

Abstract
Pulmonary hypertension (PH) is an insidious pulmonary vasculopathy with high mortality and morbidity and its underlying pathogenesis is still poorly delineated. The hyperproliferation and apoptosis resistance of pulmonary artery smooth muscle cells (PASMCs) contributes to pulmonary vascular remodeling in pulmonary hypertension, which is closely linked to the downregulation of fork-head box transcriptional factor O1 (FoxO1) and apoptotic protein caspase 3 (Cas-3). Here, PA-targeted co-delivery of a FoxO1 stimulus (paclitaxel, PTX) and Cas-3 was exploited to alleviate monocrotaline-induced pulmonary hypertension. The co-delivery system is prepared by loading the active protein on paclitaxel-crystal nanoparticles, followed by a glucuronic acid coating to target the glucose transporter-1 on the PASMCs. The co-loaded system (170 nm) circulates in the blood over time, accumulates in the lung, effectively targets the PAs, and profoundly regresses the remodeling of pulmonary arteries and improves hemodynamics, leading to a decrease in pulmonary arterial pressure and Fulton's index. Our mechanistic studies suggest that the targeted co-delivery system alleviates experimental pulmonary hypertension primarily via the regression of PASMC proliferation by inhibiting cell cycle progression and promoting apoptosis. Taken together, this targeted co-delivery approach offers a promising avenue to target PAs and cure the intractable vasculopathy in pulmonary hypertension.
Key words: Pulmonary hypertension, Pulmonary artery smooth muscle cells, Fork-head box transcriptional factor O1, Caspase 3, Co-delivery
Graphical abstract
Self-assembly paclitaxel nanocrystals embedded with Cas-3 targeting at media tunica of pulmonary artery rectify the dysregulation of FoxO 1 and promote cell apoptosis simultaneously, alleviating pulmonary vascular remodeling, improving hemodynamics and reducing pulmonary arterial pressure and Fulton's index.
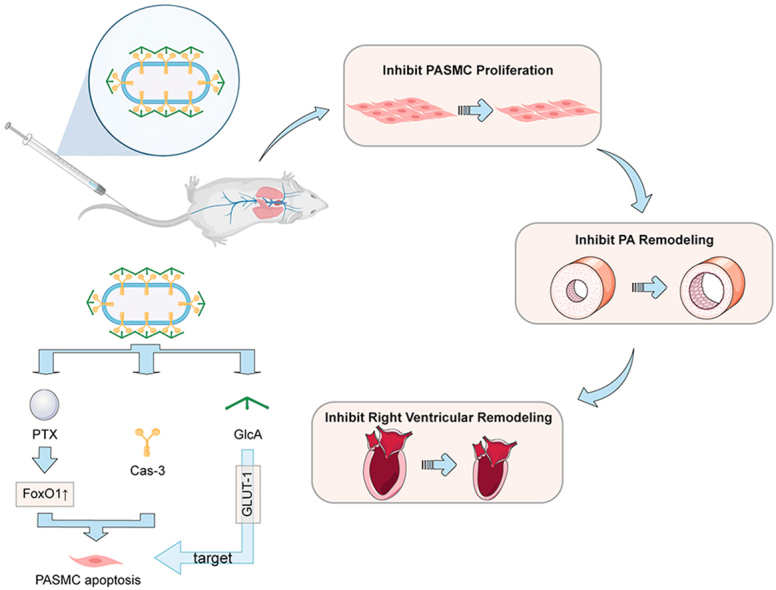
1. Introduction
PH is a group of devastating disorders characterized by progressive remodeling of pulmonary vessels and increased pulmonary vascular resistance, which leads to right heart failure and even premature death despite vigorously applied conventional interventions1. PH is categorized into five groups by the World Health Organization, including PH associated with abnormalities in the small branches of the pulmonary artery (group 1), PH induced by left heart disease (group 2), PH caused by lung diseases and/or hypoxia (group 3), PH associated with pulmonary embolus or pulmonary thrombosis (group 4), and PH due to unclear and/or multifactorial mechanisms (group 5)2. For managing PH, a comprehensive treatment strategy and multidisciplinary care are recommended by the 2022 ESC/ERS Guidelines, and of them, drug therapy is essential3. Current approved-drug treatments predominantly include prostacyclin analogs and receptor agonists, phosphodiesterase-5 (PDE-5) inhibitors, endothelin-receptor antagonists and calcium-sensitizing compounds4. Although significant advances have been made in the pharmaceutic treatment of PH5, its mortality remains very high with a 5-year survival rate of 20%–60%6. Pulmonary artery (PA) remodeling is predominantly induced by the over-proliferation of PASMCs and endothelial dysfunction plays a vital role in the progression of PH7. To this end, inhibition of PASMC proliferation represents a promising strategy to treat PH.
Among many mediators, two essential proteins, FoxO1 and Cas-3, are involved in the PASMC hyperproliferation and pathological progression of PH. FoxO1 is a crucial member of the winged-helix family of transcription factors and contains a highly conserved DNA-binding domain of approximately 110 amino acids8,9. FoxO1 is robust in regulating metabolism, cell cycle progression, apoptosis, autophagy, oxidative stress resistance, and DNA damage repair10,11. E.g., FoxO1 downregulation contributes to hyperproliferation and apoptosis resistance of PASMCs in PH12. In addition, our previous studies suggested that Cas-3, a critical apoptosis executor, was also downregulated in PASMCs, which leads to their hyperproliferation13. This evidence indicates that the downregulated FoxO1 and Cas-3 exacerbate the PASMC proliferation and the PA remodeling. As a result, co-delivery of FoxO1 stimulus and Cas-3 to PASMCs that can restore their regular proliferative activity has encouraging potential to alleviate PH.
Nanoparticles are extensively used to improve drug delivery because of their small size, which allows the drug to penetrate small capillaries and results in efficient accumulation at the target sites14, 15, 16. The therapeutic potential of nanoparticles has been proven by the clinical approval of over 85 nanomedicine products for a wide range of therapeutic purposes16. Several traditional nanoparticles, such as liposomes17, 18, 19, polymer nanoparticles (micelles)20, solid lipid nanoparticles21, and silica nanoparticles22, were developed to deliver the active compounds, i.e., prostacyclin analogue iloprost23, PDE-5 inhibitor sildenafil24, anti-inflammatory pitavastatin25 and nucleic acid drugs26, for relieving PH, demonstrating improved efficacy against the disease. Drug nanocrystals, as an essential type of clinically-relevant nanoparticle, are carrier-free solid drug particles within the nanometer size range and have crystalline characteristics14. Due to its high drug-loading capability (as high as 100%), nanocrystal technology is attracting increasing attention in drug delivery14,27. Furthermore, drug nanocrystals favor lung accumulation because of their nonspherical shape28. In this study, by using PTX-nanocrystal particles (NPs) as carriers, a targeted co-delivery system of PTX and Cas-3 was developed, in which the active protein was loaded onto the metal–phenolic network (MPN)-coated PTX NPs via the non-covalent interactions such as hydrogen bonding and π–π stacking29, followed by a coating of glucuronic acid (GlcA) for targeting the glucose transporter-1 (GLUT-1) on the PASMCs (Fig. 1A). PTX is a frequently used chemotherapeutic agent33; however, recent evidence indicated that PTX could upregulate FoxO1 and regress the proliferation of PASMCs12. MPN can stabilize the drug nanocrystals against aggregation with little toxicity30,31. Moreover, MPN is effective in associating with biopharmaceutical drugs via non-covalent bond rather than electrostatic interaction and allows for improving drug delivery with safety32. Our data in this present work demonstrated that the targeted co-delivery was able to regress the PA remodeling and improve the cardiac function and hemodynamics in an experimental PH model, which is mediated by efficiently targeting PASMCs and inhibiting their proliferation. This work provided a novel PA-targeted approach and offered a new avenue for PH treatment.
Figure 1.

Characterization of GlcA-NPplex. (A) Schematic illustration of the preparation process of GlcA-NPplex. (B) Native PAGE study of Cas-3 loaded in NPplex. (C) Influence of mass ratio of TA/Cas-3 on the particle size of NPplex (n = 3). (D) Particle size distribution of GlcA-NPlex measured by DLS. (E) TEM image of GlcA-NPlex. The scale bar is 200 nm. (F) Stability of GlcA-NPplex from optimized formulation stored in a 50% serum at 37 °C for 12 h (n = 3). In vitro release profile of (G) PTX and (H) Cas-3 from GlcA-NPplex in buffer solution at different pH conditions at 37 °C for 24 h (n = 3). The results are given as the mean ± SD.
