

ABSTRACT
The commentary by Colombo and Rich recently published in Cancer Cell provides a timely and comprehensive review of the clinical maximum tolerated doses (MTDs) of antibody−drug conjugates (ADCs) and their corresponding small molecules/chemotherapies. The authors identified similarities between their MTDs and therefore question the historic assumptions made for ADCs, namely, that they increase the MTDs of their corresponding cytotoxic molecules. However, the authors did not address the superior anti-tumor responses of ADCs compared to their corresponding chemotherapies, as reported in clinical trials. In this point of view, we propose a revised model wherein the anti-tumor activities of ADCs and consequently their therapeutic indexes (TIs) are not solely associated with changes not only in their MTDs but also in their minimal effective doses (MEDs). In addition, when using an exposure-based TI calculation method, the superior anti-tumor activities of ADCs relative to their corresponding chemotherapy can readily be explained. We discussed the clinical and preclinical data in support of lower MEDs of ADCs and generated a revised graph illustrating the TI improvements of ADCs vs chemotherapy more accurately. We believe that our revised model can provide a blueprint for future improvements in protein engineering and chemical engineering of toxins to further advance ADC research and development.
KEYWORDS: Antibody-drug conjugates; oncology drug development, linker payload development; pharmacology; protein engineering; therapeutic index; toxicology; translational studies
Introduction
Antibody−drug conjugates (ADCs) have regained interest by drug developers based on the recent clinical successes as single agents or in combination with chemotherapies or checkpoint inhibitors, resulting in improved objective response rates and durability of responses in both liquid and solid tumor indications. Overall, a total of over 12 ADC have been approved, six of which were approved in the last 2 years (reviewed in Ref. 1). Some of the key lessons learned when developing first-generation ADCs with various payloads, linkers and conjugation methods, are summarized below:
While ADCs are generally perceived to broaden the therapeutic index (TI) of their payloads, the clinical data suggest that the maximum tolerated doses (MTDs) of payloads and conjugates (when dose normalized to their payloads) remained comparable.2
The MTDs of ADCs are frequently caused by payload-mediated, non-target related, platform toxicities outside tumor tissues, which are unique for each linker-payload type. The mechanisms affecting these MTDs are under intense investigation and are known as off-target, off-tumor toxicities.3,4
On-target toxicities of ADCs can become dose limiting in the clinic when choosing a target with low to moderate expression levels on normal tissues, such as TROP2,5 EpCAM,6 EphA2,7 and other targets (reviewed in Ref. 8). These MTDs are qualified as on-target, off- tumor toxicities of ADCs.
A variety of conditional ADC approaches, designed to render target antigen binding or payload release contingent on the presence of tumor environment-specific triggers, including pH changes or protease activities, have not yet yielded clinical success (reviewed in Ref. 9).
The transfer of maleimide-based maleimidocaproyl (mc) linkers from ADCs to human serum albumin (retro-Michael reaction) can contribute to anti-tumor activities of ADCs10 (reviewed in Ref. 11).
