

Abstract
The irreversible ERBB1/2/4 inhibitor neratinib causes plasma membrane-associated K-RAS to mislocalize into intracellular vesicles liminal to the plasma membrane; this effect is enhanced by HDAC inhibitors and is now a Phase I trial (NCT03919292). The combination of neratinib and HDAC inhibitors killed pancreatic cancer and lymphoma T cells. Neratinib plus HDAC inhibitor exposure was as efficacious as (paclitaxel+gemcitabine) at killing pancreatic cancer cells. Neratinib reduced the phosphorylation of PAK1, Merlin, LATS1/2, AKT, mTOR, p70 S6K, and ERK1/2 which required expression of Rubicon, Beclin1, and Merlin. Neratinib altered pancreatic tumor cell morphology which was associated with MST4 degradation reduced Ezrin phosphorylation and enhanced phosphorylation of MAP4K4 and LATS1/2. Knockdown of the MAP4K4 activator and sensor of membrane rigidity RAP2A reduced basal LATS1/2 and YAP phosphorylation but did not prevent neratinib from stimulating LATS1/2 or YAP phosphorylation. Beclin1 knockdown prevented MST4 degradation, Ezrin dephosphorylation and neratinib-induced alterations in tumor cell morphology. Our findings demonstrate that neratinib enhances LATS1/2 phosphorylation independently of RAP2A/MAP4K4 and that MST4 degradation and Ezrin dephosphorylation may represent a universal trigger for the biological actions of neratinib.
Keywords: ERK, HDAC inhibitor, Hippo, Merlin, neratinib, PI3K, RAP2, RAS, YAP
1 |. INTRODUCTION
In prior studies we have conclusively demonstrated that the multi-kinase inhibitor neratinib can cause the rapid degradation of receptor tyrosine kinases and RAS proteins via Rubicon-dependent LC3-associated phagocytosis (LAP) (Booth, Roberts, Poklepovic et al., 2017; Booth, Roberts, Rais et al., 2017; Booth et al., 2018; Booth, Roberts, Rais et al., 2019; Booth, Roberts, & Dent, 2019; Booth, Poklepovic, & Dent, 2019; Dent et al., 2019; Dent, Booth, Poklepovic, & Hancock, 2019). Neratinib in parallel inactivates the Hippo pathway, with YAP and TAZ exiting the nucleus and being degraded (Booth, Poklepovic et al., 2019; Dent et al., 2019). Based on our collective findings combining neratinib with various histone deacetylase inhibitors in a diverse range of tumor cell types, a Phase I trial in solid tumor patients has opened combining neratinib and sodium valproate (NCT03919292).
Davis et al. (2011) published studied the kinase inhibitory properties of ~40 small molecule kinase inhibitors against the ~400 kinases within the human kinome (Davis et al., 2011). These findings were confirmed by Klaeger et al. (2017). In the Davis et al study, afatinib was shown to have very restricted substrate specificity, specifically inhibiting only members of the ERBB family receptor tyrosine kinases. Neratinib, in contrast, was shown to have many additional previously unknown targets. The majority of the newly discovered neratinib targets were serine/threonine kinases, rather than the targets of its initial development that were receptor tyrosine kinases. Kinases inhibited by neratinib with IC50 values below 100 nM included; MST2, MST3, MST4, MAP4K1, MAP4K3, MAP4K5, MAP3K4, YSK1, and YSK4. MST4 plays an important role in regulating plasma membrane—Golgi trafficking, cytoskeletal interactions with the plasma membrane, as well as being part of the Hippo pathway. In normal tissues, the Hippo pathway regulates organ size and in oncology plays a key role in promoting tumor growth, invasion, and chemotherapy resistance (Meng et al., 2015; Thompson & Sahai, 2015; Crawford, Bronner and Zbieg, 2018). Whether neratinib is an irreversible inhibitor of the MAP4K serine/threonine kinases, as it is of the receptor tyrosine kinases, is at present unknown.
Sterile 20 (Ste20) is a kinase within the pheromone-response pathway in budding yeast. A number of mammalian homologs to Ste20 are known including multiple MAP4Ks and MST1/2/3/4 (Bitra, Sistla, Mariam, Malvi, & Anand, 2017; Bae & Luo, 2018; Chen et al., 2018; Hergovich, 2013; Hergovich, 2016; Jang, Kim, & Bae, 2018; Kong, Zhao, Men, & Teng, 2015; Liu & Deng, 2019; Patel, Camargo, & Yimlamai, 2017; Rawat & Chernoff, 2015; Sharif & Hergovich, 2018). MST1/2 are considered to be core components of the Hippo Pathway. Hippo regulates organ size and organ homeostasis by controling apoptosis and proliferation. MST1/2 phosphorylate, and activate, the large tumor suppressor, LATS1/2. Essential in this process are several scaffold proteins including MOB1, MOB4, SAV1, RASSF1A, and MO25. The relative levels of MST1: MOB1 complexed versus MST4:MOB4 complexed can change pathway signaling from promoting growth or undergoing cell death. Neratinib has been claimed to variably inhibit MST1/2/3/4 but not LATS1/2 or NDR1/2; we published that neratinib activated LATS1/2 (Dent et al., 2019). Activated LATS1/2 phosphorylates the transcriptional coactivator Yes-associated protein (YAP) and its paralog TAZ. LATS1/2-mediated phosphorylation of YAP and TAZ causes the proteins to exit the nucleus. YAP and TAZ located in the cytosol are then ubiquitinated and proteolytically degraded. YAP is recognized as an oncogene that enhances the transcription of genes involved in cell proliferation and invasion by partnering with the TEAD family of transcription factors. Hence, the inactivation of YAP predisposes the cell to undergo death processes. MST1/2 are not the only kinases that phosphorylate and activate LATS1/2. Knock out of LATS1/2, but not of MST1/2, prevents regulatory YAP/TAZ phosphorylation. The kinases MAP4K1/2/3 and MAP4K4/6/7 are direct LATS1/2-activating kinases. Hence, combined deletion of the MAP4Ks and MST1/2, but neither alone, is required to completely suppress LATS1/2 and YAP/TAZ phosphorylation.
The present studies were performed to extend our understanding of the molecular biology of the FDA-approved breast cancer neo-adjuvant drug neratinib. We discovered that the key molecular mediators of neratinib action are Rubicon/LC3-associated phagocytosis and the docking protein Merlin that are required to cause secondary inactivation of AKT, mammalian target of rapamycin (mTOR), p70 S6K, and extracellular regulated kinases 1 and 2 (ERK1/2).
2 |. MATERIALS AND METHODS
2.1 |. Materials
Sodium valproate was purchased from Selleckchem (Houston, TX). Neratinib was supplied by Puma Biotechnology Inc. (Los Angeles, CA). All Materials we obtained as described in the references (Booth, Roberts, Poklepovic et al., 2017; Booth, Roberts, Rais et al., 2017; Booth et al., 2018; Booth, Roberts, Rais et al., 2019; Booth, Roberts, & Dent, 2019; Dent, Booth, Roberts et al., 2019; Dent, Booth, Poklepovic et al. 2019), Multiple control studies have been presented showing on-target specificity of antibodies to detect both total protein levels and phosphorylated levels of proteins (Figures S1 and S2).
2.2 |. Methods
All Methods used in this manuscript have been performed and described in the references (Booth, Roberts, Poklepovic et al,al., 2017; Booth, Roberts, Rais et al., 2017; Booth et al., 2018; Booth, Roberts, Rais et al., 2019; Booth, Roberts, & Dent, 2019; Booth, Poklepovic et al., 2019; Dent, Booth, Roberts et al., 2019).
2.3 |. Data analysis
Comparison of the effects of various treatments (in triplicate three times) was using one-way ANOVA and a two-tailed Student’s t-test. Differences with a p-value of <.05 were considered statistically significant. Experiments are the means of multiple individual points from multiple experiments (±SD).
3 |. RESULTS
Previously we presented evidence that clinically relevant concentrations of the breast cancer drug neratinib, that is, 50 nM with the safe Cmax at ~150 nM, killed pancreatic cancer cells (Booth, Roberts, Poklepovic et al., 2017; Booth, Roberts, Rais et al., 2017; Booth et al., 2018; Booth, Roberts, Rais et al., 2019; Booth, Roberts, & Dent, 2019; Booth, Poklepovic et al., 2019; Dent, Booth, Roberts et al., 2019). The lethality of neratinib could be enhanced in pancreatic cancer cells by clinically relevant concentrations of the irreversible ERBB1/2/4 inhibitor afatinib, by several chemically distinct histone deacetylase inhibitors, by PDE5 inhibitors, and by HMG CoA reductase inhibitors. The combination of neratinib and the HDAC inhibitor sodium valproate is now an open Phase I trial at Massey Cancer Center (NCT03919292). The third patient on the trial with Stage IV pancreatic adenocarcinoma has exhibited stable disease for 6 months.
