

Summary
Time-lapse microscopy is the only method that can directly capture the dynamics and heterogeneity of fundamental cellular processes at the single-cell level with high temporal resolution. Successful application of single-cell time-lapse microscopy requires automated segmentation and tracking of hundreds of individual cells over several time points. However, segmentation and tracking of single cells remain challenging for the analysis of time-lapse microscopy images, in particular for widely available and non-toxic imaging modalities such as phase-contrast imaging. This work presents a versatile and trainable deep-learning model, termed DeepSea, that allows for both segmentation and tracking of single cells in sequences of phase-contrast live microscopy images with higher precision than existing models. We showcase the application of DeepSea by analyzing cell size regulation in embryonic stem cells.
Keywords: Cell biology, deep learning, microscopy, cell size, live imaging, cell segmentation, cell tracking
Graphical abstract
Highlights
-
•
DeepSea is a deep-learning model for cell segmentation and tracking
-
•
DeepSea software is a user-friendly tool for quantitative analysis of live microscopy
-
•
DeepSea can accurately segment and track different types of single cells
-
•
DeepSea can be easily trained to segment and track new cell types
Motivation
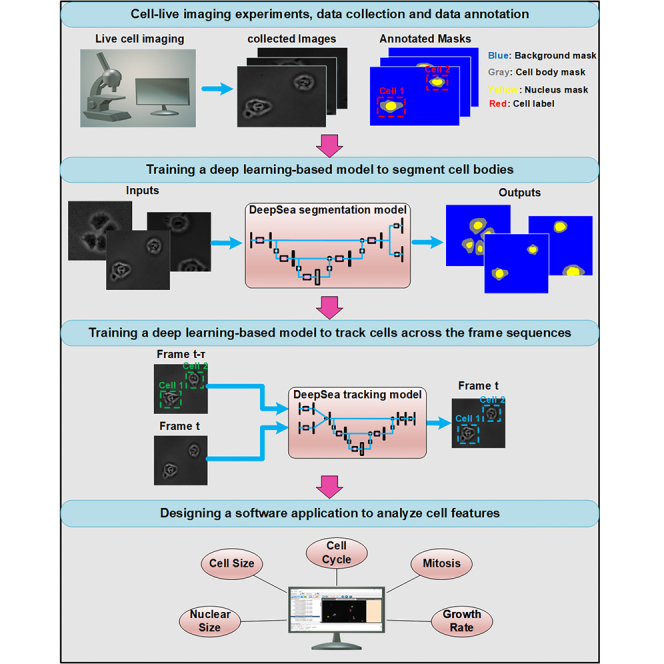
Time-lapse microscopy allows for direct observation of cell biological processes at the single-cell level with high temporal resolution. Quantitative analysis of single-cell time-lapse microscopy requires automated segmentation and tracking of individual cells over several days. Precise segmentation and tracking remain challenging because cells change their shape, divide, and show unpredictable movements. This work is motivated by recent advances in the application of deep-learning models for the analysis of microscopy images. We present a deep-learning-based model and a user-friendly software, termed DeepSea, to automate both the segmentation and tracking of individual cells in time-lapse microscopy images. We showcase the application of our software by monitoring the size of the stem cells as cells progress through the cell cycle.
Zargari et al. develop a deep-learning model to detect individual cells and track them over time in live microscopy images. The user-friendly DeepSea software allows researchers to extract quantitative information about dynamics of cell biological processes at the single-cell level.
Introduction
Cells frequently adapt their behavior in response to environmental cues to make important fate decisions, such as whether to divide or not. In addition, individual cells within a clonal population and under identical conditions display heterogeneity in response to environmental cues.1 In recent years, it has become clear that single-cell-level analysis over time is essential for revealing the dynamics and heterogeneity of individual cells.2,3
Single-cell quantitative live microscopy can directly capture both dynamics and heterogeneity of cellular decisions by continuous long-term measurements of cellular features.4,5 Widely available microscopy techniques such as label-free phase-contrast live microscopy allow for monitoring the dynamics of morphological features such as the size and shape of the cells.6 The key to the successful application of single-cell live microscopy is the scalable and automated analysis of a large dataset of images. Typical live-cell imaging of biological features of cells is a multi-day experiment that produces several gigabytes of images collected from thousands of cells.4 A major challenge for quantitative analysis of these images is the difficulty in accurately defining the borders of a cell, segmenting them, and tracking them over time. Low signal-to-noise ratio, existing non-cell small particles in the background, the proximity of cells, and unpredictable movements are among the challenges for software-based automated image analysis of live single-cell microscopy data. In addition, cells are non-rigid bodies, and thus tracking them is more challenging because they can change their shapes with time. Most critically, they divide into two new daughter cells during mitosis, which is unique and not comparable with other phenomena we encounter in conventional object-tracking applications. Solving single-cell microscopy challenges requires integrating different disciplines, such as cell biology, image processing, and machine learning.
In recent years, deep learning (DL) has outperformed conventional rule-based image processing techniques in tasks such as object segmentation and object tracking.7,8,9 Traditional image segmentation approaches often require experiment-specific parameter tuning, while DL schemes are adaptive and trainable. More recently, DL-based image processing methods have attracted attention among cell biologists and microscopists, for example, to localize single molecules in super-resolution microscopy,10 enhance the resolution of fluorescence microscopy images,11 develop an automated neurite segmentation system using a large 3D anisotropic electron microscopy image dataset,12 design a model to restore a wide range of fluorescence microscopy data,13 and train a fast model that refocuses 2D fluorescence images onto 3D surfaces within the sample.14 In particular, DL-based segmentation methods have greatly facilitated the task of cell body segmentation in microscopy images.15,16,17,18 However, the successful application of DL-based models for time-lapse microscopy depends on applying the segmentation and tracking models in one platform to automate the analysis of a large sequence of images of live cells.
Here, we developed a versatile and trainable DL model for cell body segmentation and cell tracking in time-lapse phase-contrast microscopy images of mammalian cells at the single-cell level. Using this model, we developed a user-friendly software tool, termed DeepSea, for automated and quantitative analysis of phase-contrast microscopy images. We showed that DeepSea captures dynamics and heterogeneity of cellular features such as cell cycle division and cell size in different cell types. Our analysis of cell size distribution in mouse embryonic stem cells revealed that despite their short G1 phase of the cell cycle, embryonic stem cells exhibit cell size control in the G1 phase of the cell cycle.
