

Abstract

Piperidines are frequently found in natural products and are of importance to the pharmaceutical industry. A generally useful asymmetric route to enantiomerically enriched 3-substituted piperidines remains elusive. Here we report a cross-coupling approach to enantioenriched 3-piperidines from pyridine- and sp2-hybridized boronic acids. The key step involves a Rh-catalyzed asymmetric reductive Heck reaction of aryl, heteroaryl, or vinyl boronic acids and phenyl pyridine-1(2H)-carboxylate to provide 3-substituted tetrahydropyridines in high yield and excellent enantioselectivity with a wide functional group tolerance. A three-step process involving i) partial reduction of pyridine, ii) Rh-catalyzed asymmetric carbometalation, and then iii) another reduction provides access to a wide variety of enantioenriched 3-piperidines, including clinically used materials such as Preclamol and Niraparib.
Nitrogen-containing heterocycles are prevalent in natural products and drug candidates. Specifically, piperidines are frequently found in pharmaceutically relevant molecules.1−3 For instance, Niraparib is an anticancer drug,4 Preclamol and OSU-6162 are antipsychotic agents,5,6 Tiagbine is an anticonvulsant drug,7 Pergolide has been used to treat Parkinson’s disease,6 and CP-868388 is a potential PPAR agonist from Pfizer8 (Figure 1). A number of recent papers have also uncovered further important biological activities associated with piperidines.9−12
Figure 1.

Drug candidates featuring chiral piperidine moieties.
Synthetic access to enantioenriched 3-substituted piperidines has been explored;13−15 however, a generally useful asymmetric route for the synthesis of such piperidines has not been reported. Stepwise construction of the piperidine ring suffers from lengthy synthesis and requires either stoichiometric chiral building blocks or resolution to control the absolute stereochemistry. For example, in Merck’s reported process routes for the synthesis of Niraparib, enantiopure piperidines are accessed by chiral HPLC separation or via a biocatalytic transaminase-based dynamic kinetic resolution followed by seven additional steps.16,17
A straightforward approach to 3-piperidines would rely on enantioselective reduction or functionalization of pyridine. While the dearomatization of pyridine itself is a high-energy process, a widely used strategy to circumvent this is to reduce and/or functionalize pyridiniums.18 Recently, Turner reported that a mixture of oxidase and reductase enzymes10 can be used to chemo-enzymatically dearomatize pyridiniums to access enantioenriched 3- and 4-substituted products.
The inherent reactivity of pyridiniums allows for nucleophilic addition at the 2-, 4-, or 6- positions to provide access to 2- or 4-substituted piperidines (Scheme 1A).18 However, access to 3-substituted piperidines is not viable using this approach. Rhodium-catalyzed asymmetric Suzuki–Miyaura-type coupling of aryl boronic acids to racemic piperidine-based allyl chlorides provides access to substituted tetrahydropiperidines and, after further reduction, piperidines with high enantioselectivity, though this method requires multiple steps to access the starting allyl chloride.19,20
Scheme 1. Pyridinium Functionalization and Rh-Catalyzed Carbometalation Approaches to Piperidines.

We envisioned that dihydropyridine would serve as an ideal coupling partner to access 3-substituted piperidines through a cross-coupling approach. A Rh-catalyzed asymmetric carbometalation of dihydropyridines could furnish 3-substituted tetrahydropyridines (Scheme 1B) via a reductive Heck-type process and offer access to libraries of enantioenriched 3-piperidines via a three-step process: i) partial reduction of pyridine, ii) Rh-catalyzed asymmetric carbometalation, and then iii) another reduction.
The functionalization of terminal dienes has been extensively studied. However, relatively few studies have reported the successful functionalization of internal and substituted dienes.21−23 Dihydropyridines have been used in Diels–Alder reactions to provide complex structures,24−26 but catalytic asymmetric functionalization of dihydropyridine is rare and limited to the copper-catalyzed protoborylation reported by Ito.27 To the best of our knowledge carbometalation of dienes is unknown.