We initially wished to compare and contrast in pancreatic cancer cells the lethality of (neratinib+HDAC inhibitor) versus the standard of care therapeutic drugs gemcitabine and paclitaxel. The lethality of (neratinib+sodium valproate) was enhanced by either gemcitabine or paclitaxel (Figures S3A and S3B). The combination of (gemcitabine+paclitaxel) displayed a very similar level of tumor cell killing compared with that caused by (neratinib+valproate). Thus our in vitro data suggests that (neratinib+valproate) is at least as efficacious against pancreatic cancer as standard of care therapies. Inhibition of p38 mitogen activated protein kinase (MAPK) or JNK1/2 suppressed killing, as did expression of activated AKT or activated MEK1 (Figures S4A and S4B). Previously, we demonstrated that neratinib and HDAC inhibitors could interact to kill blood cancer cells, cells that do not express ERBB family receptors, and to enhance checkpoint immunotherapeutic (I/O) antibodies against mammary tumors (Dent, Booth, Roberts et al., 2019). Cutaneous T cell lymphomas (Mycosis fungoides) are a malignant regulatory T cell (T reg) and as such, represent a putative surrogate cell type for assessing whether one component of the biology by which (neratinib+HDAC inhibitor) had enhanced immunotherapeutic efficacy might also be via the killing of activated T reg cells (Fujimura, Okuyama, Ito, & Aiba, 2008; Geskin et al., 2018). Our prior immunohistochemical data had argued that the levels of T regs in (neratinib+valproate) treated tumors had declined (Booth et al., 2017). In the present studies, we found that neratinib interacted with both sodium valproate and with vorinostat, a standard of care therapeutic in this disease, to kill cutaneous T cell lymphoma cells (Figures S4C–E). The efficacy of neratinib as a single agent was similar to that of another standard of care therapeutic for this disease at its Cmax, betamethasone (betatrex). This data further supports the hypothesis that key primary target(s) for neratinib are not ERBB family receptor tyrosine kinases.
We next performed studies to define the molecular mechanisms by which neratinib regulates tumor cell biology. We know that the tumor suppressor Merlin, also known as neurofibromatosis 2 (NF2), plays a key role, after neratinib exposure, in regulating plasma membrane/cytoskeletal function as well as signalosome formation and the Hippo/YAP pathway (Dent, Booth, Roberts et al., 2019). In our prior studies, we also demonstrated that the protein Rubicon, essential for the uptake of extracellular antigens via LC3-associated phagocytosis (LAP), was required for the neratinib-dependent degradation of growth factor receptors and plasma membrane RAS proteins.
Knockdown of Rubicon prevented neratinib and (neratinib+valproate) from causing the dephosphorylation of PAK T423 and Merlin S518 and from enhancing the phosphorylation of LATS1/2 T1079 and YAP S127 (Figure 1a). Thus, neratinib-dependent regulation of the Hippo pathway requires LAP. Treatment of PDX pancreatic cancer cells with neratinib reduced the phosphorylation of the cytoskeletal protein Ezrin, Merlin and the CDC42/RAC-regulated kinase PAK1 whereas it did not significantly alter MAP4K1 S171 phosphorylation but did enhance phospho-MAP4K4 S629 levels (Figure 1b) (Davis et al., 2011; Klaeger et al., 2017; Meng et al., 2015; Thompson & Sahai, 2015). We next attempted to link altered function/activity of Rubicon and Merlin with alterations in signaling and cell viability. The knockdown of either Merlin or Rubicon significantly reduced neratinib-dependent tumor cell killing (Figure 1c). Knockdown of Merlin or Rubicon caused neratinib-exposed cells to modestly activate AKT, mTOR, p70 S6K, and ERK1/2 (Figure 1d). This data tentatively suggests that MAP4K4 may be the MAP4K linking neratinib to LATS1/2 functionality.
FIGURE 1.

Neratinib exposure reduces the phosphorylation of PAK1, Ezrin, and Merlin, and increases the phosphorylation of MAP4K4. (a) FCC1199 GEMM and PDX human pancreatic cancer cells were transfected with scrambled siRNA molecules or with validated siRNA molecules to knock down the expression of Rubicon. Twenty-four hours after transfection, cells were treated with vehicle control, neratinib (50 nM), sodium valproate (250 μM), or the drugs in combination as indicated for 6 hr. Cells were fixed in place and immunostaining performed to determine the total expression of the indicated proteins and the phosphorylation of these proteins. The data as presented are protein phosphorylation corrected for total protein expression. (n = 3+/−SD) *p < .05 less than vehicle control; #p < .05 greater than vehicle control value. (b) PDX human pancreatic cancer cells were treated with vehicle control or with neratinib (50 nM) for 6 hr. Cells were fixed in place and immunostaining performed to determine the total expression of the indicated proteins and the phosphorylation of these proteins. The data as presented are protein phosphorylation corrected for total protein expression. (n = 3+/−SD) *p < .05 less than vehicle control; #p <.05 greater than vehicle control value. (c) PANC1 and MiaPaca2 cells were transfected with a scrambled siRNA control or with validated siRNA molecules to knock down the expression of Rubicon or Merlin. Twenty-four hours after transfection cells were treated with vehicle control or with (neratinib [50 nM]+sodium valproate [250 μM]) for 24 hr. Cells were isolated, and viability determined via a trypan blue exclusion assay (n = 3+/−SD) #p < .05 greater than the corresponding value in siSCR cells; *p < .05 less than the corresponding value in siSCR cells. (d) PANC1 and MiaPaca2 cells were transfected with a scrambled siRNA control or with validated siRNA molecules to knock down the expression of Rubicon or Merlin. Twenty-four hours after transfection cells were treated with vehicle control or with neratinib (50 nM) for 6 hr. Cells were fixed in place and immunostaining performed to determine the total expression of the indicated proteins and the phosphorylation of these proteins. The data as presented are protein phosphorylation corrected for total protein expression. (n = 3+/−SD) *p < .05 less than vehicle control value; **p < .05 less than siSCR control value; #p < .05 greater than siSCR control value. siSCR, small interfering scrambled; siRNA, small interfering RNA
When re-examining our previously published data, we noted that neratinib, in addition to causing mislocalization of K-RAS V12 – GFP and K-RAS V12 – RFP from the plasma membrane into the cytosol, was also changing the morphology of pancreatic cancer cells (Figures S5–S7) (Booth, Roberts, Rais et al., 2019; Dent, Booth, Roberts et al., 2019). Similar findings were then observed in a PDX isolate of pancreatic cancer and an established GEMM tumor cell isolate (Figures S6 and S7). Thus, neratinib treatment rapidly alters the function of multiple membranes trafficking regulatory proteins, including causing inactivation of PAK1 and activation of MAP4K4.
In PANC1 cells transiently transfected to express K-RAS V12 – GFP, knockdown of either Rubicon or Merlin suppressed the ability of neratinib to mislocalize K-RAS V12 – GFP into intracellular vesicles liminal to the plasma membrane ( Figure 2b). Whilst knockdown of Rubicon or Merlin reduced vesicle formation, it did not alter the ability of neratinib to alter tumor cell morphology from a rounded cell to a more flattened angular cell. Similar data were obtained in GEMM FC1199 cells (Figure S7). Knockdown of the macroautophagy regulatory protein Beclin1 both prevented vesicle formation and prevented the change in tumor cell morphology (Figure 2b). Thus, the neratinib-dependent mislocalization of RAS requires Rubicon and Merlin, but Rubicon and Merlin are downstream of Beclin1 in this process and also of the unknown primary neratinib effector which also controls cell morphology. That neratinib could rapidly change pancreatic tumor cell morphology in the absence of Rubicon or Merlin argues that the primary upstream targets of the drug are likely to be neratinib-inhibitable kinases, such as MST4, that regulate plasma membrane dynamics via Ezrin/ERM protein phosphorylation.
FIGURE 2.

Neratinib exposure alters morphology independently of either Rubicon or Merlin. (a) PANC1 cells were transfected with a scrambled siRNA control or with validated siRNA molecules to knock down the expression of Rubicon or Merlin. Twenty-four hours after transfection cells were treated with vehicle control or with neratinib (50 nM) for 20 min. Cells were fixed in place and immunostaining performed to determine the total expression and localization of K-RAS (a representative selection from three studies). (b) FC1199 GEMM cells were transfected with a scrambled siRNA control or with a validated siRNA molecule to knock down the expression of Beclin1. In parallel, they were transfected to express K-RAS V12 – GFP and K-RAS V12 – RFP. Twenty-four hours after transfection cells were treated with vehicle control or with neratinib (50 nM) for 1 hr. Cells were fixed in place and fluorescence microscopy performed to determine the localization of K-RAS (a representative selection from three studies). siRNA, small interfering RNA
The biological actions of MAP4K family enzymes, for example in the sensing of membrane rigidity and regulation of the Hippo pathway, have been previously described (Meng et al., 2018). The RAS-related GTPase RAP2A is a relay switch in mediating the sensing of plasma membrane stiffness as well as acting to enhance the phosphorylation of LATS1/2 in the Hippo pathway. When the membrane is less stiff, RAP2A stimulates the activity of MAP4K4, MAP4K6, MAP4K7, and ARHGAP29; this leads to increased phosphorylation of LATS1/2 and subsequently that of YAP/TAZ.