Results
Designing and training a cell segmentation and tracking model
First, we created an annotated dataset of phase-contrast live image sequences of three cell types: (1) mouse embryonic stem cells, (2) bronchial epithelial cells, and (3) mouse C2C12 muscle progenitor cells. To facilitate manual annotation of the cells, we developed a MATLAB-based software to generate a labeled training dataset, including pairs of original cell images and corresponding cell ground-truth mask images (our annotation software is available at https://deepseas.org/software/). To further generalize our model, we used image augmentation techniques to increase the size of our dataset with more variations efficiently and less expensively. In addition to six conventional image augmentation techniques with random settings such as cropping, changing the contrast and brightness, blurring, applying the vertical/horizontal flip, and adding Gaussian noise,19,20 we proposed and applied a random cell movement method as a novel image augmentation strategy to generate new cell images (with their annotated masks) that look more different than the original existing samples (Figure S1). Next, we used the annotated and augmented dataset of cell images to train our supervised DL-based segmentation model called DeepSea to detect and segment the cell bodies. To design our DeepSea segmentation model, we were inspired by the UNET model, which has been successful in different segmentation tasks.21 We made several innovative changes to make this model more suitable for single-cell live microscopy. First, we scaled down 2D UNET to considerably reduce the number of parameters and thus have a faster model processing large high-resolution images with less computational and memory costs. To do this, we modified our model with convolutional residual connections to increase the depth of the network with fewer extra parameters.22,23,24 Second, we added an auxiliary edge detection layer trained on the edge area between touched cells to enhance the learning algorithm to focus on touching cell edges and thus improve the segmentation accuracy in hard samples with high-density touched cell images (Figure S2A). In the training process, we also used a progressive learning technique (used in progressive general adversarial networks [GANs]25) to help the model generalize well for different image resolutions and generate large high-resolution masks that better separate the touching cell edges (Figure S3). The progressive learning technique makes the model first learn coarse-level features and then finer information. Table S1 shows how our proposed techniques and modifications can improve the segmentation scores for simple and crowded samples as measured by precision.
To be able to visualize the dynamics of cellular behavior over time, we added cell tracking capability to our DeepSea model. We trained a DL tracking model to localize and link single cells from one frame to the next and detect cell divisions (mitosis). As shown in Figure S2B, we used a baseline architecture similar to the DeepSea segmentation model. This model extracts the convolutional information from two consecutive image inputs (segmented cell images of times t-τ and t) to localize and detect the same target single cell or its daughter cells among the segmented cells in the current frame (time t) by generating a binary mask (Figures S4 and S5). With this model, we could monitor multiple cellular phenotypes and several cell division cycles across the microscopy image sequences to generate lineage tree structures of cells. To make our model widely accessible, we developed a DL-based software with a graphical user interface (Figure S6A) that allows researchers with no background in machine learning to automate the measurement of cellular features of live microscopy data. We added manual editing options to DeepSea software to allow researchers to correct our model outputs when needed to bring all the DeepSea detections to the highest possible accuracy and, thus, fully track the life cycle of the cells. An interesting feature of our software is that it also allows researchers to train a new model with an annotated dataset of any cell type. We provide step-by-step instructions on how to use our software and train a model with a new dataset. Our software, instructions, and cell image datasets are publicly available at https://deepseas.org/.
DeepSea performance evaluation
The trained segmentation model fits the exact boundary of the target cells and labels their pixels with different colors, helping to determine each cell’s shape and area within the input microscopy image (Figure S2A). To evaluate the performance of our segmentation model, we compared the model’s predictions with true manually segmented cell bodies at different thresholds of the standard intersection over union (IoU) metric on the test images. Next, we used the standard average precision metric, which is commonly used in pixel-wise segmentation and object detection tasks, to compare DeepSea with recently developed segmentation models. DeepSea was able to outperform existing state-of-the-art models such as CellPose,15 StarDist,16 and 2D-UNET21,26 in terms of latency and mean average precision (mAP) when trained on the same training sets and tested on the same test sets at all pre-defined IoU thresholds (Figures 1A and 1B). Notably, we observed close prediction accuracy between images with a higher density of cells with touching edges (hard cases) and images with a lower density of cells (easy) with an overall higher precision compared with the CellPose model (Figures 1C and 1D). Examples of DeepSea’s accuracy in high-density cell cultures are shown in Figure S6B. In addition, we demonstrated the generalizability of the DeepSea model performance with different cell-type test images of our dataset (Figure 1E). Three examples of the DeepSea and CellPose segmentation model’s output are compared in Figure 2. Next, we compared the performance of DeepSea with CellPose in measuring cellular phenotypes such as cell size. A comparison of cell size distribution obtained from DeepSea and CellPose showed that DeepSea obtains a median cell size that is closer to the median cell size obtained by manual segmentation (Figure 3). Together, these results indicate that DeepSea’s segmentation model works robustly across different densities of cells and different cell types in our dataset with high precision.
Figure 1.

Segmentation model evaluation on the test set images
(A) Comparing the performance of DeepSea, Cell Pose, StarDist, and 2D-UNET using the standard average precision at different IoU matching thresholds.
(B) Measuring models’ latency (per image) to compare the DeepSea efficiency with the other models.
(C and D) Comparing models’ performance in segmenting easy (sparse cell density) and hard (high cell density) test images using average precision with one standard error of the mean shown by error bars.
(E) Comparing models’ performance in segmenting different cell types of the DeepSea dataset.
Figure 2.

Three examples of segmentation outputs
DeepSea output (middle column) compared with the CellPose (right column) for different cell types. DeepSea has higher average precision (AP) compared with the CellPose model.
Figure 3.