Rh-catalyzed carbometalation reactions have been reported on strained and bicyclic alkenes.28−32 Recently, we have reported a cross-coupling approach for the asymmetric synthesis of cyclobutanes.28 The method involved Rh-catalyzed carbometalation of cyclobutenes, and we wondered if carbometalation could be used to selectively modify dihydropyridines.
Initial studies evaluated dihydropyridines as viable starting materials for Rh-catalyzed carbometalation. We attempted synthesis of dihydropyridines (1) bearing diverse protecting groups via partial reduction of in situ-generated pyridiniums (Scheme 2). Many protecting groups either were incompatible with the reduction conditions or underwent oxidation to provide pyridine. Major challenges with starting material synthesis include the isolation of mixtures of diene isomers, doubly reduced side products, and pyridine impurities. Carbamate protecting groups gave the best results in terms of synthesis and scalability. Specifically, phenyl carbamate-protected dihydropyridine (1a) was isolated as a white solid and could be recrystallized to provide high-purity starting material in 72% yield.
Scheme 2. Synthesis of Dihydropyridines.

After extensive exploratory work and optimization of the rhodium-catalyzed carbometalation of phenyl carbamate dihydropyridine (1a) with phenyl boronic acid (2a), we found that the combination of [Rh(cod)(OH)]2, (S)-Segphos, and aq. CsOH in a THP:toluene:H2O (1:1:1) solvent mixture at 70 °C provided tetrahydropyridine 3a in 81% isolated yield and 96% ee (Table 1, entry 1). Examined deviations from these conditions led to reduced yield and/or enantioselectivity. Poor conversion with recovered starting material was observed at less than 1 M concentration (Table 1, entry 2). Chiral diene- and ferrocene-based ligands provided poor reactivity, whereas other C2-symmetric bisphosphines examined give lower yields (Table 1, entries 3–5). Aqueous cesium hydroxide appears essential for high yields, as lower conversion was observed using aqueous cesium carbonate (Table 1, entry 6), and cesium carbonate in the absence of water gave only trace product (Table 1, Entry 8). The rhodium source and ligand are crucial for reactivity (Table 1, entries 9 and 10).
Table 1. Optimized Reaction Conditions: Standard Deviation Studies.

| Entry | Deviation from Standard Reaction Conditiona | Conversion (%) | ee (%) |
|---|---|---|---|
| 1 | None | 95 (81b) | 96 |
| 2 | 0.5 M instead of 1 M | 60 | 96 |
| 3 | L2 instead of L1 | 43 | 83 |
| 4 | L3 instead of L1 | 50 | 92 |
| 5 | L4 instead of L1 | 70 | 92 |
| 6 | Cs2CO3 instead of aq. CsOH | 75 | 94 |
| 7 | No aq. CsOH | 40 | 96 |
| 8 | Cs2CO3 instead of aq. CsOH; no H2O | <5 | – |
| 9 | No [Rh(cod)(OH)]2 | <5 | – |
| 10 | No Ligand | <5 | – |
Reaction conditions: 1 (0.5 mmol, 1 equiv), 2a (1.5 mmol, 3 equiv), [Rh(cod)(OH)]2 (0.015 mmol, 3 mol%), ligand (0.035 mmol, 7 mol%), aq. CsOH (1 mmol, 2 equiv), THP:Tol (1:1, 0.5 mL), H2O (0.25 mL), 70 °C, 20 h.
Isolated yield (%).
We explored this reaction with a variety of boronic acids (Scheme 3). 4- and 3-substituted phenyl boronic acids featuring diverse functional groups were tested. Electron-withdrawing (3d, 3l, and 3m) and -donating groups (3e–3h) in 4-substituted nucleophiles were well tolerated, providing the corresponding products in good yield and high enantioselectivity. Functional groups such as ethers (3e–3g), thiols (3h), halides (3i–3k), esters (3l), and nitriles (3m) are compatible with the reaction. Furthermore, 4-pyrazole phenyl boronic acid provided 3n in 58% yield and 96% ee. The pyrazole motif is featured in Niraparib, and 3n could potentially provide access to Niraparib and analogues.