Neratinib as a single agent modestly increased the phosphorylation of LATS1/2 by ~30% in PANC1 cells over a 4 hr time course. (Figure S8A). Knockdown of RAP2A significantly reduced the basal levels of LATS1/2 phosphorylation by ~30% but did not prevent neratinib from increasing LATS1/2 phosphorylation to levels observed in scrambled control transfected cells. This further argues that RAP2A is not a mediator of neratinib signaling in this context.
Knockdown of RAP2A reduced basal Merlin S518 phosphorylation by ~25% and in RAP2A knockdown cells, neratinib conversely stimulated phospho-Merlin S518 levels (Figure S8B). Knockdown of RAP2A enhanced the lethality of neratinib but only at 100 nM and higher concentrations that are above its plasma Cmax (Figure S8C). Very similar data were obtained in MiaPaca2 cells (Figure S9). Collectively, this data again argues that RAP2A is marginally involved in the regulation of neratinib biology, at least in this context.
Neratinib has recently been shown to concentrate in the pancreas, although the precise -fold increase in concentration above plasma levels was not determined in that study (Ardestani et al., 2019). We next determined whether signaling by RAP2A was required for K-RAS V12 – GFP to become mislocalized in vesicles. Knockdown of RAP2A in pancreatic cancer cells did not prevent neratinib from altering the morphology of the cells but did prevent K-RAS V12 – GFP from becoming mislocalized in vesicles liminal to the plasma membrane (Figure 3). Hence, again, it appears RAP2A is unlikely to be a key mediator of neratinib signaling into the Hippo pathway.
FIGURE 3.

Knockdown of RAP2A prevents neratinib from causing K-RAS V12 – GFP vesicularization. PANC1 and MiaPaca2 cells were transfected with a scrambled siRNA or with a siRNA to knock down RAP2A and in parallel transfected with a plasmid to express K-RAS V12 – GFP. Twenty-four hours later, cells were treated with vehicle control or with neratinib (50 nM). Cells were fixed in place after 1 hr and examined at 60× magnification
Unlike epithelial carcinoma cells that express ERBB family receptors, the originally proposed primary targets of neratinib, blood cancer express very low or no receptors whatsoever of this family. A priori, it would thus be expected for neratinib to exhibit almost no detectable biology in these liquid tumor cell types. Based on the data in Figure 2, this fact permits us to use blood cancer cells as a test-bed surrogate model system to examine neratinib effects in the absence of ERBB family receptors. The MAP4K MST4, a kinase proposed by others to be potently inhibited by neratinib at its catalytic site, is expressed in both solid and liquid tumor cells and we performed further studies in T cell lymphoma cells. Treatment of T cells with neratinib significantly reduced the total expression of MST4 as well as MST4 phosphorylation (Figure 4). One key substrate for MST4 is the cytoskeletal protein Ezrin. Phosphorylated Ezrin T567 has been shown to play key roles in metastatic spread and pseudopodium formation, stabilization of P-glycoprotein, and chemotherapy resistance (Pore et al., 2015; Yang et al., 2011; Zhang et al., 2013; Zhou et al., 2010). Although neratinib did not reduce total Ezrin expression, in agreement with prior data in pancreatic cancer cells, Jurkat T cells, and HL60 APL cells, it profoundly reduced Ezrin T567 phosphorylation in the cutaneous T cells (Figure 5) (Dent, Booth, Roberts et al., 2019). What was most noticeable at 60× magnification were the filamentous projections extending beyond the plasma membrane of the T cells, where phospho-Ezrin exhibited a punctate zebra-like staining pattern. In cells treated with neratinib, the extracellular projections disappeared and instead, Ezrin T567 phosphorylation had begun to localize in the cytosol.
FIGURE 4.
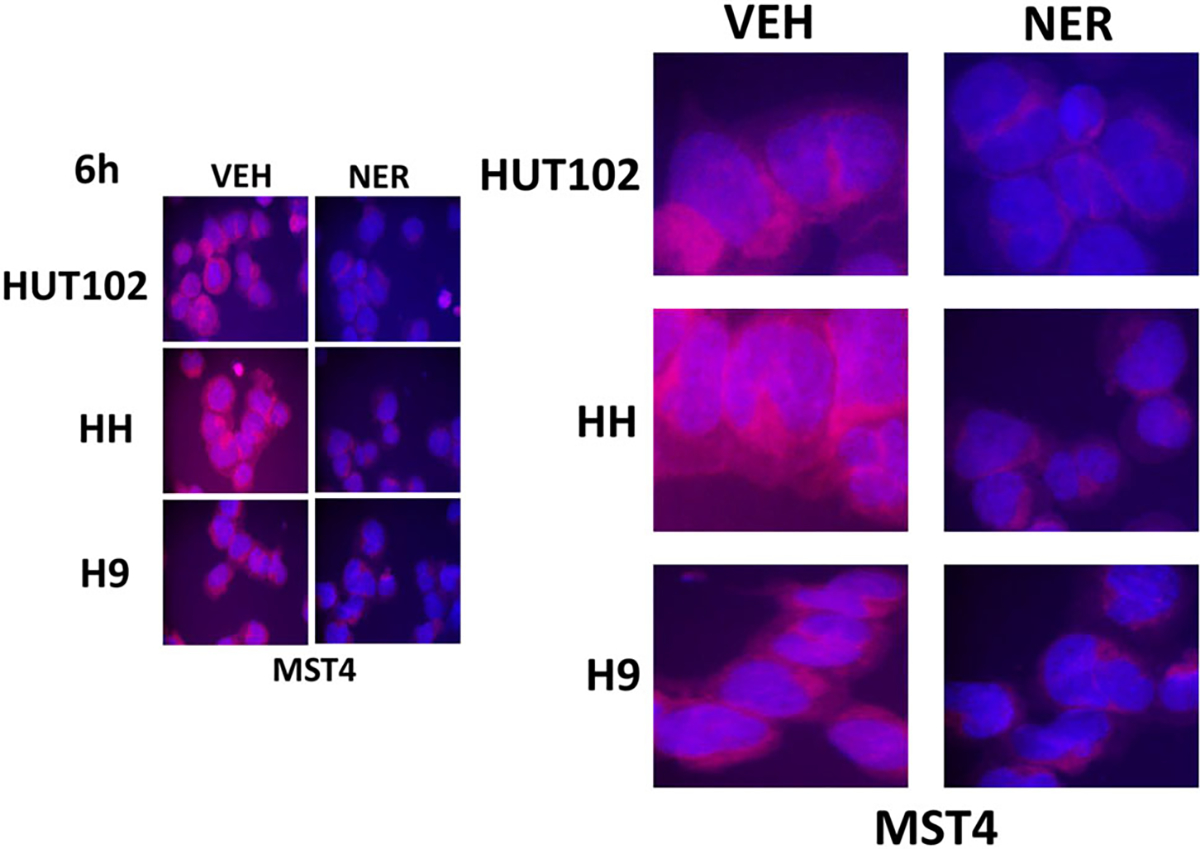
Neratinib reduces MST4 expression in T cell lymphoma cells. T cell lymphoma cells were treated with vehicle control or with neratinib (50 nM) for 6 hr. Cells were cytospun onto glass slides, air-dried and fixed. Cells were stained to determine the total expression of ERK2 and of MST4 and with DAPI. Cells were visualized at 60× magnification. ERK2 expression was invariant (not shown). Data are representative of three separate studies. DAPI, 4′,6-diamidino-2-phenylindole; ERK2, extracellular regulated kinases 2
FIGURE 5.