Cell size distribution of ground-truth test dataset compared with the distribution obtained from DeepSea and CellPose detections
The DeepSea tracking model receives the segmented target cell image at the previous time point and the segmented cell image at the current time point to generate a binary mask localizing the target cell (or its daughter cells) at the current time point (Figures S4 and S5). For the tracking model, we evaluated the model’s performance on the test set by measuring the AP of single-cell tracking from one frame to the next frame, as well as mitosis detections. We matched the binary masks obtained from the tracking model at time t to the true target cell bodies (at time t) at different matching thresholds of IoU. While our model achieved 0.98 ± 0.2 precision (@0.5 IoU threshold) for tracking single cells, the precision of our model for mitosis detection was around 0.89 ± 0.3 (@0.5 IoU threshold) (Figures 4A and 4B). Mitosis detection was particularly more challenging for stem cell images (Figure S7B). We speculated that there might be a direct relationship between the single-cell and mitosis detection results and the frame imaging intervals. Thus, we ran an experiment to measure the tracking model sensitivity to the frame sampling rate. We used the test frame sequences of bronchial epithelial cells and down-sampled the frames to make sub-sampling intervals of 5, 10, and 15 min. The results are shown in Figure S7C, confirming that the model precision is sensitive to the changes in the frame’s time distance. It shows that the higher sampling rate reduces the tracking model failures, especially for the cells that move and change quickly over time.
Figure 4.

Tracking model evaluation on the test set
We evaluated the model performance using the standard average precision at different IoU matching thresholds.
(A) Single-cell tracking precision at different IoU matching thresholds.
(B) Mitosis detection precision at different IoU matching thresholds.
Next, we systematically compared DeepSea tracking precision with some existing cell tracking tools (Table S2). As shown, some of these tools only support a part of the required process, either single-cell tracking27,28 or mitosis detection,29 and some of them are proposed to be used for both, like Trackmate.30 Similar to the DeepSea tracking pipeline, they all first need to detect and segment the cell bodies before starting the cell tracking process and frame-by-frame cell linking. The segmentation precision of all of them with our cell images is lower than 50%. Thus, we decided to use DeepSea segmentation outputs as the input for these tracking tools to obtain the best possible tracking results and compared only cell tracking performance of these tools. We assessed the tracking model of DeepSea and other tracking tools in a full cell cycle-tracking task. One example of DeepSea's full cell cycle tracking and mitosis detection is shown in Figure 5. This test uses the trained tracking model to track and label the target single-cell motion trajectories across the live-cell microscopy frame sequences from birth to division. In this evaluation process, we used MOTA (multi-object tracking accuracy), which is a widely used metric in multi-object tracking schemes and measures the precision of localizing objects over time across the frame sequences (Equation 5). We also included other commonly used tracking metrics such as IDS (identity switch), MT (mostly tracked), ML (mostly lost), and Frag (fragmentation) to provide more detailed evaluation information.7,31,32
Figure 5.

Example of the cell cycle-tracking process
It is obtained by feeding nine consecutive stem cell frames (with a sampling time of 20 min) to our trained tracking model. Daughter cells are linked to their mother cells by an underline (in the sixth and seventh frames).
Our DeepSea tracking model achieved a MOTA value of 0.94 ± 0.2 compared with the Trackmate model, which had a MOTA of 0.29 ± 0.7 (Table S3). In the evaluation process, we used 228 full ground-truth cell cycle trajectories, each including more than three consecutive frames. The Trackmate algorithm30 is one of the widely used cell tracking tools. The main factor for Trackmate’s overall low MOTA was that it frequently did not detect mitotic events leading to high false positive (FP) and false negative (FN) labels (Table S3; Equation 5). We also would like to note that rule-based tools like Trackmate are not trainable to be rapidly adapted to any specialized dataset.
Cell cycle duration is adjusted based on birth size: Showcasing the application of the DeepSea
Cells need to grow in size before they can undergo division. Different cell types maintain a fairly uniform size distribution by actively controlling their size in the G1 phase of the cell cycle.33 However, the typical G1 control mechanisms of somatic cells are altered in mouse embryonic stem cells (mESCs).34,35 mESCs have an unusually rapid cell division cycle that takes about 10 h to be completed (Figure 6A). The rapid cell cycle of mESCs is primarily due to an ultrafast G1 phase that is about 2 h compared with ∼10 in skin fibroblast cells with daughter cells born at different sizes (Figures 7A and 7B). An interesting question is whether mESCs can employ size control in their rapid G1 phase, just as most somatic cells do.
Figure 6.

Showcasing the DeepSea application
Cell size regulation in mouse embryonic stem cells.
(A) Distribution of the cell cycles.
(B) Histogram of birth size ratio of daughter cell pairs.
(C) Comparing the cell cycle duration of the cells born small with those born large.
Figure 7.

Stem cell feature analysis
(A) Cell size ratio graph of daughter cell pairs.
(B) Automated measurement of G1 duration using Fucci sensor (Geminin-GFP) that increases its activity as cells enter the S phase.
(C) Cell area versus cell volume measurement using confocal microscopy for each embryonic stem cell.
(D) One example of cell surface measurement, obtained from our confocal microscopy experiment.
Using confocal microscopy, we showed that the area of a cell is closely correlated with the cell volume, making the area a faithful measurement of cell size (Figures 7C and 7D). By measuring the size of the sister cells at birth, we showed that 42% of divisions resulted in daughter cells of different sizes (Figure 6B). We hypothesized that smaller-born cells would spend more time growing compared with their larger sister cells. In support of this hypothesis, we observed that smaller-born cells increase their cell cycle duration by about ∼2 h compared with their larger sister cells (Figure 6C).
Together, our results show that DeepSea can be applied to accurately quantify cell biological features of cells, such as cell size or cell cycle duration. In addition, our findings support the hypothesis that mESCs can adjust the cell cycle duration based on birth size, suggesting cell size control through an unknown molecular mechanism.33 Besides, this shows that DeepSea can capture cell size distribution that is closer to the ground truth as determined by manually segmented cells.
Discussion
Here, we introduced DeepSea, an efficient DL model for automated analysis of time-lapse images of cells. The segmentation and tracking of cell bodies and sub-cellular organelles from microscopy images are critical steps for nearly all microscopy-based biological analysis applications. Although phase-contrast microscopy is a non-invasive and widely used method for live-cell imaging, developing automated segmentation and tracking algorithms remains challenging. Segmentation of phase-contrast images remains difficult because of the presence of bright light artifacts such as halo at the edges of the cells and inhomogeneity in the refractive index producing noisy images. Although not unique to phase-contrast microscopy, cell tracking has its own challenges due to the unpredictable nature of cells in their movements over time, the close proximity of cells, and the division of cells. Here, we leveraged the recent advancement in DL-based image processing to address some of these challenges.
