

Abstract
Advanced bladder cancer is still an area of high unmet need even with the use of immune checkpoint inhibitors and antibody drug conjugates. Therefore, transformatively novel therapeutic approaches are needed. Xenogeneic cells are capable of inducing potent innate and adaptive immune rejection responses, which properties could turn xenogeneic cells into an immunotherapeutic agent. Here, we investigated the anti-tumor effects of intratumoral xenogeneic urothelial cell (XUC) immunotherapy alone and in combination with chemotherapy in two murine syngeneic models of bladder cancer. In both bladder tumor models, intratumoral XUC treatment suppressed tumor growth, and the efficacy was enhanced with chemotherapy. The experiments on mode of action for intratumoral XUC treatment found that the remarkable local and systemic anti-tumor effects were achieved with significant intratumoral immune cell infiltration and systemic activation of immune cell cytotoxic activity, cytokine IFNγ production and proliferation ability. The intratumoral XUC alone and combined treatment increased T cell natural killer cell infiltration into tumors. In the bilateral tumor model with intratumoral XUC monotherapy or combined therapy, the uninjected tumors at the other side also simultaneously demonstrated significant tumor growth delay. Consequently, intratumoral XUC treatment alone and the combination resulted in elevated chemokine CXCL9/10/11 levels. These data suggest that intratumoral XUC therapy may be useful in the treatment of advanced bladder cancer as a local therapy that injects xenogeneic cells into either primary or distant tumors. By exerting both local and systemic anti-tumor effects, this new treatment would complete the comprehensive cancer management along with systemic approaches.
Keywords: Bladder cancer, immunity, immunotherapy, xenogeneic urothelial cell, intratumoral
Introduction
Bladder cancer is a major cause of morbidity and mortality worldwide and it was estimated with 81,180 cases and 61,700 deaths in US in 2022 [1]. For advanced bladder cancer, five-Year relative survival rate only increased one single digit from 5 to 6% with a decade of efforts to find more treatment options [2]. For eligible patients with advanced bladder cancer, platinum-based chemotherapy: gemcitabine plus cisplatin (GC) or methotrexate, vinblastine, doxorubicin, and cisplatin (MVAC) is standard first line therapy [3]. For patients who show either response or stable disease through their full course of platinum-based first-line chemotherapy, maintenance therapy with the immune check point inhibitor (ICI), programmed cell death ligand 1 (PD-L1) inhibitor, avelumab is now recommended [4]. That was based on the randomized phase III JAVELIN Bladder 100 trial that showed avelumab maintenance therapy significantly prolonged overall survival compared with the best supportive care alone, but by more than 7 months and not all patients are eligible for maintenance therapy [5]. Pembrolizumab is a PD-1 ICI used as a second-line therapy for patients with bladder cancer who previously received chemotherapy and subsequently experienced disease progression or metastasis and is also an option for first-line treatment of patients with advanced or metastatic urothelial carcinoma who are not eligible for any platinum-containing chemotherapy [6,7]. However, in phase 3 KEYNOTE-361 trial single-agent pembrolizumab, pembrolizumab plus chemotherapy (cisplatin or carboplatin plus gemcitabine) and chemotherapy only resulted in median progress free survival (PFS) 3.9, 7.1 and 8.3 months and median overall survival (OS) 15.6, 14.3 and 17.0 months respectively, indicating that the addition of pembrolizumab to first-line platinum-based chemotherapy did not significantly improve efficacy for treatment of advanced bladder cancer [8]. Furthermore, phase 3 IMvigor130 trial failed to show that adding atezolizumab, another anti-PD-L1 ICI to chemotherapy would improve OS vs chemotherapy alone in this patient population whose disease progressed during or after treatment with a platinum-based chemotherapy (previously treated) [9]. And the phase 3 trial (IMvigor211) also missed its primary end point of improved OS in the patients with advanced cancer after progression with platinum-based chemotherapy treated with atezolizumab, compared to second-line chemotherapy [9]. DANUBE studies demonstrated that durvalumab (a PD-L1 inhibitor), with or without tremelimumab (a CTLA-4 inhibitor), did not have an overall survival benefit relative to platinum-based chemotherapy and the combination with first line chemotherapy has either slight improvement or no improvement in survival [10].
In addition, despite there is a great promise for immune checkpoint inhibitors in treating advanced bladder cancer, the beneficial response rate is limited that a large majority of patients do not respond to anti-PD(L)1 drugs monotherapy and in some cases, there are severe immune adverse events [11]. These studies highlight the challenges in treating advanced bladder cancer and the need for novel therapeutic agents that target host immune and tumor microenvironment (TME) to activate anti-tumor immunity beyond using immune checkpoint inhibitors and chemotherapeutic agents. Mechanisms of immune resistance in tumors including low tumor immunogenicity and immunosuppressive TME cause limited effect of ICI immunotherapy strategy in the treatment of cancers. Thus, these obstacles highlight the urgent need for the continuing development of new immunotherapeutics to improve anti-tumor immune responses in difficult-to-treat cancers, providing additional therapeutic options with less toxicity and more efficacy. Intratumoral immunotherapy with direct injections of immunostimulatory agents (immune receptor agonists, cytokines, bacteria and virus) into the tumor sites induces the local tumor-specific immunity and generate a systemic, durable clinical response [12-14].
Xenogeneic cells trigger innate and adaptive immune responses in the host body, exemplified by immune cell infiltration, proliferation, activation and cytokine production, serving as a powerful immune barrier to successful clinical xenotransplantation [15-18]. Nonetheless, the immunological barriers could be powerful immunological boosters in cancer immunotherapy. In our group, we have focused on harnessing xenogeneic cells to enhance anti-tumor immune responses to generate local and systemic anti-tumor immunity. We have proposed that xenogeneic cells could be used as an immunotherapeutic agent to treat cancers [18,19]. In our therapeutic hypothesis, the xenogeneic tissue-specific cells are used as an immunotherapeutic agent to elicit anti-tumor innate and adaptive, as well as humoral and cellular immunity to tumor cells of the same tissue types since orthologous xenoantigens in xenogeneic tissue-specific cells and neoantigens in tumor cells of the same tissue types share similarities [19]. Xenoantigens on xenogeneic tissue specific cells when administered locally could break tolerance to self-tumor neoantigens and convert an immunosuppressive microenvironment to an active one [20].
Our previous work has demonstrated that intravesical administration of xenogeneic urothelial cells (XUC) can enhance anti-tumor immunity and convert the immunosuppressive effect of the TME in orthotopic murine bladder tumor models in monotherapy or combination with chemotherapy [19]. In addition, intravesical injection of XUCs can act as a protective agent for the urothelium damaged by the chemotherapeutic agent [21]. And intratumoral administration of xenogeneic mammary cells and xenogeneic pancreatic cells can induce therapeutically significant anti-tumor immunity to the poorly immunogenic 4T1 and Pan18 cell graft tumors [22]. In this study, we further investigated whether the method of intratumoral injection of XUCs can drive local and systemic tumor cell-specific immune responses. We examined whether local intratumoral injection of XUCs into tumors, either as monotherapy or in combination with chemotherapy, could induce immunogenic responses and at the same time generate therapeutically significant anti-tumor immune effects. Murine MBT-2 and MB49 bladder cancer cells were used for establishing syngeneic heterotopic distant tumor models to evaluate anti-tumor effect of intratumoral injection of XUCs. Responses to treatment were assessed by measuring tumor growth and tumor weight of the tumor-bearing mice. To investigate the mechanisms of action, tumor histology and immunohistochemistry and cytokine gene expression were measured. Splenic lymphocyte proliferation, cytokine production and cytotoxicity activities were also assessed. The findings showed that intratumoral injection of XUCs in monotherapy and combination with chemotherapy inhibit tumor growth. The therapeutic efficacy of intratumoral XUCs was significantly enhanced by the addition of cytotoxic chemotherapeutic agents. Mice that received combined treatment showed maximal attenuation in tumor growth rate. The anti-tumor immunity was explained by altered immune cell infiltration in tumors and immune cell functions. Our findings demonstrate that xenogeneic urothelial cells given intratumorally, provide a potent anti-tumor effect in murine bladder tumor models by enhancing recruitment and activation of immune cells in tumors for local and systemic anti-tumor effects. Moreover, intratumoral xenogeneic cell treatment turns immunologically “cold” tumors to “hot” ones, generates systemic anti-tumor immunity, and synergizes with chemotherapy.
Materials and methods
Mice
C3H/He and C57BL/6 mice, male, 6 to 8 weeks old, were purchased from National Laboratory Animal Center (Taipei, Taiwan). All mice were maintained in a specific pathogen-free room at constant temperature and humidity (22±5°C, 60±3% humidity) with a 12 h light-dark cycle and treated according to National Institutes of Health guidelines. All mouse experiments and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) review board at the China Medical University (Approval No. CMUIACUC-2019-126).
Cell lines and reagents
The murine bladder cancer cell line MBT-2 and MB49 were maintained in DMEM and RPMI1640 respectively, supplemented with 10% FBS, penicillin and streptomycin (complete medium) at a humidified 5% CO2 atmosphere at 37°C. All reagents for cell cultures were purchased from Invitrogen (Invitrogen Co., Carlsbad, CA). Xenogeneic urothelial cells (XUC) were isolated and expanded from porcine bladder urothelium. Tissues were obtained from a local slaughterhouse. Porcine bladder urothelium were treated with Dispase II (2.4 U/mL, Gibco, Waltham, MA, USA) to strip urothelium from stroma and the urothelial sheets were minced into small pieces. The fragments were then digested in a digestion solution with collagenase Type IV (Sigma, Merck KGaA, Louis, USA) in Hanks’ balanced salt solution (enzymatic activity: 100 U/ml) to disaggregate the cells. XUC cells were isolated and grown in the 75 cm2 plastic flasks with complete DMEM/Ham’s F12 medium supplemented with 2% Antibiotic-Antimycotic and 10% FBS. XUC passage 3-10 cells were used in the experiments. These cells were found to be free of contamination by Mycoplasma by the Mycoplasma detection kit (Roche Diagnostics, Chicago, IL).
