

Abstract
Chronic elevated free fatty (FFA) levels are linked to metabolic disorders and tumorigenesis. However, the molecular mechanism by which FFAs induce cancer remains poorly understood. Here, we show that the tumor suppressor PTEN protein levels were decreased in high fat diet (HFD) fed mice. As palmitic acid (PA, C16:0) showed a significant increase in the HFD fed mice, we further investigated its role in PTEN down regulation. Our studies revealed that exposure of cells to high doses of PA induced mTOR/S6K-mediated phosphorylation of PTEN at T366. The phosphorylation subsequently enhanced the interaction of PTEN with the E3 ubiquitin ligase WW domain-containing protein 2 (WWP2), which promoted polyubiquitination of PTEN and protein degradation. Consistent with PTEN degradation, exposure of cells to increased concentrations of PA also promoted PTEN-mediated AKT activation and cell proliferation. Significantly, a higher level of S6K activation, PTEN T366 phosphorylation, and AKT activation were also observed in the livers of the HFD fed mice. These results provide a molecular mechanism by which a HFD and elevated PA regulates cell proliferation through inactivation of tumor suppressor PTEN.
Keywords: Palmitic acid, PTEN, T366 phosphorylation, mTOR, S6K
1. Introduction
Obesity is a global epidemic and associated with an increased risk of developing both metabolic disorders and cancer. Evidence from recent epidemiological studies suggests a strong correlation between dietary fat over-consumption and incidence and mortality of cancers, particularly in breast, colorectal, prostate, lung, ovarian and liver cancers[1–7]. It is well known that, with high fat diet (HFD) consumption and obesity, free fatty acid (FFA) levels are often elevated in the circulation and in tissue. Among circulating FFAs, palmitic acid (C16:0, PA) is the most abundant saturated FFAs[8]. Indeed, our recent metabolomic analysis of liver tissue of HFD fed mice also revealed a significant increase in PA level[9]. Despite its strong association with cancer[10–12], the direct effects of PA on cancer cell proliferation, as well as the underlying molecular mechanisms of action, remain largely unknown.
Tumorigenesis is a complex process involving aberrant level or impaired function of tumor suppressors and oncogenes as well as dysregulated molecular pathways. The nuclear phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a well-characterized tumor suppressor[13, 14]. PTEN achieves its tumor suppressive function primarily by antagonizing phosphatidylinositol 3-kinase (PI3K)/AKT through its lipid phosphatase activity[15]. Frequent dysregulation of PTEN, including mutations and deletions, has been found in a variety of cancers, which promotes both initiation and progression[16–18]. Thus, maintaining normal level and wild type activity of PTEN is critical for its tumor suppressive function. Mechanistically, PTEN activity and stabilization are modulated by posttranslational modifications including phosphorylation and ubiquitination[15]. It is well established that PTEN is phosphorylated at its C2 and C-tail domains, occurring at S362, T366, S370, S380, T382, T383 and S385[15, 19, 20]. The phosphorylation has been shown to lead to conformational change and attenuated PTEN activity. Specifically, T366 phosphorylation of PTEN has been suggested to destabilize PTEN via promoting PTEN ubiquitination[20]. The ubiquitin-mediated degradation of PTEN is thought to be the primary mechanism for the loss function of PTEN[21, 22]. The NEDD4-like E3 ligase WWP2 was found to interact with PTEN, leading to PTEN polyubiquitination and protein degradation[23]. Thus, targeting the WWP2-mediated polyubiquitination is critical for modulating the abundance of PTEN in cells.
In this study, we showed that whole cell extract from livers of mice fed a HFD exhibit a lower level of PTEN protein. As our previous metabolomic analysis of liver tissue from the same mice revealed that PA is the only fatty acid that showed a significant increase in the HFD fed mice [9], we proceeded to test the role of elevated PA in PTEN down regulation. Our results reveal that treatment of cells with a high concentration of PA reduced PTEN protein levels. Mechanistically, PA treatment led to phosphorylation of PTEN at T366 through activation of mammalian target of rapamycin (mTOR)/S6K pathway. Phosphorylation of PTEN in turn increased its interaction with the E3 ligase WWP2, leading to PTEN polyubiquitination and protein degradation. Importantly, our data suggest that PTEN T366 phosphorylation and protein degradation led AKT activation, which promotes cell proliferation upon PA exposure. Collectively, our studies elucidate a novel molecular mechanism by which PA regulates cancer cell proliferation under conditions of obesity and over-consumption of dietary fat.
2. Materials and methods
2.1. Reagents and antibodies
Palmitic acid (C16:0, PA) (Sigma, Burlington, MA, USA) was added to the cell culture medium as a PA-BSA complex, as described[8]. mTOR inhibitor RAD001 (20 nM, 16h) and S6K inhibitor NaSal (10 mM, 16h) were purchased from MedChemExpress (Monmouth Junction, NJ, USA) and Selleck (Houston, TX, USA), respectively. The proteasome inhibitor MG132 (20 μM, 6h) was purchased from A.G Scientific (San Diego, CA, USA). The lysosomal inhibitor leupeptin (50 μM, 16h) was purchased from Sigma (Burlington, MA, USA). The GSK3β inhibitor TWS119 (10 μM, 16h) was obtained from Santa Cruze (Dallas, Texas, USA). Cycloheximide (50 μM) was purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-PTEN, anti-Phospho-PTEN (Ser380), anti-p70S6K, anti-Phospho-p70S6K (T389) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-Phospho-PTEN (Thr366) antibody was purchased from Abcam (Cambridge, MA, USA). Anti-vinculin, anti-HA, anti-WWP2 antibodies and PTEN-siRNA (EHU106441) were purchased from Sigma (Burlington, MA, USA). WWP2-siRNA (sc-40362) and control-siRNA (sc-37007) were obtained from Santa Cruze (Dallas, Texas, USA). S6K and mTOR inhibitors were added to cells for 1 h before PA treatment.
2.2. Animals and diets
Ethics Statement: Care and treatment of animals was in accord with guidelines from and approval by the University of California Riverside Institutional Animal Care and Use Committee (AUP#20140014). All mice had ad libitum access to food and water (except during two overnight fasts as described previously[9]). At the end of the study, mice were euthanized by carbon dioxide inhalation (before noon), in accordance with stated NIH guidelines.
C57BL/6N male mice (Charles River Laboratories) were weaned at three weeks of age and fed either low-fat, normal lab chow (control) or a 40 kcal% high fat diet, enriched in coconut oil (8.7% PA) and soybean oil (10.6% PA) for 24 weeks. The animals were maintained on a 12:12 h light-dark cycle in a specific pathogen free vivarium. Details regarding diet formulation and mouse metabolic phenotype and patty acid analysis data, including body weight have been published previously[9].