The lack of a comprehensive, high-quality annotated dataset of cells prevents the full utilization of DL-based models for microscopy image analysis systems. We generated large manually annotated datasets of time-lapse microscopy images of three cell types, which are publicly available and can be used for new image analysis models. In addition, we were able to significantly increase the size of annotated data covering more variations by applying image augmentation techniques, which benefited from both conventional image augmentation techniques and a proposed random cell movements method. We expect this resource to facilitate the future application of DL-based models for the analysis of microscopy images.
To address the challenge of cell segmentation and tracking, we built a DL model, termed DeepSea, that can efficiently segment cell areas in phase-contrast microscopy images. Our segmentation model was trained on our generated dataset and achieved an IoU value of 0.90 ± 0.2 at the IoU matching threshold of 0.5. We were able to improve on existing segmentation models by incorporating (1) an auxiliary model trained on where cell edges meet to be able to separate cells that are close to each other, (2) the addition of the residual blocks to decrease the number of parameters without sacrificing the accuracy making our model efficient, and (3) a progressive learning technique to improve the generalizability of our model for images with different resolutions. Importantly, we were able to exploit the DL capabilities to automate the tracking of cells across the time-lapse microscopy image sequences. Our DeepSea tracking model was able to track the full cell cycle trajectories with a MOTA value of 0.94 ± 0.3 obtained from 228 cell cycles. We also showed that more frequent imaging of microscopy frames would increase the accuracy of tracking the full cell cycle by providing more information about the cell features right before cell division.
We showcased the application of DeepSea by investigating cell size regulation in mESCs across hundreds of cell division cycles. Our cell size analysis revealed that smaller-born mESCs regulate their size by spending more time growing in the G1 phase of the cell cycle. These findings strongly support the idea that mESCs actively monitor their size, consistent with the presence of size control mechanisms in the short G1 phase of the embryonic cell cycle.
Limitations of the study
We would like to note that our dataset and models are limited to the phase contrast 2D images of three cell types. However, researchers can train our model using their own annotated images of single cells using DeepSea software training options. A larger dataset of samples from different cell types and different imaging modalities would be useful for testing our proposed model’s generalization, reliability, and robustness. In addition, in our future work, we will investigate other deep models that have recently achieved considerable advancement in object detection and tracking tasks, such as Recurrent Yolo, TrackR-CNN, JDE, RetinaNet, and CenterPoint,7,8,9 or merge their architecture with our current models to improve the results. To reduce DeepSea’s sensitivity to frame sampling, we will also evaluate the idea of feeding more previous frames into the tracking model, including the cell images of t, t-τ, t-2τ, and t-3τ, as one of the possible solutions.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental models: Cell lines | ||
| HBEC3kt | laboratory of Harold Varmus | N/A |
| Embryonic Stem Cells | Shariati lab | https://www.novusbio.com/products/v65-mouse-embryonic-stem-cells_nbp1-41162 |
| Software and algorithms | ||
| CellPose | Stringer et al.15 | https://www.cellpose.org/ |
| StraDist | Schmidt et al.16 | https://github.com/stardist/stardist |
| 2D-UNET | Ronneberger et al.26 | N/A |
| DeepSea | This paper |
https://deepseas.org/Zenodo https://doi.org/10.5281/zenodo.7906713 |
| Trackmate | Ershov et al.30 | https://imagej.net/plugins/trackmate/ |
| CellTracker | Piccinini et al.27 | http://celltracker.website/index.html |
| MDMLM | Kazuya et al.29 | https://github.com/naivete5656/MDMLM |
| CellTracking | Kazuya et al.28 | https://github.com/ithet1007/Cell-Tracking |
Resource availability
Lead contact
Further information and requests should be addressed to and will be fulfilled by the lead contact, S. Ali Shariati (alish@ucsc.edu).
Materials availability
All unique/stable materials generated in this study are available from the lead contact upon reasonable request with a completed Materials Transfer Agreement.
Experimental model and subject details
Cell culture and microscopy
Mouse ESCs (V6.5) were maintained on 0.1% gelatin-coated cell culture dishes in 2i media (Millipore Sigma, SF016-100) supplemented with 100U/ml Penicillin-Streptomycin (Thermo Fisher, 15140122). Cells were passaged every 3–4 days using Accutase (Innovate Cell Technologies, AT104) and seeded at a density of 5,000–10,000 cells/cm2. For live imaging, between 5000 and 10,000 cells were seeded on 35mm dishes with a laminin-coated (Biolamina) 14mm glass microwell (MatTek, P35G-1.5-14-C). Cells were imaged in a chamber at 37C perfused with 5% CO2, a Zeiss AxioVert 200M microscope with an automated stage, and an EC Plan-Neofluar 5x/0.16NA Ph1objective or an A-plan 10×/0.25NA Ph1 objective. The same culture condition was used for confocal imaging, except that 24 h after seeding, the media was replaced with 2mL DMEM-F12 (Thermo Fisher, 11039047) containing 2ul CellTracker Green CMFDA dye (Thermo Fisher, C2925) and placed back in the incubator for 35 min. Next, 2 μl of CellMask Orange plasma membrane stain (Thermo Fisher, C10045) was added, and the dish was incubated for another 10 min. Dishes were washed three times with DMEM-F12, after which 2mL of fresh 2i media was added. Cells were imaged directly after the live-cell staining protocol using the Zeiss 880 Microscope using a 20x/0.4 N.A. objective and a 1μm interval through the z axis.
Immortalized human bronchial epithelial (HBEC3kt) cell line homozygous for wildtype U2AF1 at the endogenous locus was obtained as a gift from the laboratory of Harold Varmus (Cancer Biology Section, Cancer Genetics Branch, National Human Genome Research Institute, Bethesda, United States of America and Department of Medicine, Meyer Cancer Center, Weill Cornell Medicine, New York, United States of America) and cultured according to Fei et al.36 This host cell line was used for lentiviral transduction and blasticidin selection to generate a line with stable expression of KRASG12V using a lentiviral plasmid obtained as a gift from the laboratory of John D Minna (Hamon Center for Therapeutic Oncology Research, The University of Texas Southwestern Medical Center) described in.37 Cells from passage 11 were grown to 80% confluency in Keratinocyte SFM (1X) (Thermo Fisher Scientific, USA) before being re-seeded as biological duplicates at three densities: 0.3M, 0.2M, and 0.5M cells per well in 6-well plates and allowed to adhere before live-cell imaging over a 48h time period.