MBT-2 and MB49 heterotopic graft bladder tumor murine models in syngeneic immunocompetent mice and treatment
MBT-2 and MB49 heterotopic graft bladder tumor murine models were generated by implanting the tumor cells subcutaneously to form tumors. Briefly, a total of 1 × 106 MBT-2 cells or 5 × 105 MB49 cells were injected carefully underneath the skins of the backs of the mice under isoflurane anesthesia (MBT-2 cells, C3H mice; MB49 cells, C57BL/6 mice) using a 27-gauge needle to establish transplanted tumor models (6-week-old male mice were used). Tumors were measured once a week. Once the volumes of tumors were >2000 mm3, the mice were euthanized with 100% compressed CO2 gas at a flow rate of 20% chamber vol/min for 5 min. Once tumors reached 50-100 mm3, the tumor bearing mice were divided into 4 treatment groups. The tumor-bearing mice were divided into four treatment groups: (i) vehicle control, (ii) xenogeneic urothelial cell: intratumoral injection of xenogeneic urothelial cells (1 × 105 or 106 cells) twice on day 0 and day 7, (iii) gemcitabine plus cisplatin (GC) chemotherapy: intraperitoneal injection (IP) of gemcitabine (6 mg/mouse, day 1) and cisplatin (0.12 mg/mouse, day 2) once a week, for 4 weeks, and (iv) combined treatment. Treatment procedures were performed using sterile techniques. Graft tumor burdens were monitored daily and measured with calipers. Estimates of tumor volumes were calculated using the formula for a spheroid as length × width2 × 0.5. At the end of experiments, mice were sacrificed, tumors were harvested and weighed and other organs were taken for further analysis.
Assays for splenic lymphocyte cytotoxic activity
Carboxyfluorescein diacetate succinimidyl ester (CFDA-SE)-labelled MBT-2 cells or XUC cells were prepared by staining the cells for 15 min in the darkness with 5 μM CFDA-SE (Thermo Fisher Scientific, C1157, USA) in PBS and then washed. These cells were then used as the target cells for assessment of effector splenic lymphocyte cytotoxic activity. First the CFDA-SE labelled cells were cultured in monolayer for 24 hours and then co-cultured with effector lymphocytes isolated from the spleens of mice with different treatments at effector/target (E:T) ratio = 10:1. After incubation for 8-10 hours, effector cells were removed, the fluorescent intensity of remaining adherent CFDA-SE labeled targets cells were measured by a fluorimeter. The intensity of CFDA-SE labeled targets cells without co-culturing effector cells was set as the baseline. Relative cytotoxic activity of effector lymphocytes from mice of different treatment was calculated from triplicate samples as [(Baseline intensity - experimental intensity)/(Baseline intensity)] and expressed as a percentage.
Enzyme-linked immunosorbent assay (ELISA)
IFNγ level in culture medium of effector lymphocytes isolated from the tumor-bearing mice of different treated groups, stimulated by the co-culture of target tumor cells for 3 days, was evaluated using an enzyme-linked immunosorbent assay kit (Biolegend, San Diego, CA, USA) according to the manufacturer’s protocol.
Mixed lymphocyte proliferation (MLR) assay
For carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) proliferation assay, lymphocytes from spleens of tumor bearing mice with different treatments were labelled as effector cells. The assay was performed by co-culturing 5 × 104 target xenogeneic cells or tumor cells together with 5 × 105 CFDA-SE-labeled effector lymphocytes from spleens (E/T ratio of 10:1) for 3 days. The intensity of CFDA-SE fluorescence in lymphocytes was measured by using AttuneTM Acoustic Focusing Cytometer (Thermo) and analyzed with FlowJo Software.
Histochemistry and immunohistochemistry
Tumors removed from the mice of different treated groups were fixed in formalin and embedded in paraffin and paraffin sections were stained with anti-mouse Ki67 (ab16667, Abcam, Cambridge, MA, USA), CD4 (50134-R766, Sino Biological, Wayne, PA, USA), CD8 (50389-T26, Sino Biological), CD56 (108577-T08, Sino Biological) and CD11B (ab133357, Abcam) monoclonal antibody (dilution 1:100) by the standard manufacturer’s procedures using automated Leica Bond III-autostainer. DAB was applied and incubated to visualize the signals of the antibody staining. Hematoxylin was used as counterstain. The slides were observed under a light microscope. Numeration of staining positive cells was performed in 4 random high-power fields (HPF) of the tumor sections × 400 magnification, and expressed as average cell number per field.
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay
DNA fragmentation in apoptotic cells of tumor sections was detected by TUNEL assay, following the manufacturer’s protocol. (TUNEL BrightGreen Apoptosis Detection Kit, Vazyme Biotec, Nanjing, Jiangsu, China). All images were obtained using a fluorescence microscope (Nikon Eclipse 80i) with an attached CCD camera.
Quantitative RT-PCR (qRT-PCR)
For qRT-PCR analysis, total RNAs was extracted from tumors using TRIzol® (Invitrogen, CA, USA). Real time RT-PCR quantitative mRNA analyses were performed using one-step RT-PCR kit with SYBR Green and a Bio-Rad iCycler (BioRad, CA, USA). The relative quantity of gene expression was analyzed by the 2(-ΔΔCt) method with normalization to the endogenous control β-actin and the RNA level in naïve control was set to 1. The sequences of IFNγ primers: Forward primer: 5’-CAGCAACAGCAAGGCGAAAAAGG-3’ and Reverse primer: 5’-TTTCCGCTTCCTGAGGCTGGAT-3’. The sequences of CXCL9 primers: Forward primer: 5’-CCTAGTGATAAGGAATGCACGATG-3’ and Reverse primer: 5’-CTAGGCAGGTTTGATCTCCGTTC-3’. The sequences of CXCL10 primers: Forward primer: 5’-ATCATCCCTGCGAGCCTATCCT-3’ and Reverse primer: 5’-GACCTTTTTTGGCTAAACGCTTTC-3’. The sequences of CXCL11 primers: Forward primer: 5’-CCGAGTAACGGCTGCGACAAAG-3’ and Reverse primer: 5’-CCTGCATTATGAGGCGAGCTTG-3’.
NanoString analysis
Tumor samples were harvested as described above. The samples were collected and enzymatically digested (Liberase™, Roche, Solna, Sweden) before total RNA was isolated using the RNeasy® Plus RNA isolation kit (Qiagen AB, Sweden). The gene expression levels were directly measured as mRNA counts using the Mouse-Pan cancer immune-oncology kit (NanoString, Seattle, WA). Gene expression analysis was performed using nSolver Analysis software (NanoString).
In vivo XUC tracking
The XUCs were first labeled with AIE dots (LuminiCell Tracker™ 670-Cell Labeling Kit) (EMD Millipore Corporation, Temecula, CA, USA). AIE dot-labeled (1 nM) XUCs (1 × 106) were dissolved in 30 μL, which were then intratumorally injected into the tumor of mice. The Xenogen IVIS Spectrum (Xenogen, Alameda, CA, USA) was utilized to image the mice by placing the anesthetized mice on the equipped platform (λex = 500 nm, signal collection: 680 nm), exposure time = 50 ms, scans: day 0, 7, 14, and 21 post injection, respectively.
Statistical analysis
All statistical analyses were performed using GraphPad Prism software (GraphPad Software, Inc.). Data are expressed as mean ± standard deviation (SD). Differences between two groups or among multiple groups were assessed by the unpaired t-test or the one-way ANOVA test. Differences were considered significant at a P level less than 0.05. P values were as follows: *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
Results
The intratumoral XUCs exhibit in vivo anti-tumor efficacy in monotherapy and combination with chemotherapy using the murine MBT-2 and MB49 bladder graft tumor model
To evaluate the therapeutic effect of intratumoral XUC treatment for bladder cancer, we therefore tested the anti-tumor effect of intratumoral XUCs in monotherapy and in combination with chemotherapy using the murine graft tumor model. We used murine syngeneic graft tumor models using mouse bladder tumor cell line MBT-2 and MB49 derived from the C3H/He and C57BL/6 strains, respectively. Treatment of intratumoral XUCs, intraperitoneal chemotherapy or combined intratumoral XUC immunotherapy and chemotherapy was initiated when the tumor growth to 50-100 mm3. The overall experimental strategy was presented graphically as shown in Figure 1A. We performed the murine syngeneic subcutaneous heterotopic graft tumor models Hence, the tumor-bearing mice as designed in Figure 1A were assigned into: (1) vehicle control, (2) intratumoral XUCs (1 × 105 or 106 cells), (3) intraperitoneal injection (IP) chemotherapy: gemcitabine (6 mg/mouse, day 1) and cisplatin (0.12 mg/mouse, day 2), GC regime, (4) combined intratumoral XUC immunotherapy and chemotherapy. The results showed that XUCs therapy and GC chemotherapy limited tumor volume and weight. Furthermore, intratumoral injection of XUCs with 1 × 106 cells significantly inhibited tumor volume and weight compared with XUCs of 1 × 105 cells and chemotherapy. The highest anti-tumor activity to combined immunotherapy and chemotherapy in mouse bladder tumor cell line of MBT-2 (Figure 1B and 1C). Similar results were found in MB49 mouse bladder tumor cell lines with the highest anti-tumor activity in combined immunotherapy and chemotherapy (Figure 1D and 1E), indicating that both xenogeneic cell therapy and chemotherapy suppressed tumor growth and the combination could produce a better result.
Figure 1.