2.3. Cell culture, immunoblotting and immunoprecipitation
Cells were cultured with McCoy’s 5A containing 10% fetal bovine serum (Omega Scientific, CA, USA) and maintained at 37°C in a humidified atmosphere with 5% CO2. Cell transfection was performed with Lipofectamine 2000 (Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Following indicated treatments or transfection, cells were harvested and lysed with NP-40 lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40, 15 mM EDTA). The protein concentration was determined with BCA assay kit (Fisher Scientific, Waltham, MA, USA). Approximately 20 μg of total protein was separated by SDS-PAGE and subjected to immunoblot (IB) analysis.
For co-immunoprecipitation (co-IP), cell lysate was pre-cleared with Protein A beads (Fisher Scientific, Waltham, MA, USA) at 4°C for 45 min. Afterwards, indicated antibody or control IgG was added and incubated at 4°C overnight. The protein A beads were added and the samples were incubated for another 4 h at 4°C. The beads were then washed three times with NP-40 lysis buffer and then boiled with in SDS loading buffer. The co-IPed proteins were separated by SDS-PAGE and detected by IB analysis.
For tissue samples, 100 mg of mouse liver was added into the soft tissue homogenizing microtubes (OMNI International, Kennesaw, GA, USA) containing 1 ml freshly prepared NP-40 lysis buffer. The tissues were immediately homogenized using the Precellys®24 Homogenizer (Bertin, France) at 6500 rpm for 2×30 sec in the presence of Pierce™ Phosphatase Inhibitor Mini Tablets (Thermo Fisher, Rockford, IL, USA). The homogenized mix was transferred into pre-chilled 1.5 ml tubes and centrifuged at 12,000 rpm for 15 min at 4°C. The clarified supernatant was mixed with equal volume of loading buffer and subjected to SDS-PAGE followed by IB analysis.
2.4. QPCR
Total RNA was extracted using the TRIzol reagent (Life Technologies, Carlsbad, CA, USA). Reverse transcription was performed with 0.5 μg RNA using the iScript Reverse Transcription Supermix (Bio-Rad, Hercules, CA, USA). The mRNA level of PTEN was determined with iQ™ SYBR green supermix (Bio-Rad, Hercules, CA, USA). The primers used were as follows: forward, 5’- ACCCACCACAGCTAGAACTT and reverse, 5’- GGGAATAGTTACTCCCTTTTTGTC. The relative mRNA level of PTEN was normalized to GAPDH and quantified with the 2−ΔΔCq method.
2.5. Ubiquitination assay
U2OS cells were co-transfected with expression vector for His-ubiquitin and HA-PTEN. Twenty-four hours after transfection, PA was added into the medium and the cells were incubated an additional 16 h at 37°C. Cells were then collected, washed with pre-chilled phosphate-buffered saline (PBS) and lysed with pull-down lysis buffer (6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH 8.0, 5 mM imidazole and 10 mM β-mercaptoethanol) for 30 min. Cell lysate was incubated with Ni2+-NTA beads for 4 h at RT. Beads were sequentially washed once with wash buffer I (6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH 8.0), wash buffer II (8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH 8.0) and wash buffer III (8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl, pH 6.3, 0.2% Triton X-100) respectively. The ubiquitinated products were eluted by incubating the beads with 50 μl of elution buffer (500 mM imidazole, 0.15 M Tris-HCl, pH 6.7, 30% glycerol, 5% SDS and 0.72 M β-mercaptoethanol) for 10 min at RT. The ubiquitination of PTEN was detected via IB with an anti-PTEN antibody.
2.6. BrdU incorporation and Cell Counting Kit-8 (CCK8) assay
Cell proliferation was determined by incorporation of BrdU into DNA using the BrdU Cell Proliferation Assay Kit (Millipore, Temecula, CA, USA) according to the manufacturer’s instruction. Briefly, U2OS cells were seeded into the 96-well plate with the density of 1,000 cells per well in 100 μl of cell culture medium. Twenty-four hours after seeding, 20 μl of 500x diluted BrdU reagent and PA were added into the medium and incubated for 16 h. The cells were fixed for 30 min at RT. Following fixing, the plate was washed three times with wash buffer and incubated with 100 μl of pre-diluted anti-BrdU monoclonal antibody for 1 h at RT followed by peroxidase conjugated secondary antibody for 30 min at RT. The plate was then incubated with 100 μl of TMB Peroxidase Substrate for 30 min at RT in the dark and read at 450 nm. The experiment was performed in triplicate.
The cell proliferation was also detected by the Cell Counting Kit-8 (CCK-8) assay. Briefly, cells were seeded into the 96-well plate (1,000 cells/well) and treated with PA for 16 h. 10 μl of CCK-8 solution was added into the medium and cultured for 4 h at 37°C. The absorbance of each well at 450 nm was detected by the microplate reader (Bio-Rad, Hercules, CA, USA).
2.7. Statistical analysis
The data are presented as mean ± standard deviation (SD). The difference between two groups was analyzed with the Student’s t test or One-way ANOVA. The statistical analysis was performed using the GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA, USA). Values of P<0.05 were considered as statistically significant.
3. Results
3.1. Elevated palmitic acid prompts the degradation of PTEN
To explore the potential effect of elevated FFA on PTEN tumor suppressor activity, we first examined PTEN protein levels in the liver of mice fed with a high fat diet (HFD)[9]. The results show that the PTEN protein level was remarkably decreased in the liver tissue from HFD mice compared with that of the control group (Fig. 1a and Supplementary Fig. 1a). By comparison, the PTEN mRNA level did not show any significant change (Supplementary Fig. 1b). Because our previous metabolomic analysis of liver tissue from the same mice revealed that PA is the only fatty acid that showed significant difference between HFD and control mice[9], we investigated whether elevated PA is responsible for the PTEN down-regulation. As shown in Figure 1b and Supplementary Figure 2a, exposure of U2OS cells to high doses of PA significantly reduced the abundance of PTEN protein in a dose- and time-dependent manner. A similar result on PTEN protein reduction was observed in another cancer cell line HCT116 (Supplementary Fig. 2b). Furthermore, the PTEN mRNA level was not affected under those conditions (Supplementary Fig. 2c). Finally, we measured the PTEN degradation rates following PA treatment and confirmed the PA-induced protein degradation (Supplementary Fig. 2d). These data indicate that elevated PA alone is capable of negatively modulating the PTEN protein levels.
Figure 1.