2. Materials and methods
2.1. Preparation and characterization of the co-delivery system
MPN-PTX NPs were prepared via an anti-solvent precipitation–ultrasonic method using MPN as a stabilizer30. Briefly, 10 mg of PTX dissolved in 250 μL of ethanol was mixed with 9 mL of distilled water, followed by probe ultrasonication (20–25 kHz, Ningbo Scientz Biotechnology Co. Ltd. Ningbo, China) in an ice bath at 250 W for 10 min and adding 0.5 mL of tannic acid (TA) solution (2 mg/mL) and 0.25 mL of FeCl3·6H2O solution (Fe3+ 0.25 mg/mL) in sequence. The MPN-PTX NPs were purified by centrifugation and redispersion cycles three times to remove the free TA and FeIII using an ultrafiltration tube. MPN-PTX NP/Cas-3 complex (NPplex) was prepared by mixing 500 μL of Cas-3 solution (0.1 mg/mL) with an equal volume of MPN-PTX NPs (PTX 1 mg/mL) through low-speed vortexing and incubation for 30 min at room temperature. GlcA-NPplex was prepared by incubating 5 μL of GlcA solutions (15 mg/mL) with the suspension of NPplex at the various mass ratio of TA/GlcA. Dye-labeled nanoparticles were fabricated using a similar procedure. Dye-labeled nanoparticles were prepared by dissolving the dye with PTX in ethanol as the organic phase before mixing with the stabilizer solution.
The particle size, polydispersion index (PDI) and zeta-potential were determined using a 90 Plus particle size analyzer (Brookhaven Instruments, Holtsville, NY, USA) at 25 °C according to the dynamic light scattering (DLS) principle33. The nanoparticle-shape determination was performed in transmission electron microscopy (TEM, JEOL, JEM-1230 TEM, Tokyo, Japan) after staining with 2% (w/w) phosphotungstic acid for 1 min. Agarose gel electrophoresis was carried out in a Bio-Rad high-sensitivity chemiluminescence imaging system (Bio-Rad, Chemidoc XRS+, Hercules, CA, USA) to verify the loading of the active protein onto the nanoparticles. For the drug-release test, the sample was transferred into a 3.5 kDa dialysis bag, incubated in release media at different pHs and placed in an incubator (Jintan, SHA-C, Jintan, China) with a shaking speed of 100 rpm at 37 °C. Samples were withdrawn at specific time intervals. For PTX release, the collected sample was filtrated through a 0.22-μm filter and analyzed by a high-performance liquid chromatography system (HPLC, SHIMAZU LC-10AT, Kyoto, Japan) at 227 nm. The isolation was conducted in a Diamonsil C18 column (4.8 mm × 200 mm, 5 mm; Dikma Technologies, Beijing, China), using methanol/water (80/20, v/v) as a mobile phase at 1 mL/min. The release of FITC-protein was studied by testing the FITC intensity using a fluorescence spectrometer (SHIMADZU RF-5301PC, Kyoto, Japan).
Circular dichroism (CD) spectra were recorded using a circular dichroism chromatograph (Japan Spectrophotometer Co., Ltd., J-810, Tokyo, Japan) equipped with a temperature-controlling unit and a quartz cuvette. Ellipticity was expressed in millidegrees. The measurement parameters included as follows: bandwidth, 1 nm; response, 1 s; wavelength range, 260–200 nm; scanning speed, 100 nm/min; cell length, 0.1 cm; temperature, 25 °C; protein concentration, 0.1 mg/mL. The UV–Vis and Fourier-transformed infrared (FTIR) spectra were recorded in the Infrared chromatograph (Bruker, Tensor 27, Billerica, MA, USA) and UV–Vis spectrophotometer (SHIMAZU, UV-2450, Kyoto, Japan).
2.2. Cell culture
Primary PASMCs were isolated from pulmonary artery of monocrotaline (MCT)-induced PH rats, as described in a previous report13. PASMCs were cultured in RPMI 1640 medium containing 10% FBS and 1% penicillin–streptomycin and maintained at 37 °C in 5% CO2.
2.3. Flow cytometry and confocal laser scanning microscope (CLSM)
The cellular study was performed by flow cytometry (FCM, BD FACSCalibur, San Jose, CA, USA). For the uptake study, PASMCs were cultured in a 12-well plate at a density of 2 × 105 cells/mL and incubated for another 36 h. The culture medium was discarded and PASMCs were incubated with the preparations at 5 μg/mL FITC at 37 °C for 4 h. The cells were washed with PBS and trypsin and observed using CLSM, and the fluorescence intensity was measured by flow cytometry. For the study of the internalization pathway, cells were pre-incubated with uptake inhibitors, nystatin (10 μmol/L), methyl-β-cyclodextrin (M-β-CD, 2.5 mmol/L) or chlorpromazine (CPZ, 10 μg/mL), at 37 °C for 0.5 h, analyzed by CLSM and FCM. For the GLUT-1 influence study on the uptake, cells were pre-treated with the specific GLUT-1-inhibitor (BAY-876) at 2 nmol/L for 0.5 h before 4-h incubation with FITC-labeled nanoparticles at 5 μg/mL FITC, followed by CLSM observation and flow cytometry analysis. For the co-localization study, cells were cultured with FITC-labeled nanoparticles at 5 μg/mL dye for 4 h at 37 °C, stained with the caveolae marker, Alexa Fluor 488 labeled Caveolae (Cave)-1, for 3 h, washed with PBS and imaged by CLSM. The co-localization of nanoparticles with lysosomes was observed using CLSM. PASMCs were cultured with FITC-labeled nanoparticles with 5 μg/mL FITC in a serum-free culture medium for 4 h at 37 °C, stained with 1 mL of LysoTracker Red for 1 h at 37 °C, and washed three times with PBS.
2.4. Cytotoxicity and apoptosis
Cytotoxicity was investigated by the MTT assay. In brief, cells cultured in a 96-well plate at a density of 3 × 104 cells/mL for 36 h were incubated with various preparations at various drug concentrations for 48 h at 37 °C. Cytotoxicity was assessed by MTT assay at the absorbance of 570 nm. For determination of apoptosis, cells were cultured in a 12-well plate at a density of 2 × 105 cells/mL for 24 h and then were incubated with formulations at fixed drug concentration for 24 h. After that, the percentage of apoptotic cells was quantified by Annexin V-FITC/PI apoptosis kit (Beyotime Institute of Biotechnology, Haimen, China) and flow cytometry (BD FACSCalibur, San Jose, CA, USA).
2.5. Animal studies
All animal protocols were approved by the Ethics Committee of Nanjing Drum Tower Hospital for Institutional Animal Care and Use (protocol number 2019AE02017) and were in accordance with the Guide for the Care and Use of Laboratory Animals. Adult (6–8 weeks old) male Sprague–Dawley rats were purchased from Nanjing Medical University. All rats were housed under specific pathogen-free and controlled temperature conditions with a 12-h light–dark cycle at 20–22 °C. On Day 1, rats were injected subcutaneously with 60 mg/kg MCT (Sigma–Aldrich, c2401, St. Louis, Missouri, USA). MCT-injected rats randomly received the preparation or vehicle on Days 21, 25 and 29, respectively.
2.6. Pharmacokinetic studies
The preparations, free DiR and DiR-GlcA-NPplex, were injected into MCT-PH rats via the tail vein at the DiR dose of 0.5 mg/kg according to the body weight. Blood was taken from the orbit at 0.5, 1, 2, 4, 6, 8, 10, and 24 h after the administration. Plasma was collected by centrifugation at 4500 rpm (TDZ5-WS, Centrifuge Xiangyi, Changsha, China) for 10 min. The concentration of DiR was determined by the multifunctional microplate reader (BMG LABTECH, POLARstar Omega, Ortenberg, Germany). The pharmacokinetic parameters were calculated by the software DAS 2.0 (Data Analysis System 2.0, Shanghai, China).
2.7. In vivo imaging and tissue distribution
Free DiR or DiR-GlcA-NPplex was injected into MCT-PH rats via the tail vein. The rats were sacrificed 24 h after the administration by neck dissection. Then the major tissues, including the heart, liver, spleen, lung, and kidney, were dissected for ex vivo imaging (KODAK, In-Vivo FX PRO, Carestream, Canada). The excitation and emission wavelengths of the DiR channel are 748 and 780 nm, respectively.
For the biodistribution study, the MCT-PH rats (3 rats/group) were intravenously injected with DiR-labeled GlcA-NPplex via the tail vein at a DiR dose of 0.5 mg/kg, according to the rats' body weight. The co-localization of the GlcA-NPplex with PA α-SMA (α-smooth muscle actin) in the models was studied using Leica Corp. IX 7 fluorescence microscopy (Leica Microsystems Inc. IX7, Wetzlar, Germany). The rats received intravenous administration of the free IR783-labeled preparations at 0.5 mg/kg IR783. Four hours after the treatment, the rats’ lungs were isolated and embedded with OCT for further fluorescent imaging. Briefly, the lung sections were cut into 2 μm and then blocked for 15 min with 5% FBS at room temperature. After the excess-serum removal, the sections were stained for 12 h with a mouse anti-rat α-SMA monoclonal antibody at 5 μg/mL and incubated with FITC (diluted 1:200 with FITC buffer) for 1 h.
