

Abstract
Various c-mesenchymal-to-epithelial transition (c-MET) inhibitors are effective in the treatment of non-small cell lung cancer; however, the inevitable drug resistance remains a challenge, limiting their clinical efficacy. Therefore, novel strategies targeting c-MET are urgently required. Herein, through rational structure optimization, we obtained novel exceptionally potent and orally active c-MET proteolysis targeting chimeras (PROTACs) namely D10 and D15 based on thalidomide and tepotinib. D10 and D15 inhibited cell growth with low nanomolar IC50 values and achieved picomolar DC50 values and >99% of maximum degradation (Dmax) in EBC-1 and Hs746T cells. Mechanistically, D10 and D15 dramatically induced cell apoptosis, G1 cell cycle arrest and inhibited cell migration and invasion. Notably, intraperitoneal administration of D10 and D15 significantly inhibited tumor growth in the EBC-1 xenograft model and oral administration of D15 induced approximately complete tumor suppression in the Hs746T xenograft model with well-tolerated dose-schedules. Furthermore, D10 and D15 exerted significant anti-tumor effect in cells with c-METY1230H and c-METD1228N mutations, which are resistant to tepotinib in clinic. These findings demonstrated that D10 and D15 could serve as candidates for the treatment of tumors with MET alterations.
Key words: Cancer therapy, Drug design, c-MET, Proteolysis targeting chimeras (PROTACs), Drug resistance
Graphical abstract
Novel exceptionally potent and orally active c-MET degrader namely D10 and D15 were developed. D10 and D15 significantly inhibited tumor cells growth and induced potent c-MET degradation in vitro and in vivo.

1. Introduction
The c-mesenchymal-to-epithelial transition (c-MET) is a member of the transmembrane receptor tyrosine kinases (RTKs), and its ligand is hepatocyte growth factor (HGF)1. The combination of HGF to the extracellular part of c-MET leads to the receptor dimerization and autophosphorylation, which could regulate the proliferation, differentiation, metastasis and survival of cells by activating multiple signaling cascades, such as RAS-mitogen-activated protein kinase (MAPK) pathway, phosphoinositide-3 kinase (PI3K)-protein kinase B (AKT) pathway, signal transducer and activator of transcription (STAT) and nuclear factor kappa-B (NFκB) pathways2. Notably, the dysregulation of the HGF/c-MET signaling pathway is associated with various types of human malignancies including cancers of the brain, stomach, lung, kidney, and liver, etc.3. The mechanisms of aberrant activation of the c-MET pathway mainly include MET gene amplification, protein overexpression, and MET exon-14 skipping (METex14) mutation. c-MET overexpression or MET gene amplification is commonly detected in multiple solid tumors. METex14, which prevents CBL-mediated c-MET protein degradation resulting in sustaining activation of downstream signaling pathways, has been identified as a carcinogenic factor in brain cancer, non-small cell lung cancer (NSCLC) and other tumor types4,5.
c-MET targeting drugs mainly include macromolecular drugs and tyrosine kinase inhibitors (TKIs). Macromolecular drugs selectively targeting c-MET, such as monoclonal antibody and antibody–drug conjugates (ADC), have not been approved by US Food and Drug Administration (FDA). In recent years, c-MET tyrosine kinase inhibitors (TKIs) that bind to the intramembrane tyrosine kinase domain have shown promising antitumor effects in preclinical and clinical studies, and some of them have been approved by FDA. c-MET TKIs are mainly classified into three types (Fig. 1A). Type I inhibitors, binding to the active state of kinase, are classed into type Ia and Ib. Type Ia inhibitors such as crizotinib could interact with G1163 of c-MET, but type Ib such as capmatinib and foretinib could not. Type II inhibitors such cabozantinib and foretinib bind to the inactive state of c-MET. Type I and II inhibitors are both ATP competitive inhibitors, whereas type III inhibitors like tivantinib are non-ATP competitive allosteric inhibitors6. Unfortunately, despite the promising initial efficacy of c-MET inhibitors in clinical treatment, the inevitable drug resistance remains a challenge in almost all patients7. Thus, novel therapeutic strategies targeting c-MET are urgently needed to treat cancers and overcome c-MET acquired resistance.
Figure 1.

Chemical structures of several representative c-MET inhibitors of different types (A) and c-MET PROTACs (B).
Recently, the proteolysis-targeting chimeras (PROTACs) have been gaining momentum for their potential as novel therapeutics for human diseases8. PROTAC is a small bifunctional molecule consisting of three parts: target protein ligand, linker and E3 ubiquitin ligase ligand. More specifically, by hijacking the E3 ligase, PROTAC could simultaneously bind to a target protein and to an E3 ligase complex to induce the formation of a ternary complex (target protein–PROTAC–E3 ligase), and then lead to the polyubiquitination of target protein, resulting the degradation of target protein. Unlike classical inhibitors require high affinity to exert its efficacy, which are “occupancy-driven mechanism”, the degradation effects of PROTACs do not necessarily correlate with its affinity, which are the ‘‘event-driven’’ paradigm9. After a protein is degraded, PROTAC can participate in the next round of protein degradation, which is the called “catalytic property”10. Because of their special mechanism, PROTACs show some advantages over small molecules inhibitors, such as targeting undruggable targets, exhibiting catalytic property, improving pharmacological properties, decreasing potential toxicity and overcoming inhibitor resistance11,12. Up to now, several PROTACs have entered clinical trials for the treatment of multiple cancers10,13,14. Therefore, the exploration of c-MET PROTACs could enrich the modalities of c-MET-targeted treatment and potentially overcome the resistance towards c-MET inhibitors.
To date, only two studies reported that c-MET PROTACs employing foretinib, a multi-targeted kinase inhibitor targeting c-MET, VEGFR, RON, TIE-2, AXL and ROS1 etc., to synthesized von Hippel-Lindau (VHL)-recruiting and cereblon (CRBN)-recruiting c-MET PROTACs9,15. In these studies, PROTAC 1 and PROTAC 2 effectively degraded c-MET and inhibited the proliferation of tumor cells (Fig. 1B). Furthermore, they also proved PROTAC 1 could induce the internalization of transmembrane c-MET for further degradation, in which the ligand of PROTAC 1 (foretinib) binds to intracellular domain of c-MET. However, the use of foretinib as warhead, which could bind more than 100 kinases, results in the low selectivity of these PROTACs, which could degrade more than 100 proteins9. What's more, for those c-MET PROTACs, only a few biological and pharmacological studies have been conducted in vitro and none anticancer effects in vivo have been reported. Herein, we describe the design, synthesis, and extensive evaluation of a series of c-MET degraders, in which we found exceptionally potent, and orally active c-MET degraders namely D10 and D15, highlighting the potential of our study as a new therapeutic strategy for tumors with MET alterations.
2. Results and discussion
2.1. Rational design of cereblon-recruiting c-MET degraders
Tepotinib (Fig. 1A), a highly selective c-MET inhibitor, was approved by the FDA for the treatment of NSCLC, and it shows promising anti-tumor activity with good tolerance in clinical treatment16. Thus, tepotinib was selected as the warhead to obtain c-MET PROTACs. The co-crystal structure of tepotinib with c-MET (PDB ID: 4R1V) indicated that tepotinib binds to c-MET in a U-shaped conformation, with the benzonitrile and methylpiperidine groups both exposed to solvent (Fig. 2A and B). Since the methylpiperidine group showed no interaction with c-MET, we hypothesized that solvent-exposed piperidine group could be employed as the tethering site with various linkers. Thus, we removed the methyl group of the methylpiperidine to obtain compound 6 (Fig. 2C). We next evaluated their binding affinity to c-MET and antiproliferative effect on c-MET-sensitive EBC-1 NSCLC cells (c-MET overexpressed) and Hs746T gastric carcinoma cells (METex14). The competitive binding assay showed that compound 6 displayed comparable binding affinity to c-MET (Kd = 9.97 nmol/L) compared with tepotinib (Kd = 4.69 nmol/L). Additionally, compound 6 exhibited comparable anti-proliferative effect in EBC-1 and Hs746T cells compared with tepotinib (Fig. 2D and Supporting Information Fig. S1), demonstrating that compound 6 could serve as the warhead of c-MET PROTACs.
Figure 2.

