

Summary
Mass spectrometry (MS)-based immunopeptidomics is an attractive antigen discovery method with growing clinical implications. However, the current experimental approach to extract HLA-restricted peptides requires a bulky sample source, which remains a challenge for obtaining clinical specimens. We present an innovative workflow that requires a low sample volume, which streamlines the immunoaffinity purification (IP) and C18 peptide cleanup on a single microfluidics platform with automated liquid handling and minimal sample transfers, resulting in higher assay sensitivity. We also demonstrate how the state-of-the-art data-independent acquisition (DIA) method further enhances the depth of tandem MS spectra-based peptide sequencing. Consequently, over 4,000 and 5,000 HLA-I-restricted peptides were identified from as few as 0.2 million RA957 cells and a melanoma tissue of merely 5 mg, respectively. We also identified multiple immunogenic tumor-associated antigens and hundreds of peptides derived from non-canonical protein sources. This workflow represents a powerful tool for identifying the immunopeptidome of sparse samples.
Keywords: microfluidics, automation, immunopeptidome, sample preparation, mass spectrometry, data-independent acquisition
Graphical abstract
Highlights
-
•
An automated microfluidics system with enhanced sensitivity for immunopeptidomics
-
•
DIA methods are exploited for in-depth and reliable immunopeptidomics analyses
-
•
A comprehensive immunopeptidomics spectral library for DIA analyses
Motivation
Mass spectrometry (MS) is to date the gold-standard method for decoding the immunopeptidome of cell lines or clinical tumor tissues. Although the recent improvements of MS instrumentation and subsequent data analytics tools have enhanced the depth of data interpretation, the overall assay sensitivity is still largely limited by the conventional sample preparation methods. To enhance the sensitivity of the traditional sample preparation of MS-based immunopeptidomics, we introduced an automated workflow to purify immunopeptidome from scarce cell and tumor samples.
Recent improvements in mass spectrometry instrumentation and data analysis have advanced the field of immunopeptidomics. Nonetheless, the assay sensitivity is still largely limited, especially for low-input samples. Li et al. introduce an automated microfluidics workflow for sensitive immunopeptidomics and demonstrate the detection of tumor antigens from melanoma tissues.
Introduction
The research field of immunopeptidomics aims to identify the complex repertoire of short peptides presented on the cell surface by the major histocompatibility complex (MHC), also known as the human leukocyte antigen (HLA). HLA-restricted peptides play a crucial role in adaptive immunity by serving as unique epitopes for T cells to identify “self” and “non-self” antigens.1 Owing to aberrant cellular activities, cancer cells exhibit a distinct immunopeptidome that can trigger T cells’ cytotoxic reactions.1,2 Therefore, extensive studies have focused on discovering cancer-specific peptide antigens for developing targeted T cell-mediated cancer therapies, such as TCR-T therapy3,4,5,6,7 and cancer vaccines.8,9,10 Untargeted mass spectrometry (MS) is the only available and sensitive methodology to comprehensively characterize the repertoire of naturally presented immunopeptidome2,11,12 extracted from cancer cells,6,13,14 tumor tissues,15,16,17 and liquid biopsies.18 Indeed, there has been a growing interest in immunopeptidomics in recent years accelerated by improvements of MS technologies19,20 and data analytics tools16,21,22,23,24,25,26,27,28 for comprehensive peptide identification (ID) and quantification.
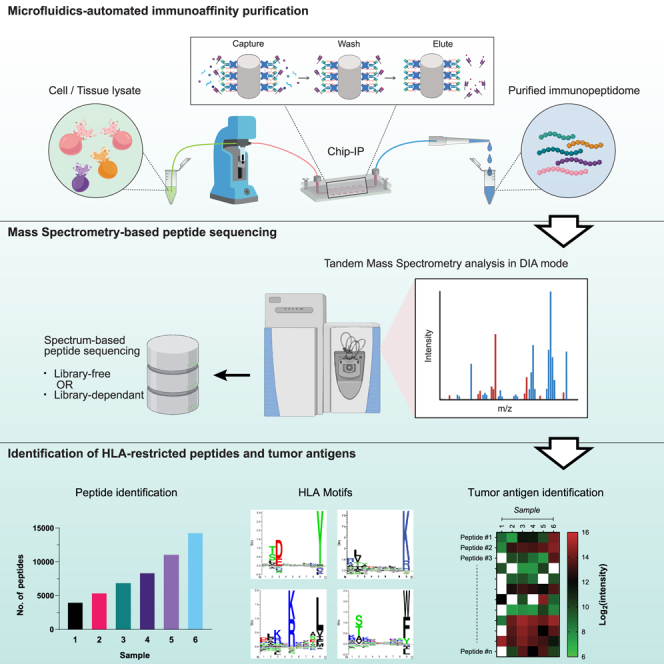
Immunoaffinity purification (IP) is the method of choice for extracting HLA-restricted peptides. Additional steps are required to further purify the peptides before them being analyzed by MS.11,12,29 Sample preparation thus remains a crucial step in the immunopeptidomics pipeline. MS acquisition methods and data-processing tools are steadily being improved. On the contrary, sample-processing optimizations to boost peptide recovery from crude samples remain scarce. Conventionally, IP is conducted in chromatography columns packed with antibody-coated microbeads, usually specific against the pan-HLA-I or pan-HLA-II complexes. When a sample (cell or tissue lysate) passes through the column, the microbeads selectively capture HLA-peptide complexes. The rest of the lysate is then washed off the column and the peptides are eluted with acid. While the antibodies ensure an excellent specificity for peptide enrichment, the entire IP procedure is laborious and requires extensive manual operations.12 Such complex workflows hinder the implementation of immunopeptidomics on large-scale clinical applications. Chong et al. have recently upgraded the column-based IP to a high-throughput format using multi-well membrane plates on a positive pressure module.30 As a result, up to 96 samples can be processed simultaneously. However, this workflow does not obviate frequent manual interventions during the IP process, such as pipetting samples to each well, stacking the plates in the correct order, and loading/unloading the plate stack on/from the positive pressure module, among many steps. Additionally, the issues of microbeads-based assays remain, such as the bulky dead volume, insufficient capture of targets and removal of impurities, and unpredictable packing conditions among multiple assays that decreases reproducibility. Some studies have shown the utility of automatic robotic liquid handling platforms (e.g., Agilent AssayMAP) for relevant purposes.31,32 Indeed, these workflows yielded high numbers of peptide identifications from cell lines in a reproducible and high-throughput format. However, such robotic platforms are extremely costly to implement (not only the device but also the vendor-specific consumables), posing a significant financial burden for most laboratories. Meanwhile, Feola et al. reported a microfluidic device for performing IP33 with lower antibody consumption than column-based systems. However, it showed poor system integration as the fluidic control was not clearly demonstrated, and the C18 cleanup steps were still done offline. Additionally, it resulted in non-typical contamination of long peptides (above 10 mers) and low HLA-I binder content, suggesting insufficient assay optimization.
We developed an inexpensive, automated, and easy-to-operate workflow for sensitive and reliable immunopeptidomics to address the aforementioned bottlenecks using advanced microfluidics technology. Microfluidics devices are known to enormously reduce the sample volume to the sub-milliliter range thanks to their ultralow internal volume,34,35,36 which is suitable for handling scarce clinical samples. In addition, we engineered a meta-array of micropillars within the device to increase the surface-to-volume ratio for efficient immunoaffinity interactions. Our microfluidics platform includes a fully automated workflow that requires minimal material consumption and enhances target purification. Our IP procedure (hereafter “chip-IP”) was driven by a programmable fluidic control system whereby the supply of all the assay reagents and the sample loading were readily cooperated. Similarly, C18 cartridges required for sample cleanup are directly integrated into the automated fluidic system via a programmable switch valve to omit the unnecessary sample transfers. We demonstrated the competitive performance of this approach with low-input samples and tumor biopsies in combination with data-independent acquisition (DIA) computational approaches, leveraging large spectral libraries for sensitive MS detection of tumor antigens.
Results
Automation of the IP workflow using microfluidics and programmable fluidic controls
Micropillar arrays are widely used in microfluidics devices to enhance the capturing efficiency of targets from biomolecules37,38,39,40 to cells.41,42 We adopted a design from a previous study38 and generated ∼250,000 micropillars in a single 50-cm-long fluidic channel on the silicon substrate in a cleanroom facility (Figures 1A, S1A, and S1B). Each pillar has a square cross-section with a side length of 20 μm and a height of 100 μm (Figures 1A and S1C). All pillars face the flow direction in a diagonal orientation to facilitate a homogeneous inter-pillar flow distribution. Some additional microstructures were also engineered in the chip to assist the in-flow reaction, such as a short filtering section and bifurcating flow distributors (Figure S1C). In addition, the chip is thermally bonded to a polymer slab for formation of a hermetic fluidic channel before use (Figures 1A and S1A).
Figure 1.

The microfluidics chip-IP system for HLA-I immunopeptidome enrichment
(A) The silicon microfluidics chip has a footprint of 58 mm × 20 mm with a 50-cm-long fluidic channel in a meandering layout (scale bar, 1 cm). The channel was enclosed by a thick polymer slab (made of polydimethylsiloxane), which can be seen from the side view. The micropillar arrays were observed in the magnified view by scanning electron microscopy imaging (scale bar, 20 μm).
(B) Schematic diagram of the automated fluidic control setup. Individual syringe pumps drove the chip-IP and the C18 peptide cleanup modules, whose flow outputs were linked by a 6-port switch valve for a streamlined workflow. The WiFi-like signs indicate the programmable communication among the labeled parts.
(C) Scheme of surface chemistry on the silicon surface and the chip-IP flow.
(B) and (C) were created with BioRender.com.
In basic microfluidics connection setups, the fluidic tubing is directly inserted into the polymer slab via needle punches. However, the long channel length of our chip and the complex lysate samples significantly elevate the flow resistance, posing a great challenge for such a simple connection scenario. Furthermore, liquid leaking from the chip inlets or the sealing interface would result in irreversible chip damage and sample loss. Therefore, clamping reinforcements were designed to enhance the chip’s mechanical robustness (Figure S1E). This mechanism housed the chip between two rigid metallic plates tightened by threaded screws along the edge. In addition, two threaded holes were drilled into the top plate above the chip inlets to provide fluidic access with standard UNF-threaded connectors. O-rings were placed under each access hole to provide watertight sealing.