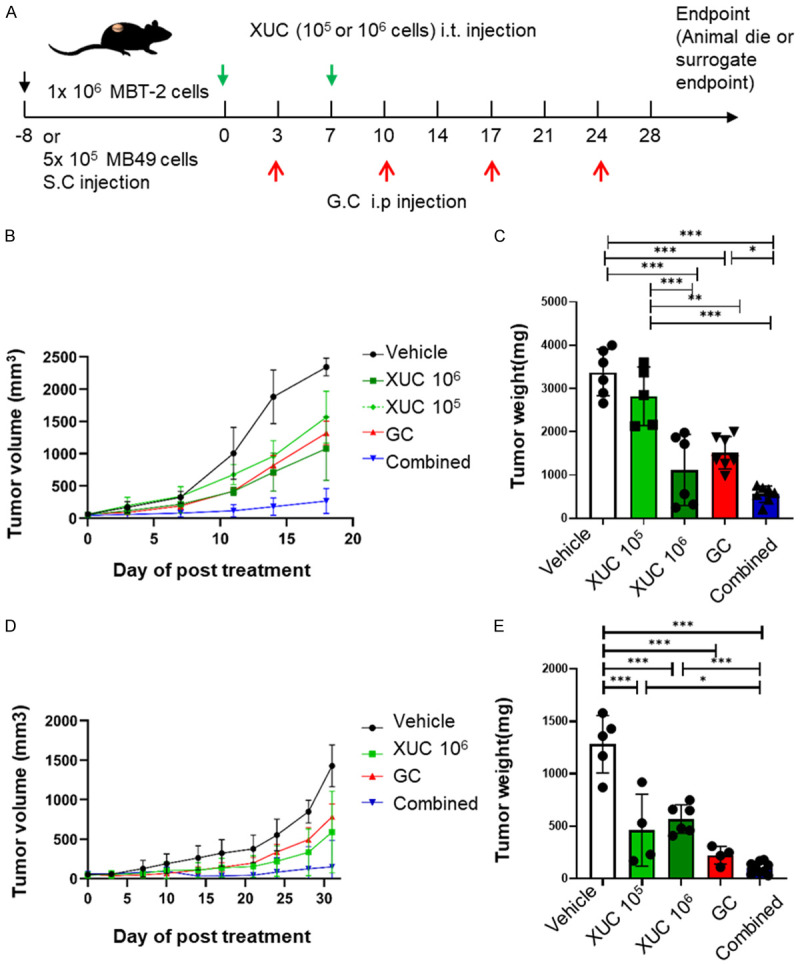
The anti-tumor effect of intratumoral xenogeneic urothelial cells in monotherapy and combination with chemotherapy using two murine bladder heterotopic graft tumor models. The mice were injected subcutaneously (s.c.) with either MBT-2 or MB49 cells into the hind flanks and when the tumor size reached 50-100 mm3, mice were randomized into 4 groups (day 0) received vehicle control, two consecutive intratumoral (i.t.) injection of xenogeneic urothelial cells (XUC) (1 × 105 or 106 cells), intraperitoneal (i.p.) injection of gemcitabine (6 mg/mouse, day 1) and cisplatin (0.12 mg/mouse, day 2) (GC), or combined treatment of XUCs plus GC. Tumor diameters were measured twice a week for 28 days. Tumor volume was determined using the formula for a spheroid: (0.5 × length × width2). A. Experimental design and treatment scheme. B. Tumor growth rate measured in volumes of MBT-2 tumor bearing mice with different treatments. C. Tumor weights from MBT-2 tumor bearing mice with different treatments. D. Tumor growth rate measured in volumes of MB49 tumor bearing mice with different treatments. E. Tumor weights from MB49 tumor bearing mice with different treatments. All tumor volumes are expressed as the mean tumor burdens ± SD (standard deviation, n = 5-10 mice/group). Tumor weights are expressed as mean ± SD (n = 6-7 per group). Significances were compared with mice that received vehicle, or mice that received either intratumoral XUCs or GC chemotherapy alone. *P<0.05; **P<0.01; ***P<0.001.
The intratumoral xenogeneic cell immunotherapy in monotherapy and combination with chemotherapy elicits immune cell cytotoxicity and IFNγ activation
T effector cells have been implicated in performing cytotoxic functions to achieve anti-tumor effects by rejecting cancer cells. In order to evaluate the cytotoxic response of intratumoral XUC immunotherapy on T effector cells. First, we used lymphocytes isolated from mouse spleens of the tumor bearing mice with different treatments as effector cells. MBT-2 cells, MB49 cells or XUCs were used as target cells. The target cells were then labeled with CFDA-SE and co-cultured with effector cells. The cells were washed after the final administration and incubation, and the fluorescence intensity of the cells was measured using the remaining CFDA-SE label. The results showed that the effector cells in the intratumoral XUC and combination treated groups enhanced the cytotoxicity to the targeted tumor cells and xenogeneic cells compared to those isolated in the vehicle treated and the chemotherapy treated groups in MBT-2 tumor model (Figure 2A). Similarly, in MB49 tumor model, the effector cells in the XUC and combination treatment groups exhibited more pronounced anti-tumor cells or xenogeneic cell cytotoxic effect than the vehicle treated and chemotherapy treated groups (Figure 2B). Interferon γ (IFNγ) is a cytokine with important roles in tissue homeostasis, immune and inflammatory responses and tumor immune surveillance. IFNγ can suppress tumors by acting directly on tumor cells that inhibit tumor proliferation, while increasing the major histocompatibility complex (MHC) by enhancing the function of tumor-infiltrating immune cells, including T helper cells, cytotoxic T lymphocytes (CTLs), and macrophages expression as well as antigen presentation [23]. Here, we assessed the role of IFNγ in immune cells in anti-tumor immunity. Lymphocytes were first isolated from the spleen of tumor-bearing mice of different treatment groups, then co-cultured with MBT-2, MB49 or XUCs cells, and finally the co-cultured conditioned medium was collected to test IFNγ activation status. IFNγ measurement in co-culture conditioned medium of effector cells and tumor cells or xenogeneic cells was performed using ELISA. The data showed that an increase in IFNγ activation in lymphocytes from XUC and combination treated MBT-2 tumor bearing mice were observed when MBT-2 or XUC cells were co-cultured (Figure 2C). Similarly, enhanced IFNγ activation in lymphocytes from MB49 tumor bearing mice were observed in both intratumoral XUC monotherapy and combination therapy groups when co-cultured with MB49 or XUC cells (Figure 2D). These data demonstrate that intratumoral XUCs and combined therapy with GC promote T cell cytotoxicity while sustaining their cytokine expression capacities.
Figure 2.
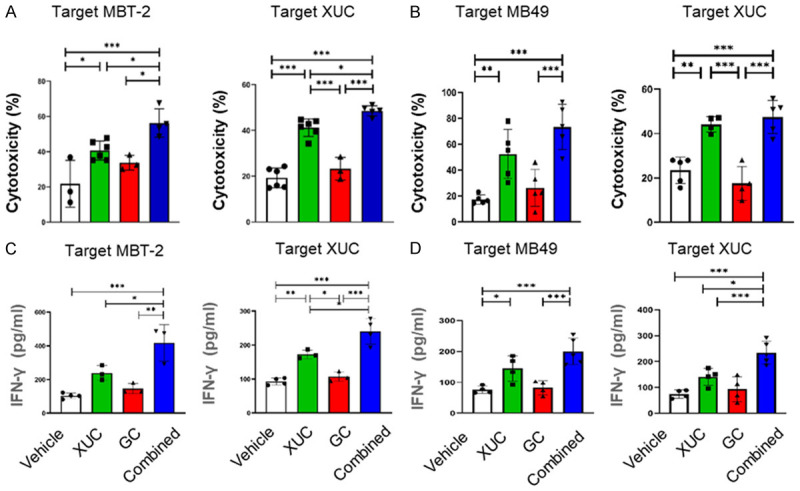
The stimulatory activity on reactive T-cell cytotoxicity and cytokine production in MBT-2-tumor and MB49-tumor-bearing mice by different treatments. Spleen lymphocytes isolated from tumor bearing mice with different treatment were tested for the ability to kill tumor cells. Effector lymphocytes (1 × 106) of different treatment groups were added into the plate seeded with 1 × 105/well of target CFDA-SE-labeled effector MBT-2 cells or XUCs and co-cultured for 8-16 h. At the end of co-culture, suspension effector cells in the wells were washed out and the intensity of CFDA-SE-labeled target cells and was measured. IFNγ level in supernatants collected after 2 days of co-culture of lymphocytes isolated from spleens of mice with different treatments with MBT-2 cells or XUCs was measured by ELISA. (A) The cytotoxicity of effector lymphocytes of MBT-2 tumor bearing mice to MBT-2 cells or XUCs. (B) The cytotoxicity of effector lymphocytes of MB49 tumor bearing mice to MB49 cells or XUCs. The relative cytotoxic activity of lymphocytes was determined following the formula with measured relative fluorescence units (RFU) of each sample: (RFUexp-RFUctrl)/RFUctrl. The intensity of CFDA-SE-labeled target cells without adding lymphocytes was set as controls. IFNγ level, measured by ELISA, in supernatants collected after 2 days of co-culture of lymphocytes isolated from spleens of MBT-2 tumor bearing mice of different treatment groups with MBT-2 cells or XUCs (C). (D) Lymphocytes isolated from spleens of MB49 tumor bearing mice under different treatments with MB49 cells or XUCs. Error bars represent SD (n≥3). *P<0.05; **P<0.01; ***P<0.001.
The intratumoral xenogeneic cell immunotherapy in monotherapy and combination with chemotherapy promote T cell proliferation
To evaluate whether intratumoral XUC monotherapy and combined therapy are capable of increase T cell proliferation, we co-cultured lymphocytes isolated from mice spleens after different treatments as effector cells and co-cultured with target tumor cells or XUCs. The results demonstrated that effector cells from XUCs-treated mice increased proliferation of targeted MBT-2 cells and exhibited more significant anti-MBT-2 cell proliferation in cells from combination treatment mice than isolated effector cells from vector-treated mice in MBT-2 tumor bearing mice. The highest proliferative activity against XUCs was observed in the group receiving combination therapy (Figure 3A and 3B). Similarly, the effector cells in MB49 tumor bearing mice receiving intratumoral XUC treatment increased the proliferation to target MB49 cells or XUCs and the effector cells from the combination exhibited most significant anti-MB49 cell and XUC cell proliferation (Figure 3C and 3D).
Figure 3.

The immune activation on reactive T-cell proliferation in MBT-2 tumor bearing mice by different treatments. The mixed lymphocyte reaction was performed using lymphocytes isolated from the spleens of MBT-2 or MB49 tumor bearing mice with different treatments as effector cells and co-cultured with target tumor cells or XUCs. A. Representative flow cytometry analysis of CFDA-SE dilution after 3 days of mixed cultures of CFDA-SE-labeled lymphocytes (effector cells) from MBT-2 tumor bearing mice when co-cultured with target MBT-2 cells or XUCs. B. Quantitation of proliferating lymphocytes responding to MBT-2 cells or XUCs. C. Representative flow cytometry analysis of CFDA-SE dilution after 3 days of mixed cultures of CFDA-SE-labeled lymphocytes (effector cells) from MB49 tumor bearing mice when co-cultured with target MB49 cells or XUCs. D. Quantitation of proliferating lymphocytes responding to MB49 cells or XUCs. Error bars represent SD. *P<0.05; **P<0.01; ***P<0.001.