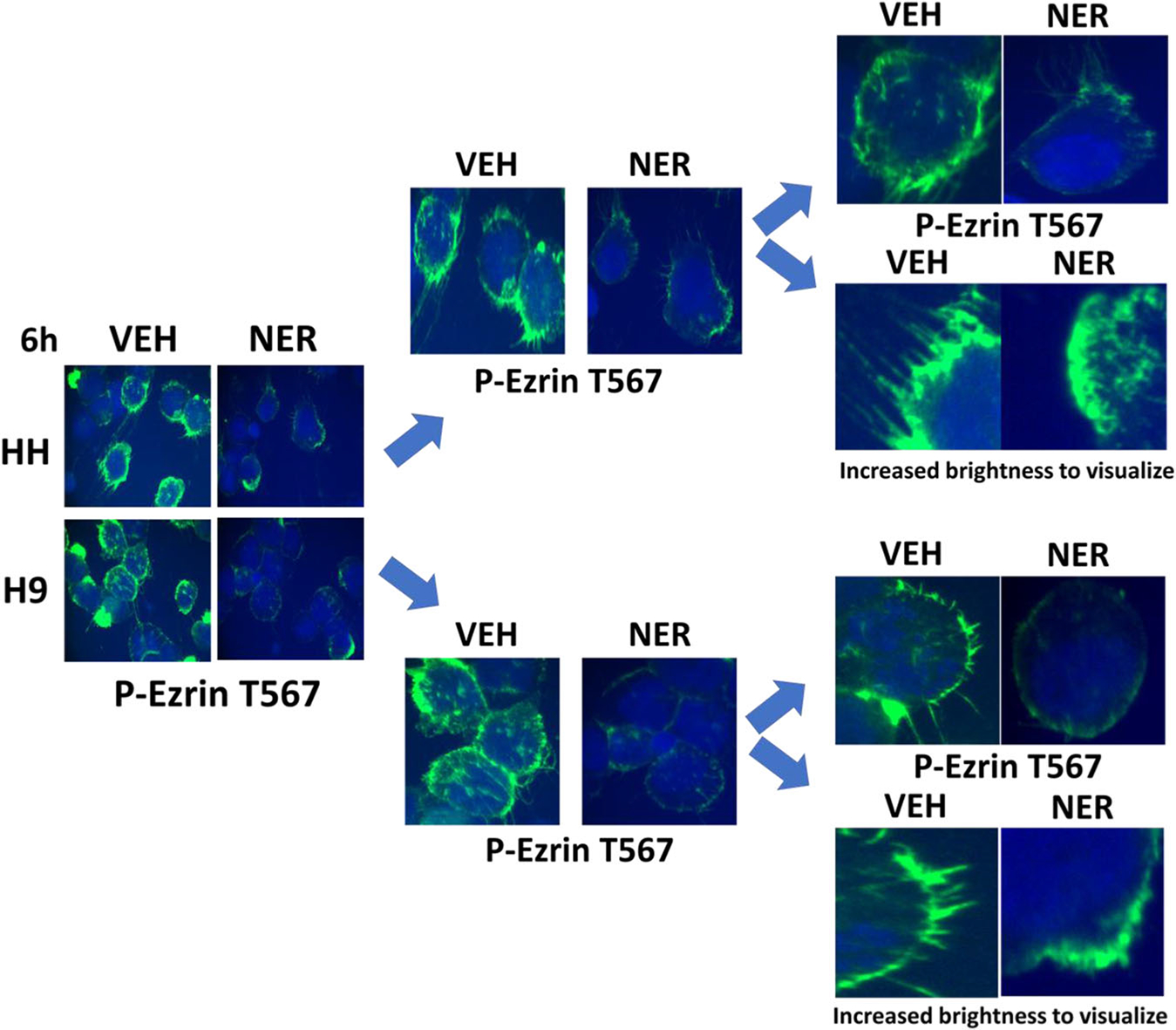
Neratinib reduces Ezrin T567 phosphorylation in T cells. T cell lymphoma cells were treated with vehicle control or with neratinib (50 nM) for 6 hr. Cells were cytospun onto glass slides, air dried, and fixed. Cells were stained to determine the total expression of Ezrin (not shown) and Ezrin T567 phosphorylation and with DAPI. The expression of Ezrin was not changed by neratinib. Cells were visualized at 60× magnification. Data are representative of three separate studies. DAPI, 4′,6-diamidino-2-phenylindole
Additional studies then defined the localization of YAP, Merlin, and LATS1/2 with respect to neratinib exposure and RAP2A functionality. Under vehicle control conditions, YAP staining was in the nucleus and the cytosol whereas YAP S127 staining was almost completely cytosolic (Figure 6, Figure S10). Neratinib exposure caused the majority of YAP protein to leave the nucleus and to significantly increase YAP S127 phosphorylation by ~35% throughout the cell. The knockdown of RAP2A did not alter YAP nuclear localization although the basal phosphorylation of YAP S127 throughout the cell was reduced by ~20%. In RAP2A knockdown cells, neratinib caused YAP to exit the nucleus that was associated with increased YAP S127 phosphorylation. Thus, RAP2A/MAP4K signaling again does not appear to be an essential component of the regulatory pathway by which neratinib regulates the Hippo pathway.
FIGURE 6.
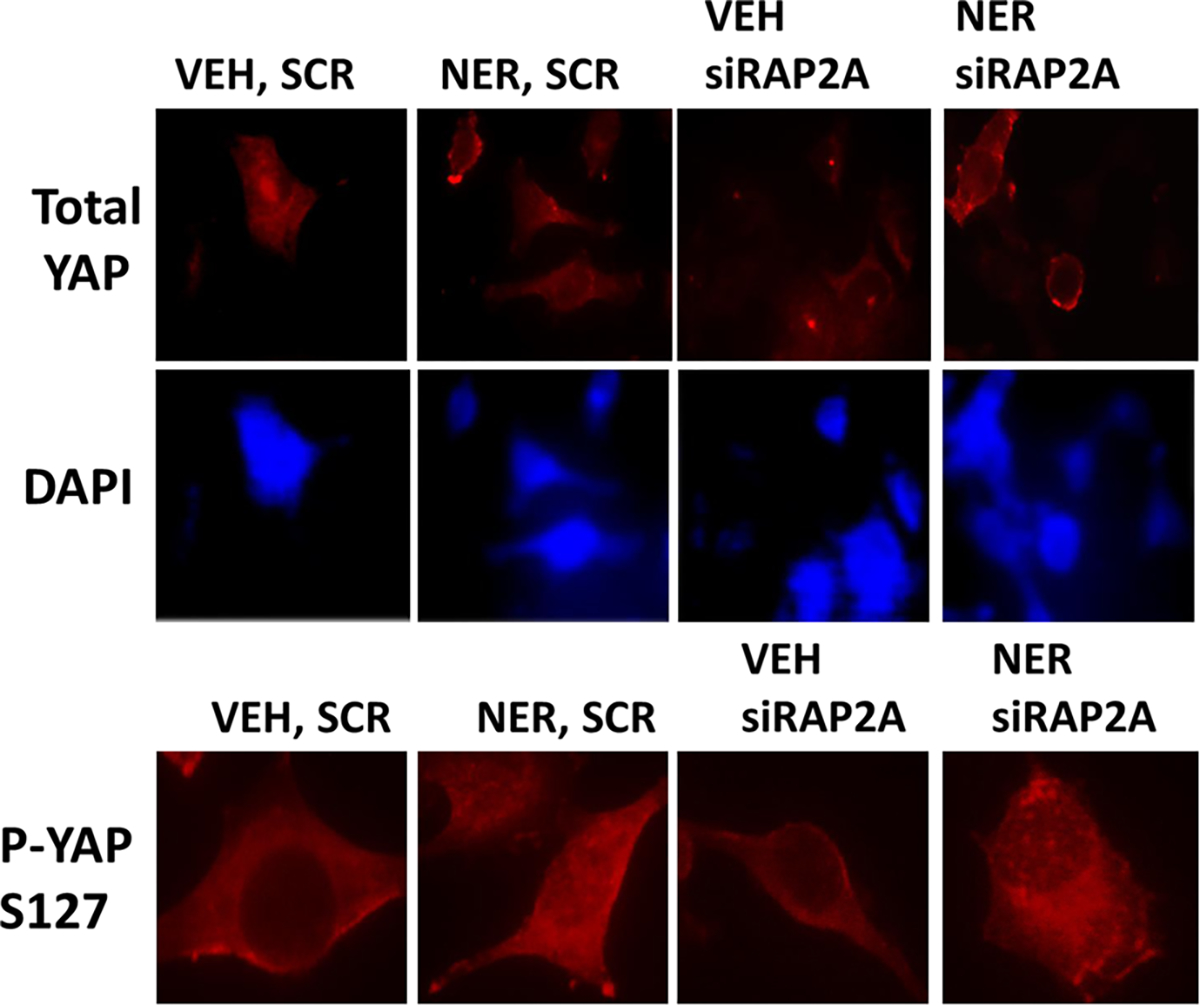
Neratinib causes YAP S127 phosphorylation to increase and YAP to exit the nucleus. PANC1 cells were transfected with a scrambled siRNA or with a siRNA to knock down RAP2A. Twenty-four hours after transfection cells were treated with vehicle control or neratinib (50 nM). Cells were fixed in place 3 hr after drug exposure and stained for total YAP expression and for YAP S127 phosphorylation followed by an examination at 60×. siRNA, small interfering RNA; YAP, Yes-associated protein
Under vehicle control conditions, a modest amount of LATS1/2 and Merlin colocalization was observed in tumor cells (Figure S11). Neratinib treatment increased plasma membrane localization of Merlin and LATS1/2, however, neratinib did not cause Merlin and LATS1/2 to colocalize. Knockdown of the stiffness-sensor RAP2A caused both membrane localization and colocalization of both Merlin and LATS1/2. In knockdown cells, Merlin, and LATS1/2 became membrane-localized after neratinib exposure, though again with only a modest level of Merlin-LATS colocalization. Over the 3 hr treatment time course, the association of Merlin with the plasma membrane increased, with the levels of colocalization between Merlin and LATS1/2 also modestly increasing (Figure 7).
FIGURE 7.
