

Abstract
Background
Amphibians, particularly anurans, display an enormous variation in genome size. Due to the unavailability of whole genome datasets in the past, the genomic elements and evolutionary causes of anuran genome size variation are poorly understood. To address this, we analyzed whole-genome sequences of 14 anuran species ranging in size from 1.1 to 6.8 Gb. By annotating multiple genomic elements, we investigated the genomic correlates of anuran genome size variation and further examined whether the genome size relates to habitat types.
Results
Our results showed that intron expansions or contraction and Transposable Elements (TEs) diversity do not contribute significantly to genome size variation. However, the recent accumulation of transposable elements (TEs) and the lack of deletion of ancient TEs primarily accounted for the evolution of anuran genome sizes. Our study showed that the abundance and density of simple repeat sequences positively correlate with genome size. Ancestral state reconstruction revealed that genome size exhibits a taxon-specific pattern of evolution, with families Bufonidae and Pipidae experiencing extreme genome expansion and contraction events, respectively. Our result showed no relationship between genome size and habitat types, although large genome-sized species are predominantly found in humid habitats.
Conclusions
Overall, our study identified the genomic element and their evolutionary dynamics accounting for anuran genome size variation, thus paving a path to a greater understanding of the size evolution of the genome in amphibians.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-023-09499-8.
Keywords: Genome size variation, Simple repeat sequences, Anurans, Transposable elements
Background
Eukaryotic genomes vary greatly in size, with existing genomes differing in size by about 200,000-fold, thus providing an open question about the evolutionary forces that shape the genome size evolution [1]. Several studies have conducted comparative genomic analyses of closely related and allopatric species to understand the evolutionary factors underlying genome size variation across multiple taxa [2–6]. One popular explanation is that the proliferation and contraction of repetitive sequences play an essential role in the evolution of eukaryotic genome size [4, 7–11]. Moreover, gene or genome duplication [12–15] and intron size changes [10, 16, 17] have also been implicated as underlying evolutionary forces causing variations in genome size. Further, a significant correlation between genome sizes and the abundance and density of simple sequence repeats (SSRs) was also reported [18, 19]. Unfortunately, the comprehensive characteristics and dynamics underlying eukaryotic genome size evolution remain contentious.
Repetitive sequences are abundantly distributed in eukaryotic genomes and can influence genome size. It can be categorized based on their sequence features or the process by which they are created. One major category consists of transposable elements (TEs), which include sequences that are dispersed throughout the entire genome and can move and multiply across the host genome [20]. The TEs are further divided into two groups [21–24], with Class I transposons (retrotransposons) which make new copies using an RNA-mediated copy-and-paste mechanism [25], and Class II transposable elements (DNA transposons) which replicate elements using a DNA-mediated cut-and-paste mechanism [26]. Studies have shown that retrotransposons occupy a significant fraction of TEs in the eukaryotic genome [11, 27]. Based on the structural features, retrotransposons are further subdivided into Long Terminal Repeat retrotransposons (LTRs), Long Interspersed Nuclear Elements (LINEs) and Short Interspersed Nuclear Elements (SINEs). Another category of repetitive sequences is the tandem repeats, which include sequences found in consecutive copies along the DNA. This group comprises minisatellites and microsatellites or simple sequence repeats (SSRs) [28, 29]. The SSRs comprise Perfect SSRs (P-SSRs), Compound SSRs (C-SSRs), and Imperfect SSRs (I-SSRs) and are distributed in eukaryotic genomes [30]. Although some studies have shown that SSRs occupy only about 1% of the genome [31, 32], others reported that SSRs occupy up to 23% of the genome [33].
Anurans (collective term for frogs and toads) are a suitable model for investigating mechanisms underlying divergence in genome size owing to their diverse genome sizes [34, 35]. With the advent of advanced sequencing technology, genomes of several anurans are now available, thus contributing ideal resources for exploring the characteristics and dynamics of genome size evolution. Although the dynamic evolution of repetitive sequences significantly contributes to eukaryotic genome size evolution, the relative contributions of these sequences to genome size evolution in anurans remained controversial [36, 37]. For instance, a recent study on cave frog (Platyplectrum ornatum) suggested that reduction of intron abundance, loss of TEs, and suppression of activity could be the primary causes of genome contraction [16]. Also, Wang et al. (2021) reported no significant relationship between TEs diversity and genome size [38]. In contrast, Haley and Mueller (2022) showed that larger genome size was associated with increased TE diversity at the superfamily level [39]. Hence, these previous studies failed to obtain consistent results on the genomic elements contributing to anuran genome size evolution. Additionally, anurans occupy diverse habitats (e.g., forests, savanna, humid ecosystems etc.) and migrate between habitats for breeding. Yet, it is unknown if anurans modify their genome size due to species-specific habitat requirements and life cycles [40, 41].
Here, we conducted comparative genomic analyses of fourteen anurans that include species ranging in genome size from Spea multiplicata (1.1GB) to Ranitomeya imitator (6.8GB) to understand the mechanism(s) underlying genome size evolution. Specifically, we seek to answer the following questions: (1) What are the exact genomic elements (TEs, SSR or both) accounting for the genome size variation in anurans? (2) How do non-coding regions (intron, exon, and intergenic regions) drive anuran genome size evolution? (3) What are the evolutionary causes of the genome size evolution in anurans? And (4) Does the anuran genome size correlate with habitat types? The results of our study offer deep insights into the genomic elements and evolutionary mechanisms underlying the evolution of anuran genome size.
Results
Repetitive sequence landscape
Repetitive sequences were more abundant in species with larger genome sizes (Supplementary Materials 1: Table S3). Furthermore, there was a significant correlation (R2 = 0.95, P = 5.733965e-09) (Fig. 1A) between the genome size and abundance of all repetitive sequences based on the result of the combined analysis using RepeatModeler, RepeatMasker, and TRF. Analyses showed that R. imitator (genome size = 6.8GB) and L. catesbeianus (genome size = 6.3GB) had higher repetitive sequences of 59.3% and 62.5%, respectively. However, only 33.7% of repetitive sequences were found in the S. multiplicata (genome size = 1.1GB).
Fig. 1.

Shows the association between genome size and repetitive sequences/transposable elements diversity index in anuran species. (A) Positive correlation between genome size and repetitive elements. The X-axis shows the genome size, the length of the repetitive elements by the Y-axis, and the 95% confidence interval by the gray region. (B) Abundance and Distribution of the Transposable elements across anuran species. (C) Genome size and transposable element diversity index (Simpson diversity index). (D) Genome size and transposable element diversity index (Shannon Diversity Index). Gray shade represents the 95% confidence interval
We observed a species-specific association of the TEs abundance with the genome size. For instance, while S. multiplicata (genome size = 1.1 Gb) exhibited a persistent reduction in multiple repetitive sequence classes (Fig. 1B), we found multiple repetitive sequences within the genomes of L. catesbeianus (genome size = 6.3 Gb) and R. imitator (genome size = 6.8 Gb). The larger genomes contain a higher proportion of repetitive sequences, suggesting the proliferation of repetitive sequences as the leading cause of genome expansion (Fig. 1B). Besides, analyses showed that although DNA transposons, LINEs and LTRs occupied a large proportion of the anuran genome, different correlations with genome size were observed (Supplementary Materials 2: Fig. S3; Supplementary Materials 1: Table S4). The linear regression model showed that LINEs had the strongest positive correlation (R2 = 0.89, P = 4.01e-07) with genome size, followed by LTRs (R2 = 0.82, P = 8.54e-06), DNA transposons (R2 = 0.55, P = 2.56e-03) and SINEs (R2 = 0.40, P = 1.53e-02). Except for R. imitator (SINE = 2.69%), the proportion of SINEs was extremely low (< 0.3%) in all species. This result suggests that SINEs have little impact on anuran genome size variation (Fig. 1B; Supplementary Materials 1: Table S4).