Design of c-MET degraders based upon c-MET inhibitor tepotinib and CRBN ligand thalidomide. (A–B) The co-crystal structure of tepotinib with c-MET (PDB ID: 4R1V). (C) Structure of tepotinib and compound 6. (D) The binding affinities (Kd) to c-MET and IC50 of tepotinib and compound 6 in EBC-1 and Hs746T cells. The Kd determinations were performed in a competitive binding assay in triplicate. Data shown are mean ± SD of triplicate measurements. (E) Design of c-MET PROTACs.
The CRBN and VHL E3 ubiquitin ligase are the mostly utilized in PROTACs for their wide expression in many types of cells10. Compared to VHL ligands, which are peptidomimetics with molecular weight (MW) > 400, CRBN ligands such as lenalidomide and thalidomide exhibit excellent physiochemical and pharmacokinetic (PK) properties with MW of approximately 25017. Thus, we employed thalidomide as the CRBN ligand of our PROTACs.
In a PROTAC, the length, type and attachment site of the linker play pivotal roles in degradation potency, substrate selectivity, and molecule kinetics10,18,19. Thus, in this research we applied different linkers (in length and types) to connect with thalidomide at different sites to obtain the optimal c-MET PROTACs (Fig. 2E).
2.2. Synthesis and evaluation of c-MET degraders for their antiproliferative effects
We firstly designed compounds D1–D8 to determine the optimal linker length, in which tepotinib was linked with thalidomide at the 4′ position by alkyl linkers with different length, and we next evaluated their antiproliferative effects in a panel of cell lines. The synthetic routes for preparing compounds D1–D8 are outlined in Scheme 1. As shown in Table 1, among compounds D1–D8, degraders with the linkers containing 4–6 methylene groups (D3–D5) led to greater antiproliferative effects in EBC-1 and Hs746T cells, which were weaker than tepotinib. Longer or shorter linkers did not improve the antiproliferative effects.
Scheme 1.

Synthesis of tepotinib-based c-MET degraders D1–D9. Reagents and conditions: (A) Synthesis of intermediate 6 and 8: (a) PPh3, DIAD, THF (dry), 0 °C, 12 h; (b) Pd(dppf)2Cl2, K3PO4, DMF/H2O, 80 °C, 10 h; (c) PPh3, CCl4, DCM, 45 °C, 12 h; (d) K2CO3, DMF, 80 °C, 10 h; (e) CF3COOH, DCM, 6 h; (f) K2CO3, DMF, 10 h; (g) CF3COOH, DCM, 6 h. (B–C): (a) THF, 55 °C, 12 h; (b) DIPEA, DMF, 24 h.
Table 1.
Antiproliferative effects of c-MET degraders D1–D8 with various linker length.
| Cpd. | Site | Linker | IC50 (μmol/L)a |
|||
|---|---|---|---|---|---|---|
| c-MET-sensitive |
c-MET-insensitive |
|||||
| EBC-1 | Hs746T | A549 | HepG2 | |||
| D1 | 4′ | 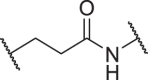 |
0.00703 ± 0.00205 | 0.07555 ± 0.0201 | 1.260 ± 0.549 | >100 |
| D2 | 4′ | 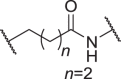 |
0.0109 ± 0.00596 | 0.0658 ± 0.0095 | 0.653 ± 0.122 | >100 |
| D3 | 4′ | 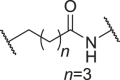 |
0.00945 ± 0.00257 | 0.0763 ± 0.0129 | 0.413 ± 0.125 | 0.399 ± 0.105 |
| D4 | 4′ | 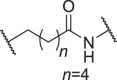 |
0.00805 ± 0.00146 | 0.0516 ± 0.0113 | 1.640 ± 0.303 | 1.843 ± 0.928 |
| D5 | 4′ | 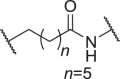 |
0.00563 ± 0.00111 | 0.0968 ± 0.0231 | 1.035 ± 0.179 | 4.137 ± 2.648 |
| D6 | 4′ | 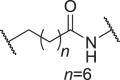 |
0.0195 ± 0.00186 | 0.159 ± 0.050 | 8.695 ± 1.959 | 20.12 ± 10.59 |
| D7 | 4′ | 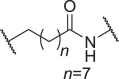 |
0.0214 ± 0.0051 | 0.103 ± 0.077 | 45.62 ± 12.62 | >100 |
| D8 | 4′ | 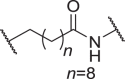 |
0.0379 ± 0.0072 | 0.226 ± 0.112 | 21.04 ± 5.55 | >100 |
| Tepotinib | – | – | 0.00132 ± 0.00039 | 0.00263 ± 0.00072 | 0.277 ± 0.059 | 0.476 ± 0.045 |

The data are averages of three independent determinations.
In addition to the linker's length, the structure and connecting site of the linker also play a role in the degradation effect. Therefore, we modified the linker with PEG and piperazine chain, and changed the linker connecting site with thalidomide to form a series of degraders (D9–D15). The synthetic routes for preparing compounds D9–D15 are outlined in Scheme 1, Scheme 2, Scheme 3. As shown in Table 2, compared to the antiproliferative effect of D6 in EBC-1 and Hs746T cells (IC50 = 0.0195 and 0.159 μmol/L, respectively), compound D13 with a PEG linker, showed higher efficiency (IC50 = 0.00585 and 0.02467 μmol/L, respectively), indicating the advantage of the PEG linker over alkyl linker. In addition, D9 with linker tethered at the 5′ position of thalidomide, was more effective in EBC-1 and Hs746T cells (IC50 = 0.00398 and 0.00721 μmol/L, respectively) than D3 (IC50 = 0.00945 and 0.0763 μmol/L, respectively) with linker tethered at the 4′ position of thalidomide, demonstrating the advantage of 4′ position connection over 5′ position connection of thalidomide. We further synthesized D10–D12 containing the PEG linker tethered at the 5′ position of thalidomide. The antiproliferative effect of D10 in EBC-1 and Hs746T cells (IC50 = 0.00334 and 0.00589 μmol/L, respectively) were comparable to tepotinib and were more effective than compounds D11 and D12. Additionally, conformational restriction is often used as a strategy to improve activity and PK property of PROTACs20,21. Next, we designed and synthesized D14 and D15 containing a conformationally restricted linker with a piperazine group. As shown in Table 2, D14 obtained an IC50 of 0.00244 μmol/L in EBC-1 cells and 0.00388 μmol/L in Hs746T cells. D15 obtained an IC50 of 0.00211 μmol/L in EBC-1 cells and 0.00350 μmol/L in Hs746T cells, which was comparable to tepotinib.
Scheme 2.

Synthesis of tepotinib-based c-MET degraders D10–D13. Reagents and conditions: (a) DIPEA, DMF, 80 °C, 10 h; (b) CF3COOH, DCM, 6 h; (c) HATU, DIPEA, DMF, 24 h.
Scheme 3.

Synthesis of tepotinib-based c-MET degraders D14–D15. Reagents and conditions: (a) DIPEA, DMF, 80 °C, 10 h; (b) CF3COOH, DCM, 6 h; (c) HATU, DIPEA, DMF, 24 h.
Table 2.
Antiproliferative effects of c-MET degraders D9–D15 with modification of the linker types and sites tethered to thalidomide.
| Cpd. | Site | Linker | IC50 (μmol/L)a |
|||
|---|---|---|---|---|---|---|
| c-MET-sensitive |
c-MET-insensitive |
|||||
| EBC-1 | Hs746T | A549 | HepG2 | |||
| D9 | 5′ | 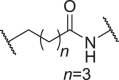 |
0.00398 ± 0.00068 | 0.00721 ± 0.00230 | 0.977 ± 0.220 | 2.47 ± 0.67 |
| D10 | 5′ | 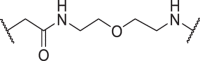 |
0.00334 ± 0.00048 | 0.00589 ± 0.00150 | 0.511 ± 0.247 | 1.54 ± 0.50 |
| D11 | 5′ | 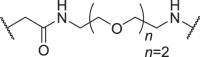 |
0.00555 ± 0.00060 | 0.00619 ± 0.00185 | 1.15 ± 0.24 | 7.47 ± 1.42 |
| D12 | 5′ | 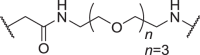 |
0.00461 ± 0.00044 | 0.00772 ± 0.00216 | 0.433 ± 0.096 | 0.672 ± 0.141 |
| D13 | 4′ | 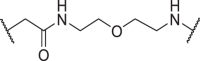 |
0.00585 ± 0.00095 | 0.0247 ± 0.0113 | 3.29 ± 0.99 | 10.74 ± 2.28 |
| D14 | 5′ | 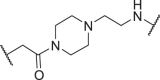 |
0.00244 ± 0.00033 | 0.00388 ± 0.00060 | 0.665 ± 0.103 | 1.95 ± 0.83 |
| D15 | 5′ | 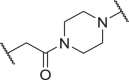 |
0.00211 ± 0.00031 | 0.00350 ± 0.00056 | 0.377 ± 0.058 | 0.543 ± 0.107 |
| Tepotinib | – | – | 0.00132 ± 0.00039 | 0.00263 ± 0.00072 | 0.277 ± 0.059 | 0.476 ± 0.045 |

