

Abstract
The gammaherpesviruses (γHVs) establish a lifelong infection in their hosts, with the cellular outcome of infection intimately regulated by target cell type. Murine gammaherpesvirus 68 (MHV68), a small animal model of γHV infection, infects macrophages in vivo, resulting in a range of outcomes, from lytic replication to latent infection. Here, we have further investigated the nature of MHV68 macrophage infection using reductionist and primary in vivo infection studies. While MHV68 readily infected the J774 macrophage cell line, viral gene expression and replication were significantly impaired relative to a fully permissive fibroblast cell line. Lytic replication only occurred in a small subset of MHV68-infected J774 cells, despite the fact that these cells were fully competent to support lytic replication following pre-treatment with interleukin-4, a known potentiator of replication in macrophages. In parallel, we harvested virally-infected macrophages at 16 hours after MHV68 infection in vivo and analyzed gene expression by single cell RNA-sequencing. Among virally infected macrophages, only rare (0.25%) cells had lytic cycle gene expression, characterized by detection of multiple lytic cycle RNAs. In contrast, ~50% of virally-infected macrophages were characterized by expression of ORF75A, ORF75B and/or ORF75C, in the absence of other detectable viral RNAs. Selective transcription of the ORF75 locus also occurred in MHV68-infected J774 cells. In total, these studies indicate that MHV68 efficiently infects macrophages, with the majority of cells characterized by an atypical state of restricted viral transcription, and only rare cells undergoing lytic replication.
Importance:
The human gammaherpesviruses Epstein-Barr virus and Kaposi’s sarcoma associated herpesvirus are DNA viruses that cause lifelong infection and are associated with multiple diseases, especially in immunocompromised individuals. Murine gammaherpesvirus 68 (MHV68) is a powerful mouse model that permits close examination of these viruses. Previous studies of MHV68 identified that macrophages are an important in vivo target of infection; how infection within these cells is regulated remains incompletely understood. Here, we demonstrate that MHV68 infection of macrophages is characterized by two divergent outcomes across a population of infected cells: while a small subset of cells undergo lytic replication, to make new virus progeny, the majority of cells are characterized by an atypical, restricted form of infection characterized by a distinct viral gene transcription program not previously reported. These studies highlight important cell-type specific outcomes of gammaherpesvirus infection and identify a potential alternate program by which these viruses usurp macrophages.
Introduction
The gammaherpesviruses (γHVs) are large double-stranded DNA tumor viruses that establish a lifelong infection in their host and are associated with a variety of diseases, particularly in immunosuppressed individuals. These viruses include the human γHVs Epstein-Barr Virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV or HHV-8), and infect multiple cell types, typically resulting in either lytic infection or latent infection on a cellular level (1). The outcome of infection at this level is influenced by multiple factors, including target cell type and cell-type specific virus-host interactions. The γHVs are associated with the development of several malignancies especially in immunocompromised individuals. (2, 3). Due to the strict host specificity of the human γHVs, we and others have used the mouse γHV, murine gammaherpesvirus 68 (MHV68, γHV68 or MuHV-4), as a genetic and phenotypically relevant model to study γHV infection and pathogenesis (4, 5).
MHV68 infection of mice is a multi-step process, from lytic infection at mucosal surfaces in epithelial cells, to dissemination to the B-cell compartment where it can establish lifelong, systemic latency (6). While B cells are a major latent reservoir for MHV68 (7–9), EBV (10) and potentially KSHV (11), MHV68 has also been shown to establish latency in lung, spleen and peritoneal macrophages (7, 12, 13). Macrophage infection may occur by multiple mechanisms, from direct virus binding and entry, to internalization of the virus via epithelial cell presentation of MHV68 (14), to potential infection through an Fc receptor-dependent mechanism (15). MHV68 has also been reported to undergo lytic replication in macrophages in vitro and in vivo, albeit at lower levels than replication within fibroblasts (16, 17). While MHV68 passage through myeloid cells has been reported to be important for the establishment of B cell latency, a process potentially involving both alveolar macrophages and marginal zone macrophages (12, 18), not all macrophage subsets are created equal: subcapsular sinus macrophages have been reported to be poorly permissive for MHV68 infection, serving an antiviral role (19). These data emphasize the complexity of MHV68-macrophage interactions and the need for further analysis of these interactions.
Here, we sought to better understand the regulation of MHV68 infection in macrophages in cell culture and primary infection compared with fibroblasts, a known target of lytic replication. We found efficient infection of both cell types, but significantly reduced DNA synthesis and gene expression in macrophages relative to fibroblasts. Lytic protein induction only occurred in rare macrophages. In our in vivo analysis, we found a rare population of macrophages undergoing lytic replication, with the major population of infected peritoneal macrophages demonstrating a unique transcriptional profile characterized by expression of ORF75A, ORF75B, and/or ORF75C, in the absence of other lytic genes. These studies suggest that macrophages are efficiently infected but characterized by atypical, restricted viral transcription that is not representative of lytic nor latent gene expression patterns described to date.
RESULTS
An in vitro model of MHV68-macrophage infection shows limited lytic replication despite robust viral entry.
The outcome of MHV68 on a cell level varies depending on host cell type and immune status (5, 20, 21). MHV68 infection of macrophages has been associated with both lytic replication and latent infection (7, 12, 16) with data suggesting that myeloid cells may be an important intermediary prior to B-cell latency (12). Despite these insights, the exact nature of myeloid cell infection remains incompletely characterized. We characterized MHV68 infection in the mouse myeloid-like cell line, J774, compared with infection of the 3T12 mouse fibroblast cell line, which fully supports lytic virus replication (22).
First, we compared the ability of MHV68 to replicate in both cell lines. As expected, MHV68 underwent robust lytic replication in 3T12s, characterized by rapid virus production with extensive cytopathic effect (CPE) by 72 hours post infection (hpi). In contrast, MHV68-infected cultures of J774 cells showed minimal evidence of infection, with no discernable CPE (Fig. 1A) and minimal viral replication (Fig. 1B).
Figure 1. MHV68-infected J774 macrophages are deficient in virus replication relative to permissive 3T12 fibroblasts, despite evidence of equivalent virus entry and LANA gene expression.