2.8. Echocardiography measurements
Echocardiography was performed by an experienced operator using an ultrasound diagnostic system (VINNO corporation, VINNO6 system, Suzhou, China) equipped with an 18 MHz transducer (VINNO corporation, X6-16 L, Suzhou, China). Images were obtained from parasternal views. In brief, rats were placed in a supine position after being anesthetized by inhalation of a mixture gas of 2.5% isoflurane in oxygen (2 L/min). Left ventricular ejection fraction (LVEF) was measured in motion mode at the parasternal long axis view. Pulmonary artery acceleration time (PAAT), as a surrogate of pulmonary arterial pressure, was tested by Pulsed-wave Doppler imaging at the parasternal short axis view. Right ventricle internal diameter (RVID) and tricuspid annular plane systolic excursion (TAPSE) were measured at the apical four-chamber view. Cardiac output (CO) depended on heart rate (HR) and stroke volume (SV), which was calculated by the following Eq. (1):
| CO (L/min) = HR (bpm) × SV (mL) × 10−3 | (1) |
2.9. Hemodynamic and RV hypertrophy measurements
On Day 35, rats were anesthetized, orotracheally intubated and ventilated with a frequency of 60 breaths per min to measure hemodynamics. Briefly, after being fixed on the platform, the left chest cavity of each rat was open for right ventricle (RV) exposure, and then the intravenous catheter (BD Insyte, Becton, Dickinson and Company, 24 GA, New Jersey, USA) flushed by heparin was inserted into the RV outflow tract. Mean pulmonary arterial pressure (mPAP) was determined after the catheter was connected to a pressure transducer of a monitor (Philips, Intelli Vue MP2, Amsterdam, The Netherlands), and the pulmonary arterial pressure waveform was stable for 1–2 min. After perfused with saline through the pulmonary circulation, the lungs and heart were explanted from the rats. The left lung was fixed in 10% neutral formalin for histology. The right lung was frozen in liquid nitrogen and stored at −80 °C. For the test of RV hypertrophy, the Fulton index [RV/(LV + S)] was calculated after the RV and left ventricle plus septum (LV + S) were isolated.
2.10. Histology
After being fixed in the neutral formalin for 24 h, the lung and other vital organs, including the liver, spleen, heart, and kidney, were dehydrated, embedded in paraffin, and cut into 3-μm sections for hematoxylin-eosin (H&E) staining. For immunohistochemistry, paraffin slides were baked at 60 °C for 2 h, then deparaffinized, hydrated, and subjected to antigen retrieval. Slides were incubated with primary antibodies at 4 °C, including anti-α-smooth muscle actin (α-SMA) (1:400, Abcam, Cambridge, England), anti-FoxO1 (1:800, CST, Boston, MA, USA), and anti-Ki67 (1:450, Abcam, Cambridge, England), anti-cleaved Caspase-3 (1:450, Abcam, Cambridge, England), anti-CyclinD1 (1:400, Abcam, Cambridge, England). After overnight incubation, the slides were washed and incubated with the respective secondary antibodies at 37 °C for 40 min. The media wall thickness of small pulmonary arteries (20–150 μm, medial wall thickness%) was measured, and the percentage of partially or fully muscularized distal pulmonary vessels (muscularized wall%) was calculated. The staining intensity of α-SMA/per artery was measured to evaluate the PASMC proliferation. The percentage of perivascular Ki67, cleaved Cas-3, and CyclinD1 positive cells was calculated. The FoxO1 expression in the lungs was also quantified by Image-Pro Plus 6.0 software (MEDIA CYBERNETICS, MD, USA). At least 10 images were randomly captured using a microscope (Olympus, DP27, Hachioji, Japan). All analyses were performed by another person who is blind to the experiment protocol. For the PASMC-apoptosis determination, the TUNEL assay was conducted with a Fluorescein (FITC) TUNEL Cell Apoptosis Detection Kit (KeyGEN BioTECH, KGA7063, Nanjing, China) according to the manufacturer's instructions. The cells with red fluorescence were calculated as apoptotic cells. The number of TUNEL-positive cells for each section of PAs was counted in at least 6 random fields ( × 400) in a blinded manner. The apoptosis rate was determined by the percentage of TUNEL-positive cells on the PASMCs.
2.11. Immunofluorescence
On Day 21 after the rats received a cervical subcutaneous injection of 60 mg/kg MCT to establish PH model, the rats received three intravenous injections of diverse formulations via the tail vein at the predetermined time points in 2 weeks at 0.4 mg/kg PTX and 0.04 mg/kg Cas-3 based on the body weight. The rats were euthanized with cervical dissection on Day 35, and the lungs were extracted and stored in a −80 °C freezer for further fluorescent imaging and Western blotting analysis.
Double immunofluorescence staining was performed with primary antibodies, α-SMA (1:300, Sigma) and FoxO1 (1:100, Abcam). After overnight incubation, the slides were washed 3 times with PBS (pH 7.4) and incubated with the respective secondary antibodies at room temperature in the dark for 50 min. Tissue sections were counterstained with nuclear DAPI (Servicebio, Wuhan, China) and quenched by AutoFluo Quencher (Servicebio, Wuhan, China). After dried slightly, the slides were mounted with an anti-fluorescence quenching mounting medium and observed under a fluorescence microscope (NIKON, Nikon Eclipse C1, Tokyo, Japan). The immunofluorescence micrographs were quantified using Image J software.
2.12. Western blotting (WB)
For WB assay, primary antibodies including anti-FoxO1 (1:5000, Abcam), anti-phospho-FoxO1 (pFoxO1) at Ser 256 (1:1000, CST), and anti-β-actin (1:10,000, Proteintech, Rosemont, IL, USA) were used. For the Western blot assay, PASMCs were lysed in cold lysis buffer followed by centrifugation at 15,000 rpm (TGL-16 B, Anting Scientific Instrument Factory, Shanghai, China) for 5 min, and the supernatants were collected and determined by a BCA kit (Beyotime Institute of Biotechnology Co. Ltd.). Proteins were separated by SDS-PAGE and then transferred to polyvinylidene fluoride (PVDF) membranes. For in vivo apoptosis mechanism determination, proteins were similarly extracted from the isolated tissues and transferred to PVDF membranes. After blocking, the membranes were incubated with primary and horseradish peroxidase (HRP) secondary antibodies. The band images were exposed to ECL detection system G:Box ChemiXR5 (Syngene, Cambridge, UK), and the expressions of proteins were quantified by ImageJ software (NIH, Bethesda, MD, USA).
2.13. Statistical analysis
Data are presented as the mean ± SD. All statistical analyses were performed using Prism (Graphpad version 8.0). The Student's t-test was used for comparisons between two groups, and ANOVA was used for multiple comparisons with P values < 0.05 considered significant.
3. Results
3.1. Preparation and characterization of the targeted co-delivery system
Three steps were involved in preparing the co-delivery system. First, the MPN-coated PTX pure nanocrystals, termed MPN-PTX NPs, were prepared by the anti-solvent precipitation–ultrasonic method.30,31. Increasing the PTX loading from 1 to 10 mg in a 10-mL MPN solution did not affect the particle size (Supporting Information Fig. S1A). The formulation containing 10 mg of PTX was selected for further study due to its high drug payload. DLS data revealed that the MPN-PTX NPs have a diameter of approximately 156 nm (Fig. S1B). Next, the MPN-PTX NP/Cas-3 complex (NPplex) was prepared by loading the active protein, Cas-3, onto the MPN-PTX NPs via non-covalent interactions29. The native PAGE results depicted that the bands of the Cas-3 protein were not visible when the mass ratio of TA (MPN)/Cas-3 was ≥4, suggesting the protein was efficiently loaded onto the nanoparticles (Fig. 1B and C). The interactions between MPN-PTX NPs and Cas-3 were confirmed by UV–Vis and FTIR spectra (Supporting Information Fig. S2). Herein, the NPplex with a TA/Cas-3 mass ratio of 20:1 was selected for further studies due to its improved stability against aggregation in serum (Supporting Information Fig. S3).
Finally, GlcA was coated onto NPplex, which could target the GLUT-1 on PASMCs59. Increasing the Cas-3/GlcA mass ratio from 0 to 1.5 led to a minimal change in nanoparticle size but an increase in zeta potential (Supporting Information Figs. S4A and S4B). The targeted co-delivery system (GlcA-NPplex) with the 20:1:1.5 mass ratio of TA/Cas-3/GlcA and a 171-nm diameter (Fig. 1D) was selected for further study due to the high GlcA loading. The GlcA-NPplex had loadings of 27.8% PTX and 2% Cas-3 and encapsulation efficacy of 91% and 68%, respectively. TEM study showed that the lead nanoparticles exhibited a rod-like shape with a 150–200 nm diameter in length (Fig. 1E). As shown in Fig. 1F, after incubation in 50% serum for 12 h, the average particle size and size distribution of GlcA-NPplex were minimally altered, suggesting the lead nanoparticles might possess good stability in physiological environments such as in the serum (Fig. 1F). The release profile of PTX and Cas-3 from GlcA-NPplex was pH-dependent and demonstrated faster release at pHs of 5.5 and 6.8 than pH 7.4. The faster drug release at lower pH from GlcA-NPplex is due to the cationic ion substitution by proton as the complexation between the TA catechol-group and metal ions can be reversed30,31. It is worth noting that less than 1% Cas-3 was released at pH 7.4 at 24 h and indicated the protein was well loaded in the nanoparticles. Moreover, the CD spectra of released Cas-3 were similar to the free protein, indicating little alteration in structure and activity (Supporting Information Fig. S5).
