

Abstract
Background
Dendrobium officinale Kimura et Migo (D. officinale) is a well-known traditional Chinese medicine with high content polysaccharides in stems. The SWEET (Sugars Will Eventually be Exported Transporters) family is a novel class of sugar transporters mediating sugar translocation among adjacent cells of plants. The expression patterns of SWEETs and whether they are associated with stress response in D. officinale remains uncovered.
Results
Here, 25 SWEET genes were screened out from D. officinale genome, most of which typically contained seven transmembrane domains (TMs) and harbored two conserved MtN3/saliva domains. Using multi-omics data and bioinformatic approaches, the evolutionary relationship, conserved motifs, chromosomal location, expression patterns, correlationship and interaction network were further analyzed. DoSWEETs were intensively located in nine chromosomes. Phylogenetic analysis revealed that DoSWEETs were divided into four clades, and conserved motif 3 specifically existed in DoSWEETs from clade II. Different tissue-specific expression patterns of DoSWEETs suggested the division of their roles in sugar transport. In particular, DoSWEET5b, 5c, and 7d displayed relatively high expression levels in stems. DoSWEET2b and 16 were significantly regulated under cold, drought, and MeJA treatment, which were further verified using RT-qPCR. Correlation analysis and interaction network prediction discovered the internal relationship of DoSWEET family.
Conclusions
Taken together, the identification and analysis of the 25 DoSWEETs in this study provide basic information for further functional verification in D. officinale.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-023-09419-w.
Keywords: Sugar transporter, Polysaccharides, Stem, Expression pattern, Biotic and abiotic stress
Introduction
Dendrobium officinale Kimura et Migo is a widely known traditional Chinese medicine in Orchidaceae [1], whose stems harbor great medicinal and economic value due to its high content of active polysaccharides [2, 3]. The 2-O-acetylglucomannan is the main form of polysaccharides in D. officinale stems [4]. Polysaccharides content was reported to gradually accumulate from vegetative growth to mature stage in stems, peaking in the 12th month after sprouting [5, 6]. In higher plants, polysaccharides are synthesized from carbohydrates produced in leaves [7]. Firstly, photosynthetic sucrose is loaded to the phloem by sugar transporters. Then, with the help of hydrolases and synthetases, sucrose is unloaded from the phloem into sink tissues for storage as different types of sugars [8]. Profiting from the development of multi-omics analysis, high quality genome of D. officinale have become available [9] and vital insights into the biosynthesis pathway and regulation of bioactive polysaccharides have been achieved [5, 6, 10–14]. However, the molecular mechanisms of how sugar is transported from leaves to stems for polysaccharides accumulation in D. officinale remain largely unknown.
Monosaccharide transporters (MSTs), sucrose transporters/sucrose carriers (SUCs/SUTs) [8, 15], and sugars will eventually be exported transporters (SWEETs) are three major types of sugar transporters. DoHT1, encoding a MST in D. officinale, exhibited the most dominant expression level in leaves [16]. Wang et al. [13] found eight SUT genes in D. officinale and most of them were expressed in flowers, indicating that DoSUTs might mainly function in the development of floral organs. The SWEET family is a new group of sugar transporters facilitating the translocation of sugars across the intracellular or plasma membranes in plants and animals [17, 18]. Different from SUTs and MSTs, SWEETs can transport not only sucrose but also monosaccharides in bi-direction [8]. The plant SWEET proteins typically contain seven TMs and harbor two conserved MtN3/saliva domain (PF03083), which are linked by the 4th TM [19]. SWEET proteins are phylogenetically divided into four clades (Clade I-IV) [17]. A number of studies have shown that SWEETs participate in various physiological activities through mediating long-distance transport of sugar [20], such as seed filling [21], fruit development [22], nectar secretion [23], pollen nutrition [24], response to abiotic and biotic stresses [25–28]. In Arabidopsis thaliana (L.) Heynh., SWEETs belonging to different clades appear selective preferences for sugar types. SWEET proteins from Clade I and II are involved in hexose translocation, Clade III SWEETs specifically transport sucrose, and Clade IV SWEETs tend to transport fructose [8]. Wang et al. [29] identified 22 SWEET family members in D. officinale genome, however, no further researches have been done to uncover more details about their structural characteristics, evolutionary relationship, expression profiles, and biological functions.
Although we have had the basic understanding of polysaccharide synthesis, little is known about the mechanisms underlying sugar transport and polysaccharide accumulation in D. officinale. In this work, based on the chromosome-scale genome of D. officinale [9], the SWEET gene family was screened and analyzed using multiple bioinformatic tools and online websites. Our findings do favor to further functional analysis and application of D. officinale SWEET family in molecular marker-assisted selection for breeding new varieties with high polysaccharides content and stress resistance.
Results
Identification and phylogenetic analysis of SWEET genes in D. officinale
To identify the SWEET gene family members in D. officinale, the conserved MtN3/saliva domain (PF03083.hmm) was used to align with the public genome database (accession number GCA_019514585.1) [9] downloaded from NCBI. In total, 25 SWEET family members were screened from the chromosome-level genome of D. officinale by bioinformatic analysis and were renamed based on the homology with AtSWEETs. The detailed basic physicochemical properties information of DoSWEETs were discovered and listed in Additional file 1, including gene name, chromosomal location, coding sequence length, and protein characteristics (size, molecular weight, isoelectric point, sub-cellular localization, number of transmembrane domains and MtN3/saliva domain). The DoSWEETs were distributed on nine chromosomes, and chromosome 11 contained the largest number of members (six DoSWEETs). The protein lengths of DoSWEETs ranged from 139 to 297 amino acids and the predicated molecular weights (Mw) were 15.83–33.11 KDa. The isoelectric points (pI) of DoSWEETs were all larger than 8.00, indicating that they were neutral or basic proteins (Additional file 1). Multiple online websites were used to predict the sub-cellular localization of DoSWEETs. Most DoSWEETs were located in cell membrane and contained seven transmembrane domains and two MtN3/saliva domains.
To investigate the evolutionary relationship of DoSWEETs, an un-rooted maximum likehood tree was constructed using the full length of 84 SWEET proteins from A. thaliana (dicot), Oryza sativa L. (monocot), Phalaenopsis equestris (Schauer) Rchb.f. (the model plant of orchidaceae species), and D. officinale. All SWEETs were classified into four clades. In all clades, DoSWEETs formed subclade with SWEETs from P. equestris, indicating the closer relationship (Fig. 1). Clade II was the the largest clade and contained 14 DoSWEETs (DoSWEET4-7) with four pairs of paralogs (DoSWEET4a-DoSWEET4b, DoSWEET5a-DoSWEET5b, DoSWEET5c-DoSWEET5d, and DoSWEET6b-DoSWEET6c), followed by clade III with eight members (Fig. 2a). The DoSWEETs (DoSWEET9-15) in clade III comprised two pairs of paralogs (DoSWEET10-DoSWEET15, and DoSWEET13-DoSWEET14). Clade I had 2 DoSWEETs (DoSWEET2a and 2b), which was also identified as a pair of paralogs (Fig. 2a). Clade IV was the smallest one, containing one DoSWEET (DoSWEET16) (Figs. 1 and 2a). The phylogenetic tree with actual branch lengths and scale was shown in Additional file 2 (Fig. S1). Except for DoSWEET2a-DoSWEET2b, the members of other paralog pairs were located on the same chromosomes, respectively (Additional file 1 and Fig. 3). To better study the evolution of DoSWEET gene family, gene duplication events were further explored (Fig. 3 and Additional file 3). Based on the close distance and high similarity, ten gene pairs (DoSWEET5a-DoSWEET5b, DoSWEET5c-DoSWEET5d, DoSWEET6b-DoSWEET6c, DoSWEET6c-DoSWEET6d, DoSWEET7a-DoSWEET7b, DoSWEET7a-DoSWEET7c, DoSWEET7b-DoSWEET7c, DoSWEET7b-DoSWEET7d, DoSWEET7c-DoSWEET7d, and DoSWEET13-DoSWEET14) were considered to be evolved from tandem duplication events (Fig. 3 and Additional file 1, 3). And the other seven duplicated gene pairs were more likely to expand through segmental duplications due to their longer physical distance (larger than 100 kb) with each other (Fig. 3 and Additional file 1, 3). To investigate the selective evolutionary pressure on DoSWEET gene divergence after duplication, the non-synonymous and synonymous substitutions per site (Ka and Ks values), and Ka/Ks ratio were calculated (Additional file 3). We found that Ka/Ks ratios of all those gene pairs were less than 1, indicating that the occurrence rate of synonymous substitutions was higher than that of non-synonymous substitutions. This results revealed that the purifying selection had the primary influence on DoSWEET gene family in the evolution.