Analysis of MHV68 infection (MOI=1 PFU/cell) comparing 3T12 fibroblasts and J774 macrophages. (A) Representative brightfield images of infected 3T12 or J774 cells. 3T12 cells show early signs of CPE, including cell rounding (24 hpi) with extensive monolayer destruction (72 hpi), whereas J774 cells show minimal CPE. (B) Virus replication as defined by plaque assay. Data depict mean +/− SEM, with limit of detection at 102 PFU/mL (t=0, 24, and 48 are from 2, 6, and 6 independent experiments with one biological sample per experiment). (C) Flow cytometric analysis of LANAβlac expression defined by cleavage of the CCF2 fluorescent beta-lactamase substrate. Data depict events that were single, viable cells, with values in the right gate defining the frequency of cells with CCF2 substrate cleavage, an indicator of LANAβlac expression. Samples infected with WT MHV68 (left panel) do not encode LANAβlac and define background cleavage. Representative flow cytometry plots were defined as samples that were closest to the mean frequency in panel D. (D) Quantification of LANAβlac + cell frequency defined by flow cytometry, showing mean +/− SEM, as in panel C. Data are from 3 independent experiments with 3 biological replicates per experiment. Data for 3T12 cells at 72 hpi was not assessed due to extensive cell destruction. Statistical analysis was performed using unpaired t test with statistically significant differences as indicated, * p<0.05, ** p<0.01, **** p<0.0001.
The limited replication of MHV68 in J774 cells could potentially be due to inefficient infection. To define infection frequency, we infected cells with WT MHV68 LANAβlac, a virus that contains a gene fusion between the ORF73 gene, an immediate early gene encoding the latency-associated nuclear antigen (LANA) and the beta-lactamase (βlac) gene (23, 24). As ORF73, and LANA, are expressed during latent and lytic infection (23), βlac activity serves as an effective indicator of multiple states of infection by flow cytometry, defined by cleavage of a fluorescent substrate. As predicted, infection of cells with WT MHV68 lacking the beta-lactamase reporter had no detectable βlac+ cells (left panel, Fig. 1C). In contrast, infection with WT MHV68 LANAβlac resulted in a high frequency of LANAβlac+ cells in both 3T12s fibroblasts (~77%) and J774 cells (~45%) at 24 hpi, that increased to ~81% LANAβlac+ cells by 72 hpi (Fig. 1C, D). These data demonstrate that J774 cells are fully susceptible to MHV68 infec1tion and are capable of initiating transcription and translation of ORF73 (an immediate early gene), despite restricted production of infectious virus.
J774 macrophages are deficient in viral DNA replication and lytic viral gene expression.
We next quantified viral DNA replication between the two cell lines. MHV68 underwent large (>2.5 log10) increases in viral DNA replication in 3T12 fibroblasts; in contrast, MHV68-infected J774 myeloid cells had minimal changes in viral DNA replication over input virus (Fig. 2A).
Figure 2. MHV68-infected J774 macrophages are deficient in viral DNA replication and viral gene expression relative to infected fibroblasts.

Analysis of MHV68 replication and gene expression (MOI=1 PFU/cell) comparing 3T12 fibroblasts and J774 macrophages at the indicated times. (A) Viral infection quantified by viral genome copies via q-PCR. (B-C) Quantitative analysis of viral RNA expression of ORF73 (immediate early gene), ORF6 (an early/late gene), and M7 (late gene) by qRT-PCR, comparing (top row) 3T12 and J774 cells or (bottom row) time-dependent changes within J774s. “Relative Difference” defined as quantification of target gene transcript in comparison to reference gene transcript (host 18S) as described in Methods. Data are from 3 independent experiments with 3 biological replicates per experiment, measured in technical triplicates. The value shown is the mean +/SEM of each experiment. Statistical analysis used unpaired t test with statistically significant differences as indicated. ** p<0.01, ****p<0.0001. ns, not statistically significant.
We next analyzed MHV68 transcription in J774 cells, quantifying immediate early (ORF73), early, early/late (ORF6), and late gene (M7) expression (25). Viral gene expression was decreased in J774 myeloid cells relative to 3T12 fibroblasts at both 16 and 24 hpi (Fig. 2B). Whereas ORF73 (and Blac) expression was reduced ~10 fold, both ORF6 and M7 transcripts were decreased >100 fold (Fig. 2B, Supp. Fig 1), consistent with greater deficits in lytic gene expression for early and late genes.
When we quantified viral gene expression within J774 cells over time, we found that ORF73 was relatively comparable between 24–48 hpi (bottom panel, Fig. 2B). In contrast, ORF6 and M7 expression levels increased ~10x between 24 hpi and 48 hpi (Fig. 2B). As ORF6 and M7 are lytic cycle transcripts, these changes are consistent with some degree of lytic replication in MHV68-infected J774 cultures.
MHV68-infected J774 macrophages contain a small subset of cells that demonstrate a lytic infection profile.
To assess lytic replication within MHV68-infected J774 cells, we used two approaches that afforded single-cell resolution of infection. First, we infected 3T12 and J774 cells with a recombinant WT MHV68 virus, MHV68.H.GFP, that encodes GFP fused to a hygromycin resistance protein, under the transcriptional control of the human cytomegalovirus IE promoter inserted between ORF27 and 29b (26). This virus robustly expresses GFP during active, lytic infection, allowing identification of cells that have initiated transcription via flow cytometry. When we infected 3T12 and J774 cells with MHV68.H.GFP virus and assessed GFP expression at 18 hpi, 3T12 cells had a much higher frequency of GFP+ cells than J774 cells (~8.2% vs. ~0.5% GFP+ respectively, Fig. 3A–B, Supp. Fig. 2). For these studies, background fluorescence was defined by infection with WT MHV68.LANAβlac, without addition of a fluorescent substrate. These data indicate that early in infection, a rare population of J774 cells initiate robust transcriptional induction from the viral genome.
Figure 3. MHV68-infected J774 macrophages contain a small subset of cells that demonstrate lytic infection profiles.

Analysis of MHV68 infection and lytic cycle profiles with either MHV68.H.GFP or MHV68.LANAblac (MOI=1 PFU/cell) comparing 3T12 fibroblasts and J774 macrophages. (A) Flow cytometric analysis of virus-expressed GFP in 3T12 or J774 cells. Data depict events that were single, viable cells, with values defining the frequency of cells with GFP expression, an indicator of MHV68.H.GFP infection, with gating strategy in Supp. Fig. 2. Samples infected with WT MHV68.LANAblac (left column), without the addition of the CCF2 fluorescent substrate, defined background fluorescence. (B) Quantification of GFP + cell frequency defined by flow cytometry. Symbols depict individual experimental values, with data showing mean +/− SEM. (C) Flow cytometric analysis of MHV68 lytic viral protein vRCA and phosphorylated host γH2AX in either 3T12 (top) or J774 (bottom) cells at the indicated timepoints, comparing mock (left) or WT MHV68.LANAblac infected cells. Data depict events that were single, viable cells, with values defining the frequency of cells with vRCA and/or phosphorylated host γH2AX, indicative of lytic MHV68 infection, with gating strategy presented in Supp. Fig. 3. Data for 3T12 cells at 48 hpi was not assessed due to extensive cell destruction. (D) Quantification of positive protein expression defined by flow cytometry, showing mean +/− SEM, as in panel C. Representative images were defined as samples that were closest to the mean frequency. Data are from 3 independent experiments with 2 biological experiments per experiment. Statistical analysis used unpaired t test with statistically significant differences as indicated. ****p<0.0001, **p<0.01.