3.2. In vitro characterization of the targeting capability of GlcA-NPplex to PASMCs and the resultant apoptosis
We characterized the targeting ability of GlcA-NPplex to PASMCs and the resultant anti-proliferation effects in a 2D cell culture. As shown in Supporting Information Fig. S6, the cellular uptake of GlcA-NPplex is time-dependent in a 4-h period. Essentially, blocking the GLUT-1 receptor with the specific GLUT-1-inhibitor BAY-876 reduced the GlcA-NPplex uptake by approximately 75% compared with non-blocking (Fig. 2A and B, P < 0.01). The data revealed that GlcA-NPplex well-targeted PASMCs. Next, we investigated the uptake pathway of GlcA-NPplex into PASMCs. As depicted in Supporting Information Fig. S7, the pre-incubation with the typical clathrin-mediated pathway blocker chlorpromazine (CPZ) did not affect the uptake of FITC-labeled GlcA-NPplex. In contrast, the uptake declined significantly when the caveolar route was suppressed by the caveolae inhibitors, nystatin or M-β-CD (P < 0.01). Also, confocal imaging displayed co-localization of the red FITC-GlcA-NPplex with the green caveolae marker Alexa Fluor 488-labeled Cave-1 in the merged image (Supporting Information Fig. S8A). These results suggested that GlcA-NPplex uptake by PASMCs was mainly mediated by the caveolin pathway. Increasing evidence indicates that the caveolae-mediated internalization process authorizes cells to ingest materials without endo-lysosome detainment16,34,35. Indeed, confocal imaging demonstrated little co-localization between the red nanoparticles and LysoTracker Red during uptake (Fig. S8B). As a result, our data suggest that GlcA-NPplex could target PASMCs and most likely was internalized primarily by a non-endo-lysosomal route, promising to improve the delivery of protein drugs to the cell cytosol.
Figure 2.

PASMC-targeting and improved apoptosis. (A and B) Influence of GLUT-1 inhibitor on GlcA-NPplex uptake, (A) qualitative and (B) quantitative analysis of cellular uptake. PASMCs were pre-treated with the GLUT-1 inhibitor BAX-876 at a 2 nmol/L concentration for 0.5 h at 37 °C and then were incubated with the FITC-labeled nanoparticles at 5 μg/mL FITC at 37 °C for 3 h. The cells without BAX-876 pretreatment were used as a control. n = 3, ∗∗P < 0.01. The scale bar is 20 μm. (C) Quantified apoptosis following preparation treatment. FCM analysis of PASMC apoptosis was performed after 24-h incubation with 0.25 μg/mL PTX or 0.025 μg/mL Cas-3. Data are represented as mean ± SD. n = 3, ∗∗P < 0.01, ∗∗∗P < 0.001 compared to saline group; ###P < 0.001 compared to GlcA-NPplex group.
Next, the PASMC apoptosis induced by various formulations was investigated using an Annexin V FITC-PI assay (Fig. 2C). The apoptosis rates from the groups treated with GlcA-NPs, PTX/Cas-3 physical mixture, and GlcA-NPplex were about 36%, 35%, and 70%, respectively. Evidently, GlcA-NPplex demonstrated a significantly higher capacity to induce PASMC apoptosis as compared to the physical mixture formulation (P < 0.01). However, GlcA-NPplex had little toxicity to normal cells at fixed PTX concentrations and suggested its potential safety after injection (Supporting Information Fig. S9). These results indicated that the targeted co-delivery of PTX and Cas-3 using GlcA-NPplex could effectively inhibit the proliferation of PASMCs.
Downregulation of FoxO1 and Cas-3 results in hyperproliferation and apoptosis resistance of PASMCs in PH36,37. Here, we studied the protein expression by WB assay after incubation. As the effectors of the apoptotic caspases, Cas-3 presents as dimeric zymogens and is activated after the proteolytic cleavage of the inter-subunit linker38,39. As a result, cleaved Cas-3 rather than Cas-3 was measured in the study. GlcA-NPplex treatment enabled 1.5- and 2-fold increase of FoxO1 and Cas-3 compared with the PTX/Cas-3 physical mixture (P < 0.05, P < 0.001), comparable to the normal PASMC-level (Supporting Information Fig. S10A, S10B and S10D). The data demonstrated that GlcA-NPplex is able to deliver the FoxO1 effector (PTX) and apoptosis activator (Cas-3) to PASMCs with high efficacy. Furthermore, the other two apoptosis-resistance proteins, phosphorylated FoxO1 and Cyclin D140, 41, 42, were downregulated by GlcA-NPplex (Fig. S10A, S10C and S10E). These data revealed that GlcA-NPplex could upregulate FoxO1 and Cas-3 while decreasing the expression of phosphorylated FoxO1 and Cyclin D1, responsible for PASMC-apoptosis enhancement.
3.3. Prolonged blood circulation and lung- and PA-targeting
The blood circulation time of the delivery system plays an essential role in targeting diseased PAs. The pharmacokinetic study of DiR-labeled nanoparticles was performed in MCT-PH rats. As shown in Supporting Information Fig. S11, GlcA-NPplex exhibited a higher plasma level in a 24-h period after intravenous injection compared to the free DiR. The pharmacokinetic parameters verified that GlcA-NPplex exhibited enhanced performance in vivo compared to free DiR, as demonstrated by the prolonged blood half-life and MRT (mean residence time), increased bioavailability (AUC, area under the time–concentration) and reduced clearance rate (CL) (Supporting Information Table S1).
Next, the distribution of free DiR and DiR-GlcA-NPplex in different organs of MCT-PH rats was studied by ex vivo imaging at 24 h after tail vein injection. Both formulations mainly accumulated in the liver, spleen and lung (Fig. 3A and B). However, the lung accumulation of DiR-GlcA-NPplex was markedly higher than free DiR (Fig. 3B, P < 0.01). Additionally, we studied the targetability of IR783-GlcA-NPplex to the PAs by merging the IR783-labeled nanoparticles (red) with the PA marker α-SMA (green) using confocal imaging after dosing. As shown in Fig. 3C and D, the IR783-GlcA-NPplex administration enabled significantly stronger overlapped yellow fluorescence than IR783-dosing, displaying a 1.9-fold increase in fluorescence intensity. α-SMA locating in the tunica media of lung vasculature is a marker that indicates PA hyperproliferating43. Therefore, GlcA-NPplex effectively targeted the PA tunica media. Taken together, the results revealed that GlcA-NPplex had prolonged blood circulation time and could efficiently target both the lung and the tunica media of lung vasculature.
Figure 3.

Targeting the axis of lung-PA-PASMCs. (A) Ex vivo images displaying tissue distribution of DiR-labeled GlcA-NPplex at 24 h after administration via tail vein. Scale bar, 2 cm. (B) Quantification of nanoparticle distribution (Data are represented as mean ± SD. n = 3, ∗∗P < 0.01 compared to free DiR group). Scale bar, 50 μm. (C) Co-localization of IR783-labeled GlcA-NPplex with PA α-SMA in MCT-PH rats. (D) Quantified fluorescence intensity for PA-targeting of IR783-labeled preparations (n = 3, ∗∗P < 0.01). The preparation was dosed at 0.5 mg/kg IR783, according to the animal body weight. The lung was isolated at 4 h post-treatment, sectioned and marked with anti-α-SMA antibody. Yellow fluorescence implies the co-localization.
3.4. Therapeutic efficacy
Due to the enhanced in vivo pharmacokinetics and lung- and PA-targeting capability, GlcA-NPplex was expected to ameliorate experimental PH. In this study, we evaluated the treatment efficacy of the co-delivery approach against PH in MCT-PH rats via multiple parameters, including pulmonary arterypressure (PAP), RV hypertrophy, hemodynamics, PA remodeling, and PASMC proliferation and apoptosis (Supporting Information Fig. S12).
We first measured the efficacy of the co-delivery approach in reducing mean PAP (mPAP) and inhibiting RV hypertrophy. The mPAP and RV hypertrophy index [Fulton's index: RV/(LV + S)] were significantly elevated on Day 35 in the rats treated with MCT compared with the control rats without MCT administration (Fig. 4A and B), demonstrating that the experimental PH model was successfully developed. The three intermittent injections of formulations including PTX (0.4 mg/kg), caspases 3 (0.04 mg/kg), the combination, or GlcA-NPs every 4 days yielded a modest effect on decreasing mPAP (Fig. 4B) and little influence on RV hypertrophy (Fig. 4C). However, the injection of GlcA-NPplex caused an 18.4% decrease in mPAP (P < 0.05) and a 21.0% decrease in Fulton's index (P < 0.05) compared with the control MCT-PH rats on Day 35.