Scheme 3. Rhodium-Catalyzed Carbometalation of Dihydropyridines with Aryl Boronic Acids.
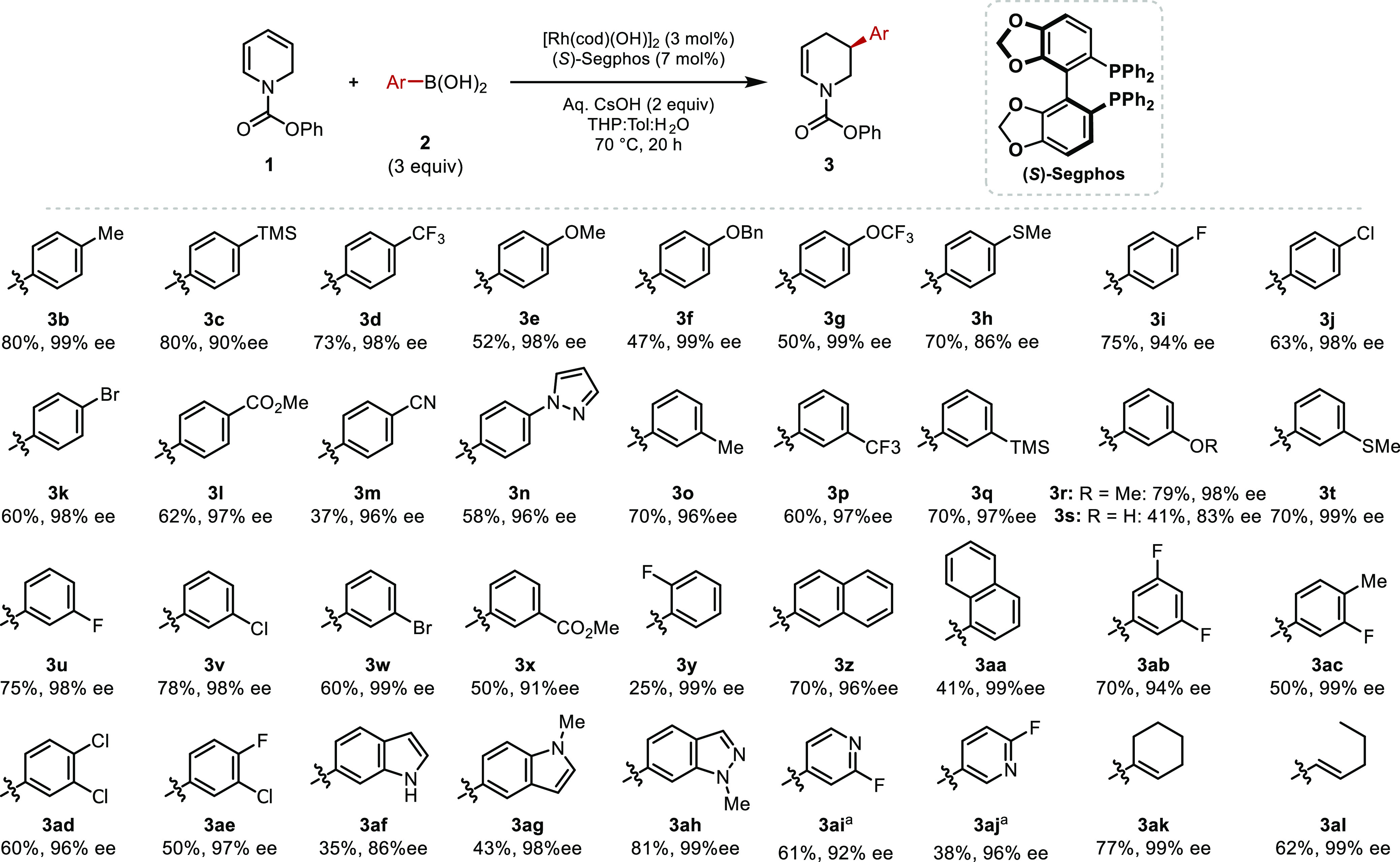
Reaction conducted with [Rh(cod)(OH)]2 (0.025 mmol, 5 mol%) and ligand (0.06 mmol, 12 mol%).