We next quantified expression of proteins expressed or modified during lytic MHV68 replication via flow cytometry (as in (27)). To do this, we measured the frequency of cells that express the viral regulator of complement activation (vRCA), a protein encoded by the ORF4 late gene, or demonstrated phosphorylated Histone H2AX (γH2AX), a host protein that undergoes virus-induced phosphorylation (16). Mock infected cells had minimal expression of either of these proteins (“Mock”, Fig. 3C), whereas ~45% of MHV68-infected 3T12 cells were either vRCA+, γH2AX+, or vRCA+γH2AX+ by 24hpi (Fig. 3C–D, Supp. Fig. 3). In contrast, MHV68-infected J774 cells only contained ~2% cells at 24hpi and ~8% at 48hpi that expressed either vRCA+, γH2AX+, or vRCA+ γH2AX+ (Fig. 3C–D). These data indicate that only a minor fraction of MHV68-infected J774 cells undergo lytic replication at these timepoints.
IL-4 pretreatment of J774 macrophages reveals a robust capacity of these cells to support lytic replication.
The minor frequency of MHV68 infected J774 cells undergoing lytic replication raised the possibility that J774 cells may poorly support lytic replication. To test whether J774 cells had the capacity to support lytic replication, we pretreated cells with IL-4, a known inducer of viral replication and reactivation from latency in primary macrophages (28, 29). First, we quantified the frequency of GFP+ cells following MHV68.H.GFP infection comparing 3T12s with untreated or IL-4 treated J774 cells, where cells were both pre-treated with IL-4 and supplied with fresh IL-4 after infection. As expected, 3T12 cells had a significant fraction of GFP+ cells with near uniform expression by 48 hpi (Fig. 4A). While untreated J774 cells had a minor frequency of GFP+ cells (~1% at 24 hpi, ~9% at 48 hpi), IL-4 treated J774 cells had a greatly enhanced frequency of GFP+ cells both early and late, with ~45% GFP+ cells by 48 hpi (Fig. 4A–B). We next quantified viral DNA replication under similar conditions, treating J774 cells with either 10 or 100 ng/mL of IL-4 for 16 hours prior to infection with fresh IL-4 provided after infection. IL-4 treatment resulted in a concentration-dependent increase in viral DNA replication by 48 hpi after treatement with either 10 or 100 ng/mL IL-4, compared to the minimal viral DNA replication observed in untreated cells, consistent with our earlier observations (Fig. 4C). These data demonstrate that J774 cells have the capacity to support lytic replication following treatment with IL-4, a known inducer of virus replication in macrophages.
Figure 4. J774 macrophages are capable of supporting lytic MHV68 replication following pretreatment with IL4, a known potentiator of virus replication in macrophages.

Analysis of the effects of cytokine IL-4 on MHV68 infection and genome replication (MOI=1 PFU/cell) in 3T12 fibroblasts and J774 macrophages. (A) Flow cytometric analysis of MHV68.H.GFP infection in 3T12 (top row), untreated J774 (middle) or IL4-treated J774 cells (10 ng/mL, 18 hour pre-treatment with fresh IL4 added post-infection; bottom row). Data depict events that were single, viable cells, with values defining the frequency of cells with GFP expression, an indicator of viral infection, transcription and translation. Representative images were defined as samples that were closest to the mean frequency. (B) Quantification of GFP+ cell frequency defined by flow cytometry, as in panel A, with individual symbols representing independent biological samples and data showing mean +/− SEM. Data are from 3 independent experiments with 2 biological replicates per experiment, measured in technical duplicates. (C) Quantitation of the impact of IL4 treatment on MHV68.LANAblac viral DNA replication in MHV68-infected J774 cells quantified by qPCR. Cells were pre-treated with 0, 10 or 100 ng/mL murine IL-4 for 16 hours, with fresh IL4 added post-infection, and cells harvested for DNA at 0, 24, or 48hpi. Data depict mean +/− SEM, with individual symbols representing independent biological samples. Data are from 3 independent experiments with 3 biological replicates per experiment measured in triplicate. Statistical analysis used (B) unpaired t test or (C) 1-way ANOVA comparing samples collected at the same timepoint untreated versus cytokine treated, with statistically significant differences as indicated, **p<0.01, ****p<0.0001.
In vivo analysis of primary MHV68 macrophage infection by single-cell RNA-seq reveals unanticipated viral gene expression.
To examine how our in vitro results reflect in vivo biology, we infected immunocompetent, C57BL/6J mice by intraperitoneal injection with WT MHV68.LANAβlac and sort purified peritoneal macrophages early after infection (16 hpi). Virally-infected peritoneal macrophages were purified based on cells that were viable, expressed the F4/80+ macrophage-specific cell surface protein and demonstrated LANAβlac+ expression by CCF-2 cleavage (as in Fig. 1). Cells were then subjected to 3’ based single-cell RNA-seq (scRNA-seq), analyzing host and viral gene expression to identify infected cell subsets and viral gene expression respectively. We identified 14 clusters of cell subsets, dominantly comprised of macrophages (Fig. 5A–C). This analysis further identified minor frequencies of: i) macrophages with a proliferative gene signature (i.e. “macrophage proliferating”), ii) macrophages with an MHV68 lytic gene signature (i.e. “macrophage lytic”), defined by expression of two canonical lytic genes ORF7 and ORF54, and infrequent iii) B cells, iv) T cells, v) dendritic cells and vi) monocytes (Fig. 5A–C, Supplemental Fig. 4A–B, Supplemental Fig. 5A–B). Data were characterized by high quality control metrics including high reads (unique molecular identifiers, UMIs) and genes per cell, with low mitochondrial reads and only “macrophage proliferating” cells characterized by a proliferative “S score” (Fig. 5D–G, Supplemental Fig. 4C-F). When we visualized (Fig. 5H) and quantified the frequency of cells with detectable viral UMIs (Fig. 5I), we found that the majority (n=7,512; 82.54%) of macrophages had detectable viral reads accounting for <0.1% of total UMIs per cell, with few (n=23; 0.25%) macrophages containing a higher proportion (>0.1%) of viral reads (Fig. 5I). Similar results were found at the cluster level, with cluster 13, identified as macrophages with lytic gene expression, characterized both by 100% viral UMI+ (including ORF7 and ORF54) and a higher proportion of viral UMIs per cell (Supplemental Fig. 4G-J). The presence of cells with no detectable viral UMIs could either be due to the depth of sequencing or to the known sparseness of scRNA-seq data (30). Because these limitations of scRNA-seq could further extend to the lack of detection of viral genes, below, subsequent discussion focuses on those genes that we can definitively detect and avoids conclusion about lack of expression.