The intratumoral xenogeneic cell immunotherapy alone and combined immunotherapy and chemotherapy lead to tumor necrosis, reduced cell proliferation and enhance tumor cell apoptosis
To understand the cellular events in tumors affected by intratumoral XUC treatment, we then investigated whether cell proliferation and apoptosis in tumors were affected by H&E, Ki67 and TUNEL staining analysis in the MBT-2 tumor model (Figure 4A). First, the HE staining was used to detect tumor cell necrosis. From the distribution in the tumor section slides, a large number of dead cells could be observed in tumors with intratumorally injected of XUCs and GC chemotherapy alone in MBT-2 tumors when compared to vehicle control treated tumors and the combination group showed the highest necrosis (Figure 4B). To further define the anti-tumor effect of intratumoral xenogeneic cell immunotherapy at the cellular level, we therefore assessed tumor cell changes in MBT-2 tumors under different treatments. At the end of the experiment, post-harvest fixed and sectioned tumors were stained for Ki67 immunohistochemistry (IHC) for examining cell proliferation, and cell death was determined by and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. Similar to the results of H&E staining, the intratumoral XUCs and GC chemotherapy all reduced the Ki67 positive staining cells in MBT-2 tumors, indicating inhibition of tumor cell proliferation (Figure 4C). Increase of apoptotic cells was also observed in tumors with intratumoral XUC and GC chemotherapy alone in MBT-2 tumors (Figure 4D). Furthermore, a significantly reduced number of proliferations and an increased number of apoptotic cells were observed in the combination treatment group compared to the monotherapy group (Figure 4C and 4D). These results indicated that intratumorally injection of XUCs and GC chemotherapy at a therapeutic dosage efficiently inhibited in vivo tumor growth in MBT-2 tumor-bearing mice, which resulted in cell cycle arrest and apoptosis.
Figure 4.
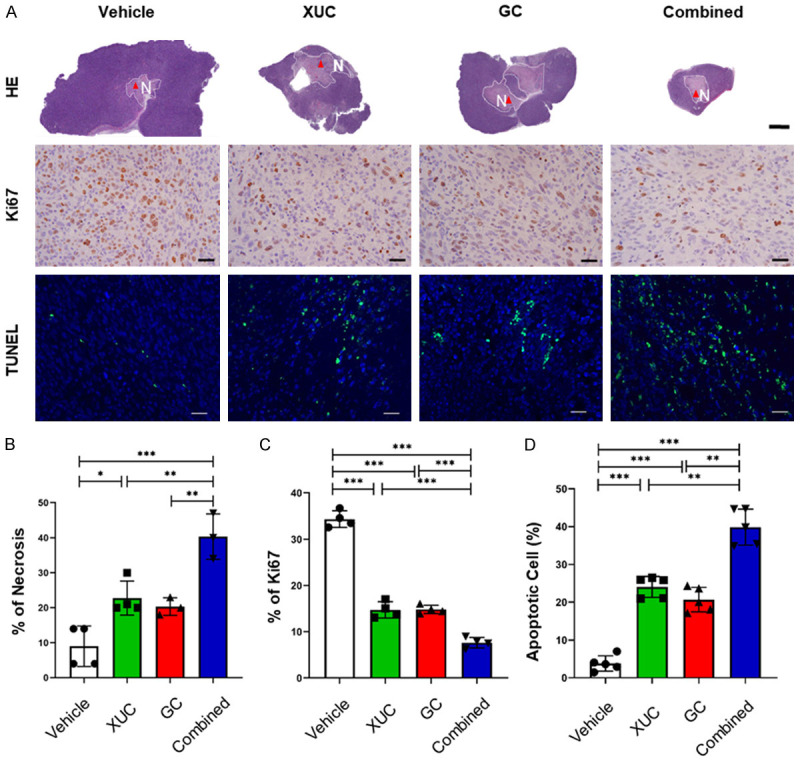
Intratumoral XUC treatment and combined therapy with GC induces necrosis, decreases the proliferation, but increases apoptosis of tumor cells. A. Representative H&E staining, Ki67 IHC and TUNEL assay of MBT-2 tumors with different treatments. Scale bar, 4 mm. B. Quantitation of the necrosis areas in MBT-2 tumors with different treatments. C. Quantitation of Ki67-positive cells on tumor sections of MBT-2 tumors with different treatments. D. Quantitation of apoptotic cells on tumor sections of MBT-2 tumors with different treatments. Scale bar, 100 μm. Data are presented as mean ± SD. *P<0.05; **P<0.01; ***P<0.001.
The intratumoral xenogeneic cell immunotherapy in monotherapy and combination with chemotherapy induce local infiltration of immune cells and altered the tumor microenvironment
The process of tumor formation and progression is influenced by two factors, genetic/epigenetic changes in tumor cells and rearrangement of the TME components through mutual and dynamic crosstalk. Immune cells are a critical component of the TME. Immune cells are divided into adaptive immune cells and innate immune cells. T cells, B cells and natural killer (NK) cells belong to the adaptive immune response. Cells that carry out an innate immune response include macrophages, neutrophils and dendritic cells [24]. Xenogeneic cells trigger innate and adaptive immune responses in the TME. Therefore, we assessed whether different treatments affected intratumoral immune cell composition. To find out the mechanisms induced by intratumoral xenogeneic therapy, we first examined infiltrating T cells in distant tumors, which were collected after various treatments to measure tumor-infiltrating CD4+ T helper cells, CD8+ cytotoxic T cells and CD56+ NK cells as well as CD11B+ myeloid derived suppressor cells (MDSC) by immunohistochemistry (IHC) (Figure 5A). These analyses showed that the intratumoral XUCs increased CD4+ T cell infiltration (Figure 5B), and both intratumoral XUC and GC chemotherapy effectively increase the tumor infiltration of CD8+ T cells (Figure 5C). CD56+ NK cell infiltration was further enhanced by intratumoral XUC immunotherapy (Figure 5D). Highest immune cell infiltration was all observed in the combination treatment group (Figure 5B-D). On the other hand, the intratumoral XUCs and GC chemotherapy effectively decreased the presence of CD11B+ MDSCs and the combined treatment group had greatest effect (Figure 5E).
Figure 5.
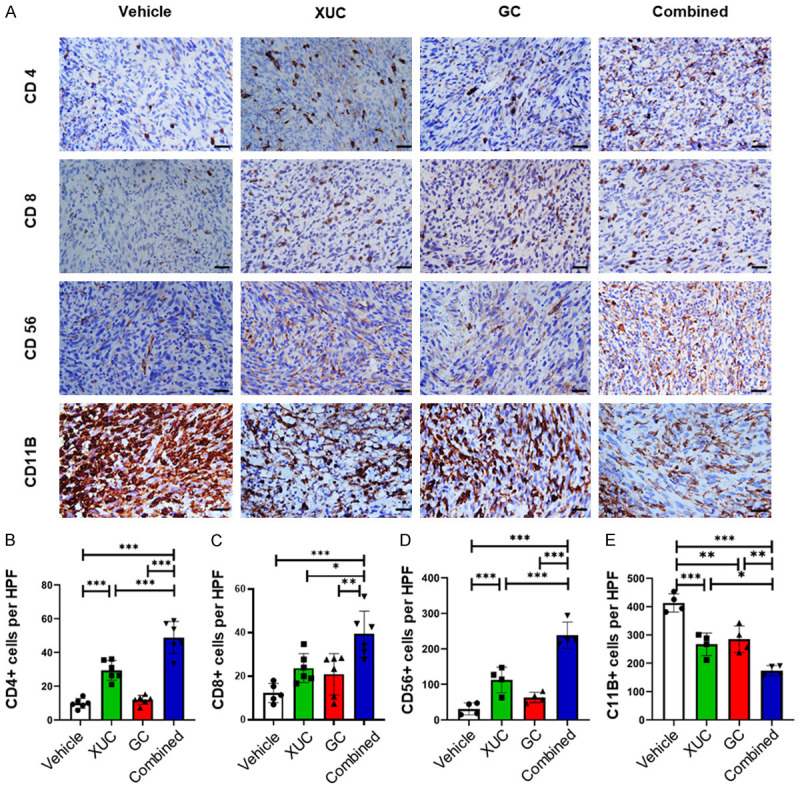
Intratumoral XUC treatment alters tumor infiltration by immune effector and suppressor cells. Combined therapy with GC and intratumoral XUCs increases the infiltration of heterotopic bladder tumors with T cells and NK cells as compared with either treatment strategy alone. Measurement of T cells (CD4+, CD8+), NK cells (CD56+), and MDSCs (CD11B+) in the tumor of tumor bearing mice by immunohistochemistry. (A) Representative images of IHC staining for CD4+, CD8+, CD56+, and CD11B+ cells. Quantitation of CD4+ (B), CD8+ (C), CD56+ (D), and CD11B+ (E) staining cells in MBT-2 tumors in each group. Scale bar, 100 μm. Immune cell infiltration was assessed by measuring intratumoral positive staining in 4 high power fields (HPF). Data are expressed as mean ± SD (n = 5). Significances were compared with mice that received vehicle control, or mice that received either intratumoral xenogeneic cell therapy or chemotherapy alone. *P<0.05; **P<0.01; ***P<0.001; **P<0.05.
The intratumoral xenogeneic cell immunotherapy in monotherapy and combination with chemotherapy exert systemic anti-tumor effect
Increased anti-tumor immune cell infiltrating immune responses by local intratumoral administration of xenogeneic cells suggest that such local therapy may have systemic effects on immunity. To analyze the systemic anti-tumor immune responses after local treatments, we used the subcutaneous bilateral graft tumor model to represent a primary and a distant tumor. MBT-2 tumor cells were implanted bilaterally into the flanks of C3H/He mice prior to randomization into treatment groups. The treatment protocol was only one tumor injected with XUCs intratumorally, and the other tumor received vehicle control. When animals with MBT-2 tumors were treated with vehicle, tumor size was larger than the one in mice treated with CG treatment (Figure 6A and 6C). Then, with one side intratumoral XUC treatment, we observed significant tumor growth suppression not only in the injected MBT-2 tumors, but also in the contralateral tumors that did not receive xenogeneic cell treatment (Figure 6B). This systemic anti-tumor effect by local intratumoral immunotherapy also enhanced the tumor suppressive effect of cytotoxic agent GC in injected (enestic) and uninjected (anenestic) tumors with tumor shrinkage in both injected and uninjected lesions in combined treatment of intratumoral XUCs plus GC for MBT-2 tumors (Figure 6D). These data demonstrate that locally administered intratumoral xenogeneic cell therapy has a systemic anti-tumor effect.