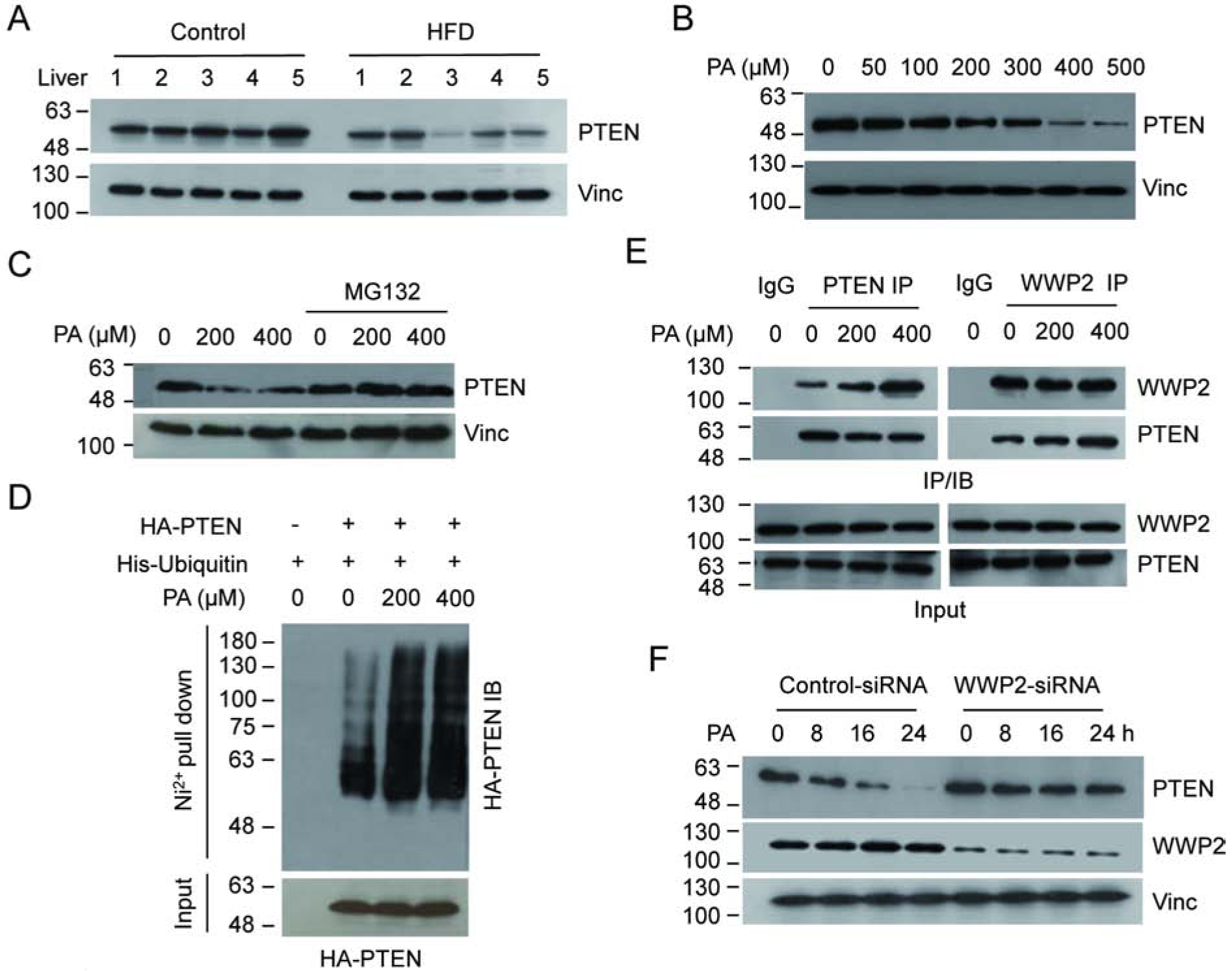
Elevated PA levels correlate with decreased PTEN. (A) The PTEN levels in whole cell extracts from five HFD fed mice and five control mice were determined by IB analysis with PTEN antibody. (B) U2OS cells were treated with increasing concentrations of PA and the protein level of PTEN was detected. (C) U2OS cells exposed to PA were treated with or without MG132 and the PTEN levels were detected. (D) U2OS cells were co-transfected with His-ubiquitin and HA-PTEN expressing vectors. The ubiquitinated products were pull downed by Ni2+-NTA beads under denaturing condition and PTEN was detected by HA antibody. (E) U2OS cells were treated with MG132 to normalize protein levels and the interaction between endogenous WWP2 and PTEN, with or without PA, was determined by reciprocal IP/IB as indicated. (F) U2OS were transfected with either WWP2 or control siRNA. The protein levels of PTEN and WWP2 were detected by IB analysis.
It has been well documented that ubiquitin proteasome-mediated protein degradation plays a critical role in regulating the protein stability of PTEN[23]. We therefore investigated whether this process plays a role in PA-induced PTEN down-regulation. As shown in Figure 1c and Supplementary Figure 2e, treatment of the proteasome inhibitor MG132, but not the lysosomal inhibitor leupeptin, blocked the PTEN reduction upon PA exposure, suggesting that the ubiquitin-proteasome pathway may play a role in the process. To test this directly, we next used a pull-down assay to examine polyubiquitination of PTEN in the presence or absence of elevated PA. Our results reveal that PA treatment indeed enhances the polyubiquitination level of PTEN (Fig. 1d). Furthermore, because the E3 ligase WWP2 contributes greatly to PTEN polyubiquitination, we examined its role in PA-induced PTEN polyubiquitination by measuring its interaction with PTEN. When cells were treated with elevated PA, endogenous PTEN-WWP2 interaction was enhanced as detected by reciprocal immunoprecipitation (Fig. 1e), which is consistent with observed PTEN polyubiquitination. To further establish the role of WWP2 in PTEN degradation, we knocked down WWP2 using specific small interfering RNA (WWP2-siRNA) and showed that PA-induced PTEN degradation was abolished under WWP2 abrogation condition (Fig. 1f). Together, these results demonstrate that treating cells with higher concentrations of PA enhances the polyubiquitination of PTEN and triggers its protein degradation via the ubiquitin-proteasome pathway.
3.2. Elevated PA leads to PTEN phosphorylation at T366
It has been demonstrated that the phosphorylation of PTEN at T366 and S380 is critical for modulating PTEN stability[20]. We thus investigated the effect of elevated PA on T366 and S380 phosphorylation (Fig. 2a). Upon PA treatment, increased PTEN phosphorylation at T366 was clearly detected while phosphorylation at S380 was only marginally affected. Importantly, increased PTEN T366 phosphorylation was also detected in the liver samples from HFD fed mice (Fig. 2b). To further confirm the role of T366 phosphorylation in PA-induced PTEN degradation, we expressed PTEN phosphorylation-defective mutant T366A or phosphorylation mimic mutant T366D and examined their response to PA treatment (Fig. 2c). While PA treatment led to the reduction of wild-type PTEN, it did not have a noticeable effect on either PTEN T366 phosphorylation mutants. These results suggest that T366 phosphorylation is required for PA-induced PTEN down-regulation
Figure 2.
