 2002), and acetylcholine (for review see Prado et al., 2017) have all been functionally linked to impaired reversal learning. These observations suggest that AIE impairs converging circuits that collectively contribute to reversal learning deficits. Here, we report that loss of DCX and decreased hippocampal cholinergic tone after AIE are associated with reduced flexibility during a reversal learning task in the Morris water maze in both sexes. Interestingly, AIE-induced impairments in reversal learning are most notable on the second day of reversal training, perhaps indicating conflicts in consolidation and retrieval of the novel platform location. The deficit in AIE-exposed rodents is most evident across a 24-h intertrial interval window as opposed to across trials within a test day, suggesting that spatial reversal learning deficits after AIE are linked to long-term memory, further implicating the hippocampus as a mediator of these deficits. Unlike males, females did exhibit a modest deficit in spatial learning. This sex-dependent effect on spatial learning is also evident in adult rats after a 4-day binge EtOH cycle (Maynard et al., 2018), suggesting this deficit in spatial acquisition in females after EtOH exposure spans developmental periods. Excitingly, not only did indomethacin restore cholinergic projections to the dorsal hippocampus, adult neurogenesis, and reduce neuroimmune gene expression, but indomethacin also restored reversal learning behavioral performance in both males and females, suggesting that these molecular circuits are mechanistic mediators of cognitive flexibility in reversal learning tasks after AIE.
In sum, the findings from the current study provide novel insights into the mechanistic role of neuroinflammation in cholinergic-hippocampal circuitry deficits after AIE and its impact on reversal learning in both male and female rats. Interestingly, molecular and behavioral deficits after AIE and reversal by indomethacin were consistent across both sexes. Excitingly, these findings further emphasize that the restoration of these molecular cascades after AIE could be responsible for the restoration of reversal learning deficits in both sexes.
Supplementary Material
ACKNOWLEDGMENTS
These studies were supported by the Neurobiology of Adolescent Drinking in Adulthood (NADIA) consortium of the NIAAA (U24 AA020024, U01 AA020023) and the Bowles Center for Alcohol Studies (P60 AA011605). Additional funding was provided by NIH training grants T32 AA007573, K99 AA030089 (VAM), and K01 AA025713 (RPV). We would like to thank Jennie Vaughn for her assistance in editing this manuscript and Jay Campbell for his assistance with animal experiments.
Funding information
National Institute on Alcohol Abuse and Alcoholism, Grant/Award Number: AA007573, AA011605, AA020023, AA020024, AA025713 and AA030089
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no competing financial interests.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
REFERENCES
- Amateau SK & McCarthy MM (2004) Induction of PGE2 by estradiol mediates developmental masculinization of sex behavior. Nature Neuroscience, 7, 643–650. Available from: 10.1038/nn1254 [DOI] [PubMed] [Google Scholar]
- Anacker C & Hen R (2017) Adult hippocampal neurogenesis and cognitive flexibility — linking memory and mood. Nature Reviews. Neuroscience, 18, 335–346. Available from: 10.1038/nrn.2017.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breder CD, Dewitt D & Kraig RP (1995) Characterization of inducible cyclooxygenase in rat brain. The Journal of Comparative Neurology, 355, 296–315. Available from: 10.1002/cne.903550208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadwater MA, Liu W, Crews FT & Spear LP (2014) Persistent loss of hippocampal neurogenesis and increased cell death following adolescent, but not adult, chronic ethanol exposure. Developmental Neuroscience, 36, 297–305. Available from: 10.1159/000362874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Magee JC & Bazan NG (2002) Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. Journal of Neurophysiology, 87, 2851–2857. Available from: 10.1152/jn.2002.87.6.2851 [DOI] [PubMed] [Google Scholar]