The data are averages of three independent determinations.
What's more, we found that EBC-1 and Hs746T cells showed high sensitivity to c-MET degraders with IC50 values of nanomolar concentration, while the IC50 values were micromolar concentrations in A549 and HepG2 cells (Table 1, Table 2). This discrepancy between these cell lines may result from their different levels of c-MET expression and c-MET autophosphorylation (Supporting Information Fig. S2A). In addition, the inhibitory effects on normal cells were also measured including LO2, 293T, HMEC and BEAS-2B cells. The result showed that all c-MET degraders exhibited no cytotoxicity up to 100 μmol/L, which was much better than tepotinib, demonstrating the low cytotoxicity of c-MET degraders in normal cells. For the reason, we hold that the low drug exposure of PROTAC in cells due to its low permeability may result in the low cytotoxicity. While low concentration of PROTACs could exert well activity in EBC-1 cells and Hs746T cells (c-MET-sensitive cancer cells) for their catalytic properties, which is consistent of above results.
2.3. D10 and D15 induced c-MET degradation and inhibited c-MET phosphorylation
After confirming the potent antiproliferative effects of D3, D10, D12, D14, and D15, their degradation effects were further evaluated by Western blotting. As shown in Table 3 and Supporting Information Fig. S3, these compounds exhibited high capability of degrading c-MET protein in EBC-1 and Hs746T cells in a dose-dependent manner. Among the five compounds, D10 and D15 showed the highest degradation rates against c-MET. D10, D14, and D15 achieved a maximum degradation (Dmax) of 99% in EBC-1 and Hs746T cells. In contrast, none of tepotinib, thalidomide, their combination (Tep + Tha) showed c-MET degradation effect.
Table 3.
Degradation effects of c-MET degraders in vitro.
| Cpd. | DC50 (nmol/L)a |
Dmax (nmol/L)b |
||
|---|---|---|---|---|
| EBC-1 | Hs746T | EBC-1 | Hs746T | |
| D3 | 3.1 ± 1.3 | 8.3 ± 1.6 | >95% | >99% |
| D10 | 0.69 ± 0.21 | 0.77 ± 0.11 | >99% | >99% |
| D12 | 1.7 ± 0.4 | 2.7 ± 0.5 | >95% | >99% |
| D14 | 0.83 ± 0.16 | 0.60 ± 0.15 | >99% | >99% |
| D15 | 0.44 ± 0.11 | 0.35 ± 0.08 | >99% | >99% |
| Tepotinib | – | – | – | – |
| Thalidomide | – | – | – | – |
| Tep + Tha | – | – | – | – |
a,bThe data are averages of three independent determinations.
–Not applicable; Tep: tepotinib; Tha: thalidomide.
Based on both antiproliferative activity and degradation rates, we selected D10 and D15 for further study. c-MET promotes cell proliferation through auto-phosphorylation, which activates downstream signaling pathways1. As shown in Fig. 3A and Supporting Information Fig. S4A, D10 and D15 effectively inhibited the levels of c-MET phosphorylation (p-c-MET) and STAT3 phosphorylation (p-STAT3) in EBC-1 and Hs746T cells in a concentration-dependent manner. We found that D10/D15 at 10 nmol/L significantly induced c-MET degradation and 100 nmol/L treatment of D10/D15 almost completely induced degradation of c-MET. Intriguingly, D10 and D15 at 10 nmol/L almost abrogated p-c-MET and significantly eliminated p-STAT3, demonstrating that D10 and D15 could significantly inhibit the downstream signal transduction of the c-MET pathway. As the thalidomide is a part of PROTAC, we also found that CRBN substrates such as IKZF1 and IKZF3 both could be degraded by D10 and D15 (Fig. S4B and S4C), which have also been observed in other PROTACs22,23.
Figure 3.

D10 and D15 showed potent degradation effects on c-MET and inhibitory effects on p-c-MET. (A) The effects of tepotinib, D10 and D15 on c-MET and its downstream signaling pathways in EBC-1 cells. Cells were treated with D10 and D15 for 48 h at the indicated concentration. (B) The effects of D10 and D15 on c-MET and c-MET phosphorylation in EBC-1 cells. Cells were treated with D10 and D15 (10, 100 nmol/L) at indicated time. (C) EBC-1 cells were pretreated with D10 and D15 at 100 nmol/L) for 24 h, then washed with PBS three times, and harvested at the indicated time for Western blot analysis. (D) EBC-1 cells were treated with D15 at 10 nmol/L) for 48 h and then collected for liquid chromatography−mass spectrometry (LC−MS) analyses. The panel showed the relative abundance of c-MET (LFQ intensity value) between the degrader-treated and control groups.
Next, we performed time-dependent experiments to study the kinetics of c-MET degradation and p-c-MET inhibition of D10 and D15. As shown in Fig. 3B and Fig. S4D, those effects were enhanced with prolonged incubation time. D10 and D15 at 10 nmol/L reduced the c-MET protein level substantially (approximately 50%) after 24 h treatment, whereas the near-complete degradation was achieved at 100 nmol/L after 12 h. Intriguingly, complete inhibition of p-c-MET was observed after 6 h treatment with D10 and D15 at 10 and 100 nmol/L, respectively, and both degraders led to a complete inhibition effect sustained for 24 h in EBC-1 and Hs746T cells.
Next, washout experiments were performed to assess the duration of c-MET degradation induced by D10 and D15. EBC-1 and Hs746T cells were pretreated with D10 and D15 for 24 h, washed and replaced with a fresh medium. As shown in Fig. 3C, and Fig. S4E, c-MET protein was degraded continuously after the medium was washed out and c-MET protein and p-c-MET were completely recovered after an additional 48 h because of the protein re-synthesis, indicating that D10 and D15 showed long-lasting degradation effects.
To understand the selectivity of D15 on the cellular proteome, we also performed the mass spectrometry (MS)-based label-free quantitative (LFQ) proteomics analysis in EBC-1 cells. As shown in Fig. 3D, we found that over 80% of c-MET protein was degraded upon D15 treatment, and 18 proteins were downregulated across all the identified 3372 proteins (P value <0.05, |Fold change (Log2) | >1.5, Supporting Information Table S1). Overall, D15 is a potent and highly selective c-MET PROTAC compared with the previous reported foretinib-based c-MET PROTACs9.
Previous study has shown that treatment with foretinib-based PROTAC induces polyubiquitination of c-MET15. Next, we investigated the mechanism underlying the degradation effects of D10 and D15 using a set of rescue assays. EBC-1 and Hs746T cells were pretreated with the E1 ubiquitin-activating enzyme inhibitor MLN4924, the proteasome inhibitor MG132, CRBN E3 ligand thalidomide, and c-MET inhibitor tepotinib, then D10 and D15 were added. As shown in Fig. 4A and B, pretreatment with MLN4924, MG132, completely blocked c-MET degradation effect of D10 and D15 in EBC-1 and Hs746T cells, indicating that c-MET degradation induced by D10 and D15 was mediated by the ubiquitin proteasome pathway. Similarly, pretreatment with thalidomide or tepotinib, which could compete with D10/D15 for binding with CRBN and c-MET, respectively, also completely blocked c-MET degradation effect of D10 and D15 (Fig. 4C and D), demonstrating the essential factor of D10/D15 binding with CRBN and c-MET in the c-MET degradation. We also found that thalidomide combined with D15 significantly reduced the antiproliferative effect compared with D15 single agent in EBC-1 and Hs746T cells as thalidomide competed with D15 for bind with CRBN (Supporting Information Fig. S5). In contrast, no difference between tepotinib + thalidomide with tepotinib single agent.
Figure 4.

C-MET degradation effects of D10 and D15 were mediated through the ubiquitin proteasome pathway and the formation of ternary complex. (A–B) EBC-1 (A) and Hs746T (B) cells were pretreated with MLN-4924 (10 μmol/L), MG132 (10 μmol/L) for 6 h, followed by treatment with D10 and D15 at 10 nmol/L for 72 h. (C–D) EBC-1 (C) and Hs746T (D) cells were pretreated tepotinib (100 nmol/L) or thalidomide (10 μmol/L) for 6 h, followed by treatment with D10 and D15 at 10 nmol/L for 72 h.
2.4. D10 and D15 significantly inhibited cell migration and invasion
C-MET plays a role in maintaining the transformed metastatic phenotype such as migration and invasion1. Next, we examined the effects of D10 and D15 on cancer cells migration and invasion. The wound-healing assay and matrigel invasion assay showed that D10 and D15 significantly inhibited the migratory (Fig. 5A–C) and invasive (Fig. 5D and E) capacity of EBC-1 and Hs746T cells at low nanomolar concentrations in a dose-dependent manner. Notably, D10 and D15 at 10 nmol/L almost abrogated the migratory and invasion abilities of EBC-1 and Hs746T cells comparable to tepotinib.
Figure 5.