Fig. 1.

Phylogenetic analysis of SWEETs proteins from D. officinale, A. thaliana, O. sativa and P. equestris. The un-rooted phylogenetic tree was constructed using Maximum Likelihood Estimate method in MEGA 11. The bootstrap value was set as 1000 replicates. Percentages of replicate trees in which the associated sequences clustered together in the bootstrap test are shown next to the branches. Clade I, II, III, and IV are marked by green, gray, blue, and purple, respectively. DoSWEETs are marked in red stars. The abbreviations of species names are as follows: Do, Dendrobium officinale; At, Arabidopsis thaliana; Os, Oryza sativa; the proteins started with PA are PeSWEETs from Phalaenopsis equestris
Fig. 2.

Phylogenetic relationship and conserved domain analysis of DoSWEETs. a Phylogenetic relationship and conserved domain distribution of DoSWEETs. An un-rooted phylogenetic tree was constructed using Maximum Likelihood Estimate method with a bootstrap analysis of 1000 replicates in MEGA 11, based on the full-length amino acid sequences of DoSWEETs. Percentages of replicate trees in which the associated sequences clustered together in the bootstrap test are shown next to the branches. Clade I, II, III, and IV are marked by green, gray, blue, and purple, respectively. TM domains are highlighted in orange boxes and MtN3/saliva domains are marked with gray frames. TM, transmembrane domain. b The conserved motifs identified by MEME tools in DoSWEETs. Motif 1, 2, 4, 5, 6, 7 were commonly presented in DoSWEETs marked with red frame ① and motif 1, 2, 3, 4, 5, 6 were commonly presented in DoSWEETs marked with red frame ②. The length of each box is proportional to the size of the motif. The sequence logos of motif 1–7 are shown
Fig. 3.
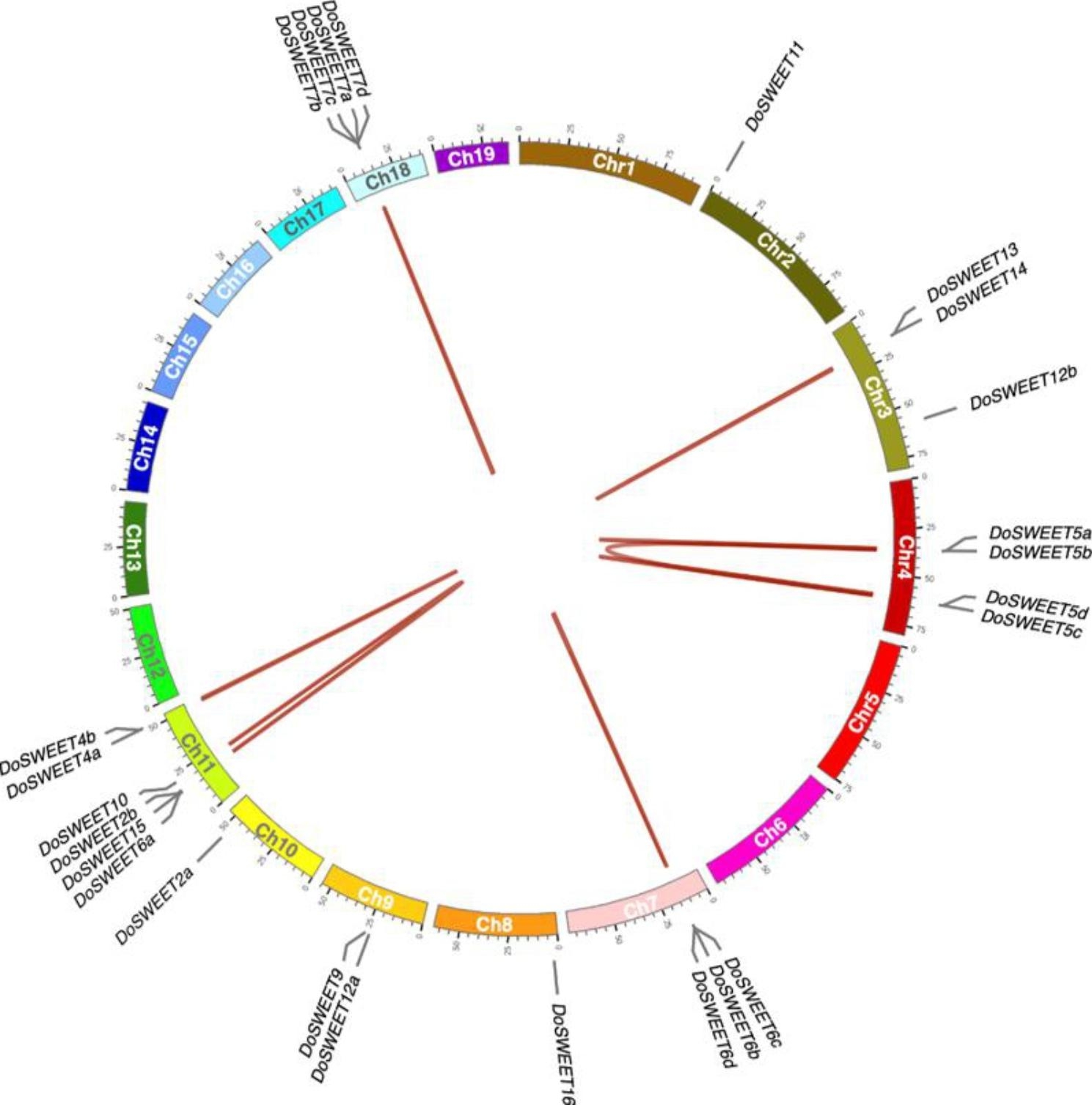
Localization and synteny of DoSWEETs in D. officinale genome. Different chromosomes are represented by boxes with different colors. The gray solid lines at the edge of the boxes indicate the location of the genes. The duplicated gene pairs, whose alignable sequence covered over 75% of the longer gene, and at the same time, the similarity of aligned regions was more than 75%, are linked with the red lines inside the circle
Conserved domain and motif analysis of DoSWEETs
All of the DoSWEETs harbor seven TMs and contain two MtN3/saliva domains, except for DoSWEET4b (Additional file 1 and Fig. 2a). Multiple alignment analysis by DNAMAN software showed that the sequences of seven TMs were conserved among DoSWEETs (Additional file 2: Fig. S2). To further analyze the conserved TMs, hydrophobicity and hydrophilicity values were estimated and the results indicated that the TM regions were more hydrophobic (Additional file 2: Fig. S3).
Ten conserved motifs were predicted by MEME (Additional file 2: Fig. S4). Motif 1, 2, 4, 5, 6 were shared in all DoSWEETs. Motif 7 existed in DoSWEETs from clade I, III and IV, while motif 3 was unique in clade II (Fig. 2), which might be related to functional divergence between clade II and the other clades. The distribution and sequence logo of seven conserved motifs are shown in Fig. 2b.