Figure 4.

The curative effect of different formulations on the pulmonary hypertension and depressed right ventricle contractility of rats induced by MCT. (A) Schematic flowcharter of animal experiment. (B) Pulmonary arterial pressure was directly reflected by mPAP on Day 35. (C) Fulton's index [RV/(LV + S)] suggested the degree of right ventricular hypertrophy on Day 35. Echocardiographic examination was conducted in MCT-PH rats receiving GlcA-NPplex or GlcA-NPs on Day 33. (D) The representative images of pulmonary arterial blood flow spectral pattern, PAAT, RVID, and TAPSE. (E–J) Quantifying the echocardiographic parameters comprising PAAT, PVR, RVID, TAPSE, CO, LVEF (%). PAAT, PVR, RVID and TAPSE demonstrate RV function. Increasing the mPAP accelerates the PA blood flow and shorts the acceleration time or PAAT. With the PH development, the RV afterload rises and the RVID increases. TAPSE indicates RV systolic function. CO represents cardiac ejection function, while LVEF reflected the systolic function of left ventricle. Data are represented as mean ± SD. ∗P < 0.05, ∗∗∗P < 0.001 compared to MCT group; ns, no significance. n = 6, unless indicated otherwise. CON: controlled rats (normal rats) received intravenous injections of 0.9% saline; MCT: rats received MCT treatment. The MCT-induced rats were dosed with preparations at 0.4 mg/kg PTX and 0.04 mg/kg Cas-3 via the tail vein. PAAT: pulmonary arterial accelerating time; PVR: pulmonary vascular resistance; RVID: right ventricle internal diameter; TAPSE: tricuspid annular plane systolic excursion; CO: cardiac output; LVEF: left ventricle ejection fraction.
We next studied the efficacy of the co-delivery approach in improving hemodynamics. Given the impact of increased pulmonary hypertension on the RV function, the transthoracic echocardiographic examination was performed in MCT-PH rats on Day 35 after receiving two formulations, GlcA-NPplex and GlcA-NPs. Representative echocardiographic images depict that the two-formulation treatment resulted in an increase of both PAAT and TAPSE and a RVID as compared to the control MCT-PH rats (Fig. 4D), wherein GlcA-NPplex led to improved changes in the three tested parameters. Furthermore, the quantitative analysis indicated that the GlcA-NPplex administration prolonged the PAAT duration from 19.7 ± 4.44 to 28.9 ± 5.44 ms (Fig. 4E, P < 0.05), reduced the PVR by 31% (Fig. 4F, P < 0.05), and effectively reversed the right ventricle dysfunction by allowing 21.9% and 51.4% increase in CO and TAPSE (Fig. 4 G–I, P < 0.05), respectively, and 34.9% decrease in RVID (P < 0.001), along with a negative effect on LVEF (Fig. 4J, P < 0.05), as compared to the control MCT-PH rats. GlcA-NPs yielded modest efficacy in the hemodynamics as compared to GlcA-NPplex. Taken together, the dosing of GlcA-NPplex could effectively enhance hemodynamics in MCT-PH rats.
We also measured the capability of the co-delivery approach to ameliorate PA remodeling. The MCT-PH rats exhibited a significant increase in the pulmonary arteriole's tunica media thickening after 35-day MCT exposure compared to the control rats, as indicated in the H&E staining image (Fig. 5A). The quantitative analysis revealed that the percentage of medial thickening of the pulmonary arterioles was approximately 3-fold higher in the MCT-PH rats than in the control rats (Fig. 5D). The treatment with the formulations, except for Cas-3, decreased the medial thickening and the muscularization of pulmonary arterioles in MCT-PH rats (Fig. 5A and E). Notably, GlcA-NPplex efficiently regressed the PA remodeling with the highest efficacy as compared with the control MCT-PH rats, manifested by around 34% reduction in medial thickening (GlcA-NPplex vs. MCT, 35.9 ± 5.08% vs. 69.1 ± 10.34%, respectively, P < 0.001) and 36% decline in the percentage of arterioles muscularization (GlcA-NPplex vs. MCT, 23.9 ± 9.38% vs. 60.5 ± 13.45%, respectively, P < 0.01).
Figure 5.

Efficacy of different formulations on the PA-remodeling regression and PASMC-hyperproliferation inhibition in MCT-PH rats. (A) H&E staining of the lung sections and (B) immunohistochemical detection of α-SMA expression. (C) Ki67 expression determined by immunohistochemistry. Semi-quantitative analysis of (D) PA medial thickness, (E) muscularization of pre-acinar arterioles, and (F) α-SMA expression relative to per artery in PAs. Semi-quantitative analysis of (G) Ki67 positive cells relative to PASMCs. Data are represented as mean ± SD. n = 4–6, ∗∗P < 0.01, ∗∗∗P < 0.001 compared to MCT group; #P < 0.05, ##P < 0.01 compared to GlcA-NPplex group; ns, no significance. Scale bar, 50 μm. CON: controlled rats (normal rats) received intravenous injections of 0.9% saline; MCT: rats received MCT treatment. The MCT-induced rats were dosed with preparations at 0.4 mg/kg PTX and 0.04 mg/kg Cas-3 via the tail vein.
α-SMA overexpression is a significant indicator of PA remodelin44. To further verify the inhibition of the PA remodeling by the formulations, we also measured the expression of α-SMA. Obviously, GlcA-NPplex downregulated α-SMA (GlcA-NPplex vs. MCT, 36.0 ± 3.42% vs. 58.9 ± 2.66%, respectively, P < 0.001) compared with the control MCT-PH rats (Fig. 5B and F). Overall, GlcA-NPplex effectively suppressed the PA remodeling in the MCT-PH models.
Finally, we tested the effect of the co-delivery approach on the hyperproliferation and apoptosis of PASMCs. Hyperproliferation and apoptosis resistance of PASMCs in the tunica media of PAs significantly contribute to PA remodeling45. The increase in Ki67-positive cells is a vital indicator of hyperproliferation12. As assayed by immunohistochemistry (Fig. 5C and G), the PAs from the MCT-PH rats displayed a significant escalation in Ki67-positive cells (%) compared to that from the normal non-diseased rats. The treatment with the drug-loaded formulations allowed for a decrease in Ki67-positive cells (P < 0.05). Of these formulations, the two preparations, GlcA-NPs and GlcA-NPplex, possess the most profound influence on this proliferative indicator, wherein GlcA-NPplex reduced Ki67-positive cells (%) by 2-fold (P < 0.001) in comparison with that from the control MCT-PH rats without any treatments. Moreover, the treatment with the PTX-loaded formulations increased TUNEL-positive apoptotic cells compared to the control MCT-PH rats. Significantly, GlcA-NPplex administration demonstrated more profound apoptosis than the treatment with GlcA-NPs or the physical mixture (P < 0.001, Supporting Information Fig. S13). Additionally, H&E staining analysis showed that the GlcA-NPplex treatment did not cause pathological injury to the vital organs in MCT-PH rats compared to saline treatment (Supporting Information Fig. S14). These results indicated that the GlcA-NPplex is potent to regress the PASMC proliferation and promote apoptosis in the remodeled PAs, along with good biocompatibility.
3.5. Mechanisms of GlcA-NPplex for alleviating PH
To study the underlying mechanism that the formulations inhibited the PA remodeling, we assayed the expression of FoxO1 and Cas-3 after dosing. First, immunofluorescence imaging was performed to examine the expression and location of FoxO1 in the PAs after incubated with GlcA-NPplex or GlcA-NPs. Evidently, the FoxO1-positive cells were upregulated noticeably by the treatment (Fig. 6A and B, GlcA-NPs vs. MCT, 9.85 ± 1.14 vs. 4.96 ± 0.759, P < 0.01; GlcA-NPplex vs. MCT, 13.5 ± 1.47 vs. 4.96 ± 0.759, P < 0.001). Next, the expression of FoxO1 and its phosphorylated form in PAs was assessed with an immunoblotting assay. The results revealed the reciprocal relationship between FoxO1 and phosphorylation of FoxO1 in rats treated with MCT for 35 days, showing a nearly 4-fold increase in the expression of phosphorylated FoxO1 in contrast to a 74% decrease in the FoxO1 expression level (Fig. 6C–E). Dosing GlcA-NPs or GlcA-NPplex significantly boosted the expression of FoxO1 and markedly reduced its phosphorylation level in the lung of the MCT-PH rats as compared to the rats without any treatments (Fig. 6D and E, P < 0.001). Of note, additional studies are needed to explore how the two formulations enable profound regression in the phosphorylation of FoxO1.
Figure 6.