Method details
Dataset
We collected phase-contrast time-lapse microscopy image sequences of three different cell types, including two in-house datasets of Mouse Embryonic Stem Cells (MESC, 31 sets, 1074 images) and Bronchial epithelial cells (7 sets, 2010 images) and one dataset of Mouse C2C12 Muscle Progenitor Cells (7 sets, 540 images) obtained from an external resource with the cell culture described in.38 Our collected datasets are publicly available at https://deepseas.org/datasets/. Some dataset statistics are shown in Table S4. We designed an annotation software in MATLAB (https://deepseas.org/software/) to manually create the ground-truth mask images corresponding to our cell images. We applied an image augmentation scheme to generate a larger dataset with more variations efficiently and less expensively, aiming to train a more generalized model. In our image augmentation scheme, in addition to conventional image transformations,19,20 we proposed moving the stem cell bodies by the random vectors of (θ,d) relative to their center points, where θ is the direction angle between 0 and 360 and d is the displacement in pixels (Figure S1). The proposed cell image augmentation method improved the model performance with unseen test images (different microscopy live imaging sets not used in the training set), confirming that it could less overfit training samples and thus help the model generalization. For each training image, we applied a pipeline of augmentation functions which were randomly selected and set.
Segmentation model
As mentioned before, our dataset samples are label-free microscopy images that are usually noisy, low contrast, hard, and high cell density samples. It is difficult for any existing instance tools (that have not seen these types of images in their training process) to segment the cell bodies of our test images. The original pre-trained version of StraDist and StarDist models achieved an average precision of around 43% and 5%, respectively, on our test sets. Figure S7A shows the CellPose and StarDist outputs compared with the ground truth mask images.
In the instance segmentation task, we proposed and built a 2D deep learning-based model called DeepSea (Figure S2A). To design our DeepSea segmentation model, we were inspired by the UNET model.21,26 Since we needed a fast segmentation and tracking model to be used in our DeepSea software, we decided to reduce the number of parameters and make a scaled-down version of 2D UNET. By reducing the model size, we could feed larger high-resolution images into the model and get more accurate results39,40 with less computational and memory costs. However, to compensate for the model compression and also avoid the model from underfitting the training data, we modified the scaled-down 2D-UNET model with the convolutional residual connections. It has been proved that the residual connections can increase the depth of the network with fewer extra parameters. They also can accelerate the speed of the training of the deep network, reduce the effect of the vanishing Gradient Problem, and potentially obtain higher accuracy in network performance.22,23,24 Our DeepSea segmentation model involves only 1.9 million parameters, which is considerably smaller than typical instance segmentation models such as UNET,26 PSPNET,41 and SEGNET.42
During the training process, we started training the model with the low-resolution images 95x128, then increased it to 191x256, and finished it with 384x512, as described in Figure S3. Our learning algorithm started with the lowest resolution part and then progressively added the other high-resolution blocks until the desired image size and full DeepSea model were achieved. The progressive learning technique (as used in progressive GANs25) can help the model generalize well for different image resolutions and generate large-high-resolution masks that better separate the touching cell edges. Also, when adding the higher resolution part to the training process, our learning algorithm reduces the learning rate of previously trained parts, making the different parts of the model learn information from different resolutions independently.
The auxiliary edge representations (highlighting the edge area between touching cells) and the auxiliary training loss value (Equation 2) also encouraged the learning algorithm to spend more computational budget and time to separate the touching cells. They thus improved the model performance, especially for hard samples where we have high-density touching cells. We also artificially increased and repeated the hard cell images in our training dataset to make the model see them more during the training process (almost the same number as non-touching samples). This also helps the learning algorithm balance the loss functions (Equation 2). To create each touching cell edge mask, we first created a weight map from the ground truth cell masks according to Equation 1:
| (Equation 1) |
where x is pixels in the image, d1 is the distance to the border of the nearest cell, d2 is the distance to the border of the second nearest cell, and w0 and σ were set to 10 and 25, respectively. Then we make a binary image by replacing all pixel values above a determined threshold (=1.0) with 1s and setting all other pixels to 0s.
In the training process, we used the early stopping technique to stop training when the validation score stopped improving. We also took advantage of batch normalization and dropout techniques to improve the model’s speed, performance, and stability.43 Besides, the image augmentation pipeline we designed (Figure S1) could help the model see more variations during the training process and then process the unseen test samples more confidently. We chose the RMSprop optimization function with the learning rate scheduler of the OneCycleLR method (LR = 1e-3) to optimize model weights and minimize the proposed loss function (Equation 2). Our loss function is a linear combination of cross-entropy (CE) loss and Dice loss (DL) functions,44 as well as auxiliary loss functions (EdgeCE and EdgeDL) for the touching cell edge representations. CE takes care of pixel-wise prediction accuracy, while DL helps the learning algorithm increase the overlap between true area and predicted area, which is essentially needed where the number of image background pixels is much higher than foreground pixels (object area pixels).