The microfluidics chip was integrated into a programmable fluidic control system for an automated assay operation (Figures 1B and S1D). The syringe pumps driving the liquid flow were controlled by Python programming via a serial connection to accommodate different flow rates in various steps (Table S1). A 12-port programmable distribution valve was installed on the syringe pump to enable injections of multiple reagents in a sequential way (Figure S1D). This combination proved very useful for an IP experiment that involves tedious washing processes. A similar fluidic control system was also applied to the post-IP peptide cleanup using C18 materials. The C18 cartridges were mounted to the fluidic tubing via a Luer-to-UNF adapter on another fluidic control setup. Notably, the C18 cartridge and the chip-IP device were linked via an automated 6-port switch valve (Figures 1B and S1D). By tuning the serial commands, the switch valve could rotate to different positions corresponding to a decoupled mode whereby the C18 cartridge is individually preconditioned while the IP is ongoing, and a coupled mode whereby the eluted peptides directly flow into the conditioned C18 cartridge (Figure S1F). This new assay strategy formed a streamlined workflow for purifying HLA immunopeptidome in a fully automatic manner.
We exploited established functionalization chemistry on the silicon surface to facilitate a specific and robust immunoassay in the chip (Figure 1C). The silicon micropillars were first thermally treated to form an oxidation layer of approximately 100 nm (Figure S1C). This thin layer provided a chemically active silicon oxide surface for grafting silane molecules carrying protein-binding groups, such as a chlorosilane agent with a functional N-hydroxysuccinimide (NHS) ester group. The NHS esters can readily react with the primary amine groups for covalent protein immobilization, representing an easy and fast protein-coating strategy. An intermediate layer of protein A/G was coated on the surface before coating the antibodies to ensure an optimal antibody orientation for antigen binding. The antibody-protein A/G link was reinforced by a crosslinking reaction, which reduced the coelution of antibodies in the resulting peptide samples and enabled the reusing of coated chips.
The automatic IP workflow enriches immunopeptidome from low numbers of human B cells
To evaluate the performance of the novel chip-IP workflow, we enriched HLA class I (HLA-I) bound peptides from different numbers of RA957 cells, an immortalized human B cell line30 (Table S2). The IP was done with a low range of cell populations (0.2, 1, 5, and 10 million cells) to demonstrate the sensitive enrichment of immunopeptidome using the microfluidics device. For comparison, IP in microbeads-packed columns was also done in parallel with the same numbers of cells as the control method. The resulting peptides were analyzed in a Q Exactive HF-X MS instrument with the DIA method. Library-directed DIA computational workflow has been widely proven to provide a more comprehensive landscape of proteome and immunopeptidome.22,43,44,45,46,47 Therefore, to compare the peptide recovery from the column-IPs and the chip-IPs, we built a representative RA957-specific spectral library in Spectronaut with all the data-dependent acquisition (DDA) and DIA data from chip- and column-IPs that were generated during the project. Over 4,000 and 10,000 peptides were identified from 0.2 million to 1 million cells, respectively, by applying a 1% false discovery rate (Figure 2A and Table S3). The chip-IP gained a 1.5-fold increase in peptide ID below 5 million cells (Figure S2A). The percentage of HLA-I peptides predicted to bind the respective HLA alleles was above 90% in all conditions, and the peptide length distribution exhibited an ideal 9mer-dominant profile that is typical for HLA-I peptides (Figures 2B and 2C). Moreover, the peptides identified in each chip-IP condition were mapped to the four main HLA-I alleles of RA957 cells in a constant ratio regardless of the cell numbers (Figure 2D), showing an unbiased on-chip enrichment of immunopeptidome.
Figure 2.

Overall comparisons of HLA-I immunopeptidome enrichment from RA957 cells between the new chip-IP and the conventional column-IP
(A–C) The immunopeptidome of each IP method was characterized by the number of identified peptides (A), the percentage of predicted HLA-I binder peptides (B), and the peptide length distribution (C; aa, amino acid). The RA957 cell pellets with different numbers of cells were processed individually for all the IP assays, which were performed in duplicates. The “Rep” bar represents the number of unique peptides derived from each replicate that was plotted as mean ± SD. The “Whole” bar represents the number of peptides merged from the duplicates.
(D) The chip-IP immunopeptidome of each cell group was deconvoluted and clustered to the four main HLA-I alleles of RA957 cells.
The drastic difference in peptide ID between chip- and column-IP inspired us to dig deeper into the various properties of the results. We first examined the relations of peptides identified in each method. Across the four cell population groups of RA957 cells, over 50% of the column-IP-identified peptides were shared with the chip-IP, while the latter revealed more unique peptides (Figure 3A). Next, we extracted the MS2 signals of the common peptides and quantified their tendencies toward either method. In three out of the four groups, most of the common peptides were enriched with higher intensities on the chip than in the column (indicated by the offset of the density center toward the chip axis), implying augmented on-chip quantifications (Figure 3B). Unique peptides identified by each method were consistent in length distribution (Figure 3C). Nonetheless, they depicted other distinct characteristics. Strikingly, the chip-IP-derived unique peptides were found to be more hydrophilic, as they have overall lower GRAVY (grand average of hydropathy) indices48 (Figure 3D). During the reverse-phase high-performance liquid chromatography, hydrophilic peptides are typically eluted earlier than hydrophobic ones, meaning they manifest lower retention time in the chromatogram. To this end, we analyzed the distribution of retention times of the peptides uniquely identified in each cell group with different IP methods. Indeed, the chip-IP unique peptides showed an earlier elution profile than the column unique peptides, especially during 6–18 min (Figure 3E). In terms of the entire population, the chip-IP also showed “busier” patterns in the lower range of the liquid chromatography (LC) gradient (Figure S2B): more peptides were detected in the chip than in the column. Additionally, although the unique identifications in the chip-IP were less abundant (lower MS2 intensities) than in the column-IP unique peptides (Figure S2C), they exhibited a higher identification score with lower number of cells (i.e., 0.2 and 1 million cells, Figure S2D). This phenomenon might imply that the microstructured chip could potentially enrich more efficiently less abundant peptides. The peptides identified by both IP methods also obtained better scores in the chip, further verifying the excellent quality of the chip-IP approach (Figure S2D).
Figure 3.

Characterization of the shared and distinct peptides between the two IP methods
(A) Venn diagrams depict the relations of the immunopeptidome between the two IP methods.
(B) Density plots for the MS2 intensities of the shared peptides. Each point represents a peptide whose intensities from each method are projected on the corresponding axes. Therefore, the dots below the dashed line (a “bisector” line “y = x”) have higher MS2 intensities in the chip than in the column; otherwise, lower intensities in the chip for those above the line.
(C–E) For the unique peptides, multiple properties were analyzed: the peptide length distributions (C), the peptide GRAVY scores (D), and the retention time (RT) distributions (E).
Enrichment of immunopeptidome from needle-biopsy-sized human tumor tissues
Once we demonstrated the utility of chip-IP for enhanced identification of immunopeptidome from low numbers of human cells, we tested this new workflow with small clinical tumor samples. The conventional column-IP has an insufficient sensitivity for enriching immunopeptidome from a low amount of tissues.30,49 Nonetheless, there is often limited availability of patient-derived clinical samples. Therefore, a more sensitive IP workflow that can enrich the immunopeptidome from needle-biopsy-sized tumor samples is in urgent need for clinical immunopeptidomics studies.
We collected six tissues in varying sizes (5–40 mg) from a malignant melanoma tumor and performed chip-IP to enrich the HLA-I immunopeptidome. The peptides from each tissue sample were analyzed in the same LC-tandem MS (MS/MS) setup used for the DIA method. As already mentioned, a library-based search offers better performance for DIA analysis. The spectral library is typically generated from DDA and DIA data acquired from multiple replicates of the investigated samples for comprehensive data coverage. This represents an obvious challenge when there is an insufficient sample supply. On the contrary, a library-independent (coined “directDIA”) approach is an interesting alternative that has not yet been applied to immunopeptidomics. Therefore, we started from a library-free directDIA search for immunopeptidome of these small tissues (Figure 4A). Over 3,700 peptides were identified in all the small tissues (Table S4). More than 90% of the peptides were predicted as HLA-I binders in four out of six tissues and above 85% in the remaining two tissues. 9mer-peptides still dominated the entire population, revealing the excellent purification performance of the chip-IP. In comparison, with a library-based “hybridDIA” search that leveraged available DDA and DIA measurements of larger tumor tissues (500–900 mg) from the same patient, we observed a significant boost in the numbers of peptide ID and predicted HLA-I binders (Table S4). The ID for each small tissue specimen doubled or tripled with the spectral library, and the fraction of binders was above 90% (Figures 4B and 4C). Importantly, 12 tumor-associated antigenic (TAA) peptides were found in the specimens (Figure 5A). Most of them are derived from the MAGEA (melanoma-associated antigen) gene family, while two are from the TYR (tyrosinase) gene. Both genes are known to be associated with melanoma.50,51 In particular, peptides “EVDPASNTY,” “EVDPIGHLY,” “EVDPIGHVY,” and “SAYGEPRKL” have been found to be immunogenic by experimental validations in respective studies by Kobayashi et al.,52,53,54,55,56,57,58 Podaza et al.,52,53,54,55,56,57,58 Schultz et al.,52,53,54,55,56,57,58 Benlalam et al.,52,53,54,55,56,57,58 Kula et al.,52,53,54,55,56,57,58 van der Bruggen et al.,52,53,54,55,56,57,58 and Chaux et al.52,53,54,55,56,57,58 The comprehensive library search revealed a clear trend whereby the tissue weights correlated positively with the number of identified peptides (Figures 4B and 4C).
Figure 4.