- Clelland CD, Choi M, Romberg C, Clemenson GD, Fragniere A, Tyers P et al. (2009) A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science, 325, 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman L, He J, Lee J, Styner M & Crews FT (2011) Adolescent binge drinking alters adult brain neurotransmitter gene expression, behavior, brain regional volumes, and neurochemistry in mice. Alcoholism, Clinical and Experimental Research, 35, 671–688. Available from: 10.1111/j.1530-0277.2010.01385.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman LG, Liu W, Oguz I, Styner M & Crews FT (2014) Adolescent binge ethanol treatment alters adult brain regional volumes, cortical extracellular matrix protein and behavioral flexibility. Pharmacology, Biochemistry, and Behavior, 116, 142–151. Available from: 10.1016/j.pbb.2013.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conejero-Goldberg C, Davies P & Ulloa L (2008) Alpha7 nicotinic acetylcholine receptor: a link between inflammation and neurodegeneration. Neuroscience and Biobehavioral Reviews, 32, 693–706. Available from: 10.1016/j.neubiorev.2007.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Kuhn CM, Winkler J & Kuhn HG (2004) Decreased neurogenesis after cholinergic forebrain lesion in the adult rat. Journal of Neuroscience Research, 77, 155–165. Available from: 10.1002/jnr.20116 [DOI] [PubMed] [Google Scholar]
- Crews FT, Fisher R, Deason C & Vetreno RP (2021) Loss of basal forebrain cholinergic neurons following adolescent binge ethanol exposure: recovery with the cholinesterase inhibitor Galantamine. Frontiers in Behavioral Neuroscience, 15, 32. Available from: 10.3389/fnbeh.2021.652494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Mdzinarishvili A, Kim D, He J & Nixon K (2006) Neurogenesis in adolescent brain is potently inhibited by ethanol. Neuroscience, 137, 437–445. Available from: 10.1016/j.neuroscience.2005.08.090 [DOI] [PubMed] [Google Scholar]
- Crews FT, Robinson DL, Chandler LJ, Ehlers CL, Mulholland PJ, Pandey SC et al. (2019) Mechanisms of persistent neurobiological changes following adolescent alcohol exposure: NADIA consortium findings. Alcoholism, Clinical and Experimental Research, 43, 1806–1822. Available from: 10.1111/acer.14154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-A A, Dannenhoffer CA, Elton A, Lee S-H, Ban W, Shih Y-YI et al. (2021) Altered Cortico-subcortical network after adolescent alcohol exposure mediates behavioral deficits in flexible decision-making. Frontiers in Pharmacology, 12, 778884. Available from: 10.3389/fphar.2021.778884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo A, Brigman JL, Radke AK, Rudebeck PH & Holmes A (2017) The neural basis of reversal learning: an updated perspective. Neuroscience, 345, 12–26. Available from: 10.1016/j.neuroscience.2016.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G (2002) Why new neurons? Possible functions for adult hippocampal neurogenesis. The Journal of Neuroscience, 22, 635–638. Available from: 10.1523/JNEUROSCI.22-03-00635.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp DJ & Crews FT (1999) Induction of Cyclooxygenase-2 in brain during acute and chronic ethanol treatment and ethanol withdrawal. Alcoholism, Clinical and Experimental Research, 23, 633–643. Available from: 10.1111/j.1530-0277.1999.tb04165.x [DOI] [PubMed] [Google Scholar]
- López DE & Ballaz SJ (2020) The role of brain Cyclooxygenase-2 (cox-2) beyond neuroinflammation: neuronal homeostasis in memory and anxiety. Molecular Neurobiology, 57, 5167–5176. Available from: 10.1007/s12035-020-02087-x [DOI] [PubMed] [Google Scholar]
- Macht V, Crews FT & Vetreno RP (2020) Neuroimmune and epigenetic mechanisms underlying persistent loss of hippocampal neurogenesis following adolescent intermittent ethanol exposure. Current Opinion in Pharmacology, 50, 9–16. Available from: 10.1016/j.coph.2019.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macht V, Elchert N & Crews F (2020) Adolescent alcohol exposure produces protracted cognitive-behavioral impairments in adult male and female rats. Brain Sciences, 10, 785. Available from: 10.3390/brainsci10110785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macht V, Vetreno R, Elchert N & Crews F (2021) Galantamine prevents and reverses neuroimmune induction and loss of adult hippocampal neurogenesis following adolescent alcohol exposure. Journal of Neuroinflammation, 18, 212. Available from: 10.1186/s12974-021-02243-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macht V, Vetreno RP & Crews