Based on the identification of tandem repeat sequences in the genomes of 14 species, our results reveal that satellite DNA constitutes a proportion ranging from 0.0022 to 0.43% (63,538 − 6,275,648 bp) of the genome. At the same time, SSRs account for a proportion ranging from 0.47 to 2.11% (21,272,043 − 67,891,025 bp) (Supplementary Materials 1: Table S8). Additionally, we found no correlation between genome size and the total length (R2 = 0, P = 0.9001969) and number (R2 = 0.14, P = 0.1945149) of satellite sequences (Supplementary Materials 2: Fig. S6). However, a significant correlation was observed between genome size and the total length (R2 = 0.55, P = 0.002242437) and number (R2 = 0.71, P = 0.0001458194) of simple repeats (Supplementary Materials 2: Fig. S6).
TEs community diversity and dynamics
We observed no strong positive correlation between genome size and TEs diversity (Simpson Diversity Index R2 = 0.07, P = 0.3774581, Shannon Diversity Index R2 = 0.27 P = 0.05701796) (Fig. 1C and D; Supplementary Materials 1: Table S2). However, a weak positive correlation between the Shannon diversity index of TEs and genome size was seen.
The divergence curves showed an L-shaped distribution for X. tropicalis and R. marina with a divergence of below 5%, which suggests a recent burst event (Fig. 2; Supplementary Materials 2: Fig. S8). Other species, including H. boettgeri, S. multiplicata, X. laevis, L. catesbeianus, R. temporaria, B. gargarizans, N. parkeri, Q. spinosa, L. dumerilii, R. imitator, E. coqui, and E. pustulosus, exhibited a bimodal or multi-peaked distribution. In this case, there were two or more peaks, with the first peak occurring at approximately 0–10% divergence and the second or third peak occurring at about 30% divergence, indicating a recent accumulation of TEs and a lack of deletion of more ancient TEs (Fig. 2; Supplementary Materials 2: Fig. S8). Although we observed two or more burst patterns, our result showed that the accumulation and lack of deletion of more ancient TE activity are mainly responsible for anuran genome size variation.
Fig. 2.

Transposable element age distribution landscapes of anuran genome sizes. The Y-axis shows the genomic coverage of different types of TEs, and the X-axis shows the Kimura substitution level as a percentage from 0 to 40. The Y-axis represents TE abundance as a proportion of the genome (e.g., 1.0 = 1% of the genome). The distribution landscape of TE divergence is categorized as follows: L-shaped distribution (TE divergence peak less than or equal to 5%), bimodal distribution (two peaks occur), or multi-peaked distribution (more than two peaks occur)
Intact LTR-RTs amplification
We observed species-specific amplification of intact LTR-RTs in anuran genomes, with different insertion abundances across species (Supplementary Materials 2: Fig. S4). Species with larger genome sizes, such as L. catesbeianus and R. imitator, had relatively low insertion abundances. In contrast, species with medium genome sizes, such as B. gragraizans and R. temporaria, had the highest insertion abundance (Supplementary Materials 2: Fig. S4). In particular, intact LTR-RTs in B. gragraizans displayed high abundance and a recent insertion abundance (< 1 MYA) compared to other species (Supplementary Materials 2: Fig. S4). Our results indicate that the large genome size is attributed to older LTR activity, which implies a higher probability that the LTR elements inserted earlier in the genome will not be intact.
Microsatellite sequence landscape
The percentage of SSRs in anuran genomes ranges from approximately 2.84–5.63% (79,217,477 bp − 231,473,092 bp), with the highest rate (5.63%, 231,473,092 bp) found in the genome of R. temporaria and the lowest (2.84%, 79,217,477 bp) in E. coqui (Supplementary Materials 1: Table S5). Our analysis revealed a positive correlation between genome size and the total length and number of SSRs (R2 = 0.83 P = 5.83299e− 06, R2 = 0.89 P = 3.96004e− 07) (Fig. 3A and B). As an implication, the positive correlation between genome size and the total length and number of SSRs suggests that SSRs experienced expansion within the anuran genome and contributed to the genome size. Nevertheless, genome size did not correlate with the relative abundance (I-SSRs: R2 = 0.09 P = 0.2916664, P-SSRs: R2 = 0.19 P = 0.1231148) and relative density of SSRs (I-SSRs: R2 = 0.08 P = 0.3329214, P-SSRs: R2 = 0.15 P = 0.1692003) (Fig. 3C and D). We also observed that the six categories of SSRs had different abundances in I-SSRs and P-SSRs (Supplementary Materials 2: Fig. S2). Specifically, Mononucleotide, Dinucleotide and Tetranucleotide were the most abundant types of SSRs among the P-SSRs in the 14 anuran genomes (Supplementary Materials 2: Fig. S2B). In contrast, Mononucleotide, Dinucleotide and Trinucleotide were the three most abundant types of I-SSRs (Supplementary Materials 2: Fig. S2A). These results suggest that Mononucleotide, Dinucleotide, Trinucleotide, and Tetranucleotide are the main categories of SSRs that shape the genomes of the 14 anuran species.
Fig. 3.

Relationship between anuran genome size and exons, introns, intergenic regions and Simple Sequence Repeats, respectively. (Gray shade represents the 95% confidence interval) Figures A, B, C, D, E, F, G, and H represent the relationship between genome size and length of SSRs, number of SSRs, relative abundance of SSRs, relative density of SSRs, intron length, average intron length, average exon length, and intergenic region length for 14 anuran species
Distribution of the non-coding regions of the genome
We found no correlation between genome size and intron length (R2 = 0, P = 0.8973065) (Fig. 3E; Supplementary Materials 2: Fig. S1). To further verify this, we compared the average intron and exon lengths in the genomes. Similarly, genome size failed to correlate strongly with the average intron length (R2 = 0.01, P = 0.7217873) and average exon length (R2 = 0.08, P = 0.3282922) (Fig. 3F and G). Our result further showed an increase in the length of the intergenic region as the genome size increases (Fig. 3H; Supplementary Materials 2: Fig. S1B). Additionally, the exon abundance of the other 13 species was found to be lower when compared to X. laevis (Supplementary Materials 2: Fig. S1B).
Phylogenetic estimation and ancestral state reconstruction
To understand the evolutionary relationship between anuran genome size and TEs abundance, we classified the evolutionary branches based on anuran families: Pipidae, Ranidae, Bufonidae, Dendrobatidae, Dicroglossidae, Eleutherodactylidae, Leptodactylidae, Myobatrachidae and Pelobatidae (Fig. 4). Our results indicate that the common ancestor of all species (node 1) had a small genome size (genome size = 1.92Gb) and a low TE abundance (node 1*) (proportion of TEs = 40.39%). However, we discovered that the ancestral genome size increased as the species diverged (Fig. 4). In the family Bufonidae, genome size tends to increase gradually as the species continued to diverge (node 9 to node 13) (although there were occasional intermediate cases of smaller genomes), reaching a peak at the ancestral nodes (Genome size = 3.89Gb) for some species (e.g., R.marina and B.gargarizans) with the highest TEs proportion (TEs = 67.32%). In contrast, the family with the smallest ancestral genome (e.g., Pipidae) experienced continuous genome contraction as species diverged (From node 2 to node 5) (genome size = 2.73Gb, 2.62Gb and 2.24Gb). However, for some species, e.g., R. imitator and L. catesbeianus, a sudden genome size increase following divergence from the ancestral nodes was observed (nodes 9 and 6, respectively). In contrast, in families Ranidae and Dicroglossidae, the TEs of ancestral nodes experienced a single event of expansion and contraction (node 6* through node 10*) (proportion of TEs = 58.34%, 61.05%, and 57.51%) (Fig. 4).