D10 and D15 significantly inhibited cell migration and invasion. Wound healing assay (A–C) and Transwell assay (D–E) of EBC-1 and Hs746T cells treated with tepotinib, D10, D15 and vehicle control (DMSO) treatment at the indicated concentration for 12h. Histograms show the relative cell migration and cell invasion (bottom). Data are mean ± SD, n = 3, ∗∗P < 0.01 (t test). Scale bars: 100 mm.
2.5. D10 and D15 induced apoptosis and G1 cell cycle arrest in cancer cells
To investigate the mechanism underlying the antiproliferative activity of D10 and D15, we explored the ability of D10 and D15 to induce EBC-1 and Hs746T cell apoptosis and cell cycle transition. As shown in Fig. 6A and B, the apoptosis rates produced by D10 at 10 and 100 n mol/L in EBC-1 cells were 43.6% and 62.5%, respectively, while the apoptosis rates of D15 at 10 and 100 nmol/L in EBC-1 cells were 41.7% and 68.0%, respectively. These rates were slightly higher than that produced by tepotinib (39.3% and 59.4%, respectively). In Hs746T cells, D10 and D15 at 10 and 100 nmol/L also significantly induced apoptosis comparable to tepotinib. Additionally, as depicted in Fig. 6C and D, D10 and D15 induced cell cycle arrest at the G0/G1 phase in a concentration-dependent manner in EBC-1 and Hs746T cells; moreover, the activity was comparable to that of tepotinib at 10 and 100 nmol/L. In short, D10 and D15 induced apoptosis and G1 cell cycle arrest in cancer cells comparable to tepotinib, which correlates with their antiproliferative activity.
Figure 6.

D10 and D15 induced cell apoptosis and G1 cell cycle arrest. Representative images of flow cytometry analysis of apoptosis (A–B) and cell cycle distributions (C–D) of EBC-1 and Hs746T cells treated with tepotinib, D10, D15 and vehicle control (DMSO) at the indicated dose for 48 h. Histograms show the relative cell percentage of apoptosis and the cell cycle distributions of cell cycle phase of each group in EBC-1 and Hs746T cells (bottom). Data are mean ± SD, n = 3, ∗∗P < 0.01 (t test).
2.6. Degradation of c-MET exerted pivotal role in the anticancer effect of D15
We next investigated the degradation and inhibition effect in the anticancer effect of D15 (Fig. 4A). Previous studies have shown that an additional methyl group on the glutarimide moiety of CRBN ligands that significantly reduced its affinity for CRBN E3 ligase thus blocking the degradation effects9,15, and we next designed analogue D16 (Fig. 7A). Synthetic route for preparing compounds D16 is outlined in Scheme 4. We validated that D16 had no degradation effect on c-MET protein and showed weaker effect on p-c-MET than D15 (Fig. 7B, Supporting Information Fig. S6A).
Figure 7.

Degradation of c-MET exerted pivotal role in the anticancer effect of D15. (A) Chemical structures of D15 and the negative control D16. (B) The effects of c-MET and p-c-MET by D16 in EBC-1 cells. (C) The binding affinities (Kd) to c-MET and antiproliferative effects of D15 and D16 in EBC-1 and Hs746T cells. The Kd determinations were performed in a competitive binding assay in triplicate. Data shown are mean ± SD of triplicate measurements. (D) Representative images of flow cytometry analysis of apoptosis in EBC-1 cells treated with D15, D16 and vehicle control (DMSO) at the indicated dose for 48 h. Histograms show the relative cell percentage of apoptosis in EBC-1 cells (right). Data are mean ± SD, n = 3, ∗∗P < 0.01 (t test). (E, F) Wound healing assay (E) and Transwell assay (F) in EBC-1 cells treated with D15, D16 and vehicle control (DMSO) at the indicated dose for 12 h. Histograms show the relative cell migration and cell invasion (right). Data are mean ± SD, n = 3, ∗∗P < 0.01 (t test). Scale bars: 100 mm.
Scheme 4.

Synthesis of negative control compounds D16. Reagents and conditions (a) DIPEA, DMF, 80 °C, 10 h; (b) CF3COOH, DCM, 6 h; (c) HATU, DIPEA, DMF, 24 h.
We next measured the binding affinities (Kd) of D15 and D16 to c-MET and their antiproliferative effect of in EBC-1 and Hs746T cells. We found that D15 showed low binding affinity to c-MET with Kd value of 169.7 nmol/L in contrast to its inhibitory and degradation effects at low nanomolar concentration in EBC-1 and Hs746T cells (Fig. 7C, Fig. S6B), highlighting the fact that high degradation effects can be achieved through weak binding affinity. Although the binding affinity of D16 (Kd = 344.9 nmol/L) was 2-fold weaker than that of D15, it was approximately 10-fold weaker in inhibiting EBC-1 and Hs746T cells proliferation (Fig. 7C and Fig. S6C and D). Moreover, D16 showed a much weaker effect on inducing cell apoptosis, inhibiting cell migration and invasion than that of D15 (Fig. 7D–F, Fig. S6E–G). The comparison of D15 with D16 demonstrate that degradation of c-MET may mainly contributes to the antiproliferative effect of D15.
2.7. D10 and D15 significantly inhibited tumor growth in xenograft models
Given the exceptionally potent antiproliferative and degradation effects of D10 and D15 in the in vitro, we further conducted in vivo studies to examine their efficacy in BALB/c nude mice bearing EBC-1 and Hs746T xenograft tumors. As shown in Fig. 8A–C, compared with vehicle, D10 at dose of 10 mg/kg by intraperitoneal (i.p.) administration significantly inhibited EBC-1 tumor growth with tumor growth inhibition (TGI%) rates of 68.2%, while D15 led to TGI% rates of 85.3%, which was higher than tepotinib (71.9%). The final tumor weight was measured to further validate the efficacy of D10 and D15 (Fig. 8D). Furthermore, no obvious body weight loss and no other obvious toxic signs had been represented in nude mice, indicating that D10 and D15 were well tolerated in the nude mice (Fig. 8E).
Figure 8.

D10 and D15 significantly repressed EBC-1 tumor growth in vivo by i.p. administration. (A) Treatment schedule for the EBC-1 xenograft tumors model treated with vehicle control, tepotinib (10 mg/kg), D10 (10 mg/kg) or D15 (10 mg/kg). (B) The change of tumor volume was measured every 2 days. (C) Tumors' picture. (D) Tumor weight of all mice in each group (n = 6). Data are mean ± SD, ns (no significance), ∗∗P < 0.01 (one-way ANOVA). (E) The change of body weight of all mice was measured every 2 days.
We also explored the oral anticancer efficacy of D10 and D15 in vivo (Fig. 9A). Daily oral administration (per os, p.o.) of D10 and D15 at a dose of 20 mg/kg led to a TGI% values of 71.4% and 91.6%, respectively, in Hs746T xenograft tumors. Notably, higher doses (40 mg/kg) of D10 and D15 resulted in marked Hs746T xenograft tumor regression with TGI% values of 88.5% and 99.2%, respectively (Fig. 9B and C). Meanwhile, D10 (p.o., 40 mg/kg) and D15 (p.o., 40 mg/kg) was well tolerated in the nude mice, and no significant weight loss was observed during the treatment period (Fig. 9D).
Figure 9.

