

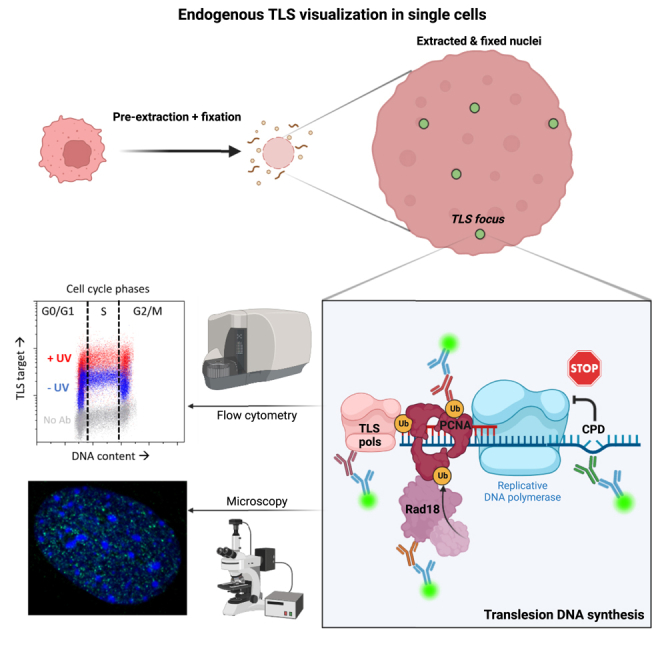
Summary
Translesion DNA synthesis (TLS) is an evolutionarily conserved process that cells activate to tolerate DNA damage. TLS facilitates proliferation under DNA damage conditions and is exploited by cancer cells to gain therapy resistance. It has been so far challenging to analyze endogenous TLS factors such as PCNAmUb and TLS DNA polymerases in single mammalian cells due to a lack of suitable detection tools. We have adapted a flow cytometry-based quantitative method allowing detection of endogenous, chromatin-bound TLS factors in single mammalian cells, either untreated or exposed to DNA-damaging agents. This high-throughput procedure is quantitative, accurate, and allows unbiased analysis of TLS factors’ recruitment to chromatin, as well as occurrence of DNA lesions with respect to the cell cycle. We also demonstrate detection of endogenous TLS factors by immunofluorescence microscopy and provide insights into TLS dynamics upon DNA replication forks stalled by UV-C-induced DNA damage.
Keywords: DNA damage tolerance, PCNAmUb, TLS pols, XP-V, flow cytometry, chromatin, replication stress, oxidative stress, UV-C, CPDs
Graphical abstract
Highlights
-
•
Tools for monitoring endogenous translesion synthesis (TLS) factors
-
•
Detection of endogenous PCNAmUb and Y-family TLS pols in single mammalian cells
-
•
Monitoring chromatin-bound TLS factors during cell cycle phases
-
•
PCNAmUb and not TLS polη co-localizes with sites of replication forks stalled by CPDs
Motivation
Detection of chromatin-bound TLS factors in single cells, such as PCNAmUb and TLS pols, has typically depended on either western blotting or detection of ectopically expressed, tagged proteins. Western blotting has the limitation of only providing information on the average level of proteins in a cell population, while localization studies using ectopically expressed, tagged proteins may not faithfully represent the behavior of endogenous proteins. We help to address this problem by developing flow cytometry and immunofluorescence methods to detect endogenous, chromatin-bound PCNAmUb as well as Y-family TLS pols in single mammalian cells. We applied these methods to study DNA replication and TLS dynamics in HCT116 colon cancer cells exposed to UV-C irradiation and provide evidence for uncoupling of PCNAmUb from Polη-dependent TLS at replication forks stalled by UV-C lesions.
The DNA damage tolerance pathway involving translesion DNA synthesis (TLS) allows cells to proliferate in the presence of DNA lesions and is widely studied in the context of cancer. Detection of TLS factors has often relied on ectopically tagged proteins. Here, Egger and colleagues report methods for analyzing endogenous TLS in single cells by flow cytometry and immunofluorescence.
Introduction
Translesion synthesis (TLS) constitutes a branch of the cellular DNA damage tolerance pathway involving DNA lesions bypassed by specialized DNA polymerases, known as TLS pols. Thanks to a catalytic site that is more open than that of replicative DNA polymerases, these enzymes can accommodate damaged DNA bases and facilitate DNA replication under DNA damage conditions. However, TLS pols have lower fidelity than replicative DNA polymerases and are therefore mutagenic.1 By facilitating proliferation under DNA damage and increasing genetic diversity, TLS is exploited by cancer cells to adapt to therapy, thus escaping apoptosis, and has recently drawn much attention as a pathway to target so as to sensitize cancer cells to therapy.2,3 Y-family TLS pols (η, ι, κ, and Rev1) are implicated in rescuing replication forks arrested by DNA damage. Their recruitment onto DNA lesions primarily depends upon monoubiquitination of the replication-associated protein PCNA (PCNAmUb), catalyzed by the Rad6(E2)/Rad18(E3) ubiquitin ligase complex,4,5 and is dependent upon formation of excess single-stranded (ss)DNA produced by enzymatic uncoupling of replication forks stalled by DNA lesions.6,7 So far, it has been difficult to study endogenous PCNAmUb and TLS pols recruitment due to a lack of specific tools and detection methods in single cells. In particular, detection of endogenous nuclear PCNAmUb in single cells by immunostaining has been challenging mainly due to the lack of a specific antibody to detect PCNAmUb. Ectopically expressed PCNAmUb was previously detected in chicken DT40 cells by fluorescence resonance energy transfer (FRET), using a fluorescently tagged version of both ubiquitin and PCNA.8 Although PCNAmUb and TLS pols recruitment can be analyzed by western blotting in total or nuclear extracts, this rather crude method only provides an indication of the average level of recruitment in a large number of cells. Further, this method can be difficult to apply in cell lines that are sensitive to the extraction procedure. Furthermore, analysis of their recruitment in respect to the cell cycle involves synchronization procedures that can induce bias in the interpretation of the experiment. It has also been challenging to analyze recruitment of endogenous Y-family TLS pols onto damaged chromatin in single cells, presumably because of their low expression level. Current methods involve ectopic expression of epitope-tagged versions, such as fluorescent proteins, followed by detection of natural fluorescence in live or fixed cells.9,10,11,12,13 These methods can also induce bias since they involve TLS pols overexpression. In addition, the presence of the tag may affect the chromatin-binding affinity of the protein under study. Finally, upon transfection, the level of ectopically expressed TLS pols can be variable from cell to cell.
With this in mind, we sought to develop a procedure to visualize endogenous PCNAmUb as well as Y-family TLS pols in single cells by both flow cytometry and immunofluorescence. The quantitative cytometry-based method described here is simple: it allows monitoring of the dynamics of PCNAmUb and TLS pols, as well as that of Rad18 in individual cells in a quantitative fashion and requires fewer cells than in western blot. Using this method, TLS pols recruitment to chromatin can be analyzed in a cell population with great accuracy and in relation to the cell cycle phases. TLS factors binding to chromatin can be analyzed quantitatively and with respect to DNA lesions, DNA damage markers, and DNA synthesis. We have also applied this procedure to visualize both endogenous PCNAmUb and TLS pols bound to chromatin by immunofluorescence and show that PCNAmUb and TLS pols can be detected in single untreated cells or following exposure to DNA-damaging agents.