Tissue-specific expression pattern of DoSWEETs
To investigate the tissue-specific expression patterns of DoSWEETs, the FPKM values were downloaded from OrchidBase 4.0 [30] and were visualized in the heatmap (Fig. 4a and Additional file 4). Eighteen DoSWEETs were detected in at least one of the ten tissues. All of the detected DoSWEETs displayed tissue-specific expression patterns (Fig. 4a). DoSWEET5c was the most abundant one in all of the ten tissues (Fig. 4a and Additional file 4). The most of DoSWEETs (72.2%) were expressed in roots, stems, leaves and flower buds from D. officinale (Fig. 4b). DoSWEET4b and 11 were highly expressed in the pollinia, whereas DoSWEET6a, 7d, 9, and 14 showed the opposite expression patterns, with the lowest level in the pollinia (Fig. 4a). Stem, rich in polysaccharides, is the medicinal part of D. officinale. DoSWEET5b, DoSWEET5c, and DoSWEET7d displayed relatively high expression levels in the stems (FPKM > 100) (Fig. 4c), which were determined using RT-qPCR (Fig. 4d), indicating their potential roles in polysaccharide accumulation in this tissue. In addition, DoSWEET2b, DoSWEET5b, and DoSWEET5c were highly expressed in the roots and green root tips (FPKM > 100). Especially DoSWEET2b, the expression level was dominant in the roots compared to other tissues (Fig. 4c and Additional file 4).
Fig. 4.

Expression patterns of DoSWEETs in different tissues. a A heatmap displaying the FPKM values of DoSWEETs in different tissues of D. officinale. One biological replicate per sample. b A venn diagram showing the expression of DoSWEETs in the roots, stems, leaves, and flower buds. The majority of DoSWEETs (72.2%) were expressed in the four tissues analyzed. FPKM values greater than 1 were counted. c The highly expressed DoSWEETs (FPKM > 100) in the roots, stems, leaves, and flower buds. d Expression detection of DoSWEET5b, DoSWEET5c, and DoSWEET7d in the roots, stems, and leaves by RT-qPCR. Bars represent means ± SD (n = 3). Different letters above each column indicate the significant differences based on Duncan’s test (P < 0.05)
DoSWEET2b and DoSWEET16 were regulated by low temperature, drought stress and MeJA treatment
SWEETs have been reported to participate in stress responses [31]. In order to identify the cis-acting elements on the promoters of DoSWEETs, the 2000 bp upstream sequences from the translational initial codon were extracted and analyzed using the online website PlantCARE, (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/). Eighteen types of cis-acting elements were identified (Additional file 5) and were further classified into three categories, including stress response, tissue-specificity, and progress-specificity. Each DoSWEET harbored at least five types of cis-acting elements on its promoter (Fig. 5a).
Fig. 5.

Stress-responsive analysis of DoSWEETs. a Cis-acting elements analysis of DoSWEETs promoters using PlantCARE. The 2000 bp upstream sequences from the translational initiation codon of DoSWEETs were analyzed. The identified elements were divided into three classes, including stress-response, tissue-specificity, and progress-specificity. Dot color and size indicate the type and number of the elements, respectively. b DoSWEET2b and DoSWEET16 were regulated by low temperature, drought stress, and MeJA treatment. Gray and red represent down- and up-regulation, respectively. The downloaded RNA-seq datasets of low temperature and MeJA treatment contained three replicates, while the datasets of drought stress had only one biological replicate. Log2(mean FPKM of experimental group/control group) values were used to draw the heatmap. c The log2 fold change of DoSWEETs expression under different stresses detected by RT-qPCR. PEG treatment was used to simulate the drought stress conditions. Bars represent means ± SD (n = 3). The symbols above each column indicate the significant differences compared with control based on Student’s t-test (**P < 0.01; *P < 0.05; ns. represented no significance)
To further explore the responses of DoSWEETs under different stresses, the transcriptome sequencing data under low temperature [32], drought stress [33], cadmium stress [34] and MeJA treatment [35] were obtained from NCBI and the FPKM values were calculated to assess DoSWEETs expression levels. Only DoSWEET2b and DoSWEET16 were found to respond to stresses and the change trends were similar with each other (Fig. 5b and Additional file 6). After treatment with 0℃ for 20 h, DoSWEET2b and DoSWEET16 were down-regulated by 0.84 and 1.10 fold, respectively (Fig. 5b and Additional file 6). Drought stress also caused the down-regulation of the two genes, and their expression increased after re-watering (Fig. 5b). When treated with exogenous MeJA, DoSWEET2b was induced by 0.90 fold and DoSWEET16 expression was 1.80-fold higher than the control (Fig. 5b and Additional file 6). The result is consistent with the MeJA-responsive elements predicted on their promoters (Fig. 5a). Furthermore, the responsive intensity was positively related to the number of MeJA-responsive elements (Fig. 5a and b). No DoSWEETs were found to respond to cadmium stress. To verify the responses of DoSWEET2b and DoSWEET16 under the above stresses, RT-qPCR analysis was conducted. The results showed that the expression changes of DoSWEET2b and DoSWEET16 under MeJA treatments were consistent with that in transcriptome data, while the expression under cold stress exhibited the opposite trend (Fig. 5b and c). PEG treatment was used to simulate the drought stress conditions. RT-qPCR confirmed the decrease of DoSWEET16 expression after PEG treatment. However, no significant change of was detected for DoSWEET2b (Fig. 5b and c).
Potential SWEET dimers formed when functioning
Function of SWEET sugar transporter requires a pore consisting of at least two SWEETs [36]. To find out the potential SWEET dimers in D. officinale, the correlation analysis and interaction network prediction were performed. Correlation analysis indicated that DoSWEET4a-DoSWEET5c, DoSWEET4b-DoSWEET7a, DoSWEET4b-DoSWEET11, DoSWEET6b-DoSWEET14, and DoSWEET7a-DoSWEET11 were co-expressed with highly positive correlation coefficients of 0.87, 0.88, 0.98, 0.85, and 0.91, respectively. By contrast, DoSWEET4b-DoSWEET6a and DoSWEET6a-DoSWEET11 showed negative correlations with correlation coefficients of -0.83 and − 0.80, respectively (Fig. 6). To analyze the functional and physical interaction of DoSWEETs, STRING software was used to draw the interaction network map. Multiple interrelationships were found among DoSWEETs. For example, more than three types of interaction evidence were identified between DoSWEET11 and other DoSWEETs, including DoSWEET5d, DoSWEET6d, DoSWEET7d, DoSWEET9, DoSWEET12b, and DoSWEET13 (Fig. 7). All those DoSWEET pairs were potential to form dimers when functioning. In addition, DoSWEET4b, DoSWEET9, DoSWEET10, DoSWEET11, DoSWEET12b, DoSWEET13, DoSWEET14, DoSWEET15, and DoSWEET16 were related to SUC2, a sucrose transport protein in A. thaliana [15] (Fig. 7), indicating their potential function in sugar translocation.
Fig. 6.

Correlation analysis of DoSWEETs using corrplot R package. The co-expression relationship of DoSWEETs were analyzed using corrplot R package based on the FPKM values in different tissues and under different stresses. The correlation coefficients are shown in the upper right portion of the diagram and visualized in the bottom left portion. Bigger dot size represents higher correlation coefficient. Red and gray dots represent the positive and negative correlationship, respectively
Fig. 7.

Interaction network prediction of DoSWEETsusing STRING. The interactions were predicted based on the homologous genes of DoSWEETs in A. thaliana. The lines with different colors represent different types of interaction evidence
Discussion
As an important energy source for plant growth and development, sugar is synthesized in the leaves and transported into non-photosynthetic sink tissues via symplastic pathway and apoplastic pathway [37]. Over the past 30 years, the molecular mechanisms of sugar apoplastic transport have been uncovered in model plants [17, 38]. Polysaccharide is one of the most valuable medicinal ingredients in D. officinale stems, it is meaningful to screen candidate SWEET genes in D. officinale for clarification of their roles in polysaccharide accumulation.