Fig. 4.

Reconstruction ancestral state of anuran genome size(left) and proportion of Transposable Elements(right) across 14 anuran species. Branching colors represent values reconstructed from phylogenetic relationships. The names and values on the nodes represent ancestral names and ancestral values for the branches, including Pipidae, Ranidae, Bufonidae, Dendrobatidae, Dicroglossidae, Eleutherodactylidae, Leptodactylidae, Myobatrachidae and Pelobatidae. In the figure, the nodes in the size branch of the genome (left) are represented by “node + number” (e.g., node 1), and the nodes in the TE branch are represented by “node + number*” (e.g., node 1*)
In addition, we observed the species with large genome sizes do not have the highest percentages of TEs in their ancestral nodes. For instance, although R. imitator and L. catesbeianus had the largest genome size compared to other species, we do not observe high TEs in their ancestral nodes (node 9* and node 6*, respectively) (Fig. 4). However, ancestral nodes (From node 2 to node 9) of other families (Pipidae, Ranidae, Bufonidae, Dendrobatidae, Dicroglossidae, Eleutherodactylidae, Leptodactylidae, Myobatrachidae and Pelobatidae) had moderately large genome sizes and TEs proportions (Fig. 4).
Relationship between genome size and habitat types
Our results showed no correlation (R2 = 0, P = 0.8274032) between genome size and habitat types (Supplementary Materials 2: Fig. S5). However, we observed that species with genome sizes ranging from 2.6-4.5Gb had the highest ecological niche width or ecological tolerance (Supplementary Materials 2: Fig. S5; Supplementary Materials 1: Table S6). Additionally, large genome-sized species (e.g., L. catesbeianus and R. imitator) tend to inhabit humid habitats. In contrast, species with smaller genomes (e.g., S. multiplicata) prefer more arid habitats (Supplementary Materials 1: Table S7).
Discussion
In this study, we performed comparative analyses of 14 anuran genomes to systematically explore the mechanisms underlying genome size evolution. Our result was consistent with an earlier study, which suggested that TEs amplification drives the expansion of genome size and that genome size is positively correlated with TEs numbers [6]. Our result further showed a strong positive correlation between the quantity of SSRs and the anuran genome size. Although previous studies based on a few available genomes indicated that SSRs occupy only 1.00% of the genome in most species [31], our result showed that the SSRs occupy ~ 2.84–5.63% (79,217,477 − 231,473,092 bp) of the entire anuran genomes. This observation is consistent with an earlier study in penaeid shrimp [33], which reported SSR proportions higher than 1%, and the roles SSR plays in genome plasticity and adaptive evolution. It is apparent that SSRs underwent expansion within the anuran genome, contributing to genome size evolution. However, the evolutionary triggers of increased SSRs in anurans with larger genome sizes are unknown, and additional research on this may provide further insight.
Our results showed no correlation between intron length and anuran genome size. This result was not in tandem with previous studies highlighting intron reduction as one of the leading causes of reduced genome size in vertebrates, including frogs [16, 42]. The introns in vertebrate genomes are influenced by a variety of factors, such as life history [43], metabolic rate [44] and life span [45], which in turn may affect anuran genome size. Also, our result showed no correlation of the intron or exon size with the genome size, except for the intergenic regions that showed a strong positive association with the anuran genome size. In addition, some species with less than 3 Gb of genomes (e.g., X. laevis, X. tropicalis, R. marina, and N. parkeri, etc.) had a 1:1 ratio of introns to intergenic regions. Previous studies have suggested that genome size changes are in a balance between insertions and deletions [46] and that the same ratio of introns to intergenic regions may be due to TE insertions, where the rate of intron amplification is the same as that of intergenic regions [47]. Nevertheless, further research integrating multiple genomic tools is required to explicitly understand the effects of the non-coding region on the genome size evolution across diverse vertebrates.
Our study showed species-specific intact LTR-RTs amplification in the anuran genome. The reduction in LTR-RTs length is mainly induced by a continuous decrease in the number of intact LTR-RTs [48]. However, our results suggest that intact LTRs are rare in large genome-sized species, and their total LTR-RTs length is not reduced, perhaps due to the accumulation of unpaired LTRs (solo-LTRs and truncated LTR-RTs). In addition, we found that the expansion of TEs in the anuran genome is species-specific, and species may have produced different genome expansion patterns after diverging from a common ancestor. Although an early study found that TEs expansion events occurred within lineage rather than from a few shared ancestral bursts [8], a recent study observed that the expansion history of TEs existed both in ancestral burst events and in recent transposition bursts [49]. Based on TEs expansion history or TEs age analysis, our study showed that the persistent accumulation of TEs with two or more burst events was the primary factor determining the size of the anuran genome. These results suggest that the effect of amplification or contraction of TEs on the size of the anuran genome is mainly a continuous process.
Our study revealed that the common ancestor of all the studied species had a small genome and a low level of TEs proportion and further experienced a Brownian motion (jump in genome size) occurring after divergence to acquire a larger genome size [50]. The pattern of change in genome size and TEs appear taxon-specific. Our study showed a disparity in the proportions of TEs states in species and their ancestors. For instance, large genome-sized species (e.g., R. imitator and L. catesbeianus) and their ancestors had the lowest percentage of TEs state, with smaller genome-sized species (e.g., R. marina and B. gargarizans) and their ancestors having the most abundant TEs proportions. Recent studies suggest that low levels of piRNA silencing mechanisms may reduce the inhibition of TEs, leading to a burst of TEs [51]. We predict that the rate and accumulation of TEs expansion among the anuran genomes are closely related to the silencing mechanism of piRNAs; however, this requires further study.
Our results indicated no relationship between anuran genome size and habitat types, although large genome-sized species preferred humid habitats, whereas small genome-sized species frequently occur in arid habitats. Previous studies have shown that cell size and replication rates are influenced by genome size, leading to longer developmental times in species with larger genomes [40, 52–54]. For instance, spadefoot toads with smaller genomes exhibit significantly shorter development times. Subsequent studies have revealed a correlation between anuran habitats and their life history traits [50]. Factors such as temperature and high drought conditions can impact developmental cycles. Moreover, the genome size of anurans is closely linked to their developmental rate, with species possessing smaller genomes exhibiting shorter development periods [55, 56]. This can be attributed to the need for organisms under water shortage stress to complete their development quickly. The results we observed in this study are consistent with their conclusions [50].
In addition, previous studies have demonstrated a correlation between genome size and ecological breadth, whereby species with larger genomes tend to occupy a broader range within their ecological niche [57]. However, our analysis cannot test the hypothesis that ecological breadth contributes to genome size variation in anurans. Therefore, further studies are required to investigate the extent to which the ecological breadth of anurans can influence their genome size.