Peptide identification from small melanoma tumor tissues by chip-IP
The identification was performed by different approaches of DIA analysis: the library-free directDIA with UniProt database (A), the library-dependent hybridDIA with UniProt database (B), and the hybridDIA with nuORF database (C). General aspects of the immunopeptidome were evaluated for each approach: the number of identified peptides, the percentage of HLA-I binders, the peptide length distribution, and the clustering of HLA-I binding motifs. The unique peptides identified in each sample’s technical replicates during the MS analysis were merged to represent the peptidome. Each small tissue was processed and analyzed individually. The spectral library for hybridDIA analyses was built with DDA and DIA runs acquired from the small tissues (processed by chip-IP) and some bigger tissues sectioned from the same patient (processed by column-IP).
Figure 5.

Chip-IP identified the tumor-associated antigens and nuORF-derived antigens in the small tumor tissues
(A) Identification of tumor-associated antigen (TAA) peptides via chip-IP. Information about the source gene, peptide sequence, and immunogenicity validation level as annotated in the Immune Epitope Database is provided. The immunogenicity level was labeled for experimentally validated immunogenic peptides. The immunogenicity level of “Predicted” indicates that the peptide has not been validated experimentally but is derived from a typical TAA gene.
(B) Enumeration of nuORF-derived peptides. The percentage of predicted HLA-I binders is labeled in red above the bar of binder peptides.
(C and D) Length distribution (C) and HLA-I motif clustering (D) of nuORF peptides.
(E) Types of nuORFs for the 554 non-canonical peptides.
Identification of peptides derived from novel non-canonical sources
Recent studies have shown that some actionable antigenic peptides are derived from non-canonical sources originating from aberrant cellular activities at various levels: genomic, epigenomic, transcriptomic, translational, and antigen-processing levels.2,16,24,25,59 Standard protein reference databases usually contain only canonically expressed proteins translated from annotated protein-coding genes. However, Ouspenskaia et al. have lately generated an expanded database of high-confidence cryptic peptide antigens by ribosome profiling, covering proteins translated from novel or unannotated open reading frames (nuORFs) in malignant and healthy samples.60 Therefore, we constructed a spectral library with this nuORF database for the DIA data of our small tissues to explore the non-canonical space. The overall quality of the identified immunopeptidome (ID, HLA-I binders, length distribution, and so forth) remained as before (Figure 4C), and 554 peptides derived from non-canonical sources were identified (Figure 5B), out of which 93% were predicted as HLA-I binders with the typical length distribution (Figures 5B and 5C; Table S4). The nuORF-derived peptides covered various non-canonical protein sources, including 3′ dORF (downstream ORF), 5′ uORF (upstream ODF), and long non-coding RNAs (lncRNAs) (Figure 5E). Interestingly, they preferably bind three HLA allotypes: HLA-A0301, HLA-B0801, and HLA-B5701 (Figure 5D).
A similar utility of the chip-IP for HLA-I immunopeptidome discovery has also been demonstrated with small tumor tissues collected from the liver metastasis of another malignant melanoma patient. Again, immunopeptidome from canonical and non-canonical sources were identified (Figures S3 and S4; Table S5).
A generic spectral library enables immunopeptidome discovery from small tumor tissues
Although library-directed DIA analyses can boost the identification of immunopeptidome and improve data reproducibility, it is not straightforward to build such a sample-specific spectral library for both basic studies and clinical investigations. On the one hand, one needs to collect a large number of biological samples for comprehensive immunopeptidome coverage. On the other hand, it would be time consuming and laborious to process these samples, conduct the MS analyses, and generate the library in a specific software environment. To overcome these limitations, we explored the application of a sample-independent generic spectral library with hundreds of available MS raw files that have been measured with the same instrumentation in our laboratory in recent years,14,15,16,22,30,61,62,63,64 which we coined the “Lausanne-Lib” (Table S6). The Lausanne-Lib consists of data from more than 400 immunopeptidomics measurements of various cancer cell lines and tissues, and benign samples from human donors. Intentionally, we did not include the exact tumor samples we analyzed above, as we hypothesized that the Lausanne-Lib’s extensive sample coverage should contain HLA allotypes with binding specificities similar to those expressed in our samples, and thus could benefit in a broader immunopeptidome identification than directDIA. Moreover, we also generated the Lausanne-Lib with both the UniProt database for routine canonical peptide search and the nuORF database (nuORFdb) for identifying non-canonical antigens, containing 343,427 and 333,125 HLA-I peptides, respectively.
Lausanne-Lib-based hybridDIA analyses enhanced the peptide identification from tissues weighing more than 20 mg in comparison with the directDIA approach (Figure 6A and Table S6). Because each tissue section was collected independently, a certain level of variance was expected in the peptide identification. The largest tissue of 40 mg indeed contained a large number of unique peptides (Figure S5). Despite the disparities in peptide identifications, the performance in enriching predicted HLA-I binders (Figure 6B) and the main HLA-binding motifs (Figures 6C and 6D) did not contrast significantly between the two Lausanne-Libs. The generic library has indeed largely expanded the ensemble peptidome discovered in the entire set of tumor tissues, with 90% of the directDIA-identified peptides contained in the Lausanne-Lib identifications (Figure 6E). Meanwhile, several TAAs and hundreds of nuORF-derived peptides were identified (Figures 6F–6H) with this generic spectral library. Hence, these results demonstrated the benefits of using the Lausanne-Lib for DIA analysis when resources are limited for creating a sample-specific spectral library.
Figure 6.

DIA analyses of melanoma tissues with the Lausanne-Lib
(A–D) The peptide identifications (A) and the percentage of predicted HLA-I binders (B) were revealed from small melanoma tissues using different DIA analysis methods. The directDIA was done with UniProt database and the Lausanne-Lib-based hybridDIA was done with either the UniProt database (Lausanne-Lib-UniProt) or the nuORF database (Lausanne-Lib-nuORF). The clustering of HLA-I binding motifs was deconvoluted for the Lausanne-Lib-UniProt (C) and the Lausanne-Lib-nuORF (D) analyses.
(E) A Venn diagram shows the intersections of peptides identified from different DIA analyses.
(F) The TAA peptides discovered by Lausanne-Lib-UniProt.
(G) The TAA peptides discovered by Lausanne-Lib-nuORF.
(H) Enumeration of nuORF-derived peptides. The percentage of predicted HLA-I binders is labeled in red above the bar of binder peptides.
Discussion
We have presented a novel microfluidics-based platform for enriching HLA-I immunopeptidome, which proved highly sensitive and automatable. We designed and manufactured dedicated microfluidics chips with silicon substrates because it is the standard material in the semiconductor industry, with established and reproducible manufacturing techniques. This technology creates microengineered devices that can handle the minute quantity of liquid in a controllable manner. Furthermore, we incorporated a dense array of micropillars to expand the surface-to-volume ratio for enhanced molecular interactions, such as antibody-antigen binding. Notably, the micropillars were fixed in regular orders, leaving identical gaps among all the pillars. Unlike the random packing of microbeads in the columns, the micropillar arrays allow efficient in-flow binding of specific targets and removal of irrelevant substances.
The improved efficiency also benefits in reduced use of antibodies. In conventional column-based IPs, milligrams of antibodies are used per test.12 Unless the antibodies are produced in-house by growing hybridoma cells, such a large consumption of commercial antibodies (€400–500/100 μg) imposes a major financial challenge on research laboratories. Therefore, the IP enabled by microfluidics technology facilitates more economical assays.
We applied this concept to establish a new sample preparation process for HLA immunopeptidome discoveries. In particular, both the IP and C18 peptide cleanup steps were integrated into a streamlined workflow where all intermediate sample transfers were omitted. Thus, a great deal of sample loss was avoided. In the future, spiking of recombinant heavy-isotope-coded peptide MHCs and multiplexed isotope tagging65 as normalization standards into HLA-bound peptides purified from a commonly used cell line would enable accurate quantification of peptide losses and inter-laboratory benchmarking of IP efficiency. Chip-IP showed a more elevated peptide ID at lower cell numbers, which was attributed to the improved assay efficiency in the microfluidics device. Indeed, compared with the densely packed microbeads in columns, the regular distribution of micropillars offers improved hydrodynamics,66,67 whereby a more homogeneous fluidic flow enhances the antibody-antigen interactions and the removal of impurities.
We tested this new workflow for decoding the immunopeptidome from a low number of human cells and needle-biopsy-sized tumor tissues. We obtained excellent in-depth immunopeptidome compared with the conventional format of microbeads-packed columns. In this study, we were mostly interested to compare the IP efficiency of the two methods using the same starting material. Therefore, using a standard cell line was not necessary. The samples we used are representative of a typical melanoma tissue and a B cell line; however, in future studies a common cell line with a defined HLA expression could be used for method benchmarking. While a significant portion of the immunopeptidome was shared between the two methods, the chip-IP-specific peptides exhibited some unique properties. With a lower number of cells (i.e., 0.2 million and 1 million cells), the chip-specific peptides exhibited a higher identification score (Figure S2D). Also, for the shared peptides between the two IP methods, those identified by the chip demonstrated a certain extent of advantage. Another interesting property was shown by an elevated hydrophilicity. It is not straightforward to explain why the chip-IP favors the enrichment of more hydrophilic peptides. A possible reason could be the chemistry on the micropillar surface. As already mentioned, a chlorosilane layer was coated onto the silicon pillars to facilitate the coating of essential proteins for the IP. The chrolosilane molecules have a PEGylated backbone that is highly hydrophilic to repel non-specific protein binding. While providing a non-fouling surface for clean IP, it might also render the capture of HLA-I complexes carrying more hydrophilic peptides. Furthermore, the acid elution and C18 cleanup scenarios were significantly different. The new workflow integrated these steps online with chip-IP, which might also result in this phenomenon. However, as yet the exact reason needs to be interrogated.