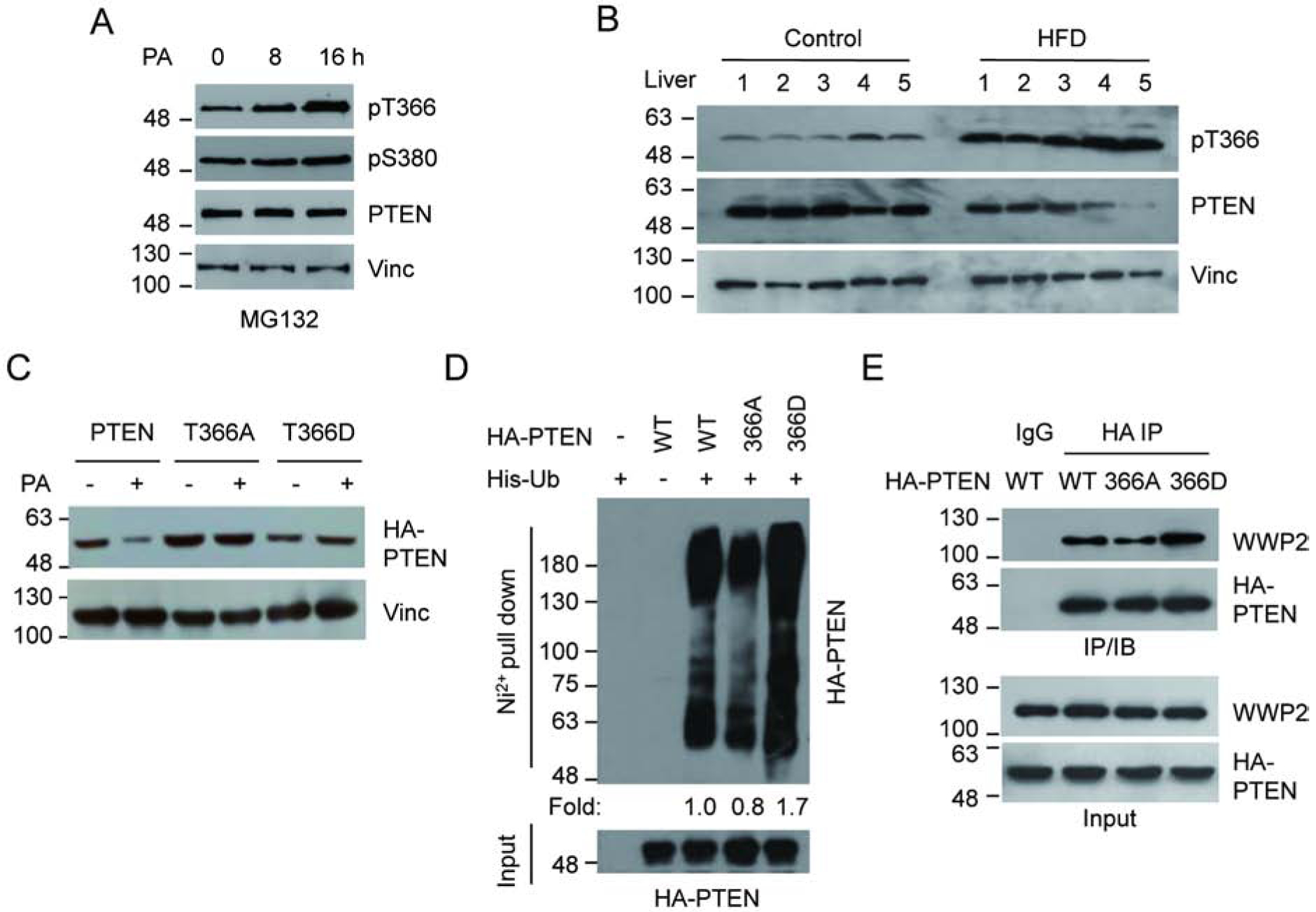
PA promotes the phosphorylation of PTEN at T366. (A) U2OS cells were treated with 400 mM PA in the presence of MG132, and the level of PTEN phosphorylation was examined. (B) PTEN T366 phosphorylation of liver samples from five HFD fed mice and five controls were determined by IB analysis using corresponding antibodies. (C) U2OS cells were transfected with HA-PTEN, HA-T366A or HA-T366D expressing vectors, treated with PA or control, and the PTEN protein level was determined by IB analysis. (D) U2OS cells were transfected with His-ubiquitin together with wild-type or mutant PTEN. Ubiquitinated PTEN was pull downed by Ni2+-NTA beads and detected by HA antibody (E) The interaction between WWP2 and PTEN mutants was examined in U2OS cells by IP with anti HA antibody, followed by IB with WWP2 antibody after transfected with wild-type or mutant HA-PTEN expressing vectors.
To provide further evidence for the involvement of T366 phosphorylation in PTEN stability, we compared polyubiquitination levels of the T366A and T366D mutants in U2OS cells (Fig. 2d). Consistent with the protein levels observed in the cell (Fig. 2c), the T366A mutant showed a reduced level of polyubiquitylation compared to wild-type PTEN while the T366D mutant showed an increased level of polyubiquitylation. The T366A mutant also showed decreased interaction with the E3 ligase WWP2 compared to wild-type PTEN while the T366D mutant showed an increased interaction (Fig. 2e). Of note, the WWP2 interacting domain has been mapped within 100–187aa on PTEN. Thus T366 phosphorylation is likely to cause a conformational change of the protein, thus affecting its protein-protein interaction. These data support the finding that the phosphorylation of PTEN at T366 facilitates the PTEN-WWP2 interaction, which consequently triggers the polyubiquitination and subsequent degradation of PTEN.
3.3. Activation of the mTOR/S6K pathway is required for PA-induced PTEN degradation
It has been reported that PTEN phosphorylation is regulated by the mTOR/S6K pathway[8]. To investigate the role of mTOR/S6K in PA-induced PTEN phosphorylation and down-regulation, we first examined the effect of PA on S6K activation by measuring S6K T389 phosphorylation (pS6K). The results clearly indicated an increased S6K phosphorylation upon PA treatment. Furthermore, treatment with the specific inhibitor of mTOR RAD001 blocks the increased phosphorylation (Fig. 3a). Importantly, the PA-induced PTEN T366 phosphorylation was also blocked in the presence of the mTOR inhibitor RAD001 as well as the S6K inhibitor NaSal (Fig. 3c). Consistent with these observations, the PA-induced PTEN down-regulation (Fig. 3c) and PTEN-WWP2 interaction (Fig. 3d) were also blocked by the mTOR and S6K inhibitors. Since it was reported that GSK3β also phosphorylated T366 of PTEN[24, 25], we treated cells with the GSK3β inhibitor TWS119 and found that PA-induced PTEN T366 phosphorylation and protein down regulation were not affected (Fig. 3c). Importantly, increased S6K phosphorylation levels were also detected in the liver tissue from HFD mice (Fig. 3b). Together, the data indicate that the mTOR/S6K pathway is an important upstream regulator for PA-induced PTEN phosphorylation and down-regulation.
Figure 3.