Figure 6.
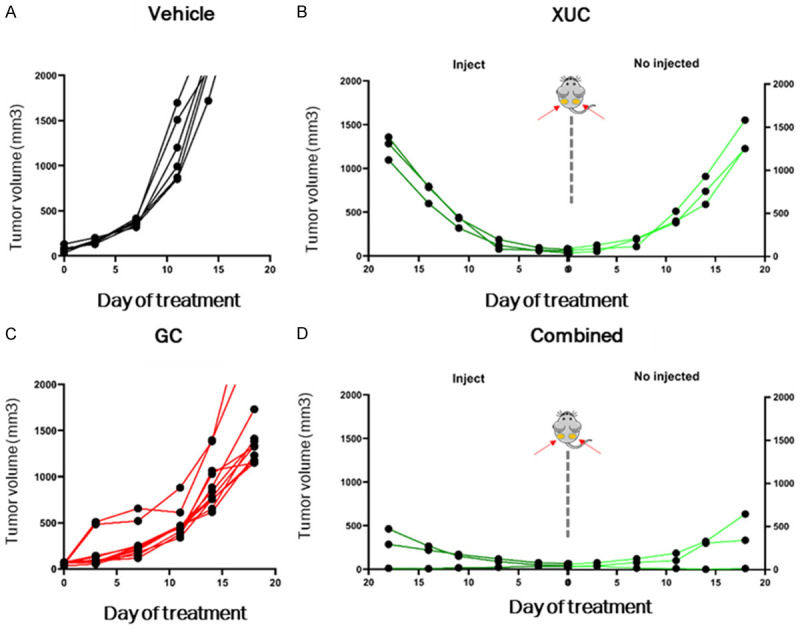
Intratumoral XUC treatment and combined therapy display systemic anti-tumor responses in bilateral MBT-2 tumor models. MBT-2 cells (1 × 106 for each side) were inoculated in both sides of mice. The MBT-2 tumor-bearing mice were either untreated or treated with intratumoral XUCs into tumors on one side but not the other side. Tumor growth on the left (injected) and right (not injected) sides was monitored and average tumor volume was determined. Tumor volume follow-up of the injected and distant tumors in the different groups of treatment (A) Vehicle, (B) Intratumoral XUC treatment, (C) GC treatment, (D) Combined therapy with GC and intratumoral XUCs.
NanoString gene expression profiling reveals immune-related genes regulated by xenogeneic monotherapy and combination with chemotherapy
Response rates to xenogeneic and combination correlate with the extent of tumor immune infiltrate, but the mechanisms underlying the recruitment of T cells following therapy are poorly characterized. A greater understanding of these processes may see the development of therapeutic interventions that enhance T-cell recruitment and, consequently, improved animal model outcomes. We therefore investigated the factors essential for immune cell recruitment and subsequent therapeutic efficacy of these immunotherapies. To identify immune-related transcriptional targets of xenogeneic cell monotherapy and combination with chemotherapy, we subjected RNA from MBT-2 tumors with XUCs and combination with GC to NanoString profiling using the nCounter PanCancer Mouse immunology-related genes (Figure 7A). Interestingly, the data show that immune response genes of CXCL9, CXCL10, and CXCL11 were up-regulated by XUCs and combination with GC (Figure 7B). The relative fold-change regulations are depicted in Figure 7C and 7D by NanoString or qRT PCR. These data suggest a role for CXCL9, CXCL10, and CXCL11 in the TME and immune responses. Chemokines are proteins which induce chemotaxis, promote differentiation of immune cells, and cause tissue extravasation. Given these properties, their role in anti-tumor immune responses in the cancer environment is of great interest. Although immunotherapy has shown clinical benefit for some cancer patients, other patients do not respond. One of the mechanisms of resistance to checkpoint inhibitors may be chemokine signaling. The CXCL9, -10, -11/CXCR3 axis regulates immune cell migration, differentiation, and activation, leading to tumor suppression (paracrine axis). However, there are some reports that show involvements of this axis in tumor growth and metastasis (autocrine axis). Thus, a better understanding of CXCL9, -10, -11/CXCR3 axis is necessary to develop effective cancer control. In this article, we summarize recent evidence regarding CXCL9, CXCL10, CXCL11/CXCR3 axis in the immune system and discuss their potential role in cancer treatment [25]. Therefore, xenogeneic cell monotherapy and combination with chemotherapy modulate the expression of many genes related to immunology and tumor immune microenvironment.
Figure 7.

CXCL9, CXCL10, and XCL11/CXCR3 axis for immune activation is involved in intratumoral XUC treatment. Four tumor samples per treatment group were collected for analysis. All four groups were run on the NanoString nCounter PanCancer Mouse Immune Profiling gene expression platform. A. NanoString immune-related gene expression heat map of 13 differentially expressed genes in xenogeneic and combined treatments compared to vehicle group. Red and green color represent up-regulated and down-regulated genes, respectively. B. Immune response categories as defined by Nanostring annotation. C. Comparisons of CXCL9, CXCL10 and CXCL11 gene expression level in NanoString assay. D. Quantitative RT-PCR analysis of CXCL9, CXCL10 and CXCL11 mRNA expression in MBT-2 tumors from 4 group mice after treatment. Data are presented as cDNA copies of indicated gene per copy of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Data are mean ± SD; *P<0.05.
Intratumorally injected XUCs remain in the injected sites
To evaluate the destination of injected cells, the distribution of intratumoral injections of XUCs was then further assessed. XUCs were labelled with LuminiCell Trackers™, which are biocompatible organic fluorescent nanoparticles based on aggregation induced emission (AIE) dot technology. AIE fluorogens are non-emissive in solutions but become highly fluorescent upon aggregate formation with long signal duration, low background auto-fluorescence and minimal signal quenching. Labelled XUCs were injected intratumorally, and mice were sacrificed at indicated time points, and distribution was assessed within the body by in vivo imaging and tumor sections through fluorescence microscopy. Injected cells remained in the tumor on days 7 and 14, but disappeared at day 21 (Figure 8A and 8B). Residues of xenogeneic cells were tested by the presence of porcine mitochondrial cytochrome-b (Cyt-b) gene in tumors. The results showed that porcine mitochondrial DNA was still detectable on day 21 after the last injection, while the normal tissues not detected (Figure 8C). These results demonstrated the safety and retention of XUCs when administered intratumorally.
Figure 8.
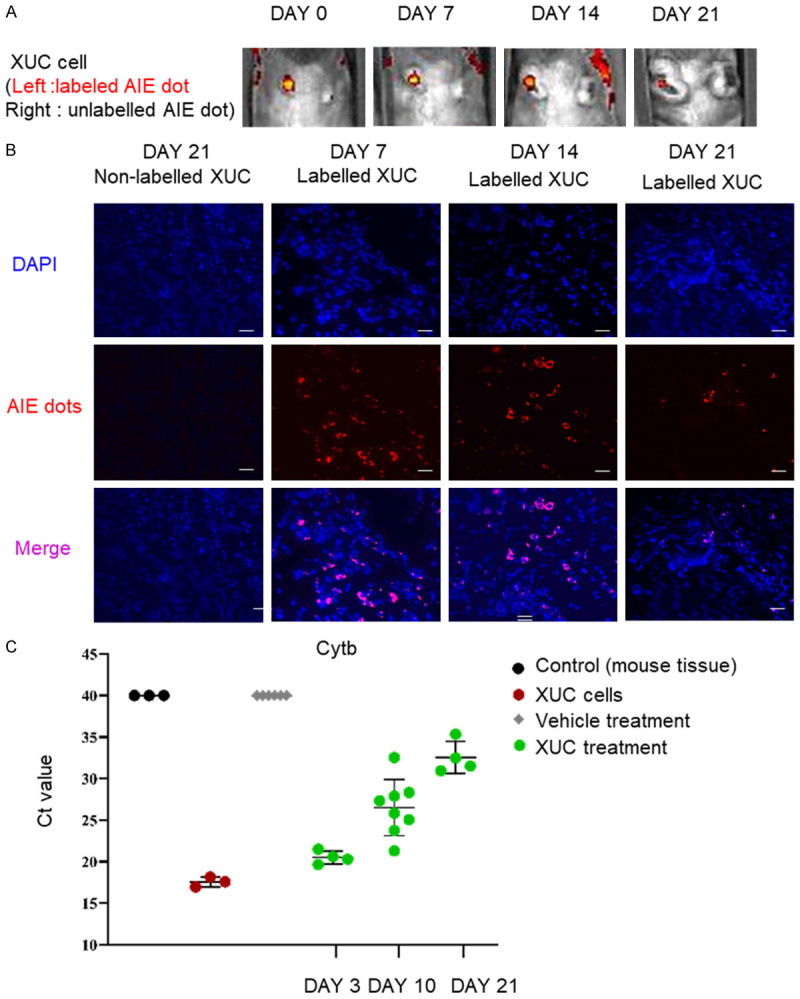
Xenogeneic urothelial cells (XUC) remain in the injected tumor site after intratumoral injection. LuminiCell labelled XUCs or non-labelled XUCs were intratumorally injected into MBT-2 tumors. A. Representative images of IVIS AIE dot labeled (red) XUCs at 0, 7, 14, and 21 days after the intratumoral injection of XUC cells with or without labelling. B. Representative section images of the injected site at 7, 14, and 21 days after injection of XUC cells. C. The presence of porcine mitochondrial DNAs in MBT-2 tumors was detected different time points after injection. DNA was isolated from tumors with vehicle control or intratumoral XUC treatment for real-time qPCR with primers specific for porcine mitochondrial cytochrome-b (Cytb) gene. Threshold cycles (Ct) were determined and plotted.