In 2010, Chen et al. [17] first identified SWEET1 as a membrane-located glucose uniporter in A. thaliana, mediating glucose efflux across the membrane. From then on, benefiting from sequencing technologies and bioinformatic tools, the SWEET family have been widely identified in many other plants [18]. In this study, 25 SWEET members were found in D. officinale genome (Additional file 1). Similarly, other species in monocots generally have more than 20 members in the SWEET family. For example, there are 21 members in O. sativa [39], 24 in Zea mays (L.) [21], 23 in Sorghum bicolor (L.) Moench [40], 25 in Musa acuminata Colla [41], 39 in Ananas comosus (L.) Merr. [22], and 22 in Saccharum spontaneum (L.) [42]. Specially, in P. equestris, another orchid species, only 16 putative SWEET genes were discovered [29]. Compared to clade I, III and IV, DoSWEETs from clade II specifically harbored the conserved motif 3 and showed obvious gene expansion (Figs. 2 and 3), which is similar to the SWEET family in Dendrobium chrysotoxum Lindl. [43]. In A. thaliana, members of this clade (SWEET4-8) tend to transport monosaccharides [8]. These findings suggest that members of DoSWEET4-7 subfamily may be related to monosaccharides translocation and abundant polysaccharides content in D. officinale stems. In addition, the conserved motif 3 is likely to be involved in functional divergence during evolution. Duplication is one of the primary driving forces to facilitate the gene expansion and genome evolution [44, 45]. Here, more pairs of tandem duplication DoSWEETs were identified than segmental duplication ones, Therefore, tandem duplication is considered to be the main reason for the expansion of the SWEET family in D. officinale, resulting in the high content of polysaccharides in the stems.
The whole process of sugar transport in plants involves the cooperation of SUTs, MSTs, and SWEETs [31]. The functions and interactions of these sugar transporters are relatively well studied in A. thaliana. Therefore, the interaction network of DoSWEETs was predicted based on their homologous SWEETs in A. thaliana. Notably, seven of the eight DoSWEETs from clade III were found to interact with SUC2 by textmining and co-expression (Fig. 7). SUT/SUC2 has been identified as a H+-sucrose symporter mediating phloem apoplast loading in a previous study [15]. In addition, SWEETs belonging to clade III have been reported to mediate sucrose translocation in A. thaliana [8]. In that case, clade III DoSWEETs are most likely to be involved in sucrose loading in the phloem. Apart from DoSWEETs from clade III, DoSWEET4b was also predicted to interact with SUC2 (Fig. 7). Considering the mechanism of SWEETs on sugar transport and the specificity of DoSWEET4b in conserved domain (only harbored one MtN3/saliva domain), we assume that DoSWEET4b may form oligomers with other DoSWEETs when functioning. Correlation analysis showed that DoSWEET4b had extremely positive correlation (value = 0.98) with DoSWEET11 (Fig. 6) which was also predicted to be co-expressed with SUC2 (Fig. 7). In A. thaliana, sucrose is synthesized in the leaves and transported to the apoplast by AtSWEET11 and AtSWEET12, and then is loaded into the phloem for long-distance transport by SUT1/SUC2 [20]. OsSWEET11 in rice is also associated with sucrose transport [20]. Taken together, we conjecture that DoSWEET4b and DoSWEET11 may form oligomers to participate in sucrose transport in source tissues, which remains to be confirmed by experiments.
The different expression patterns of SWEET members indicate the differentiation of their roles in plant growth and development. DoSWEET5c was constitutively expressed in D. officinale, with extremely high level in the stems (Fig. 4a and Additional file 4). In model plants, function of stem-expressed SWEETs is hardly characterized due to the low sucrose content in their stems, except for S. bicolor. To know more about the potential role of DoSWEET5c, we screened homologous genes of DoSWEET5c in S. bicolor genome using the online website Phytozome ver.13 (https://phytozome-next.jgi.doe.gov/blast-search). Five orthologs (SbSWEET3-6, SbSWEET4-1, SbSWEET4-3, SbSWEET9-3, and SbSWEET4-2) in S. bicolor were found with high bitscores (Additional file 7). SbSWEET4-3 was uniquely expressed in S. bicolor stems and was potentially responsible for sucrose unloading from the phloem into the stem apoplast [40]. It has been reported that orthologs with high sequence conservation appear to be functionally similar to each other [46]. So, it is speculated that DoSWEET5c might also be involved in sucrose phloem unloading in D. officinale stems. In addition, DoSWEET5b, the paralogous gene of DoSWEET5c, exhibited the similar expression pattern with DoSWEET5c (Fig. 4a and Additional file 4) and was highly homologous with SbSWEET4-3 (Additional file 7). Therefore, we consider that DoSWEET5b and DoSWEET5c are important for sugar transport and function redundantly in sucrose unloading and polysaccharide accumulation in D. officinale stems. DoSWEET13 was expressed relatively high in the flower buds (Fig. 4 and Additional file 4) and was classified into the same clade with OsSWEET11. OsSWEET11 is reported to be essential for reproductive development of rice [47, 48]. Thus, it is likely that DoSWEET13 might play a role in the differentiation and development of D. officinale flower buds. These findings help to narrow down the range of candidate genes for sugar transport in D. officinale. In addition, seven DoSWEETs were not detected in the ten examined tissues. There are three explanations for this finding. Firstly, these DoSWEETs are likely to express in other specific tissues or development stages. DoSLR1-1 was only expressed in the seed [49]. SbSWEET2-1 and SbSWEET7-1 were specifically expressed in the panicle from the start of heading to 36 days afterward [40]. Secondly, the seven DoSWEETs may be stress-responsive genes. For example, DcSUVH2a was only detected under high temperature [50]. Thirdly, these unexpressed DoSWEETs may have lost their functions during evolution [49]. However, more direct evidences should be provided by subsequent experiments to illustrate the specific function of DoSWEETs.
D. officinale is an epiphytic orchid and grows in the barks, the branches or the crack in the rocks. Its growth and development usually suffer from biotic and abiotic stresses. Sugar is not only the main energy source necessary for life processes, but also takes part in the responses to diverse stresses. In this study, we mainly focused on the cold, drought, and MeJA-responsive DoSWEET genes. Cis-acting elements prediction also help us to understand more about DoSWEET gene family in stress response. Consistent with the drought-responsive elements found on the promoter, DoSWEET16 expression decreased under drought stress and then increased after rewatering (Fig. 5b). Although the expression of DoSWEET2b also decreased after drought treatment, no relevant elements were predicted on its promoter. Similarly, although no cold-responsible element was found, DoSWEET2b and DoSWEET16 were down-regulated under cold stress. On one hand, the inconsistency may result from the inaccuracy of the prediction. On the other hand, in addition to stresses, gene expression is closely related to many other factors, such as specific tissues and growth stages. Samples of the stress-related RNA-seq data used in this work were all leaves, while the responses of DoSWEETs may not be limited to this tissue. To further clarify the roles of DoSWEETs in stress response, it is of great significance to detect their expression changes in other tissues of D. officinale. DoSWEET2b, highly expressed in root (Fig. 4), is orthologous to AtSWEET2, OsSWEET2a, and OsSWEET2b. AtSWEET2 showed dominant expression level in the tonoplast of roots and was regulated by both pathogen (Pythium irregulare) infection and biotic stresses [51]. In addition, symbioses are often formed between orchids and soil mycorrhiza fungi, which is beneficial to plant growth. DoSWEETs with high level in the roots may also be related to pathogen nutrition in symbiotic relationship construction. DoSWEET16, whose expression decreased in response to cold stress (Fig. 5), is the ortholog of OsSWEET16, AtSWEET17, and AtSWEET16 from clade IV (Fig. 1). AtSWEET16 was located in the vacuole membrane to transport sugar and was down-regulated under cold stress. Plants overexpressing AtSWEET16 failed to accumulate fructose under cold treatment and showed improved cold tolerance [25]. These results suggest that AtSWEET16 is involved in strengthening stress resistance through sugar efflux regulation. In this study, although the responses of DoSWEET2b and DoSWEET16 under cold, drought, and MeJA treatment have been confirmed by RT-qPCR, more experiments should be done to uncover the detailed functions and mechanisms.