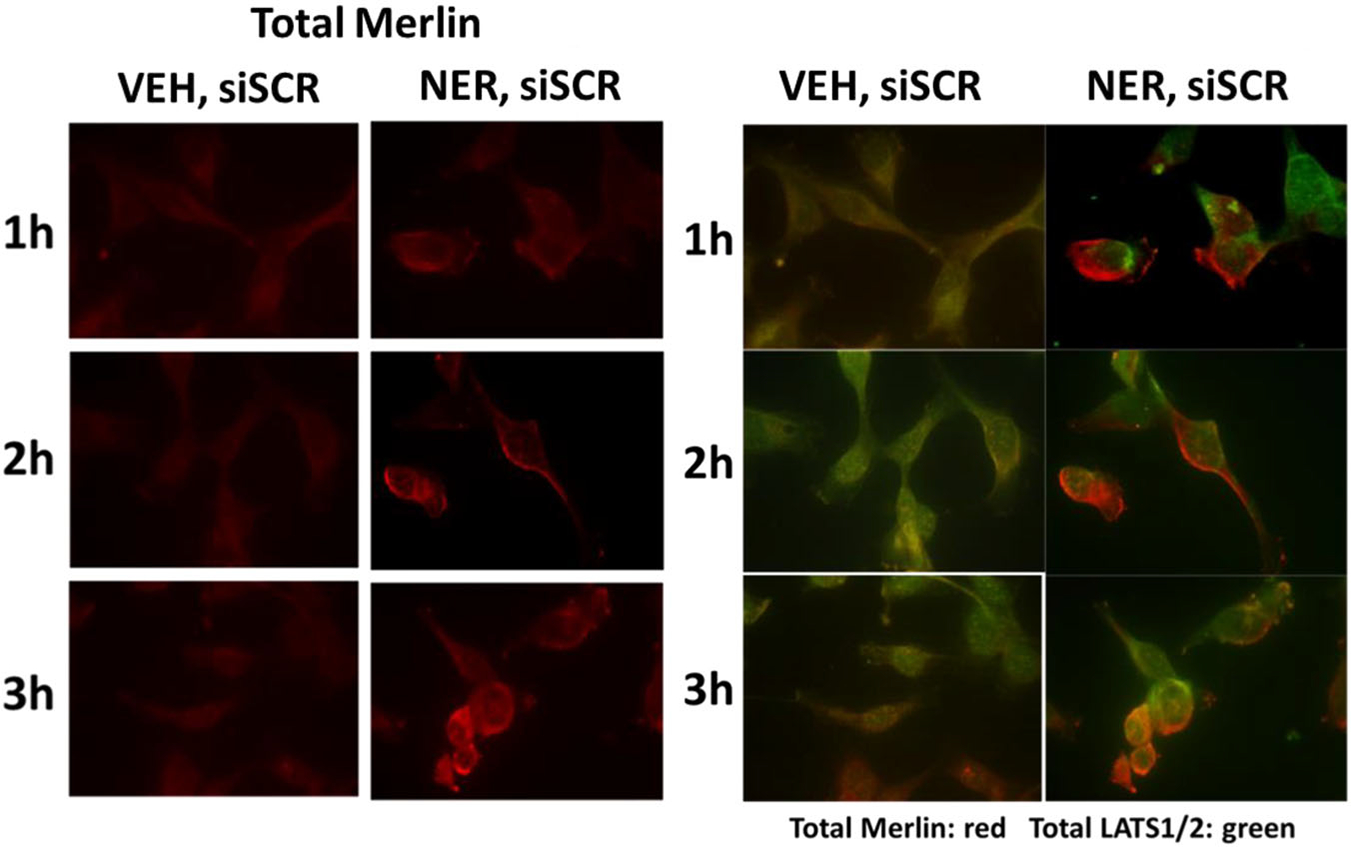
Neratinib causes Merlin and LATS1/2 to partially co-localize after 3 hr of exposure. PANC1 cells were transfected with a scrambled siRNA. Twenty-four hours after transfection cells were treated with vehicle control or neratinib (50 nM). Cells were fixed in place 1–3hr after drug exposure and stained for total Merlin expression and total LATS1/2 expression followed by an examination at 60×
In T cells that lack ERBB family member expression, MST4 levels declined after cells were exposed to neratinib and similar data in pancreatic tumor cells have also been published by ourselves (Dent Booth, Roberts et al., 2019). Knockdown of Beclin1 prevented neratinib reducing MST4 expression in pancreatic cancer cells and prevented neratinib altering tumor cell morphology ( Figure 8a,b). Upon closer examination of the images, it became evident that neratinib caused MST4 staining to change from being relatively uniform across the cell to becoming more gravelly/punctate in appearance (Figure 8c; Figure S12). In neratinib-treated cells, lacking the Beclin1 expression, an even greater number of discrete punctate vesicles were present. This data suggests that the earliest stages of neratinib-induced MST4 vesicularization do not require Beclin1 and that in the absence of Beclin1 and autophagic digestion, the numbers of MST4 containing vesicles become elevated.
FIGURE 8.
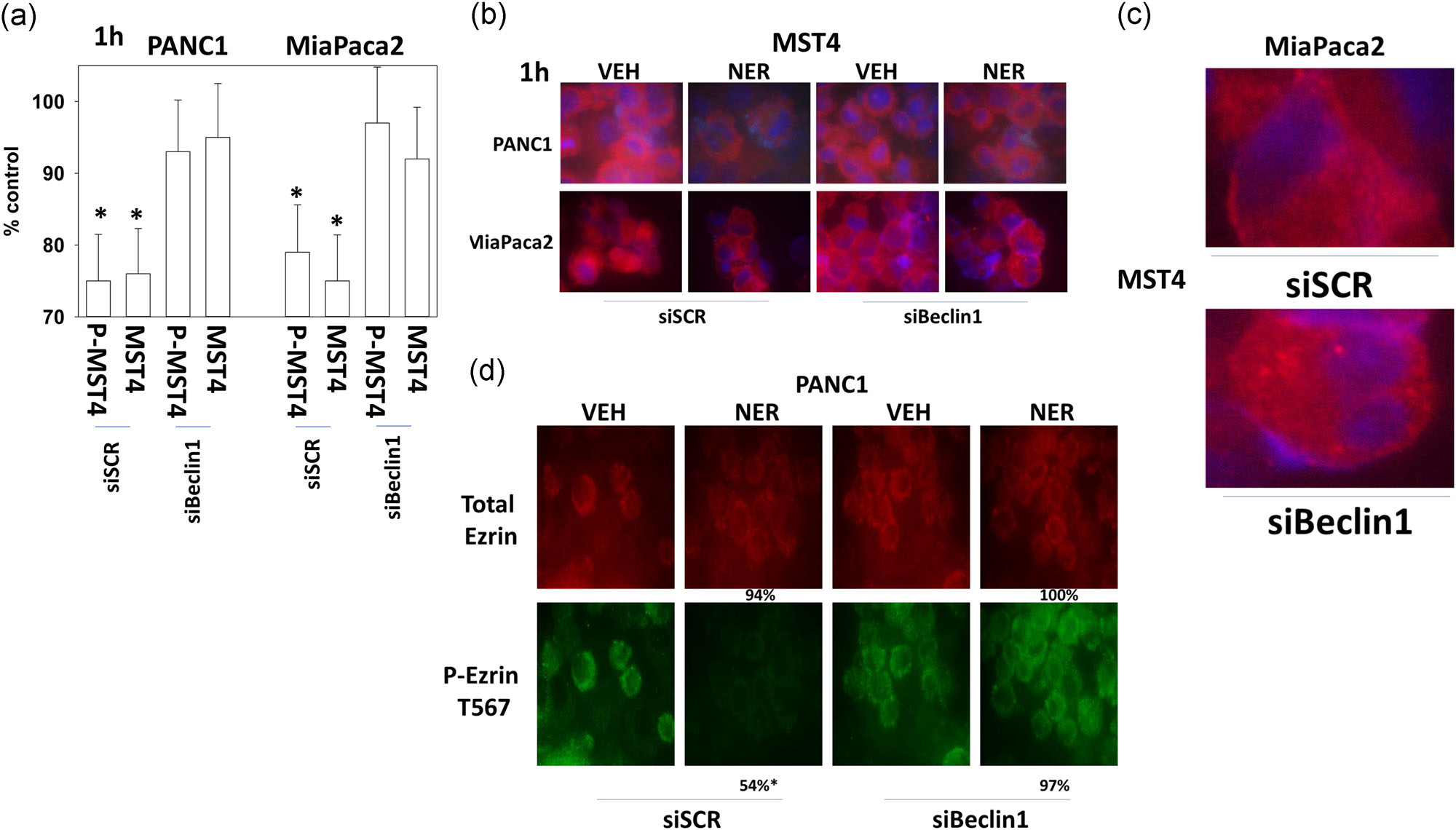
Knockdown of Beclin1 prevents MST4 degradation and maintains Ezrin T567 phosphorylation. (a,b) PANC1 and MiaPaca2 cells were transfected with a scrambled siRNA or with a siRNA to knock down Beclin1. Twenty-four hours later, cells were treated with vehicle control or with neratinib (50 nM). Cells were fixed in place after 1 hr and stained with an anti-ERK2 antibody and an anti-MST4 antibody. Stained cells were examined at 10× and at 60× magnification; the total expression of Ezrin did not alter under any treatment condition (not shown). The graphical data as presented are as MST4 expression compared with vehicle control expression corrected for total protein expression of invariant ERK2. (n = 3+/−SD) *p < .05 less than vehicle control value. (c) Enlarged image data from MiaPaca2 cells treated with neratinib, with either scrambled control transfection or transfection to knockdown Beclin1. (d) PANC1 cells were transfected with a scrambled siRNA or with a siRNA to knock down Beclin1. Twenty-four hours later, cells were treated with vehicle control or with neratinib (50 nM). Cells were fixed in place after 1 hr and stained with an anti-Ezrin antibody and an anti-phospho-Ezrin T567 antibody. Stained cells were examined at 10× and at 60× magnification. The numeric data as presented are expression/phosphorylation compared with vehicle control expression/phosphorylation levels. (n = 3+/−SD) *p < .05 less than vehicle control value. ERK2, extracellular regulated kinases 2; siRNA, small interfering RNA
Finally, we determined the impact of knocking down Beclin1 on the neratinib-dependent dephosphorylation of Ezrin T567. Neratinib exposure again did not alter the total expression of Ezrin protein but did significantly decrease Ezrin T567 phosphorylation (Figure 8d; Figure S13). Knockdown of Beclin1 prevented the dephosphorylation of Ezrin T567, and as we have noted, prevented MST4 degradation. Thus, even though neratinib has been proposed to be a low nanomolar IC50 catalytic inhibitor of MST4, the primary mechanism by which neratinib regulates Ezrin T567 phosphorylation is instead via the autophagic degradation of MST4. And hence, in the meantime, the precise primary target(s) of neratinib still remains elusive.