D10 and D15 significantly repressed Hs746T tumor growth in vivo by p.o. administration. (A) Treatment schedule for the Hs746T cells xenograft tumors model treated with vehicle, tepotinib, D10 and D15. (B–D) BALB/c mice transplanted with Hs746T cells were orally administrated with vehicle control, tepotinib, D10, or D15 with single doses of 20, 40 mg/kg. The change of Hs746T tumor volume (B), tumor weight (C) and change of body weight (D) of all mice in each group were shown (n = 7). Data are mean ± SD, ∗∗P < 0.01 (one-way ANOVA). (E) Representative images of H&E, c-MET, p-c-MET, and cleaved-PARP immunohistochemical (IHC) staining in harvested tumors from each group in (B) are shown. Scale bars represent 50 mm.
To elucidate the molecular changes upon in vivo administration of D10 and D15 in tumors, we next assessed hematoxylin and eosin (H&E), c-MET, p-c-MET and cleaved-poly ADP-ribose polymerase (PRAP)-stained immunohistochemical sections of the tumors (Fig. 9E). In consistent with the in vitro studies, the number of tumor foci in the tepotinb, D10 and D15 groups was much lower than that in the vehicle group. Additionally, tepotinb, D10 and D15 groups significantly reduced the percentage of p-c-MET-positive tumor cells and increased the percentage of cleaved PARP-positive tumor cells with single doses of 20 and 40 mg/kg. Notably, single doses of 20 and 40 mg/kg of D15 dramatically reduced the levels of c-MET, whereas only high doses of D10 (40 mg/kg) resulted in significant inhibition of c-MET, demonstrating that D15 exhibited more efficient degradation effect than D10 in vivo.
2.8. PK properties of D10 and D15in vivo
Since the anticancer assays in vivo were conducted, the PK properties of D10 and D15 were further explored. Due to their bifunctional nature, PROTACs are expected to face challenges about absorption, distribution, metabolism, excretion (ADME) owing to their large and flexible structures, which limits the development for orally active PROTACs24, and the investigations on their PK properties are only beginning to emerge25. Therefore, we evaluated the concurrent PK characteristics of D10 and D15 to deepen the understanding of PROTAC ADME behaviors associated with their efficacy. The average plasma concentration-versus-time profiles of D10, D15 and tepotinib after p.o. administration (20 mg/kg for tepotinib, 40 mg/kg for D10 and D15), i.p. administration (10 mg/kg), and intravenous (i.v.) administration (1 mg/kg) in rats are shown in Fig. 10A. Corresponding PK parameters are shown in Fig. 10B. The PK curves and PK parameters of tepotinib via i.v. and p.o. administration in the present study was close to previously reported values, with Vss of 19 and 20 L/kg, CL of 4.7 and 4.2 L/h/kg, and F of 55% and 56%, respectively.
Figure 10.

PK properties of D10 and D15in vivo. (A) The mean plasma concentrations vs time in rats after administrated with D10, D15 and tepotinib via different routes (mean ± SD, n = 4). (B) The mean pharmacokinetic parameters in rats after administrated with D10, D15 and tepotinib via different routes (mean ± SD, n = 4). ∗P < 0.05, ∗∗P < 0.01 compared with tepotinib group via corresponding routes. ##P < 0.01 compared with D10 group via corresponding routes.
The PK characters of tepotinib, D10 and D15 after i.v. and i.p. dosing did not show showed obvious difference. While obvious differences between PROTACs and tepotinib were observed after p.o. dosing, D10 and D15 significantly decreased exposure compared to tepotinib. The comparison of the value of Cmax, AUC and F between PROTACs and tepotinib showed that D10 and D15 were exposed to much lower concentrations in blood after p.o. dosing, which could explain why D10 (40 mg/kg, p.o.) and D15 (40 mg/kg, p.o.) were less effective than tepotinib (10 mg/kg, p.o. and 20 mg/kg, p.o.). Regarding D10 and D15, the PK properties (such as Cmax, AUC and F) of D15 were better than D10, which could explain why D15 (40 mg/kg, p.o.) was more efficient than D10 (40 mg/kg, p.o.) in xenograft models. And the better PK properties of D15 than D10 may result from its conformational restriction by a piperazine linker. Though D15 exhibited low exposures and bioavailability, D15 showed potent inhibitory effect of in EBC-1 and Hs746T xenograft tumor, highlighting the catalytic-mechanism characteristic of PROTACs. Previous study has manifested that no linear correlation between PK and pharmacodynamic (PD) for PROTAC, and PD efficacy of PROTACs extend beyond the detectable PK presence, which could also support the rationality our results26. Nevertheless, in view of the poor PK properties of our PROTACs, some approaches such as employing diverse rigid linkers and dosage forms are needed for further optimization.
2.9. D10 and D15 exhibited antiproliferative effects on acquired type Ib c-MET TKIs resistance
One of the advantages of PROTAC is its potential to overcoming drug resistance27. Previous study has shown that MET mutations (e.g., D1228N and Y1230H) confer resistance to type Ib inhibitors in c-MET-addicted SNU-638 gastric cancer and Hs746T cells28. We next constructed c-METY1230H and c-METD1228N mutations in EBC-1 and Hs746T cells and compared the antiproliferative effects of tepotinib, D10, and D15 in each cell line (Fig. S7A). As shown in Fig. 11A and Fig. S7B and C, the antiproliferative effects of D10/D15 were almost 10-fold stronger than tepotinib in EBC1Y1230H or EBC1D1228N cells; this was similar in Hs746T cells. Furthermore, Western blot analysis showed that tepotinib at 1000 nmol/L partially inhibited p-c-MET level in Y1230H- or D1228N-EBC1 and Hs746T cells (Fig. 11B and C). In contrast, D10 and D15 at 100 nmol/L partially inhibited p-c-MET level, and complete c-MET degradation and p-c-MET inhibition were observed after treatment with D10 and D15 at 1000 nmol/L. These data illustrated that D10 and D15 could exhibit antiproliferative effect on clinically relevant form of acquired type Ib c-MET TKIs resistance.
Figure 11.

c-MET degradation overcame acquired type Ib c-MET TKIs resistance. (A) Antiproliferative effects of tepotinib, D10 and D15 in EBC-1 and Hs746T cells with Y1230H and D1228N mutation, respectively. Data shown are mean ± SD of triplicate measurements. (B) The effects of c-MET and p-c-MET by tepotinib, D10 and D15 in EBC-1 and Hs746T cells with Y1230H and D1228N mutation, respectively. Cells were treated with tepotinib, D10 and D15 for 48 h at the indicated dose.
2.10. Synergy of tepotinib and D10/D15 significantly inhibited cells growth
PROTACs exhibit complete and long-lasting pharmacological activity than classical molecules via catalytic property of protein degradation15,26. However, due to the event-driven mechanism of PROTACs, it takes a period to degrade proteins. In contrast, small molecule inhibitors could exert rapid pharmacological activity for their occupancy-driven mechanism. The binding and dissociation of tepotinib as well as D10/D15 with c-MET are dynamic processes as their reversible non-covalent binding mode with c-MET. Thus, drug combination of tepotinib and D10/D15 could take the advantage of the rapid effect of tepotinib and long-lasting, complete effect of D10/D15, to get complete p-c-MET inhibition, which may result in the synergistic effects.
Next, the synergistic effects of tepotinib with D10/D15 were explored, as determined by the combination index (CI) using the Chou–Talalay method. As shown in Fig. 12A–C and Supporting Information Fig. S8, the dose–response curve for tepotinib combined with D10 or D15 revealed high potency and strong synergistic effect in EBC-1 and Hs746T cells (CI < 1). The cell proliferation assays also showed that tepotinib combined with D10/D15 exhibited profound inhibitory effect than single agent (Fig. 12D and E and Supporting Information Fig. S9). In addition, as shown in Fig. 12F, though tepotinib could compete with D10/D15 for binding to c-MET, which blocked c-MET degradation effect of D10/D15, tepotinib combined with D10/D15 exhibited more effective potency in inhibiting p-c-MET and p-STAT3 than mono-drug treatment. Taken together, these data demonstrated the synergistic effects of tepotinib and D10/D15 in inhibited cells growth.
Figure 12.

