

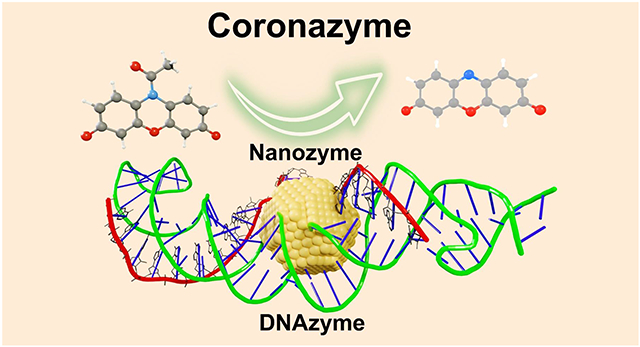
Abstract
Artificial enzymes such as nanozymes and DNAzymes are economical and stable alternatives to natural enzymes. By coating Au nanoparticles (AuNP) with DNA corona (AuNP@DNA), we amalgamated nanozyme and DNAzyme into a new artificial enzyme with catalytic efficiency five times higher than AuNP nanozymes, ten times higher than other nanozymes, and significantly greater than most of the DNAzymes on the same oxidation reaction. The AuNP@DNA demonstrates excellent specificity as its reactivity on a reduction reaction does not change. Single-molecule fluorescence and force spectroscopies and DFT simulation indicate a long-range oxidation reaction initiated by radical production on the AuNP surface, followed by radical transportation to the DNA corona, where the binding and turnover of substrates take place. The AuNP@DNA is named coronazyme because of its close natural enzyme mimicking through the well-orchestrated structures and synergetic functions. By incorporating different nanocores and various corona materials beyond DNA, we anticipate the coronazymes represent generic enzyme mimics to carry out versatile reactions in harsh environments.
Graphical Abstract
Introduction
Enzymes are natural catalysts with exceptional reactivity and substrate selectivity. However, their applications are limited since enzymes suffer from insufficient stability under operating conditions, narrow substrate scope, and high costs. Over the past decade, artificial enzymes such as nanozymes1-5 and DNAzymes6-10 have been developed to overcome the limitations of natural enzymes. While nanozymes usually comprise stable and affordable metals or metal oxides as core materials, DNAzymes employ versatile and programmable DNA fragments as essential components to catalyze enzymatic reactions. Chimera enzymes have been assembled using the structures of nanozymes and DNAzymes11-12. However, these chimera assemblies only demonstrate incremental improvement in catalytic functions without synergistically integrating the core activities of respective artificial enzymes. Recently biomolecules, e.g., DNA, have been coated on nanozymes to enhance biocompatibility and control substrate accessibility11, 13-14. But they do not directly participate in catalytic reactions.
In contrast, catalytic reactions are carried out by DNA templates in DNAzymes, which are most promising among all biomaterials because of their fully programmable structure and versatile interactions with many ligands8-9, 11, including metals. Similar to the well-known protein corona, the decoration of DNA on nanoparticles readily forms a DNA corona15-23. The non-catalytic roles of the DNA corona24-25 during nanozyme catalysis remain unclear since controversial observations exist among DNA-based biohybrid nanozymes: some studies suggest that DNAs inhibit the catalytic activities of parent nanoparticles because of their hindrance to surface sites26-27. Meanwhile, other studies suggest that DNAs could enhance catalytic reactivity by facilitating substrate adsorption28-29.
In this work, we discovered that DNA corona decorating a 5 nm AuNP directly catalyzes an oxidation reaction per se (Fig. 1a, and Supporting Information Section 3), an unprecedented phenomenon that breaks the boundary between DNAzymes and nanozymes. Despite being the most stable element on the periodic table, gold surprisingly mimics a wide range of natural enzymes such as oxidase, peroxidase, catalase, and nuclease. Consequently, AuNP was most intensively investigated as a nanozyme material. Here, a DNA hairpin consisting of two poly(adenosine) internal loops in its stem was prepared, which could strongly bind to AuNP to form the DNA corona. To avoid the ubiquitous heterogeneity in single particles, single-molecule spectroscopy30 was used to probe fluorogenic reactions occurring on the AuNP@DNA. We detailed the reaction mechanism by dissecting the two components, DNA corona, and AuNP nanocore, using single-molecule kinetics. Due to the presence of DNA corona, the AuNP@DNA exhibited better catalytic reactivity (, is the number of reaction sites) and superior reaction selectivity (, the Michaelis constant) compared to pristine 5 nm AuNP or other nanozymes. Furthermore, its efficiency () is higher than most of the DNAzymes working on the same Amplex Red (AR) substrate. Using DFT calculation and single-molecule force spectroscopy, we revealed the role and function of the DNA corona during biohybrid nanozyme catalysis. Our results indicate that an enzyme mimicking reactivity is initiated at the nanocore of the AuNP@DNA while carried out remotely within the DNA corona, hence the name coronazyme. Such a remote catalysis mechanism is widely applicable for more efficient designs of artificial enzymes by separately optimizing structures and functions of the nanocores and corona phases.
Figure 1.

Single-molecule fluorogenic reaction on AuNP@DNA. (a) Schematic of AuNP@DNA. A DNA hairpin binds to a 5 nm Au nanosphere through the two poly(adenosine) strands in the internal loop of its stem (see Supporting Information 3.1 for details). (b) N-deacetylation of Amplex Red (AR) generates a fluorescent product Resorufin (RF, emission ~583 nm), in a 50 mM pH 7.4 sodium phosphate buffer. (c) Scheme of the single-molecule reactivity measurement. Nanozymes are immobilized on the surface with reactants constantly flowing over. A 532 nm laser is configured in total internal reflection geometry to excite RFs generated on the nanozymes. An ultra-sensitive sCMOS camera captures the emission signals.