Discussion
In the present study, we demonstrated the use of xenogeneic cells as an immuno-oncology intratumoral immunotherapeutic agent that can potentiate T-cell responses and reverse the immunosuppressive TME in a therapeutically favorable manner. The intratumoral administration of xenogeneic urothelial cells in two syngeneic murine heterotopic graft bladder tumor models resulted in a reduction in bladder cancer tumor cell engraftment tumors and prolonged survival in mice (Figure 1). Increased cytotoxicity and IFNγ activation in lymphocytes from XUCs-treated mice could be observed when cells were co-cultured with murine bladder cancer cells or XUCs in the intratumoral XUC treatment and combination therapy groups, suggesting that intratumoral xenogeneic cell immunotherapy would turn on the body rejection and anti-tumor immune responses at the same time (Figures 2 and 3). Our findings demonstrate that through anti-tumor immunity, intratumoral xenogeneic cell therapy can effectively confine tumors by enhancing tumor necrosis (Figure 4A and 4B), repressing cell proliferation (Figure 4A and 4C), and increasing apoptosis (Figure 4A and 4D). The chemotherapy combinations offer a new therapeutic direction, demonstrating synergistic anti-tumor activity and improved tumor control with combination therapy. Therefore, this localized immuno-oncology approach could be a new option for immunotherapy combined with chemotherapy. Intratumoral xenogeneic cell immunotherapy resulted in changes in the TME, which enhanced the infiltration of intratumoral CD8+ and CD4+ T cells, CD56+ NK cells with the decrease of CD11B+ MDSC cells (Figure 5A-E). Furthermore, intratumoral administration of xenogeneic urothelial cells alters the tumor environment and increases systemic anti-tumor immunity, thus inhibiting the growth of injected and distant uninjected tumors (Figure 6). Here, we further searched for the immune response genes of the immune-related genes CXCL9, CXCL10 and CXCL11 induced by intratumoral xenogeneic cell therapy (Figure 7). For cold tumors, improved T-lymphocyte entrance can be achieved by induction of chemokines such as CXCL9, CCL10, or CCL11 that recruit T cells [26]. Based on the above research results, intratumoral administration of xenogeneic urothelial cells combined with chemotherapy could provide a new treatment strategy for advance bladder cancer.
Cancer immunotherapy is associated with recognition of tumor-associated antigens by patient immune cells [27]. Cancer immune elimination also involves innate immune components such as NK cells and NK cell-mediated antibody-dependent cytotoxicity and the release of pro-inflammatory cytokines such as tumor necrosis factor-alpha and interferon-gamma [28,29]. Over the past decade, the field of oncology has been revolutionized by the advent of ICIs, such as CTLA-4, PD-1, and PD-L1. Combining anti-PD-L1 antibodies with chemotherapy still retains the systemic toxicity of chemotherapy but also increases efficacy and survival. However, the median duration of response obtained with anti-PD-L1 plus chemotherapy was close to chemotherapy alone, indicating that anti-PD-L1 has shorter median duration of response [30]. In addition, anti-PD-L1 was combined with anti-CTLA-4 antibody, which improved response rates obtained with anti-PD-L1 alone, increased response durability and effectivity with increasing doses of CTLA-4, but also produced immune-related adverse event responses [31-34]. By contrast, intratumoral xenogeneic cell immunotherapy provides enhanced local efficacy and reduced systemic toxicity by achieving high bioavailability at the injected tumor site while limiting systemic exposure. Because multiple image-guided intratumoral injections of immunostimulatory agents in patients with advanced solid tumors, including deep and visceral organ locations is feasible [35], incorporating standardized intratumoral injection delivery technology would optimize the efficacy of intratumoral immunotherapy as a precision medicine to precisely administer treatment into tumors with right immune responses toward tumor cells.
The cancer immune cycle is defined as that required by the immune system to effectively control cancer growth [36]. The release of neoantigens due to genomic instability during the process is initiated. Cancer-associated antigens are captured by dendritic cells, which prime and activate tumor specific cytotoxic CD8+ T cells after migrating to the lymph node. These effector cells migrate and infiltrate the tumor stroma and identify and eliminate cancer cells. T cell mediated cytotoxic responses release new tumor antigens, thereby promoting cancer immune cycling [37,38]. It has been shown in mouse cancer analyses that persistent loss of cancer cells expressing T cell targets may enable cancers to evolve to avoid attack [39]. Anti-PD1/PD-L1 therapy provides little benefit to many patients, whom could be stratified as insensitive or exhibiting primary or acquired resistance [40,41]. Negative regulation of T cell responses that escape lymphoid organ checkpoints and tumor immune regulatory functions has been shown in human cancers to be the cause of failure of immune protection in patients [41-43]. Under anti-PD-1/PD-L1 therapy, multiple biological factors contribute to treatment resistance, including lack of T cell recognized cancer antigens, impaired cancer antigen presentation and impaired cancer specific T cell activation and T cell to tumor transfer. Poor invasiveness and accumulation of immunosuppressive factors and cells in the TME [37]. The synchronized blockade of immunosuppressive mechanisms may be combined with other conventional strategies to further overcome immune tolerance and promote tumor regression through the immunosuppressive nature of TME induced by autologous tumor cells [44]. Therefore, a new therapeutic approach is needed to enhance tumor immunogenicity and immune cell infiltration by reducing immunosuppression in the TME. Intratumoral xenogeneic cell immunotherapy has multiple mechanisms of action, including innate NK cell and adaptive T cell responses and TME non self-cell humoral immune responses to generate systemic immune responses. Xenografts can generate strong T- and B-cell responses to solid organ and cellular xenografts, and T-cell responses play a major role in xenogeneic rejection. Tumors are eliminated by the host immune system recognizing xenoantigens on xenogeneic cells [45-48]. Using bladder tumor cells co-cultured with XUC, increased activation and cytotoxicity of immune cells to cancer cells in mouse lymphocytes was observed (Figures 2 and 3). The xenogeneic cell immune rejection can ease tumor neoantigen tolerance, thereby promoting TME responses to immune cell repertoire and immune gene expression (Figure 7), thereby inhibiting tumor progression. It is possible that xenogeneic cellular immunotherapy can also be used as an in situ vaccine to directly generate a systemic immune response at the tumor site, and use epitope spreading to generate anti-tumor immunity. The host body immune system recognizes xenoantigen on xenogeneic cells and develops strong responses to eliminate xenogeneic cells [45-48]. Thus, various xenoantigens, which can engage with antibodies and immune cells may help break tolerance to tumor neoantigens and reverse immunosuppressive TME on immune cell profile and immune gene expression (Figures 5A, 5B and 7) to improve cancer outcomes. Xenogeneic DNA vaccination approaches that use xenoantigens-orthologous proteins from other species which differ in several amino acids can enhance the immunogenicity and overcome the immune tolerance to tumor antigens to elicit a cross-reactive anti-tumor immune response [49-51]. Xenogeneic DNA vaccination of dogs with advanced malignant melanoma has been demonstrated as a safe and potentially therapeutic modality [52]. With thousands of xenoantigens in xenogeneic cells, xenogeneic cell immunotherapy could induce more intense immune responses in tumor sites as an in situ vaccination and induce epitope spreading for more efficacious anti-tumor immunity. We have previously demonstrated that anti-tumor effects of intratumoral xenogeneic mammary cells for breast cancer and xenogeneic pancreatic cells for pancreatic cancer specifically [22]. Here we showed that intratumoral xenogeneic urothelial cells for bladder cancer also inhibited tumor progression, which tumor specificity could contribute to the expression of orthologous tissue specific proteins in xenogeneic tissue cells, which activate anti-tumor immune responses to the homologous proteins expressed in tumor cells of the same tissue type as xenogeneic DNA vaccine works [20].
Overt immunological responses and zoonotic infection risks are the major safety concerns for clinical application of xenogeneic cell therapy. From the clinical experiences on xenogeneic cell therapy using porcine islets in human clinical trials with native cells or encapsulated cells, no complications and porcine endogenous retrovirus infection were reported on the recipients with or without immunosuppression [53-55], demonstrating the safety of implanting xenogeneic cells into the human body. Xenogeneic cell therapy is regulated by FDA and EMA, which require quality control measures for isolation, expansion and well characterization of xenogeneic cells from safe donor animals and studies showing significant and durable efficacy and long-term safety of xenogeneic cell products [20]. Therefore, as small molecule drugs and protein drugs, in order to be used in clinics, xenogeneic cell therapy must go through a rigorous evaluation of safety, quality, and effectiveness before this new therapeutic modality can be utilized to treat diseases.
Antibody-drug conjugates (ADC) have played an increasingly prominent role in the treatment of advanced bladder cancer. Two such agents, enfortumab vedotin (EV) and sacituzumab govitecan (SG), are currently indicated for patients with advanced bladder cancer following platinum-based and immune checkpoint inhibitor therapy. EV is a Nectin-4-directed antibody-drug conjugate comprised of a fully human mAb conjugated to the microtubule-disrupting agent, monomethyl auristatin E (MMAE), via a protease-cleavable linker, which in phase III EV-301 trial showed longer OS (12.88 vs 8.97 months) than chemotherapy (docetaxel, paclitaxel, or vinflunine) as well as longer PFS (5.55 vs 3.71 months) than chemotherapy [56]. The incidence of grade ≥3 AEs was similar in both ADC and chemotherapy groups, suggesting no less toxicities for ADCs [56]. SG is a TROP-2-directed antibody-drug conjugate with an SN-38 payload that has also shown preliminary activity in advanced bladder cancer with most common toxicities of neutropenia and diarrhea [57]. Furthermore, EV+pembrolizumab (EV+P) combination vs gemcitabine+cisplatin or carboplatin in patients with previously untreated advanced bladder cancer and are ineligible for cisplatin have been currently being investigated, but substantial toxicity was observed [58]. Thus, there is a great unmet need to further improve outcomes by combining therapies which would need to be an effective, non-cross resistant therapy with non-overlapping toxicity to systemic immunotherapy and chemotherapy. Intratumoral delivery of immunotherapeutics is being tested in clinical trials to promote superior anti-tumor immune activity in the context of limited systemic toxicity [59]. Our novel intratumoral XUC immunotherapy could trigger local and systemic immunologic responses via direct injection to induce rejection of xenoantigens and then spread to tumor-derived antigens and subsequent activation of tumor-specific effector T cells, which would an ideal intratumoral therapeutic agent. Intratumoral XUC immunotherapy represents a novel intratumoral immunotherapy strategy by locally administering immune-activating xenogeneic cells into the tumor itself to break immunosuppressive shields and furthermore, the local immune activation in injected tumors can further lead to systemic anti-tumor immunity in noninjected tumors. These unique features of intratumoral XUC treatment grant its advantages to combine with other bladder cancer therapeutic modalities such as systemic cytotoxic chemotherapeutic agents or ADCs with cytotoxic payloads without adding further toxicities caused by cytotoxic agents. In addition, local treatment nature and multiple MOAs of intratumoral XUC treatment could induce less immune-related adverse events than ICIs, whose are driven by ICI abilities to destruct self-tolerance and activate self-reactive T cells as well as break the mechanisms of adaptive immune resistance to ICIs [60,61]. Thus, intratumoral XUC immunotherapy could also be used in combination with ICIs on ICI resistant cancers to achieve high rate of disease control and durable responses.