Figure 5. Single cell RNA-seq analysis of primary MHV68-infected LANAβlac+ peritoneal cells harvested 16 hours post infection in vivo.
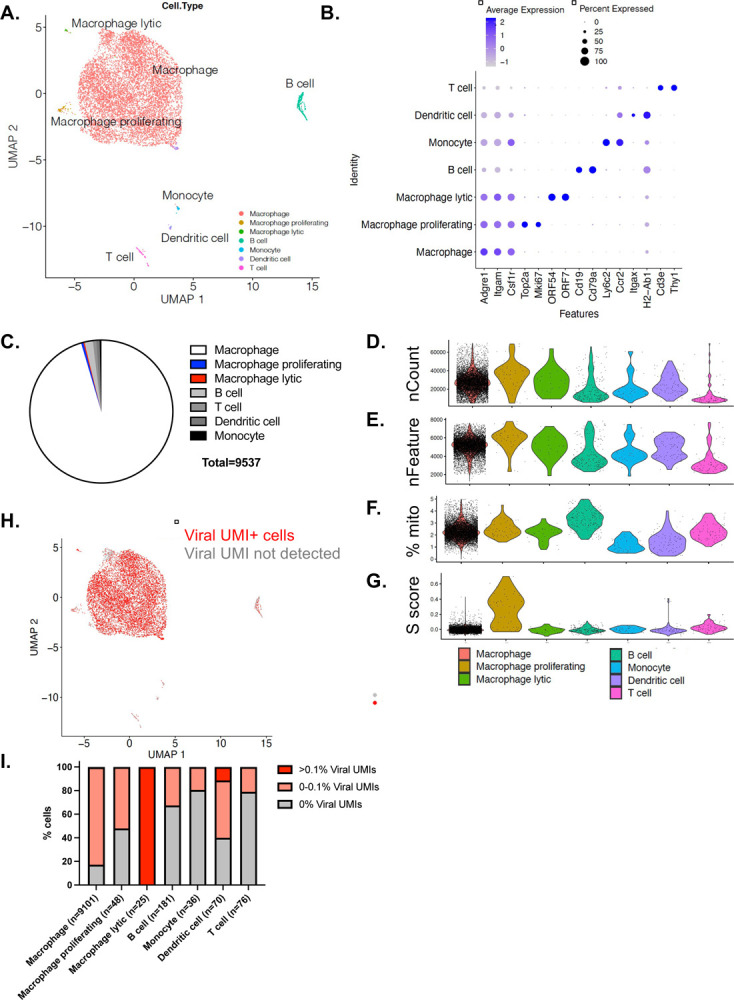
C57BL/6J mice were infected with 1×106 PFU of WT MHV68.LANAblac virus, with MHV68-infected, LANAblac+ peritoneal macrophages harvested and sort-purified at 16 hpi and subjected to scRNA-seq analysis. Data focus on cell types identified based on Seurat-designated clusters as outlined in Supplemental Figures 4–5. (A-B) Visualization of cell types present within scRNA-seq dataset, with cell identities based on lineage marker genes depicted in panel B (C) Quantification of cell subsets defined by scRNA-seq identifies macrophages as the dominant population. (D-F) Quality control metrics across cell subsets, including UMI Count (D), Features (E), % mitochondrial reads (F), and S score, a proliferationassociated gene signature (G). (H) UMAP visualization of cells that contain at least 1 viral UMI per cell identified by red. (I) % of cell subsets stratified by extent of viral UMIs detected per cell, including cells with >0.1% total reads that are viral UMIs (bright red), cells with detectable viral UMIs accounting for <0.1% of total reads (pale red), or no viral UMIs detected (gray). In total, 56 cells had >0.1% of total UMIs contributed by viral UMIs, including 23 of 9101 macrophages (0.25%), 25 of 25 macrophages with lytic gene expression (100%) and 8 of 70 dendritic cells (11.43%). Data are from 9,537 cells, with cell subsets containing between 36–9,101 cells (as in panel I).
Next, we analyzed viral gene expression within macrophages, quantifying the frequency of cells that expressed each viral gene, comparing the dominant macrophage population (n=9101 cells), with minor macrophage populations with either a proliferative gene signature (n=48 cells) or lytic gene expression (n=25 cells). While we detected ORF73-βlac mRNA, the transcript encoding the LANAβlac reporter by which cells were purified, in 12.89% of macrophages, there were equivalent or higher frequencies of cells that expressed 3 other viral genes: ORF75C (30.75%), ORF75B (27.83%) and ORF75A (12.54%) (Fig. 6A). Additional transcripts were detected in a lower frequency of cells: ORF72 (5.3%), ORF58 (3.5%), with 1–3% cells containing detectable RNA for M3, ORF7, ORF50, ORF57, ORF61, ORF74, M12, M13, or M14 (Fig. 6A). In contrast, the remaining viral genes were only sporadically detected in less than 1% of cells. A similar hierarchy, albeit reduced in magnitude, was also observed in the proliferating macrophage cell subset (Fig. 6B).
Figure 6. scRNA-seq analysis of MHV68 transcription within LANAβlac+ peritoneal macrophages harvested ex vivo 16 hours post-infection.