Experimental Section
Synthesis of poly(A) functionalized AuNPs.
The sequences of oligonucleotides are listed in Supporting Information Table 1. The synthesis of poly(A)-DNA template contained three major steps (Supporting Information Section 3): (1) the synthesis of oligonucleotides (ppd1.1 – ppd1.5); (2) pairing up ppd1.2 - ppd1.3, and ppd1.4 - ppd1.5; and (3) ligation of ppd1.1, annealed ppd1.2 - ppd1.3 and ppd1.4 - ppd1.5.
The citrate-capped Au colloidal solution was mixed with poly(A)-DNA in the 1:10 mole ratio. The mixture was kept at −80°C for ~15 min, during which a ligand exchange process occurred by replacing citrate with adenine of the DNA. The mixture was then rapidly thawed at room temperature to redistribute particles in the solution. UV-Vis spectrum and TEM images of the as-prepared sample are shown in Supporting Information Fig. S2 and S3. The surface-bound DNA corona prevents aggregation and well-suspends the AuNP@DNA in solution.
Ensemble catalytic reactions.
Two fluorogenic reactions were used in this work to determine the catalytic performances of nanozymes. They are: (1) the N-deacetylation of AR oxidation by H2O2 to generate the high fluorescence product resorufin (RF), acetate, and water; and (2) the N-deoxygenation of resazurin (RZ) reduction by NH2OH to generate RF and nitrite. Ensemble catalysis was carried out to confirm the effective catalytic conversions on AuNPs by measuring the fluorescence spectra and absorption of RF (Supporting Information Section 4).
Single-molecule fluorescence microscopy and data analyses.
Detailed procedures of single-molecule catalysis and data analyses are in Supporting Information Sections 6 and 7. Briefly, nanozymes were immobilized within a homemade microfluidic chamber, which was assembled between a microscope slide and a piece of coverslip. This chamber was connected to a syringe pump for the continuous supply of reaction mixture (20 μL/min) and placed under a TIR fluorescence microscope during reaction (Supporting Information Fig. S10). 0.1-1.8 μM AR and 60 mM H2O2 were dispersed in 50 mM pH 7.4 phosphate buffer for the AR oxidation reaction, while 0.05-0.6 μM RZ and 1 mM NH2OH were dispersed in 50 mM pH 7.4 phosphate buffer for the RZ reduction reaction. A 532 nm continuous wave laser beam (DragonLaser) of ~8 mW was focused onto an area of ~35 × 35 μm2 to excite the fluorescence of the product RF. The fluorescence emission signals were collected by a 100x N.A. 1.49 oil immersion objective (UAPON 100xOTIRF, Olympus), filtered by a longpass (Chroma, ET542lp) and a bandpass (Chroma ET575/50m) filter. The resulting signals were captured with an sCMOS camera (Photometrics Prime 95B) through the Olympus CellSens Dimension software at a frame rate of 33 fps. The images were then analyzed using ThunderSTORM31-32, an ImageJ plugin program, in combination with a home-written Matlab program to localize the positions of individual fluorescent product molecules (Supporting Information Section 7.1 and Supporting Information Fig. 11). 40 nm AuNP were visible because of their photoluminescence signals (Supporting Information Fig. S14B), therefore an additional background subtraction step was taken before further analysis.
Single-molecule mechanochemical experiments and data analysis.
The DNA-AuNP and DNA-reaction substrates interactions were investigated in a dual-trap laser-tweezers instrument33, as described in Supporting Information Section 5. The laser-tweezer setup (Supporting Information Fig. S8) contained a ligated 1558 bp dsDNA handle to the left and a 2391 bp dsDNA handle to the right of the synthesized DNA hairpin. To evaluate the interaction between poly(A)-DNA and AuNP, the AuNP@DNA construct was tethered between the two beads after bringing the DNA-conjugated streptavidin-coated polystyrene bead close to the anti-dig antibody-coated bead. To evaluate the interaction between poly(A)-DNA and reaction substrates, the DNA construct was first tethered, and then AR, RZ or RF with desired concentrations was supplied to the chamber. Tension on the DNA construct was developed when the two optically trapped beads were moved apart by steering one of the trapping lasers. Upon moving two beads apart, the tethered DNA was stretched. The tension produced on the DNA-AuNP/substrates conjugate was calculated based on the spring constant of each trap and the displacement of the bead from the center of the trap. The FX curves were recorded through a LabView program (National Instruments, Austin, TX) at 1 kHz with a loading rate of 5.5 pN/s (in the 10-30 pN force range), and data treatment was performed using Matlab (The MathWorks) and Igor (WaveMetrics) programs.
Theoretical calculations.
The interactions between DNA and substrate molecules were investigated using DFT calculations. Calculation procedures are detailed in Supporting Information Section 10. Briefly, all the calculations were performed with the Gaussian16 package at B3LYP-D3BJ/6-31++G** level. Frequency calculations were performed at the same theoretical level to guarantee all the structures obtained were local minima (no imaginary frequencies) on the potential energy surfaces. An SMD model described the solvation effects with the dielectric constant Ɛ = 2.4. The visual illustration of the interactions between the substrate and the bases was produced with the method of independent gradient model based on Hirshfeld partition (IGMH) implemented in the Multiwfn 3.8 program package.
Results and discussion
Single-molecule peroxidase kinetics of DNA-AuNP nanozyme.