Results
Detection of endogenous PCNAmUb by flow cytometry
We modified a protocol from a previous method employed to detect chromatin-bound RPA by flow cytometry,14 to allow detection of poorly expressed, loosely chromatin-bound proteins, such as PCNAmUb and TLS pols. In this procedure, cells are briefly pre-extracted with detergent, before their rapid fixation, so as to freeze them in their natural position within the cell cycle. We optimized the detection of chromatin-bound PCNAmUb by trying various combinations of detergent-based pre-extraction and fixation steps (STAR Methods; Figures S1A–S1D). Using an antibody that specifically recognizes PCNAmUb in western blot (Figure S1E; Thakar et al.,15 Swain et al.,16 and Despras et al.17), we could detect chromatin-bound PCNAmUb by flow cytometry, which, in this assay, is scored as an increase of the fluorescence signal compared with the background signal of the control sample (no antibody; Figure 1A). PCNAmUb chromatin binding was observed in untreated HCT116 colon cancer cells, which further increased upon exposure to genotoxic doses of either UV-C irradiation or hydrogen peroxide (H2O2), but not to camptothecin (CPT) as expected (Figures 1A and S1F), because CPT generates mainly DNA double-strand breaks and limited ssDNA (Recolin et al.,18 for review). Consistent with this notion, upon exposure to UV light, the increase in PCNAmUb fluorescence correlated with an increase in RPA fluorescence (Figure S2A), supporting previous observations.14,19 In parallel, PCNAmUb chromatin binding was confirmed by analysis of cellular fractions by western blot (Figure S2B). As expected, Rad18 downregulation by small interfering RNA (siRNA) decreased PCNAmUb fluorescence in two different UV-irradiated cell lines, as expected (Figure S2C). A similar result was obtained upon expression of a PCNA mutant that cannot be monoubiquitinated (K164R; Figure S2D), demonstrating the specificity of the signal. Further, upon exposure to UV light, a specific increase in PCNAmUb fluorescence and not total PCNA, whose level remained unchanged, was observed (Figures S2E and S2F). By plotting the integrated PCNAmUb fluorescence intensity against that of DAPI (DNA content), PCNAmUb chromatin binding could be further scored in relation to cell cycle (Figure 1B). After UV irradiation, PCNAmUb increased in all cell cycle phases, while it was mainly restricted to G1 and G2/M phases upon exposure to H2O2 (Figure 1B). Analysis of PCNAmUb by both western blot and flow cytometry during a time course following exposure to UV radiation shows a tight correlation between the two methods (Figures 1C–1E). Western blot analysis shows an increase in the total level of PCNAmUb with time, reaching a maximum at 8.5 h after irradiation in this experiment (Figure 1C). A very similar increase was also seen by flow cytometry (Figure 1D), and the kinetics of the two detection methods very closely overlapped (Figure 1E). PCNAmUb could also be detected by flow cytometry in other cell lines (Figure S3A), although at different levels. The intensity of PCNAmUb fluorescence correlated with both the amount of cells in S phase (Figure S3B) and PCNAmUb abundance, as determined by western blot (Figure S3C). Altogether, these results show that chromatin recruitment of PCNAmUb can be reliably detected by flow cytometry and can be correlated with the cell cycle stages where it occurs.
Figure 1.

Detection of endogenous PCNAmUb recruitment to chromatin by flow cytometry
(A) Detection of endogenous PCNAmUb in HCT116 cells either untreated (UT, blue) or exposed to 20 J/m2 of UV-C light (UV, red), 1 mM hydrogen peroxide (H2O2, orange), or 1 μM camptothecin (CPT, green) by flow cytometry. A sample devoid of primary antibody (No Ab) was included as a control. Data were plotted as PCNAmUb fluorescence intensity versus the total cells count. A.U., arbitrary units. n = 3.
(B) In this panel, PCNAmUb fluorescence intensity was plotted against the DAPI fluorescence that counterstains the DNA (DNA content), thus giving the cell cycle profile. n = 3.
(C and D) Time course of PCNAmUb analyzed by either western blot (C) or flow cytometry (D) in HCT116 cells UT or exposed to 20 J/m2 UV. Samples were taken at the indicated times after UV irradiation (red arrow). The increase in the deepness of the red color indicates the increase in time. Data of (D) are plotted as in (A) and (B). No Ab was included as a control. n = 2.
(E) Quantification of PCNAmUb time courses of (C) and (D). The western blot signals of PCNAmUb were normalized to the total PCNA level (blue line). The geometric mean (G-Mean) of cells computed by flow cytometry is plotted in green. n = 2.
Detection of endogenous TLS pols by flow cytometry
We next applied the same protocol to detect chromatin recruitment of endogenous Y-family TLS pols by flow cytometry using specific antibodies validated in western blot (see STAR Methods and Figures S4A–S4C, S6C, and S6D). As can be seen in Figures 2A, 2B, and S5, we could detect chromatin binding of at least two TLS pols, Polη and Polι, as well as Rad18 (Figure 2C). Their association with chromatin was confirmed in parallel by western blotting (Figures S2B and S4C). Notwithstanding, differences in the fluorescence intensity were observed among TLS pols. In particular, upon UV irradiation, increased Polη and Polι fluorescence was clearly detectable, while this was much less evident for Polκ (Figure S5). Although these results are consistent with the notion that both Polη and Polι, and not Polκ, are involved in TLS of UV damage, at this stage, we cannot exclude that the observed differences are due to the relative abundance of TLS pols, the strength of the antibodies used, or both. Polη and Polι were found to be chromatin bound at all cell cycle stages, while Rad18 increased in a DNA replication-dependent manner (Figures 2C and S5). As for Rev1, we failed to detect a significant signal with currently available antibodies. At the same time, by flow cytometry, we could also clearly detect cyclobutane pyrimidine dimer (CPD) UV photoproducts, using a specific antibody (see STAR Methods), which were mainly distributed in the S phase upon UV irradiation (Figures 2D and S5). Importantly, we found that detection of different TLS factors by flow cytometry is strictly dependent upon the fixation method, which can be different for each protein target (Figure 2E). Altogether, these results show that at least two endogenous Y-family TLS pols can be detected by flow cytometry, as well as Rad18 and CPDs, and that their binding to chromatin can be observed in relation to the cell cycle in single cells without the use of synchronization methods.
Figure 2.

Detection of endogenous Rad18 and TLS pols chromatin recruitment by flow cytometry
(A–C) Detection of endogenous TLS pols (A and B) or Rad18 (C) in HCT116 cells either untreated (UT, blue) or exposed to 20 J/m2 of UV-C light (UV, red) by flow cytometry. A sample devoid of primary antibody (No Ab) was included as a control. Data are plotted as in Figures 1A and 1B.
(D) Detection of UV-induced cyclobutane pyrimidine dimers (CPDs) by flow cytometry. No Ab was included as a control. Data are plotted as in Figures 1A and 1B.
(E) Table describing the different fixation methods to detect chromatin-bound proteins related to the TLS pathway in human cells. n = 3. For more details see Egger et al.20
Detection of nuclear PCNAmUb by immunofluorescence in single cells
Using the same extraction and fixation procedure, we attempted to detect PCNAmUb by indirect immunofluorescence in mammalian cells. We observed clear PCNAmUb nuclear foci in untreated HCT116 cells that increased following exposure to UV radiation and whose extent was strongly reduced either upon Rad18 downregulation by siRNA (Figures 3A and 3B; STAR Methods) or upon expression of the pcnaK164R mutant that cannot be monoubiquitinated (Figure S6A). The PCNAmUb background level persisting in cells treated by siRad18 could be either due to incomplete Rad18 depletion or to the activity of the CRL4Cdt2 (E3) ligase.21 PCNAmUb signal was detected as discrete nuclear foci, co-localizing with total PCNA even in untreated cells (Figure 3C), which likely represent PCNAmUb induced by endogenous replication stress, consistent with the flow cytometry data shown in Figure 1. PCNAmUb foci were co-localized with total PCNA (Figure 3C) and the ssDNA binding protein RPA (Figure 3D), in line with the notion that ssDNA is essential for PCNAmUb, although not all RPA foci co-localized with PCNAmUb, and vice versa. As expected, the intensity of both the PCNAmUb and RPA fluorescence increased upon UV irradiation (Figure 3E). We also observed PCNAmUb foci in cells treated with different DNA-damaging agents such as cisplatin and H2O2, but to a much lesser extent with CPT (Figures 4A and 4B), consistent with flow cytometry data (Figures 1A, 1B, and S1). Altogether, these results show that PCNAmUb can be detected in single HCT116 cells by indirect immunofluorescence, enabling its observation at the sub-nuclear level and its co-localization with diverse factors implicated in DNA metabolism.
Figure 3.