| (Equation 2) |
In the test phase, we used the IoU index, a value between 0 and 1 and known as the Jaccard index as well45 (Equation 3), to match the segmentation model predictions to the ground truth annotated masks:
| (Equation 3) |
In each test image, we labeled each detected cell body whose IoU index was higher than a pre-defined threshold value as a valid match and so True Positive (TP) prediction. Also, the ground truth cell body masks with no valid match were categorized into the False Negative (FN) set, and the predictions with no valid ground truth masks were labeled as the False Positive (FP) cases (non-cell objects). Then using Equation 4, we calculated the average precision (AP) value for each image in the test set, used by the other state-of-the-art methods in cell body segmentation tasks:15
| (Equation 4) |
Tracking model
Our tracking model aimed to localize and link the same target single-cell bodies from one frame to the next and also detect cell divisions (mitosis). We used a baseline architecture similar to the DeepSea segmentation model (as a fast and accurate enough architecture) but with multiple images, two inputs, and one output (Figures S2B, S4, and S5). The first input is the target cell image at the previous frame (previous time point t-τ), the second input is the segmented cell image at the current frame (current time point t), and the output is a binary mask at the current frame. This model extracts the convolutional information from the input images to localize and find the target single cell or its daughter cells among the segmented cells on the current frame by generating a binary mask (Figures S4B and S5B). To increase the accuracy of the tracking model, we limited our search space in x and y coordinates to a small square with the size of 5 times the target cell size centered at the previous frame target cell’s centroids (Figures S4A and S5A). Since cells move slowly through space, the cell’s previous location presents a good guess of where the model should expect to find it in the current frame. To validate the tracking model’s output, we used the IoU (Intersection over Union, Equation 3) as a validation score. We matched the tracking model binary mask to each segmented cell body on the current frame and measured the IoU value (Figures S4C and S5C). In the validation process, if the IoU score of a segmented cell body on the current frame was higher than a pre-specified IoU matching threshold value (e.g., IoU_thr = 0.5), we labeled it as a positive detection and valid link. Then, we categorized them into true or false positive detections by comparing them with ground truth cell labels aiming to measure the average precision (AP metric introduced in Equation 4) of the tracking model in tracking the target cell bodies from one frame to the next and detecting cell divisions.
The number of the DeepSea tracking model parameter is only 2.1 million, while the other deep tracking models, such as ROLO,46 DeepSort,47 and TrackRCNN,48 which are mostly used in other object tracking applications, involve more than 20 million parameters, confirming that we have an efficient model in the tracking process as well. Also, since the number of cell division events is naturally much fewer than single-cell tracking events, we artificially repeated and increased the cell division events fifty times more than single-cell tracking events in our training set. This helped the model see a balanced number of both single-cell links and cell divisions during the training process and thus reduced the risk of overfitting the most repeated category. The train optimization function and hyperparameters are the same as the segmentation model training process.
To evaluate our tracking model in a continuous cell trajectory tracking process during an entire cell life cycle from birth to division, we used MOTA (Multiple Object Tracking Accuracy, Equation 5), which is widely used in multi-object tracking challenges.7,31,32 To our knowledge, this is the first time that this metric has been used to evaluate a cell tracking model performance. We also used other commonly used tracking metrics, as follows, to give more detailed evaluation information.
IDS: Identity Switch is the number of times a cell is assigned a new label in its track.
MT: Mostly Tracked is the number of target cells assigned the same label for at least 80% of the video frames.
ML: Mostly Lost is the number of target cells assigned the same label for at most 20% of the video frames.
Frag: Fragmentation is the number of times a cell is lost in a frame but then redetected in a future frame (fragmenting the track).
| (Equation 5) |
where n is the frame number. A perfect tracking model achieves MOTA = 1.
Designed software tools
We designed two software tools for the Deepsea project, including 1) Manual annotation software and 2) DeepSea cell segmentation and tracking software. The step-by-step instruction with examples of how to use them is uploaded to the page at https://deepseas.org/software/. The manual annotation software is a MATLAB-based tool that we designed and used to manually segment and label the cells of the raw dataset of microscopy images we collected. This tool helped us provide the required ground truth dataset that we needed for training the cell segmentation and tracking models. It can also be used for manually annotating any other image datasets.
Also, DeepSea software (Figure S6A) is a user-friendly and automated software designed to enable researchers to 1) load and explore their phase-contrast cell images in a high-contrast display, 2) detect and localize cell bodies using the pre-trained DeepSea segmentation model, 3) track and label cell lineages across the frame sequences using the pre-trained DeepSea tracking model, 4) manually correct the DeepSea models' outputs using user-friendly editing options, 5) train a new model with a new cell type dataset if needed, 6) save the results and cell label and feature reports on the local system. It employs our latest trained DeepSea models in the segmentation and tracking processes.
Quantification and statistical analysis
After each successful training of the DeepSea model, we evaluated our model's segmentation and tracking performance using a set of test images. To test the reproducibility of our results, we repeated all the experiments with the cross-validation method. We chose five random sets of training/validation/testing from the generated dataset to report the mean and variance of the performance metrics.
Acknowledgments
This work was supported by the NIGMS/NIH through a Pathway to Independence Award K99GM126027/R00GM126027 (S.A.S.), a start-up package of the University of California, Santa Cruz (S.A.S), R01HD098722 (L.H.), and the James H. Gilliam Fellowships for Advanced Study program (S.R.). We acknowledge core support from the UCSC Institute for the Biology of Stem Cells (IBSC), IBCS’s imaging facility (SCR_021135), CIRM Shared Stem Cell Facilities (CL1-00506-1,2), and CIRM Major Facilities (FA1-00617-1).
Author contributions
A.Z. and S.A.S. conceived the project and wrote the manuscript. S.A.S. performed live microscopy imaging. A.K. and N.M. developed the DeepSea software and website and implemented the machine-learning code. A.B. and E.H.-R. contributed to the generation of bronchial epithelial cell images. A.K., N.C., N.M., C.W.N., and K.A. contributed to generating a manually annotated dataset of images. G.L., N.C., R.H., and S.K. performed user quality control of DeepSea segmentation and tracking functions and provided detailed feedback on the performance of the software. G.L., S.R., and L.H. contributed to the confocal microscopy images and provided feedback to improve our manuscript.
Declaration of interests
The authors declare no competing interests.
Published: June 12, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100500.
Supplemental information
Data and code availability
-
•
Our collected datasets of images of segmented cells and cells masks are publicly available at https://deepseas.org/datasets/.