The levels of released, circulatory payloads/toxins can contribute to both platform toxicities and anti-tumor activities of ADCs.2
ADCs, in general, induce superior anti-tumor activities, both clinically and preclinically, compared to their corresponding small molecules/toxins when administered at equimolar levels.12,13
To provide more context on the relationship between MTDs and TIs of ADCs, we would like to point out that while both MTD and minimum effective dose (MED) are generally used to graphically highlight the magnitude of the TI improvements of ADCs, they are not part of the recommended methods used to calculate TIs, for neither ADCs nor small molecules.14 By definition, TI calculations are based on ADC exposure levels and represent the ratio of the highest exposure to the drug, resulting in minimal toxicity, relative to the drug exposure that produces the desired anti-tumor effects.15
When comparing small-molecule chemotherapeutics with their corresponding ADCs, it is important to understand the difference in the exposure kinetics between the two. For example, the systemic exposure of cytotoxic drugs is measured in minutes to hours, while the exposure of ADCs is measured in days. Hence, for an ADC with a half-life of 4 to 5 days, the systemic exposure of the conjugated toxin is much higher than the corresponding, small-molecule chemotherapy, irrespective of the notion that the administered dose, after standardization to the small-molecule compound, is roughly equivalent.2
Small-molecule chemical payloads distribute preferentially into highly perfused normal tissues and tumors during their short exposure time window, causing systemic toxicity, which often requires prolonged recovery periods of several weeks, known as drug holidays, during repeat dose cycles. Drug holidays reduce the efficacy of anticancer drugs and can lead to treatment resistance. A narrow focus on MTDs between ADCs and their small-molecule cytotoxins disregards some of these fundamental differences between both modalities. In this context, it is not surprising that the improved anti-tumor activities of ADCs compared to their corresponding cytotoxic drugs do not correlate with their MTDs, and thus, other mechanisms account for the differences in their efficacy and TIs.
We propose that an exposure-based model to calculate TIs is more suitable not only to explain the TI differences between both modalities but also to better understand the improvements in ADC platforms and to inform the selection of clinical ADC candidates. In this model, the pharmacokinetic and pharmacodynamic (PK/PD) parameters are based on drug exposure levels over time, e.g., area under the curve (AUC) and maximum drug concentration (Cmax), as opposed to MTDs or minimal effective doses (MEDs).14,16 In addition, inter-individual variability in drug exposure caused by factors such as genetic polymorphisms of drug metabolizing enzymes, efflux pumps, drug−drug interactions, differences in target antigen expression, body weights, disease, or environmental factors is better accounted for in an exposure-based TI calculation model. Finally, different regimens of a given dose can produce different TIs for the same ADC. For instance, a single dose can be divided into multiple lower doses that yield the same cumulative dose per cycle. An example is the dose fractionation method that was successfully applied for the CD33-ADC Mylotarg in acute myeloid leukemia (AML) patients, where the same total dose administered in three smaller doses improved overall responses and reduced platform toxicities markedly,17 leading to a higher TI value. Such fractionated dose schedules result in a similar AUC and a lower Cmax and therefore improve the TIs of ADCs with Cmax-driven toxicities,18 which cannot be explained by ADC dose levels alone.
In summary, when applying exposure-based TI calculation methods, the superior anti-tumor activities of ADCs relative to their corresponding small molecules can readily be explained by higher intratumoral exposure levels of the toxins as a result of the more tumor-specific uptake of the toxin when delivered in the ADC format. In addition to such payload-mediated anti-tumor activities, the antibody moiety can provide additional anti-tumor activities via activation of immune effector cells such as macrophages (antibody-dependent cellular phagocytosis), dendritic- and natural killer cells (antibody-dependent cellular toxicity) or complement activation (complement-dependent cytotoxicity) (reviewed in Ref. 19), resulting in lower MEDs and improved TIs relative to the corresponding chemotherapy, as illustrated in Figure 1.
Figure 1.
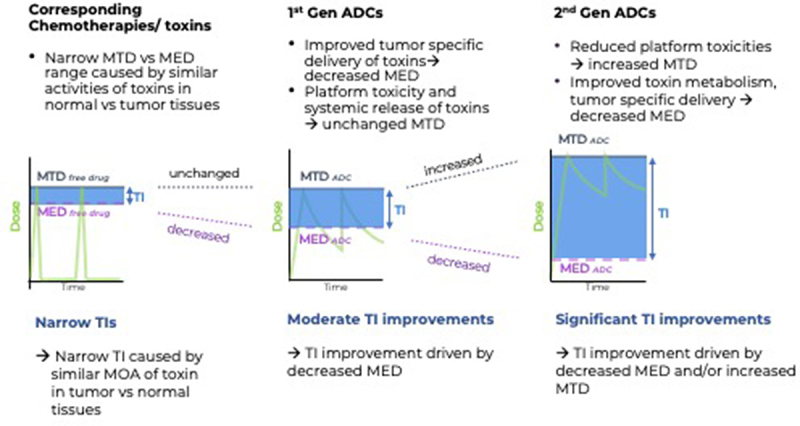
Revised model to illustrate TI improvements of ADCs vs their corresponding chemotherapies.