Detection of nuclear PCNAmUb in single cells by immunofluorescence
(A) HCT116 cells untreated (UT), or exposed to 20 J/m2 of UV-C (UV, red), treated with either siRNA control (siCtrl) or an siRNA targeting Rad18 (siRad18). Cells were stained with the PCNAmUb antibody and visualized by indirect immunofluorescence. DNA was counterstained with DAPI. Insets: magnification of single cells (indicated by a white arrow). Right: quantification of PCNAmUb foci with CellProfiler software (see STAR Methods). A.U., arbitrary units. Stars indicate significant differences, ∗∗∗p < 0.001 (non-parametric Mann Whitney test). n = 3.
(B) Western blot of HCT116 cells of the experiment shown in (A), treated with the indicated siRNA, exposed (+UV) or not (−UV) to 20 J/m2 of UV-C. Proteins were detected with the indicated antibodies. The anti-PCNA antibody detects both unmodified and PCNAmUb, n = 3.
(C) Top: HCT116 cells UT or exposed to UV-C (UV) were co-stained with both PCNAmUb and total PCNA and viewed by indirect immunofluorescence. DNA was visualized with DAPI. Middle: magnification of a nucleus from a single cell of each panel (indicated by a white arrow). Cross-sections were drawn with Zen Blue software to quantify the co-localized relative intensities of PCNAmUb (green) and PCNA (red) fluorescence. Bottom: quantification of the relative intensity of the cross-section for both PCNA and PCNAmUb labeling of each nucleus of the middle panel. n = 2. Scale bar: 10 μm.
(D) HCT116 cells UT or exposed to UV-C (UV) were co-stained with both PCNAmUb and RPA2 antibodies and viewed by indirect immunofluorescence. DNA was visualized with DAPI. Far top right: quantification of the relative intensity of the cross-section here below for both PCNA and RPA2 labeling of the nucleus indicated by a white arrow. The cross-section was drawn with ImageJ software to visualize co-localization of PCNAmUb (green) with RPA2 (red) fluorescence. n = 2. Scale bar: 10 μm.
(E) Quantification of either PCNAmUb (left) or RPA2 (right) of experiment shown in (D). Stars indicate significant differences, ∗∗∗p < 0.001. ns, non-significant (non-parametric Mann Whitney test).
Figure 4.

PCNAmUb detection by immunofluorescence in HCT116 cells exposed to different DNA-damaging agents
(A) Left: wide-field images of HCT116 cells untreated (UT) or exposed to either 30 μM cisplatin (CisPt) or 1 μM camptothecin (CPT) stained with the anti-PCNAmUb antibody and counterstained with DAPI to visualize nuclei. Insets: magnification of individual nuclei. Right: quantification of PCNAmUb immunofluorescence mean intensity of nuclei assessed with CellProfiler. Scale bar: 20 μm. n = 2.
(B) Left: wide-field images of HCT116 cells UT or exposed to hydrogen peroxide (H2O2), stained with the anti-PCNAmUb antibody and counterstained with DAPI to visualize nuclei. Right: quantification of PCNAmUb of the left panel. Scale bar: 20 μm. n = 2.
Detection of chromatin-bound TLS Polη and Polι by immunofluorescence in single cells
As done for PCNAmUb, we next attempted to detect Y-family TLS pols in single cells by indirect immunofluorescence. We were able to detect endogenous Polη (Figures 5A and 5B) and Polι (Figure 5C) in nuclei of HCT116 cells. Polη was not detected in HCT116 cells upon downregulation by siRNA, nor in XP30RO fibroblasts harboring a homozygous mutation in the Polη gene (Figures S6C and S6D).22 In contrast, Polη was detectable in XP30RO cells complemented with either wild-type Polη or GFP-Polη.13 In the latter, most of the Polη foci detected by the Polη antibody also co-localized with the GFP fluorescence, although a fraction of them did not. Equally, the Polι signal was strongly reduced upon inhibition of its expression by siRNA (Figure 5C), demonstrating the specificity of the signal. As a comparison, we also transfected HCT116 cells with EGFP-Polη. As can be seen in Figure 5B, detection of endogenous Polη by immunofluorescence gives a better and more comprehensive landscape of Polη distribution in isolated cells compared with ectopic transfection, which provides poor information and is limited to the fraction of cells that were successfully transfected. Detailed analysis of the fluorescence signals generated by the Polη antibody shows that endogenous Polη forms discrete nuclear foci in both untreated and UV-irradiated cells (Figure 5B), supporting two previous observations.23,24 This is different from what has been observed in cells transfected with GFP-tagged Polη in which only a small fraction of the cells form nuclear foci in unperturbed conditions, while in the remaining population, the protein remains uniformly distributed in the cell.13,25 Quantification shows that upon UV irradiation, or exposure to H2O2, the fluorescence intensity markedly increased, suggesting recruitment to damaged chromatin (Figure 5A). In conclusion, these results show that both endogenous TLS Polη and Polι can be detected by immunofluorescence in isolated mammalian cells.
Figure 5.

Detection of nuclear Polη in HCT116 cells treated with different DNA-damaging agents
(A) Left: detection of chromatin-bound Polη in HCT116 cells untreated (UT) or exposed to either 20 J/m2 of UV-C light (UV), or 1 mM hydrogen peroxide (H2O2). Scale bar: 20 μm. Insets: magnification of the nuclei indicated by a white arrow. Right: quantification of Polη mean intensity in the indicated samples. A.U., arbitrary units. n = 3. Stars indicate significant differences. ∗∗∗p < 0.001 (non-parametric Mann Whitney test). UV, n = 3; H2O2, n = 2.
(B) Left detection of either endogenous or ectopically expressed EGFP-Polη chromatin bound in HCT116 cells exposed to 20 J/m2 of UV-C. Right: quantification of Polη foci intensity in the indicated samples. The percentage of Polη+ cells (blue gate) is indicated. Scale bar: 20 μm. Stars indicate significant differences. ∗∗p < 0.001 (non-parametric Mann Whitney test). n = 3.
(C) Left: detection of Polι by indirect immunofluorescence in HCT116 cells treated with either control siRNA (Ctrl) or Polι-specific siRNA, UT or exposed to 20 J/m2 UV. Right: quantification of Polι foci per nucleus shown in (A). ∗∗∗p < 0.001 (non-parametric Mann Whitney test). n = 2.
Getting insights into TLS activation by UV damage during ongoing DNA synthesis
As an application of this procedure to study TLS dynamics in proliferating cells, we wished to analyze the localization of PCNAmUb and Polη with respect to DNA lesions induced by UV irradiation (CPDs) and to sites of DNA synthesis. For this purpose, we exposed cells to UV light to generate DNA lesions, followed by a short pulse with the nucleotide analog EdU to label ongoing replication forks (Figure 6A). Cells were sampled post-UV irradiation and triple stained with antibodies for either PCNAmUb (red, Figure 6) or Polη (red, Figure 7), EdU (green), and CPDs (blue). Figure 6B shows that in untreated (UT) cells, nuclear PCNAmUb foci were visible in both EdU-negative cells, representing cells in either G1 or G2/M cell cycle phases, and in EdU-positive cells (green, S-phase cells), consistent with results obtained by flow cytometry (Figures 1A and 1B). Co-localization of PCNAmUb and EdU was already visible immediately after the EdU pulse (Figures 6C and 6E, t = 0) and increased shortly after (t = 0.5 h). White spots (merge of the three colors, see color table in Figure 6A) could also be clearly visible at this time point, showing co-localization of PCNAmUb and EdU at sites of DNA lesions (CPDs; Figure 6E, right). With time, the intensity of the CPD and PCNAmUb fluorescence increased, giving rise to a magenta color (merge of red and blue). Of note, CellProfiler quantification shows that the number of CPDs foci per nucleus decreased with time, while their intensity increased, suggesting clustering of CPD lesions (Figure S6E). Because the CPD signal intensity stalled at 2.5 h and only slightly decreased at 5 h, this suggested that CPD clustering might represent sites of active DNA repair (e.g., nucleotide excision repair, NER). In support of this possibility, proteins involved in NER have been previously observed forming discrete nuclear foci in mammalian cells.26 Further, these kinetics are consistent with a previous study showing that CPDs are still relatively abundant 5 h after irradiation.27 At later time points (2.5–5 h), the magenta color was predominant (merge of red and blue), indicating that PCNAmUb was mostly located onto CPDs, moving away from EdU incorporation sites. These observations suggest that following UV irradiation, PCNAmUb transiently co-localizes with sites of DNA synthesis stalled by UV-induced DNA lesions. CellProfiler quantification (Figure 6D) shows that PCNAmUb occurred first in EdU-positive cells (EdU+; 0.5 to 1 h time point) while at later time points (2.5–5 h) EdU-negative cells (EdU−) also started to show increased PCNAmUb. These latter events may correspond to G1 cells entering into S phase in the presence of UV-induced DNA lesions since PCNAmUb increased during replication of both untreated and UV-irradiated cells (Figure 1B).