Limitations of this study
Although this study provides valuable insights into the molecular mechanisms underlying anuran genome size evolution, certain limitations should be noted. First, the quality of genome assembly can influence the estimation of TE content and abundance. While most of the 14 genomes were assembled at the chromosomal level with genomic completeness exceeding 90.0%, there are a few species, such as E. pustulosus, L. catesbeianus, and E. coqui, with relatively lower assembly quality (completeness of only 75.7%, 46.5%, and 76.4%, respectively) perhaps due to technological limitations as at the time of genome assembly [58]. Analyses showed that the anuran genome sizes used in our study do not negatively impact the genome completeness (Supplementary Materials 2: Fig. S7). Secondly, different evaluation methods or technical approaches used in genome assembly can lead to inconsistent genome size estimation. For example, in previous studies, the genome size of R. marina was assessed using either densitometry or flow cytometry analysis of stained nuclei in erythrocytes, hepatocytes and renal cells, resulting in a range of estimates from 3.98 to 5.65 Gb [59–66]. However, more recent studies on R. marina used short-read k-mer distributions and quantitative PCR (qPCR) of single-copy genes to evaluate the genome size resulting in size estimates ranging from 1.98 to 2.38 Gb [67]. These discrepancies highlight that the variability in genome size estimation could depend on the chosen methodology of genome assembly, thus underscoring the importance of careful consideration and validation when assessing genome size. While we took several steps to include high-quality genome assembly in our study and the robustness of our analyses, the patterns revealed in our study may, to some extent, reflect the genomic and ecological correlates of anuran genome size variation. This study advances our understanding of amphibian genome size evolution and sets the stage for further investigations.
Additionally, there may be some limitations in studying SSRs using whole-genome approaches. The low quality of a genome, such as the presence of gaps or assembly errors, can potentially result in the loss of simple sequence repeats (SSRs). Past studies have indicated that SSRs are present in both protein-coding and non-coding regions of eukaryotic genomes [68, 69]. Therefore, assembly errors or the presence of gaps can decrease the detection rate of SSRs. Based on the results presented above, although three species in this study had low genome assembly quality, the genomes of the remaining 11 species exhibited good assembly quality. Therefore, this study can provide a reliable conclusion. The identification and profiling of repeats can be affected by sequencing errors, misalignments, or insufficient coverage, leading to the potential misidentification of repetitive sequences by the algorithms [70–74]. The absence of haplotype information in genome assemblies poses a challenge in distinguishing between simple repeat polymorphisms occurring within haplotypes and between haplotypes. Studying simple repeats using genome assemblies instead of raw reads can yield valuable insights into repeat evolution and function. However, it is important to exercise caution when interpreting the results, considering the possible presence of assembly errors and incomplete representation.
Methods
Genome retrieval and quality assessment
We obtained published whole genome assemblies of anurans from the National Center of Biotechnology and Information, DRYAD, gigaDB and GSA databases (Table 1). In the case of NCBI genome assemblies, we searched using the taxonomy database (keywords “anurans” and “amphibians”). In sum, we retrieved whole genomes of 14 anuran species (Bufo gargarizans, Eleutherodactylus coqui, Engystomops pustulosus, Hymenochirus boettgeri, Limnodynastes dumerilii, Lithobates catesbeianus, Nanorana parkeri, Rana temporaria, Xenopus laevis, Xenopus tropicalis, Quasipaa spinosa, Rhinella marina, Spea multiplicata, and Ranitomeya imitator) belonging to nine families. Genome-wide data versions, release dates and database information are available in the supplementary material (Supplementary Materials 1: Table S1). To ensure the accuracy of our datasets, following the methods of Ibrahim et al. [75], we collected the information on the genome completeness data of 14 anuran species from the NCBI and relevant citations (referring to Benchmarking Universal Single-Copy Orthologs (BUSCO) assessment results [76]). This data includes information such as the number of scaffolds, scaffold N50, contigs, contig N50, genome assembly level, and genome size (referring to the assembly size) (Supplementary Materials 1: Table S1). For species without BUSCO assessment results, e.g., Lithobates catesbeianus, we conducted genome completeness using BUSCO v5.4.4 [76] based on the vertebrata_odb10 (Version 2021-02-19) database. Finally, we assessed if the genome size could negatively impact the genome completeness.
Table 1.
Sample information, including genome size and data sources
| Species | Genome size | References | Accession number | |
|---|---|---|---|---|
| Bufo gargarizans | 4.5 | [96] | GCF_014858855.1 | |
| Eleutherodactylus coqui | 2.8 | [97] | GCA_019857665.1 | |
| Engystomops pustulosus | 2.6 | [97] | GCA_019512145.1 | |
| Hymenochirus boettgeri | 3.2 | [97] | GCA_019447015.1 | |
| Limnodynastes dumerilii | 2.4 | [98] | GCA_011038615.1 | |
| Lithobates catesbeianus | 6.3 | [58] | GCA_002284835.2 | |
| Nanorana parkeri | 2.1 | [36] | GCA_000935625.1 | |
| Rana temporaria | 4.1 | [99] | GCA_905171775.1 | |
| Xenopus laevis | 2.7 | [100] | GCA_017654675.1 | |
| Xenopus tropicalis | 1.5 | [101] | GCA_000004195.4 | |
| Quasipaa spinosa | 2.6 | [102] | https://datadryad.org/stash/landing/show?id=doi%3A10.5061%2Fdryad.ghx3ffbpw | |
| Rhinella marina | 2.6 | [67] | http://gigadb.org/dataset/view/id/100483 | |
| Spea multiplicata | 1.1 | [103] | https://gsajournals.figshare.com/articles/dataset/Supplemental_Material_for_Seidl_et_al_2019/8303672?file=15558683 | |
| Ranitomeya imitator | 6.8 | [104] | GCA_905332335.1 | |
Repetitive element classification
We annotated the repetitive sequences using a de novo and homology-based method. First, we created a species-specific consensus sequence library using RepeatModeler v2.0.2 [77]. Next, we downloaded the Repbase [78] sequence library and the Dfam [79] dataset, merged the data generated by RepBase, Dfam, and RepeatModeler, and created the final repetitive sequence library. We then used RepeatMasker v4.1.2 [80] to identify the TEs and perform annotation. After that, we used linear regression analysis of annotations of the genome of the 14 species to estimate the level of association between the abundance of repetitive sequences and the genome size. To identify the tandem repeats, we used Tandem Repeats Finder v4.09 (default parameter) [81]. To further understand the relationship between simple sequence repeats (SSRs) and genome size, we used Krait [28] to identify SSRs in 14 anura genomes.
Dynamics and diversity analysis of the active TE
The diversity of TE in each species was measured using the Simpson’s and Shannon diversity indices. In Simpson’s diversity index, D = 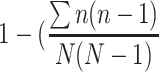 )[82], n represents the proportion of the genome occupied by each TE, and N represents the ratio of the sum of TEs to the size of the genome. In the Shannon diversity index
)[82], n represents the proportion of the genome occupied by each TE, and N represents the ratio of the sum of TEs to the size of the genome. In the Shannon diversity index 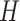 =
= 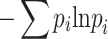 [83],
[83], 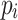 is the proportion of sequences belonging to TEs type i. This is similar to the case in ecological community diversity assessment where
is the proportion of sequences belonging to TEs type i. This is similar to the case in ecological community diversity assessment where 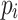 represents the number of individuals belonging to the same species. In the equations above, TE diversity was assessed at the level of TE subclasses, namely LTRs, LINEs, SINEs, and DNA transposons, considered as species. Finally, we analyzed the relationship between genome size and diversity indices using linear regression equations [84].
represents the number of individuals belonging to the same species. In the equations above, TE diversity was assessed at the level of TE subclasses, namely LTRs, LINEs, SINEs, and DNA transposons, considered as species. Finally, we analyzed the relationship between genome size and diversity indices using linear regression equations [84].