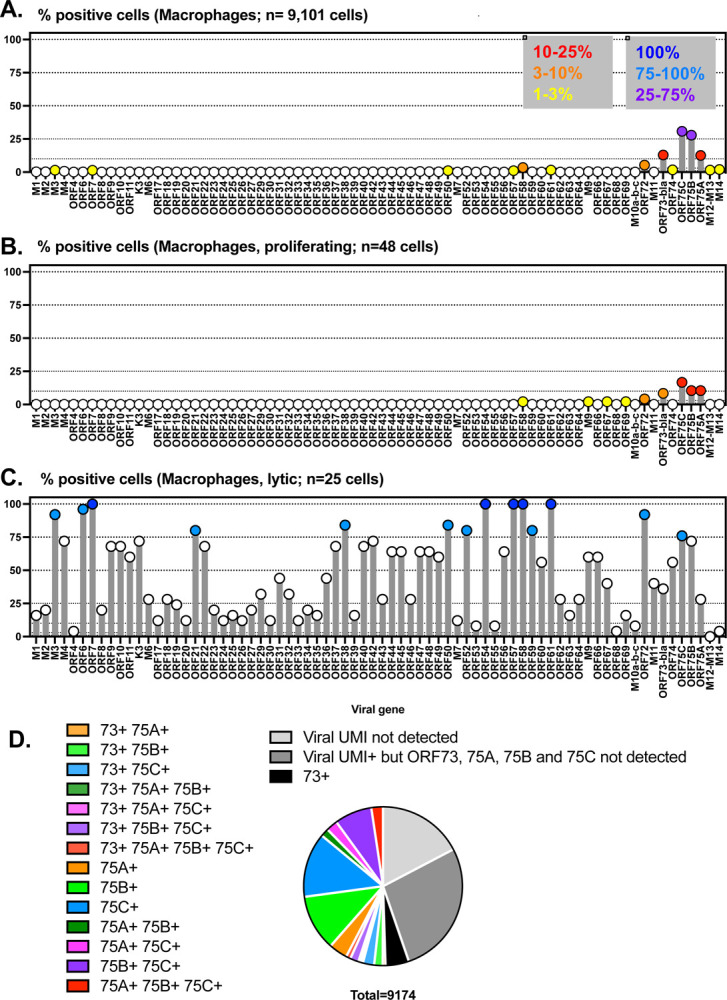
C57BL/6J mice were infected with 1×106 PFU of WT MHV68.LANAblac virus, with MHV68-infected, LANAblac+ peritoneal macrophages harvested and sort-purified at 16 hpi and subjected to scRNA-seq analysis. Analysis focuses on macrophage populations identified in Figure 5. (A-C) Frequency of cells with detectable reads mapping to MHV68 genes, comparing (A) macrophages, (B) macrophages with proliferative gene signature, or (C) macrophages with lytic gene expression, with number of cells for each population indicated. Viral genes are indicated on the x axis, arranged from left to right based on the gene arrangement for the published MHV68 genome, with the frequency of cells positive for each viral gene depicted on the y axis. To facilitate comparison of gene expression across the viral genome and cell populations, circles are color-coded to identify values that are in the indicated ranges (as defined in the key, panel A). Dotted lines indicate different threshholds for viral gene positivity, with these threshold values arbitrarily based on data. For panel C, only genes with >75% positivity were color-coded, given the limited number of events in this cell subset. (D) Frequency of macrophages based on viral gene expression, with a focus on the inter-relationship between ORF73, ORF75A, ORF75B, and ORF75C expression within single cells. Data in panel D shows data from 9,174 macrophages including both proliferating and lytic subsets. The frequency of viral UMI+ events differed between cell subsets in panels A-C (macrophages, 82.79% viral UMI+; macrophage proliferating (52.08% viral UMI+; macrophage lytic, 100% viral UMI+).
In contrast to the dominant macrophage subset, rare lytic macrophages had a distinct viral gene signature. Here, we detected multiple lytic cycle-associated genes including ORF7, ORF54, ORF57, ORF58, and ORF61 in 100% of these rare cells (dark blue circles, Fig. 6C). Further, >75% of cells contained detectable RNAs for M3, ORF6, ORF21, ORF38, ORF50, ORF52, ORF59, ORF72 and ORF75C (light blue circles, Fig. 6C). The high frequency of cells that expressed multiple, canonical lytic cycle transcripts is consistent these cells initiating lytic gene transcription in vivo.
We next sought to understand the high frequency detection of ORF75 RNAs within the dominant macrophage population (Fig. 6A). Previous studies of the ORF75 locus have revealed multiple transcriptional units, including a polycistronic RNA that spans from the 5’ end of ORF75A through the 3’ end of ORF75C (31). Notably, this polycistronic RNA would map to ORF75C based on polyA-based, oligo-dT primed scRNA-seq, and would not generate sequencing reads that would map to ORF75B or ORF75A. We also considered whether a high-degree of homology at the 3’ ends of the ORF75 genes might complicate mapping of sequencing reads to each individual ORF75. Though these genes likely arose from gene duplication, these genes have limited nucleotide identity within the final 150 nucleotides of each gene: using blastn (32), there is no significant similarity between the 3’ ends of either ORF75A and ORF75B or ORF75A and ORF75C. The closest homology across the final nucleotides of ORF75B and ORF75C remained extremely low, with only a stretch of 22 of 26 nucleotides aligning across the final 150 nucleotides of ORF75B and ORF75C (data not shown). This nucleotide divergence at the 3’ end of these genes strongly suggests that UMIs mapping to each gene were not due to errors in mapping. When we examined the intersection of ORF73, ORF75A, ORF75B, and ORF75C detection within single cells, considering all combinations of expression for these 4 viral genes, we found that 50.89% of macrophages had detectable expression of ORF75A, ORF75B and/or ORF75C; in many of these cells, we did not detect ORF73 (Fig. 6D). In total, these studies identify a high frequency of macrophages expressing transcripts from the ORF75 locus, and further corroborate that a low frequency of primary infected peritoneal macrophages are characterized by lytic transcription early after primary infection.
MHV68-infected J774 macrophages show expression of the ORF75 locus in the absence of full lytic gene transcription.
We next tested analyzed the expression of the ORF75 genes in MHV68-infected J774 cells. ORF75A, ORF75B and ORF75C were readily detected in MHV68-infected J774 cells from 16–48 hpi, albeit lower than in MHV68-infected 3T12 cells (Fig. 7A). Among these genes, we observed two different patterns. ORF75A expression was relatively comparable to ORF73 at all timepoints measured, with minimal change in expression between 24 and 48 hpi, suggesting constitutive transcription for both ORF73 and ORF75A (Fig. 7A). In contrast, ORF75B and ORF75C transcript levels were moderately lower than ORF73 at 16 and 24 hpi and showed a ~4 and ~22 fold increase between 24 and 48 hpi, respectively (right panel, Fig. 7A). The induction of these transcripts suggested that these transcripts may be at least, in part, induced during lytic replication in J774 cells (akin to the increased signal observed for ORF6, M7, Fig. 2B).
Figure 7. MHV68-infected J774 macrophages express multiple ORF75 genes with ORF73 and ORF75A characterized by similar expression characteristics.

Analysis of MHV68.LANAblac gene expression (MOI=1 PFU/cell) comparing 3T12 fibroblasts and J774 macrophages. (A) Quantitation of viral RNAs for ORF73 (immediate early), ORF75A (immediate early/early-late), ORF75B (early) and ORF75C (late) genes during MHV68 infection by qRT-PCR, comparing expression in 3T12 and J774 cells at the indicated timepoints. Data are from 3 independent experiments with 3 biological replicates per experiment (depicted by individual symbols), measured in technical triplicates, with data showing mean +/− SEM for each timepoint and sample. Statistical analysis was done using unpaired t test with statistically significant differences as indicated. ** p<0.01,***p<0.001 ****p<0.0001. (B) Quantitative RNA analysis of indicated genes in 3T12 and J774 cells, with or without treatment using a viral inhibitor (CDV or PAA). For panels A-B, “Relative Difference” defined as quantification of target gene transcript in comparison to reference gene transcript (host 18S) as described in Methods. Data for 3T12 cell line are from 1 independent experiment, J774 cell line data are from 2 independent experiments, with three biological replicates per experiment, measured in technical triplicates.