Synergy of tepotinib with D10/D15 significantly inhibited cells growth. (A–C) Effects of tepotinib with D10/D15 as single agents or drug combinations in EBC-1 (A) and Hs746T (B) cells. Data are mean ± SD, n = 3. CI was calculated by the Chou–Talalay equation using multiple doses and response points, and the data are averages of three independent determinations. CI values of three different indicated fraction affect (Fa) are shown (C). (D–E) The antiproliferative effects of tepotinib, D10 and D15 as single agents or drug combinations in EBC-1 cells at the indicated concentration. Data are mean ± SD, n = 3. ∗∗P < 0.01 (one-way ANOVA). (F) The effects of c-MET and p-c-MET by tepotinib with D10 and D15 as single agents or drug combinations in EBC-1 and Hs746T cells at the indicated concentration.
3. Conclusions
In this study, we described the design, synthesis, and evaluation of c-MET PROTACs using tepotinib and thalidomide. Through rational structure optimization, we presented for the first time the discovery of highly potent and orally active c-MET degraders exemplified by D10 and D15. Compared with reported c-MET-PROTACs, D15 has some advantages as follow. Firstly, only a few biological and pharmacological studies have been conducted in vitro and none anticancer effects in vivo have been reported. Herein, we found that D15 inhibited cell growth with low nanomolar IC50 values and achieved picomolar DC50 values and >99% of maximum degradation (Dmax) in EBC-1 and Hs746T cells, which exhibited higher pharmacological activities than reported c-MET PROTACs. Oral administration of D15 induced approximately complete tumor suppression (TGI% = 99.2%) in the Hs746T xenograft model with well-tolerated dose-schedules. Secondly, the warhead of the reported c-MET-PROTACs is foretinib, a multi-targeted kinase inhibitor, while the warhead of D10/D15 is highly selective c-MET inhibitor tepotinib. The global proteomic profiling of D15 showed that only 18 proteins were significantly downregulated (P value < 0.05, |Fold change (Log2) | > 1.5) (Fig. 3D), exhibiting higher selectivity compared with the previous reported foretinib-based c-MET PROTACs, which could significantly downregulate more than 100 proteins. Compared with tepotinib, D15 has some advantages as follow. Firstly, the antiproliferative effect of D10/D15 was almost 10-fold stronger than tepotinib in type Ib c-MET TKIs resistant c-METY1230H or c-METD1228N mutations cells. Secondly, D10/D15 exhibited no cytotoxicity up to 100 μmol/L in normal cell lines including LO2, 293T, HMEC and BEAS-2B cells, which was much better than tepotinib, demonstrating the low cytotoxicity effects of c-MET degraders in normal cells.
Based on the excellent performance of D10 and D15 in vitro and in vivo, they can be exploited as the candidate drugs for the treatment tumors with MET alterations.
4. Experimental
4.1. General information
Unless otherwise mentioned, all solvents and reagents are commercially available without further purification. TCL was used to determine the endpoint of reaction. Compounds were separated by silica gel column. 1H NMR (600 MHz) and 13C NMR (151 MHz) were recorded on Bruker spectrometer 600. Chemical shifts are corrected by the tetramethylsilane (TMS) and the unit of chemical shifts is ppm. The molecular weight of final compounds is recorded on ESI-HRMS. The purity of final compounds was determined by HPLC. HPLC was performed on an Agilent HPLC workstation equipped with a Diamonsil C18 (5 μm, 4.6 mm × 150 mm) column.
The synthesis and characterization of compounds are shown in the Supporting Information.
4.2. Biological assays
4.2.1. Cell lines and reagents
In this study, all cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). A549 cells were cultured in Roswell Park Memorial Institute (RPMI) (Hyclone, Logan, UT, USA). EBC-1 cells were cultured in Minimum Essential Medium (MEM) (Hyclone, USA). Hs746T, LO2, 293T and HepG2 cells were cultured in Dulbecco's modified Eagle medium (DMEM; Hyclone, USA) with 25 mmol/L glucose (Invitrogen, Carlsbad, CA, USA). All cell lines were cultured in medium containing 10% fetal bovine serum (FBS) (Hyclone, USA), 1% penicillin (Invitrogen, USA) and 1% streptomycin (Invitrogen, USA). All these cells were cultured at 37 °C and 5% CO2.
The antibodies were purchased from different sources. rabbit anti-GAPDH (#10494-1-AP), anti-Ki67(#27309-1-AP) was purchased from Proteintech, Rosemont, IL, USA; Rabbit anti-c-MET (D1C2) (#8198), rabbit anti-phos-c-MET (Tyr1234/1235) (#3077), rabbit anti-STAT3 (D3Z2G) (#12640), rabbit anti-phos-STAT3 (Tyr705) (#9145) and rabbit anti-Cleaved PARP (Asp214) (#5625) were purchased from Cell Signaling Technology, Boston, MA, USA. Crizotinib (#T1661) and tepotinib (#T6121) were purchased from Targetmol, USA.
4.2.2. Plasmids and lentivirus infection
EBC-1 and Hs746T cells lines that stably overexpressing c-MET (Y1230H) and c-MET (D1228N) were established using pCDH plasmid (System Biosciences, Palo Alto, CA, USA) with the following primers: 5′-CCAACTTTGTGCCAACCGGTCGCCACCATGAAGGCCCCCGCTGTGCTTGCACCTG-3′ (forward) and 5′-AATGCCAACTCTGAGCTTTGATGTCTCCCAGAAGGAGGCTGGTCG-3′ (reverse) by lentiviral transduction. For lentivirus infection, lipofectamine 3000 reagent was used for transfection of plasmids according to the manufacturer's instructions (Invitrogen, USA). Lentivirus was generated the 293T cells transfected with the lentiviral packing vector mix (System Biosciences, USA). After 48 h, lentivirus was collected and used to infect EBC-1 and Hs746T cells. After 48 h infection, puromycin (3 μg/mL) was utilized to select stable cell lines and pooled clones were screened by WB with anti-c-MET.
4.2.3. Cell proliferation inhibition assay
Various cell lines were seeded in 96-well plates (100 μL per well) at the density of 4000 cells/well to adhere overnight. The cells were treated with medium containing various concentrations of compounds (100 μL) and incubated for 72 h. The antiproliferative ability of compounds was determined by cell counting kit-8 (CCK-8 kit) according to the manufacturer's instructions (Dojindo Laboratories, mamoto Ken, Japan).
4.2.4. Drug synergy assays
For drug synergy assays, synergy effect of drug pairs was quantitative defined by the Chou–Talalay equation29. EBC-1 and Hs746T cells were treated with different concentrations of single drug and combinational treatment for 72 h, respectively, and cell viability was validated with CCK-8 kit.
4.2.5. Enzyme binding assay
Compounds including tepotinib, 6, D15 and D16 were diluted with 100% DMSO. Tepotinib and compound 6 were tested from 0.1 μmol/L, 3-fold dilution for 8 points. D15 and D16 were tested from 5 μmol/L, 2-fold dilution for 9 points. The c-MET binding affinity (Kd) of compounds was determined by ADP-Glo™ Kinase Assay (Promega, WI, USA) according to the manufacturer's instructions. The kinase reaction contained 2 ng/μL c-MET, 0.5 mmol/L substrate-biotin, 1 mmol/L ATP, 2 mmol/L DTT, 0.2 mg/mL Poly (4:1 Glu,Tyr) Peptide. Kinase react and stop incubate at 30 °C for specified period. The luminescence was measured with a plate-reading luminometer to read relative light unit (RUL) as Eq. (1):
| % Enzyme activity= (RLUSample–RLUBlank)/(RLU1%DMSO–RLUBlank) × 100 | (1) |
4.2.6. Cell apoptosis and cell cycle assay
EBC-1 and Hs746T cells (1 × 106 cells/well) were cultured in 6-well dishes to adhere overnight and were then treated with medium containing various concentrations of compounds for 48 h. For cell apoptosis analysis, the apoptosis rates of cells were detected by Annexin V-FITC apoptosis detection kit (Abcam, MA, USA). Samples were analyzed by a FACS calibur Flow Cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). For cell cycle analysis, cells were fixed in 75% ethanol overnight at −20 °C, and then washed with PBS. Then cells were treated with 100 μL RNase A (0.2 mg/mL) in PBS for 30 min at 37 °C and propidium iodide (PI) was added. Samples were analyzed by a FACS calibur Flow Cytometer.
4.2.7. Cell migration and invasion
For cell migration assay, EBC-1 and Hs746T cells were mechanically scratched using a 200 μL pipette tip. The debris were washed with PBS and treated various concentrations of compounds in medium without FBS accompanied with mitomycin C (1 μmol/L) treatment. The relative migration rates of cells were counted based on images at 0 h and 12 h in the same place. For cell invasion assay, 10 μL liquid Matrigel (BD Biosciences) was added to the upper surface of the Transwell chamber (Corning, NY, USA). Cells were washed with PBS and 10,000 cells were added to each well with various concentrations of compounds with mitomycin C (1 μmol/L) treatment. After 24 h, 4% invaded cells were fixed with paraformaldehyde and stained with crystal violet. The number of the invaded cells was counted after taking photographs.
4.2.8. Western blot