4 |. DISCUSSION
The present studies were performed to increase our understanding of the molecular mechanisms by which neratinib regulates tumor cell biology. The key findings are (a) without Rubicon-dependent LC3-associated phagocytosis and without the enhanced structural and complex forming function of Merlin, neratinib-induced inactivation of AKT, ERK1/2, mTOR, and p70 S6K does not occur; (b) neratinib rapidly reduces the expression of MST4 and the phosphorylation of its substrate Ezrin T567 that results in altered cell morphology, an event that requires Beclin1, but is independent of Rubicon or Merlin; (c) the membrane stiffness-sensor RAP2A plays an essential role in maintaining the basal phosphorylation of LATS1/2 in pancreatic cancer cells but that RAP2A neither mediates neratinib-induced LATS1/2 phosphorylation or YAP/TAZ phosphorylation. The ability of neratinib to alter membrane rigidity and morphology via destruction of MST4 concomitant with the regulation of downstream signaling into the Hippo pathway we wish to call Eklusis; from the Latin for a gradual loss of focus (Figure S14).
As it was originally developed, neratinib was claimed to be a specific irreversible inhibitor of ERBB family receptor tyrosine kinases. In our first publication using this drug, we discovered it could cause the downregulation of the ERBB1 expression via ubiquitination and autophagy, of ERBB3 to which it associates but does not chemically modify and of other receptor tyrosine kinases who localize in quaternary structures alongside ERBB family receptors in the plasma membrane (Claus et al., 2018; Dent, Booth, Roberts et al., 2019). RAS proteins, located on the inner leaflet of the plasma membrane, were also shown to be rapidly downregulated by the drug. Chemical biology data from several groups indicated that neratinib has inhibitory properties against multiple MAP4K serine/threonine kinase targets and our initial work centered on MST1/2, MST3, and MST4 (Dent, Booth, Roberts et al., 2019). In particular that the MST4 target Ezrin became dephosphorylated after neratinib exposure that was associated with reduced MST4 expression and phosphorylation in both pancreatic cancer cells and in blood cancer cells (Figure S14). The data presented in this manuscript has provided additional mechanistic information and further focused on the directions of any putative future studies. Our data delineating a RAP2A-dependent pathway by which membrane stiffness is sensed and how it signals to the Hippo pathway is the same as described by others (Meng et al., 2018). And, our initial findings with RAP2A and MAP4K4 also initially suggested a simple route from neratinib to the regulation of the Hippo pathway, yet as we subsequently demonstrated, this hypothesis was proven to be false.
Neratinib causes Ezrin T567 dephosphorylation that correlates with a reduction in membrane stiffness and altered cell morphology. However, our prior and present data strongly argue that Ezrin T567 dephosphorylation requires degradation of MST4, and not per se its catalytic inhibition. Thus, although some aspects of the chemical biology data and our own work appear congruent, the actual mechanism by which neratinib regulates MST4-Ezrin biology, autophagy, diverges from the chemical biology claims. So, we now have to ask ourselves the obvious question; how does neratinib reduce MST4 expression, bearing in mind that MST4 degradation occurs in the absence of ERBB receptor family expression? As of December 2019, there are 71 Medline-listed manuscripts in which MST4 has been studied. The scaffold protein MO25 plays a key role in facilitating MST4 activation as well as activation of the AMP-dependent protein kinase (AMPK); neratinib increases MO25 expression, activates the AMPK and elevates LATS1/2 phosphorylation (Dent, Booth, Roberts et al., 2019). As neratinib rapidly alters cellular morphology before any possibility of altered protein transcription being responsible for the changes in MST4 biology, the altered MO25 expression may also not be a component of the key neratinib signaling mechanism.
There are multiple MAP4K family enzymes in a tumor cell. Our present findings show that neratinib increased MAP4K4 phosphorylation but this was not required for the neratinib-dependent enhancement of either LATS1/2 or YAP/TAZ phosphorylation. Neratinib reduced Merlin S518 phosphorylation that causes increased Merlin plasma membrane localization and this correlates with the observation that AKT was unable to phosphorylate either mTOR or p70 S6K. that is, we are altering the formation/destruction of quaternary signaling complexes liminal to the plasma membrane. Signaling by MAP4K4 is known to activate the p38 MAPK and JNK pathways, effects that were also observed in our studies (Rawat & Chernoff, 2015). Expression of dominant-negative p38 MAPK or use of the JNK inhibitory peptide significantly reduced (neratinib+valproate) lethality. This suggests one component of immediate early death signaling by MAP4K4 may be mediated by the p38 and JNK pathways.
MAP4K4 plays multiple roles in a variety of cell types. It can act to suppress skeletal muscle differentiation and knockdown of MAP4K4 can suppress systemic inflammation, including the production of IL-6 (Aouadi et al., 2009; Huang et al., 2014; Wang et al., 2013). Perhaps more significantly, based on our prior data showing that (neratinib+valproate) can enhance the antitumor actions of anti-PD1 and anti-CTLA4 antibodies, is that MAP4K4 plays an essential role in promoting T cell proliferation and activation and in parallel preventing regulatory T cell generation (Aouadi et al., 2009; Booth, Roberts, Poklepovic et al., 2017; Booth, Roberts, & Dent et al., 2019; Chuang, Wang, & Tan, 2016; Huang et al., 2014; Jin et al., 2016; Tesz et al., 2007; Wang et al., 2013). Although RAP2A-MAP4K4 did not appear to be involved in neratinib effects, it is known that MAP4K4 also plays a key role regulating cytoskeletal remodeling and motility, potentially linking its activation to neratinib-effects on Rubicon and LAP (Ma & Baumgartner, 2013; Ma & Baumgartner, 2014; Vitorino et al., 2015). Our data demonstrating that neratinib and HDAC inhibitors combined to kill multiple malignant T regulatory cell lines also suggests that this drug combination could have antitumor efficacy in cutaneous hematological cancers (Dent, Booth, Roberts et al., 2019).
Neratinib reduced Ezrin T567, PAK1 T423, and Merlin S518 phosphorylation in a Rubicon-dependent fashion that collectively implies that neratinib-enhanced Merlin association with the plasma membrane is a secondary effect which requires the initiation of LAP. Merlin-deficient human meningioma cells and in arachnoidal cells with Merlin knockdown display constitutive mTORC1 activation (James et al., 2009). Rho A/PAK-dependent phosphorylation of Merlin S518 causes the protein to mislocalize into the cytoplasm from the plasma membrane and this effect can be recapitulated by a phosphorylation site mimetic (S518D) (Surace, Haipek, & Gutmann, 2004). Mutant Merlin S518D does not suppress tumor growth or invasion, exhibits reduced binding to the plasma membrane glycoprotein CD44 and which is also associated with profound changes in cell morphology and organization of the actin cytoskeleton (Feng et al., 2016; Prolo et al., 2019; Stamenkovic & Yu, 2010; Takahashi, Haga, & Tanihara, 2015; Tanaka et al., 2017). Further studies will be required to link LAP, macro-autophagy, MST4 expression, and Merlin plasma membrane localization.