To test the impact of viral DNA replication, a key process required for late gene transcription, we treated MHV68-infected J774 or 3T12 cells with cidofovir (CDV), an inhibitor of the viral DNA polymerase (33, 34). In parallel, we treated cells with the viral DNA synthesis inhibitor phosphonoacetic acid (PAA) at 200 μg/mL (35); this treatment was toxic to J774 cells (data not shown). CDV and PAA would inhibit lytic cycle transcription of both early/late and late genes with minimal impact on gene expression during latent (or restricted) infection. As expected, infected 3T12 cells had higher baseline expression for ORF73, ORF75A, ORF75B, ORF75C than infected J774 cells (Fig. 7B). When 3T12 cells were treated with CDV or PAA, there was a pronounced reduction across viral genes compared to untreated samples, ranging from ~10-fold reduction in CDV- or PAA-treated cultures for ORF73 and ORF75A, to ~1000-fold reduction for ORF75C and M7 (Fig. 7B). In contrast, the impact of CDV on gene expression in J774 cells was much more modest, with ~2-fold reduction in RNA detected for ORF73 and ORF75A in CDV treated cultures relative to untreated cultures (Fig. 7B), compared with ~5–10 fold reduction in ORF75B, ORF75C and M7 RNAs, consistent with these transcripts being induced, in part, in a lytically-replicating subset of cells. These data are consistent with expression of the ORF75 locus in MHV68-infected J774 cells and suggest that at least the ORF75A gene shows comparable regulation to that observed for ORF73.
DISCUSSION
The cross-talk between MHV68 and macrophages has been a significant area of ongoing investigation, with early studies identifying macrophages as a significant latent reservoir beyond B cells (7, 13). Since then, macrophages have been identified as an early target of MHV68 infection after both intraperitoneal (36) and intranasal infection (12, 19), where a significant fraction of virus passes through macrophages (as defined by cre-dependent lineage tracing studies) (12). Whether MHV68 directly infects macrophages, or macrophage infection is expedited by intercellular handoff from infected epithelial cells (14) or by antibody-dependent entry through Fc receptors (15) would likely be influenced by the route of infection, stage of infection and host immune status. Regardless, there is now clear evidence that MHV68 can undergo replication within a subset of macrophages, a process revealed by lineage-tracing studies (12), lytic antigen expression within these cells (e.g. (16, 17)), and the studies presented here. MHV68 replication in macrophages can further be enhanced in the context of certain parasitic infections that both antagonize IFNγ-dependent suppressive mechanisms and induce expression of the Th2 cytokines IL-4 or IL-13, STAT6-inducing cytokines that can directly transactivate the gene 50 promoter, to promote lytic replication (29).
By examining the heterogeneity of infection at single cell resolution, through both flow cytometric analysis for markers of entry and LANA gene expression, lytic replication and single-cell RNA sequencing, our studies provide a refined perspective of MHV68 infection in macrophages. These studies unequivocally identify that MHV68 robustly infects macrophages and is capable of initiating transcription and translation of at least the ORF73 gene, with only a minor fraction of these cells showing hallmarks of lytic replication. Whether the majority of LANA+ cells reflect a bona fide latent population or reflect a previously undescribed, restricted form of infection remains an open question. Although ORF75 expression has not been previously observed during MHV68 macrophage infection, the ORF75 genes have been detected in the MHV68-positive latently-infected S11E B cell lymphoma (37). KSHV ORF75 transcription has also been detected in Kaposi’s sarcoma lesions in the absence of many lytic genes, both as a bicistronic K15/ORF75 transcript and as monocistronic ORF75 (38, 39). ORF75 expression in KS tumors did not clearly correspond with expression from the ORF73 locus (39), suggesting undiscovered modes of transcriptional regulation of the ORF75 genes that may span across at least MHV68 and KSHV.
Broadly our findings are consistent with various aspects of published literature. On the one hand, the magnitude of MHV68 lytic replication within bone marrow macrophages or myeloid cells is relatively modest compared to fibroblasts (17, 40), and lytic antigen expression is reported to occur within a subset of bone marrow-derived macrophages and RAW264.7 macrophages (15, 28, 29). Despite the relatively modest lytic replication of MHV68 in macrophages, MHV68 encodes multiple mechanisms that facilitate this process, including ORF4 which encodes the vRCA and promotes replication in a complement-independent manner (40) and ORF36 which encodes a viral protein kinase that phosphorylates H2AX and antagonizes HDAC1 and HDAC2 (16, 41). Conversely, multiple inhibitory mechanisms have been reported to limit lytic replication in macrophages, including IFNγ-dependent inhibition of the gene 50 promoter (42), and cell-intrinsic mechanisms mediated in part by HDACs and NCoR (41, 43) MHV68 lytic replication in macrophages is further regulated by cellular modulators of metabolism, constrained both by Liver X receptor- and Low-Density Lipoprotein Receptor-dependent mechanisms (44, 45). We postulate that these inhibitory mechanisms may affect the relative proportion of cells initiating lytic replication with MHV68, may constrain the magnitude of virus produced by lytic cells or may fundamentally alter the susceptibility of infection across all cells. Future studies will require single cell-based investigation of MHV68 to differentiate between these two potentially distinct modes of regulation.
Despite major inroads from the above studies, in vitro models of macrophage infection to date have fallen short of defining the dominant outcome of infection described here as being truly latent, an important future question that will only be resolved by focused analysis on cells lacking lytic protein expression. We anticipate that this could be done by purifying infected macrophages that lack lytic gene expression, followed by characterization of the state of the viral genome (i.e. episomal or linear) and the ability of these cells to enter productive virus replication (i.e. reactivation from latency). Alternatively, it is possible that this analysis may identify alternate regulation of the viral genome, or a failure to produce virus particles, which would suggest a restricted form of infection. Ultimately, these studies should seek to afford single-cell resolution, potentially combined with temporal fate-mapping strategies, to better differentiate between heterogeneous states of infection that may exist across a population of macrophages, and how the nature of infection may evolve over time. This is particularly relevant in the context of in vivo infection, where IFNγ constrains MHV68 reactivation from latency within peritoneal macrophages (46) and CD4 T cells constrain chronic macrophage infection in the lung (17). Recent studies of parasitic infection have further revealed that parasite-dependent expansion of the large peritoneal macrophage compartment, a primary target of MHV68 infection (36), can dramatically enhance the pool of MHV68-infected macrophages (47) indicating that MHV68 infection of macrophages is subject to multiple positive and negative regulatory pathways.