Figure 6.

Nuclear PCNAmUb co-localization with UV-C-induced DNA lesions and sites of DNA synthesis in single cells
(A) Schematic drawing of the experimental procedure. A color table is included to facilitate the interpretation of the results.
(B) First row: wide-field images of HCT116 cells untreated (UT −UV) or exposed to UV-C (+UV), followed by pulse labeling with the nucleotide analog EdU. Antibodies were used to detect PCNAmUb (red) and CPDs (blue), and EdU (green) was detected by click reaction (see STAR Methods) at the indicated times after UV-C exposure and viewed by indirect immunofluorescence. DNA was visualized with DAPI (gray). Other rows: magnification of single-cell nuclei from each wide field corresponding to each time point. Scale bar: 10 μm.
(C) Quantification of the relative intensity of the cross sections of the nuclei magnified in the top panel.
(D) Quantification of relative EdU and PCNAmUb levels at the indicated time points post-UV irradiation. The black dashed line discriminates EdU− from EdU+ cells (i.e., cells that were in S phase during the EdU pulse). The blue dashed line discriminates PCNAmUb-negative from PCNAmUb-positive cells (arbitrary gates). n = 2.
(E) Quantification of PCNAmUb-EdU (left), PCNAmUb-CPD (middle, and PCNAmUb-CPD-EdU (right) co-localization. Stars indicate significant differences, ∗p < 0.05, ∗∗ p < 0.01, ∗∗∗p < 0.001, ns, non-significant (non-parametric Mann Whitney test).
Figure 7.

Nuclear Polη co-localization with UV-C-induced DNA lesions and sites of DNA synthesis by immunofluorescence in single cells
(A) Schematic drawing of the experimental procedure. A color table is included to facilitate the interpretation of the results.
(B) First row: wide-field images of HCT116 cells untreated (UT −UV) or exposed to UV-C light (+UV), followed by pulse labeling with the nucleotide analog EdU. Antibodies were used to detect Polη (red) and CPDs (blue), and EdU (green) was detected by click reaction at the indicated times after UV-C exposure and viewed by indirect immunofluorescence. DNA was visualized with DAPI (gray). Other rows: magnification of nuclei of single cells from each wide field corresponding to each time point. Scale bar: 10 μm.
(C) Quantification of the relative intensity of the cross-sections of the nuclei magnified in the top panel.
(D) Quantification of Polη-EdU (left), Polη-CPD (middle), and Polη-CPD-EdU (right) co-localization. Stars indicate significant differences, ∗p < 0.05; ∗∗∗p < 0.001; ns, non-significant (non-parametric Mann Whitney test).
As for Polη, the picture was surprisingly different (Figure 7). Consistent with flow cytometry data (Figure 2), Polη foci could be observed in untreated cells that were not co-localizing with sites of ongoing DNA synthesis (Figures 7B–7D, EdU, green) but were close to them (white arrows). At early time points post-UV irradiation (t = 0.5 h), and in contrast to PCNAmUb foci, Polη foci were still observed close to EdU foci but not completely overlapping. At later time points (1–5 h), Polη foci were close but clearly separated from EdU foci, with only rare foci showing co-localization. Notably, 1 h post-UV irradiation, Polη foci were often co-localizing (magenta color) or in close proximity to the CPD foci, while at later time points, Polη foci were clearly separated from CPDs. Unlike PCNAmUb, the intensity of the Polη foci increased immediately following UV irradiation, but it did not increase further with time, as determined by CellProfiler quantification (Figure S6F), while the CPD signal followed a trend similar to that observed in Figures 6 and S6E. Taken together, these observations suggest that upon UV irradiation, formation of PCNAmUb and Polη foci is spatially distinct, suggesting a two-step process for TLS, such as activation at stalled forks (PCNAmUb) and slow bypass by Polη (see discussion). Altogether, these results show that this protocol allows studying dynamics of PCNAmUb and Polη foci in single cells, in relation to DNA lesions and DNA synthesis sites.
Discussion
Failure to detect both endogenous PCNAmUb and Y-family TLS pols in single cells has been a major hurdle to study TLS. This has been mainly due to lack of a specific antibody able to detect PCNAmUb and probably to the low expression level of TLS pols. We have succeeded in detecting endogenous PCNAmUb and TLS pols in single cells by both flow cytometry and immunofluorescence, suggesting that failure to detect them was not only a problem of abundance but also a matter of developing an optimized detection protocol. This simple and fast method now makes it possible to analyze endogenous TLS in single cells without the use of synchronization procedures that would introduce bias in the analysis of the results.
Results obtained in this article show that, upon exposure of HCT116 cells to DNA-damaging agents, recruitment of both PCNAmUb and TLS pols onto chromatin occurs in all cell cycle phases, although mainly in S phase and with some important differences depending on the type of DNA damage. In cells exposed to UV radiation, PCNAmUb chromatin binding was detected in all cell cycle stages, with a slight increase in G1 and G2/M phases. A similar pattern was observed for at least two Y-family TLS pols (η and ι). These observations are consistent with the notion that UV-induced DNA lesions stall replication forks and that TLS can also occurs outside S phase.25,28,29,30,31,32 When cells were treated with H2O2, the pattern of PCNAmUb fluorescence observed by flow cytometry was rather different, being more restricted to the G1 and G2/M phases. This result can be explained by the observation that 8-oxodG, the main DNA lesion generated by H2O2, does not interfere much with replicative polymerases33 and therefore limits the extent of PCNAmUb in S phase. These lesions are actively repaired by a base excision repair-based process, leading to formation of a gapped ssDNA intermediate that stimulates PCNAmUb. Notwithstanding, different observations were reported about the cell cycle phase when the gap filling process occurs,28,29 which could be explained by the use of a different cell line and different methods of cell synchronization. The non-invasive and quantitative method presented here provides a clear picture, showing that in cells treated with H2O2, PCNAmUb occurs mainly in G1, in line with an early report,31 but can also occur in S phase.
Observation of PCNAmUb by immunofluorescence shows that it forms discrete nuclear foci in untreated cells, as well as upon exposure to DNA-damaging agents. As expected, these foci were co-localized with PCNA, and to some extent with RPA, although they did not always overlap, while some other foci were clearly distinct from each other, suggesting ssDNA-dependent and -independent recruitment. The method described in this article also allowed detection of endogenous TLS Polη and Polι by immunofluorescence. Interestingly, in unperturbed cells, Polη formed discrete nuclear foci in virtually all cells, which is different from observations made using cells transfected with epitope-tagged Polη. The distribution of endogenous Polη as nuclear foci in all cells is consistent with several observations showing that Polη participates in DNA synthesis to assist the canonical replisome when encountering difficult-to-replicate DNA regions, such as repetitive DNA and common fragile sites.17,34,35,36 Hence, it cannot be excluded that the observed differences may be due to ectopic overexpression of epitope-tagged Polη. Thus, detection of endogenous Polη with this protocol provides a more comprehensive picture compared with cells transfected with tagged versions of it. Importantly, Polη foci were found not co-localizing with sites of DNA synthesis (EdU) but were often in close proximity to them. This result is again different from previous observations using ectopically expressed GFP-Polη, in which co-localization was observed with sites of BrdU incorporation, although in as few as 15% of the cells,13 which could be explained as forced recruitment of GFP-Polη upon overexpression.