Upregulated FoxO1 expression in the media tunica of PAs in MCT-PH rats. (A) The representative immunofluorescent images displaying the expression and location of FoxO1 in the pulmonary vessels. α-Smooth muscle actin (α-SMA, Red), FoxO1 (Green), and the merge of α-SMA and FoxO1 (Yellow), with nucleus stain (DAPI). The scale bar is 50 μm. (B) Quantification of FoxO1 positive cells in the media tunica. (C) The expression of FoxO1 and phosphorylated FoxO1 in the lungs of rats examined by Western blotting. The representative changes of FoxO1 and phosphorylation status was shown with the same result in three separable experiments. (D and E) Semi-quantitative analysis. Data are represented as mean ± SD, n = 4. ∗∗P < 0.01, ∗∗∗P < 0.001 compared to MCT ; #P < 0.05 compared to GlcA-NPplex group; ns, no significance. Scale bar, 50 μm. CON: controlled rats (normal rats) received intravenous injections of 0.9% saline; MCT: rats received MCT treatment. The MCT-induced rats were dosed with preparations at 0.4 mg/kg PTX and 0.04 mg/kg Cas-3 via the tail vein.
Next, the pathways for restraining the hyperproliferation of PASMCs in the lung tissues treated with various formulations were studied by immunohistochemistry assay. GlcA-NPplex elevated FoxO1- and Cas-3-positive cells (%) by 3- and 4.5-fold (P < 0.001) with the highest efficacy among the preparations compared with that from the control MCT-PH rats without any treatments (Fig. 7A, B and D). FoxO1 negatively affects the PASMC proliferation via downregulation of cell cycle protein (cyclin D1)46. Inhibition of cyclin D1 expression was evident in MCT-PH rats receiving GlcA-NPplex (Fig. 7A and C). GlcA-NPplex administration demonstrated elevated suppression of cyclin D1 compared to other drug-loading preparations; however, the suppression efficacy was not intense, probably due to administration-dose effects. Overall, these results revealed that GlcA-NPplex constrained PASMC hyperproliferation by restricting cell cycle progression and promoting apoptosis.
Figure 7.

Upregulated FoxO1 expression in the lung tissue and pathways of regressing the proliferated PAs in MCT-PH rats. (A) The expression of FoxO1, caspase 3, and cyclin D1 examined with the immunohistochemical assay. Scale bar, 50 μm. Quantified assay of (B) FoxO1-, (C) cyclin D1- (D) Cas-3-positive cells in the media tunica. Data are represented as mean ± SD. n = 6, ∗∗∗P < 0.001 compared to MCT group; ###P < 0.001 compared to GlcA-NPplex group; ns, no significance. CON: controlled rats (normal rats) received intravenous injections of 0.9% saline; MCT: rats received MCT treatment; PTX/Cas-3: combination of PTX and Cas-3. The MCT-induced rats were dosed with preparations at 0.4 mg/kg PTX and 0.04 mg/kg Cas-3 via the tail vein.
4. Discussion
Given the considerable advancement in drug delivery, targeted drug delivery to the lung and PAs remains a significant challenge. The study found that the rod-like GlcA-NPplex efficiently accumulated in the lung and targeted the PAs after intravenous administration. To our best knowledge, few reports demonstrated the targeting ability of the drug delivery system (DDS) to the active axis of lung–PA13. A previous report indicated that conventional DDS, such as liposomes, had a poor capability of lung–PA-targeting; in contrast, the GlcA decoration could improve the liposome PA-targeting, yet accompanying modest co-localization with the PA tunica media13. Whereas, GlcA-NPplex allowed profound co-localization with the PA tunica media. The improved rod-like nanoparticle targetability to the lung–PA axis is likely due to the increased surface area for the adhesion to the PAs under flow conditions compared to the spherical nanoparticles47, 48, 49. Furthermore, the PTX nanocrystals in GlcA-NPplex are stiff and can not be deformed. Therefore, the increased stiffness of rod-shaped GlcA-NPplex compared with the soft liposomes might contribute to the PA accumulation via a rocket-like membrane penetration model that could tear the cell membrane50. The discovery may inspire the design of drug delivery to the lung and PAs.
Over-proliferation of PASMCs is a central contributor to PA remodeling51. However, the current use of therapeutic regimes, such as phosphodiesterase-5 inhibitors, prostacyclin analogues or receptor agonists, endothelin-receptor antagonists, and calcium channel blockers5, has a little inhibition effect on the proliferation of PASMCs, though they can lessen the symptoms of PH. Moreover, few effective treatments were reported to block the proliferation of PASMCs in PH52. This study found that effectively delivering the FoxO1 activator (PTX) using 170-nm GlcA-NPs inhibited PASMC proliferation, promoted the apoptosis and regressed PA remodeling with high efficacy compared with free-PTX administration. The findings demonstrated the potential of PTX-based delivery systems for PH treatment.
CyclinD1 plays a crucial role in cell cycle progression and PASMC proliferation in pulmonary hypertension53. The precise mechanism of upregulating checkpoint molecule-cyclinD1 in PH remains elusive. Previous findings indicated that FoxO1-induced cell cycle arrest was partially mediated by the enhanced transcription and expression of cyclin-dependent kinase (CDK) inhibitor p27kip154. Nevertheless, accumulating evidence showed that FoxO1 upregulation led to a reduced expression of cyclin D1, and was associated with an impaired capacity of CDK4 to phosphorylate and inactivate the S-phase repressor pRb55. Consistently, the results from the Pullamsett's team suggested that deletion of FoxO1 was responsible for the augmented cyclin D1 expression in PASMCs12; in contrast, increased FoxO1 expression by PTX contributed to cyclin D1 downregulation and cell cycle arrest of PASMCs, promoting regression of PA remodeling12. In this study, we found the FoxO1 expression was strengthened by co-delivery of PTX and Cas-3 using GlcA-NPs or GlcA-NPplex, facilitating suppression of cyclin D1 expression (Fig. 7 and Fig. S10). The results implied that the cyclin D1 synthesis could be affected by Cas-3.
Significantly, targeted co-delivery of the FoxO1 activator (PTX) and apoptosis effector (Cas-3) to PASMCs using GlcA-NPplex could inhibit the PA remodeling and effectively recover the cardiac functions and hemodynamics, allowing promising long-term benefits for PH patients. The treatment enhancement resulted from the factors as follows: (i) extremely high payload capacity in GlcA-NPplex (30% w/w vs. 3%–5% in conventional drug carriers), (ii) prolonged blood circulation time, (iii) efficient targetability to the lung–PA axis and PASMCs (iii) synergistic effect between PTX and Cas-3. We demonstrated that, through inhibiting cell cycle progression and stimulating apoptosis, the proliferation of PASMCs could be significantly regressed. The findings represent a route to explore a new therapy regime to combat PH. Also, the findings offer initial evidence for the combinatorial use of PTX and apoptosis effectors such as imatinib in clinical practices to treat PH.
We proved that GlcA-NPplex alleviated MCT-induced PH. Of note, PH is classified into various phenotypes according to pathophysiological mechanisms, hemodynamic features, clinical demonstration and therapeutic management, including pulmonary arterial hypertension such as idiopathic PH and heritable PH, PH due to left heart disease, PH induced by lung diseases such as chronic lung disease or hypoxia, PH due to chronic blood clots in the lungs, and PH induced by risk factors encompassing sarcoidosis, sickle cell anemia, and chronic hemolytic anemia56. The current animal models cannot completely represent the clinical observations. For instance, the MCT model enables us to understand the pulmonary vascular remodeling progression and its pathophysiology and is the most commonly used model for new therapeutic interventions; the low oxygen-induced model helps us know the molecular processes involved in the vascular remodeling caused by chronic pulmonary disease and oxygen deprivation57; and bleomycin-induced model is beneficial to understand the pulmonary inflammation and fibrosis on PH, always associating parenchymal lung disease58. As such, further experiments are desired to prove its treatment efficacy in other PH models, e.g., low oxygen-induced PH and bleomycin-induced PH.
5. Conclusions
In summary, targeted co-delivery of FoxO1 activator and apoptotic protein Cas-3 to the PAs using the developed GlcA-NPplex enables efficient inhibition of PASMC hyperproliferation, which in turn suppresses PA remodeling and improves the cardiac functions and hemodynamics. Moreover, we found that the proliferation of PASMCs could be repressed by inhibiting cell cycle progression and stimulating apoptosis. Co-delivery of FoxO1 stimulus and apoptotic activator represents an effective pathway to fight against hyperproliferation of PASMCs and alleviate PA remodeling. Hyperproliferation of PASMCs and the resultant remodeling of the PAs commonly occur in all phenotypes of PH. We believe that the therapy regimen developed in this work represents a promising platform strategy for treating different forms of PH.