This study has important limitations that constrain our interpretations. First, many of the studies focus on infection of the J774 mouse myeloid cell line, a system we have leveraged as a reductionist in vitro system to study MHV68-macrophage interactions. Second, our studies of primary in vivo infection focus on a single, early timepoint of macrophage infection. Thus, it remains possible that a time course of in vivo macrophage infection may reveal a delayed form of lytic cycle transcription. Conservatively, we can conclude at least that the high frequency of ORF75 genes (ORF75A, B, and/or C) within primary macrophages is not explained by either the current paradigm of lytic or latent transcription (e.g. exemplified by (25)). The expression of individual ORF75 gene products, as defined by polyA-based scRNA-seq suggests that there are additional mechanisms of transcriptional regulation within the ORF75 locus than previously identified in lytically infected fibroblasts (31). These studies may further identify macrophage-specific regulation of the ORF75 locus and/or unique roles for the ORF75 gene products within macrophages. The ORF75 genes are known to encode proteins with a number of functions that could disproportionally impact macrophage infection.
The frequent expression of ORF75 genes within macrophages suggests that ORF75-targeted interventions (e.g. vaccination against ORF75 gene products, which contain known T cell epitopes (48–50) may selectively disrupt macrophage infection over infection in other cell types. Additionally, the distinct gene expression patterns of infected macrophages indicates variable susceptibility to treatment with current antivirals.
In total, our studies demonstrate both heterogeneity of MHV68 infection outcomes within macrophages and reveal an alternate transcriptional state at least early after primary macrophage infection in vivo. These studies further suggest a previously unappreciated role for the ORF75 locus as a potentially important regulator of macrophage infection, or a marker of an alternate outcome of infection that is neither lytic nor latent.
METHODS
Viruses and Tissue Culture
Mouse 3T12 fibroblasts (ATCC CCL-164) and mouse J774A.1 macrophages (subsequently referred to as “J774”; ATCC TIB-67) were cultured in complete DMEM (cDMEM): Dulbecco’s modified Eagle medium (DMEM; Life Technologies) supplemented with 5% and 10% fetal bovine serum (FBS; Atlanta Biologicals) respectively, 2 mM L-glutamine, 10 U/mL penicillin, and 10 μg/mL streptomycin sulfate. Cells were cultured at 37°C with 5% CO2. Wild-type (WT) MHV68, MHV68.LANA::βlac, and MHV68.H.GFP viruses were grown and prepared as described previously (23, 24, 26), with all virus titers determined from at least three independent plaque assays.
Infection
All in vitro infections were based on live cell counts with the TC20 Automated Cell Counter (Bio-Rad) with Trypan Blue dye (Bio-Rad, Cat. No. 145–0021). Viral stock was diluted with cDMEM for viral inoculum with a multiplicity of infection (MOI) of 1 PFU/cell. Cells were incubated at 37°C with 5% CO2 for 1 hour, with rocking every 15 min, before viral inoculum was removed and replaced with 1mL of cDMEM. For qPCR analysis of viral DNA replication and for beta-lactamase staining and flow cytometric analysis of infected cells, viral inoculum was left on cells after 1 hr infection time. For IL-4 treatments, media containing recombinant mouse IL-4 (PeproTech, Cat. No. 21414) was added to cells 16 hours before infection. Following infection, media containing 0, 10 or 100ng/mL of fresh IL-4 containing media were added respectively. Samples were harvested at the indicated hours post-infection (hpi) through either cell scraping (for DNA isolation) or through incubation in TRIzol (for RNA isolation).
Plaque Assay
Plaque assay quantification of viral titer was performed using 3T12 cells. Cells were plated in 12-well plates at 8.5×104 cells per well one day prior to infection. Viral samples were diluted 10-fold in 5% cDMEM. An internal standard was included for each infection to ensure reproducible sensitivity for each plaque assay. Cells were incubated with virus for 1 h at 37°C at 5% CO2. Plates were rocked every 15 min. Cells were then covered with an overlay composed of a 1:1 mix of 10% cDMEM and carboxymethyl cellulose (CMC; Sigma, Cat. No. C- 4888) supplemented with Gibco™ Amphotericin B (Thermo Fisher Scientific, Cat. No. 15290018). Cells were incubated for 8 days before staining with 0.5% methylene blue and plaques were counted.
RT-qPCR
RNA was isolated from infected cells harvested at indicated times by 10-minute incubation in TRIzol® reagent (Thermo Fisher Scientific, Cat. No. 15596026), followed by TURBO™ DNase (Invitrogen, Cat. No. AM2238) treatment according to manufacturer’s protocols. RNA amplification and removal of DNA was confirmed by PCR amplification of the control host gene, 18S, in the presence or absence of reverse transcriptase. RNA presence and absence of DNA was confirmed with RT-PCR and PCR respectively. RT-PCR was performed using the OneStep RT-PCR kit (Qiagen, Cat. No. 210212) with the following conditions: (i) 50°C for 30 min, (ii) 95°C for 15 min, (iii) 40 cycles of 94°C for 30 sec, 52°C for 30 sec, and 72°C for 30 sec, (iv) 72°C for 10 min, and (v) hold at 4°C. PCR was performed using Taq DNA polymerase (Qiagen, Cat No. 201205) with the following conditions: (i) 95°C for 5 min, (ii) 40 cycles of 94°C for 30 sec, 52°C for 30 sec, and 72°C for 30 sec, (iii) 72°C for 10 min, and (iv) hold at 4°C. RNA samples that showed no product following PCR amplification were deemed DNAfree, and then converted to cDNA using random primers (250ng/uL) (Invitrogen, Cat. No. 48190011) and SuperScript II reverse transcriptase (Invitrogen, Cat. No. 18064014) following the manufacturer’s protocol. 100 nanograms of cDNA was used for qPCR analysis of the identified genes (Quantitect Primer Assay, Qiagen) using the iQ SYBR green supermix (BioRad Cat. No.1708880) with the following conditions: (i) 95°C for 3 min, (ii) 40 cycles of 95°C for 15 sec, 60°C for 1 min, and (iii) 95°C for 15 sec, 60°C for 1 min, and 95°C for 15 sec. Amplification of viral genes were normalized to murine 18S expression to calculate the relative difference of target gene expression using the Pfaffl method, as previously described (51, 52) : Target primer efficienc^TargetΔCt/18S primer efficienc^18SΔCt. A single product for each target was confirmed by melt curve analysis. PCR primers are listed in Table 1.