FT (2022) Cholinergic and neuroimmune signaling interact to impact adult hippocampal neurogenesis and alcohol pathology across development. Frontiers in Pharmacology, 13, 849997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard ME, Barton EA, Robinson CR, Wooden JI & Leasure JL (2018) Sex differences in hippocampal damage, cognitive impairment, and trophic factor expression in an animal model of an alcohol use disorder. Brain Structure & Function, 223, 195–210. Available from: 10.1007/s00429-017-1482-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain JA, Hayes DM, Morris SA & Nixon K (2011) Adolescent binge alcohol exposure alters hippocampal progenitor cell proliferation in rats: effects on cell cycle kinetics. The Journal of Comparative Neurology, 519, 2697–2710. Available from: 10.1002/cne.22647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrachi T, Vaknin-Dembinsky A, Brenner T & Treinin M (2021) Neuroinflammation modulation via α7 nicotinic acetylcholine receptor and its chaperone, RIC-3. Molecules, 26, 6139. Available from: 10.3390/molecules26206139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapel P, Leanza G, Kokaia M & Lindvall O (2005) Forebrain acetylcholine regulates adult hippocampal neurogenesis and learning. Neurobiology of Aging, 26, 939–946. Available from: 10.1016/j.neurobiolaging.2004.07.015 [DOI] [PubMed] [Google Scholar]
- Nixon K & Crews FT (2002) Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. Journal of Neurochemistry, 83, 1087–1093. Available from: 10.1046/j.1471-4159.2002.01214.x [DOI] [PubMed] [Google Scholar]
- Pascual M, Blanco AM, Cauli O, Miñarro J & Guerri C (2007) Intermittent ethanol exposure induces inflammatory brain damage and causes long-term behavioural alterations in adolescent rats. The European Journal of Neuroscience, 25, 541–550. Available from: 10.1111/j.1460-9568.2006.05298.x [DOI] [PubMed] [Google Scholar]
- Patrick ME & Terry-McElrath YM (2019) Prevalence of high-intensity drinking from adolescence through young adulthood: National Data from 2016–2017. Substance Abuse: Research and Treatment, 13, 117822181882297. Available from: 10.1177/1178221818822976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov VA, Parrish WR, Rosas-Ballina M, Ochani M, Puerta M, Ochani K et al. (2009) Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain, Behavior, and Immunity, 23, 41–45. Available from: 10.1016/j.bbi.2008.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov VA, Wang H, Czura CJ, Friedman SG & Tracey KJ (2003) The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Molecular Medicine, 9, 125–134. Available from: 10.1007/BF03402177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G & Watson C (1986) The rat brain in stereotaxic coordinates, 2nd edition. San Diego, CA: Academic Press Inc. [Google Scholar]
- Prado VF, Janickova H, Al-Onaizi MA & Prado MAM (2017) Cholinergic circuits in cognitive flexibility. Neuroscience, Cognitive Flexibility: Development, Disease, and Treatment, 345, 130–141. Available from: 10.1016/j.neuroscience.2016.09.013 [DOI] [PubMed] [Google Scholar]
- Reardon C (2016) Neuro-immune interactions in the cholinergic anti-inflammatory reflex. Immunology Letters, 178, 92–96. Available from: 10.1016/j.imlet.2016.08.006 [DOI] [PubMed] [Google Scholar]
- Reitz NL, Nunes PT & Savage LM (2021) Adolescent binge-type ethanol exposure in rats mirrors age-related cognitive decline by suppressing cholinergic tone and hippocampal neurogenesis. Frontiers in Behavioral Neuroscience, 15, 1–13. Available from: 10.3389/fnbeh.2021.772857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar AJ, Vetreno RP, Zhang H, Kokare DM, Crews FT & Pandey SC (2016) A role for histone acetylation mechanisms in adolescent alcohol exposure-induced deficits in hippocampal brain-derived neurotrophic factor expression and neurogenesis markers in adulthood. Brain Structure & Function, 221, 4691–4703. Available from: 10.1007/s00429-016-1196-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Rooke J, Kolmogorova D, Melanson B, Mallet J-F, Matar C et al. (2018) Sex differences in the peripheral and central immune responses following lipopolysaccharide treatment in pubertal and adult CD-1 mice. International Journal of Developmental Neuroscience, 71, 94–104. Available from: 10.1016/j.ijdevneu.2018.07.012 [DOI] [PubMed] [Google Scholar]