A similar selectivity trend was observed for a range of 3-substituted phenyl boronic acids, where broad functional group tolerance was observed, including with an unprotected phenol (3s), albeit with reduced yield and ee. The corresponding products (3o–3x) were isolated in high yields and enantioselectivities. 3r and 3s are potential intermediates in syntheses of Preclamol, and they could also serve as precursors for the synthesis of CP-868388 (Figure 1). Ortho-substituted phenyl boronic acids such as 2-methyl phenyl boronic acid and 2-chloro phenyl boronic acid showed poor reactivity; however, we were able to use 2-fluoro phenyl boronic acid and 1-naphthyl boronic acid and isolated 3y and 3aa in 25% yield, 99% ee and 41% yield, 99% ee, respectively. Disubstituted phenyl boronic acids were also tested, and 3ab–3ad were isolated in high yields and enantioselectivities (Scheme 3).
Transition metal catalysis with heterocycles is challenging due to catalyst poising. We were delighted to observe that the carbometalation proceeded with boronic acids featuring indole (3af and 3ag) and indazole motifs (3ah). Furthermore, some heterocyclic boronic acids are prone to undergo rapid protodeborylation.33 We were able to add 2-fluoro-pyridine boronic acids and isolated 3ai and 3aj with high levels of stereoinduction, though a higher (10 mol% Rh) catalyst loading was required for good conversion. Addition of cyclohexenyl and pentenyl boronic acids proceeded smoothly to provide 3ak and 3al in high yields and excellent enantioselectivities (Scheme 3).
Next, we tried addition to dihydropyridines featuring other carbamate groups, such as 4-methoxyphenyl carbamate, methyl carbamate, isopropyl carbamate, and benzyl carbamate, that may allow for selective protecting group manipulation, if required, which is relevant to natural product synthesis and drug discovery. The dihydropyridines featuring alkyl carbamates were freshly prepared for the best results. Addition of phenyl boronic acid provided 3am, 3an, 3ao, and 3ap in high yields and enantioselectivities (Scheme 4).
Scheme 4. Evaluation of Other Dihydropyridines and Dihydroquinolines.

Reaction conducted at room temperature for 48 h.
We next examined whether the method developed above was suitable for the addition of other diene–amine partners. However, as anticipated, the synthesis for substituted dihydropyridines is more challenging than starting from the parent pyridine itself, but we were able to prepare suitably protected 2- and 4-methyl dihydropyridines. Asymmetric addition to 2-methyl dihydropyridine gave 3aq (57%, 91% ee, Scheme 4). However, attempts to use 4-methyl dihydropyridine did not provide 3ar, even at higher temperatures. Addition to dihydroquinoline provided the asymmetric arylated product (3as), though poor enantioselectivity was observed using our standard conditions, and we note that these substrates are qualitatively more reactive than dihydropyridines. Performing reactions on dihydroquinolines at 30 °C, however, allows 3as to be isolated in 85% yield with 90% ee after 48 h. Substituted dihydroquinolines were also successfully employed, and 3at to 3av could be isolated with >80% yield and ∼85% ee (Scheme 4). We attempted addition to 7-membered ring (azepine) featuring diene–amino carbamate, though no desired product (3aw) was observed.
To test the scalability of the method, we performed the reaction on a 5 mmol scale, and 1.10 g of 3a was isolated (81% yield, 96% ee, Scheme 5A). Multiply substituted dihydropyridines can also potentially be accessed by manipulating tetrahydropyridine products 3 obtained in the reductive Heck reaction. As a proof of concept, we treated 3a with NBS in the presence of methanol to give trisubstituted piperidine 4 in 86% yield in a 4:1 diastereomeric ratio (Scheme 5A).
Scheme 5. Synthesis of Enantioenriched Piperidines.