EBC-1 and Hs746T cells were seeded in 6-well plates to adhere and were treated with different concentrations of compounds for indicated time points. Subsequently cells were washed with PBS and lysed in RIPA buffer. The protein concentration was quantified and total protein lysates were separated by 10% SDS-PAGE and transferred to nitrocellulose membrane. Membranes were sequentially probed with indicated primary and secondary antibodies and were imaged by the Imaging system (Bio-Rad, Hercules, CA, USA).
4.2.9. Animal models for tumor growth
Animal research has been approved by the Animal Care Committee of the Beijing Institute of Biotechnology. All operations were following the Animal Care and Use Committee Guidelines of China. 6-Week-old BALB/c nude mice were purchased from SiPeiFu company, Beijing, China, and housed in a specific pathogen free (SPF) animal facility. EBC-1 and Hs746T cells (5 × 106) were injected subcutaneously into the dorsal flank of nude mice. Nude mice were divided into four groups including tepotinib, D10, D15, and vehicle groups once the tumor volume reached about 80 mm3. For EBC-1 nude mice xenograft models, nude mice were treated with D10 and D15 (10 mg/kg, i.p./qd) and vehicle control (10% DMSO + 10% PEG300 + 5% Tween 80 + 75% H2O, i.p./qd) for 10 days. For Hs746T nude mice xenograft models, nude mice were treated with tepotinib (10, 20 mg/kg, p.o./qd), D10 (20, 40 mg/kg, p.o./qd), (20, 40 mg/kg, p.o./qd) and vehicle control (10% DMSO + 10% PEG300 + 5% Tween 80 + 75% H2O, p.o./qd) for 12 days. The tumor volume of mice was measured every 2 days using calipers, The tumor volume was calculated with Eq. (2):
| V = (Longest diameter × Shortest diameter2)/2 | (2) |
Tumor growth inhibition (TGI) was utilized to identify the inhibitory strength of drugs on tumor growth as Eq. (3):
| TGI (%) = (Vc–Vt)/(Vc–V0) × 100 | (3) |
where Vc is the median volume of control, and Vt is the median volume of treated groups at the end of the study, and V0 is median volume of control at the start of the study. The body weight of mice was measured every 2 days. The experiment was terminated when the maximum tumor size reached approximately 1.5 cm in diameter. Euthanasia was performed after deep anesthesia. Subsequently, tumors were isolated from the animals, weighed, and photographed.
4.2.10. The pharmacokinetic (PK) study in vivo
Pharmacokinetic experiments in rats. All the animal experiments were performed in the Beijing Center for Drug Safety Evaluation approved by the Institutional Animal Care and Use Committee of the Center, and animal experiments were complied with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Male Sprague–Dawley (SD) rats (180–200 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). The animals were bred in a constant temperature and humidity environment with a 12 h light/dark cycle. They were fasted for 12 h but free access to water prior to oral administration. Thirty-six male SD rats were randomly divided into nine groups (4 rats per group). D10, D15 and tepotinib were single dosed via i.v. (1 mg/kg), i.p. (10 mg/kg) and p.o. routes (40 mg/kg for D10 and D15, 20 mg/kg for tepotinib), respectively, to obtain the pharmacokinetic behaviors. The pharmacokinetic parameters through different routes were calculated and compared parallelly. Blood samples (0.1 mL) were harvested into heparin tubes at pre-dose and 2 (for i.v. and i.p.), 5, 15, 30 min, and 1, 2, 4, 6, 8, 12, and 24 h post dose for i.v., i.p. and p.o. dosing. All plasma samples were separated by centrifugation and stored in a −20 °C freezer until being retrieved for analysis. A 20 μL aliquot of each sample was added with 20 μL acetonitrile and 160 μL acetonitrile containing IS (5 ng/mL propranolol hydrochloride) to precipitate protein. The mixture was vortex-mixed for 1 min and centrifuged at 15,000×g for 10 min. The upper layer was collected, diluted 2-fold in water, and determined using a LC–MS/MS approach as below mentioned.
Bioanalysis method. The plasma concentrations of D10, D15 and tepotinib were simultaneously assayed using a LC–MS/MS system consisting of a LC instrument (LC-20AD, Shimadzu) coupled with 8060 triple quadrupole mass spectrometer detector (Simadzu, Japan). For chromatography, a Phenomenex C18 column (3.0 mm × 50 mm, 2.6 μm, USA) was utilized. Mobile phase consisted of 0.1% formic acid in water (v/v, mobile phase A) and 0.1% formic acid in acetonitrile (v/v, mobile phase B) with an optimized flow rate of 0.6 mL/min in a 4-min run. Gradient elution of the target analyte was as follows: 0–0.5 min, 5% B; 0.5–2.0 min, from 5% to 95% B; 2.0–2.5 min, kept at 95% B; 2.6 min, returned to 5% B; 2.6–4.0 min, 5% B. The analytes and internal standard were detected by positive ion spray in the multiple-reaction-monitoring modes (MRM) and injection volume was 5 μL. The MRM transitions of analytes and IS were 879.60/498.35 for D10, 861.55/480.35 for D15, 493.00/112.00 for tepotinib, and 260.1/116.0 for IS. Calibration curve ranges were 0.5–1000 ng/mL for the three compounds.
Data analysis. Phoenix WinNonlin 9.0 (Pharsight, CA, USA) was applied to estimate the pharmacokinetic parameters of D10, D15 and tepotinib with noncompartmental analysis. Of these parameters, the area under the plasma concentration–time curve (AUC), half-life in terminal phase (t1/2), and mean retention time (MRT) were calculated from all the dosing routes; the maximal plasma concentration (Cmax) and time to reach the peak (Tmax) were obtained from extravascular dosing routes; the volume of distribution at steady state (Vss) and systemic clearance (CL) were obtained from i.v. injection administration. Absolute bioavailability was evaluated as Eq. (4):
| Bioavailability (F, %) = [(AUCextra-venous route /AUCiv) × doseiv/doseextra-venous route)] × 100 | (4) |
Statistical analysis was conducted by Student t test between different groups for major pharmacokinetic parameters (Cmax, Tmax, AUC, Vss, and CL). A P-value <0.05 was considered statistically significant.
4.2.11. MS-based proteomic analysis
Sample preparation. EBC-1 cells were treated with DMSO or 10 nmol/L D15 for 48 h, then cells were washed by PBS for 3 times and lysed with 8M UA (8M urea, 100 mmol/L Tris-HCl, pH 8.0) containing complete protease inhibitor tablets. Subsequently, the supernatant was collected after lysed cells were centrifuged at 14,000 rpm at 4 °C for 15 min, followed by reduction with 10 mmol/L TCEP at RT for 30 min, and cysteine alkylation with 50 mmol/L CAA at RT for 30 min. The denatured proteins were digested overnight at 37 °C by trypsin (at a 1:50 ratio of enzyme to protein). Reactions were quenched by adding FA to a final concentration of 0.1%, followed by desalted on reversed-phase C18. The amount of the purified peptides was determined using Nanodrop (Thermo Fisher Scientific, MA, USA).
LC–MS/MS analysis. The peptides mixture was analyzed with an EASY-nLC 1200 ultra-high pressure liquid chromatography system (Thermo Fisher Scientific) using a homemade 30 cm C18 column (ID 150 μm, 1.9 μm, 100 Å). Peptides separation was conducted through a 150-min gradient at a constant flow rate of 600 nL/min: 7%–12% B in 18 min, 12%–32% B in 91 min, 32%–45% B in 30 min, 45%–95% B in 2 min, then held at 95% B for 9 min (Buffer A was 0.1% FA and buffer B was 0.1% FA in 80% ACN). The peptides mixture was analyzed with an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) coupled to a nanoelectrospray ion source (Thermo Fisher Scientific). Spray voltage was set to 2200 V and heating capillary at 320 °C. MS data were acquired with data-dependent acquisition (DDA) method, and the dynamic exclusion duration was set to 25 s. For the MS1 scan, mass spectra were acquired in the positive-ion mode over the range of 300–1400 m/z, with a maximum ion injection time of 50 ms and a resolution of 120,000 at m/z 200. Fragmentation of precursor ions was performed by higher-energy collision dissociation (HCD) with a normalized collision energy of 35%. The MS2 spectra was acquired with an automatic gain control target value of 1.0e4 and a maximum injection time of 35 ms.
MS data analysis. MS raw files were first analyzed by MaxQuant (version 2.0.3.0) against the UniProt Human database (downloaded on Sep 2022, counting 20,398 entries). All 6 raw data (two conditions, three replicates each) were analyzed simultaneously and tagged with a unique experiment label based on treatment conditions. The protease was set as trypsin/P with a maximum of two missed cleavages. Carbamidomethyl (C) was set as fixed modification, and Oxidation (M) and Acetyl (Protein N-term) were set as variable modifications. The first search tolerance was 20 ppm, and the main search tolerance was 4.5 ppm. The false discovery rate (FDR) was set as ≤0.01 at the spectra, protein, and modification levels. Other parameters are kept as default. The match between run function was disabled and label-free relative quantification (LFQ) was enabled using default settings. The protein groups results file from MaxQuant were then analyzed in R (version 4.2.1). All proteins identified from the contaminated and reversed database were filtered out. The LFQ intensities were extracted from the proteinGroups.txt file to represent the expression matrix, then the matrix was subjected to median normalization of each protein group across all samples, followed by the log2-transformation. Proteins quantified in at least two replicates with an intensity CV of less than 0.3 were retained for further evaluation. Significance was assessed by Student's t-test, and the resulting P values were adjusted by the Benjamini–Hochberg method.