A two-step process for TLS?
Current models in yeast suggest that following PCNAmUb, TLS pols are immediately recruited to bypass the lesion and facilitate passage of the replication fork (TLS on the fly37), a situation that may be different in vertebrates. By investigating the dynamics of both PCNAmUb and Polη by immunofluorescence in single HCT116 cells, during a time course of UV irradiation, we have observed that PCNAmUb co-localizes with UV-induced DNA lesions at sites of DNA synthesis (CPDs), while Polη did not, but was clearly located very close to EdU-positive sites. These results may suggest that in HCT116 cells, the signal required for TLS activation is generated at stalled replication forks, while Polη recruitment is a later event. A possible explanation of these observations is that the two processes, PCNAmUb and Polη recruitment, are spatially distinct, similar to a recent observation reported in the yeast S. cerevisiae38 and previous reports in vertebrate cells.39,40,41 At late time points post-UV irradiation (5 h), Polη foci were close to EdU foci but clearly excluded from them, suggesting Polη recruitment at post-replicative gaps left behind the forks. Meanwhile, we cannot exclude that the Polη foci we observed correspond to TLS on the lagging strand and that TLS on the leading strand (on the fly) occurs too quick to be detected in fixed cells. Alternatively, on the leading strand, resumption of DNA synthesis downstream of a DNA lesion is assured by repriming by Primpol,42 leaving gaps filled in post-replication in a Polη-dependent process. As a caveat, it cannot be excluded that the difference between PCNAmUb and Polη localization may be due to the ability of Polη to bypass only CPDs, one of the two main lesions generated by UV-C irradiation. More detailed analysis of TLS dynamics in other cell lines and using super-resolution microscopy would be important to clarify this point. Isolated PCNAmUb foci observed in untreated cells might represent sites of endogenous replication stress where replication forks stall frequently and therefore incorporate very few EdU. We have also been able to observe formation of discrete CPD foci in HCT116 cells, whose size increased with time following irradiation with UV-C light, suggesting clustering. These may represent sites of DNA repair in which NER factors may nucleate, thus opening the possibility to study NER factors recruitment to CPDs in single cells.
In conclusion, the procedures reported in this work now open a new avenue for the analysis of endogenous TLS pols, as well as of PCNAmUb in virtually all cell types by either flow cytometry or immunofluorescence microscopy. This procedure might now also allow to study endogenous TLS activation in the context of somatic immunoglobulin gene hypermutation and maintenance of hematopoietic stem cells (Sale,43 for review). In principle, the procedures described here will now make it possible to use TLS pols as well as PCNAmUb as predictive markers for cancer resistance to therapeutic treatments (such as in BRCA-mutated and colon cancer3,44 among others). Along this line, the use of PCNAmUb and/or TLS pol staining could be useful to set up screening strategies for the identification of chemical inhibitors, which could spin the development of TLS inhibitors. In this context, it is reasonable to expect the discovery of new synthetic lethal interactions that may be implemented to the current tool belt of chemo- or immunotherapeutic regimens used in cancer therapy.
Limitations of the study
The methods described in this article can, in principle, be applied to detect PCNAmUb and TLS pols in any cell. A limitation in detecting these factors is their relative abundance, which depends upon the cell line, the proportion of S-phase cells, and the degree of endogenous replication stress. Another limitation is the availability of a suitable antibody to detect TLS pols, whose specificity must be first tested in cells depleted of the target. Insertion of a tag into the endogenous gene of interest by current CRISPR-Cas9 technology may help to circumvent this problem, although the possibility that the tag could change the affinity of the protein for chromatin has to be taken into account. We believe that this method will be very useful to study TLS dynamics at DNA replication forks stalled by diverse bulky DNA lesions and can be extended to TLS pols not included in our study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ubiquityl-PCNA (Lys164) | Cell Signaling Technology | RRID:AB_2798219 |
| PCNA | Millipore | RRID:AB_11203836 |
| α-tubulin | Sigma-Aldrich | RRID:AB_477583 |
| Rad18 | Abcam | RRID:AB_1603946 |
| CHK1 | Santa Cruz Biotechnology | RRID:AB_627257 |
| CHK1pS345 | Cell Signaling Technology | RRID:AB_331212 |
| γH2AX | Cell Signaling Technology | RRID:AB_2118009 |
| H2AX | Cell Signaling Technology | RRID:AB_10694556 |
| Polη | Abcam | RRID:AB_2756352 |
| Polκ | Atlas Antibodies | RRID:AB_2668606 |
| Polι | Proteintech | RRID:AB_2167009 |
| RPA | Abcam | RRID:AB_302873 |
| CPDs | Cosmo Bio | RRID:AB_1962813 |
| USP1 | Proteintech | RRID:AB_2214314 |
| Histone H2B | Abcam | RRID:AB_302612 |
| Goat Anti-Rabbit Alexa 488 | Thermo Fisher Scientific | RRID:AB_2576217 |
| Goat Anti-Rabbit Alexa 568 | Thermo Fisher Scientific | RRID:AB_10563566 |
| Goat anti-Mouse Alexa 660 | Thermo Fisher Scientific | RRID:AB_2535722 |
| Goat anti-Mouse Alexa 660 | Thermo Fisher Scientific | RRID:AB_2535722 |
| Goat anti-Rabbit HRP | Cell Signaling Technology | RRID:AB_2099233 |
| Goat anti-Mouse HRP | Cell Signaling Technology | RRID:AB_330924 |
| Chemicals, peptides and recombinant proteins | ||
| Camptothecin | Sigma-Aldrich | C9911 |
| Cisplatin | Sigma-Aldrich | P4394 |
| DAPI | Sigma-Aldrich | D9542 |
| Formaldehyde | Thermo Fisher | 28908 |
| Hydrogen Peroxyde | Sigma-Aldrich | 216763 |
| Sodium fluoride | Sigma-Aldrich | 201154 |
| β-Glycerophosphate | Sigma-Aldrich | G9422 |
| Halt Proteases and Phosphatases inhibitors | Thermo Fisher Scientific | 78440 |
| Perm/Wash buffer | BD biosciences | 554723 |
| Prolong Diamond AntiFade | Thermo Fisher | P36961 |
| RNAse A | EMD Millipore | 70856 |
| Benzonase | Sigma-Aldrich | E1014-5KU |
| BSA | Sigma-Aldrich | A2153 |
| Critical commercial assays | ||
| BCA Protein Assay Kit | Thermo Fisher Scientific | 23225 |
| Click-iT EdU 488 | Thermo Fisher Scientific | C10337 |
| ECL Crescendo | Millipore | WBLUR0500 |
| Experimental models: Cell lines | ||
| HCT166 | ATCC | CCL-247 |
| NCI-H1299 | ATCC | CRL-5803 |
| HEK293T | ATCC | CRL-3216 |
| T98G | ATCC | CRL-1690 |
| XP30RO | Masutani et al.22 | N/A |
| XP30RO:Polη | Kannouche et al.13 | N/A |
| XP30RO:GFP-Polη | Kannouche et al. 13 | N/A |
| MEFs wild-type | Langerak et al.45 | N/A |
| MEFs pcnaK164R | Langerak al.45 | N/A |
| Oligonucleotides | ||
| Control siRNA (Luciferase) | 5′-CACGUACGCGG AAUACUUCGATT-3′ |
N/A |
| Polη siRNA | Durando et al.46 | N/A |
| Polι siRNA | Somyajit et al.23 | N/A |
| Polκ siRNA | Bétous et al.47 | N/A |
| Rad18 siRNA | Kermi et al.48 | N/A |
| Recombinant DNA | ||
| pEGFP:Polη | Kannouche et al.13 | N/A |
| pCDNA3-HA-FLAGpcnaK164R | This paper | N/A |
| Software and algorithms | ||
| Cell Profiler 4.2.1 | Stirling et al.49 | N/A |
| Kaluza | Beckman Coulter | N/A |
| Zen Blue | Zeiss | N/A |
| Prism | GraphPad | N/A |
| Other | ||
| 4–15% Criterion™ TGX™ Precast Midi Protein Gel, | BioRad | 5671085 |
| Tris/Glycin Buffer | BioRad | 1610771EDU |
| Trans-Blot Turbo Transfer System | BioRad | 1704150 |
| ChemiDoc | Biorad | N/A |
| Poly-D-Lysine | Gibco | A38904-01 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Domenico Maiorano (domenico.maiorano@igh.cnrs.fr).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact upon reasonable request with a completed Materials Transfer Agreement.