As described above, simple (monosubstituted) 3-substituted piperidines exhibit a vast array of important bioactivities. To access these targets, a simple hydrogenation/deprotection strategy was used. Palladium-on-carbon-mediated hydrogenation followed by carbamate deprotection using aqueous potassium hydroxide in methanol led to piperidines 5 and 6 in 76% and 72% yields, respectively (over two steps). 6 is a known precursor to (−)-Preclamol (Scheme 5B). The absolute configuration of the products was assigned by comparing the optical rotation of 5(34) and 6(34−36) to literature values. Wilkinson’s catalyst-mediated hydrogenation of 3k and basic deprotection gave 7, a Niraparib precursor,16,17 in 68% yield over two steps (Scheme 5C).
To get a preliminary mechanistic understanding of the reaction, we conducted deuterium labeling experiments (Scheme 6A). Reaction with d5-phenyl boronic acid (d5-2a) led to 8, with no deuterium incorporation observed in the tetrahydropyridine ring. Next, we replaced the aqueous cesium hydroxide with cesium carbonate and conducted the reaction in D2O to produce tetrahydropyridine 9 (Scheme 6A), featuring exclusive deuterium incorporation in the 4-position. In prior reports of Rh-catalyzed carbometalations of bicyclic strained alkenes,29−31 and in our prior work on the carbometalation on cyclobutene, 1,4-Rh-shifts were often observed after carbometalation,28 but here no 1,4-Rh shift is observed for dihydropyridines.
Scheme 6. Mechanistic Hypothesis and Studies.

A prior report of Rh-catalyzed carbometalation with chromenes did note 1,4-Rh shifts, and the noticeable difference in the kinetics and enantioselectivity that we observed with dihydropyridine and dihydroquinoline substrates inspired us to conduct further experiments on 1h. Interestingly, 10 was observed when D2O was used, featuring deuterium incorporation exclusively on the benzene ring, clearly indicating that 1,4-Rh shifts occur with the dihydroquinolines (Scheme 6A).
Based on these deuterium studies, we propose two different mechanistic pathways, depending on the starting material. First, coordination of L1 with Rh(cod)(OH)]2 provides I. Transmetalation with phenyl boronic acid gives Rh-complex II. Carbometalation of 1a or 1h provides complex III. We postulate that the dihydropyridine-derived complex III undergoes regioselective protodemetalation in the presence of water to provide 3a and regenerates I (Scheme 6B, Mechanism Type 1). Complexes of type III that are dihydroquinoline derived, however, undergo a 1,4-Rh shift to give V, possibly via IV. Finally, protodemetalation in the presence of water leads to product 3as and regenerates I (Scheme 6B, Mechanism Type II).
We present a new synthetic strategy to access enantioenriched piperidines involving highly regio- and enantioselective Rh-catalyzed carbometalation of dihydropyridine to provide 3-substituted tetrahydropyridines. The method has broad functional group tolerance and can be performed on a gram scale, and the products are valuable precursors to enantioenriched piperidines. The utility of the reaction was demonstrated by the formal syntheses of Preclamol and Niraparib. Further, the tetrahydropyridine products contain useful functional groups which should allow them to serve as precursors to complex synthetic targets, for example by producing heavily functionalized trisubstituted piperidines such as 4.
Acknowledgments
The authors thank the UK Engineering and Physical Sciences Research Council (EP/W007363/1) for financial support. S.M. is grateful to The European Union’s Horizon 2020 research and innovation program for the Marie Skłodowska-Curie fellowship (890680) for funding.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c05044.
Experimental procedures, compound synthesis and characterization, and supporting discussion (PDF)
The authors declare the following competing financial interest(s): SM and SF are named as inventors on a UK priority patent application (13320698) filed by Oxford University Innovation Limited.
Supplementary Material
References
- Baumann M.; Baxendale I. R. An Overview of the Synthetic Routes to the Best Selling Drugs Containing 6-Membered Heterocycles. Beilstein J. Org. Chem. 9265 2013, 9 (1), 2265–2319. 10.3762/bjoc.9.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57 (24), 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- Taylor R. D.; Maccoss M.; Lawson A. D. G. Rings in Drugs. J. Med. Chem. 2014, 57 (14), 5845–5859. 10.1021/jm4017625. [DOI] [PubMed] [Google Scholar]
- Search Results, Clinical Trials of Niraparib. https://clinicaltrials.gov/ct2/results?term=niraparib (accessed June 14, 2023).