AuNP is a peroxidase mimic. Hence, we used AR, a well-known fluorogenic substrate for peroxidase, to determine its catalytic performances (Fig. 1b, inset, and Supporting Information Section 4.1). In the presence of H2O2 and 5 nm AuNP, the nonfluorescent AR readily converts to a highly fluorescent molecule RF with its emission at ~583 nm (Fig. 1b). Single-molecule catalysis was carried out inside a microfluidic chamber, in which nanozymes were pre-immobilized (Fig. 1c, and Supporting Information Section 6). The formation of reaction product RF in a 50 mM sodium phosphate buffer (pH 7.4) at 25 °C was seen as reoccurring fluorescence bursts at the same location because RF was only generated on the surface of the nanozyme one at a time. There were multiple nanozymes in the ~40 μm × 40 μm field of view, so super-localization imaging was performed to ensure the correct assignment of RF molecules to their parent nanozymes. Briefly, the centroids of each emission spot within a frame were fitted to two-dimensional Gaussians to achieve tens of nanometer resolution (Fig. 2a). The nearest-neighbor searching was performed across consecutive frames to assign centroids of RF to different nanozymes (Supporting Information Section 7.1). The waiting time, , between two consecutive RF formations from the same nanozyme depicts a single reaction turnover time (Fig. 2b). The statistical values of many on the same nanozyme reflect the catalytic properties of the nanozyme.
Figure 2.

Single-molecule AR oxidation. (a) Localizing an RF molecule by fitting its emission signal to a two-dimensional Gaussian function. The localization error is 6.5 nm. (b) Extraction of reaction waiting time for a single catalyst. Nanozymes are identified by grouping the RFs in consecutive frames using the nearest-neighbor method. The dark time before the generation of a fresh RF is the waiting time for a single turnover. (c) Dependence of the single-catalyst turnover rate on the AR concentration for the nanozyme catalyzed reaction. [H2O2] = 60 mM. Error bars depict standard errors of the mean. Each data point is an average of more than 80 nanozymes. (d) Comparison of the catalytic efficiency for 5 nm AuNP against the AuNP@DNA.
The average turnover rate ( denotes averaging) at 5 nm AuNP increases with increasing AR concentration until its saturation point (Fig. 2c, black). The [AR]-dependent reactivity well matches modified Michaelis-Menten kinetics:
| (Eq 1) |
In which is the rate constant for a single reactive site, the number of reactive sites within one nanozyme, the Michaelis-Menten constant, and the substrate concentration (Supporting Information Section 7.5). For 5 nm AuNP, and are 0.031 ± 0.001 s−1 and 0.21 ± 0.02 μM, respectively. The catalytic efficiency equals 0.15 ± 0.01 s−1 μM−1, lower than that of natural peroxidase HRP34 (~2.84 s−1 μM−1). Surprisingly, adding DNA corona to AuNP significantly enhanced the peroxidase reactivity (Fig. 2c, red; see SI and reference23 for detailed preparation). The saturation reactivity for AuNP@DNA is higher than that of the original 5 nm AuNP. Fitting the curve with Eq1, the and are 0.0360 ± 0.0003 s−1 and 0.048 ± 0.002 μM, respectively. A significant decrease of indicates that the AR interacts with the AuNP@DNA much stronger than the 5 nm AuNP. Moreover, the AuNP@DNA exhibited the similar activity heterogeneity as its parent 5 nm AuNP35 (Supporting Information Section 7.6). Overall, the catalytic efficiency () has a five-time increase to 0.75 ± 0.03 s−1 μM−1, suggesting that the AuNP@DNA is a much more efficient catalyst than its parent AuNP (Fig. 2d). To the best of our knowledge, this is one of the highest catalytic efficiencies among all the reported nanozymes for AR oxidation.
Long-range oxidation of AR within the DNA corona.
It is worth noting that the reactivity in a single AuNP@DNA fluctuates over time instead of being steady (Supporting Information Section 8). The fluctuating reactivity for a single nanozyme is known as "dynamic disorders"36-39, a process hidden from conventional measurements. Interestingly, such fluctuations are not random. A catalytic "memory effect" seen as the autocorrelation decays for the waiting time series reflects the underlying surface restructuring of the nanoparticle during catalysis35-36 (Fig. 3a). The product of decay constant and average turnover time provides the correlation time for the nanozymes, which is related to the time scale for the surface restructuring. This correlation time is dependent on the reaction turnover for 5 nm AuNP because of the adsorbate-surface interactions40: high substrate concentration simultaneously results in rapid reaction turnover and faster surface restructuring; hence a linear dependence is seen (Fig. 3b, black). Strikingly, the turnover rate-dependent surface restructuring no longer exists for the AuNP@DNA (Fig. 3b, red). The lack of turnover rate dependence indicates that the adsorbate-surface interactions do not exist in AuNP@DNA. This constant reactivity fluctuation seen in AuNP@DNA is likely due to the conformational dynamics of the surrounding DNA since the fluctuation rate matches with the previously reported values for the conformational changes41-42.
Figure 3.

Oxidation reaction occurs in the DNA corona phase. (a) Autocorrelation function of the microscopic reaction time from single AuNP@DNA catalyzing the oxidation of 800 μM AR. The x-axis is a conversion from the turnover index to reaction time using the average turnover time of each nanozyme and averaged over >80 nanozymes. Solid curves are single exponential fits with decay constants of 163 ± 25 s, and 269 ± 22 s for 5 nm AuNP and AuNP@DNA, respectively. (b) Dependence of the reactivity fluctuation rates on the turnover rates of 5 nm AuNP and AuNP@DNA in AR oxidation. The y-axis is the inverse of the reactivity fluctuation correlation time obtained from . Solid lines are linear fits reflecting the adsorbate-surface interactions. (c) AR (400 μM) oxidation by 40 nm AuNP@DNA. 40 nm AuNPs are visible because of their photoluminescence signals. Red crosses are the centroids of generated RFs by fitting their emission signals with two-dimensional Gaussian functions. (d) Intensity versus time trajectory for the single 40 nm AuNP@DNA marked by the arrow in (c). Fluorescence intensity is integrated after subtracting the background emission signal from 40 nm AuNP.