Apart from microfluidics engineering, the state-of-the-art DIA technique was adopted for acquiring the MS data, which has demonstrated remarkable power for reproducible and comprehensive immunopeptidomics. Chip-IP can capture the lowly abundant peptides and enrich the immunopeptidome from low-input samples. However, the traditional DDA method often induces a biased peptide discovery for its intensity-based precursor ion selection for MS/MS. Therefore, the more comprehensive and reproducible DIA method should be an ideal MS strategy for our chip-IP platform. We compared two ways to analyze DIA data. One is a library-free search (directDIA) that does not need a pre-built spectral library, mimicking a clinical setup when only the tiny core needle biopsies are available. The other is the hybridDIA approach, which maps the MS2 spectra to a pre-built spectral library from the DIA and available DDA data in case additional immunopeptidomics measurements of the autologous tumors are available. The latter has been proved to provide a more in-depth immunopeptidome for peptide identification. In the case where no sample-specific spectral library is available, we notably demonstrated the feasibility of using a sample-independent spectral library to harness the advantage of library-based DIA analysis. A spectral library was generated with published DDA and DIA runs from multiple studies, which we named the Lausanne-Lib. Applying a hybridDIA analysis with this library could reveal a broader immunopeptidome than the directDIA approach. Conversely, in our case, the directDIA analyses already exhibited an excellent depth of peptide search for the two smallest tissues below 15 mg. Some differences also exist between the generic Lausanne-Lib and the sample-specific counterpart. By looking into the composition of HLA-binding motifs (Figures 4B and 4C vs. Figures 6C and 6D), we found that the Lausanne-Lib-based hybridDIA results contained significantly fewer peptides binding to the HLA-B∗57:01 allele than those of the sample-specific library. Indeed, this HLA allele was rarely included in the relevant publications to which we referred for generating the Lausanne-Lib. However, this finding offers a hint for improving our strategy of using the Lausanne-Lib for DIA analysis, which is to include more diverse samples expressing, for example, various HLA-binding motifs. We demonstrated that generating a spectral library from available MS data will be a helpful resource for those who plan to implement DIA analysis in their MS-based immunopeptidomics pipeline.
Non-canonical HLA-bound peptides are invaluable sources of potential tumor-specific neoantigens for effective immunotherapies. Nevertheless, it demands extensive experimental and informatics efforts to construct a specific protein database with expansion in the cryptic unannotated protein-coding regions. We directly adopted a recently published database, nuORFdb, which has an expanded coverage of proteins translated from the unannotated ORFs. Hundreds of nuORF-derived HLA-restricted peptides could be identified in our chip-IP results. This nuORFdb thus represents a powerful tool for immunopeptidomics studies to discover cryptic tumor antigens hidden in the “dark” zones.
It might be of interest to cut the assay cost if one chip could be used for multiple individual IPs without noticeable cross-contamination. We did not engage much regarding the reusability of the chip-IP platform. However, given that the antibodies have been crosslinked on the intermediate layer of protein A/G (which has also been immobilized covalently on the silicon surface), it would be feasible to regenerate the chip surface for a second or third use. This is also the reason why 0.1 N acetic acid was used in this study, as it did not induce too much harm to the bioactivity of antibodies. We have tried regenerating the chip with more acid washes after the first use. The peptide identification from the second and third chip-IP did not differ significantly from the first use, although a slight decrease was apparent (Figure S6). A less harmful regeneration strategy thus needs to be investigated for potential chip reusability.
Limitations of the study and future developments
Currently, expertise in microengineering and cleanroom operations is crucial for establishing the chip-IP method in other immunopeptidomics laboratories. A broader implementation of this technology would require outsourcing the manufacturing to a qualified provider. Coupling the chip-IP with the downstream sample introduction to MS via the electrospray tip could potentially further help to reduce sample loss. However, this would mandate the use of pre-columns to clean the acidic eluate prior to sample loading on the analytical column, and the feasibility of this scenario must be carefully tested. Further packaging steps and software developments can enclose the fluidic control parts and the chip as an assay module. With a well-packaged assay module, the chip-IP workflow can be expanded to enrich the HLA-II peptides or specific HLA alleles in a high-throughput format. The user would only need to coat the relevant antibodies on the chip and operate the IP according to their protocols. Sequential purifications can also be performed for the same sample. Meanwhile, given the limited system scale in this proof-of-concept study, we could not perform multiple chip-IPs simultaneously for a comprehensive study of the reproducibility and reusability. According to the current data, an upper limit of detection appeared at 5 million RA957 cells, but this does not represent the true capability of the chip because the abundance of HLA molecules varies among different cells. A larger test scale would be necessary to address the system’s dynamic range of detection.
New modalities of MS detection that offer enhanced sensitivity and data completeness, such as timsTOF SCP,68 as well as other advanced machine-learning-based computational tools for MS/MS prediction and rescoring (such as MS2Rescore,69 Prosit,70 and MSBooster71) would improve the detection of immunopeptidomes purified with chip-IP, which is especially important for low-input samples. By obtaining the promising results presented in this report, we anticipate offering a low-cost yet high-throughput and reproducible sample preparation workflow for future immunopeptidomics research.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| W6/32 pan anti-human HLA-I antibody | in-house produced by hybridoma cells | N/A |
| Biological samples | ||
| Human malignant melanoma tissues | University Hospital of Lausanne (CHUV, Lausanne, Switzerland) | https://www.chuv.ch/fr/pathologie/ipa-home/pathologie-clinique/banque-de-tissus/ |
| Human liver metastasis lesions of melanoma | University Hospital of Lausanne (CHUV, Lausanne, Switzerland) | https://www.chuv.ch/fr/pathologie/ipa-home/pathologie-clinique/banque-de-tissus/ |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI 1640 Medium, GlutaMAX™ Supplement | Life Technologies | Cat#61870010 |
| Fetal bovine serum | Dominique Dutscher | Cat#35-079-CV |
| Penicillin/Streptomycin | BioConcept | Cat#4-01F00-H |
| protein-A Sepharose™ 4B beads | Invitrogen | Cat#101042 |
| Dimethyl pimelimidate dihydrochloride | Sigma-Aldrich | Cat#D8388 |
| Boric acid | Sigma-Aldrich | Cat#B6768 |
| Sodium tetraborate | Sigma-Aldrich | Cat#221732 |
| Ethanolamine | Sigma-Aldrich | Cat#398136 |
| Sodium azide | Sigma-Aldrich | Cat#S2002 |
| (CH3)2SiCl(CH2)4COONHS | Pro-Chimia Surfaces | Cat#SI 002-m4-0.1 |
| Recombinant protein A/G | Pierce™, Thermo Fisher | Cat#21186 |
| Octyl β-D-glucopyranoside | Sigma-Aldrich | Cat#O8001 |
| Sodium deoxycholate | Sigma-Aldrich | Cat#30970 |
| Iodoacetamide | Sigma-Aldrich | Cat#I6125 |
| UltraPure™ 0.5M EDTA, pH 8.0 | Invitrogen™ | Cat#15575020 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | P8340 |
| Phenylmethylsulfonylfluoride | Roche | Cat#70500620 |
| Acetonitrile | Sigma-Aldrich | Cat#1.00029 |
| Trifluoroacetic acid | Sigma-Aldrich | Cat#1.08262 |
| Formic Acid | Pierce™ | Cat#85178 |
| ReproSil-Pur Basic C18 | Dr. Maisch, High Performance LC GmbH | Cat#r119.b9 |
| Critical commercial assays | ||
| iRT Kit | Biognosys | Cat#Ki-3002-2 |
| Deposited data | ||
| UniProt, June 2020 | The UniProt Consortium | https://www.uniprot.org/ |
| nuORF database | Ouspenskaia et al.60 | https://www.nature.com/articles/s41587-021-01021-3 |
| The Cancer Genome Atlas (TCGA) database | TCGA Research Network | https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga |
| The Geno-type-Tissue Expression (GTEx) database | The Geno-type-Tissue Expression (GTEx) Project | https://gtexportal.org/home/ |
| The Immune Epitope Database (IEDB) | Vita et al.72 | https://www.iedb.org/ |
| Cancer Antigenic Peptide Database | The Brussels branch of Ludwig Cancer Research and/or at the de Duve Institute | https://caped.icp.ucl.ac.be/ |
| The mass spectrometry immunopeptidomics data and the spectral libraries | This paper | PRIDE: PXD040858 |
| Experimental models: Cell lines | ||
| human B-cell line RA957 | A gift from Pedro Romero, Ludwig Cancer Research Lausanne | N/A |
| W6/32 hybridoma cells | ATCC® | HB-95™; RRID:CVCL_7872 |
| Software and algorithms | ||
| KLayout 0.26.5 | KLayout project | https://www.klayout.de/build.html |
| Spectronaut® 16.2 | Biognosys | https://biognosys.com/software/spectronaut/ |
| NewAnce | Chong et al.16 | https://github.com/bassanilab/NewAnce |
| GraphPad Prism 9.1.0 | GraphPad Software, LLC | https://www.graphpad.com/ |
| Adobe Illustrator 27.4 | Adobe | https://www.adobe.com/products/illustrator.html |
Resource availability
Lead contact
Further information and requests should be addressed to and will be fulfilled by the lead contact, Michal Bassani-Sternberg (michal.bassani@chuv.ch).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell line and tissue samples
The human B-cell line RA957 (a gift from Pedro Romero, Ludwig Cancer Research Lausanne) was cultured in RPMI 1640 medium (GlutaMAX supplemented, Life Technologies, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Dominique Dutscher, Brumath, France) and 1% Penicillin/Streptomycin (BioConcept, San Diego, CA). Cells were grown to high confluency, washed three times with ice-cold PBS, and pelleted by centrifugation at 300 rcf for 5 min to the required amount. The cell pellets were snap-frozen on dry ice and stored dry at −80°C until use. In particular, we prepared individual frozen cell pellets with each corresponding to the desired number of cells (i.e., 0.2, 1, 5, and 10 million). Two pellets were used for each number of cells during the following IP process.
Snap-frozen tumor tissues were obtained from patients with malignant melanoma and liver metastasis lesions from the University Hospital of Lausanne (CHUV, Lausanne, Switzerland). Informed consent of the participants was obtained following the institutional review board’s requirements (Ethics Commission, CHUV). Small tissues were cut arbitrarily from different regions of the tumor samples and stored at −80°C before the downstream analyses. For the malignant melanoma tissues, six individual sections with varying sizes were collected: 5, 12, 21, 24, 25, and 40 mg; while three individual sections with varying sizes were collected from the liver metastasis lesions: 5, 10, and 20 mg. Information about the patients, such as gender, race, and age are not available in this study. However, it should not influence the discoveries reported in our study as all the tissues involved in the same assay were collected from the same patient.
High-resolution 4-digit HLA-I typing of the cells and tumor tissues was performed at the Laboratory of Diagnostics, Service of Immunology and Allergy, CHUV, Lausanne (Table S2).