Furthermore, the targeted co-delivery system has promising translation perspective. PTX has been reported its efficacy in reversing pulmonary vascular remodeling in MCT-PH rodents mediated by upregulating FoxO1 and suppressing endoplasmic reticulum stress. However, a high PTX dose is needed to combat this pathological process with the likelihood ofincreased adverse reactions. The co-delivery system renders the synergistic effect of upregulating FoxO1 expression and promoting cell apoptosis, therefore, minimizing the dose of PTX. In addition, PTX, as one of the widely applied anti-neoplasm agents in the clinic, is easily accessible to expand its therapeutic regime for PH patients. Nevertheless, this drug delivery strategy supports its significant potential in the regression of pulmonary vasculature of MCT-PH rodents, the clinical study is warranted to testify its effect on human PH secondary to heterogeneous pathogenesis.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81872823, 82073782, and 82170063), the Shanghai Science and Technology Committee (19430741500, China), and the Key Laboratory of Modern Chinese Medicine Preparation of Ministry of Education of Jiangxi University of Traditional Chinese Medicine (zdsys-202103, China), the Medical Science and Technique Development Foundationof Nanjing Municipal Government (QRX17013, China), the Key Project from Medical Science and Technique Development Foundationof Nanjing Municipal Government (ZKX20017, China), and the Science Foundationof Ministry of Health of Jiangsu Province in China (ZDA2020016).
Author contributions
Bingbing Li and Wei He conceived and designed the research work. Wei He and Bingbing Li supervised the work. Xiaohong Jiang, Chao Teng, Huiling Yu, Xuyang Xing, Qi Jiang and Chenshi Lin carried out the experiments and performed data analysis. Wei He, Bingbing Li, Chao Teng, Huiling Yu and Xiaohong Jiang wrote the manuscript. Wei He, Ruifeng Zhang and Zongmin Zhao commented and corrected the manuscript. All of the authors have read and approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2022.12.002.
Contributor Information
Ruifeng Zhang, Email: zrf2000@163.com.
Wei He, Email: weihe@cpu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Southgate L., Machado R.D., Gräf S., Morrell N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat Rev Cardiol. 2020;17:85–95. doi: 10.1038/s41569-019-0242-x. [DOI] [PubMed] [Google Scholar]
- 2.Simonneau G., Montani D., Celermajer D.S., Denton C.P., Gatzoulis M.A., Krowka M., et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53 doi: 10.1183/13993003.01913-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Humbert M., Kovacs G., Hoeper M.M., Badagliacca R., Berger R.M., Brida M., et al. ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43:3618–3731. doi: 10.1093/eurheartj/ehac237. 2022. [DOI] [PubMed] [Google Scholar]
- 4.Bisserier M., Pradhan N., Hadri L. Current and emerging therapeutic approaches to pulmonary hypertension. Rev Cardiovasc Med. 2020;21:163–179. doi: 10.31083/j.rcm.2020.02.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spiekerkoetter E., Kawut S.M., de Jesus Perez V.A. New and emerging therapies for pulmonary arterial hypertension. Annu Rev Med. 2019;70:45–59. doi: 10.1146/annurev-med-041717-085955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lau E.M., Giannoulatou E., Celermajer D.S., Humbert M. Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol. 2017;14:603–614. doi: 10.1038/nrcardio.2017.84. [DOI] [PubMed] [Google Scholar]
- 7.Tuder R.M., Archer S.L., Dorfmüller P., Erzurum S.C., Guignabert C., Michelakis E., et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D4–D12. doi: 10.1016/j.jacc.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lam E.W.-F., Brosens J.J., Gomes A.R., Koo C.-Y. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer. 2013;13:482–495. doi: 10.1038/nrc3539. [DOI] [PubMed] [Google Scholar]
- 9.Weigel D., Jürgens G., Küttner F., Seifert E., Jäckle H. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell. 1989;57:645–658. doi: 10.1016/0092-8674(89)90133-5. [DOI] [PubMed] [Google Scholar]
- 10.Kitamura T. The role of FOXO1 in β-cell failure and type 2 diabetes mellitus. Nat Rev Endocrinol. 2013;9:615–623. doi: 10.1038/nrendo.2013.157. [DOI] [PubMed] [Google Scholar]
- 11.Li Y., Ma Z., Jiang S., Hu W., Li T., Di S., et al. A global perspective on FOXO1 in lipid metabolism and lipid-related diseases. Prog Lipid Res. 2017;66:42–49. doi: 10.1016/j.plipres.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 12.Savai R., Al-Tamari H.M., Sedding D., Kojonazarov B., Muecke C., Teske R., et al. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat Med. 2014;20:1289–1300. doi: 10.1038/nm.3695. [DOI] [PubMed] [Google Scholar]
- 13.Li B., He W., Ye L., Zhu Y., Tian Y., Chen L., et al. Targeted delivery of sildenafil for inhibiting pulmonary vascular remodeling. Hypertension. 2019;73:703–711. doi: 10.1161/HYPERTENSIONAHA.118.11932. [DOI] [PubMed] [Google Scholar]
- 14.Wang X., Mohammad I.S., Fan L., Zhao Z., Nurunnabi M., Sallam M.A., et al. Delivery strategies of amphotericin B for invasive fungal infections. Acta Pharm Sin B. 2021;11:2585–2604. doi: 10.1016/j.apsb.2021.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi Z., Li Q., Mei L. pH-Sensitive nanoscale materials as robust drug delivery systems for cancer therapy. Chin Chem Lett. 2020;31:1345–1356. [Google Scholar]
- 16.He W., Xing X., Wang X., Wu D., Wu W., Guo J., et al. Nanocarrier-mediated cytosolic delivery of biopharmaceuticals. Adv Funct Mater. 2020;30 [Google Scholar]
- 17.Gupta V., Gupta N., Shaik I.H., Mehvar R., McMurtry I.F., Oka M., et al. Liposomal fasudil, a rho-kinase inhibitor, for prolonged pulmonary preferential vasodilation in pulmonary arterial hypertension. J Control Release. 2013;167:189–199. doi: 10.1016/j.jconrel.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keshavarz A., Alobaida A., McMurtry I.F., Nozik-Grayck E., Stenmark K.R., Ahsan F. CAR, a homing peptide, prolongs pulmonary preferential vasodilation by increasing pulmonary retention and reducing systemic absorption of liposomal fasudil. Mol Pharm. 2019;16:3414–3429. doi: 10.1021/acs.molpharmaceut.9b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rashid J., Nahar K., Raut S., Keshavarz A., Ahsan F. Fasudil and DETA NONOate, loaded in a peptide-modified liposomal carrier, slow PAH progression upon pulmonary delivery. Mol Pharm. 2018;15:1755–1765. doi: 10.1021/acs.molpharmaceut.7b01003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ravikumar P., Menon J.U., Punnakitikashem P., Gyawali D., Togao O., Takahashi M., et al. Nanoparticle facilitated inhalational delivery of erythropoietin receptor cDNA protects against hyperoxic lung injury. Nanomedicine. 2016;12:811–821. doi: 10.1016/j.nano.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McLendon J.M., Joshi S.R., Sparks J., Matar M., Fewell J.G., Abe K., et al. Lipid nanoparticle delivery of a microRNA-145 inhibitor improves experimental pulmonary hypertension. J Control Release. 2015;210:67–75. doi: 10.1016/j.jconrel.2015.05.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silva Filho P.M., Paz I.A., Nascimento N.R.F., Santos C.F., Araújo V.R., Aquino C.P., et al. Incorporation of nitroprusside on silica nanoparticles—a strategy for safer use of this NO donor in therapy. Mol Pharm. 2019;16:2912–2921. doi: 10.1021/acs.molpharmaceut.9b00110. [DOI] [PubMed] [Google Scholar]
- 23.Jain P.P., Leber R., Nagaraj C., Leitinger G., Lehofer B., Olschewski H., et al. Liposomal nanoparticles encapsulating iloprost exhibit enhanced vasodilation in pulmonary arteries. Int J Nanomed. 2014;9:3249–3261. doi: 10.2147/IJN.S63190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beck-Broichsitter M., Hecker A., Kosanovic D., Schmehl T., Gessler T., Weissmann N., et al. Prolonged vasodilatory response to nanoencapsulated sildenafil in pulmonary hypertension. Nanomedcine. 2016;12:63–68. doi: 10.1016/j.nano.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Ichimura K., Matoba T., Koga J-i, Nakano K., Funamoto D., Tsutsui H., et al. Nanoparticle-mediated targeting of pitavastatin to small pulmonary arteries and leukocytes by intravenous administration attenuates the progression of monocrotaline-induced established pulmonary arterial hypertension in rats. Int Heart J. 2018;59:1432–1444. doi: 10.1536/ihj.17-683. [DOI] [PubMed] [Google Scholar]