Conclusions
In this study, we identified 25 DoSWEETs in D. officinale and analyzed their characterizations, including evolution relationship, conversed domains, chromosomal localization, and expression patterns, to explore their potential functions. In particular, it was found that DoSWEET5b, 5c and 7d were enriched in the stems (the site of polysaccharides accumulation). DoSWEET2b and 16 responded to different abiotic stresses, which expanded our understandings on the biological functions of DoSWEETs. In addition, the potential DoSWEET dimers formed when functioning were predicted. These findings are beneficial to the functional analysis of DoSWEETs in the future.
Materials and methods
Genome-wide identification of SWEET gene family in D. officinale
The chromosome-level genome of D. officinale was downloaded from NCBI with accession number GCA_019514585.1 (https://www.ncbi.nlm.nih.gov/assembly/GCA_019514585.1, accessed on March 5, 2022) [9]. The hidden Markov model (HMM) of the MtN3/saliva domain (PF03083.hmm) was built from the seed alignment file PF03083.alignment.seed obtained from Pfam database (https://www.ebi.ac.uk/interpro/entry/pfam/PF03083/, accessed on March 5, 2022). And then HMMER 3.1 (https://www.ebi.ac.uk/Tools/hmmer/, accessed on March 6, 2022) was used to screen putative SWEETs in the local pep file of D. officinale using PF03083.hmm as a query with a default E-value. All obtained SWEET candidates were submitted to the online website SMART (https://smart.embl.de/, accessed on March 8, 2022) and InterPro (https://www.ebi.ac.uk/interpro/search/sequence/, accessed on March 8, 2022) to confirm the MtN3/saliva domain. The identified DoSWEETs were renamed based on the homology with SWEETs from A. thaliana.
Conserved domain and motif analysis of DoSWEETs
For conserved domain analysis, the position information of the transmembrane domain (TM) and MtN3/saliva domain on DoSWEETs was obtained using InterPro (https://www.ebi.ac.uk/interpro/search/sequence/, accessed on March 8, 2022), and then visualized on EvolView (http://www.evolgenius.info/evolview/#/treeview, accessed on March 9, 2022). To identify the conserved motifs, the full length protein sequences of DoSWEETs were analyzed by MEME (https://meme-suite.org/meme/tools/meme) with the following parameters: site distribution being zero or one occurrence per sequence, the number of motifs to find being 10, and the optimum motif width between 6 and 50. All other parameters were set as the default values.
Physicochemical properties analysis and sub-cellular localization prediction of DoSWEETs
The ExPASy Server (https://web.expasy.org/compute_pi/, accessed on April 25, 2022) [52] was used to compute molecular weight (Mw) and isoelectric point (pI) of DoSWEETs. The Hydrophobicity Profile and Hydrophilicity Profile in DNAMAN 7.0 (accessed on April 23, 2022) were used to compute hydrophobicity and hydrophilicity of DoSWEETs. Plant-mPLoc (http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/, accessed on April 25, 2022) [53], WoLF PSORT (https://wolfpsort.hgc.jp, accessed on March 14, 2023) [54], and ProtComp 9.0 (http://linux1.softberry.com/berry.phtml?topic=protcomppl&group=programs&subgroup=proloc, accessed on March 14, 2023) were employed to predict sub-cellular localization of DoSWEETs.
Phylogenetic tree construction of DoSWEETs
The amino acid sequences of SWEETs from A. thaliana (the model plant in dicots) and Oryza sativa L. (the model plant in monocots) were obtained from a previous study [39] and from the online websites (https://www.arabidopsis.org and http://rice.uga.edu, accessed on April 15, 2022). The protein file of Phalaenopsis equestris (Schauer) Rchb.f. (another species in orchidaceae) was downloaded from Orchidstra 2.0 (http://orchidstra2.abrc.sinica.edu.tw, accessed on April 18, 2022) [55] and SWEET proteins were screened by HMMER 3.1 (https://www.ebi.ac.uk/Tools/hmmer/, accessed on March 6, 2022) based on PF03083.hmm. Multiple sequence alignment of all SWEET protein sequences, including DoSWEETs, were performed by ClustalW in MEGA 11 (https://www.megasoftware.net, accessed on May 6, 2022) [56]. And then the sequence alignment file was used to construct the un-rooted phylogenetic tree using the Maximum Likelihood Estimate (MLE) method with the Poisson model and a bootstrap analysis of 1000 replicates in MEGA 11. ‘Use all sites’ was chosen for gaps treatment. The sequence alignment file for phylogenetic tree construction is available in Zenodo (10.5281/zenodo.7947133).
Chromosome localization of DoSWEETs and Ka/Ks calculation
The sequences of DoSWEETs were mapped to D. officinale reference genome using ncbi-blast-2.13.0+ (https://www.ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST, accessed on March 15, 2022) to obtain the position information. The duplicated gene pairs of DoSWEETs were also identified using ncbi-blast-2.13.0+. Genes matched the following conditions were defined as duplicated gene pairs: a, a length of alignable sequence covered > 75% of the longer gene; b, similarity of aligned regions > 75% [57]. And the duplicated gene pairs with physical distance within 100 kb were considered as tandem duplication gene pairs, otherwise [58]. Then ClustalW (http://www.genome.jp/tools/clustalw/, accessed on March 15, 2022) was employed to align the coding sequences of DoSWEET pairs. And the results were used to calculate the non-synonymous substitutions (Ka), synonymous substitutions (Ks) values, and Ka/Ks ratio between each of the gene pairs using KaKs_Calculator 3.0 (accessed on May 12, 2022) with Nei-Gogobori method [59].
The chromosome localization of DoSWEETs as well as the tandem duplication gene pairs were visualized using Circos-0.69-6 (accessed on July 12, 2022) [60].
Tissue-specific expression pattern and stress response analysis of DoSWEETs
The tissue-specific expression data of DoSWEETs were downloaded from OrchidBase 4.0 (http://orchidbase.itps.ncku.edu.tw/est/FPKM.aspx?projectname=Dendrobium, accessed on May 23, 2022) [30]. Each tissue only contained one biological replicate in the database. The RNA-seq data of D. officinale under low temperature (PRJNA314400) [32], drought stress (PRJNA432825) [33], cadmium stress (PRJNA561268) [34] and MeJA treatment (PRJNA732289) [35] were downloaded from NCBI (https://www.ncbi.nlm.nih.gov, accessed on May 20, 2022). The datasets of low temperature, cadmium stress, and MeJA treatment contained three replicates, while drought stress had only one biological replicate. Data processing and analysis were performed as Hao et al. reported [61]. Briefly, after filtering the adaptor and low-quality reads by trimmomatic 0.39 (accessed on April 15, 2022) [62], the clean data were mapped to D. officinale reference genome using HISAT2 2.2.0 (accessed on April 15, 2022) [63]. StringTie 2.2.1 (http://ccb.jhu.edu/software/stringtie/, accessed on April 16, 2022) [64] was employed to calculate fragments per kilobase per million (FPKM) values to assess the gene expression levels. Then, the change of gene expression was calculated as log2(mean FPKM of experimental group/control group). The heatmaps in Figs. 4a and 5b were generated from the FPKM values and log2(mean FPKM of experimental group/control group) values, respectively, using GraphPad Prism 9 (accessed on March 22, 2022).