-
•
All the implemented methods are publicly available as Python scripts and can be downloaded from https://github.com/abzargar/DeepSea. The Manual annotation software and the DeepSea cell segmentation and tracking software with step-by-step instructions are uploaded to the page at https://deepseas.org/software/. The DOI is in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Fiorentino J., Torres-Padilla M.-E., Scialdone A. Measuring and modeling single-cell heterogeneity and fate decision in mouse embryos. Annu. Rev. Genet. 2020;54:167–187. doi: 10.1146/annurev-genet-021920-110200. [DOI] [PubMed] [Google Scholar]
- 2.Bogdan P., Deasy B.M., Gharaibeh B., Roehrs T., Marculescu R. Heterogeneous structure of stem cells dynamics: statistical models and quantitative predictions. Sci. Rep. 2014;4:4826. doi: 10.1038/srep04826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Semrau S., Goldmann J.E., Soumillon M., Mikkelsen T.S., Jaenisch R., van Oudenaarden A. Dynamics of lineage commitment revealed by single-cell transcriptomics of differentiating embryonic stem cells. Nat. Commun. 2017;8:1096. doi: 10.1038/s41467-017-01076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skylaki S., Hilsenbeck O., Schroeder T. Challenges in long-term imaging and quantification of single-cell dynamics. Nat. Biotechnol. 2016;34:1137–1144. doi: 10.1038/nbt.3713. [DOI] [PubMed] [Google Scholar]
- 5.Chessel A., Carazo Salas R.E. From observing to predicting single-cell structure and function with high-throughput/high-content microscopy. Essays Biochem. 2019;63:197–208. doi: 10.1042/ebc20180044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zatulovskiy E., Zhang S., Berenson D.F., Topacio B.R., Skotheim J.M. Cell growth dilutes the cell cycle inhibitor Rb to trigger cell division. Science. 2020;369:466–471. doi: 10.1126/science.aaz6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciaparrone G., Luque Sánchez F., Tabik S., Troiano L., Tagliaferri R., Herrera F. Deep learning in video multi-object tracking: a survey. Neurocomputing. 2020;381:61–88. doi: 10.1016/j.neucom.2019.11.023. [DOI] [Google Scholar]
- 8.Yun S., Kim S. 2019. Recurrent YOLO and LSTM-Based IR Single Pedestrian Tracking; pp. 94–96. [Google Scholar]
- 9.Zhou X., Wang D., Krähenbühl P. Objects as points. arxiv. 2019 doi: 10.48550/arXiv.1904.07850. Preprint at. [DOI] [Google Scholar]
- 10.Ouyang W., Aristov A., Lelek M., Hao X., Zimmer C. Deep learning massively accelerates super-resolution localization microscopy. Nat. Biotechnol. 2018;36:460–468. doi: 10.1038/nbt.4106. [DOI] [PubMed] [Google Scholar]
- 11.Wang H., Rivenson Y., Jin Y., Wei Z., Gao R., Günaydın H., Bentolila L.A., Kural C., Ozcan A. Deep learning enables cross-modality super-resolution in fluorescence microscopy. Nat. Methods. 2019;16:103–110. doi: 10.1038/s41592-018-0239-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beier T., Pape C., Rahaman N., Prange T., Berg S., Bock D.D., Cardona A., Knott G.W., Plaza S.M., Scheffer L.K., et al. Multicut brings automated neurite segmentation closer to human performance. Nat. Methods. 2017;14:101–102. doi: 10.1038/nmeth.4151. [DOI] [PubMed] [Google Scholar]
- 13.Weigert M., Schmidt U., Boothe T., Müller A., Dibrov A., Jain A., Wilhelm B., Schmidt D., Broaddus C., Culley S., et al. Content-aware image restoration: pushing the limits of fluorescence microscopy. Nat. Methods. 2018;15:1090–1097. doi: 10.1038/s41592-018-0216-7. [DOI] [PubMed] [Google Scholar]
- 14.Wu Y., Rivenson Y., Wang H., Luo Y., Ben-David E., Bentolila L.A., Pritz C., Ozcan A. Three-dimensional virtual refocusing of fluorescence microscopy images using deep learning. Nat. Methods. 2019;16:1323–1331. doi: 10.1038/s41592-019-0622-5. [DOI] [PubMed] [Google Scholar]
- 15.Stringer C., Wang T., Michaelos M., Pachitariu M. Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods. 2021;18:100–106. doi: 10.1038/s41592-020-01018-x. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt U., Weigert M., Broaddus C., Myers G. In: Cell Detection with Star-Convex Polygons. held in Cham, 2018//. A.F. Frangi. Schnabel J.A., Davatzikos C., Alberola-López C., Fichtinger G., editors. Springer International Publishing; 2018. pp. 265–273. [Google Scholar]
- 17.Liu L., Ouyang W., Wang X., Fieguth P., Chen J., Liu X., Pietikäinen M. Deep learning for generic object detection: a survey. arxiv. 2019 doi: 10.48550/arXiv.1809.02165. Preprint at. [DOI] [Google Scholar]
- 18.Minaee S., Boykov Y., Porikli F., Plaza A., Kehtarnavaz N., Terzopoulos D. Image segmentation using deep learning: a survey. arxiv. 2020 doi: 10.48550/arXiv.2001.05566. Preprint at. [DOI] [PubMed] [Google Scholar]
- 19.Khan A., Nawaz U., Ulhaq A., Robinson R.W. Real-time plant health assessment via implementing cloud-based scalable transfer learning on AWS DeepLens. PLoS One. 2020;15:e0243243. doi: 10.1371/journal.pone.0243243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mumuni A., Mumuni F. Data augmentation: a comprehensive survey of modern approaches. Array. 2022;16:100258. doi: 10.1016/j.array.2022.100258. [DOI] [Google Scholar]
- 21.Siddique N., Paheding S., Elkin C.P., Devabhaktuni V. U-net and its variants for medical image segmentation: a review of theory and applications. IEEE Access. 2021;9:82031–82057. doi: 10.1109/ACCESS.2021.3086020. [DOI] [Google Scholar]
- 22.He K., Zhang X., Ren S., Sun J. Deep residual learning for image recognition. arxiv. 2015 doi: 10.48550/arXiv.1512.03385. Preprint at. [DOI] [Google Scholar]
- 23.Liu T., Chen M., Zhou M., Du S.S., Zhou E., Zhao T. Towards understanding the importance of shortcut connections in residual networks. arxiv. 2019 doi: 10.48550/arXiv.1909.04653. Preprint at. [DOI] [Google Scholar]
- 24.Shafiq M., Gu Z. Deep residual learning for image recognition: a survey. Appl. Sci. 2022;12:8972. doi: 10.3390/app12188972. [DOI] [Google Scholar]