Method details
Fabrication of the microfluidics chip with micropillar arrays
The chip fabrication and assembling were done in the Center of MicroNanoTechnology (CMi) facility at EPFL in Lausanne. The microfluidics structures were designed using the open-source KLayout tool and transferred to a 4-inch silicon wafer by a photolithography procedure (Figure S1A). A photomask was prepared with mirrored structures patterned on a 5-inch chromium glass mask. To start the process, the surface of 4-inch silicon wafers was cleaned and primed by HMDS treatment, followed by spin coating a 1.5 μm-thick positive photoresist AZ ECI 3007 on an automated coat/develop platform (ACS200 Gen3, Süss MicroTec, Germany). The wafers were then aligned with the photomask and exposed in a mask aligner (MA6/BA6 Gen 3 Mask Aligner, Süss MicroTec, Germany) with a short pulse of UV radiation of 20 mW/cm2 for 7 seconds. After an automated development process on ACS200, the exposed wafers underwent a plasma etching process (on AMS 200 SE, Alcatel) to remove the unexposed photoresist and reveal the microstructures.
To facilitate the covalent protein coating on a silicon surface, a thin oxide layer of about 100 nm was created through a dry oxidation process (Figures S1A and S1C). Each wafer was diced into four rectangular chips measuring 58 mm long and 20 mm wide. The chip accommodates a single fluidic channel (L 50 cm × W 650 μm × H 100 μm) filled with the micropillar arrays. Finally, the fluidic channel was enclosed by a 2 mm-thick PDMS (polydimethylsiloxane) slab. Two access holes were pierced through the slab as the flow inlet and outlet (Figure 1A). Moreover, a pair of stainless steel clamps (Figure S1E) was designed to reinforce the hermeticity of the fluidic connections (manufactured by Nice Rapid Prototyping, Zhongshan, China).
Automated fluidic control system
We applied an automated microfluidics instrumentation (Advanced Microfluidics, Switzerland) for handling the on-chip reactions: antibody coating/crosslinking, IP, and peptide cleanup in C18 cartridges (Figure S1C). To minimize the cross-contamination, separate syringe pumps conducted the chip-IP and the peptide cleanup processes. A 12-port distribution valve was installed on the syringe pumps. One port was reserved for liquid waste disposal, one for introducing reagents into the chip, and the rest for other buffer solutions required in each workflow. A 6-port rotary valve linked the flow between the chip-IP and peptide cleanup steps. By tuning the valve positions, we created two flow scenarios: one for separating the IP (until the acid elution step) from the C18 cartridge conditioning and the other for sending the IP elution immediately into the C18 cartridge (Figure S1E). In particular, Python scripts operated all the above steps in an automatic sequence, where one can readily customize each solution’s volume and flow rate according to different protocols. A detailed protocol used in this report for antibody coating, chip-IP, and peptide cleanup is described in Table S1.
Purification of pan-HLA antibody and preparation of antibody-crosslinked beads
W6/32 monoclonal antibodies were purified using protein-A Sepharose 4B beads (Invitrogen, Carlsbad, CA) from the supernatant of HB95 (ATCC® HB-95™) cells grown in CELLLine CL-1000 flasks (Sigma-Aldrich, St. Louis, MI). The antibody quantity was determined on a NanoDrop One spectrophotometer (Thermo Fisher Scientific). For the conventional column-based IP, 5 mg of purified antibodies were mixed with 1 ml of protein-A beads for 1 hour at room temperature, followed by the addition of 20 mM dimethyl pimelimidate (DMP, Sigma-Aldrich) in 0.2 M sodium borate buffer (pH 9, Sigma-Aldrich) for 30 min. The reaction was finally quenched by 0.2 M ethanolamine (pH 8, Sigma-Aldrich), and the crosslinked beads were kept in 0.02% sodium azide (Sigma-Aldrich) at 4°C until use.
Antibody immobilization and crosslinking on-chip
The oxidized silicon surface of the chip was primed by incubating in 1 mM NHS-silane solution ((CH3)2SiCl(CH2)4COONHS, ProChimia Surfaces, Poland) for 2 hours at room temperature, resulting in the formation of a self-assembled monolayer exposing NHS-ester groups. The primary amine groups on protein molecules can readily replace the NHS ester groups, forming covalent protein immobilization on the surface. Then 100 μL of 1 mg/mL recombinant protein A/G (Pierce™, Thermo Fisher) was introduced to form an intermediate layer for grafting the Fc domains of antibodies. 100 μL of 3 mg/mL pan-HLA antibodies were subsequently introduced. To avoid a co-elution of antibodies in the final peptide sample, we crosslinked the antibodies on the protein A/G layer by 20 mM dimethyl pimelimidate dihydrochloride (DMP, Sigma-Aldrich) dissolved in 0.2 M sodium borate buffer (pH 9, Sigma-Aldrich). The reaction was quenched by 0.2 M ethanolamine (pH 8, Sigma-Aldrich). The crosslinked chips were stored in 0.02% sodium azide solution at 4°C until use.
Immunoaffinity purification of HLA-I complexes in the microfluidics chip or chromatography column
The lysis buffer was freshly prepared in PBS containing 1% octyl-beta-d glucopyranoside (Sigma-Alrich), 0.25% sodium deoxycholate (Sigma-Aldrich), 0.2 mM iodoacetamide (Sigma-Aldrich), 1 mM EDTA, 1:200 Protease Inhibitors Mixture (Sigma-Aldrich), and 1 mM Phenylmethylsulfonylfluoride (Roche). The needle biopsy-sized tumor tissues were first disrupted in lysis buffer on a Tissue Lyser bead mill (Qiagen) for 1 min at 30 Hz. Then, the lysis of frozen cell pellets or disrupted tumor tissues was done in ice-cold lysis buffer at 4°C on a tube roller mixer for 1 h. We lysed cell pellets and tumor tissues below 20 mg in 100 μL of lysis buffer and tumor tissues above this weight in 200 μL of lysis buffer. Finally, the lysates were cleared by centrifugation at 21,300 rcf for 30 min at 4°C, and the supernatant was transferred to lo-bind Eppendorf tubes for the following IP procedure. We extracted HLA-I peptides from individual cell pellets corresponding to each number of cells as described above; this was performed in duplicates (biological replicates). Regarding the tumor tissues, one tissue section was collected for each weight indicated.
For chip-IP, the cleared lysate supernatant was loaded into an antibody-coated microfluidics chip at the lowest flow rate to maximize the immunoaffinity reactions. The following steps are shown in Table S1. The column-IP was performed following the previously established protocols.16,22,30 Briefly, each column (Bio-Spin® disposable chromatography columns, Bio-Rad) contained 50 μL of antibody-coated microbeads. The lysate supernatant passed through the microbeads in a gravity-driven flow. Then the beads were extensively washed by high salt, low salt and Tris-HCl buffers before the HLA-peptides were eluted with 0.1 N acetic acid. The acid elution was cleaned in C18 cartridges (Micro SpinColumns, Havard Apparatus). Next, 25% acetonitrile (ACN; Sigma Aldrich) in 0.1% TFA (trifluoroacetic acid) eluted the HLA-I peptides off the C18 material. Before analyzing the peptides in LC-MS/MS, the excessive ACN was removed by vacuum centrifugation (Concentrator plus, Eppendorf) at 60°C for 3 hours. The dried peptides were stored at -20°C before MS analysis.
Liquid chromatography – Tandem mass spectrometry (LC-MS/MS) analyses
All samples were analyzed with our LC-MS/MS system composed of an Easy-nLC 1200 HPLC system (Thermo Fisher Scientific) coupled online to a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific). The samples were ionized by a nano-electrospray ion source (Nanospray Flex™ Ion Sources, Thermo Fisher Scientific) equipped with a column oven (PRSO-V1, Sonation, Germany). The dried HLA-I peptides were resuspended in 8 μL of 2% ACN in 0.1% FA (formic acid) supplemented with 1:10 iRT peptides (Biognosys, Zurich). Twice of 3 μL (technical replicates) was taken for MS analysis. The two replicates underwent DIA analysis for the chip-IP method with both cell and tissue samples. For the column-IP method, one replicate underwent DIA while the other DDA analysis for enriching the cell-specific spectral library. The analytical columns were fabricated in-house. We first pulled the nanospray tip on a fused silica tubing (360 μm OD × 75μm ID, BGB Analytik, Switzerland) using a laser puller (P-2000, Sutter instrument). Then, a 50 cm-long part was cut and packed with ReproSil-Pur C18 materials (1.9 μm diameter, 120 Å pore size, Dr. Maisch GmbH, Ammerbuch, Germany). During the sample injection, mounted analytical columns were kept at 50°C. We used a mixture of 0.1% FA (Solvent A) and 0.1% FA in 95% ACN (Solvent B) to perform the following 60 min-gradient at 250 nL/min: 0–52 min (2 - 25% B); 52–54 min (25–35% B); 54–55 min (35–100% B); 55–60 min (100% B).
For data-independent acquisition (DIA), a full MS1 scan was acquired from 300 to 1650 m/z with a resolution of 60,000 (m/z = 200) and the ion accumulation time of 60 ms, which was followed by 22 DIA MS2 scans with a resolution of 30,000 (m/z = 200) and an automatic gain control (AGC) of 3×106. The fixed first mass was 200 m/z, and the overlap between consecutive MS2 scans was 1 m/z. A stepped normalized collision energy (25.5, 27, and 30) was employed, and the maximum ion accumulation was set to auto.
For data-dependent acquisition (DDA), a full MS1 scan was conducted from 300 to 1650 m/z with a resolution of 60,000 (m/z = 200) and an automatic gain control (AGC) value of 3×106 ions. A “Top 20” strategy was used to isolate the twenty most abundant precursors within a 1.2 m/z isolation window, which undergo a fragmentation by higher-energy collision dissociation (HCD) at a normalized collision energy of 27. The MS2 scans were performed with a resolution of 30,000 (m/z = 200) and an ion-accumulation time of 120 ms. Peptides with charge states of four and above were exempt from fragmentation.