To understand the dynamics and activity of TEs in each species, we used a Perl script parseRM.pl (parameter -l 50,1 -v) [85] to parse the output file (.fa.out) from RepeatMasker. We then calculated the percentage of divergence of TEs consensus sequences. As TE proliferation may influence genome size evolution, we used Kimura distance as a measure to evaluate the historical dynamics of TE expansion in 14 species to uncover the process of genome size variation. Finally, we used histograms to show the percentage of divergence of consensus sequences belonging to the same TEs types.
Identification of the LTR-RTs
We implemented LTRharvest software [86] to identify intact LTR-RTs and analyze their impact on genome size and evolution. We run LTRharvest (parameter values -minlenltr 100 -maxlenltr 7000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis 1 -similar 85 -vic 10 -seed 20 -seqids yes) after creating the necessary files by Suffixerator (parameter values -tis -suf -lcp -des -ssp -sds -dna). The output file from LTRharvest was then inputted in LTR_retriever v 2.9.0 [87] using default settings for further analysis. As per the method described by Jukes et al. [88], we calculated the insertion time of LTR-RTs as T = k/2r, where k represents the divergence rate and a neutral mutation rate of 1.38 × 10^(-8) per site per year [89].
Microsatellite sequence analysis
Given that compound SSRs are more complex and derived from the recombination of homologous SSRs [90], we only considered P-SSRs and I-SSRs in our study. To identify and classify SSRs in the genome of each species, we used Krait v 1.3.3 [28] with default settings. After that, we calculated the relative abundance (total SSRs/total valid length), density (total SSR length/total valid length), and percentage of sequences covered by SSRs. Using linear regression equations, we further determined the relationship between genome size and the aforementioned indices [84].
Estimating the distribution of the non-coding regions of the genome
We used the methods of Francis and Worheide [47] to calculate the exonic and genic sequences in the genome of each species. We used a Python script (available at https://bitbucket.org/wrf/sequences/src/master/gtfstats.py) that takes genome and annotation files as input files. We then tallied the number of introns, exons, intronic gaps, intergenic regions, and intergenic gaps in each genome and estimated the average length of introns and exons. Finally, we employed linear regression [84] to evaluate the relationship between genome size and the number of exonic and genic sequences.
Phylogenetic relationships and ancestral state reconstruction
We estimated the phylogenetic relationships between the 14 anuran species using OrthoFinder v 2.5.4 [91]. OrthoFinder assigned 522,457 genes, accounting for 93.2% of the total, to 27,105 orthogroups. Among the identified orthogroups, 5338 contained genes from all species, and 9 of these orthogroups consisted solely of single-copy genes. Protein-coding sequences were then used to infer the species trees, with analysis performed using the Species Tree inference from All Genes (STAG) method [92] utilizing the 5338 orthogroups identified using OrthoFinder.The resulting trees were visualized using iTOL [93]. To better understand the evolution of ancestral genome size, we reconstructed the ancestral state of genome size and TE proportion using the Maximum Likelihood approach and the fastAnc function in the R package phytools [94]. To evaluate the relationships between the species-specific genome size and TEs, we obtained the evolutionary branch information on the clade of each species using Lifemap [95] and combined this information with the ancestral state. We finally visualized the trees using iTOL.
Evaluate the relationship between genome size and habitat
To understand the potential correlation between genome size and habitat diversity, we obtained habitat type data for each species from the IUCN archives (www.iucnredlist.org). Species with no detailed habitat type were removed in this analysis. We counted each species’ habitat type (Supplementary Materials 1: Table S6) and used a linear regression model [84] to assess the potential correlation between genome size and habitat diversity. In this analysis, habitat diversity is defined as the species’ niche width.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We are very grateful to Ya-li Wang for the technical services to support this study.
Abbreviations
- TEs
Transposable Elements
- LTRs
Long Terminal Repeat retrotransposons
- LINEs
Long Interspersed Nuclear Elements
- SINEs
Short Interspersed Nuclear Elements
- SSRs
Simple Repeat Sequences
- P-SSRs
Perfect SSRs
- C-SSRs
Compound SSRs
- I-SSRs
Compound SSRs
- C-SSRs
Imperfect SSRs
- MAY
million years ago
- IUCN
Species Tree inference from All Genes
- STAG
International Union for Conservation of Nature
- BUSCO
Benchmarking Universal Single-Copy Orthologs
Authors’ contributions
Conceptualization, Y-B.S.; methodology, B.Z., and Y-B.S.; validation, Y-B.S.; formal analysis, B.Z.; investigation, B.Z.; data curation, B.Z.; writing—original draft preparation, B.Z.; writing—review and editing, L.M.N. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Key Research Development Program of China (#2022YFF0802300), Yunnan Fundamental Research Projects (grant number 202101AV070009), and Natural Science Foundation of China (31871275).
Data Availability
The data and materials that support the findings of this study are available in the main text and supplementary material of this article.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gregory TR. Coincidence, coevolution, or causation? DNA content, cell size, and the C-value enigma. Biol Rev. 2001;76(1):65–101. doi: 10.1017/S1464793100005595. [DOI] [PubMed] [Google Scholar]
- 2.Gall JG. Chromosome structure and the C-value paradox. J Cell Biol. 1981;91(3 Pt 2):3s–14s. doi: 10.1083/jcb.91.3.3s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hahn MW, Wray GA. The g-value paradox. Evol Dev. 2002;4(2):73–5. doi: 10.1046/j.1525-142X.2002.01069.x. [DOI] [PubMed] [Google Scholar]
- 4.Elliott TA, Gregory TR. Do larger genomes contain more diverse transposable elements? Bmc Evol Biol 2015, 15. [DOI] [PMC free article] [PubMed]
- 5.Petrov DA. Evolution of genome size: new approaches to an old problem. Trends Genet. 2001;17(1):23–8. doi: 10.1016/S0168-9525(00)02157-0. [DOI] [PubMed] [Google Scholar]
- 6.Canapa A, Barucca M, Biscotti MA, Forconi M, Olmo E. Transposons, genome size, and Evolutionary Insights in animals. Cytogenet Genome Res. 2015;147(4):217–39. doi: 10.1159/000444429. [DOI] [PubMed] [Google Scholar]
- 7.Vitte C, Panaud O, Quesneville H. LTR retrotransposons in rice (Oryza sativa, L.): recent burst amplifications followed by rapid DNA loss. BMC Genomics. 2007;8(1):218. doi: 10.1186/1471-2164-8-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heckenhauer J, Frandsen PB, Sproul JS, Li Z, Paule J, Larracuente AM, Maughan PJ, Barker MS, Schneider JV, Stewart RJ et al. Genome size evolution in the diverse insect order Trichoptera. Gigascience 2022, 11. [DOI] [PMC free article] [PubMed]
- 9.Petrov DA, Sangster TA, Johnston JS, Hartl DL, Shaw KL. Evidence for DNA loss as a determinant of genome size. Science. 2000;287(5455):1060–2. doi: 10.1126/science.287.5455.1060. [DOI] [PubMed] [Google Scholar]
- 10.Neafsey DE, Palumbi SR. Genome size evolution in pufferfish: a comparative analysis of diodontid and tetraodontid pufferfish genomes. Genome Res. 2003;13(5):821–30. doi: 10.1101/gr.841703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun C, Shepard DB, Chong RA, Lopez Arriaza J, Hall K, Castoe TA, Feschotte C, Pollock DD, Mueller RL. LTR retrotransposons contribute to genomic gigantism in plethodontid salamanders. Genome Biol Evol. 2012;4(2):168–83. doi: 10.1093/gbe/evr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontdevila A. The dynamic genome: a darwinian Approach. Oxford University Press; 2011.