We have demonstrated the ability of intratumoral xenogeneic urothelial cell immunotherapy to inhibit tumor growth in syngeneic heterotopic bladder tumor models. Furthermore, our studies have shown that intratumoral xenogeneic cell treatment can effectively provide systemic anti-tumor effects. We have also demonstrated that intratumoral xenogeneic cell immunotherapy generates local and systemic anti-tumor immunity by altering the immune TME and activating immune cells in the lymphatic system. These data suggest that intratumoral XUC immunotherapy can be used as a new and promising approach for the treatment of advanced bladder cancer. Our study reveals new prospects for the anti-tumor activity of intratumoral XUC immunotherapy, which is worthy of further study in the clinic.
Acknowledgements
This work was supported by the NSTC grant (110-2314-B-039-026-MY3-) and CMUH grants (DMR-111-212, DMR-112-202 and DMR-CELL-1807).
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 3.von der Maase H, Sengelov L, Roberts JT, Ricci S, Dogliotti L, Oliver T, Moore MJ, Zimmermann A, Arning M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 2005;23:4602–4608. doi: 10.1200/JCO.2005.07.757. [DOI] [PubMed] [Google Scholar]
- 4.Flaig TW, Spiess PE, Abern M, Agarwal N, Bangs R, Boorjian SA, Buyyounouski MK, Chan K, Chang S, Friedlander T, Greenberg RE, Guru KA, Herr HW, Hoffman-Censits J, Kishan A, Kundu S, Lele SM, Mamtani R, Margulis V, Mian OY, Michalski J, Montgomery JS, Nandagopal L, Pagliaro LC, Parikh M, Patterson A, Plimack ER, Pohar KS, Preston MA, Richards K, Sexton WJ, Siefker-Radtke AO, Tollefson M, Tward J, Wright JL, Dwyer MA, Cassara CJ, Gurski LA. NCCN guidelines® insights: bladder cancer, version 2.2022. J Natl Compr Canc Netw. 2022;20:866–878. doi: 10.6004/jnccn.2022.0041. [DOI] [PubMed] [Google Scholar]
- 5.Powles T, Park SH, Voog E, Caserta C, Valderrama BP, Gurney H, Kalofonos H, Radulović S, Demey W, Ullén A, Loriot Y, Sridhar SS, Tsuchiya N, Kopyltsov E, Sternberg CN, Bellmunt J, Aragon-Ching JB, Petrylak DP, Laliberte R, Wang J, Huang B, Davis C, Fowst C, Costa N, Blake-Haskins JA, di Pietro A, Grivas P. Avelumab maintenance therapy for advanced or metastatic urothelial carcinoma. N Engl J Med. 2020;383:1218–1230. doi: 10.1056/NEJMoa2002788. [DOI] [PubMed] [Google Scholar]
- 6.Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L, Vogelzang NJ, Climent MA, Petrylak DP, Choueiri TK, Necchi A, Gerritsen W, Gurney H, Quinn DI, Culine S, Sternberg CN, Mai Y, Poehlein CH, Perini RF, Bajorin DF KEYNOTE-045 Investigators. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376:1015–1026. doi: 10.1056/NEJMoa1613683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vuky J, Balar AV, Castellano D, O’Donnell PH, Grivas P, Bellmunt J, Powles T, Bajorin D, Hahn NM, Savage MJ, Fang X, Godwin JL, Frenkl TL, Homet Moreno B, de Wit R, Plimack ER. Long-term outcomes in KEYNOTE-052: phase II study investigating first-line pembrolizumab in cisplatin-ineligible patients with locally advanced or metastatic urothelial cancer. J. Clin. Oncol. 2020;38:2658–2666. doi: 10.1200/JCO.19.01213. [DOI] [PubMed] [Google Scholar]
- 8.Powles T, Csőszi T, Özgüroğlu M, Matsubara N, Géczi L, Cheng SY, Fradet Y, Oudard S, Vulsteke C, Morales Barrera R, Fléchon A, Gunduz S, Loriot Y, Rodriguez-Vida A, Mamtani R, Yu EY, Nam K, Imai K, Homet Moreno B, Alva A KEYNOTE-361 Investigators. Pembrolizumab alone or combined with chemotherapy versus chemotherapy as first-line therapy for advanced urothelial carcinoma (KEYNOTE-361): a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:931–945. doi: 10.1016/S1470-2045(21)00152-2. [DOI] [PubMed] [Google Scholar]
- 9.Powles T, Duran I, van der Heijden MS, Loriot Y, Vogelzang NJ, De Giorgi U, Oudard S, Retz MM, Castellano D, Bamias A, Flechon A, Gravis G, Hussain S, Takano T, Leng N, Kadel EE 3rd, Banchereau R, Hegde PS, Mariathasan S, Cui N, Shen X, Derleth CL, Green MC, Ravaud A. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2018;391:748–757. doi: 10.1016/S0140-6736(17)33297-X. [DOI] [PubMed] [Google Scholar]
- 10.Powles T, van der Heijden MS, Castellano D, Galsky MD, Loriot Y, Petrylak DP, Ogawa O, Park SH, Lee JL, De Giorgi U, Bögemann M, Bamias A, Eigl BJ, Gurney H, Mukherjee SD, Fradet Y, Skoneczna I, Tsiatas M, Novikov A, Suárez C, Fay AP, Duran I, Necchi A, Wildsmith S, He P, Angra N, Gupta AK, Levin W, Bellmunt J DANUBE study investigators. Durvalumab alone and durvalumab plus tremelimumab versus chemotherapy in previously untreated patients with unresectable, locally advanced or metastatic urothelial carcinoma (DANUBE): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2020;21:1574–1588. doi: 10.1016/S1470-2045(20)30541-6. [DOI] [PubMed] [Google Scholar]
- 11.Rouanne M, Roumiguie M, Houede N, Masson-Lecomte A, Colin P, Pignot G, Larre S, Xylinas E, Roupret M, Neuzillet Y. Development of immunotherapy in bladder cancer: present and future on targeting PD(L)1 and CTLA-4 pathways. World J Urol. 2018;36:1727–1740. doi: 10.1007/s00345-018-2332-5. [DOI] [PubMed] [Google Scholar]
- 12.Champiat S, Tselikas L, Farhane S, Raoult T, Texier M, Lanoy E, Massard C, Robert C, Ammari S, De Baère T, Marabelle A. Intratumoral immunotherapy: from trial design to clinical practice. Clin Cancer Res. 2021;27:665–679. doi: 10.1158/1078-0432.CCR-20-0473. [DOI] [PubMed] [Google Scholar]
- 13.Aznar MA, Tinari N, Rullan AJ, Sanchez-Paulete AR, Rodriguez-Ruiz ME, Melero I. Intratumoral delivery of immunotherapy-act locally, think globally. J Immunol. 2017;198:31–39. doi: 10.4049/jimmunol.1601145. [DOI] [PubMed] [Google Scholar]
- 14.Hong WX, Haebe S, Lee AS, Westphalen CB, Norton JA, Jiang W, Levy R. Intratumoral immunotherapy for early-stage solid tumors. Clin Cancer Res. 2020;26:3091–3099. doi: 10.1158/1078-0432.CCR-19-3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coe TM, Markmann JF, Rickert CG. Current status of porcine islet xenotransplantation. Curr Opin Organ Transplant. 2020;25:449–456. doi: 10.1097/MOT.0000000000000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hawthorne WJ, Cowan PJ, Buhler LH, Yi S, Bottino R, Pierson RN 3rd, Ahn C, Azimzadeh A, Cozzi E, Gianello P, Lakey JRT, Luo M, Miyagawa S, Mohiuddin MM, Park CG, Schuurman HJ, Scobie L, Sykes M, Tector J, Tonjes RR, Wolf E, Nunez JR, Wang W. Third WHO global consultation on regulatory requirements for xenotransplantation clinical trials, Changsha, Hunan, China December 12-14, 2018: “The 2018 Changsha Communique” The 10-Year Anniversary of The International Consultation on Xenotransplantation. Xenotransplantation. 2019;26:e12513. doi: 10.1111/xen.12513. [DOI] [PubMed] [Google Scholar]
- 17.Galsky MD, Arija JÁA, Bamias A, Davis ID, De Santis M, Kikuchi E, Garcia-Del-Muro X, De Giorgi U, Mencinger M, Izumi K, Panni S, Gumus M, Özgüroğlu M, Kalebasty AR, Park SH, Alekseev B, Schutz FA, Li JR, Ye D, Vogelzang NJ, Bernhard S, Tayama D, Mariathasan S, Mecke A, Thåström A, Grande E IMvigor130 Study Group. Atezolizumab with or without chemotherapy in metastatic urothelial cancer (IMvigor130): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2020;395:1547–1557. doi: 10.1016/S0140-6736(20)30230-0. [DOI] [PubMed] [Google Scholar]
- 18.Huang CP, Chen CC, Shyr CR. Xenogeneic cell therapy provides a novel potential therapeutic option for cancers by restoring tissue function, repairing cancer wound and reviving anti-tumor immune responses. Cancer Cell Int. 2018;18:9. doi: 10.1186/s12935-018-0501-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang CP, Wu CC, Shyr CR. Combination of novel intravesical xenogeneic urothelial cell immunotherapy and chemotherapy enhances anti-tumor efficacy in preclinical murine bladder tumor models. Cancer Immunol Immunother. 2021;70:1419–1433. doi: 10.1007/s00262-020-02775-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang CP, Yang CY, Shyr CR. Utilizing xenogeneic cells as a therapeutic agent for treating diseases. Cell Transplant. 2021;30:9636897211011995. doi: 10.1177/09636897211011995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang CP, Chen CC, Tsai YT, Wu CC, Shyr CR. Intravesical administration of xenogeneic porcine urothelial cells attenuates cyclophosphamide-induced cystitis in mice. Cell Transplant. 2019;28:296–305. doi: 10.1177/0963689718822773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang CP, Liu LC, Chang CC, Wu CC, Shyr CR. Intratumoral xenogeneic tissue-specific cell immunotherapy inhibits tumor growth by increasing antitumor immunity in murine triple negative breast and pancreatic tumor models. Cancer Lett. 2022;545:115478. doi: 10.1016/j.canlet.2021.10.044. [DOI] [PubMed] [Google Scholar]