Based on the constant fluctuation rate and the decreased for AuNP@DNA, we proposed a long-range reaction mechanism that occurs within the DNA corona (Supporting Information Section 7.3). The oxidation of AR starts with a reversible homolytic O-O bond cleavage of H2O2 on AuNP to generate •OH radicals. These radicals oxidize the DNA bases at their proximity, converting them into radical cations. Subsequent charge (hole) transfer steps occur after the initial base oxidation43-46 until the charge reaches the DNA-bound AR. Lastly, AR converts to RF through two consecutive one-electron transfer processes, reducing the DNA base to its original state. To act as an effective catalyst, AuNP serves as the radical initiation center, while the DNA corona functions as a radical transporter and a substrate binder that can be distally located away from the AuNP core. Compared to a natural enzyme such as HRP, the AuNP mimics the ferric center, while the DNA corona mimics the surrounding peptide. Unlike HRP, where only one ferric center and one reactive site exist, the nanocore provides multiple sites to generate •OH radicals, whereas the charge transfer within the DNA corona converts multiple remote bases into reactive centers. Therefore, the DNA and AuNP synergistically contribute to the enhanced reactivity of the AuNP@DNA.
Another direct evidence supporting the long-range catalysis within the AuNP@DNA is the fluorescence quenching on 40 nm AuNP. The 40 nm AuNP also has peroxidase reactivity by catalyzing AR oxidation. Although its reactivity is apparent at the ensemble level, no fluorescence spot is seen at the single-molecule level because fluorophore RF are quenched at the surface47-49 (Supporting Information Section 9). Surrounding the 40 nm AuNP with DNA corona, however, restores the fluorescence emission of RF (Fig. 3c-d), suggesting that the RF formation occurs within the DNA corona rather than on the AuNP surface. The residence times of RF on 5 nm AuNP@DNA and 40 nm AuNP@DNA are nearly the same (Fig. S14), further supporting the direct formation of RF within the DNA corona as it is known that RF desorption from Au surface is dependent on the size of nanoparticles50. Previously, to facilitate sensing or chemical catalysis, complicated procedures have been designed to avoid the fluorescence quenching on larger AuNP without much success51-54. The use of DNA corona provides a facile approach to achieve this function.
Selectivity enhancement within DNA corona.
The fact that AuNP can catalyze various enzymatic reactions indicates that a pristine AuNP lacks substrate specificity because different substrate molecules can adsorb and be activated on AuNP surface. To determine the impact of DNA corona on reaction specificity, we performed a dehydrogenase-mimicking reaction in comparison to the AR oxidation. RZ, a nonfluorescent phenoxazine dye, is catalytically converted to the same RF in the presence of nanozyme and NH2OH35, 55 (Fig. 4a inset). Using the same single-molecule microscopy, we found that the [RZ]-dependent reactivity also followed the Michaelis-Menten kinetics in a 50 mM sodium phosphate buffer (pH 7.4). and for 5 nm AuNP are 0.047 ± 0.002 s−1 and 0.31 ± 0.02 μM, respectively, giving a catalytic efficiency () at 0.15 ± 0.01 s−1 μM−1. As a nanozyme, the 5 nm pristine AuNP exhibits almost identical catalytic efficiency in RZ reduction and AR oxidation, thus lacking specificity. After adding DNA corona to the AuNP, the and become 0.019 ± 0.001 s−1 and 0.10 ± 0.01 μM, respectively, giving a catalytic efficiency at 0.18 ± 0.02 s−1 μM−1. Unlike the AR oxidation, no catalytic efficiency enhancement is seen, suggesting that the DNA corona does not favor the RZ reduction.
Figure 4.

DNA corona phase does not participate in the dehydrogenase-mimicking reaction. (a) Dependence of the single-catalyst turnover rate on [RZ] for the nanozyme catalyzed reaction in a 50 mM pH 7.4 sodium phosphate buffer at 25 °C. 195 bare AuNPs and 167 DNA@AuNPs were measured; [NH2OH] = 1 mM. Inset: chemical equation of the N-deoxygenation RZ reduction. (b) Dependence of the reactivity fluctuation rates on the turnover rates of 5 nm AuNP and AuNP@DNA in RZ reduction.
Notably, the reactivity fluctuation analyses show a clear dependence on the turnover rate during the RZ reduction (Fig. 4b). The surface restructuring rate for both 5 nm AuNP and AuNP@DNA increases linearly with an increasing turnover rate. This result strongly suggests that the RZ reduction only occurs on the AuNP surface. Since DNA corona does not directly participate in the reaction, its presence blocks the available surface sites on AuNP, leading to decreased reactivity. Likewise, the lower surface restructuring rate across the concentration range is due to the surface passivation by adsorbed DNA corona (Supporting Information Section 7.4).
Origin of reactivity and selectivity enhancement within DNA corona.