- 13.Lu J, Peatman E, Tang H, Lewis J, Liu Z. Profiling of gene duplication patterns of sequenced teleost genomes: evidence for rapid lineage-specific genome expansion mediated by recent tandem duplications. BMC Genomics. 2012;13(1):246. doi: 10.1186/1471-2164-13-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van De Peer Y, Mizrachi E, Marchal K. The evolutionary significance of polyploidy. Nat Rev Genet. 2017;18(7):411–24. doi: 10.1038/nrg.2017.26. [DOI] [PubMed] [Google Scholar]
- 15.Ohno S. Evolution by gene duplication. Berlin, New York: Springer-Verlag; 1970. [Google Scholar]
- 16.Lamichhaney S, Catullo R, Keogh JS, Clulow S, Edwards SV, Ezaz T. A bird-like genome from a frog: mechanisms of genome size reduction in the ornate burrowing frog, Platyplectrum ornatum. Proc Natl Acad Sci U S A 2021, 118(11). [DOI] [PMC free article] [PubMed]
- 17.Malmstrom M, Britz R, Matschiner M, Torresen OK, Hadiaty RK, Yaakob N, Tan HH, Jakobsen KS, Salzburger W, Ruber L. The most developmentally truncated fishes show extensive hox gene loss and miniaturized genomes. Genome Biol Evol. 2018;10(4):1088–103. doi: 10.1093/gbe/evy058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song X, Yang T, Zhang X, Yuan Y, Yan X, Wei Y, Zhang J, Zhou C. Comparison of the microsatellite distribution patterns in the Genomes of Euarchontoglires at the taxonomic level. Front Genet. 2021;12:622724. doi: 10.3389/fgene.2021.622724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding S, Wang S, He K, Jiang M, Li F. Large-scale analysis reveals that the genome features of simple sequence repeats are generally conserved at the family level in insects. BMC Genomics. 2017;18(1):848. doi: 10.1186/s12864-017-4234-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sotero-Caio CG, Platt RN, Suh A, Ray DA. Evolution and diversity of transposable elements in Vertebrate Genomes. Genome Biol Evol. 2017;9(1):161–77. doi: 10.1093/gbe/evw264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finnegan DJ. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989;5(4):103–7. doi: 10.1016/0168-9525(89)90039-5. [DOI] [PubMed] [Google Scholar]
- 22.Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8(12):973–82. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 23.Kapitonov VV, Jurka J. A universal classification of eukaryotic transposable elements implemented in repbase. Nat Rev Genet. 2008;9(5):411–2. doi: 10.1038/nrg2165-c1. [DOI] [PubMed] [Google Scholar]
- 24.Piégu B, Bire S, Arensburger P, Bigot Y. A survey of transposable element classification systems – A call for a fundamental update to meet the challenge of their diversity and complexity. Mol Phylogenet Evol. 2015;86:90–109. doi: 10.1016/j.ympev.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Beauregard A, Curcio MJ, Belfort M. The take and give between retrotransposable elements and their hosts. Annu Rev Genet. 2008;42:587–617. doi: 10.1146/annurev.genet.42.110807.091549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–68. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev. 1999;9(6):657–63. doi: 10.1016/S0959-437X(99)00031-3. [DOI] [PubMed] [Google Scholar]
- 28.Du L, Zhang C, Liu Q, Zhang X, Yue B, Hancock J. Krait: an ultrafast tool for genome-wide survey of microsatellites and primer design. Bioinformatics. 2018;34(4):681–3. doi: 10.1093/bioinformatics/btx665. [DOI] [PubMed] [Google Scholar]
- 29.Du L, Liu Q, Zhao K, Tang J, Zhang X, Yue B, Fan Z. PSMD: an extensive database for pan-species microsatellite investigation and marker development. Mol Ecol Resour. 2020;20(1):283–91. doi: 10.1111/1755-0998.13098. [DOI] [PubMed] [Google Scholar]
- 30.Song XH, Yang TB, Yan XH, Zheng FK, Xu XQ, Zhou CQ. Comparison of microsatellite distribution patterns in twenty-nine beetle genomes. Gene 2020, 757. [DOI] [PubMed]
- 31.Oliveira EJ, Padua JG, Zucchi MI, Vencovsky R, Vieira MLC. Origin, evolution and genome distribution of microsatellites. Genet Mol Biol. 2006;29(2):294–307. doi: 10.1590/S1415-47572006000200018. [DOI] [Google Scholar]
- 32.Zhang X, Yuan J, Sun Y, Li S, Gao Y, Yu Y, Liu C, Wang Q, Lv X, Zhang X, et al. Penaeid shrimp genome provides insights into benthic adaptation and frequent molting. Nat Commun. 2019;10(1):356. doi: 10.1038/s41467-018-08197-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan J, Zhang X, Wang M, Sun Y, Liu C, Li S, Yu Y, Gao Y, Liu F, Zhang X, et al. Simple sequence repeats drive genome plasticity and promote adaptive evolution in penaeid shrimp. Commun Biol. 2021;4(1):186. doi: 10.1038/s42003-021-01716-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olmo E, Morescalchi A. Genome and cell sizes in frogs: a comparison with salamanders. Experientia. 1978;34(1):44–6. doi: 10.1007/BF01921891. [DOI] [PubMed] [Google Scholar]
- 35.Seidl F, Levis NA, Schell R, Pfennig DW, Pfennig KS, Ehrenreich IM. Genome of Spea multiplicata, a rapidly developing, phenotypically plastic, and Desert-Adapted Spadefoot Toad. G3 (Bethesda) 2019;9(12):3909–19. doi: 10.1534/g3.119.400705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun YB, Xiong ZJ, Xiang XY, Liu SP, Zhou WW, Tu XL, Zhong L, Wang L, Wu DD, Zhang BL, et al. Whole-genome sequence of the tibetan frog Nanorana parkeri and the comparative evolution of tetrapod genomes. P Natl Acad Sci USA. 2015;112(11):E1257–62. doi: 10.1073/pnas.1501764112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun YB, Zhang Y, Wang K. Perspectives on studying molecular adaptations of amphibians in the genomic era. Zoological Res. 2020;41(4):351–64. doi: 10.24272/j.issn.2095-8137.2020.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J, Itgen MW, Wang H, Gong Y, Jiang J, Li J, Sun C, Sessions SK, Mueller RL. Gigantic genomes provide empirical tests of transposable element Dynamics Models. Genomics Proteom Bioinf. 2021;19(1):123–39. doi: 10.1016/j.gpb.2020.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haley AL, Mueller RL. Transposable element diversity remains high in gigantic genomes. J Mol Evol. 2022;90(5):332–41. doi: 10.1007/s00239-022-10063-3. [DOI] [PubMed] [Google Scholar]