Co-expression relationship of DoSWEETs was analyzed using corrplot R package in RStudio 2022.02.1 + 461 (https://www.rstudio.com, accessed on April 22, 2022) with R 4.1.3 (https://www.r-project.org, accessed on April 22, 2022) based on the FPKM values in different tissues and under different stresses.
Cis-acting elements prediction on DoSWEETs promoters
To identify the potential cis-acting elements on the promoters, the 2000 bp upstream sequences from the translational initiation codon of DoSWEETs were obtained from D. officinale genome and subsequently submitted to PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/, accessed on July 6, 2022) [65] to predict the cis-regulated elements using the default parameters. The number, distribution, and classification of the elements were further analyzed and visualized using ggplot2 package in RStudio 2022.02.1 + 461 (https://www.rstudio.com, accessed on April 22, 2022) with R 4.1.3 (https://www.r-project.org, accessed on April 22, 2022).
Interaction relationship prediction of DoSWEETs
The protein sequences of DoSWEETs were submitted to STRING (https://cn.string-db.org/, accessed on May 5, 2022) [66] to identify protein-protein functional interactions. All identified interaction partners were gathered and searched using A. thaliana as the reference organism with the default parameters.
Plant materials and stress treatment
The tissue-cultured D. officinale plantlets were grown on Murashige and Skoog (MS) medium [67] containing 30 g·L–1 sucrose, 7 g·L–1 Agar, 0.4 mg·L–1 6-benzylaminopurine (6-BA) and 0.1 mg·L–1 1-naphthylacetic acid (NAA) with pH 5.8 at 24 ± 1 °C under a set photoperiod of 16-h light /8-h dark. The plants were transferred to fresh medium every 8 weeks.
After 6 weeks of culture, the plantlets with the same growth status were used for sampling or stress treatment. For tissue-specific expression determination, the roots, stems, and leaves of D. officinale were collected. For low temperature treatment, the D. officinale plantlets were subjected to 0℃ for 20 h, 25 °C was used as the control. For simulating drought stress, D. officinale plantlets were treated with or without 20% (w/v) PEG6000 for 24 h. For MeJA treatment, the plantlet leaves were sprayed with 0.25% ethanol solution with or without 1 mM MeJA for 4 h. The leaves of stress-treated plantlets were collected and quickly frozen in liquid nitrogen for RNA extraction and detection of gene expression. All experiments were repeated for three times.
RNA extraction and RT-qPCR
Total RNA was isolated using the EASYspin Plus Complex Plant RNA Kit (Aidlab, Beijing, China), followed by reverse-transcription using the TRUEscript 1st Strand cDNA Synthesis Kit (Onestep gDNA Removal). RNA quantity was evaluated by NanoDrop 2000 (Thermo Fisher Scientific, Wilmington, DE, USA). RT-qPCR was performed using the 2× Sybr Green qPCR Mix (Low ROX) (Aidlab, Beijing, China) with QuantStudio 3 (ABI, California, USA) in a total reaction volume of 20 µL. The PCR cycles were as follows: 95 °C for 2 min, followed by 40 cycles of 95 C for 15 s and 60 °C for 30 s. DoGAPDH was used as an internal control gene [68]. The relative expression levels of DoSWEETs were calculated using the 2−ΔΔCt method [69]. ΔΔCt = (Ct, Target gene − Ct, DoGAPDH) − (Ct, Target gene − Ct, DoGAPDH)Max. Fold changes of DoSWEETs under stresses were calculated as 2−ΔΔCtTreatment/2−ΔΔCtControl and the values of log2 fold change were used to draw Fig. 5c. All experiments included three independent biological replicates. The primers used in this study are listed in Additional file 8.
Statistical analysis
Visualization of RT-qPCR was performed using GraphPad Prism 9 (accessed on March 22, 2022). The tissue-specific expression data of DoSWEETs (Fig. 4d) were analyzed by Duncan’s multiple range test using one-way ANOVA program of SPSS 25 (P < 0.05) (accessed on March 22, 2022). Significance analysis of DoSWEETs expression under different stresses compared to the control (Fig. 5c) was determined by two-tailed Student’s t-test in SPSS 25.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We sincerely thank all researchers who unselfishly shared genomic data and RNA-seq data.
Abbreviations
- MeJA
Methyl jasmonate
- SWEET
Sugars will eventually be exported transporters
- TM
Transmembrane domain
Authors’ contributions
LH and YZ designed experiments; LH analyzed experimental data; XS, SWQ, JHD, HS performed some experiments; LH and YHW performed some samples collection, LH, YHW and YZ wrote the manuscript, YZ revised the manuscript.
Funding
This work was supported by the Sichuan Science and Technology Department (23MZGC0219) and the Talent Engineering Scientific Research Project of Chengdu University (2081923037).
Data Availability
The datasets generated and/or analyzed during the current study are available in the NCBI Genome database with accession number GCA_019514585.1 [9] and SRA database with accession number PRJNA314400 [32], PRJNA432825 [33], PRJNA561268 [34] and PRJNA732289 [35]. The sequence alignment file for phylogenetic tree construction is available in Zenodo (10.5281/zenodo.7947133).
Declarations
Ethics approval and consent to participate
The Dendrobium officinale Kimura et Migo collected in this study were identified by professor Yuehua Wang of Chengdu University. A voucher specimen of this material has been deposited in the herbarium of Institute of Botany, Chinese Academy of Sciences with deposition number PE02032867. The tissue-cultured D. officinale was conserved in Chengdu University and the permission to use has been obtained from Chengdu University. All experiments involving the plant and its material complied with relevant institutional, national, and international guidelines and legislation, and the IUCN Policy Statement on Research Involving Species at Risk of Extinction and the Convention on the Trade in Endangered Species of Wild Fauna and Flora.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
7/20/2023
The ORCID given for Yi Zhang was incorrect. The article has been updated to rectify the error.
Contributor Information
Li Hao, Email: haoli@cdu.edu.cn.
Xin Shi, Email: 925219365@qq.com.
Shunwang Qin, Email: 13408062337@163.com.
Jiahong Dong, Email: 3138192704@qq.com.
Huan Shi, Email: 1940852693@qq.com.
Yuehua Wang, Email: 1961689636@qq.com.
Yi Zhang, Email: zhangyi1@cib.ac.cn.