- Macchia M.; Cervetto L.; Demontis G. C.; Longoni B.; Minutolo F.; Orlandini E.; Ortore G.; Papi C.; Sbrana A.; Macchia B. New N-n-Propyl-Substituted 3-Aryl- and 3-Cyclohexylpiperidines as Partial Agonists at the D4 Dopamine Receptor. J. Med. Chem. 2003, 46 (1), 161–168. 10.1021/jm021019a. [DOI] [PubMed] [Google Scholar]
- Tedroff J.; Ekesbo A.; Sonesson C.; Waters N.; Carlsson A. Long-Lasting Improvement Following (-)-OSU6162 in a Patient with Huntington’s Disease. Neurology 1999, 53 (7), 1605–1605. 10.1212/WNL.53.7.1605. [DOI] [PubMed] [Google Scholar]
- Schachter S. C. Tiagabine. Epilepsia 1999, 40 (s5), s17–s22. 10.1111/j.1528-1157.1999.tb00915.x. [DOI] [PubMed] [Google Scholar]
- Bagley S. W.; Brandt T. A.; Dugger R. W.; Hada W. A.; Hayward C. M.; Liu Z.. Phenyl Substituted Piperidine Compounds for Use as PPAR Activators. WO2004048334A1, 2003.
- Wu J.; Chen Z.; Barnard J. H.; Gunasekar R.; Pu C.; Wu X.; Zhang S.; Ruan J.; Xiao J. Synthesis of Chiral Piperidines from Pyridinium Salts via Rhodium-Catalysed Transfer Hydrogenation. Nat. Catal. 2022, 5 (11), 982–992. 10.1038/s41929-022-00857-5. [DOI] [Google Scholar]
- Harawa V.; Thorpe T. W.; Marshall J. R.; Sangster J. J.; Gilio A. K.; Pirvu L.; Heath R. S.; Angelastro A.; Finnigan J. D.; Charnock S. J.; Nafie J. W.; Grogan G.; Whitehead R. C.; Turner N. J. Synthesis of Stereoenriched Piperidines via Chemo-Enzymatic Dearomatization of Activated Pyridines. J. Am. Chem. Soc. 2022, 144 (46), 21088–21095. 10.1021/jacs.2c07143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan A. L.; Confair D. N.; Kim K.; Barros-Álvarez X.; Rodriguiz R. M.; Yang Y.; Kweon O. S.; Che T.; McCorvy J. D.; Kamber D. N.; Phelan J. P.; Martins L. C.; Pogorelov V. M.; DiBerto J. F.; Slocum S. T.; Huang X.-P.; Kumar J. M.; Robertson M. J.; Panova O.; Seven A. B.; Wetsel A. Q.; Wetsel W. C.; Irwin J. J.; Skiniotis G.; Shoichet B. K.; Roth B. L.; Ellman J. A. Bespoke Library Docking for 5-HT2A Receptor Agonists with Antidepressant Activity. Nature 2022, 610 (7932), 582–591. 10.1038/s41586-022-05258-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Zhang X.; Nagib D. A. Chiral Piperidines from Acyclic Amines via Enantioselective, Radical-Mediated δ C–H Cyanation. Chem 2019, 5 (12), 3127–3134. 10.1016/j.chempr.2019.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizgys G. J.; Jung H. H.; Floreancig P. E. Stereoselective Piperidine Synthesis through Oxidative Carbon–Hydrogen Bond Functionalizations of Enamides. Chem. Sci. 2012, 3 (2), 438–442. 10.1039/C1SC00670C. [DOI] [Google Scholar]
- Källström S.; Leino R. Synthesis of Pharmaceutically Active Compounds Containing a Disubstituted Piperidine Framework. Bioorg. Med. Chem. 2008, 16 (2), 601–635. 10.1016/j.bmc.2007.10.018. [DOI] [PubMed] [Google Scholar]