Experimental model and subject details
Cell culture
Cells were cultured in DMEM-GlutaMAX (ThermoFisher #10566-016) supplemented with 10% FBS (Eurobio, #CVFSVF00-01) at 37°C, 5% CO2. Cells were maintained in exponential growth phase and passaged 2–3 times a week for 20–25 passages before being discarded. Cells were tested negative for mycoplasma at thawing. Before any drug treatments, cells were seeded at an equivalent density and allowed to attach overnight. For the UV irradiation, cells were washed in PBS and exposed to 20 J/m2 of UV-C light using a Stratalinker (Stratagene) and released in fresh medium for 5 h (unless stated otherwise) before being processed for western blot, immunofluorescence or flow cytometry, as described below. For cisplatin and camptothecin treatments, cells were exposed respectively to either 30 μM or 1 μM for 5 h. For the hydrogen peroxide treatment, cells were exposed to 1 mM for 30 min. Transfection of HEK293T cells with pCDNA3-HA-flag-pcnaK164R was performed using Lipofectamine™ 2000 (Invitrogen, 11668027) according to manufacturer instructions. For Polɩ and Polƞ silencing by siRNA, HCT116 cells were transfected with Lipofectamine RNAi MAX (Thermofisher Scientific, 13778100).
Method details
Samples preparation for flow cytometry
1 × 106 cells were seeded in 6-well plates. After drug treatments, cells were trypsinized, washed with ice-cold PBS and pelleted at 400 x g for 5 min. The extraction, fixation and immunodetection of targets were performed with optimized revisions of a previous protocol.14 The extraction was performed for 5–10 min on ice in 100 μL of a PBS-0,2 to 0.5% Triton X-100 buffer (depending on the target, see Figure 2E and Egger et al.20), and stopped by addition of 500 μL of PBS containing 1 mg/mL BSA. Nuclei were pelleted at 500 x g for 5 min and fixed by gentle resuspension in PBS containing either 2% formaldehyde for 30 min at room temperature or in pre-chilled PBS containing 90% methanol, for 10 min at −20°C, depending on the target proteins to be detected (see Figure 2E). The fixation was stopped with 1 mL 1 x Perm/Wash buffer (BD Biocience). Nuclei were pelleted at 750 x g for 5 min and washed in 1 x Perm/Wash buffer once. Target proteins were detected using the indicated antibodies diluted at 1/100 in 50 μL of 1 x Perm/Wash buffer overnight at 4°C in gentle rotation motion. Nuclei were washed by adding 500 μL of 1 x Perm/Wash buffer and pelleted at 750 x g for 5min. Nuclei were then incubated 1 h at RT with the indicated secondary antibodies, diluted at 1/250 in 1 x Perm/Wash buffer before being washed by the addition of 500 μL of 1 x Perm/Wash buffer. Nuclei were pelleted at 750 x g for 5 min and dissolved in 300 μL of PBS, 1 mg/mL BSA, 1 μg/mL DAPI, 100 μg/mL RNase A. Preparations were incubated at 37°C for 30 min before being analyzed on a Gallios Flow Cytometer (Beckman Coulter). Forward Scatter/Side Scatter-based debris exclusion was set up, and the doublets were excluded using the DAPI Height/DAPI Area graphs. 20,000 cells were analyzed per sample on the Kaluza dedicated software.
Western blotting
Cells were cultured in 6-well plates. Typically, 0,5-1 X 106 cells were seeded per well. At 70–80% confluence, cells were harvested by trypsinization, rinsed in ice-cold 1 x PBS. For whole cell extracts (WCE), proteins were extracted in 100μL of lysis buffer (10 mM HEPES-KOH pH 7.5, 200 mM NaCl, 0.5% NP-40, 2 mM MgCl2, 1 mM DTT, 1 mM PMSF) containing 1 mM sodium fluoride and 1 mM β-Glycerophosphate, completed with 1/1000 Benzonase nuclease. Cells were lysed in lysis buffer for 15 min at room temperature (with agitation, 1000 rpm). Debris were pelleted at 13,000 x g, 20 min, 4°C. Proteins were quantified using the BCA Protein Assay Kit.
For cell fractionation (chromatin/soluble), cells were lysed in 100 μL of PBS-0.2% Triton X-100 containing Halt Proteases and Phosphatases inhibitors for 10 min on ice. The chromatin fractions were then pelleted (3200 rpm, 3 min), while the supernatants were isolated (soluble fractions) and quantified with the BCA Protein Assay Kit. Laemmli buffer was added to the soluble fraction to a final concentration of 1 x. The chromatin pellets were then washed in the same lysis buffer for 10 min on ice before being pelleted again (3200 rpm, 3 min). Supernatants were discarded and chromatin pellets were dissolved in 100 μL of lysis buffer containing 1 x Laemmli buffer.
In both cases (WCE and cell fractionation), equivalent amounts of proteins were loaded in 4–15% Criterion™ TGX™ Precast Midi Protein Gel, 26 well. Gels were run at 150 V in 1 x Tris/Glycin Buffer. Proteins were transferred onto 0.2 μm nitrocellulose membranes (BioRad, #1704271) using the Trans-Blot Turbo Transfer System set on the mixed molecular weight program. Total proteins were stained with Ponceau S and membranes were saturated in TBS-0.1% Tween 20, 5% non-fat dry milk for 1 h at room temperature. Target proteins were immunodetected overnight at 4°C in TBS-0.1% Tween 20, 5% BSA. Membranes were rinsed 3 × 10 min in TBS-0.1% Tween 20 and incubated for 1 h at room temperature in a TBS-0.1% Tween 20, 5% non-fat dry milk containing the HRP-conjugated secondary antibodies diluted at 1/3000. Membranes were rinsed 3 × 10min in TBS-0.1% Tween 20 and revealed using ECL Crescendo using a ChemiDoc device.
Immunofluorescence
Cells were grown on Poly-D-Lysine-coated 14 mm glass coverslips in 6-well plates. Cell were rinsed in PBS and the cytoplasm was extracted with PBS-0.2% Triton X-100 solution on ice for 3 to 10 min and Egger et al.20. Addition of 200 mM sucrose to the extraction buffer improves TLS pols detection. Nuclei were then immediately fixed in either 2% formaldehyde (room temperature for 30min) or by adding 90% methanol in PBS dropwise (−20°C, 10 min), depending on the target proteins to be detected (See Figure 2E). Nuclei were rinsed in PBS containing 1 mg/mL BSA. Nuclei were then saturated for 1 h in 1 x Perm/Wash buffer. Target proteins were detected using the indicated antibodies diluted at 1/100 in 1 x Perm/Wash buffer overnight at 4°C in a humid chamber. Coverslips were rinsed 3 × 5 min in 1 x Perm/Wash buffer and incubated 1h at room temperature with secondary antibodies (Alexa Fluor), diluted at 1/250 in 1 x Perm/Wash buffer. Coverslips were rinsed 3 × 5min in 1 x Perm/Wash buffer and nuclei were counterstained with DAPI (1 μg/mL) for 5 min at room temperature. Coverslips were mounted in Prolong Diamond AntiFade and observed on a Zeiss Axio Imager with a X63 objective and the Apotome engaged. Pictures were saved and processed using the Zen Blue dedicated software, with the same exposure times. Equivalent display settings were used in the related panels. The cross sections (line scan intensity profile) were performed using ImageJ.
Fluorescent labeling of ongoing DNA synthesis
EdU detection by Click reaction (Click-iT EdU 488 Thermo Fisher Scientific, C10337) was performed as recommended by the manufacturer.