To investigate the interaction mechanism between DNA corona and substrate molecules, we performed DFT calculations at the B3LYP-BJD3 level (Supporting Information Section 10). Electrostatic potential (ESP) calculation indicates that AR strongly interacts with all four bases (Supporting Information Fig. S16). The binding energies for AR on base pair A-T, C-G, and the gold binding bases A-A are −38.6, −46.1, and −31.0 kcal/mol, respectively (Fig. 5 and Supporting Information Fig. S17 and S18), suggesting that AR strongly tends to adsorb on DNA corona. Symmetry-adapted perturbation theory (SAPT) indicates that the A-T base pair adopts a planar structure under normal conditions. However, its planarity would undergo a topographic change after AR binding (Supporting Information Fig. S19). The significant binding affinities of AR to DNA bases justify our single-molecule observation in which the 5 nm Au surface becomes less preferred by the AR molecules during catalysis. As a comparison, the RZ-DNA corona interactions are substantial but noticeably weaker than AR. The binding energies on base pairs A-T, C-G, and A-A are −24.2, −35.4, and −17.7 kcal/mol, respectively (Fig. 5 and Supporting Information Fig. S17 and S18). The relatively lower binding energies for RZ justify the enhanced reaction selectivity among AR and RZ: DNA corona is more favorable towards the AR adsorption, and the reaction occurs directly on DNA bases. Hence, stronger binding is favorable for enriching the reactant AR around reactive sites, resulting in enhanced oxidation efficiency. On the contrary, the DNA corona acts as a physical filter for RZ reduction, preventing the RZ substrate from accessing the reactive sites. The product RF has binding energies on base pairs A-T, C-G, and A-A at −25.8, −33.6, and −21.2 kcal/mol, respectively. Therefore, a significant binding energy loss exists for the AR→RF oxidation: the loss-of-affinity principle facilitates the product displacement56. No significant binding energy change exists for the RZ reduction to RF. Thus, the catalytic efficiency for the RZ reduction remains the same in the presence of the DNA corona.
Figure 5.

DFT calculation of the binding between DNA base pairs and reaction substrates (AR, RZ, and RF). Optimized structure and IGMH (independent gradient model based on Hirshfeld partition) map (dginter = 0.05 a.u., isovalue = 0.004 a.u.) for AR-CG, AR-AT, RZ-CG, RZ-AT, RF-CG, and RF-AT. Values shown are binding energies in kcal/mol.
To confirm these substrate-DNA corona interactions, we carried out single-molecule force spectroscopy using a dual-trap optical-tweezers setup (Fig. 6a).57 The DNA structure unfolds when force is increased (Fig. 6b-c, red), while it refolds after the force is released (Fig. 6b-c, black), giving a reversible force-extension (FX) curve. The sudden change in FX curves marks the unfolding of the DNA structure. Among the 17 tested DNAs, 29% feature an apparent hysteresis after the AR adsorption (Fig. 6d). In comparison, without any ligand (Control, Fig. 6g), hysteresis is insignificant (5%). This hysteresis suggests that AR can bind to the DNA structure, which delays the refolding of DNA. In another comparison, neither RZ nor RF features the same hysteresis (Fig. 6e-f), suggesting their binding affinities to DNAs are weaker. To probe the location of the AR binding, we randomized AuNP binding single-stranded poly(adenosine) to scrambled sequences, CAACATATCAACCTCAAGGAG, and GAATCACTCTAACTATACAAC. Hysteresis was again observed only for AR (Fig. 6c&h-k), suggesting the binding occurs at the duplex region of the DNA structure (Fig. 6a). These results aligned well with the DFT calculation (Fig. 5), which showed preferential binding of the AR to DNA base pairs instead of unpaired bases.
Figure 6.

Determining the interactions between reaction substrates and DNA corona using single-molecule force spectroscopy. (a) Schematic of an optical-tweezer setup. The DNA corona connects to two DNA handles which are tethered by 1064 nm laser-trapped beads through the biotin/streptavidin links (see Supporting Information and reference23 for details). (b-c) Examples of FX curves for the AR and DNA hairpin interactions. DNA unfolds as the tensile force accumulates (red traces) and refolds after the force is released (black traces). A hysteresis may exist as the DNA-bound substrate delays the refolding (see blowup insets). Percentages of molecules that show hysteresis in FX curves for various substrates interacting with the DNA construct with poly(A) (d-g) and a random sequence (h-k) in internal loops. A total of 17 DNAs were measured. Control means no substrate.
Conclusion
Based on the experimental and simulation results, we discover that AuNP@DNA demonstrates much increased catalytic efficiency and substrate specificity relative to pristine AuNPs. Its efficiency (0.75 s−1 μM−1) is also higher than natural HRP enzymes (average 0.28 s−1 μM−1), most DNAzymes (averaged at 0.63 s−1 μM−1), and >10 times higher than nanozymes (averaged at 0.06 s−1 μM−1, Supporting Information Table S3) catalyzing the AR→RF reaction. The pivotal component in the AuNP@DNA composite is the DNA corona which serves as the scaffold for catalytic transformation. A DNA corona selectively filters substrates in the reaction mixture, offering high binding affinities to desired molecules. Individual bases on the DNA serve as reactive sites after the substrate binding, while the DNA strand serves as the media to transport radicals generated remotely on the surface of a nanocore. Moreover, the DNA corona features binding energy loss after product formation, accelerating the reaction turnovers. Such orchestrated structure and synergistic function closely mimic those of enzymes; therefore, we name this new device "coronazyme". Unlike previous redox catalysis (enzyme/nanozyme/DNAzyme) in which a cascade of redox pairs is closely coupled at the reaction site, coronazyme decouples the cascade by a long-range hole transfer in the corona phase. The separate locations relieve the requirement for a single reaction site with multiple functionalities. This, therefore, facilitates modular evolvement of better catalysts by adopting the best material for each function instead of using a single material with multiple but subpar properties. We envision that optimizing the morphology of the nanoparticle core and the structure of the DNA corona will further improve the performances of coronazymes.