Left panel: Narrow TIs caused by low tumor/normal tissue selectivity of toxins. Examples of clinical toxicities caused by chemotherapies: Tubulin Inhibitors: neutropenia. Topo1 inhibitors: myelosuppression and GI-tox. Topo2 inhibitors: cardiac- and GI (gastrointestinal) tox. DNA Alkylators/PBD: Bone marrow and kidney toxicity. Calicheamicin: thrombocytopenia
Middle panel: Moderate TI improvements by first-generation ADCs caused by platform toxicities, on-target, off-tumor toxicity or suboptimal PK of ADCs. Examples of MTD caused by ADC platform toxicities: Examples of on-target, off-tumor toxicities caused by low tumor to normal expression of tumor antigens: TROP2, EphA2. Examples for suboptimal PK properties of ADC limiting the MTDs: stochastic lysine conjugation of Calicheamicin and FcRn interference, retro-Michael reaction of maleimide-based cysteine conjugates.
Right panel: Significant TI improvements caused by protein engineering and chemical engineering across ADC platforms: Examples: (1) linker engineering to minimize hydrophobicity of payloads, (2) chemical engineering of payloads toward less toxic compounds after release in circulation, (3) antibody engineering to enable site-specific conjugation to reduce off-tumor linker cleavage, (4) reducing ADC hydrophobicity, interference with FcRn recycling and Fcγ receptor binding and PK optimization, and (5) conditional ADC platforms with the goal of increasing tumor-specific activities of ADCs.
In contrast, administration of a small-molecule chemotherapeutic results in a higher Cmax, with rapid clearance and short elimination half-life (minutes to hours), as shown in the left panel in Figure 1. The more controlled and targeted delivery of the payload to the tumor by ADCs leads to prolonged tumor exposure of the small-molecule drug, taking on the kinetics of the large molecule, translating into decreased MEDs (middle and right panels in Figure 1). Conceptually, the MED of the payload leading to tumor cell death cannot be lowered without delivering more of the same payload into tumors. Such increased tumor-specific delivery of cytotoxic payloads via ADCs is one of the many reasons for their improved efficacy compared to their free payloads and explains their use in early-line treatments of solid tumors, replacing standard-of-care chemotherapy.
Importantly, the potential for ADCs to lower the MEDs of their corresponding free toxins is not discussed in the paper by Colombo and Rich. Since TI improvements can result in either decreased MEDs or increased MTDs, a lower MED of ADCs seems to be the more likely explanation for their superior anti-tumor activities in face of the similarities in their MTDs, as identified by the authors.
There is an abundance of preclinical evidence in support of lower MEDs of ADCs. In preclinical efficacy studies in tumor-bearing mice, inferior anti-tumor responses were observed consistently in control groups where an equivalent dose of toxins was administered, compared to the corresponding ADCs.12,13 The same principle likely applies in cancer patients. However, when reviewing the literature for MEDs of ADCs, we noticed a lack of standard methods to report MEDs.
This situation will likely change in the future consistent with FDA’s Project Optimus initiative (www.fda.gov/about-fda/oncology-center-excellence/project-optimus), but because of the current lack of consistency in MED reporting for ADCs in the clinic and the abundance of evidence for lower MEDs of ADCs in pre-clinical studies, we posit that the most appropriate explanation for the superior anti-tumor efficacy of ADCs and TIs is their relative lower MEDs compared to their corresponding chemotherapies. Therefore, we propose to revise the standard model used to illustrate TI improvement of ADCs vs chemotherapy as outlined in Figure 1. The revised figure displays similar MTD levels between chemotherapy and the first-generation ADC, reflective of the findings by Colombo and Rich. To account for the TI improvements of ADCs and their improved anti-tumor activities compared to the corresponding chemotherapies dosed at similar MTDs, we display lower MED levels for first-generation ADCs in the revised model.