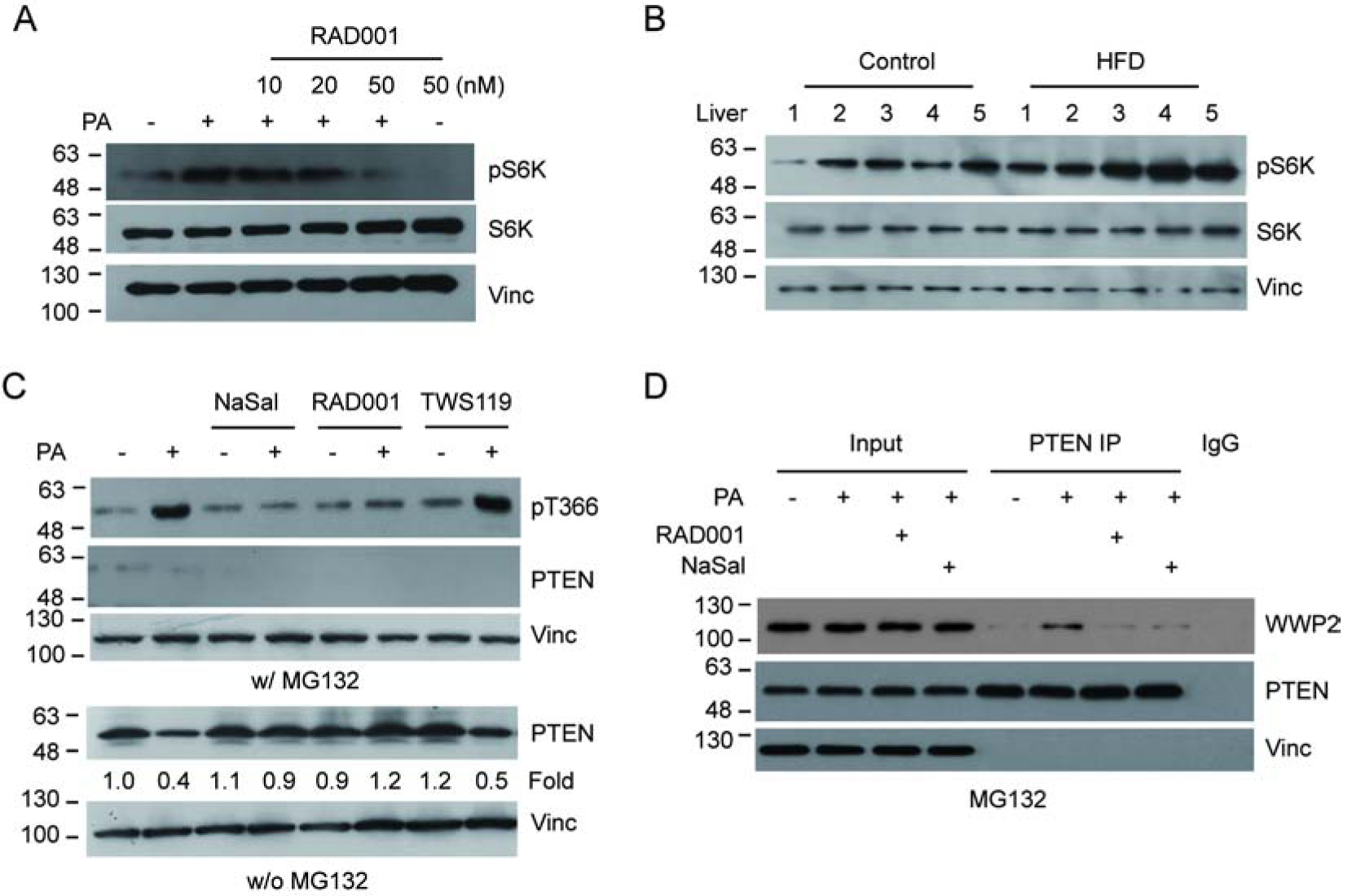
PA induces PTEN T366 phosphorylation via the mTOR/S6K pathway. (A) U2OS cells were treated with PA in the presence or absence of the mTOR inhibitor RAD001 as indicated. Activation of S6K was detected using S6K pT389 antibody (pS6K). (B) S6K phosphorylation of liver samples from five HFD fed mice and five controls were determined by IB analysis. (C) U2OS cells were treated with PA in the presence of MG132, RAD001, the S6K inhibitor NaSal or the GSK3β inhibitor TWS119. PTEN protein and T366 phosphorylation levels were detected by IB with the corresponding antibodies as indicated. (D) U2OS cells were treated with PA, with or without NaSal or RAD001. The interaction between endogenous WWP2 and PTEN was examined as indicated.
3.4. Elevated PA activates the Akt pathway and promotes cancer cell proliferation
PTEN has been identified as a key negative regulator of the Akt pathway. As elevated PA down-regulates PTEN, we next investigated whether this down-regulation led to Akt activation. Upon PA treatment, our analysis indeed showed increased Akt activation as indicated by Akt S473 phosphorylation (pAkt), and importantly, restoration of PTEN level attenuated the PA-induced Akt activation (Fig. 4a). To further validate the role of PTEN down-regulation in PA-induced Akt activation, we showed that treating PTEN−/− cells with elevated level of PA did not significantly activate Akt (Fig. 4b). Significantly, consistence with the cell culture data, the increased pAkt was also observed in the liver samples from HFD fed mice compared with that of the control group (Fig. 4c).
Figure 4.