- Angle S. R.; Breitenbucher J. G. Recent Progress in the Synthesis of Piperidine and Indolizidine Alkaloids. Stud. Nat. Prod. Chem. 1995, 16 (PJ), 453–502. 10.1016/S1572-5995(06)80060-8. [DOI] [Google Scholar]
- Chung C. K.; Bulger P. G.; Kosjek B.; Belyk K. M.; Rivera N.; Scott M. E.; Humphrey G. R.; Limanto J.; Bachert D. C.; Emerson K. M. Process Development of C-N Cross-Coupling and Enantioselective Biocatalytic Reactions for the Asymmetric Synthesis of Niraparib. Org. Process Res. Dev. 2014, 18 (1), 215–227. 10.1021/op400233z. [DOI] [Google Scholar]
- Wallace D. J.; Baxter C. A.; Brands K. J. M.; Bremeyer N.; Brewer S. E.; Desmond R.; Emerson K. M.; Foley J.; Fernandez P.; Hu W.; Keen S. P.; Mullens P.; Muzzio D.; Sajonz P.; Tan L.; Wilson R. D.; Zhou G.; Zhou G. Development of a Fit-for-Purpose Large-Scale Synthesis of an Oral PARP Inhibitor. Org. Process Res. Dev. 2011, 15 (4), 831–840. 10.1021/op2000783. [DOI] [Google Scholar]
- Kratena N.; Marinic B.; Donohoe T. J. Recent Advances in the Dearomative Functionalisation of Heteroarenes. Chem. Sci. 2022, 13 (48), 14213–14225. 10.1039/D2SC04638E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer P.; Palacin T.; Sidera M.; Fletcher S. P. Asymmetric Suzuki-Miyaura Coupling of Heterocycles via Rhodium-Catalysed Allylic Arylation of Racemates. Nat. Commun. 2017, 8 (1), 15762. 10.1038/ncomms15762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González J.; van Dijk L.; Goetzke F. W.; Fletcher S. P. Highly Enantioselective Rhodium-Catalyzed Cross-Coupling of Boronic Acids and Racemic Allyl Halides. Nat. Protoc. 2019, 14 (10), 2972–2985. 10.1038/s41596-019-0209-8. [DOI] [PubMed] [Google Scholar]
- Herrmann N.; Vogelsang D.; Behr A.; Seidensticker T. Homogeneously Catalyzed 1,3-Diene Functionalization – A Success Story from Laboratory to Miniplant Scale. ChemCatChem 2018, 10 (23), 5342–5365. 10.1002/cctc.201801362. [DOI] [Google Scholar]
- Perry G. J. P.; Jia T.; Procter D. J. Copper-Catalyzed Functionalization of 1,3-Dienes: Hydrofunctionalization, Borofunctionalization, and Difunctionalization. ACS Catal. 2020, 10 (2), 1485–1499. 10.1021/acscatal.9b04767. [DOI] [Google Scholar]
- McNeill E.; Ritter T. 1,4-Functionalization of 1,3-Dienes With Low-Valent Iron Catalysts. Acc. Chem. Res. 2015, 48 (8), 2330–2343. 10.1021/acs.accounts.5b00050. [DOI] [PubMed] [Google Scholar]
- Berti F.; Menichetti A.; Di Bussolo V.; Favero L.; Pineschi M. Synthesis of Bicyclic Tetrahydropyridine Enamides and Enecarbamates by Hetero-Cope Rearrangement of Nitroso Cycloadducts. Chem. Heterocycl. Compd. 2018, 54 (4), 458–468. 10.1007/s10593-018-2289-8. [DOI] [Google Scholar]
- Zhou Y.; Lu Y.; Hu X.; Mei H.; Lin L.; Liu X.; Feng X. Highly Diastereo- and Enantioselective Synthesis of Spirooxindole-Cyclohexaneamides through N,N′-Dioxide/Ni(II)-Catalyzed Diels–Alder Reactions. Chem. Commun. 2017, 53 (12), 2060–2063. 10.1039/C6CC10125A. [DOI] [PubMed] [Google Scholar]