Database search
The Spectronaut® software package (version 16.2, Biognosys, Switzerland) was used for analyzing the MS files. For library-free peptide identification, the raw DIA MS files were directly loaded in the directDIA workflow and searched against the UniProt database (human-reviewed sequences with isoforms, 42,348 entries, June 2020). Some parameters were modified in the default search settings: ‘Digest Type’ was ‘unspecific’; ‘Min Peptide Length' was 8, and the ‘Max' was 15; ‘Variable modifications’ contained ‘Oxidation (M)’ and ‘Acetylation (Protein N-term), no ‘Fixed modifications’; ‘PSM’ and ‘Peptide’ FDR were set to 0.01 with no protein FDR. For library-based peptide identification, we first used the DDA and DIA raw files to generate a hybrid spectral library powered by the built-in tool ‘Pulsar' before the targeted peptide search. The default settings were used for Pulsar search and library generation except for the following parameters. The ‘Protein FDR' was set to 1, while the FDR for peptide and PSM was 0.01. ‘Digestion rule' was left vacant, and the ‘Digest Type' was changed to ‘Unspecific'. Once the library was established, we performed the peptide search with a modified default setting. For ‘Identification', the DIA raw files were matched to the spectral library with a ‘Qvalue Cutoff’ of 0.01 and 1 for precursor and protein, respectively. The ‘Corss-run normalization’ was switched off. Results were exported in a peptide-centric format with the columns of ‘PG.Genes’, ‘PEP.StrippedSequence’, and ‘PEP.Quantity’ selected for data analyses. To reveal the retention time of certain peptides, the column of ‘EG.ApexRT’ was included.
The same UniProt database was used for a library-based search of canonical immunopeptidomics. To reveal the non-canonical space, we generated the spectral library against a nuORF (novel or unannotated open reading frame) database.60 The identified peptides were labeled with their corresponding protein group information, especially the canonical (PC) and non-canonical (NC) annotations. When one peptide has multiple annotations of protein sources, we consider it derived from nuORFs only when the protein group does not contain the ‘PC’ assignment. With a preset global FDR of 1% for the PSMs, the FDR for NC-annotated PSMs was calculated after the analysis, which was around 4%.
To ensure a stringent and precise identification of nuORF-derived peptides, we further aligned the NC-annotated peptides against a non-redundant BLAST database (845,586 protein entries, downloaded on 26/08/2020 via BLAST+, v2.10.1) using an algorithm built in NewAnce tool.16 Leucine and isoleucines were regarded as equal as they are not distinguishable by MS. The peptides mapped to known proteins in this database were thus discarded from the NC peptide pool.
Finally, the unique peptides identified in the two technical replicates for MS analysis were merged to represent the immunopeptidome for each sample.
HLA-I binding prediction, clustering, and other properties for MS-identified peptides
NetMHCpan 4.1 prediction software74 was used to evaluate the binding affinity of all the MS-identified peptides (8-15 amino acids) to the respective HLA-I alleles. Peptides with no more than 2% rank were considered HLA-I binders. The alignment and clustering of peptide sequences (binding motif deconvolution) were performed using the GibbsCluster 2.0 software,75 which was performed with the default settings except for the following modifications: the number of maximum clusters was set to 6 motifs; clustering moves at each iteration; ‘Max deletion and insertion length’ was set to 4 and 1, respectively; ‘Number of seeds for initial conditions’ was 5; trash cluster was used to remove outliers. Upon completion, the clustered motifs were manually assigned to the cell’s or patients’ HLA allotypes. To calculate the grand average of hydropath (GRAVY) values, the peptide list was submitted to the online ‘GRAVY calculator’ (https://www.gravy-calculator.de/). The Venn diagrams were created with Intervene.72
Listing of TAA genes and relevant peptide antigens
In order to list cancer-specific TAA genes, we obtained gene expression information for all genes expressed in 33 cancer types from the Cancer Genome Atlas (TCGA) database (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). First, the 99th percentile of genes with at least a 2.5 TPM value (transcript per million) was prioritized. Then those that are expressed in any healthy tissues (except for testis and skin) listed in the Genotype-Tissue Expression (GTEx) database (https://gtexportal.org/home/, 90th percentile with more than 1.0 TPM) were discarded to result in a sensitive and stringent list of cancer-specific TAA genes. Finally, information was curated from the literature and with the IEDB (https://www.iedb.org/)76 and CAPED (https://caped.icp.ucl.ac.be/) databases to include immunogenicity information. In addition, new short peptides (8-22 mers) were generated artificially from these cancer-specific TAA genes and were annotated as ‘predicted’.
Finally, the MS-identified peptides from small tumor specimens by chip-IP were matched to the above list to highlight the TAA epitopes. If the peptides have been tested ‘immunogenic’, the immunogenicity levels were given as ‘high’ (detected in MS and validated in IEDB with at least two T cell assays) or ‘medium’ (detected in MS and validated in one T cell assay). Otherwise, they were labeled ‘predicted’.
Generation of a public spectral library with published raw MS files
477 raw files (465 acquired in DDA and 12 in DIA) were retrieved from previous publications on human immunopeptidomics.14,15,16,22,30,61,62,63,64 An initial spectral library was generated with these files in Spectronaut® using the built-in Pulsar search engine described above, which served as a search archive for future uses. The newly acquired MS raw files were used together with the search archive to create a hybrid spectral library for better peptide identification. For example, twelve DIA raw files were acquired from the small melanoma tissues. We then generated a hybrid library with these twelve runs plus the search archive for the subsequent DIA analyses. A full list of the 477 raw files with their files names and publications can be found in Table S6.
Quantification and statistical analysis
Statistical analysis
Data were analyzed using GraphPad Prism (version 9.1.0). Figures were prepared with Adobe Illustrator (version 27.4, Adobe). Statistical analysis of Figures 3D, S2C, and S2D was conducted by a two-tailed, unpaired, and nonparametric Student’s t-test. Error bars in all plots represent mean ± standard deviation with the number of replicates indicated in the figure legends.
Acknowledgments
This work was financially supported by the Ludwig Institute for Cancer Research, the Swiss Cancer Research Foundation grant KFS-4680-02-2019 (M.B.-S.), and the Swiss National Science Foundation, PRIMA grant PR00P3_193079 (M.B.-S.). We are highly thankful to our patients for their cooperation. We also thank the Center of MicroNanoTechnology (CMi) facility at EPFL for their technical assistance during the microfluidics chip fabrication. The results published here are in part based upon data generated by the TCGA Research Network( https://www.cancer.gov/tcga) and the Geno-type-Tissue Expression (GTEx) Project (https://gtexportal.org/home/).
Author contributions
Conceptualization, X.L. and M.B.-S.; methodology, X.L.; investigation, X.L. and H.S.P.; software, F.H.; data curation, X.L., H.S.P., and E.R.A.; resources, J.M. and M.T.-C.; visualization, X.L. and M.B.-S.; supervision, M.B.-S.; funding acquisition, M.B.-S.; writing, X.L. and M.B.-S., with contributions from all authors.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: May 19, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100479.
Supplemental information
A list of the various DIA and DDA raw files was given, which were used for generating the spectral library that was searched against the UniProt reference.
Data were analyzed with direct DIA against the UniProt reference and with hybrid DIA against the UniProt and nuORF references.
Data were analyzed with direct DIA against the UniProt reference and with hybrid DIA against the UniProt and nuORF references.
Data were analyzed with hybrid DIA with the Lausanne-Lib searched against the UniProt and nuORF references.