Neratinib has been claimed, via chemical biology assessments, to be a catalytic inhibitor of MST4 with an IC50 in the low nanomolar range; however, as noted previously and in the present studies, neratinib also rapidly reduced the total expression of MST4 via autophagy. Knockdown of Beclin1 not only prevented MST4 degradation but also prevented neratinib from causing Ezrin T567 dephosphorylation and altering the morphology of pancreatic tumor cells. These findings imply that regardless of its inhibitory effect on MST4 kinase activity, which appeared to be negligible in the transfected cells, reduced protein expression of MST4 is essential to promote changes in cell morphology. Whether reduced signaling by “MST family kinases” and Ezrin dephosphorylation is responsible for the primary neratinib-dependent induction of LAP will require studies beyond the scope of the present manuscript (Martinez, 2018; Wong, Sil, & Martinez, 2018).
In the absence of Rubicon or Merlin, the ability of neratinib to rapidly cause K-RAS V12 – GFP to mislocalize into the cytoplasm as intense staining vesicles became reduced. However, knockdown of Rubicon or of Merlin did not prevent neratinib from altering tumor cell morphology, with vehicle-treated cells appearing as oval or round balls and neratinib-treated cells appearing flattened and multi-sided. These events correlated with phosphorylation of MAP4K4 S629 and the dephosphorylation of Ezrin T567 and Merlin S518. Ezrin, Rubicon, and MAP4K4 all play important roles in regulating plasma membrane and Golgi dynamics, with some studies arguing MAP4K4 promotes the epithelial–mesenchymal transition with invasion and others arguing it prevents the EMT (Ma & Baumgartner, 2013; Ma & Baumgartner, 2014; Vitorino et al., 2015). Based on a literature search, the putative interactions between LAP and Merlin or MAP4K4 have yet to be studied (Martinez, 2018; Wong et al., 2018). The possible interactions between Merlin and MAP4K4 also have yet to be explored. It is known that MAP4K4 signaling can cause c-MET to become internalized, and we have previously observed neratinib reducing the levels of c-MET and c-KIT (Booth, Roberts, Poklepovic et al., 2017; Tripolitsioti et al., 2018). Dephosphorylation of Merlin S518 promotes its liminal association with the plasma membrane, forming new complexes of signaling proteins (Hill, Wrobel, & Rubinsztein, 2019; James et al., 2009; Kissil, Johnson, Eckman, & Jacks, 2002; Rong, Surace, Haipek, Gutmann, & Ye, 2004; Tripolitsioti et al., 2018).
Neratinib has been claimed by several groups to inhibit MAP4K and MAP3K enzymes. The most potently inhibited IC50 serine/threonine kinases are MAP4K5 0.65 nM, MAP4K3 7.7 nM, MST3 6.6 nM, MST4 7.4 nM, YSK1 12 nM, YSK4 16 nM, MINK 29 nM, and MAP3K4 39 nM. In contrast, serine/threonine kinases whose IC50 inhibition is in the 100 nM range include MAP4K1 93 nM, PAK2 79 nM, MST2 58 nM, SLK 66 nM, BLK 85 nM, MAP3K3 130 nM, TNIK 140 nM, and SNARK 140 nM. In the 200 nM and above IC50 range are PAK1 210 nM, MAP4K2 220 nM, and MAP4K4 330 nM. RAP2A has been proposed to regulate the activities of MAP4K4, MAP4K6, and MAP4K7. Thus, “in theory,” neratinib-induced MAP4K4 activation would be downstream of RAP2A in mediating signals to LATS1/2. However, RAP2A signaling was not required for neratinib to increase the phosphorylation of either LATS1/2, YAP, or TAZ. Knockdown of Rubicon prevented neratinib from causing the dephosphorylation of Merlin S518 and from enhancing the phosphorylation of LATS1/2 T1079 and YAP S127 but knockdown of Rubicon did not prevent neratinib from altering cell morphology. Thus, the biology of Rubicon, Beclin1, MST4, and Ezrin will very likely play key roles in mediating the anticancer effects of neratinib. Many additional studies, beyond the scope of this manuscript, will be required to fully define the plasma membrane-localized molecular targets and signaling pathways regulated by neratinib.
At present, the Phase I trial combining ([neratinib+valproate) (NCT03919292) is recruiting patients, with one pancreatic cancer patient at the lowest dose level exhibiting stable disease for 6 months associated with a dramatic increase in appetite (Booth, Roberts, Poklepovic et al., 2017; Booth, Poklepovic, & Dent et al., 2019; Dent, Booth, Roberts et al., 2019). Future in vivo studies exploring the scheduling and safety of administering statins and sildenafil as alternate inhibitors of RAS function alongside (neratinib+valproate) are being planned.
ACKNOWLEDGMENTS
Support for the present study was funded from philanthropic funding from Massey Cancer Center and Universal Inc. Chair in Signal Transduction Research. PD acknowledges funding by the Commonwealth Health Research Board (CHRB) of Virginia (grant 236-02-18). PD has received funding support from Puma Biotechnology for these studies.
Abbreviations:
- ERK
extracellular regulated kinase
- ca
constitutively active
- dn
dominant negative
- AMPK
AMP-dependent protein kinase
- mTOR
mammalian target of rapamycin
- MAPK
mitogen activated protein kinase
- si
small interfering
- VEH
vehicle
- NER
neratinib
- VAL
sodium valproate
Footnotes
CONFLICTS OF INTEREST
PD has received funding support from Puma Biotechnology for these studies. AP is the PI of a clinical trial combining neratinib with sodium valproate; no payment is being paid by Puma to AP. No additional conflicts of interest are present for any of the other authors.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
DATA AVAILABILITY STATEMENT
Data will be available upon an appropriate request.
REFERENCES
- Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E, … Czech MP (2009). Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature, 458, 1180–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardestani A, Li S, Annamalai K, Lupse B, Geravandi S, Dobrowolski A, … Maedler K (2019). Neratinib protects pancreatic beta cells in diabetes. Nature Communications, 10, 5015. 10.1038/s41467-019-12880-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae SJ, & Luo X (2018). Activation mechanisms of the Hippo kinase signaling cascade. Bioscience Reports, 38, 10.1042/BSR20171469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitra A, Sistla S, Mariam J, Malvi H, & Anand R (2017). Rassf proteins as modulators of Mst1 kinase activity. Scientific Reports, 7, 45020. 10.1038/srep45020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth L, Roberts JL, Poklepovic A, Avogadri-Connors F, Cutler RE, Lalani AS, & Dent P (2017). HDAC inhibitors enhance neratinib activity and when combined enhance the actions of an anti-PD-1 immunomodulatory antibody in vivo. Oncotarget, 8, 90262–90277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth L, Roberts JL, Rais R, Kirkwood J, Avogadri-Connors F, Cutler RE Jr, … Dent P. (2017). [Neratinib+Valproate] exposure permanently reduces ERBB1 and RAS expression in 4T1 mammary tumors and enhances M1 macrophage infiltration. Oncotarget, 9, 6062–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth L, Roberts JL, Sander C, Lalani AS, Kirkwood J, Hancock JF, … Dent P (2018). Neratinib and Entinostat combine to rapidly reduce the expression of K-RAS, N-RAS, Gαq and Gα11 and kill uveal melanoma cells. Cancer Biology & Therapy, 20, 700–710. 10.1080/15384047.2018.1551747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth L, Roberts JL, Rais R, Cutler RE Jr, Diala I, Lalani AS, … Dent P. (2019). Neratinib augments the lethality of [regorafenib+sildenafil]. Journal of Cellular Physiology, 234, 4874–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth L, Roberts JL, & Dent P (2019). The role of cell signaling in the crosstalk between autophagy and apoptosis in the regulation of tumor cell survival in response to sorafenib and neratinib. Seminars in Cancer Biology, 10.1016/j.semcancer.2019.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth L, Poklepovic A, & Dent P (2019). Not the comfy chair! Cancer drugs that act against multiple active sites. Expert Opinion on Therapeutic Targets, 23, 893–901. 10.1080/14728222.2019.1691526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Zhang H, Shi Z, Li Y, Zhang X, Gao Z, … Zhou Z (2018). The MST4-MOB4 complex disrupts the MST1-MOB1 complex in the Hippo-YAP pathway and plays a pro-oncogenic role in pancreatic cancer. Journal of Biological Chemistry, 293, 14455–14469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HC, Wang X, & Tan TH (2016). MAP4K Family Kinases in Immunity and Inflammation. Advances in Immunology, 129, 277–314. [DOI] [PubMed] [Google Scholar]