Supplementary Material
Acknowledgement
We thank the Kent State University for the financial support. H. M. acknowledges the National Science Foundation (NSF) (No. CBET1904921) and National Institute of Health (NIH) (No. R01 CA236350) for the grant support. Z. C acknowledges the National Natural Science Foundation of China (No. 22072064) for the grant support.
Footnotes
Supporting Information
Detailed procedure of coronazyme synthesis; fluorogenic probing reactions; optical tweezer setup for force spectroscopy; single-molecule fluorescence detection; single-molecule kinetic analyses; reactivity fluctuation; reactivity of 40 nm AuNP@DNA; and computational studies.
References
- 1.Fan K; Xi J; Fan L; Wang P; Zhu C; Tang Y; Xu X; Liang M; Jiang B; Yan X; Gao L, In vivo guiding nitrogen-doped carbon nanozyme for tumor catalytic therapy. Nature Communications 2018, 9 (1), 1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ji S; Jiang B; Hao H; Chen Y; Dong J; Mao Y; Zhang Z; Gao R; Chen W; Zhang R; Liang Q; Li H; Liu S; Wang Y; Zhang Q; Gu L; Duan D; Liang M; Wang D; Yan X; Li Y, Matching the kinetics of natural enzymes with a single-atom iron nanozyme. Nature Catalysis 2021, 4 (5), 407–417. [Google Scholar]
- 3.Wu J; Wang X; Wang Q; Lou Z; Li S; Zhu Y; Qin L; Wei H, Nanomaterials with enzyme-like characteristics (nanozymes): next-generation artificial enzymes (II). Chemical Society Reviews 2019, 48 (4), 1004–1076. [DOI] [PubMed] [Google Scholar]
- 4.Huang Y; Ren J; Qu X, Nanozymes: Classification, Catalytic Mechanisms, Activity Regulation, and Applications. Chemical Reviews 2019, 119 (6), 4357–4412. [DOI] [PubMed] [Google Scholar]
- 5.Liang M; Yan X, Nanozymes: from new concepts, mechanisms, and standards to applications. Accounts of chemical research 2019, 52 (8), 2190–2200. [DOI] [PubMed] [Google Scholar]
- 6.Borggräfe J; Victor J; Rosenbach H; Viegas A; Gertzen CGW; Wuebben C; Kovacs H; Gopalswamy M; Riesner D; Steger G; Schiemann O; Gohlke H; Span I; Etzkorn M, Time-resolved structural analysis of an RNA-cleaving DNA catalyst. Nature 2022, 601 (7891), 144–149. [DOI] [PubMed] [Google Scholar]
- 7.Hollenstein M; Hipolito C; Lam C; Dietrich D; Perrin DM, A Highly Selective DNAzyme Sensor for Mercuric Ions. Angewandte Chemie International Edition 2008, 47 (23), 4346–4350. [DOI] [PubMed] [Google Scholar]
- 8.Li W; Li Y; Liu Z; Lin B; Yi H; Xu F; Nie Z; Yao S, Insight into G-quadruplex-hemin DNAzyme/RNAzyme: adjacent adenine as the intramolecular species for remarkable enhancement of enzymatic activity. Nucleic Acids Research 2016, 44 (15), 7373–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang J; Hou L; Tang D; Zhang B; Zhou J; Chen G, Hemin/G-quadruplex-based DNAzyme concatamers as electrocatalysts and biolabels for amplified electrochemical immunosensing of IgG1. Chemical Communications 2012, 48 (66), 8180–8182. [DOI] [PubMed] [Google Scholar]
- 10.Travascio P; Li Y; Sen D, DNA-enhanced peroxidase activity of a DNA aptamer-hemin complex. Chemistry & Biology 1998, 5 (9), 505–517. [DOI] [PubMed] [Google Scholar]
- 11.Sun D; Lin X; Lu J; Wei P; Luo Z; Lu X; Chen Z; Zhang L, DNA nanotetrahedron-assisted electrochemical aptasensor for cardiac troponin I detection based on the co-catalysis of hybrid nanozyme, natural enzyme and artificial DNAzyme. Biosensors and Bioelectronics 2019, 142, 111578. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L; Chu M; Ji C; Tan J; Yuan Q, Preparation, applications, and challenges of functional DNA nanomaterials. Nano Research 2022. DOI: 10.1007/s12274-022-4793-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu X; Mao X; Wang Z; Feng C; Chen G; Li G, Fabrication of nanozyme@DNA hydrogel and its application in biomedical analysis. Nano Research 2017, 10 (3), 959–970. [Google Scholar]
- 14.Zeng C; Lu N; Wen Y; Liu G; Zhang R; Zhang J; Wang F; Liu X; Li Q; Tang Z; Zhang M, Engineering Nanozymes Using DNA for Catalytic Regulation. ACS Applied Materials & Interfaces 2019, 11 (2), 1790–1799. [DOI] [PubMed] [Google Scholar]
- 15.Chen X; Wang Y; Dai X; Ding L; Chen J; Yao G; Liu X; Luo S; Shi J; Wang L; Nechushtai R; Pikarsky E; Willner I; Fan C; Li J, Single-Stranded DNA-Encoded Gold Nanoparticle Clusters as Programmable Enzyme Equivalents. Journal of the American Chemical Society 2022, 144 (14), 6311–6320. [DOI] [PubMed] [Google Scholar]
- 16.Mirkin CA; Letsinger RL; Mucic RC; Storhoff JJ, A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382 (6592), 607–609. [DOI] [PubMed] [Google Scholar]