Importantly, we added a third panel to Figure 1 for the TI improvements of second-generation ADCs, with increased MTDs and/or lower MEDs relative to first-generation ADCs. An example for a second-generation ADC is a site-specific conjugate, where conjugation to carefully selected sites on the heavy and light chain of the antibody resulted in an almost complete elimination of the main platform toxicity, neutropenia. This was associated with increased ADC exposure levels and improved ADC/Ab ratios in sera of non-human primates, compared to the conventional, non-site specific conjugate.20 When using an exposure-based TI calculation model, the selection of 4 optimized conjugation sites, two on the light chain and two on the heavy chain, translated to an over 10-fold TI improvement compared to the random conjugation of the same linker payload to 4 cysteines on the same antibody.16,20 While publication of the final results of the Phase 1 dose-escalation study including patients with HER2-positive breast and gastroesophageal tumors progressing on several lines of prior treatment is awaited, the preliminary objective response rate (54% objective response rate (ORR)) and the MTD (>3 mg/kg)21 compare favorably to a conventional Her2 conjugate (RC-48 ADC, 30.8% ORR, MTD 2.0 mg/kg), with a very similar linker payload.22 Another example of TI-improved ADCs is based on chemical engineering of the topoisomerase 1 (Topo1i) payload exatecan and its cleavable linker, resulting in the Her2-DXd conjugate Enhertu (trastuzumab deruxtecan), with enhanced payload release in tumor cells and decreased payload toxicities in normal tissues, following its release in the circulation.23 Of note, potential changes in MED or MTD of DXd- conjugates could not be captured in the study by Columbo et al, because the DXd toxins/chemotherapies have not yet been tested independently in clinical trials. However, in a head-to-head Phase 3 trial in Her2+ mBC (DESTINY-Breast03), this second-generation ADC significantly improved progression-free survival (PFS) over the first-generation trastuzumab emtansine, with an ORR of 79.9% vs 34.2%, respectively.
In conclusion, by modifying the standard illustration for TI improvements of ADCs over chemotherapy, as shown in Figure 1, and employing an exposure-based TI calculation approach, we can readily explain the improvement in anti-tumor activities of TI improved ADCs and capture the emerging success of second-generation ADCs in the clinic, including Enhertu.23 We also believe that the revised illustration and the exposure-based TI calculation method will help to guide engineering principles for future generations of ADCs with the goal of further improving their TIs to optimize the benefit of cancer patients.
Acknowledgments
We would like to thank Deepa Pakianathan, Morris Rosenberg and Suzy Jones for the comments and suggestions for the manuscript.