- Claus J, Patel G, Autore F, Colomba A, Weitsman G, Soliman TN, … Parker PJ (2018). Inhibitor-induced HER2-HER3 heterodimerisation promotes proliferation through a novel dimer interface. eLife, 7, e32271. 10.7554/eLife.32271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JJ, Bronner SM, & Zbieg JR (2018). Hippo pathway inhibition by blocking the YAP/TAZ-TEAD interface: A patent review. Expert Opinion on Therapeutic Patents, 28, 867–873. 10.1080/13543776.2018.1549226. [DOI] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, … Zarrinkar PP (2011). Comprehensive analysis of kinase inhibitor selectivity. Nature Biotechnology, 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Dent P, Booth L, Roberts JL, Liu J, Poklepovic A, Lalani AS, … Hancock JF (2019). Neratinib inhibits Hippo/YAP signaling, reduces mutant K-RAS expression, and kills pancreatic and blood cancer cells. Oncogene, 38, 5890–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent P, Booth L, Poklepovic A, & Hancock JF (2019). Signaling alterations caused by drugs and autophagy. Cellular Signalling, 64, 109416. [DOI] [PubMed] [Google Scholar]
- Feng XJ, Pan Q, Wang SM, Pan YC, Wang Q, Zhang HH, … Zhang SH (2016). MAP4K4 promotes epithelial-mesenchymal transition and metastasis in hepatocellular carcinoma. TUMOUR BIOLOGY, 37, 11457–67. [DOI] [PubMed] [Google Scholar]
- Fujimura T, Okuyama R, Ito Y, & Aiba S (2008). Profiles of Foxp3+ regulatory T cells in eczematous dermatitis, psoriasis vulgar is and mycosis fungoides. British Journal of Dermatology, 158, 1256–63. [DOI] [PubMed] [Google Scholar]
- Geskin LJ, Akilov OE, Kwon S, Schowalter M, Watkins S, Whiteside TL, … Falo LD Jr. (2018). Therapeutic reduction of cell-mediated immunosuppression in mycosis fungoides and Sézary syndrome. Cancer Immunology and Immunotherapy, 67, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergovich A (2013). Regulation and functions of mammalian LATS/NDR kinases: Looking beyond canonical Hippo signalling. Cell & Bioscience, 3, 32. 10.1186/2045-3701-3-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergovich A (2016). The roles of NDR protein kinases in Hippo signalling. Genes (Basel), 7, 21. 10.3390/genes7050021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SM, Wrobel L, & Rubinsztein DC (2019). Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death and Differentiation, 26, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Tang Q, Chu H, Jiang J, Zhang H, Hao W, & Wei X (2014). MAP4K4 deletion inhibits proliferation and activation of CD4(+) T cell and promotes T regulatory cell generation in vitro. Cellular Immunology, 289, 15–20. [DOI] [PubMed] [Google Scholar]
- James MF, Han S, Polizzano C, Plotkin SR, Manning BD, Stemmer-Rachamimov AO, … Ramesh V (2009). NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Molecular and Cellular Biology, 29, 4250–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang JW, Kim MK, & Bae SC (2018). Reciprocal regulation of YAP/TAZ by the Hippo pathway and the Small GTPase pathway. Small GTPases, 20, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Chu H, Li Y, Tao X, Cheng Z, Pan Y, … Wei X (2016). MAP4K4 deficiency in CD4(+) T cells aggravates lung damage induced by ozone-oxidized black carbon particles. Environmental Toxicology and Pharmacology, 46, 246–254. [DOI] [PubMed] [Google Scholar]
- Kissil JL, Johnson KC, Eckman MS, & Jacks T (2002). Merlin phosphorylation by p21-activated kinase 2 and effects of phosphorylation on merlin localization. Journal of Biological Chemistry, 277, 10394–10399. [DOI] [PubMed] [Google Scholar]
- Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig PA, … Kuster B (2017). The target landscape of clinical kinase drugs. Science, 358, eaan4368. 10.1126/science.aan4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D, Zhao Y, Men T, & Teng CB (2015). Hippo signaling pathway in liver and pancreas: The potential drug target for tumor therapy. Journal of Drug Targeting, 23, 125–33. [DOI] [PubMed] [Google Scholar]
- Liu Y, & Deng J (2019). Ubiquitination-deubiquitination in the Hippo signaling pathway. Oncology Reports, 41, 1455–1475. [DOI] [PubMed] [Google Scholar]
- Ma M, & Baumgartner M (2013). Filopodia and membrane blebs drive efficient matrix invasion of macrophages transformed by the intracellular parasite Theileria annulata. PLoS One, 8, e75577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, & Baumgartner M (2014). Morphed and moving: TNFα-driven motility promotes cell dissemination through MAP4K4-induced cytoskeleton remodeling. Microbial Cell, 1, 154–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J (2018). LAP it up, fuzz ball: A short history of LC3-associated phagocytosis. Current Opinion in Immunology, 55, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, Mottier-Pavie V, Plouffe SW, Hansen CG, Hong AW, … Guan KL (2015). MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nature Communications, 6, 8357. 10.1038/ncomms9357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Qiu Y, Lin KC, Kumar A, Placone JK, Fang C, … Guan KL (2018). RAP2 mediates mechanoresponses of the Hippo pathway. Nature, 560, 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SH, Camargo FD, & Yimlamai D (2017). Hippo signaling in the liver regulates organ size, cell fate, and carcinogenesis. Gastroenterology, 152, 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pore D, Bodo J, Danda A, Yan D, Phillips JG, Lindner D, … Gupta N (2015). Identification of Ezrin-Radixin-Moesin proteins as novel regulators of pathogenic B-cell receptor signaling and tumor growth in diffuse large B-cell lymphoma. Leukemia, 29, 1857–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prolo LM, Li A, Owen SF, Parker JJ, Foshay K, Nitta RT, … Grant GA (2019). Targeted genomic CRISPR-Cas9 screen identifies MAP4K4 as essential for glioblastoma invasion. Scientific Reports, 9, 14020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat SJ, & Chernoff J (2015). Regulation of mammalian Ste20 (Mst) kinases. Tr. Biochem Sci, 40, 149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong R, Surace EI, Haipek CA, Gutmann DH, & Ye K (2004). Serine 518 phosphorylation modulates merlin intramolecular association and binding to critical effectors important for NF2 growth suppression. Oncogene, 23, 8447–54. [DOI] [PubMed] [Google Scholar]
- Sharif AAD, & Hergovich A (2018). The NDR/LATS protein kinases in immunology and cancer biology. Seminars in Cancer Biolog, 48, 104–114. [DOI] [PubMed] [Google Scholar]
- Stamenkovic I, & Yu Q (2010). Merlin, a “magic” linker between extracellular cues and intracellular signaling pathways that regulate cell motility, proliferation, and survival. Current Protein & Peptide Science, 11, 471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surace EI, Haipek CA, & Gutmann DH (2004). Effect of merlin phosphorylation on neurofibromatosis 2 (NF2) gene function. Oncogene, 23, 580–7. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Osada H, Murakami-Tonami Y, Horio Y, Hida T, & Sekido Y (2017). Statin suppresses Hippo pathway-inactivated malignant mesothelioma cells and blocks the YAP/CD44 growth stimulatory axis. Cancer Letters, 385, 215–224. [DOI] [PubMed] [Google Scholar]
- Takahashi E, Haga A, & Tanihara H (2015). Merlin regulates epithelial-to-mesenchymal transition of ARPE-19 cells via TAK1-p38MAPK-mediated activation. Investigative Ophthalmology and Visual Science, 56, 2449–58. [DOI] [PubMed] [Google Scholar]
- Tesz GJ, Guilherme A, Guntur KV, Hubbard AC, Tang X, Chawla A, & Czech MP (2007). Tumor necrosis factor alpha (TNFalpha) stimulates Map4k4 expression through TNFalpha receptor 1 signaling to c-Jun and activating transcription factor 2. Journal of Biological Chemistry, 282, 19302–12. [DOI] [PubMed] [Google Scholar]
- Thompson BJ, & Sahai E (2015). MST kinases in development and disease. Journal of Cell Biology, 210, 871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripolitsioti D, Kumar KS, Neve A, Migliavacca J, Capdeville C, Rushing EJ, … Baumgartner M (2018). MAP4K4 controlled integrin β1 activation and c-Met endocytosis are associated with invasive behavior of medulloblastoma cells. Oncotarget, 9, 23220–23236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitorino P, Yeung S, Crow A, Bakke J, Smyczek T, West K, … Ye W (2015). MAP4K4 regulates integrin-FERM binding to control endothelial cell motility. Nature, 519, 425–30. [DOI] [PubMed] [Google Scholar]
- Wang M, Amano SU, Flach RJ, Chawla A, Aouadi M, & Czech MP (2013). Identification of Map4k4 as a novel suppressor of skeletal muscle differentiation. Molecular and Cellular Biology, 33, 678–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SW, Sil P, & Martinez J (2018). Rubicon: LC3-associated phagocytosis and beyond. FEBS Journal, 285, 1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang MH, Zhao MY, Wang Z, Kang R, He YL, Yin XC, … Cao LZ (2011). WAVE1 regulates P-glycoprotein expression via Ezrin in leukemia cells. Leukemia and Lymphoma, 52, 298–309. [DOI] [PubMed] [Google Scholar]
- Zhou B, Leng J, Hu M, Zhang L, Wang Z, Liu D, … Zhang Q (2010). Ezrin is a key molecule in the metastasis of MOLT4 cells induced by CCL25/CCR9. Leukemia Research, 34, 769–76. [DOI] [PubMed] [Google Scholar]
- Zhang L, Xiao R, Xiong J, Leng J, Ehtisham A, Hu Y, … Hang Q (2013). Activated ERM protein plays a critical role in drug resistance of MOLT4 cells induced by CCL25. PLoS One, 8, e52384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be available upon an appropriate request.