Quantification and statistical analysis
The apotome images were converted (Aptome RAW convert, Zen Blue) and imported into Cell Profiler 4.2.1. The Metadata of each fluorescent channel were extracted and nuclei were identified (Identify Primary Objects: Blue channel, Size 70–200 pixels, border events exclusion = yes, Threshold strategy: Global, Thresholding method: Manual, Threshold: 0.01, Threshold smoothing scale: 1.2, distinguish clumped objects: Shape, draw dividing lines: propagate). The mean intensity in the green and red targets were computed for each nucleus (previously detected) and presented using GraphPad Prism 5. For foci counting, complementary Identify Primary Objects steps were added for each target to be quantified. (Identify Primary Objects: Green/Red channels, Size 2–20 pixels, border events exclusion = yes, Threshold strategy: Global, Thresholding method: Otsu). Foci were related to their respective parent object (nuclei) and the number of foci in each nucleus was plotted using GraphPad Prism 5. The colocalization pipeline used to generate the graphs (Figures 6E and 7D) is available upon request. Briefly, it calculates the percentage of colocalizing foci per nucleus. Metadata (i.e. the conditions) were extracted based on the name of the Apotome RAW converted files (.czi, Zeiss, Zen Blue 2.3). Channels (Hoescht C = 0, Alexa Fluor 488 C = 1, AlexFluor 568 C = 2, Alexa Fluor 647 C = 3) were identified in the NamesAndTypes section. Automated nuclei detection and segmentation was performed using the IdentifyPrimaryObject function with the Otsu (Two classes) thresholding method. Nuclear foci of each target proteins were detected in a similar way, after an enhancement step (EnhanceOrSupressFeatures, speckles), using the manual thresholding method, before finally being related to their parent nucleus (RelatedObject function). Colocalizing objects were identified as the overlapping parts of different colors foci and related to their parent nuclei using the same strategy. The percentage of colocalizing foci per nucleus was then calculated as the ratio of the number of colocalized foci versus the total foci count of a defined color in individual nuclei, i.e:
, per nucleus
Quality controls of nuclei segmentation and foci identification were systematically performed using the GrayToColor and OverlayOutlines functions, reconstructing all analyzed images and outlying idenfitied nuclei and foci of each color.
Statistical analysis was performed on GraphPad Prism 5. For the Cell Profiler analyses, non-parametric Mann-Whitney tests were performed: ∗: p < 0.05; ∗∗: p < 0.01; ∗∗∗: p < 0.001; ns, non-significant. “n” indicated in each figure legends refers to the number of times the experiment was replicated and data shown are representative of the “n” experiments performed with consistent and reproducible results.
Acknowledgments
We thank members of Maiorano’s team for technical help and useful discussions, P. Kannouche for the XP30RO cell lines and the EGFP-Polη vector, H. Jacobs for the MEFs pcnaK164R, and J.-S. Hoffmann and A. Cordonnier for critical reading of the manuscript. We acknowledge the imaging facility MRI-IGH, member of the France-BioImaging national infrastructure supported by the French National Research Agency (ANR-10-INBS-04, “Investments for the future”). This work was supported by Fondation ARC (project no. PJA 20191209363 to A.A.), “Fondation MSD Avenir,” World Wide Cancer Research to D.M. (grant 20-0120), and a “Prématuration” grant from Région Occitanie to D.M.
Author contributions
Conceptualization, D.M. and T.E.; methodology, D.M. and T.E.; investigation, T.E. and A.A.; writing – original draft, D.M.; writing – review & editing, D.M., T.E., and A.A.; funding acquisition, D.M.; resources, D.M, T.E., and A.A.; supervision, D.M.
Declaration of interests
The authors declare no competing interests.
Published: June 14, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100501.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Powers K.T., Washington M.T. Eukaryotic translesion synthesis: choosing the right tool for the job. DNA Repair. 2018;71:127–134. doi: 10.1016/j.dnarep.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nayak S., Calvo J.A., Cantor S.B. Targeting translesion synthesis (TLS) to expose replication gaps, a unique cancer vulnerability. Expert Opin. Ther. Targets. 2021;25:27–36. doi: 10.1080/14728222.2021.1864321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russo M., Crisafulli G., Sogari A., Reilly N.M., Arena S., Lamba S., Bartolini A., Amodio V., Magrì A., Novara L., et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science. 2019;366:1473–1480. doi: 10.1126/science.aav4474. [DOI] [PubMed] [Google Scholar]
- 4.Kannouche P.L., Wing J., Lehmann A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe K., Tateishi S., Kawasuji M., Tsurimoto T., Inoue H., Yamaizumi M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004;23:3886–3896. doi: 10.1038/sj.emboj.7600383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byun T.S., Pacek M., Yee M.C., Walter J.C., Cimprich K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang D.J., Lupardus P.J., Cimprich K.A. Monoubiquitination of proliferating cell nuclear antigen induced by stalled replication requires uncoupling of DNA polymerase and mini-chromosome maintenance helicase activities. J. Biol. Chem. 2006;281:32081–32088. doi: 10.1074/jbc.M606799200. [DOI] [PubMed] [Google Scholar]
- 8.Batters C., Zhu H., Sale J.E. Visualisation of PCNA monoubiquitination in vivo by single pass spectral imaging FRET microscopy. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tissier A., Kannouche P., Reck M.P., Lehmann A.R., Fuchs R.P.P., Cordonnier A. Co-localization in replication foci and interaction of human Y-family members, DNA polymerase pol eta and REVl protein. DNA Repair. 2004;3:1503–1514. doi: 10.1016/j.dnarep.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 10.Bergoglio V., Bavoux C., Verbiest V., Hoffmann J.S., Cazaux C. Localisation of human DNA polymerase kappa to replication foci. J. Cell Sci. 2002;115:4413–4418. doi: 10.1242/jcs.00162. [DOI] [PubMed] [Google Scholar]
- 11.Bi X., Slater D.M., Ohmori H., Vaziri C. DNA polymerase kappa is specifically required for recovery from the benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. J. Biol. Chem. 2005;280:22343–22355. doi: 10.1074/jbc.M501562200. [DOI] [PubMed] [Google Scholar]
- 12.Kannouche P., Fernández de Henestrosa A.R., Coull B., Vidal A.E., Gray C., Zicha D., Woodgate R., Lehmann A.R. Localization of DNA polymerases eta and iota to the replication machinery is tightly co-ordinated in human cells. EMBO J. 2003;22:1223–1233. doi: 10.1093/emboj/cdf618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kannouche P., Broughton B.C., Volker M., Hanaoka F., Mullenders L.H., Lehmann A.R. Domain structure, localization, and function of DNA polymerase eta, defective in xeroderma pigmentosum variant cells. Genes Dev. 2001;15:158–172. doi: 10.1101/gad.187501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forment J.V., Walker R.V., Jackson S.P. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytometry A. 2012;81:922–928. doi: 10.1002/cyto.a.22155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thakar T., Leung W., Nicolae C.M., Clements K.E., Shen B., Bielinsky A.-K., Moldovan G.-L. Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nat. Commun. 2020;11:2147. doi: 10.1038/s41467-020-16096-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swain U., Friedlander G., Sehrawat U., Sarusi-Portuguez A., Rotkopf R., Ebert C., Paz-Elizur T., Dikstein R., Carell T., Geacintov N.E., et al. TENT4A non-canonical poly(A) polymerase regulates DNA-damage tolerance via multiple pathways that are mutated in endometrial cancer. Int. J. Mol. Sci. 2021;22:6957. doi: 10.3390/ijms22136957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Despras E., Sittewelle M., Pouvelle C., Delrieu N., Cordonnier A.M., Kannouche P.L. Rad18-dependent SUMOylation of human specialized DNA polymerase eta is required to prevent under-replicated DNA. Nat. Commun. 2016;7 doi: 10.1038/ncomms13326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Recolin B., van der Laan S., Tsanov N., Maiorano D. Molecular mechanisms of DNA replication checkpoint activation. Genes. 