Funding Statement
The author(s) reported that there is no funding associated with the work featured in this article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Abbreviations
- ADC
Antibodydrug conjugates
- MTD
Maximum tolerated dose
- TI
Therapeutic index
- MED
Minimal effective dose
- PK
Pharmacokinetic
- PD
Pharmacodynamic
- Cmax
Maximum drug concentration
- AUC
Area under the curve
- Ab
Antibody
- ORR
Objective response rate
- DXd
Deruxtecan
- Mbc
Metastatic breast cancer
- PFS
Progression-free survival
- Topo1
Topoisomerase 1
- GI
Gastrointestinal
- PBD
Pyrrolobenzodiazepine
- FcRn
Neonatal Fc receptor
- FcγR
Fc-gamma receptor
References
- 1.Fuentes-Antras J, Genta S, Vijenthira A, Siu LL.. Antibody–drug conjugates: in search of partners of choice. Trends Cancer. 2023;9(4):339–4. doi: 10.1016/j.trecan.2023.01.003. PMID: 36746689. [DOI] [PubMed] [Google Scholar]
- 2.Colombo R, Rich JR. The therapeutic window of antibody drug conjugates: a dogma in need of revision. Cancer Cell. 2022;40(11):1255–63. doi: 10.1016/j.ccell.2022.09.016. PMID: 36240779. [DOI] [PubMed] [Google Scholar]
- 3.Mahalingaiah PK, Ciurlionis R, Durbin KR, Yeager RL, Philip BK, Bawa B, Mantena SR, Enright BP, Liguori MJ, Van Vleet TR. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacology & Therapeutics. 2019;200:110–25. doi: 10.1016/j.pharmthera.2019.04.008. PMID: 31028836. [DOI] [PubMed] [Google Scholar]
- 4.Saber H, Leighton JK. An FDA oncology analysis of antibody-drug conjugates. Regul Toxicol Pharmacol. 2015;71:444–52. doi: 10.1016/j.yrtph.2015.01.014. PMID: 25661711. [DOI] [PubMed] [Google Scholar]
- 5.King GT, Eaton KD, Beagle BR, Zopf CJ, Wong GY, Krupka HI, Hua SY, Messersmith WA, El-Khoueiry AB. A phase 1, dose-escalation study of PF-06664178, an anti-Trop-2/Aur0101 antibody-drug conjugate in patients with advanced or metastatic solid tumors. Invest New Drugs. 2018;36(5):836–47. doi: 10.1007/s10637-018-0560-6. PMID: 29333575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munz M, Murr A, Kvesic M, Rau D, Mangold S, Pflanz S, Lumsden J, Volkland J, Fagerberg J, Riethmuller G, et al. Side-by-side analysis of five clinically tested anti-EpCAM monoclonal antibodies. Cancer Cell Int. 2010;10(1):44. doi: 10.1186/1475-2867-10-44. PMID: 21044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Annunziata CM, Kohn EC, LoRusso P, Houston ND, Coleman RL, Buzoianu M, Robbie G, Lechleider R. Phase 1, open-label study of MEDI-547 in patients with relapsed or refractory solid tumors. Invest New Drugs. 2013;31(1):77–84. doi: 10.1007/s10637-012-9801-2. PMID: 22370972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Criscitiello C, Morganti S, Curigliano G. Antibody–drug conjugates in solid tumors: a look into novel targets. J Hematol Oncol. 2021;14(1):20. doi: 10.1186/s13045-021-01035-z. PMID: 33509252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Nguyen AW, Maynard JA. Engineering antibodies for conditional activity in the solid tumor microenvironment. Curr Opin Biotechnol. 2022;78:102809. doi: 10.1016/j.copbio.2022.102809. PMID: 36182870. [DOI] [PubMed] [Google Scholar]
- 10.Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, Senter PD. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem. 2008;19(3):759–65. doi: 10.1021/bc7004329. PMID: 18314937. [DOI] [PubMed] [Google Scholar]