- Siddiqui A & Romeo RD (2019) Sex differences and similarities in hippocampal cellular proliferation and the number of immature neurons during adolescence in rats. Developmental Neuroscience, 41, 132–138. Available from: 10.1159/000502056 [DOI] [PubMed] [Google Scholar]
- Sircar R & Sircar D (2005) Adolescent rats exposed to repeated ethanol treatment show lingering behavioral impairments. Alcoholism, Clinical and Experimental Research, 29, 1402–1410. Available from: 10.1097/01.alc.0000175012.77756.d9 [DOI] [PubMed] [Google Scholar]
- Sternfeld M, Ming G, Song H, Sela K, Timberg R, Poo M et al. (1998) Acetylcholinesterase enhances neurite growth and synapse development through alternative contributions of its hydrolytic capacity, core protein, and variable C termini. The Journal of Neuroscience, 18, 1240–1249. Available from: 10.1523/JNEUROSCI.18-04-01240.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartzwelder HS, Healey KL, Liu W, Dubester K, Miller KM & Crews FT (2019) Changes in neuroimmune and neuronal death markers after adolescent alcohol exposure in rats are reversed by donepezil. Scientific Reports, 9, 12110. Available from: 10.1038/s41598-019-47039-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey KJ (2002) The inflammatory reflex. Nature, 420, 853–8 59. Available from: 10.1038/nature01321 [DOI] [PubMed] [Google Scholar]
- Vetreno RP, Bohnsack JP, Kusumo H, Liu W, Pandey SC & Crews FT (2020) Neuroimmune and epigenetic involvement in adolescent binge ethanol-induced loss of basal forebrain cholinergic neurons: restoration with voluntary exercise. Addiction Biology, 25, e12731. Available from: 10.1111/adb.12731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP & Crews FT (2012) Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and toll-like receptors in the adult prefrontal cortex. Neuroscience, 226, 475–488. Available from: 10.1016/j.neuroscience.2012.08.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP & Crews FT (2015) Binge ethanol exposure during adolescence leads to a persistent loss of neurogenesis in the dorsal and ventral hippocampus that is associated with impaired adult cognitive functioning. Frontiers in Neuroscience, 9, 35. Available from: 10.3389/fnins.2015.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP & Crews FT (2018) Adolescent binge ethanol-induced loss of basal forebrain cholinergic neurons and neuroimmune activation are prevented by exercise and indomethacin. PLoS One, 13, e0204500. Available from: 10.1371/journal.pone.0204500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Lawrimore CJ, Rowsey PJ & Crews FT (2018) Persistent adult neuroimmune activation and loss of hippocampal neurogenesis following adolescent ethanol exposure: blockade by exercise and the anti-inflammatory drug indomethacin. Frontiers in Neuroscience, 12, 200. Available from: 10.3389/fnins.2018.00200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vore AS, Barney TM, Gano A, Varlinskaya EI & Deak T (2021) Adolescent intermittent ethanol (AIE) produces sex specific alterations in adult neuroimmune gene expression and ethanol sensitivity that are independent of ethanol metabolism. Neuropharmacology, 195, 108635. Available from: 10.1016/j.neuropharm.2021.108635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vore AS, Doremus-Fitzwater T, Gano A & Deak T (2017) Adolescent ethanol exposure leads to stimulus-specific changes in cytokine reactivity and hypothalamic-pituitary-adrenal axis sensitivity in adulthood. Frontiers in Behavioral Neuroscience, 11, 78. Available from: 10.3389/fnbeh.2017.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Zang S, Li S-Z, Guo J-R, Wang J-F, Wang D et al. (2019) HMGB1-mediated differential response on hippocampal neurotransmitter disorder and neuroinflammation in adolescent male and female mice following cold exposure. Brain, Behavior, and Immunity, 76, 223–235. Available from: 10.1016/j.bbi.2018.11.313 [DOI] [PubMed] [Google Scholar]
- Yang H & Chen C (2008) Cyclooxygenase-2 in synaptic signaling. Current Pharmaceutical Design, 14, 1443–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J-Y & Kim M-S (2016) Deficits in decision-making and reversal learning in college students who participate in binge drinking. Neuropsychiatry, 6, 321–330. Available from: 10.4172/Neuropsychiatry.1000156 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.