- 17.Nel AE; Mädler L; Velegol D; Xia T; Hoek EMV; Somasundaran P; Klaessig F; Castranova V; Thompson M, Understanding biophysicochemical interactions at the nano–bio interface. Nature Materials 2009, 8 (7), 543–557. [DOI] [PubMed] [Google Scholar]
- 18.Ohta S; Glancy D; Chan WCW, DNA-controlled dynamic colloidal nanoparticle systems for mediating cellular interaction. Science 2016, 351 (6275), 841–845. [DOI] [PubMed] [Google Scholar]
- 19.Tan LH; Xing H; Lu Y, DNA as a Powerful Tool for Morphology Control, Spatial Positioning, and Dynamic Assembly of Nanoparticles. Accounts of Chemical Research 2014, 47 (6), 1881–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang F; Zuo X; Fan C; Zhang X-E, Biomacromolecular nanostructures-based interfacial engineering: from precise assembly to precision biosensing. National Science Review 2018, 5 (5), 740–755. [Google Scholar]
- 21.Perrin DM; Garestier T; Hélène C, Bridging the Gap between Proteins and Nucleic Acids: A Metal-Independent RNAseA Mimic with Two Protein-Like Functionalities. Journal of the American Chemical Society 2001, 123 (8), 1556–1563. [DOI] [PubMed] [Google Scholar]
- 22.Landry MP; Vuković L; Kruss S; Bisker G; Landry AM; Islam S; Jain R; Schulten K; Strano MS, Comparative Dynamics and Sequence Dependence of DNA and RNA Binding to Single Walled Carbon Nanotubes. The Journal of Physical Chemistry C 2015, 119 (18), 10048–10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pokhrel PR, Kehao; Shen, Hao; Mao, Mechanical Stability of DNA Corona Phase on Gold Nanospheres. Langmuir 2022, 28 (44), 13569–13576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bisker G; Dong J; Park HD; Iverson NM; Ahn J; Nelson JT; Landry MP; Kruss S; Strano MS, Protein-targeted corona phase molecular recognition. Nature Communications 2016, 7 (1), 10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park M; Salem DP; Parviz D; Gong X; Silmore KS; Lew TTS; Khong DT; Ang MC-Y; Kwak S-Y; Chan-Park MB; Strano MS, Measuring the Accessible Surface Area within the Nanoparticle Corona Using Molecular Probe Adsorption. Nano Letters 2019, 19 (11), 7712–7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng X; Liu Q; Jing C; Li Y; Li D; Luo W; Wen Y; He Y; Huang Q; Long YT, Catalytic gold nanoparticles for nanoplasmonic detection of DNA hybridization. Angewandte Chemie 2011, 123 (50), 12200–12204. [DOI] [PubMed] [Google Scholar]
- 27.Park KS; Kim MI; Cho DY; Park HG, Label-free colorimetric detection of nucleic acids based on target-induced shielding against the peroxidase-mimicking activity of magnetic nanoparticles. Small 2011, 7 (11), 1521–1525. [DOI] [PubMed] [Google Scholar]
- 28.Hizir MS; Top M; Balcioglu M; Rana M; Robertson NM; Shen F; Sheng J; Yigit MV, Multiplexed activity of perAuxidase: DNA-capped AuNPs act as adjustable peroxidase. Analytical chemistry 2016, 88 (1), 600–605. [DOI] [PubMed] [Google Scholar]
- 29.Liu B; Liu J, Accelerating peroxidase mimicking nanozymes using DNA. Nanoscale 2015, 7 (33), 13831–13835. [DOI] [PubMed] [Google Scholar]
- 30.Sauer M; Hofkens J; Enderlein J, Handbook of fluorescence spectroscopy and imaging: from ensemble to single molecules. John Wiley & Sons: 2010, ISBN: 978-3-527-31669-4. [Google Scholar]
- 31.Huang B; Wang W; Bates M; Zhuang X, Three-Dimensional Super-Resolution Imaging by Stochastic Optical Reconstruction Microscopy. Science 2008, 319 (5864), 810–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ovesný M; Křížek P; Borkovec J; Švindrych Z; Hagen GM, ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 2014, 30 (16), 2389–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mao H; Luchette P, An integrated laser-tweezers instrument for microanalysis of individual protein aggregates. Sensors and Actuators B: Chemical 2008, 129 (2), 764–771. [Google Scholar]
- 34.Gorris HH; Walt DR, Mechanistic Aspects of Horseradish Peroxidase Elucidated through Single-Molecule Studies. Journal of the American Chemical Society 2009, 131 (17), 6277–6282. [DOI] [PubMed] [Google Scholar]
- 35.Xu W; Kong JS; Yeh Y-TE; Chen P, Single-molecule nanocatalysis reveals heterogeneous reaction pathways and catalytic dynamics. Nature Materials 2008, 7 (12), 992–996. [DOI] [PubMed] [Google Scholar]
- 36.Lu HP; Xun L; Xie XS, Single-Molecule Enzymatic Dynamics. Science 1998, 282 (5395), 1877–1882. [DOI] [PubMed] [Google Scholar]
- 37.Min W; English BP; Luo G; Cherayil BJ; Kou SC; Xie XS, Fluctuating Enzymes: Lessons from Single-Molecule Studies. Accounts of Chemical Research 2005, 38 (12), 923–931. [DOI] [PubMed] [Google Scholar]