- 25.Karras T., Aila T., Laine S., Lehtinen J. Progressive growing of GANs for improved quality, stability, and variation. arxiv. 2018 doi: 10.48550/arXiv.1710.10196. Preprint at. [DOI] [Google Scholar]
- 26.Ronneberger O., Fischer P., Brox T. U-net: convolutional networks for biomedical image segmentation. arxiv. 2015 doi: 10.48550/arXiv.1505.04597. Preprint at. [DOI] [Google Scholar]
- 27.Piccinini F., Kiss A., Horvath P. CellTracker (not only) for dummies. Bioinformatics. 2016;32:955–957. doi: 10.1093/bioinformatics/btv686. [DOI] [PubMed] [Google Scholar]
- 28.He T., Mao H., Guo J., Yi Z. Cell tracking using deep neural networks with multi-task learning. Image Vis Comput. 2017;60:142–153. doi: 10.1016/j.imavis.2016.11.010. [DOI] [Google Scholar]
- 29.Nishimura K., Bise R. Spatial-temporal mitosis detection in phase-contrast microscopy via likelihood map estimation by 3DCNN. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2020;2020:1811–1815. doi: 10.1109/embc44109.2020.9175676. [DOI] [PubMed] [Google Scholar]
- 30.Ershov D., Phan M.-S., Pylvänäinen J.W., Rigaud S.U., Le Blanc L., Charles-Orszag A., Conway J.R.W., Laine R.F., Roy N.H., Bonazzi D., et al. TrackMate 7: integrating state-of-the-art segmentation algorithms into tracking pipelines. Nat. Methods. 2022;19:829–832. doi: 10.1038/s41592-022-01507-1. [DOI] [PubMed] [Google Scholar]
- 31.Bo W., Nevatia R. 2006. Tracking of Multiple, Partially Occluded Humans Based on Static Body Part Detection; pp. 951–958. [Google Scholar]
- 32.Ristani E., Solera F., Zou R., Cucchiara R., Tomasi C. Performance measures and a data set for multi-target, multi-camera tracking. arxiv. 2016 doi: 10.48550/arXiv.1609.01775. Preprint at. [DOI] [Google Scholar]
- 33.Zatulovskiy E., Skotheim J.M. On the molecular mechanisms regulating animal cell size homeostasis. Trends Genet. 2020;36:360–372. doi: 10.1016/j.tig.2020.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boward B., Wu T., Dalton S. Concise review: control of cell fate through cell cycle and pluripotency networks. Stem Cell. 2016;34:1427–1436. doi: 10.1002/stem.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu L., Michowski W., Kolodziejczyk A., Sicinski P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 2019;21:1060–1067. doi: 10.1038/s41556-019-0384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fei D.L., Motowski H., Chatrikhi R., Prasad S., Yu J., Gao S., Kielkopf C.L., Bradley R.K., Varmus H. Wild-type U2AF1 antagonizes the splicing program characteristic of U2AF1-mutant tumors and is required for cell survival. PLoS Genet. 2016;12:e1006384. doi: 10.1371/journal.pgen.1006384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato M., Larsen J.E., Lee W., Sun H., Shames D.S., Dalvi M.P., Ramirez R.D., Tang H., DiMaio J.M., Gao B., et al. Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations. Mol. Cancer Res. 2013;11:638–650. doi: 10.1158/1541-7786.Mcr-12-0634-t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ker D.F.E., Eom S., Sanami S., Bise R., Pascale C., Yin Z., Huh S.-i., Osuna-Highley E., Junkers S.N., Helfrich C.J., et al. Phase contrast time-lapse microscopy datasets with automated and manual cell tracking annotations. Sci. Data. 2018;5:180237. doi: 10.1038/sdata.2018.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thambawita V., Strümke I., Hicks S.A., Halvorsen P., Parasa S., Riegler M.A. Impact of image resolution on deep learning performance in endoscopy image classification: an experimental study using a large dataset of endoscopic images. Diagnostics. 2021;11:2183. doi: 10.3390/diagnostics11122183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sabottke C.F., Spieler B.M. The effect of image resolution on deep learning in radiography. Radiol. Artif. Intell. 2020;2:e190015. doi: 10.1148/ryai.2019190015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao H., Shi J., Qi X., Wang X., Jia J. Pyramid scene parsing network. arxiv. 2017 doi: 10.48550/arXiv.1612.01105. Preprint at. [DOI] [Google Scholar]
- 42.Badrinarayanan V., Kendall A., Cipolla R. SegNet: a deep convolutional encoder-decoder architecture for image segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 2017;39:2481–2495. doi: 10.1109/TPAMI.2016.2644615. [DOI] [PubMed] [Google Scholar]
- 43.Ioffe S., Szegedy C. Vol. 37. 2015. Batch normalization: accelerating deep network training by reducing internal covariate shift. (Proceedings of the 32nd International Conference on International Conference on Machine Learning). JMLR.org. [Google Scholar]
- 44.Jadon S. IEEE; 2020. A Survey of Loss Functions for Semantic Segmentation. [Google Scholar]
- 45.Rezatofighi H., Tsoi N., Gwak J., Sadeghian A., Reid I., Savarese S. Generalized intersection over union: a metric and A loss for bounding box regression. arxiv. 2019 doi: 10.48550/arXiv.1902.09630. Preprint at. [DOI] [Google Scholar]
- 46.Ning G., Zhang Z., Huang C., He Z., Ren X., Wang H. Spatially supervised recurrent convolutional neural networks for visual object tracking. arxiv. 2016 doi: 10.48550/arXiv.1607.05781. Preprint at. [DOI] [Google Scholar]
- 47.Wojke N., Bewley A., Paulus D. Simple online and realtime tracking with a deep association metric. arxiv. 2017 doi: 10.48550/arXiv.1703.07402. Preprint at. [DOI] [Google Scholar]
- 48.Voigtlaender P., Krause M., Osep A., Luiten J., Sekar B.B., Geiger A., Leibe B. MOTS: multi-object tracking and segmentation. arxiv. 2019 doi: 10.48550/arXiv.1902.03604. Preprint at. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Our collected datasets of images of segmented cells and cells masks are publicly available at https://deepseas.org/datasets/.
-
•
All the implemented methods are publicly available as Python scripts and can be downloaded from https://github.com/abzargar/DeepSea. The Manual annotation software and the DeepSea cell segmentation and tracking software with step-by-step instructions are uploaded to the page at https://deepseas.org/software/. The DOI is in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.