- 23.Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18:545–558. doi: 10.1038/s41577-018-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, Kolahian S, Javaheri T, Zare P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal. 2020;18:59. doi: 10.1186/s12964-020-0530-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, McSkane M, Baba H, Lenz HJ. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - a target for novel cancer therapy. Cancer Treat Rev. 2018;63:40–47. doi: 10.1016/j.ctrv.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu YT, Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics. 2021;11:5365–5386. doi: 10.7150/thno.58390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuster M, Nechansky A, Kircheis R. Cancer immunotherapy. Biotechnol J. 2006;1:138–147. doi: 10.1002/biot.200500044. [DOI] [PubMed] [Google Scholar]
- 28.Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008;27:5932–5943. doi: 10.1038/onc.2008.267. [DOI] [PubMed] [Google Scholar]
- 29.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 30.Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro G Jr, Psyrri A, Basté N, Neupane P, Bratland Å, Fuereder T, Hughes BGM, Mesía R, Ngamphaiboon N, Rordorf T, Wan Ishak WZ, Hong RL, González Mendoza R, Roy A, Zhang Y, Gumuscu B, Cheng JD, Jin F, Rischin D KEYNOTE-048 Investigators. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394:1915–1928. doi: 10.1016/S0140-6736(19)32591-7. [DOI] [PubMed] [Google Scholar]
- 31.Sharma P, Siefker-Radtke A, de Braud F, Basso U, Calvo E, Bono P, Morse MA, Ascierto PA, Lopez-Martin J, Brossart P, Rohrberg K, Mellado B, Fischer BS, Meadows-Shropshire S, Abdel Saci, Callahan MK, Rosenberg J. Nivolumab alone and with ipilimumab in previously treated metastatic urothelial carcinoma: CheckMate 032 nivolumab 1 mg/kg plus ipilimumab 3 mg/kg expansion cohort results. J. Clin. Oncol. 2019;37:1608–1616. doi: 10.1200/JCO.19.00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ready N, Hellmann MD, Awad MM, Otterson GA, Gutierrez M, Gainor JF, Borghaei H, Jolivet J, Horn L, Mates M, Brahmer J, Rabinowitz I, Reddy PS, Chesney J, Orcutt J, Spigel DR, Reck M, O’Byrne KJ, Paz-Ares L, Hu W, Zerba K, Li X, Lestini B, Geese WJ, Szustakowski JD, Green G, Chang H, Ramalingam SS. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J. Clin. Oncol. 2019;37:992–1000. doi: 10.1200/JCO.18.01042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, Ferrucci PF, Smylie M, Hogg D, Hill A, Márquez-Rodas I, Haanen J, Guidoboni M, Maio M, Schöffski P, Carlino MS, Lebbé C, McArthur G, Ascierto PA, Daniels GA, Long GV, Bastholt L, Rizzo JI, Balogh A, Moshyk A, Hodi FS, Wolchok JD. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381:1535–1546. doi: 10.1056/NEJMoa1910836. [DOI] [PubMed] [Google Scholar]
- 34.Ascierto PA, Del Vecchio M, Robert C, Mackiewicz A, Chiarion-Sileni V, Arance A, Lebbé C, Bastholt L, Hamid O, Rutkowski P, McNeil C, Garbe C, Loquai C, Dreno B, Thomas L, Grob JJ, Liszkay G, Nyakas M, Gutzmer R, Pikiel J, Grange F, Hoeller C, Ferraresi V, Smylie M, Schadendorf D, Mortier L, Svane IM, Hennicken D, Qureshi A, Maio M. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2017;18:611–622. doi: 10.1016/S1470-2045(17)30231-0. [DOI] [PubMed] [Google Scholar]
- 35.Sheth RA, Murthy R, Hong DS, Patel S, Overman MJ, Diab A, Hwu P, Tam A. Assessment of image-guided intratumoral delivery of immunotherapeutics in patients with cancer. JAMA Netw Open. 2020;3:e207911. doi: 10.1001/jamanetworkopen.2020.7911. [DOI] [PubMed] [Google Scholar]
- 36.Chen Daniel S, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 37.Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure) Ann Oncol. 2016;27:1492–1504. doi: 10.1093/annonc/mdw217. [DOI] [PubMed] [Google Scholar]
- 38.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 39.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 40.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer. 2016;16:121–126. doi: 10.1038/nrc.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PD-L1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71–81. doi: 10.1016/j.ctrv.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 43.Mullard A. New checkpoint inhibitors ride the immunotherapy tsunami. Nat Rev Drug Discov. 2013;12:489–492. doi: 10.1038/nrd4066. [DOI] [PubMed] [Google Scholar]
- 44.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scalea J, Hanecamp I, Robson SC, Yamada K. T-cell-mediated immunological barriers to xenotransplantation. Xenotransplantation. 2012;19:23–30. doi: 10.1111/j.1399-3089.2011.00687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Griesemer A, Yamada K, Sykes M. Xenotransplantation: immunological hurdles and progress toward tolerance. Immunol Rev. 2014;258:241–258. doi: 10.1111/imr.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ekser B, Li P, Cooper DKC. Xenotransplantation: past, present, and future. Curr Opin Organ Transplant. 2017;22:513–521. doi: 10.1097/MOT.0000000000000463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooper DKC, Ekser B, Tector AJ. Immunobiological barriers to xenotransplantation. Int J Surg. 2015;23:211–216. doi: 10.1016/j.ijsu.2015.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan J, Ku GY, Adamow M, Mu Z, Tandon S, Hannaman D, Chapman P, Schwartz G, Carvajal R, Panageas KS, Houghton AN, Wolchok JD. Immunologic responses to xenogeneic tyrosinase DNA vaccine administered by electroporation in patients with malignant melanoma. J Immunother Cancer. 2013;1:20. doi: 10.1186/2051-1426-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Srinivasan R, Wolchok JD. Tumor antigens for cancer immunotherapy: therapeutic potential of xenogeneic DNA vaccines. J Transl Med. 2004;2:12. doi: 10.1186/1479-5876-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perales MA, Blachere NE, Engelhorn ME, Ferrone CR, Gold JS, Gregor PD, Noffz G, Wolchok JD, Houghton AN. Strategies to overcome immune ignorance and tolerance. Semin Cancer Biol. 2002;12:63–71. doi: 10.1006/scbi.2001.0397. [DOI] [PubMed] [Google Scholar]
- 52.Bergman PJ, McKnight J, Novosad A, Charney S, Farrelly J, Craft D, Wulderk M, Jeffers Y, Sadelain M, Hohenhaus AE, Segal N, Gregor P, Engelhorn M, Riviere I, Houghton AN, Wolchok JD. Long-term survival of dogs with advanced malignant melanoma after DNA vaccination with xenogeneic human tyrosinase: a phase I trial. Clin Cancer Res. 2003;9:1284–1290. [PubMed] [Google Scholar]
- 53.Groth CG, Korsgren O, Tibell A, Tollemar J, Möller E, Bolinder J, Ostman J, Reinholt FP, Hellerström C, Andersson A. Transplantation of porcine fetal pancreas to diabetic patients. Lancet. 1994;344:1402–1404. doi: 10.1016/s0140-6736(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 54.Valdés-González RA, Dorantes LM, Garibay GN, Bracho-Blanchet E, Mendez AJ, Dávila-Pérez R, Elliott RB, Terán L, White DJ. Xenotransplantation of porcine neonatal islets of Langerhans and Sertoli cells: a 4-year study. Eur J Endocrinol. 2005;153:419–427. doi: 10.1530/eje.1.01982. [DOI] [PubMed] [Google Scholar]
- 55.Matsumoto S, Abalovich A, Wechsler C, Wynyard S, Elliott RB. Clinical benefit of islet xenotransplantation for the treatment of type 1 diabetes. EBioMedicine. 2016;12:255–262. doi: 10.1016/j.ebiom.2016.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Powles T, Rosenberg JE, Sonpavde GP, Loriot Y, Durán I, Lee JL, Matsubara N, Vulsteke C, Castellano D, Wu C, Campbell M, Matsangou M, Petrylak DP. Enfortumab vedotin in previously treated advanced urothelial carcinoma. N Engl J Med. 2021;384:1125–1135. doi: 10.1056/NEJMoa2035807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tagawa ST, Balar AV, Petrylak DP, Kalebasty AR, Loriot Y, Fléchon A, Jain RK, Agarwal N, Bupathi M, Barthelemy P, Beuzeboc P, Palmbos P, Kyriakopoulos CE, Pouessel D, Sternberg CN, Hong Q, Goswami T, Itri LM, Grivas P. TROPHY-U-01: a phase II open-label study of sacituzumab govitecan in patients with metastatic urothelial carcinoma progressing after platinum-based chemotherapy and checkpoint inhibitors. J. Clin. Oncol. 2021;39:2474–2485. doi: 10.1200/JCO.20.03489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heijden MSVD, Gupta S, Galsky MD, Derleth CL, Lee S, Kataria RS, Powles T. Study EV-302: a two-arm, open-label, randomized controlled phase 3 study of enfortumab vedotin in combination with pembrolizumab versus chemotherapy in previously untreated advanced urothelial carcinoma (aUC) (trial in progress) 2022;40:TPS589. [Google Scholar]
- 59.Humeau J, Le Naour J, Galluzzi L, Kroemer G, Pol JG. Trial watch: intratumoral immunotherapy. Oncoimmunology. 2021;10:1984677. doi: 10.1080/2162402X.2021.1984677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khoja L, Day D, Wei-Wu Chen T, Siu LL, Hansen AR. Tumour- and class-specific patterns of immune-related adverse events of immune checkpoint inhibitors: a systematic review. Ann Oncol. 2017;28:2377–2385. doi: 10.1093/annonc/mdx286. [DOI] [PubMed] [Google Scholar]
- 61.Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ Book. 2019;39:147–164. doi: 10.1200/EDBK_240837. [DOI] [PubMed] [Google Scholar]