- 26.Crosswhite P., Chen K., Sun Z. AAV delivery of tumor necrosis factor-alpha short hairpin RNA attenuates cold-induced pulmonary hypertension and pulmonary arterial remodeling. Hypertension. 2014;64:1141–1150. doi: 10.1161/HYPERTENSIONAHA.114.03791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y., Teng C., Azevedo H.S., Yin L., He W. Cocrystallization-like strategy for the codelivery of hydrophobic and hydrophilic drugs in a single carrier material formulation. Chin Chem Lett. 2021;32:3071–3075. [Google Scholar]
- 28.He W., Xin X., Li Y., Han X., Qin C., Yin L. Rod-shaped drug particles for cancer therapy: the importance of particle size and participation of caveolae pathway. Part Part Syst Char. 2017;34 [Google Scholar]
- 29.Guo J., Tardy B.L., Christofferson A.J., Dai Y., Richardson J.J., Zhu W., et al. Modular assembly of superstructures from polyphenol-functionalized building blocks. Nat Nanotechnol. 2016;11:1105–1111. doi: 10.1038/nnano.2016.172. [DOI] [PubMed] [Google Scholar]
- 30.Huang F., Jiang X., Sallam A.M., Zhang X., He W. A nanocrystal platform based on metal-phenolic network wrapping for drug solubilization. AAPS PharmSciTech. 2022;23:76. doi: 10.1208/s12249-022-02220-0. [DOI] [PubMed] [Google Scholar]
- 31.Magar K.T., Boafo G.F., Zoulikha M., Jiang X., Li X., Xiao Q., et al. Metal phenolic network-stabilized nanocrystals of andrographolide to alleviate macrophage-mediated inflammation in-vitro. Chin Chem Lett. 2023;34:107453. [Google Scholar]
- 32.Chung J.E., Tan S., Gao S.J., Yongvongsoontorn N., Kim S.H., Lee J.H., et al. Self-assembled micellar nanocomplexes comprising green tea catechin derivatives and protein drugs for cancer therapy. Nat Nanotechnol. 2014;9:907–912. doi: 10.1038/nnano.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du X., Hou Y., Huang J., Pang Y., Ruan C., Wu W., et al. Cytosolic delivery of the immunological adjuvant Poly I:C and cytotoxic drug crystals via a carrier-free strategy significantly amplifies immune response. Acta Pharm Sin B. 2021;11:3272–3285. doi: 10.1016/j.apsb.2021.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teng C., Lin C., Huang F., Xing X., Chen S., Ye L., et al. Intracellular codelivery of anti-inflammatory drug and anti-miR 155 to treat inflammatory disease. Acta Pharm Sin B. 2020;10:1521–1533. doi: 10.1016/j.apsb.2020.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh P., Borgstrom P., Witkiewicz H., Li Y., Borgstrom B.J., Chrastina A., et al. Live dynamic imaging of caveolae pumping targeted antibody rapidly and specifically across endothelium in the lung. Nat Biotechnol. 2007;25:327–337. doi: 10.1038/nbt1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paulin R., Michelakis E.D. Addressing complexity in pulmonary hypertension: the FoxO1 case. Circ Res. 2015;116:1732–1735. doi: 10.1161/CIRCRESAHA.115.305773. [DOI] [PubMed] [Google Scholar]
- 37.Barman S.A., Li X., Haigh S., Kondrikov D., Mahboubi K., Bordan Z., et al. Galectin-3 is expressed in vascular smooth muscle cells and promotes pulmonary hypertension through changes in proliferation, apoptosis, and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2019;316:L784–L797. doi: 10.1152/ajplung.00186.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y., Gao W., Shi X., Ding J., Liu W., He H., et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 39.White K., Dempsie Y., Caruso P., Wallace E., McDonald R.A., Stevens H., et al. Endothelial apoptosis in pulmonary hypertension is controlled by a microRNA/programmed cell death 4/caspase-3 axis. Hypertension. 2014;64:185–194. doi: 10.1161/HYPERTENSIONAHA.113.03037. [DOI] [PubMed] [Google Scholar]
- 40.Li F., Xie P., Fan Y., Zhang H., Zheng L., Gu D., et al. C terminus of Hsc70-interacting protein promotes smooth muscle cell proliferation and survival through ubiquitin-mediated degradation of FoxO1. J Biol Chem. 2009;284:20090–20098. doi: 10.1074/jbc.M109.017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kavurma M.M., Khachigian L.M. Sp1 inhibits proliferation and induces apoptosis in vascular smooth muscle cells by repressing p21WAF1/Cip 1 transcription and cyclin D1-Cdk4-p21WAF1/Cip1 complex formation. J Biol Chem. 2003;278:32537–32543. doi: 10.1074/jbc.M305650200. [DOI] [PubMed] [Google Scholar]
- 42.Zhang P., Zheng C., Ye H., Teng Y., Zheng B., Yang X., et al. MicroRNA-365 inhibits vascular smooth muscle cell proliferation through targeting cyclin D1. Int J Med Sci. 2014;11:765–770. doi: 10.7150/ijms.8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frid M.G., Brunetti J.A., Burke D.L., Carpenter T.C., Davie N.J., Reeves J.T., et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–669. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones R., Jacobson M., Steudel W. Alpha-smooth-muscle actin and microvascular precursor smooth-muscle cells in pulmonary hypertension. Am J Respir Cell Mol Biol. 1999;20:582–594. doi: 10.1165/ajrcmb.20.4.3357. [DOI] [PubMed] [Google Scholar]
- 45.Maron B.A., Leopold J.A. Emerging concepts in the molecular basis of pulmonary arterial hypertension: part II: neurohormonal signaling contributes to the pulmonary vascular and right ventricular pathophenotype of pulmonary arterial hypertension. Circulation. 2015;131:2079–2091. doi: 10.1161/CIRCULATIONAHA.114.006980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sengupta A., Kalinichenko V.V., Yutzey K.E. FoxO1 and FoxM1 transcription factors have antagonistic functions in neonatal cardiomyocyte cell-cycle withdrawal and IGF1 gene regulation. Circ Res. 2013;112:267–277. doi: 10.1161/CIRCRESAHA.112.277442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kolhar P., Anselmo A.C., Gupta V., Pant K., Prabhakarpandian B., Ruoslahti E., et al. Using shape effects to target antibody-coated nanoparticles to lung and brain endothelium. Proc Natl Acad Sci U S A. 2013;110:10753–10758. doi: 10.1073/pnas.1308345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooley M., Sarode A., Hoore M., Fedosov D.A., Mitragotri S., Gupta A.S. Influence of particle size and shape on their margination and wall-adhesion: implications in drug delivery vehicle design across nano-to-micro scale. Nanoscale. 2018;10:15350–15364. doi: 10.1039/c8nr04042g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu N., Becton M., Zhang L., Wang X. Mechanism of coupling nanoparticle stiffness with shape for endocytosis: from rodlike penetration to wormlike wriggling. J Phys Chem B. 2020;124:11145–11156. doi: 10.1021/acs.jpcb.0c08089. [DOI] [PubMed] [Google Scholar]
- 50.Guignabert C., Tu L., Le Hiress M., Ricard N., Sattler C., Seferian A., et al. Pathogenesis of pulmonary arterial hypertension: lessons from cancer. Eur Respir Rev. 2013;22:543–551. doi: 10.1183/09059180.00007513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoeper M.M., McLaughlin V.V., Dalaan A.M., Satoh T., Galie N. Treatment of pulmonary hypertension. Lancet Respir Med. 2016;4:323–336. doi: 10.1016/S2213-2600(15)00542-1. [DOI] [PubMed] [Google Scholar]
- 52.Michelakis E.D., McMurtry M.S., Wu X.C., Dyck J.R., Moudgil R., Hopkins T.A., et al. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation. 2002;105:244–250. doi: 10.1161/hc0202.101974. [DOI] [PubMed] [Google Scholar]
- 53.Li H., Wang Y., Chen L., Han L., Li L., He H., et al. The role of MIF, cyclinD1 and ERK in the development of pulmonary hypertension in broilers. Avian Pathol. 2017;46:202–208. doi: 10.1080/03079457.2016.1245409. [DOI] [PubMed] [Google Scholar]
- 54.Medema R.H., Kops G.J., Bos J.L., Burgering B.M. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt M., Fernandez de Mattos S., van der Horst A., Klompmaker R., Kops G.J.L., Lam E.W.-F., et al. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol. 2002;22:7842–7852. doi: 10.1128/MCB.22.22.7842-7852.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simonneau G., Gatzoulis M.A., Adatia I., Celermajer D., Denton C., Ghofrani A., et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–D41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 57.Maarman G., Lecour S., Butrous G., Thienemann F., Sliwa K. A comprehensive review: the evolution of animal models in pulmonary hypertension research; are we there yet? Pulm Circ. 2013;3:739–756. doi: 10.1086/674770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dignam J.P., Scott T.E., Kemp-Harper B.K., Hobbs A.J. Animal models of pulmonary hypertension: getting to the heart of the problem. Br J Pharmacol. 2022;179:811–837. doi: 10.1111/bph.15444. [DOI] [PubMed] [Google Scholar]
- 59.Teng C., Li B., Lin C., Xing X., Huang F., Yang Y., et al. Targeted delivery of baicalein-p53 complex to smooth muscle cells reverses pulmonary hypertension. J Control Release. 2022;341:591–604. doi: 10.1016/j.jconrel.2021.12.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.