- Stout D. M.; Meyers A. I. Recent Advances in the Chemistry of Dihydropyridines. Chem. Rev. 1982, 82 (2), 223–243. 10.1021/cr00048a004. [DOI] [Google Scholar]
- Kubota K.; Watanabe Y.; Hayama K.; Ito H. Enantioselective Synthesis of Chiral Piperidines via the Stepwise Dearomatization/Borylation of Pyridines. J. Am. Chem. Soc. 2016, 138 (13), 4338–4341. 10.1021/jacs.6b01375. [DOI] [PubMed] [Google Scholar]
- Goetzke F. W.; Hell A. M. L.; van Dijk L.; Fletcher S. P. A Catalytic Asymmetric Cross-Coupling Approach to the Synthesis of Cyclobutanes. Nat. Chem. 2021, 13 (9), 880–886. 10.1038/s41557-021-00725-y. [DOI] [PubMed] [Google Scholar]
- Bexrud J.; Lautens M. A Rhodium IBiox[(-)-Menthyl] Complex as a Highly Selective Catalyst for the Asymmetric Hydroarylation of Azabicyles: An Alternative Route to Epibatidine. Org. Lett. 2010, 12 (14), 3160–3163. 10.1021/ol101067d. [DOI] [PubMed] [Google Scholar]
- Menard F.; Lautens M. Chemodivergence in Enantioselective Desymmetrization of Diazabicycles: Ring-Opening versus Reductive Arylation. Angew. Chemie Int. Ed. 2008, 47 (11), 2085–2088. 10.1002/anie.200704708. [DOI] [PubMed] [Google Scholar]
- So C. M.; Kume S.; Hayashi T. Rhodium-Catalyzed Asymmetric Hydroarylation of 3-Pyrrolines Giving 3-Arylpyrrolidines: Protonation as a Key Step. J. Am. Chem. Soc. 2013, 135 (30), 10990–10993. 10.1021/ja406169s. [DOI] [PubMed] [Google Scholar]
- Dian L.; Marek I. Rhodium-Catalyzed Arylation of Cyclopropenes Based on Asymmetric Direct Functionalization of Three-Membered Carbocycles. Angew. Chemie Int. Ed. 2018, 57 (14), 3682–3686. 10.1002/anie.201713324. [DOI] [PubMed] [Google Scholar]
- Cox P. A.; Leach A. G.; Campbell A. D.; Lloyd-Jones G. C. Protodeboronation of Heteroaromatic, Vinyl, and Cyclopropyl Boronic Acids: PH-Rate Profiles, Autocatalysis, and Disproportionation. J. Am. Chem. Soc. 2016, 138 (29), 9145–9157. 10.1021/jacs.6b03283. [DOI] [PubMed] [Google Scholar]
- Colpaert F.; Mangelinckx S.; De Kimpe N. Asymmetric Synthesis of Chiral N -Sulfinyl 3-Alkyl- and 3-Arylpiperidines by α-Alkylation of N -Sulfinyl Imidates with 1-Chloro-3-Iodopropane. J. Org. Chem. 2011, 76 (1), 234–244. 10.1021/jo1020807. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Tay D. W.; Chen J.; Leung G. Y. C.; Yeung Y. Y. Enantioselective Synthesis of 2-Substituted and 3-Substituted Piperidines through a Bromoaminocyclization Process. Chem. Commun. 2013, 49 (39), 4412–4414. 10.1039/C2CC36578B. [DOI] [PubMed] [Google Scholar]
- Amat M.; Cantó M.; Llor N.; Escolano C.; Molins E.; Espinosa E.; Bosch J. Dynamic Kinetic Resolution of Racemic γ-Aryl-δ-Oxoesters. Enantioselective Synthesis of 3-Arylpiperidines. J. Org. Chem. 2002, 67 (15), 5343–5351. 10.1021/jo025894f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.