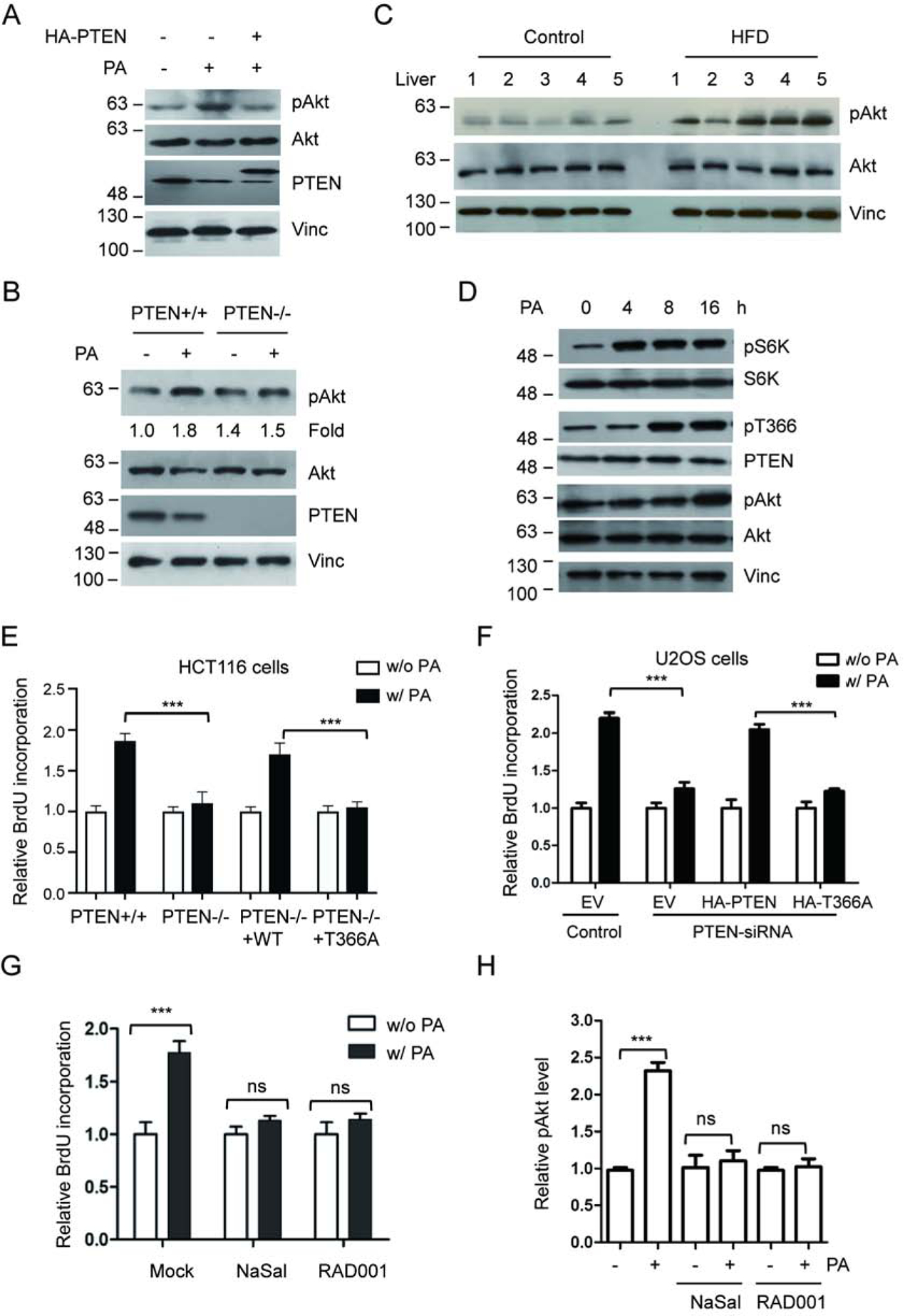
PA treatment promotes cancer cell proliferation. (A) U2OS cells were treated with PA, with or without PTEN overexpression, and activation of Akt was detected using anti-Akt p-S473 antibody (pAkt). (B) HCT116 PTEN+/+ or PTEN−/− cells were exposed to PA and Akt, pAkt and PTEN were detected with corresponding antibodies. (C) Activation of Akt in liver samples for five HFD-fed mice and five control mice was determined with anti-Akt pS473 antibody (pAkt). (D) U2OS cells were treated with 400 mM PA. Following the treatment the phosphorylation levels of S6K, PTEN, or Akt were detected by IB with corresponding antibodies at the indicated times. (E) HCT116 PTEN+/+ or PTEN−/− cells were treated with control or 400 mM PA, transfected with wild type PTEN or T366A, and cell proliferation was determined by measuring the incorporation of BrdU into DNA. (F) U2OS cells, transfected with PTEN-siRNA, wild type PTEN or T366A as indicated, were treated with control or 400 mM PA. Cell proliferation was determined by measuring the incorporation of BrdU into DNA. (G, H) U2OS cells were treated with control or 400 mM PA in the presence or absence of RAD001 or NaSal. Cell proliferation was measured by incorporation of BrdU (G) and Akt phosphorylation levels were determined by IB and normalized to Akt protein levels (H). ***P<0.001, One-way ANOVA.
As the activation of Akt pathway positively regulated the cell growth, we looked for the influence of PA on cell proliferation. Upon treating cell with PA, analysis of HCT116 PTEN+/+ cells by BrdU incorporation (Fig. 4e) and by CCK8 assay (Supplementary Fig. 3a) showed increased cell proliferation compared to control cells, while analysis of HCT116 PTEN−/− cells only showed a marginal response to PA treatment. Importantly, re-introduction of wild type PTEN, but not T366 phosphorylation mutant (T366A), into PTEN−/− cells restored PA-induced cell proliferation. Similarly, upon treating U2OS cell with PA, analysis of cell growth showed increased cell proliferation (Fig. 4f and Supplementary Fig. 3b). Knocking down of PTEN using PTEN-siRNA (Supplementary Fig. 3c) significantly reduced the PA-induced cell proliferation (Fig. 4f and Supplementary Fig. 3b). Re-introduction of wild type PTEN, but not T366A mutant, into PTEN knocking down cells restored PA-induced cell proliferation (Fig. 4f, Supplementary Fig. 3b).
To establish the role of mTOR/S6K in PA-induced cell proliferation, we first treated U2OS cells with the mTOR inhibitor RAD001 or S6K inhibitor NaSal and found that the treatments clearly blocked PA-induced Akt phosphorylation (Fig. 4h) and cell proliferation (Fig. 4g). Next, we analyzed the sequential effects of PA on S6K, PTEN and Akt. Our results demonstrate that, upon PA treatment, U2OS cells first display S6K activation (at 4h), then PTEN T366 phosphorylation (at 8h), and finally Akt activation (at 16h) (Fig. 4d). The sequential event further supports our finding that elevated PA promotes cell proliferation in part through inducing mTOR/S6K-mediated PTEN phosphorylation and inactivation.
4. Discussion
The over-consumption of dietary fat has been implicated in the development of metabolic disorders[26], leading to increased risk of cardiovascular diseases and malignancy[27–30]. However, the molecular mechanisms by which PA contributes to the pathogenesis of those diseases, particularly cancer, have not been extensively investigated. Here, we show that elevated PA hinders PTEN tumor suppressor activity in cancer cells and liver. Further mechanistic analysis revealed that elevated PA enhanced the phosphorylation of PTEN at T366 through the mTOR-S6K pathway, leading to the ubiquitin proteasome-dependent degradation of PTEN. This identification of PTEN repression by elevated PA contributes to the growing understanding that over-consumption of dietary fat can increase the risk of cancer.
Interestingly, treating HepG2 cells with elevated oleic acid (OA) has also been shown to down-regulate PTEN and promote hepatoma proliferation through up-regulation of microRNA-21[31, 32]. Thus, it is likely that PTEN protein level is regulated by both saturated and unsaturated FFAs via different mechanisms. However, because our metabolomic analysis of liver tissue revealed that PA is the only fatty acid that showed significant difference between HFD and control mice[9], we were unable to evaluate the effect of elevated OA on PTEN down-regulation in our studies. To support the role of PA in liver cell proliferation, we have analyzed our RNA-Seq data from the livers of mice fed the same diet [34]. The analysis indeed revealed several proliferation-related genes that are dysregulated in HFD mice (Supplementary Fig. 4), supporting the notion that, like OA, PA is capable of promoting liver cell proliferation. Clearly, it will be intriguing to further assess the contribution of saturated and unsaturated FFAs in line with over-consumption of dietary fat in cancers growth in the future.