- 38.Ye R; Mao X; Sun X; Chen P, Analogy between Enzyme and Nanoparticle Catalysis: A Single-Molecule Perspective. ACS Catalysis 2019, 9 (3), 1985–1992. [Google Scholar]
- 39.Roeffaers MBJ; De Cremer G; Uji-i H; Muls B; Sels BF; Jacobs PA; De Schryver FC; De Vos DE; Hofkens J, Single-molecule fluorescence spectroscopy in (bio)catalysis. Proceedings of the National Academy of Sciences 2007, 104 (31), 12603–12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imbihl R; Ertl G, Oscillatory kinetics in heterogeneous catalysis. Chemical Reviews 1995, 95 (3), 697–733. [Google Scholar]
- 41.Long X; Parks JW; Bagshaw CR; Stone MD, Mechanical unfolding of human telomere G-quadruplex DNA probed by integrated fluorescence and magnetic tweezers spectroscopy. Nucleic Acids Research 2013, 41 (4), 2746–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee M; Kim SH; Hong S-C, Minute negative superhelicity is sufficient to induce the B-Z transition in the presence of low tension. Proceedings of the National Academy of Sciences 2010, 107 (11), 4985–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Genereux JC; Barton JK, Mechanisms for DNA Charge Transport. Chemical Reviews 2010, 110 (3), 1642–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Neil MA; Barton JK, DNA Charge Transport: Conformationally Gated Hopping through Stacked Domains. Journal of the American Chemical Society 2004, 126 (37), 11471–11483. [DOI] [PubMed] [Google Scholar]
- 45.Renaud N; Berlin YA; Lewis FD; Ratner MA, Between Superexchange and Hopping: An Intermediate Charge-Transfer Mechanism in Poly(A)-Poly(T) DNA Hairpins. Journal of the American Chemical Society 2013, 135 (10), 3953–3963. [DOI] [PubMed] [Google Scholar]
- 46.Ratner M, Electronic motion in DNA. Nature 1999, 397 (6719), 480–481. [DOI] [PubMed] [Google Scholar]
- 47.Cannone F; Chirico G; Bizzarri AR; Cannistraro S, Quenching and Blinking of Fluorescence of a Single Dye Molecule Bound to Gold Nanoparticles. The Journal of Physical Chemistry B 2006, 110 (33), 16491–16498. [DOI] [PubMed] [Google Scholar]
- 48.Dulkeith E; Morteani AC; Niedereichholz T; Klar TA; Feldmann J; Levi SA; van Veggel FCJM; Reinhoudt DN; Möller M; Gittins DI, Fluorescence Quenching of Dye Molecules near Gold Nanoparticles: Radiative and Nonradiative Effects. Physical Review Letters 2002, 89 (20), 203002. [DOI] [PubMed] [Google Scholar]
- 49.Dulkeith E; Ringler M; Klar TA; Feldmann J; Muñoz Javier A; Parak WJ, Gold Nanoparticles Quench Fluorescence by Phase Induced Radiative Rate Suppression. Nano Letters 2005, 5 (4), 585–589. [DOI] [PubMed] [Google Scholar]
- 50.Zhou X; Xu W; Liu G; Panda D; Chen P, Size-Dependent Catalytic Activity and Dynamics of Gold Nanoparticles at the Single-Molecule Level. Journal of the American Chemical Society 2010, 132 (1), 138–146. [DOI] [PubMed] [Google Scholar]
- 51.Abadeer NS; Brennan MR; Wilson WL; Murphy CJ, Distance and Plasmon Wavelength Dependent Fluorescence of Molecules Bound to Silica-Coated Gold Nanorods. ACS Nano 2014, 8 (8), 8392–8406. [DOI] [PubMed] [Google Scholar]
- 52.Schneider G; Decher G; Nerambourg N; Praho R; Werts MHV; Blanchard-Desce M, Distance-Dependent Fluorescence Quenching on Gold Nanoparticles Ensheathed with Layer-by-Layer Assembled Polyelectrolytes. Nano Letters 2006, 6 (3), 530–536. [DOI] [PubMed] [Google Scholar]
- 53.Zhou X; Andoy NM; Liu G; Choudhary E; Han K-S; Shen H; Chen P, Quantitative super-resolution imaging uncovers reactivity patterns on single nanocatalysts. Nature Nanotechnology 2012, 7 (4), 237–241. [DOI] [PubMed] [Google Scholar]
- 54.Andoy NM; Zhou X; Choudhary E; Shen H; Liu G; Chen P, Single-Molecule Catalysis Mapping Quantifies Site-Specific Activity and Uncovers Radial Activity Gradient on Single 2D Nanocrystals. Journal of the American Chemical Society 2013, 135 (5), 1845–1852. [DOI] [PubMed] [Google Scholar]
- 55.Nigra MM; Arslan I; Katz A, Gold nanoparticle-catalyzed reduction in a model system: Quantitative determination of reactive heterogeneity of a supported nanoparticle surface. Journal of Catalysis 2012, 295, 115–121. [Google Scholar]
- 56.Gluhacevic von Krüchten D; Roth M; Seitz O, DNA-Templated Reactions with High Catalytic Efficiency Achieved by a Loss-of-Affinity Principle. Journal of the American Chemical Society 2022, 144 (24), 10700–10704. [DOI] [PubMed] [Google Scholar]
- 57.Koirala D; Dhakal S; Ashbridge B; Sannohe Y; Rodriguez R; Sugiyama H; Balasubramanian S; Mao H, A single-molecule platform for investigation of interactions between G-quadruplexes and small-molecule ligands. Nat Chem 2011, 3 (10), 782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.