2014;5:147–175. doi: 10.3390/genes5010147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toledo L.I., Altmeyer M., Rask M.B., Lukas C., Larsen D.H., Povlsen L.K., Bekker-Jensen S., Mailand N., Bartek J., Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155:1088–1103. doi: 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 20.Egger T., Aze A., Maiorano D. Protocol to analyze endogenous translesion DNA synthesis in single mammalian cells. STAR Protocols. 2023 doi: 10.1016/j.xpro.2023.102361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terai K., Abbas T., Jazaeri A.A., Dutta A. CRL4(Cdt2) E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol. Cell. 2010;37:143–149. doi: 10.1016/j.molcel.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masutani C., Kusumoto R., Yamada A., Dohmae N., Yokoi M., Yuasa M., Araki M., Iwai S., Takio K., Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 23.Somyajit K., Spies J., Coscia F., Kirik U., Rask M.-B., Lee J.-H., Neelsen K.J., Mund A., Jensen L.J., Paull T.T., et al. Homology-directed repair protects the replicating genome from metabolic assaults. Dev. Cell. 2021;56:461–477.e7. doi: 10.1016/j.devcel.2021.01.011. [DOI] [PubMed] [Google Scholar]
- 24.Andersen P.L., Xu F., Ziola B., McGregor W.G., Xiao W. Sequential assembly of translesion DNA polymerases at UV-induced DNA damage sites. MBoC. 2011;22:2373–2383. doi: 10.1091/mbc.e10-12-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsanov N., Kermi C., Coulombe P., Van der Laan S., Hodroj D., Maiorano D. PIP degron proteins, substrates of CRL4Cdt2, and not PIP boxes, interfere with DNA polymerase η and κ focus formation on UV damage. Nucleic Acids Res. 2014;42:3692–3706. doi: 10.1093/nar/gkt1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jagirdar K., Yin K., Harrison M., Lim W., Muscat G.E.O., Sturm R.A., Smith A.G. The NR4A2 nuclear receptor is recruited to novel nuclear foci in response to UV irradiation and participates in nucleotide excision repair. PLoS One. 2013;8 doi: 10.1371/journal.pone.0078075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng Y., Pao A., Adair G.M., Tang M. Cyclobutane pyrimidine dimers and bulky chemical DNA adducts are efficiently repaired in both strands of either a transcriptionally active or promoter-deleted APRT gene. J. Biol. Chem. 2001;276:16786–16796. doi: 10.1074/jbc.M010973200. [DOI] [PubMed] [Google Scholar]
- 28.Yang Y., Durando M., Smith-Roe S.L., Sproul C., Greenwalt A.M., Kaufmann W., Oh S., Hendrickson E.A., Vaziri C. Cell cycle stage-specific roles of Rad18 in tolerance and repair of oxidative DNA damage. Nucleic Acids Res. 2013;41:2296–2312. doi: 10.1093/nar/gks1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zlatanou A., Despras E., Braz-Petta T., Boubakour-Azzouz I., Pouvelle C., Stewart G.S., Nakajima S., Yasui A., Ishchenko A.A., Kannouche P.L. The hMsh2-hMsh6 complex acts in concert with monoubiquitinated PCNA and Pol eta in response to oxidative DNA damage in human cells. Mol. Cell. 2011;43:649–662. doi: 10.1016/j.molcel.2011.06.023. [DOI] [PubMed] [Google Scholar]
- 30.Ogi T., Limsirichaikul S., Overmeer R.M., Volker M., Takenaka K., Cloney R., Nakazawa Y., Niimi A., Miki Y., Jaspers N.G., et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell. 2010;37:714–727. doi: 10.1016/j.molcel.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Nakajima S., Lan L., Kanno S.i., Usami N., Kobayashi K., Mori M., Shiomi T., Yasui A. Replication-dependent and -independent responses of RAD18 to DNA damage in human cells. J. Biol. Chem. 2006;281:34687–34695. doi: 10.1074/jbc.M605545200. [DOI] [PubMed] [Google Scholar]
- 32.Karras G.I., Jentsch S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell. 2010;141:255–267. doi: 10.1016/j.cell.2010.02.028. [DOI] [PubMed] [Google Scholar]
- 33.Shibutani S., Takeshita M., Grollman A.P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 34.Bergoglio V., Boyer A.S., Walsh E., Naim V., Legube G., Lee M.Y.W.T., Rey L., Rosselli F., Cazaux C., Eckert K.A., et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013;201:395–408. doi: 10.1083/jcb.201207066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Twayana S., Bacolla A., Barreto-Galvez A., De-Paula R.B., Drosopoulos W.C., Kosiyatrakul S.T., Bouhassira E.E., Tainer J.A., Madireddy A., Schildkraut C.L. Translesion polymerase eta both facilitates DNA replication and promotes increased human genetic variation at common fragile sites. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2106477118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lo Furno E., Busseau I., Aze A., Lorenzi C., Saghira C., Danzi M.C., Zuchner S., Maiorano D. Translesion DNA synthesis-driven mutagenesis in very early embryogenesis of fast cleaving embryos. Nucleic Acids Res. 2022;50:885–898. doi: 10.1093/nar/gkab1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guilliam T.A., Yeeles J.T.P. Reconstitution of translesion synthesis reveals a mechanism of eukaryotic DNA replication restart. Nat. Struct. Mol. Biol. 2020;27:450–460. doi: 10.1038/s41594-020-0418-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong R.P., García-Rodríguez N., Zilio N., Hanulová M., Ulrich H.D. Processing of DNA polymerase-blocking lesions during genome replication is spatially and temporally segregated from replication forks. Mol. Cell. 2020;77:3–16.e4. doi: 10.1016/j.molcel.2019.09.015. [DOI] [PubMed] [Google Scholar]
- 39.Edmunds C.E., Simpson L.J., Sale J.E. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol. Cell. 2008;30:519–529. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 40.Diamant N., Hendel A., Vered I., Carell T., Reissner T., de Wind N., Geacinov N., Livneh Z. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res. 2012;40:170–180. doi: 10.1093/nar/gkr596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tirman S., Quinet A., Wood M., Meroni A., Cybulla E., Jackson J., Pegoraro S., Simoneau A., Zou L., Vindigni A. Temporally distinct post-replicative repair mechanisms fill PRIMPOL-dependent ssDNA gaps in human cells. Mol. Cell. 2021;81:4026–4040.e8. doi: 10.1016/j.molcel.2021.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cortez D. Replication-coupled DNA repair. Mol. Cell. 2019;74:866–876. doi: 10.1016/j.molcel.2019.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sale J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013;5 doi: 10.1101/cshperspect.a012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nayak S., Calvo J.A., Cong K., Peng M., Berthiaume E., Jackson J., Zaino A.M., Vindigni A., Hadden M.K., Cantor S.B. Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci. Adv. 2020;6:eaaz7808. doi: 10.1126/sciadv.aaz7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langerak P., Nygren A.O.H., Krijger P.H.L., van den Berk P.C.M., Jacobs H. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J. Exp. Med. 2007;204:1989–1998. doi: 10.1084/jem.20070902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Durando M., Tateishi S., Vaziri C. A non-catalytic role of DNA polymerase eta in recruiting Rad18 and promoting PCNA monoubiquitination at stalled replication forks. Nucleic Acids Res. 2013;41:3079–3093. doi: 10.1093/nar/gkt016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bétous R., Pillaire M.J., Pierini L., van der Laan S., Recolin B., Ohl-Séguy E., Guo C., Niimi N., Grúz P., Nohmi T., et al. DNA polymerase kappa-dependent DNA synthesis at stalled replication forks is important for CHK1 activation. EMBO J. 2013;32:2172–2185. doi: 10.1038/emboj.2013.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kermi C., Prieto S., van der Laan S., Tsanov N., Recolin B., Uro-Coste E., Delisle M.B., Maiorano D. RAD18 is a maternal limiting factor silencing the UV-dependent DNA damage checkpoint in Xenopus embryos. Dev. Cell. 2015;34:364–372. doi: 10.1016/j.devcel.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 49.Stirling D.R., Swain-Bowden M.J., Lucas A.M., Carpenter A.E., Cimini B.A., Goodman A. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinf. 2021;22:433. doi: 10.1186/s12859-021-04344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.