As a direct antagonist of the PI3K pathway, the activation and stability of PTEN is involved in modulating cell growth, stress response and the pathogenesis of human diseases[33]. Notably, the phosphorylation of PTEN has been suggested as the major regulatory mechanism of PTEN. In the present study, we show elevated level of PA triggers PTEN T366 but not S380 phosphorylation in cancer cell lines. In addition, liver tissue from HFD-fed mice also exhibited higher levels of PTEN T366 phosphorylation and protein down-regulation. Interestingly, our previous work indicated that elevated levels of PA increases PTEN S380, but not T366, phosphorylation in normal human umbilical vein endothelial and human aortic endothelial cells[8], which mediated PA-induced endothelial oxidative stress by activating tumor suppressor p53. This may be consistent with PTEN’s function to reduce endothelial oxidative stress in umbilical vein and aortic endothelial cells. To support this view, PA-induced AKT activation was not observed in the endothelial cells. In contrast, in mouse liver and cancer cells, AKT activation was clearly observed, supporting the role of PTEN in cell proliferation. While the mechanism underlying the differential impact of PA is not fully understood, it is clear that the influence of PA on PTEN is cell-type dependent.
Given that over-consumption of dietary fat has become a serious public problem world wide, our finding that livers of HFD-fed mice exhibited PTEN inhibition and Akt over-activation has several implications. First, we have previously shown that, in mice, HFD feeding can lead to obesity[9, 34], which is a major risk factor for the development of hepatocellular carcinoma (HCC)[35], one of the most common and lethal cancers worldwide[36] and in the U.S.[37]. Second, PTEN deficiency or mutations are well documented in HCC[38, 39] and targeted PTEN deletion in hepatocytes has been shown to lead to spontaneous development of HCC in mice[40]. Furthermore, there is a growing body of evidence suggesting that down-regulation of PTEN, rather than PTEN mutations or deletions, contributes to HCC metastasis[41]. In addition, liver cancer cells clearly revealed reduction in PTEN protein levels compared to adjacent normal liver cells[42], suggesting that maintaining wild type PTEN levels is critical for the prevention of HCC. Our data indicate that PA levels play an important role in the maintaining of PTEN function and suggest that dietary intervention to reduce PA levels might be worthwhile in mitigating the effects of dietary fat on HCC.
Supplementary Material
Elevated palmitic acid reduces tumor suppressor PTEN in high fat diet fed mice
PA-induced PTEN reduction was mediated by PTEN T366 phosphorylation and protein degradation
Elevated PA promotes cell growth through PTEN phosphorylation and protein degradation
Acknowledgements
We thank all members of our laboratories for valuable suggestions and helpful discussion. This work was supported by NIH grant R01CA075180 to X.L., NIH grant R01DK053892 to F.M.S., American Heart Association postdoctoral fellowship POST3530033 to Y.W., NIEHS T32 Training Grant (T32ES018827) to Y.N. and P.D., and Crohn’s Colitis Foundation Career Development Award (454808) to P.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
These authors declared no conflicts of interests.
References
- [1].Qin CJ, Zhao LH, Zhou X, Zhang HL, Wen W, Tang L, Zeng M, Wang MD, Fu GB, Huang S, Huang WJ, Yang Y, Bao ZJ, Zhou WP, Wang HY, Yan HX, Inhibition of dipeptidyl peptidase IV prevents high fat diet-induced liver cancer angiogenesis by downregulating chemokine ligand 2, Cancer letters, 420 (2018) 26–37. [DOI] [PubMed] [Google Scholar]
- [2].Gong Y, Dou LJ, Liang J, Link between obesity and cancer: role of triglyceride/free fatty acid cycling, Eur Rev Med Pharmacol Sci, 18 (2014) 2808–2820. [PubMed] [Google Scholar]
- [3].Liu J, Mazzone PJ, Cata JP, Kurz A, Bauer M, Mascha EJ, Sessler DI, Serum free fatty acid biomarkers of lung cancer, Chest, 146 (2014) 670–679. [DOI] [PubMed] [Google Scholar]
- [4].Przybytkowski E, Joly E, Nolan CJ, Hardy S, Francoeur AM, Langelier Y, Prentki M, Upregulation of cellular triacylglycerol - free fatty acid cycling by oleate is associated with long-term serum-free survival of human breast cancer cells, Biochemistry and cell biology = Biochimie et biologie cellulaire, 85 (2007) 301–310. [DOI] [PubMed] [Google Scholar]
- [5].Yin R, Yang T, Su H, Ying L, Liu L, Sun C, Saturated fatty acids as possible important metabolites for epithelial ovarian cancer based on the free and esterified fatty acid profiles determined by GC-MS analysis, Cancer Biomark, 17 (2016) 259–269. [DOI] [PubMed] [Google Scholar]
- [6].Hopkins MM, Meier KE, Free fatty acid receptor (FFAR) agonists inhibit proliferation of human ovarian cancer cells, Prostaglandins, leukotrienes, and essential fatty acids, 122 (2017) 24–29. [DOI] [PubMed] [Google Scholar]
- [7].Hopkins MM, Meier KE, Free Fatty Acid Receptors and Cancer: From Nutrition to Pharmacology, Handbook of experimental pharmacology, 236 (2017) 233–251. [DOI] [PubMed] [Google Scholar]
- [8].Wu Y, Zhou H, Wu K, Lee S, Li R, Liu X, PTEN phosphorylation and nuclear export mediate free fatty acid-induced oxidative stress, Antioxidants & redox signaling, 20 (2014) 1382–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Deol P, Fahrmann J, Yang J, Evans JR, Rizo A, Grapov D, Salemi M, Wanichthanarak K, Fiehn O, Phinney B, Hammock BD, Sladek FM, Omega-6 and omega-3 oxylipins are implicated in soybean oil-induced obesity in mice, Scientific reports, 7 (2017) 12488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pillon NJ, Azizi PM, Li YE, Liu J, Wang C, Chan KL, Hopperton KE, Bazinet RP, Heit B, Bilan PJ, Lee WL, Klip A, Palmitate-induced inflammatory pathways in human adipose microvascular endothelial cells promote monocyte adhesion and impair insulin transcytosis, American journal of physiology. Endocrinology and metabolism, 309 (2015) E35–44. [DOI] [PubMed] [Google Scholar]
- [11].Rezende LP, Galheigo MRU, Landim BC, Cruz AR, Botelho FV, Zanon RG, Goes RM, Ribeiro DL, Effect of glucose and palmitate environment on proliferation and migration of PC3-prostate cancer cells, Cell biology international, (2018). [DOI] [PubMed] [Google Scholar]
- [12].Cersosimo E, Xu X, Upala S, Triplitt C, Musi N, Acute insulin resistance stimulates and insulin sensitization attenuates vascular smooth muscle cell migration and proliferation, Physiological reports, 2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R, PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer, Science, 275 (1997) 1943–1947. [DOI] [PubMed] [Google Scholar]
- [14].Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV, Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers, Nature genetics, 15 (1997) 356–362. [DOI] [PubMed] [Google Scholar]