- 40.Lertzman-Lepofsky G, Mooers AO, Greenberg DA. Ecological constraints associated with genome size across salamander lineages. P Roy Soc B-Biol Sci 2019, 286(1911). [DOI] [PMC free article] [PubMed]
- 41.Hardie DC, Hebert PDN. Genome-size evolution in fishes. Can J Fish Aquat Sci. 2004;61(9):1636–46. doi: 10.1139/f04-106. [DOI] [Google Scholar]
- 42.Waltari E, Edwards SV. Evolutionary dynamics of intron size, genome size, and physiological correlates in archosaurs. Am Nat. 2002;160(5):539–52. doi: 10.1086/342079. [DOI] [PubMed] [Google Scholar]
- 43.Gregory TR. Genome size and developmental complexity. Genetica. 2002;115(1):131–46. doi: 10.1023/A:1016032400147. [DOI] [PubMed] [Google Scholar]
- 44.Wright NA, Gregory TR, Witt CC. Metabolic ‘engines’ of flight drive genome size reduction in birds. P Roy Soc B-Biol Sci 2014, 281(1779). [DOI] [PMC free article] [PubMed]
- 45.Weber JA, Park SG, Luria V, Jeon S, Kim HM, Jeon Y, Bhak Y, Jun JH, Kim SW, Hong WH, et al. The whale shark genome reveals how genomic and physiological properties scale with body size. P Natl Acad Sci USA. 2020;117(34):20662–71. doi: 10.1073/pnas.1922576117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petrov DA. Mutational equilibrium model of genome size evolution. Theor Popul Biol. 2002;61(4):531–44. doi: 10.1006/tpbi.2002.1605. [DOI] [PubMed] [Google Scholar]
- 47.Francis WR, Worheide G. Similar ratios of introns to intergenic sequence across animal genomes. Genome Biol Evol. 2017;9(6):1582–98. doi: 10.1093/gbe/evx103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lyu HM, He ZW, Wu CI, Shi SH. Convergent adaptive evolution in marginal environments: unloading transposable elements as a common strategy among mangrove genomes. New Phytol. 2018;217(1):428–38. doi: 10.1111/nph.14784. [DOI] [PubMed] [Google Scholar]
- 49.Cong Y, Ye X, Mei Y, He K, Li F. Transposons and non-coding regions drive the intrafamily differences of genome size in insects. iScience. 2022;25(9):104873. doi: 10.1016/j.isci.2022.104873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liedtke HC, Gower DJ, Wilkinson M, Gomez-Mestre I. Macroevolutionary shift in the size of amphibian genomes and the role of life history and climate. Nat Ecol Evol. 2018;2(11):1792–9. doi: 10.1038/s41559-018-0674-4. [DOI] [PubMed] [Google Scholar]
- 51.Liu X, Majid M, Yuan H, Chang H, Zhao L, Nie Y, He L, Liu X, He X, Huang Y. Transposable element expansion and low-level piRNA silencing in grasshoppers may cause genome gigantism. BMC Biol. 2022;20(1):243. doi: 10.1186/s12915-022-01441-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olmo E, Capriglione T, Odierna G. Genome size evolution in vertebrates: trends and constraints. Comp Biochem Physiol B. 1989;92(3):447–53. doi: 10.1016/0305-0491(89)90115-6. [DOI] [PubMed] [Google Scholar]
- 53.Pagel M, Johnstone RA. Variation across species in the size of the Nuclear Genome supports the Junk-DNA explanation for the C-Value Paradox. P Roy Soc B-Biol Sci. 1992;249(1325):119–24. doi: 10.1098/rspb.1992.0093. [DOI] [PubMed] [Google Scholar]
- 54.Jockusch EL. An evolutionary correlate of genome size change in Plethodontid Salamanders. Proceedings: Biol Sci. 1997;264(1381):597–604. [Google Scholar]
- 55.Wyngaard GA, Rasch EM, Manning NM, Gasser K, Domangue R. The relationship between genome size, development rate, and body size in copepods. Hydrobiologia. 2005;532:123–37. doi: 10.1007/s10750-004-9521-5. [DOI] [Google Scholar]
- 56.White MM, McLaren IA. Copepod development rates in relation to genome size and 18S rDNA copy number. Genome. 2000;43(5):750–5. doi: 10.1139/g00-048. [DOI] [PubMed] [Google Scholar]
- 57.dos Santos EV, Martinez PA, Souza G, Jacobina UP. Genome size drives ecological breadth in Pomacentridae reef fishes. J Exp Mar Biol Ecol 2021, 540.
- 58.Hammond SA, Warren RL, Vandervalk BP, Kucuk E, Khan H, Gibb EA, Pandoh P, Kirk H, Zhao YJ, Jones M et al. The north american bullfrog draft genome provides insight into hormonal regulation of long noncoding RNA. Nat Commun 2017, 8. [DOI] [PMC free article] [PubMed]
- 59.Vinogradov AE. Genome size and GC-percent in vertebrates as determined by flow cytometry: the triangular relationship. Cytometry. 1998;31(2):100–9. doi: 10.1002/(SICI)1097-0320(19980201)31:2<100::AID-CYTO5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 60.Bachmann K. Specific nuclear DNA amounts in toads of the genus Bufo. Chromosoma. 1970;29(3):365–74. doi: 10.1007/BF00325949. [DOI] [PubMed] [Google Scholar]
- 61.Bachmann K. Nuclear DNA and developmental rate in Frogs. Q J Fla Acad Sci. 1972;35(4):225–31. [Google Scholar]
- 62.Chipman AD, Khaner O, Haas A, Tchernov E. The evolution of genome size: what can be learned from anuran development? 2001, 291(4):365–74. [DOI] [PubMed]
- 63.Griffin CS, Scott D, Papworth DG. The influence of DNA content and nuclear volume on the frequency of radiation-induced chromosome aberrations in Bufo species. Chromosoma. 1970;30(2):228–49. doi: 10.1007/BF00282003. [DOI] [PubMed] [Google Scholar]
- 64.Goin OB, Goin CJ, Bachmann K. DNA and Amphibian Life History. Copeia. 1968;1968(3):532–40. doi: 10.2307/1442021. [DOI] [Google Scholar]
- 65.MacCulloch RD, Upton DE, Murphy RW. Trends in nuclear DNA content among amphibians and reptiles. Comp Biochem Phys B. 1996;113(3):601–5. doi: 10.1016/0305-0491(95)02033-0. [DOI] [Google Scholar]
- 66.Camper J, Ruedas L, Bickham J, Dixon JJG. The relationship of genome size with developmental rates and reproductive strategies in five families of neotropical bufonoid frogs. 1993, 12:79–87.