- 11.Shen X, Liu X, Li T, Chen Y, Chen Y, Wang P, Zheng L, Yang H, Wu C, Deng S, et al. Recent advancements in serum albumin-based nanovehicles toward potential cancer diagnosis and therapy. Front Chem. 2021;9:746646. doi: 10.3389/fchem.2021.746646. PMID: 34869202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haratani K, Yonesaka K, Takamura S, Maenishi O, Kato R, Takegawa N, Kawakami H, Tanaka K, Hayashi H, Takeda M, et al. U3-1402 sensitizes HER3-expressing tumors to PD-1 blockade by immune activation. J Clin Invest. 2020;130:374–88. doi: 10.1172/JCI126598. PMID: 31661465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharkey MS, Lizee G, Gonzales MI, Patel S, Topalian SL. CD4(+) T-cell recognition of mutated B-RAF in melanoma patients harboring the V599E mutation. Cancer Research. 2004;64(5):1595–99. https://www.ncbi.nlm.nih.gov/pubmed/14996715. PMID: 14996715. [DOI] [PubMed] [Google Scholar]
- 14.Haddish-Berhane N, Shah DK, Ma D, Leal M, Gerber HP, Sapra P, Barton HA, Betts AM. On translation of antibody drug conjugates efficacy from mouse experimental tumors to the clinic: a PK/PD approach. J Pharmacokinet Pharmacodyn. 2013;40(5):557–71. doi: 10.1007/s10928-013-9329-x. PMID: 23933716. [DOI] [PubMed] [Google Scholar]
- 15.Muller PY, Milton MN. The determination and interpretation of the therapeutic index in drug development. Nat Rev Drug Discov. 2012;11(10):751–61. doi: 10.1038/nrd3801. PMID: 22935759. [DOI] [PubMed] [Google Scholar]
- 16.Betts A, Clark T, Jasper P, Tolsma J, van der Graaf P H, Graziani EI, Rosfjord E, Sung M, Ma D, Barletta F, et al. Use of translational modeling and simulation for quantitative comparison of PF-06804103, a new generation HER2 ADC, with Trastuzumab-DM1. J Pharmacokinet Pharmacodyn. 2020;47(5):513–26. doi: 10.1007/s10928-020-09702-3. PMID: 32710210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taksin AL, Legrand O, Raffoux E, de Revel T, Thomas X, Contentin N, Bouabdallah R, Pautas C, Turlure P, Reman O, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66–71. doi: 10.1038/sj.leu.2404434. PMID: 17051246. [DOI] [PubMed] [Google Scholar]
- 18.Liao MZ, Lu D, Kagedal M, Miles D, Samineni D, Liu SN, Li C. Model-Informed therapeutic dose optimization strategies for antibody–drug conjugates in oncology: what can we learn from us food and drug administration–approved antibody–drug conjugates? Clin Pharmacol Ther. 2021;110(5):1216–30. doi: 10.1002/cpt.2278. PMID: 33899934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat Rev Drug Discov. 2017;16(5):315–37. doi: 10.1038/nrd.2016.268. PMID: 28303026. [DOI] [PubMed] [Google Scholar]
- 20.Graziani EI, Sung M, Ma D, Narayanan B, Marquette K, Puthenveetil S, Tumey LN, Bikker J, Casavant J, Bennett EM, et al. PF-06804103, a site-specific anti-HER2 antibody–drug conjugate for the treatment of HER2-expressing breast, gastric, and lung cancers. Mol Cancer Ther. 2020;19(10):2068–78. doi: 10.1158/1535-7163.MCT-20-0237. PMID: 32747418. [DOI] [PubMed] [Google Scholar]
- 21.Meric-Bernstam F, Calvo E, Moreno V, Chung HC, Park YH, Bang Y-J, Rosen LS, Mita MM, Garrido-Laguna I, Leung ACF, et al. A phase I dose escalation study evaluating the safety and tolerability of a novel anti-HER2 antibody-drug conjugate (PF-06804103) in patients with HER2-positive solid tumors. JCO. 2020;38(15_suppl):1039–1039. doi: 10.1200/JCO.2020.38.15_suppl.1039. [DOI] [Google Scholar]
- 22.Wang J, Liu Y, Qingyuan Z, Jifeng F, Fang J, Chen X, Han Y, Li Q, Zhang P, Yuan P, et al. RC48-ADC, a HER2-targeting antibody-drug conjugate, in patients with HER2-positive and HER2-low expressing advanced or metastatic breast cancer: a pooled analysis of two studies. JCO. 2021;39(15_suppl):1022–1022. doi: 10.1200/JCO.2021.39.15_suppl.1022. [DOI] [Google Scholar]
- 23.Ogitani Y, Aida T, Hagihara K, Yamaguchi J, Ishii C, Harada N, Soma M, Okamoto H, Oitate M, Arakawa S, et al. DS-8201a, a Novel HER2-Targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin Cancer Res. 2016;22:5097–108. doi: 10.1158/1078-0432.CCR-15-2822. PMID: 27026201. [DOI] [PubMed] [Google Scholar]