Data and code availability
-
•
The mass spectrometry immunopeptidomics data have been deposited at the ProteomeXchange Consortium via the PRIDE73 partner repository with the dataset identifier PXD040858.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Pishesha N., Harmand T.J., Ploegh H.L. A guide to antigen processing and presentation. Nat. Rev. Immunol. 2022;22:751–764. doi: 10.1038/s41577-022-00707-2. [DOI] [PubMed] [Google Scholar]
- 2.Chong C., Coukos G., Bassani-Sternberg M. Identification of tumor antigens with immunopeptidomics. Nat. Biotechnol. 2022;40:175–188. doi: 10.1038/s41587-021-01038-8. [DOI] [PubMed] [Google Scholar]
- 3.Creelan B.C., Wang C., Teer J.K., Toloza E.M., Yao J., Kim S., Landin A.M., Mullinax J.E., Saller J.J., Saltos A.N., et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat. Med. 2021;27:1410–1418. doi: 10.1038/s41591-021-01462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearlman A.H., Hwang M.S., Konig M.F., Hsiue E.H.-C., Douglass J., DiNapoli S.R., Mog B.J., Bettegowda C., Pardoll D.M., Gabelli S.B., et al. Targeting public neoantigens for cancer immunotherapy. Nat. Cancer. 2021;2:487–497. doi: 10.1038/s43018-021-00210-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leidner R., Sanjuan Silva N., Huang H., Sprott D., Zheng C., Shih Y.-P., Leung A., Payne R., Sutcliffe K., Cramer J., et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N. Engl. J. Med. 2022;386:2112–2119. doi: 10.1056/NEJMoa2119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dargel C., Bassani-Sternberg M., Hasreiter J., Zani F., Bockmann J.-H., Thiele F., Bohne F., Wisskirchen K., Wilde S., Sprinzl M.F., et al. T cells engineered to express a T-cell receptor specific for glypican-3 to recognize and kill hepatoma cells in vitro and in mice. Gastroenterology. 2015;149:1042–1052. doi: 10.1053/j.gastro.2015.05.055. [DOI] [PubMed] [Google Scholar]
- 7.Marcu A., Bichmann L., Kuchenbecker L., Kowalewski D.J., Freudenmann L.K., Backert L., Mühlenbruch L., Szolek A., Lübke M., Wagner P., et al. HLA Ligand Atlas: a benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy. J. Immunother. Cancer. 2021;9:e002071. doi: 10.1136/jitc-2020-002071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sellars M.C., Wu C.J., Fritsch E.F. Cancer vaccines: building a bridge over troubled waters. Cell. 2022;185:2770–2788. doi: 10.1016/j.cell.2022.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lybaert L., Lefever S., Fant B., Smits E., De Geest B., Breckpot K., Dirix L., Feldman S.A., van Criekinge W., Thielemans K., et al. Challenges in neoantigen-directed therapeutics. Cancer Cell. 2023;41:15–40. doi: 10.1016/j.ccell.2022.10.013. [DOI] [PubMed] [Google Scholar]
- 10.Lang F., Schrörs B., Löwer M., Türeci Ö., Sahin U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 2022;21:261–282. doi: 10.1038/s41573-021-00387-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bassani-Sternberg M., Coukos G. Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr. Opin. Immunol. 2016;41:9–17. doi: 10.1016/j.coi.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Pandey K., Ramarathinam S.H., Purcell A.W. Isolation of HLA bound peptides by immunoaffinity capture and identification by mass spectrometry. Curr. Protoc. 2021;1:e92. doi: 10.1002/cpz1.92. [DOI] [PubMed] [Google Scholar]
- 13.Demmers L.C., Kretzschmar K., Van Hoeck A., Bar-Epraïm Y.E., van den Toorn H.W.P., Koomen M., van Son G., van Gorp J., Pronk A., Smakman N., et al. Single-cell derived tumor organoids display diversity in HLA class I peptide presentation. Nat. Commun. 2020;11:5338. doi: 10.1038/s41467-020-19142-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bassani-Sternberg M., Pletscher-Frankild S., Jensen L.J., Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteomics. 2015;14:658–673. doi: 10.1074/mcp.M114.042812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bassani-Sternberg M., Bräunlein E., Klar R., Engleitner T., Sinitcyn P., Audehm S., Straub M., Weber J., Slotta-Huspenina J., Specht K., et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016;7:13404. doi: 10.1038/ncomms13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chong C., Müller M., Pak H., Harnett D., Huber F., Grun D., Leleu M., Auger A., Arnaud M., Stevenson B.J., et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 2020;11:1293. doi: 10.1038/s41467-020-14968-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim G.B., Fritsche J., Bunk S., Mahr A., Unverdorben F., Tosh K., Kong H., Maldini C.R., Lau C., Srivatsa S., et al. Quantitative immunopeptidomics reveals a tumor stroma–specific target for T cell therapy. Sci. Transl. Med. 2022;14:eabo6135. doi: 10.1126/scitranslmed.abo6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bassani-Sternberg M., Barnea E., Beer I., Avivi I., Katz T., Admon A. Soluble plasma HLA peptidome as a potential source for cancer biomarkers. Proc. Natl. Acad. Sci. 2010;107:18769–18776. doi: 10.1073/pnas.1008501107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klaeger S., Apffel A., Clauser K.R., Sarkizova S., Oliveira G., Rachimi S., Le P.M., Tarren A., Chea V., Abelin J.G., et al. Optimized liquid and gas phase fractionation increases HLA-peptidome coverage for primary cell and tissue samples. Mol. Cell. Proteomics. 2021;20:100133. doi: 10.1016/j.mcpro.2021.100133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stopfer L.E., Conage-Pough J.E., White F.M. Quantitative consequences of protein carriers in immunopeptidomics and tyrosine phosphorylation MS2 analyses. Mol. Cell. Proteomics. 2021;20:100104. doi: 10.1016/j.mcpro.2021.100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sricharoensuk C., Boonchalermvichien T., Muanwien P., Somparn P., Pisitkun T., Sriswasdi S. Unsupervised mining of HLA-I peptidomes reveals new binding motifs and potential false positives in the community database. Front. Immunol. 2022;13:847756. doi: 10.3389/fimmu.2022.847756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pak H., Michaux J., Huber F., Chong C., Stevenson B.J., Müller M., Coukos G., Bassani-Sternberg M. Sensitive immunopeptidomics by leveraging available large-scale multi-HLA spectral libraries, data-independent acquisition, and MS/MS prediction. Mol. Cell. Proteomics. 2021;20:100080. doi: 10.1016/j.mcpro.2021.100080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Racle J., Michaux J., Rockinger G.A., Arnaud M., Bobisse S., Chong C., Guillaume P., Coukos G., Harari A., Jandus C., et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 2019;37:1283–1286. doi: 10.1038/s41587-019-0289-6. [DOI] [PubMed] [Google Scholar]
- 24.Cai Y., Lv D., Li D., Yin J., Ma Y., Luo Y., Fu L., Ding N., Li Y., Pan Z., et al. IEAtlas: an atlas of HLA-presented immune epitopes derived from non-coding regions. Nucleic Acids Res. 2023;51:D409–D417. doi: 10.1093/nar/gkac776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scull K.E., Pandey K., Ramarathinam S.H., Purcell A.W. Immunopeptidogenomics: harnessing RNA-seq to illuminate the dark immunopeptidome. Mol. Cell. Proteomics. 2021;20:100143. doi: 10.1016/j.mcpro.2021.100143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gfeller D., Bassani-Sternberg M., Schmidt J., Luescher I.F. Current tools for predicting cancer-specific T cell immunity. OncoImmunology. 2016;5:e1177691. doi: 10.1080/2162402X.2016.1177691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xin L., Qiao R., Chen X., Tran H., Pan S., Rabinoviz S., Bian H., He X., Morse B., Shan B., Li M. A streamlined platform for analyzing tera-scale DDA and DIA mass spectrometry data enables highly sensitive immunopeptidomics. Nat. Commun. 2022;13:3108. doi: 10.1038/s41467-022-30867-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarkizova S., Klaeger S., Le P.M., Li L.W., Oliveira G., Keshishian H., Hartigan C.R., Zhang W., Braun D.A., Ligon K.L., et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020;38:199–209. doi: 10.1038/s41587-019-0322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sirois I., Isabelle M., Duquette J.D., Saab F., Caron E. Immunopeptidomics: isolation of mouse and human MHC class I- and II-associated peptides for mass spectrometry analysis. J. Vis. Exp. 2021:e63052. doi: 10.3791/63052. [DOI] [PubMed] [Google Scholar]
- 30.Chong C., Marino F., Pak H., Racle J., Daniel R.T., Müller M., Gfeller D., Coukos G., Bassani-Sternberg M. High-throughput and sensitive immunopeptidomics platform reveals profound interferonγ-mediated remodeling of the human leukocyte antigen (HLA) ligandome. Mol. Cell. Proteomics. 2018;17:533–548. doi: 10.1074/mcp.TIR117.000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L., McAlpine P.L., Heberling M.L., Elias J.E. Automated ligand purification platform accelerates immunopeptidome analysis by mass spectrometry. J. Proteome Res. 2021;20:393–408. doi: 10.1021/acs.jproteome.0c00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pollock S.B., Rose C.M., Darwish M., Bouziat R., Delamarre L., Blanchette C., Lill J.R. Sensitive and quantitative detection of MHC-I displayed neoepitopes using a semiautomated workflow and TOMAHAQ mass spectrometry. Mol. Cell. Proteomics. 2021;20:100108. doi: 10.1016/j.mcpro.2021.100108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feola S., Haapala M., Peltonen K., Capasso C., Martins B., Antignani G., Federico A., Pietiäinen V., Chiaro J., Feodoroff M., et al. PeptiCHIP: a microfluidic platform for tumor antigen landscape identification. ACS Nano. 2021;15:15992–16010. doi: 10.1021/acsnano.1c04371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitesides G.M. The origins and the future of microfluidics. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 35.Jung W., Han J., Choi J.-W., Ahn C.H. Point-of-care testing (POCT) diagnostic systems using microfluidic lab-on-a-chip technologies. Microelectron. Eng. 2015;132:46–57. doi: 10.1016/j.mee.2014.09.024. [DOI] [Google Scholar]
- 36.Ding Y., Howes P.D., deMello A.J. Recent advances in droplet microfluidics. Anal. Chem. 2020;92:132–149. doi: 10.1021/acs.analchem.9b05047. [DOI] [PubMed] [Google Scholar]
- 37.Stejskal K., Op de Beeck J., Dürnberger G., Jacobs P., Mechtler K. Ultrasensitive NanoLC-MS of subnanogram protein samples using second generation micropillar array LC technology with orbitrap exploris 480 and FAIMS PRO. Anal. Chem. 2021;93:8704–8710. doi: 10.1021/acs.analchem.1c00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dainese R., Gardeux V., Llimos G., Alpern D., Jiang J.Y., Meireles-Filho A.C.A., Deplancke B. A parallelized, automated platform enabling individual or sequential ChIP of histone marks and transcription factors. Proc. Natl. Acad. Sci. 2020;117:13828–13838. doi: 10.1073/pnas.1913261117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tähkä S., Sarfraz J., Urvas L., Provenzani R., Wiedmer S.K., Peltonen J., Jokinen V., Sikanen T. Immobilization of proteolytic enzymes on replica-molded thiol-ene micropillar reactors via thiol-gold interaction. Anal. Bioanal. Chem. 2019;411:2339–2349. doi: 10.1007/s00216-019-01674-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu A.R., Hiatt J.B., Lu R., Attema J.L., Lobo N.A., Weissman I.L., Clarke M.F., Quake S.R. Automated microfluidic chromatin immunoprecipitation from 2,000 cells. Lab Chip. 2009;9:1365–1370. doi: 10.1039/B819648F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu X., He R., Li S., Cai B., Zhao L., Liao L., Liu W., Zeng Q., Wang H., Guo S.-S., Zhao X.Z. Magneto-controllable capture and release of cancer cells by using a micropillar device decorated with graphite oxide-coated magnetic nanoparticles. Small. 2013;9:3895–3901. doi: 10.1002/smll.201300169. [DOI] [PubMed] [Google Scholar]