- [15].Worby CA, Dixon JE, Pten, Annual review of biochemistry, 83 (2014) 641–669. [DOI] [PubMed] [Google Scholar]
- [16].Benitez JA, Ma J, D’Antonio M, Boyer A, Camargo MF, Zanca C, Kelly S, Khodadadi-Jamayran A, Jameson NM, Andersen M, Miletic H, Saberi S, Frazer KA, Cavenee WK, Furnari FB, PTEN regulates glioblastoma oncogenesis through chromatin-associated complexes of DAXX and histone H3.3, Nature communications, 8 (2017) 15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lester A, Rapkins R, Nixdorf S, Khasraw M, McDonald K, Combining PARP inhibitors with radiation therapy for the treatment of glioblastoma: Is PTEN predictive of response?, Clinical & translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico, 19 (2017) 273–278. [DOI] [PubMed] [Google Scholar]
- [18].Wang Y, Dai B, PTEN genomic deletion defines favorable prognostic biomarkers in localized prostate cancer: a systematic review and meta-analysis, Int J Clin Exp Med, 8 (2015) 5430–5437. [PMC free article] [PubMed] [Google Scholar]
- [19].Miller SJ, Lou DY, Seldin DC, Lane WS, Neel BG, Direct identification of PTEN phosphorylation sites, FEBS letters, 528 (2002) 145–153. [DOI] [PubMed] [Google Scholar]
- [20].Maccario H, Perera NM, Davidson L, Downes CP, Leslie NR, PTEN is destabilized by phosphorylation on Thr366, The Biochemical journal, 405 (2007) 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, Tempst P, Chi SG, Kim HJ, Misteli T, Jiang X, Pandolfi PP, Ubiquitination regulates PTEN nuclear import and tumor suppression, Cell, 128 (2007) 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lee MS, Jeong MH, Lee HW, Han HJ, Ko A, Hewitt SM, Kim JH, Chun KH, Chung JY, Lee C, Cho H, Song J, PI3K/AKT activation induces PTEN ubiquitination and destabilization accelerating tumourigenesis, Nature communications, 6 (2015) 7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Maddika S, Kavela S, Rani N, Palicharla VR, Pokorny JL, Sarkaria JN, Chen J, WWP2 is an E3 ubiquitin ligase for PTEN, Nature cell biology, 13 (2011) 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tamguney T, Stokoe D, New insights into PTEN, Journal of cell science, 120 (2007) 4071–4079. [DOI] [PubMed] [Google Scholar]
- [25].Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T, Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta, The Journal of biological chemistry, 280 (2005) 35195–35202. [DOI] [PubMed] [Google Scholar]
- [26].Laws A, Hoen HM, Selby JV, Saad MF, Haffner SM, Howard BV, Differences in insulin suppression of free fatty acid levels by gender and glucose tolerance status. Relation to plasma triglyceride and apolipoprotein B concentrations. Insulin Resistance Atherosclerosis Study (IRAS) Investigators, Arteriosclerosis, thrombosis, and vascular biology, 17 (1997) 64–71. [DOI] [PubMed] [Google Scholar]
- [27].Wang DD, Hu FB, Dietary Fat and Risk of Cardiovascular Disease: Recent Controversies and Advances, Annual review of nutrition, 37 (2017) 423–446. [DOI] [PubMed] [Google Scholar]
- [28].Lamarche B, Dietary sources of saturated fat may influence cardiovascular disease risk, The Canadian nurse, 109 (2013) 19. [PubMed] [Google Scholar]
- [29].Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr., Cellular fatty acid metabolism and cancer, Cell Metab, 18 (2013) 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Guasch-Ferre M, Babio N, Martinez-Gonzalez MA, Corella D, Ros E, Martin-Pelaez S, Estruch R, Aros F, Gomez-Gracia E, Fiol M, Santos-Lozano JM, Serra-Majem L, Bullo M, Toledo E, Barragan R, Fito M, Gea A, Salas-Salvado J, Investigators PS, Dietary fat intake and risk of cardiovascular disease and all-cause mortality in a population at high risk of cardiovascular disease, The American journal of clinical nutrition, 102 (2015) 1563–1573. [DOI] [PubMed] [Google Scholar]
- [31].Vinciguerra M, Sgroi A, Veyrat-Durebex C, Rubbia-Brandt L, Buhler LH, Foti M, Unsaturated fatty acids inhibit the expression of tumor suppressor phosphatase and tensin homolog (PTEN) via microRNA-21 up-regulation in hepatocytes, Hepatology, 49 (2009) 1176–1184. [DOI] [PubMed] [Google Scholar]
- [32].Vinciguerra M, Carrozzino F, Peyrou M, Carlone S, Montesano R, Benelli R, Foti M, Unsaturated fatty acids promote hepatoma proliferation and progression through downregulation of the tumor suppressor PTEN, Journal of hepatology, 50 (2009) 1132–1141. [DOI] [PubMed] [Google Scholar]
- [33].Chen CY, Chen J, He L, Stiles BL, PTEN: Tumor Suppressor and Metabolic Regulator, Frontiers in endocrinology, 9 (2018) 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Deol P, Evans JR, Dhahbi J, Chellappa K, Han DS, Spindler S, Sladek FM, Soybean Oil Is More Obesogenic and Diabetogenic than Coconut Oil and Fructose in Mouse: Potential Role for the Liver, PloS one, 10 (2015) e0132672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chen Y, Wang X, Wang J, Yan Z, Luo J, Excess body weight and the risk of primary liver cancer: an updated meta-analysis of prospective studies, Eur J Cancer, 48 (2012) 2137–2145. [DOI] [PubMed] [Google Scholar]
- [36].Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F, Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012, International journal of cancer. Journal international du cancer, 136 (2015) E359–386. [DOI] [PubMed] [Google Scholar]
- [37].Gadalla SM, Widemann BC, Editorial: US Cancer Statistics of Survival: Achievements, Challenges, and Future Directions, Journal of the National Cancer Institute, 109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chalhoub N, Zhu G, Zhu X, Baker SJ, Cell type specificity of PI3K signaling in Pdk1- and Pten-deficient brains, Genes & development, 23 (2009) 1619–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chalhoub N, Baker SJ, PTEN and the PI3-kinase pathway in cancer, Annual review of pathology, 4 (2009) 127–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, Mizuno K, Hasegawa G, Kishimoto H, Iizuka M, Naito M, Enomoto K, Watanabe S, Mak TW, Nakano T, Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas, The Journal of clinical investigation, 113 (2004) 1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sze KM, Wong KL, Chu GK, Lee JM, Yau TO, Ng IO, Loss of phosphatase and tensin homolog enhances cell invasion and migration through AKT/Sp-1 transcription factor/matrix metalloproteinase 2 activation in hepatocellular carcinoma and has clinicopathologic significance, Hepatology, 53 (2011) 1558–1569. [DOI] [PubMed] [Google Scholar]
- [42].Zhou J, Li X, Association of PTEN expression with liver function and inflammatory changes in patients with liver cancer after chemotherapy, Oncol Lett, 16 (2018) 6633–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.