Data availability. All the mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE30 partner repository with the dataset identifier PXD038071.
4.2.12. Statistical analysis
All in vitro experiments were performed in triplicate. Differences between variables were assessed by two-tailed Student's t test or one-way analysis of variance (ANOVA). All statistical analyses were calculated by SPSS 13.0 or GraphPad Prism 8.0. The statistical data were expressed as the mean ± SD. In all assays, P value < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by Major New Drugs Innovation and Development (2018ZX09J18102-002, China). We would like to thank Editage (www.editage.cn) for English language editing.
Author contributions
Zhibing Zheng, Junhai Xiao, Xiaomei Zhuang and Song Li conceived and directed the project. Zhibing Zheng, Pengyun Li, Changkai Jia, and Junhai Xiao contributed to experimental design and program strategy. Changkai Jia, Pengyun Li, Xiaotong Hu, Wenjuan Zhang and Shiyang Sun contributed to design and chemical synthesis of compounds. Pengyun Li, Zhiya Fan, Haoxin Guo, Ning Yang and Maoxiang Zhu contributed to the in vitro biology experiments and data analysis. Pengyun Li, Shiyang Sun, Haoxin Guo, Ning Yang contributed to the in vivo biology experiments and data analysis. Zhiya Fan contributed to MS-based proteomic experiments and data analysis. Pengyun Li and Changkai Jia wrote the manuscript. Zhibing Zheng and Xiaomei Zhuang reviewed and edited the paper. Xiaomei Zhuang and Ke Liu contributed to the pharmacokinetic (PK) study in vivo and data analysis. All authors read and approved the final manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2023.01.014.
Contributor Information
Xiaomei Zhuang, Email: xiaomeizhuang@163.com.
Junhai Xiao, Email: xiaojunhai@139.com.
Zhibing Zheng, Email: zzbcaptain@aliyun.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article.
References
- 1.Comoglio P.M., Trusolino L., Boccaccio C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer. 2018;18:341–358. doi: 10.1038/s41568-018-0002-y. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y., Xia M., Jin K., Wang S., Wei H., Fan C., et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer. 2018;17:45. doi: 10.1186/s12943-018-0796-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Recondo G., Che J., Jänne P.A., Awad M.M. Targeting MET dysregulation in cancer. Cancer Discov. 2020;10:922–934. doi: 10.1158/2159-8290.CD-19-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drusbosky L.M., Dawar R., Rodriguez E., Ikpeazu C.V. Therapeutic strategies in METex14 skipping mutated non-small cell lung cancer. J Hematol Oncol. 2021;14:129. doi: 10.1186/s13045-021-01138-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang C., Zou Q., Liu H., Qiu B., Li Q., Lin Y., et al. Management of non-small cell lung cancer patients with MET exon 14 skipping mutations. Curr Treat Options Oncol. 2020;21:33. doi: 10.1007/s11864-020-0723-5. [DOI] [PubMed] [Google Scholar]
- 6.Fujino T., Suda K., Mitsudomi T. Emerging MET tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Expet Opin Emerg Drugs. 2020;25:229–249. doi: 10.1080/14728214.2020.1791821. [DOI] [PubMed] [Google Scholar]
- 7.Fernandes M., Jamme P., Cortot A.B., Kherrouche Z., Tulasne D. When the MET receptor kicks in to resist targeted therapies. Oncogene. 2021;40:4061–4078. doi: 10.1038/s41388-021-01835-0. [DOI] [PubMed] [Google Scholar]
- 8.Nalawansha D.A., Crews C.M. PROTACs: an emerging therapeutic modality in precision medicine. Cell Chem Biol. 2020;27:998–1014. doi: 10.1016/j.chembiol.2020.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bondeson D.P., Smith B.E., Burslem G.M., Buhimschi A.D., Hines J., Jaime-Figueroa S., et al. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem Biol. 2018;25:78–87. doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Békés M., Langley D.R., Crews C.M. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. 2022;21:181–200. doi: 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dale B., Cheng M., Park K.S., Kaniskan H.Ü., Xiong Y., Jin J. Advancing targeted protein degradation for cancer therapy. Nat Rev Cancer. 2021;21:638–654. doi: 10.1038/s41568-021-00365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bond M.J., Crews C.M. Proteolysis targeting chimeras (PROTACs) come of age: entering the third decade of targeted protein degradation. RSC Chem Biol. 2021;2:725–742. doi: 10.1039/d1cb00011j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J., Ma J., Liu Y., Xia J., Li Y., Wang Z.P., et al. PROTACs: a novel strategy for cancer therapy. Semin Cancer Biol. 2020;67:171–179. doi: 10.1016/j.semcancer.2020.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Zeng S., Huang W., Zheng X., Liyan c, Zhang Z., Wang J., et al. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: recent progress and future challenges. Eur J Med Chem. 2021;210 doi: 10.1016/j.ejmech.2020.112981. [DOI] [PubMed] [Google Scholar]
- 15.Burslem G.M., Smith B.E., Lai A.C., Jaime-Figueroa S., McQuaid D.C., Bondeson D.P., et al. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem Biol. 2018;25:67–77. doi: 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markham A. Tepotinib: first approval. Drugs. 2020;80:829–833. doi: 10.1007/s40265-020-01317-9. [DOI] [PubMed] [Google Scholar]
- 17.Edmondson S.D., Yang B., Fallan C. Proteolysis targeting chimeras (PROTACs) in 'beyond rule-of-five' chemical space: recent progress and future challenges. Bioorg Med Chem Lett. 2019;29:1555–1564. doi: 10.1016/j.bmcl.2019.04.030. [DOI] [PubMed] [Google Scholar]
- 18.Gadd M.S., Testa A., Lucas X., Chan K.H., Chen W., Lamont D.J., et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13:514–521. doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nowak R.P., DeAngelo S.L., Buckley D., He Z., Donovan K.A., An J., et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol. 2018;14:706–714. doi: 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiang W., Zhao L., Han X., Qin C., Miao B., McEachern D., et al. Discovery of ARD-2585 as an exceptionally potent and orally active PROTAC degrader of androgen receptor for the treatment of advanced prostate cancer. J Med Chem. 2021;64:13487–13509. doi: 10.1021/acs.jmedchem.1c00900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han X., Zhao L., Xiang W., Qin C., Miao B., Xu T., et al. Discovery of highly potent and efficient PROTAC degraders of androgen receptor (AR) by employing weak binding affinity VHL E3 ligase ligands. J Med Chem. 2019;62:11218–11231. doi: 10.1021/acs.jmedchem.9b01393. [DOI] [PubMed] [Google Scholar]
- 22.Wu H., Yang K., Zhang Z., Leisten E.D., Li Z., Xie H., et al. Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J Med Chem. 2019;62:7042–7057. doi: 10.1021/acs.jmedchem.9b00516. [DOI] [PubMed] [Google Scholar]
- 23.Zorba A., Nguyen C., Xu Y., Starr J., Borzilleri K., Smith J., et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci U S A. 2018;115:7285–7292. doi: 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pike A., Williamson B., Harlfinger S., Martin S., McGinnity D.F. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discov Today. 2020;25:1793–1800. doi: 10.1016/j.drudis.2020.07.013. [DOI] [PubMed] [Google Scholar]
- 25.Cantrill C., Chaturvedi P., Rynn C., Schaffland J., Walter I., Wittwer M. Fundamental aspects of DMPK optimization of targeted protein degraders. Drug Discov Today. 2020;25:969–982. doi: 10.1016/j.drudis.2020.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Mares A., Miah A.H., Smith I.E.D., Rackham M., Thawani A.R., Cryan J., et al. Extended pharmacodynamic responses observed upon PROTAC-mediated degradation of RIPK2. Commun Biol. 2020;3:140. doi: 10.1038/s42003-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobrovolsky D., Wang E.S., Morrow S., Leahy C., Faust T., Nowak R.P., et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood. 2019;133:952–961. doi: 10.1182/blood-2018-07-862953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engstrom L.D., Aranda R., Lee M., Tovar E.A., Essenburg C.J., Madaj Z., et al. Glesatinib exhibits antitumor activity in lung cancer models and patients harboring MET exon 14 mutations and overcomes mutation-mediated resistance to type I MET inhibitors in nonclinical models. Clin Cancer Res. 2017;23:6661–6672. doi: 10.1158/1078-0432.CCR-17-1192. [DOI] [PubMed] [Google Scholar]
- 29.Chou T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 30.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:442–450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.