References
- 1.Ng TB, Liu J, Wong JH, Ye X, Wing Sze SC, Tong Y, et al. Review of research on Dendrobium, a prized folk medicine. Appl Microbiol Biotechnol. 2012;93(5):1795–803. doi: 10.1007/s00253-011-3829-7. [DOI] [PubMed] [Google Scholar]
- 2.Yang J, Meng H, Yang S, Zhang W, Zha Y, Wen G. Correlation between soluble polysaacharide and sucrosemetabolic enzymes in Dendrobium officinale. J West China Forestry Sci. 2012;41(2):62–7. [Google Scholar]
- 3.Hu X, Guo Y, Xiang J, Du Y. Research advances in polysaccharides of Dendrobium officinale. J Anhui Agri Sci. 2015;43(15):78–8084. [Google Scholar]
- 4.Hua YF, Zhang M, Fu CX, Chen ZH, Chan GY. Structural characterization of a 2-O-acetylglucomannan from Dendrobium officinale stem. Carbohydr Res. 2004;339(13):2219–24. doi: 10.1016/j.carres.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 5.He C, Zhang J, Liu X, Zeng S, Wu K, Yu Z, et al. Identification of genes involved in biosynthesis of mannan polysaccharides in Dendrobium officinale by RNA-seq analysis. Plant Mol Biol. 2015;88(3):219–31. doi: 10.1007/s11103-015-0316-z. [DOI] [PubMed] [Google Scholar]
- 6.Jiang M, Li S, Zhao C, Zhao M, Xu S, Wen G. Identification and analysis of sucrose synthase gene family associated with polysaccharide biosynthesis in Dendrobium catenatum by transcriptomic analysis. PeerJ. 2022;10:e13222. doi: 10.7717/peerj.13222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porter HK. Synthesis of polysaccharides of higher plants. Annu Rev Plant Physiol. 1962;13(1):303–26. doi: 10.1146/annurev.pp.13.060162.001511. [DOI] [Google Scholar]
- 8.Eom JS, Chen LQ, Sosso D, Julius BT, Lin IW, Qu XQ, et al. SWEETs, transporters for intracellular and intercellular sugar translocation. Curr Opin Plant Biol. 2015;25:53–62. doi: 10.1016/j.pbi.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Niu Z, Zhu F, Fan Y, Li C, Zhang B, Zhu S, et al. The chromosome-level reference genome assembly for Dendrobium officinale and its utility of functional genomics research and molecular breeding study. Acta Pharm Sinica B. 2021;11(7):2080–92. doi: 10.1016/j.apsb.2021.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang GQ, Xu Q, Bian C, Tsai WC, Yeh CM, Liu KW, et al. The Dendrobium catenatum Lindl. Genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci Rep. 2016;6(1):19029. doi: 10.1038/srep19029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, He C, Wu K, Teixeira da Silva JA, Zeng S, Zhang X, et al. Transcriptome analysis of Dendrobium officinale and its application to the identification of genes associated with polysaccharide synthesis. Front Plant Sci. 2016;7:5. doi: 10.3389/fpls.2016.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He C, Wu K, Zhang J, Liu X, Zeng S, Yu Z, et al. Cytochemical localization of polysaccharides in Dendrobium officinale and the involvement of DoCSLA6 in the synthesis of mannan polysaccharides. Front Plant Sci. 2017;8:173. doi: 10.3389/fpls.2017.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Chen Y, Wei Q, Wan H, Sun C. Phylogenetic relationships of sucrose transporters (SUTs) in plants and genome-wide characterization of SUT genes in Orchidaceae reveal roles in floral organ development. PeerJ. 2021;9:e11961. doi: 10.7717/peerj.11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Tong Y, Adejobi OI, Wang Y, Liu A. Research advances in multi-omics on the traditional chinese herb Dendrobium officinale. Front Plant Sci. 2022;12:808228. doi: 10.3389/fpls.2021.808228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lalonde S, Wipf D, Frommer WB. Transport mechanisms for organic forms of carbon and nitrogen between source and sink. Annu Rev Plant Biol. 2004;55(1):341–72. doi: 10.1146/annurev.arplant.55.031903.141758. [DOI] [PubMed] [Google Scholar]
- 16.Li YM, Chen Y, Liu AP, Zhang N, Hei XB, Zhang G, et al. Isolation and expression analysis of a hexose transporter gene DoHT1 in Dendrobium officinale. Zhongguo Zhong Yao Za Zhi. 2018;43(6):1124–30. doi: 10.19540/j.cnki.cjcmm.20180110.003. [DOI] [PubMed] [Google Scholar]
- 17.Chen LQ, Hou BH, Lalonde S, Takanaga H, Hartung ML, Qu XQ, et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature. 2010;468(7323):527–32. doi: 10.1038/nature09606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen LQ, Cheung LS, Feng L, Tanner W, Frommer WB. Transport of sugars. Annu Rev Biochem. 2015;84(1):865–94. doi: 10.1146/annurev-biochem-060614-033904. [DOI] [PubMed] [Google Scholar]
- 19.Hamada M, Wada S, Kobayashi K, Satoh N. Ci-Rga, a gene encoding an MtN3/saliva family transmembrane protein, is essential for tissue differentiation during embryogenesis of the ascidian Ciona intestinalis. Differentiation. 2005;73(7):364–76. doi: 10.1111/j.1432-0436.2005.00037.x. [DOI] [PubMed] [Google Scholar]
- 20.Chen LQ, Qu XQ, Hou BH, Sosso D, Osorio S, Fernie AR, et al. Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science. 2012;335(6065):207–11. doi: 10.1126/science.1213351. [DOI] [PubMed] [Google Scholar]
- 21.Sosso D, Luo D, Li QB, Sasse J, Yang J, Gendrot G, et al. Seed filling in domesticated maize and rice depends on SWEET-mediated hexose transport. Nat Genet. 2015;47(12):1489–93. doi: 10.1038/ng.3422. [DOI] [PubMed] [Google Scholar]
- 22.Guo C, Li H, Xia X, Liu X, Yang L. Functional and evolution characterization of SWEET sugar transporters in Ananas comosus. Biochem Biophys Res Commun. 2018;496(2):407–14. doi: 10.1016/j.bbrc.2018.01.024. [DOI] [PubMed] [Google Scholar]
- 23.Lin IW, Sosso D, Chen LQ, Gase K, Kim SG, Kessler D, et al. Nectar secretion requires sucrose phosphate synthases and the sugar transporter SWEET9. Nature. 2014;508(7497):546–9. doi: 10.1038/nature13082. [DOI] [PubMed] [Google Scholar]
- 24.Sun MX, Huang XY, Yang J, Guan YF, Yang ZN. Arabidopsis RPG1 is important for primexine deposition and functions redundantly with RPG2 for plant fertility at the late reproductive stage. Plant Reprod. 2013;26(2):83–91. doi: 10.1007/s00497-012-0208-1. [DOI] [PubMed] [Google Scholar]
- 25.Klemens PA, Patzke K, Deitmer J, Spinner L, Le Hir R, Bellini C, et al. Overexpression of the vacuolar sugar carrier AtSWEET16 modifies germination, growth, and stress tolerance in Arabidopsis. Plant Physiol. 2013;163(3):1338–52. doi: 10.1104/pp.113.224972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chong J, Piron MC, Meyer S, Merdinoglu D, Bertsch C, Mestre P. The SWEET family of sugar transporters in grapevine: VvSWEET4 is involved in the interaction with Botrytis cinerea. J Exp Bot. 2014;65(22):6589–601. doi: 10.1093/jxb/eru375. [DOI] [PubMed] [Google Scholar]
- 27.Cohn M, Bart RS, Shybut M, Dahlbeck D, Gomez M, Morbitzer R, et al. Xanthomonas axonopodis virulence is promoted by a transcription activator-like effector-mediated induction of a SWEET sugar transporter in cassava. Mol Plant Microbe Interact. 2014;27(11):1186–98. doi: 10.1094/MPMI-06-14-0161-R. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Wang Y, Zhang H, Zhang Q, Zhai H, Liu Q, et al. The plasma membrane-localized sucrose transporter IbSWEET10 contributes to the resistance of sweet potato to Fusarium oxysporum. Front Plant Sci. 2017;8:197. doi: 10.3389/fpls.2017.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang T, Song Z, Meng WL, Li LB. Identification, characterization, and expression of the SWEET gene family in Phalaenopsis equestris and Dendrobium officinale. Biol Plant. 2018;62(1):24–32. doi: 10.1007/s10535-017-0750-7. [DOI] [Google Scholar]