- 67.Edwards RJ, Tuipulotu DE, Amos TG, O’Meally D, Richardson MF, Russell TL, Vallinoto M, Carneiro M, Ferrand N, Wilkins MR et al. Draft genome assembly of the invasive cane toad, Rhinella marina. Gigascience 2018, 7(9). [DOI] [PMC free article] [PubMed]
- 68.Morgante M, Hanafey M, Powell W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat Genet. 2002;30(2):194–200. doi: 10.1038/ng822. [DOI] [PubMed] [Google Scholar]
- 69.Li YC, Korol AB, Fahima T, Nevo E. Microsatellites within genes: structure, function, and evolution. Mol Biol Evol. 2004;21(6):991–1007. doi: 10.1093/molbev/msh073. [DOI] [PubMed] [Google Scholar]
- 70.Thiel T, Michalek W, Varshney RK, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L) Theor Appl Genet. 2003;106(3):411–22. doi: 10.1007/s00122-002-1031-0. [DOI] [PubMed] [Google Scholar]
- 71.Kofler R, Schlotterer C, Lelley T. SciRoKo: a new tool for whole genome microsatellite search and investigation. Bioinformatics. 2007;23(13):1683–5. doi: 10.1093/bioinformatics/btm157. [DOI] [PubMed] [Google Scholar]
- 72.Faircloth BC. MSATCOMMANDER: detection of microsatellite repeat arrays and automated, locus-specific primer design. Mol Ecol Resour. 2008;8(1):92–4. doi: 10.1111/j.1471-8286.2007.01884.x. [DOI] [PubMed] [Google Scholar]
- 73.Du LM, Li YZ, Zhang XY, Yue BS. MSDB: a user-friendly program for reporting distribution and building databases of microsatellites from genome sequences. J Hered. 2013;104(1):154–7. doi: 10.1093/jhered/ess082. [DOI] [PubMed] [Google Scholar]
- 74.Wang XW, Wang L. GMATA: an Integrated Software Package for genome-scale SSR mining, marker development and VIewing. Front Plant Sci 2016, 7. [DOI] [PMC free article] [PubMed]
- 75.Ibrahim MA, Al-Shomrani BM, Simenc M, Alharbi SN, Alqahtani FH, Al-Fageeh MB, Manee MM. Comparative analysis of transposable elements provides insights into genome evolution in the genus Camelus. BMC Genomics. 2021;22(1):842. doi: 10.1186/s12864-021-08117-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 77.Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF. RepeatModeler2 for automated genomic discovery of transposable element families. P Natl Acad Sci USA. 2020;117(17):9451–7. doi: 10.1073/pnas.1921046117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1–4):462–7. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 79.Hubley R, Finn RD, Clements J, Eddy SR, Jones TA, Bao WD, Smit AFA, Wheelers TJ. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016;44(D1):D81–9. doi: 10.1093/nar/gkv1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics / editoral board, Andreas D Baxevanis [] 2009, Chap. 4:Unit 4.10. [DOI] [PubMed]
- 81.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Simpson EH. Measurement of Diversity. Nature. 1949;163(4148):688–8. doi: 10.1038/163688a0. [DOI] [Google Scholar]
- 83.Shannon CE. A Mathematical Theory of Communication. 1948, 27(3):379–423.
- 84.Li JF, Miao BB, Wang SX, Dong W, Xu HS, Si CC, Wang W, Duan SQ, Lou JC, Bao ZW et al. Hiplot: a comprehensive and easy-to-use web service for boosting publication-ready biomedical data visualization. Brief Bioinform 2022, 23(4). [DOI] [PubMed]
- 85.Kapusta A, Suh A, Feschotte C. Dynamics of genome size evolution in birds and mammals. Proc Natl Acad Sci U S A. 2017;114(8):E1460–9. doi: 10.1073/pnas.1616702114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ellinghaus D, Kurtz S, Willhoeft U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics. 2008;9:18. doi: 10.1186/1471-2105-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ou S, Jiang N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat Retrotransposons. Plant Physiol. 2018;176(2):1410–22. doi: 10.1104/pp.17.01310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jukes T, Cantor C. Evolution of protein molecules. In Munro, H. N, editor. In., vol. 3; 1969: 21–132.
- 89.Ma J, Bennetzen JL. Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci U S A. 2004;101(34):12404–10. doi: 10.1073/pnas.0403715101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ledenyova ML, Tkachenko GA, Shpak IM. Imperfect and compound microsatellites in the Genomes of Burkholderia pseudomallei strains. Mol Biol + 2019;53(1):127–37. doi: 10.1134/S0026893319010084. [DOI] [PubMed] [Google Scholar]
- 91.Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20(1):238. doi: 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Emms DM, Kelly S. STAG: Species Tree Inference from All Genes. 2018:267914.
- 93.Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–6. doi: 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Revell LJ. Phytools: an R package for phylogenetic comparative biology (and other things) Methods Ecol Evol. 2012;3(2):217–23. doi: 10.1111/j.2041-210X.2011.00169.x. [DOI] [Google Scholar]
- 95.de Vienne DM. Lifemap: exploring the entire tree of life. PLoS Biol. 2016;14(12):e2001624. doi: 10.1371/journal.pbio.2001624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lu B, Jiang J, Wu H, Chen X, Song X, Liao W, Fu J. A large genome with chromosome-scale assembly sheds light on the evolutionary success of a true toad (Bufo gargarizans) Mol Ecol Resour. 2021;21(4):1256–73. doi: 10.1111/1755-0998.13319. [DOI] [PubMed] [Google Scholar]
- 97.Bredeson J, Mudd A, Medina S, Mitros T, Smith O, Miller K, Lyons J, Batra S, Park J, Berkoff K et al. Conserved chromatin and repetitive patterns reveal slow genome evolution in frogs; 2021. [DOI] [PMC free article] [PubMed]
- 98.Li Q, Qunfei G, Zhou Y, Tan H, Bertozzi T, Zhu Y, Li J, Donnellan S, Zhang G. A draft genome assembly of the eastern banjo frog Limnodynastes dumerilii dumerilii. Anura: Limnodynastidae); 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Streicher JW. Wellcome Sanger Institute Tree of Life p, Wellcome Sanger Institute Scientific Operations DNAPc, Tree of Life Core Informatics c, Darwin Tree of Life C: the genome sequence of the common frog, Rana temporaria Linnaeus 1758. Wellcome Open Res. 2021;6:286. doi: 10.12688/wellcomeopenres.17296.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Session AM, Uno Y, Kwon T, Chapman JA, Toyoda A, Takahashi S, Fukui A, Hikosaka A, Suzuki A, Kondo M, et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature. 2016;538(7625):336–43. doi: 10.1038/nature19840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mitros T, Lyons JB, Session AM, Jenkins J, Shu S, Kwon T, Lane M, Ng C, Grammer TC, Khokha MK, et al. A chromosome-scale genome assembly and dense genetic map for Xenopus tropicalis. Dev Biol. 2019;452(1):8–20. doi: 10.1016/j.ydbio.2019.03.015. [DOI] [PubMed] [Google Scholar]
- 102.Hu XX, Jiang ZY, Ming Y, Jian JB, Jiang SJ, Zhang DD, Zhang JY, Zheng SJ, Fang XD, Yang YL, et al. A chromosomal level genome sequence for Quasipaa spinosa (Dicroglossidae) reveals chromosomal evolution and population diversity. Mol Ecol Resour. 2022;22(4):1545–58. doi: 10.1111/1755-0998.13560. [DOI] [PubMed] [Google Scholar]
- 103.Seidl F, Levis NA, Schell R, Pfennig DW, Pfennig KS, Ehrenreich IM. Genome of Spea multiplicata, a rapidly developing, phenotypically plastic, and Desert-Adapted Spadefoot Toad. G3-Genes Genom Genet. 2019;9(12):3909–19. doi: 10.1534/g3.119.400705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stuckert AMM, Chouteau M, McClure M, LaPolice TM, Linderoth T, Nielsen R, Summers K, MacManes MD. The genomics of mimicry: gene expression throughout development provides insights into convergent and divergent phenotypes in a Müllerian mimicry system. 2021, 30(16):4039–61. [DOI] [PMC free article] [PubMed] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and materials that support the findings of this study are available in the main text and supplementary material of this article.