- 42.Wang Z., Xu D., Wang X., Jin Y., Huo B., Wang Y., He C., Fu X., Lu N. Size-matching hierarchical micropillar arrays for detecting circulating tumor cells in breast cancer patients’ whole blood. Nanoscale. 2019;11:6677–6684. doi: 10.1039/C9NR00173E. [DOI] [PubMed] [Google Scholar]
- 43.Lou R., Tang P., Ding K., Li S., Tian C., Li Y., Zhao S., Zhang Y., Shui W. Hybrid spectral library combining DIA-MS data and a targeted virtual library substantially deepens the proteome coverage. iScience. 2020;23:100903. doi: 10.1016/j.isci.2020.100903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinitcyn P., Hamzeiy H., Salinas Soto F., Itzhak D., McCarthy F., Wichmann C., Steger M., Ohmayer U., Distler U., Kaspar-Schoenefeld S., et al. MaxDIA enables library-based and library-free data-independent acquisition proteomics. Nat. Biotechnol. 2021;39:1563–1573. doi: 10.1038/s41587-021-00968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Midha M.K., Kusebauch U., Shteynberg D., Kapil C., Bader S.L., Reddy P.J., Campbell D.S., Baliga N.S., Moritz R.L. A comprehensive spectral assay library to quantify the Escherichia coli proteome by DIA/SWATH-MS. Sci. Data. 2020;7:389. doi: 10.1038/s41597-020-00724-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenberger G., Koh C.C., Guo T., Röst H.L., Kouvonen P., Collins B.C., Heusel M., Liu Y., Caron E., Vichalkovski A., et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data. 2014;1:140031. doi: 10.1038/sdata.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caron E., Espona L., Kowalewski D.J., Schuster H., Ternette N., Alpízar A., Schittenhelm R.B., Ramarathinam S.H., Lindestam Arlehamn C.S., Chiek Koh C., et al. An open-source computational and data resource to analyze digital maps of immunopeptidomes. Elife. 2015;4:e07661. doi: 10.7554/eLife.07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 49.Klatt M.G., Kowalewski D.J., Schuster H., Di Marco M., Hennenlotter J., Stenzl A., Rammensee H.-G., Stevanović S. Carcinogenesis of renal cell carcinoma reflected in HLA ligands: a novel approach for synergistic peptide vaccination design. OncoImmunology. 2016;5:e1204504. doi: 10.1080/2162402X.2016.1204504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brasseur F., Rimoldi D., Liénard D., Lethé B., Carrel S., Arienti F., Suter L., Vanwijck R., Bourlond A., Humblet Y. Expression of MAGE genes in primary and metastatic cutaneous melanoma. Int. J. Cancer. 1995;63:375–380. doi: 10.1002/ijc.2910630313. [DOI] [PubMed] [Google Scholar]
- 51.Rad H.H., Yamashita T., Jin H.-Y., Hirosaki K., Wakamatsu K., Ito S., Jimbow K. Tyrosinase-related proteins suppress tyrosinase-mediated cell death of melanocytes and melanoma cells. Exp. Cell Res. 2004;298:317–328. doi: 10.1016/j.yexcr.2004.04.045. [DOI] [PubMed] [Google Scholar]
- 52.Kobayashi T., Lonchay C., Colau D., Demotte N., Boon T., van der Bruggen P. New MAGE-4 antigenic peptide recognized by cytolytic T lymphocytes on HLA-A1 tumor cells. Tissue Antigens. 2003;62:426–432. doi: 10.1034/j.1399-0039.2003.00123.x. [DOI] [PubMed] [Google Scholar]
- 53.Podaza E., Carri I., Aris M., von Euw E., Bravo A.I., Blanco P., Ortiz Wilczyñski J.M., Koile D., Yankilevich P., Nielsen M., et al. Evaluation of T-cell responses against shared melanoma associated antigens and predicted neoantigens in cutaneous melanoma patients treated with the CSF-470 allogeneic cell vaccine plus BCG and GM-CSF. Front. Immunol. 2020;11:1147. doi: 10.3389/fimmu.2020.01147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schultz E.s., Zhang Y., Knowles R., Tine J., Traversari C., Boon T., Van Der Bruggen P. A MAGE-3 peptide recognized on HLA-B35 and HLA-A1 by cytolytic T lymphocytes. Tissue Antigens. 2001;57:103–109. doi: 10.1034/j.1399-0039.2001.057002103.x. [DOI] [PubMed] [Google Scholar]
- 55.Benlalam H., Linard B., Guilloux Y., Moreau-Aubry A., Derré L., Diez E., Dreno B., Jotereau F., Labarrière N. Identification of five new HLA-B∗3501-restricted epitopes derived from common melanoma-associated antigens, spontaneously recognized by tumor-infiltrating lymphocytes. J. Immunol. 2003;171:6283–6289. doi: 10.4049/jimmunol.171.11.6283. [DOI] [PubMed] [Google Scholar]
- 56.Kula T., Dezfulian M.H., Wang C.I., Abdelfattah N.S., Hartman Z.C., Wucherpfennig K.W., Lyerly H.K., Elledge S.J. T-scan: a genome-wide method for the systematic discovery of T cell epitopes. Cell. 2019;178:1016–1028.e13. doi: 10.1016/j.cell.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Bruggen P., Szikora J.P., Boël P., Wildmann C., Somville M., Sensi M., Boon T. Autologous cytolytic T lymphocytes recognize a MAGE-1 nonapeptide on melanomas expressing HLA-Cw∗1601. Eur. J. Immunol. 1994;24:2134–2140. doi: 10.1002/eji.1830240930. [DOI] [PubMed] [Google Scholar]
- 58.Chaux P., Luiten R., Demotte N., Vantomme V., Stroobant V., Traversari C., Russo V., Schultz E., Cornelis G.R., Boon T., van der Bruggen P. Identification of five MAGE-A1 epitopes recognized by cytolytic T lymphocytes obtained by in vitro stimulation with dendritic cells transduced with MAGE-A1. J. Immunol. 1999;163:2928–2936. [PubMed] [Google Scholar]
- 59.Ruiz Cuevas M.V., Hardy M.-P., Hollý J., Bonneil É., Durette C., Courcelles M., Lanoix J., Côté C., Staudt L.M., Lemieux S., et al. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Rep. 2021;34:108815. doi: 10.1016/j.celrep.2021.108815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ouspenskaia T., Law T., Clauser K.R., Klaeger S., Sarkizova S., Aguet F., Li B., Christian E., Knisbacher B.A., Le P.M., et al. Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat. Biotechnol. 2022;40:209–217. doi: 10.1038/s41587-021-01021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Forlani G., Michaux J., Pak H., Huber F., Marie Joseph E.L., Ramia E., Stevenson B.J., Linnebacher M., Accolla R.S., Bassani-Sternberg M. CIITA-transduced glioblastoma cells uncover a rich repertoire of clinically relevant tumor-associated HLA-II antigens. Mol. Cell. Proteomics. 2021;20:100032. doi: 10.1074/mcp.RA120.002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bassani-Sternberg M., Chong C., Guillaume P., Solleder M., Pak H., Gannon P.O., Kandalaft L.E., Coukos G., Gfeller D. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLoS Comput. Biol. 2017;13:e1005725. doi: 10.1371/journal.pcbi.1005725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Newey A., Griffiths B., Michaux J., Pak H.S., Stevenson B.J., Woolston A., Semiannikova M., Spain G., Barber L.J., Matthews N., et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J. Immunother. Cancer. 2019;7:309. doi: 10.1186/s40425-019-0769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gfeller D., Guillaume P., Michaux J., Pak H.-S., Daniel R.T., Racle J., Coukos G., Bassani-Sternberg M. The length distribution and multiple specificity of naturally presented HLA-I ligands. J. Immunol. 2018;201:3705–3716. doi: 10.4049/jimmunol.1800914. [DOI] [PubMed] [Google Scholar]
- 65.Stopfer L.E., Mesfin J.M., Joughin B.A., Lauffenburger D.A., White F.M. Multiplexed relative and absolute quantitative immunopeptidomics reveals MHC I repertoire alterations induced by CDK4/6 inhibition. Nat. Commun. 2020;11:2760. doi: 10.1038/s41467-020-16588-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vangelooven J., Desmet G. Theoretical optimisation of the side-wall of micropillar array columns using computational fluid dynamics. J. Chromatogr. A. 2010;1217:8121–8126. doi: 10.1016/j.chroma.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 67.Kim K., Koo J., Moon S., Lee W.G. Role of micropillar arrays in cell rolling dynamics. Analyst. 2016;142:110–117. doi: 10.1039/C6AN01506A. [DOI] [PubMed] [Google Scholar]
- 68.Brunner A.-D., Thielert M., Vasilopoulou C., Ammar C., Coscia F., Mund A., Hoerning O.B., Bache N., Apalategui A., Lubeck M., et al. Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol. Syst. Biol. 2022;18:e10798. doi: 10.15252/msb.202110798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Declercq A., Bouwmeester R., Hirschler A., Carapito C., Degroeve S., Martens L., Gabriels R. MS2Rescore: data-driven rescoring dramatically boosts immunopeptide identification rates. Mol. Cell. Proteomics. 2022;21:100266. doi: 10.1016/j.mcpro.2022.100266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gessulat S., Schmidt T., Zolg D.P., Samaras P., Schnatbaum K., Zerweck J., Knaute T., Rechenberger J., Delanghe B., Huhmer A., et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods. 2019;16:509–518. doi: 10.1038/s41592-019-0426-7. [DOI] [PubMed] [Google Scholar]
- 71.Yang K.L., Yu F., Teo G.C., Demichev V., Ralser M., Nesvizhskii A.I. MSBooster: improving peptide identification rates using deep learning-based features. bioRxiv. 2022 doi: 10.1101/2022.10.19.512904. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khan A., Mathelier A. Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinf. 2017;18:287. doi: 10.1186/s12859-017-1708-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reynisson B., Alvarez B., Paul S., Peters B., Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48:W449–W454. doi: 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Andreatta M., Lund O., Nielsen M. Simultaneous alignment and clustering of peptide data using a Gibbs sampling approach. Bioinformatics. 2013;29:8–14. doi: 10.1093/bioinformatics/bts621. [DOI] [PubMed] [Google Scholar]
- 76.Vita R., Mahajan S., Overton J.A., Dhanda S.K., Martini S., Cantrell J.R., Wheeler D.K., Sette A., Peters B. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2019;47:D339–D343. doi: 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A list of the various DIA and DDA raw files was given, which were used for generating the spectral library that was searched against the UniProt reference.
Data were analyzed with direct DIA against the UniProt reference and with hybrid DIA against the UniProt and nuORF references.
Data were analyzed with direct DIA against the UniProt reference and with hybrid DIA against the UniProt and nuORF references.
Data were analyzed with hybrid DIA with the Lausanne-Lib searched against the UniProt and nuORF references.
Data Availability Statement
-
•
The mass spectrometry immunopeptidomics data have been deposited at the ProteomeXchange Consortium via the PRIDE73 partner repository with the dataset identifier PXD040858.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.