- 30.Hsiao YY, Fu CH, Ho SY, Li CI, Chen YY, Wu WL, et al. OrchidBase 4.0: a database for orchid genomics and molecular biology. BMC Plant Biol. 2021;21(1):371. doi: 10.1186/s12870-021-03140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ji J, Yang L, Fang Z, Zhang Y, Zhuang M, Lv H, et al. Plant SWEET family of sugar transporters: structure, evolution and biological functions. Biomolecules. 2022;12(2):205. doi: 10.3390/biom12020205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu ZG, Jiang W, Chen SL, Mantri N, Tao ZM, Jiang CX. Insights from the cold transcriptome and metabolome of Dendrobium officinale: global reprogramming of metabolic and gene regulation networks during cold acclimation. Front Plant Sci. 2016;7:1653. doi: 10.3389/fpls.2016.01653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zou LH, Wan X, Deng H, Zheng BQ, Li BJ, Wang Y. RNA-seq transcriptomic profiling of crassulacean acid metabolism pathway in Dendrobium catenatum. Sci Data. 2018;5(1):180252. doi: 10.1038/sdata.2018.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang W, Wu Z, Wang T, Mantri N, Huang H, Li H, et al. Physiological and transcriptomic analyses of cadmium stress response in Dendrobium officinale seedling. Plant Physiol Biochem. 2020;148:152–65. doi: 10.1016/j.plaphy.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Li C, Shen Q, Cai X, Lai D, Wu L, Han Z et al. JA signal-mediated immunity of Dendrobium catenatum to necrotrophic southern blight pathogen. BMC Plant Biol. 2021;21(1). [DOI] [PMC free article] [PubMed]
- 36.Xuan YH, Hu YB, Chen LQ, Sosso D, Ducat DC, Hou BH, et al. Functional role of oligomerization for bacterial and plant SWEET sugar transporter family. Proc Natl Acad Sci U S A. 2013;110(39):E3685–94. doi: 10.1073/pnas.1311244110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braun DM, Wang L, Ruan YL. Understanding and manipulating sucrose phloem loading, unloading, metabolism, and signalling to enhance crop yield and food security. J Exp Bot. 2013;65(7):1713–35. doi: 10.1093/jxb/ert416. [DOI] [PubMed] [Google Scholar]
- 38.Chandran D, Reinders A, Ward JM. Substrate specificity of the Arabidopsis thaliana sucrose transporter AtSUC2. J Biol Chem. 2003;278(45):44320–5. doi: 10.1074/jbc.M308490200. [DOI] [PubMed] [Google Scholar]
- 39.Yuan M, Wang S. Rice MtN3/Saliva/SWEET family genes and their homologs in cellular organisms. Mol Plant. 2013;6(3):665–74. doi: 10.1093/mp/sst035. [DOI] [PubMed] [Google Scholar]
- 40.Mizuno H, Kasuga S, Kawahigashi H. The sorghum SWEET gene family: stem sucrose accumulation as revealed through transcriptome profiling. Biotechnol Biofuels. 2016;9(1):127. doi: 10.1186/s13068-016-0546-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miao H, Sun P, Liu Q, Miao Y, Liu J, Zhang K, et al. Genome-wide analyses of SWEET family proteins reveal involvement in fruit development and abiotic/biotic stress responses in banana. Sci Rep. 2017;7(1):3536. doi: 10.1038/s41598-017-03872-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu W, Hua X, Zhang Q, Wang J, Shen Q, Zhang X, et al. New insights into the evolution and functional divergence of the SWEET family in Saccharum based on comparative genomics. BMC Plant Biol. 2018;18(1):270. doi: 10.1186/s12870-018-1495-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Zhang GQ, Zhang D, Liu XD, Xu XY, Sun WH, et al. Chromosome-scale assembly of the Dendrobium chrysotoxum genome enhances the understanding of orchid evolution. Hortic Res. 2021;8(1):183. doi: 10.1038/s41438-021-00621-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore RC, Purugganan MD. The early stages of duplicate gene evolution. Proc Natl Acad Sci U S A. 2003;100(26):15682–7. doi: 10.1073/pnas.2535513100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cannon SB, Mitra A, Baumgarten A, Young ND, May G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004;4:10. doi: 10.1186/1471-2229-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Forslund K, Pekkari I, Sonnhammer EL. Domain architecture conservation in orthologs. BMC Bioinformatics. 2011;12(1):326. doi: 10.1186/1471-2105-12-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chu Z, Yuan M, Yao J, Ge X, Yuan B, Xu C, et al. Promoter mutations of an essential gene for pollen development result in disease resistance in rice. Genes Dev. 2006;20(10):1250–5. doi: 10.1101/gad.1416306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang B, Sugio A, White FF. Os8N3 is a host disease-susceptibility gene for bacterial blight of rice. Proc Natl Acad Sci U S A. 2006;103(27):10503–8. doi: 10.1073/pnas.0604088103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeng X, Ling H, Chen X, Guo S. Genome-wide identification, phylogeny and function analysis of GRAS gene family in Dendrobium catenatum (Orchidaceae) Gene. 2019;705:5–15. doi: 10.1016/j.gene.2019.04.038. [DOI] [PubMed] [Google Scholar]
- 50.Chen DH, Qiu HL, Huang Y, Zhang L, Si JP. Genome-wide identification and expression profiling of SET DOMAIN GROUP family in Dendrobium catenatum. BMC Plant Biol. 2020;20(1):40. doi: 10.1186/s12870-020-2244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen HY, Huh JH, Yu YC, Ho LH, Chen LQ, Tholl D, et al. The Arabidopsis vacuolar sugar transporter SWEET2 limits carbon sequestration from roots and restricts Pythium infection. Plant J. 2015;83(6):1046–58. doi: 10.1111/tpj.12948. [DOI] [PubMed] [Google Scholar]
- 52.Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531–52. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 53.Chou KC, Shen HB. Plant-mPLoc: a top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS ONE. 2010;5(6):e11335. doi: 10.1371/journal.pone.0011335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;W585–7. [DOI] [PMC free article] [PubMed]
- 55.Chao YT, Yen SH, Yeh JH, Chen WC, Shih MC. Orchidstra 2.0-A transcriptomics resource for the Orchid family. Plant Cell Physiol. 2017;58(1):e9. doi: 10.1093/pcp/pcw220. [DOI] [PubMed] [Google Scholar]
- 56.Tamura K, Stecher G, Kumar S, Battistuzzi FU. MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol. 2021;38(7):3022–7. doi: 10.1093/molbev/msab120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vatansever R, Koc I, Ozyigit II, Sen U, Uras ME, Anjum NA, et al. Genome-wide identification and expression analysis of sulfate transporter (SULTR) genes in potato (Solanum tuberosum L) Planta. 2016;244:1167–83. doi: 10.1007/s00425-016-2575-6. [DOI] [PubMed] [Google Scholar]
- 58.Wang LQ, Guo K, Li Y, Tu YY, Hu HZ, Wang BR, et al. Expression profiling and integrative analysis of the CESA/CSL superfamily in rice. BMC Plant Biol. 2010;10:282. doi: 10.1186/1471-2229-10-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Z. KaKs_calculator 3.0: calculating selective pressure on coding and non-coding sequences. Genomics Proteom Bioinf. 2022;S1672-0229(21)00259-X. [DOI] [PMC free article] [PubMed]
- 60.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hao L, Zhang Y. Genome-wide analysis of miR159 gene family and predicted target genes associated with environmental stress in Dendrobium officinale: a bioinformatics study. Genes. 2022;13(7):1221. doi: 10.3390/genes13071221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–15. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lescot M, Dehais P, Thijs G, Marchal K, Moreau Y, Van de Peer Y, et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30(1):325–7. doi: 10.1093/nar/30.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2012;41(D1):D808–15. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murashige T, Skoog F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant. 1962;15:473–97. doi: 10.1111/j.1399-3054.1962.tb08052.x. [DOI] [Google Scholar]
- 68.Jiang M, Xu SZ, Wen GS, Zhao CL. Validation of seven housekeeping genes as reference ones for qRT-PCR normalization in Dendrobium catenatum. Russ J Plant Physiol. 2017;64(4):497–508. doi: 10.1134/S1021443717040069. [DOI] [Google Scholar]
- 69.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Method. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available in the NCBI Genome database with accession number GCA_019514585.1 [9] and SRA database with accession number PRJNA314400 [32], PRJNA432825 [33], PRJNA561268 [34] and PRJNA732289 [35]. The sequence alignment file for phylogenetic tree construction is available in Zenodo (10.5281/zenodo.7947133).