Table 1:
Oligonucleotides for RT-PCR, PCR, and qPCR analysis
| Name | Accession number and Genome Position * | Sequence | Purpose |
|---|---|---|---|
| gB Forward Primer | MHV68: 17873…17892 | 5’—GGC CCA AAT TCA ATT TGC CT—3’ | TaqMan qPCR for MHV68 genome |
| gB Reverse Primer | MHV68: 17924…17943 | 5’—CCC TGG ACA ACT CCT CAA GC—3’ | TaqMan qPCR for MHV68genome |
| gB Probe | MHV68: 17896…17921 | 5’—ACA AGC TGA CCA CCA GCG TCA ACA AC—3’ | TaqMan qPCR for MHV68genome |
| 18S Forward Primer | Accession Number: NR_003278 | 5’— AGA TCA AAA CCA ACC CGG TGA—3’ | RT-PCR and PCR for murine 18S gene |
| 18S Reverse Primer | Accession Number: NR_003278 | 5’—GGT AAG AGC ATC GAG GGG GC—3’ | RT-PCR and PCR for murine 18S gene |
| ORF73 SYBR G – Forward Primer | MHV68: 104092…104073 | 5’— GAG CCC CCT ACA GAG CCC CC —3’ | SYBR Green qPCR for MHV68ORF73 gene |
| ORF73 SYBR G - Reverse Primer | MHV68: 103981…104000 | 5’— CAC CTT GCT CTG CAC CGG CA —3’ | SYBR Green qPCR for MHV68ORF73 gene |
| ORF6 SYBR G – Forward Primer | MHV68: 11699…11783 | 5’— ATG TTG TCA GCA CCC ATG AA—3’ | SYBR Green qPCR for MHV68ORF6 gene |
| ORF6 SYBR G – Reverse Primer | MHV68: 11802…11783 | 5’— AAG GGC AGT AGC TGG TCA GA—3’ | SYBR Green qPCR for MHV68ORF6 gene |
| M7 SYBR G – Forward Primer | MHV68: 70109…70128 | 5’—GCT CCT GCT GAC ACA TCA GA—3’ | SYBR Green qPCR for MHV68M7 gene |
| M7 SYBR G – Reverse Primer | MHV68: 70185…70166 | 5’—TGG GGT TTG GAC TCT GTA GG—3’ | SYBR Green qPCR for MHV68M7 gene |
| MHV68 ORF75A iQ SYBR - Forward Primer | MHV68: 114964…114945 | 5’—ACC TGA CAC CCC AAG AAC AG—3’ | SYBR Green qPCR for MHV68ORF75A gene |
| MHV68 ORF75A iQ SYBR - Reverse Primer | MHV68: 114834…114853 | 5’—GAG CAC TTT TGG TGG AGA GC—3’ | SYBR Green qPCR for MHV68ORF75A gene |
| MHV68 ORF75B iQ SYBR - Forward Primer | MHV68: 113267…113248 | 5’—CAG CCT CTC AAC CTT TCC AG—3’ | SYBR Green qPCR for MHV68ORF75B gene |
| MHV68 ORF75B iQ SYBR - Reverse Primer | MHV68: 113093…113112 | 5’—ATA GGA GCC ACC GTT GAT TG—3’ | SYBR Green qPCR for MHV68.ORF73βla ORF75B |
| MHV68 ORF 75C iQ SYBR - Forward Primer | MHV68: 109930…109911 | 5’—AAG ACA CAG AGG CTG GGA GA—3’ | SYBR Green qPCR for MHV68ORF75C gene |
| MHV68 ORF75C iQ SYBR - Reverse Primer | MHV68: 109758…109777 | 5’—GAC GGG TAG ACG TGG TGA CT—3’ | SYBR Green qPCR for MHV68ORF75C gene |
| BLAC SYBR G – Forward Primer | MHV68.ORF73βla: 104867…104886 | 5’—GCT ATG TGG CGC GGT ATT AT—3’ | SYBR Green qPCR for MHV68.ORF73βla Blac gene |
| BLAC SYBR G – Reverse Primer | MHV68.ORF73βla: 104701…104720 | 5’—AAG TTG GCC GCA GTG TTA TC—3’ | SYBR Green qPCR for MHV68.ORF73βla Blac gene |
MHV68 genome coordinates refer to NC_001826. In the case of MHV68.ORF73βla Bla gene, sequences correspond to NCBI GenBank: MH636806.1, the complete genome sequence of MHV68 containing the ORF73bla gene fusion.
Quantitative PCR for quantification of viral DNA replication
Infected cells were harvested by cell scraping at time (hpi) indicated. Harvested cells underwent three freeze/thaw cycles and DNA was isolated using the DNeasy Blood and Tissue Kit (Qiagen, Cat. No. 69504), with an overnight proteinase K incubation and heat inactivation at 56°C. DNA was normalized to a concentration of 20 ng/uL in molecular grade water. qPCR analysis was done on 100ng of DNA using a LightCycler 480 Probe Master-Mix kit (Roche, Cat. No. 04707494001) and a primer and probe set specific to MHV68 gB to quantify the number of viral genome copies. Primers and probes listed in Table 1. A glycoprotein B (gB) standard curve (53) was generated using a gB plasmid dilution series ranging from 102 to 1010 copies diluted in background DNA, with a limit of detection (LOD) of 100 copies.
Ethics Statement
All animal studies were performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Studies were conducted in accordance with the University of Colorado Denver/Anschutz Institutional Animal Use and Care Committee under the Animal Welfare Assurance of Compliance policy (assurance no. D16–00171). All procedures were performed under isoflurane anesthesia, and all efforts were made to minimize suffering.
Single-cell RNA Sequencing
To characterize MHV68 transcription during primary, acute peritoneal macrophage infection in vivo, C57BL/6 mice were infected with 1×106 PFU by intraperitoneal injection. Peritoneal cells were harvested 16 hours post-infection, stained with the live/dead discrimination dye Zombie Near-IR (Zombie NIR™, BioLegend, Cat. No. 423105), F4/80-AlexaFluor 674 (clone BM8, 1:200 dilution) in the presence of Fc receptor blockade (clone 2.4G2), and CCF2-AM (Thermo Fisher, Cat. No. K1023). Cells were sorted on a MoFlo Astrios, purifying live, single, F4/80+ cells that either had CCF-2 cleavage (indicating virus infection, defined by expression of the LANA::blac fusion protein) or lacking CCF-2 cleavage (indicating cells that were not infected or failed to express the LANA::blac fusion protein). Cells were subjected to standard 3’-based single cell RNA-seq analysis (10x Genomics), with cell processing and sequencing done in the University of Colorado Cancer Center Genomics Shared Resource (RRID: SCR_021984). Resulting data were processed as follows: Cell Ranger (v6.0.1) (54) was used to process the fastq files to cell and gene count tables using unique molecule identifiers (UMIs) with the include-introns parameter. Because of the difficulties counting viral reads, this was performed in a two-pass manner. In the first pass, reads were aligned to a chimeric genome of mouse mm10 (GENCODE M23 gene annotations) and MH636806.1 (no gene annotations, only positive and negative strand alignment). The viral counts were stored in metadata and removed from the counts matrix. Reads mapping to MH636806.1 in step 1 were aligned to the same chimeric genome but in this case, the viral transcriptome as well as specific intergenic regions were annotated (data not shown). The intergenic regions were determined as the strand-specific sequences between genes with 1 base pair of padding on both ends. To minimize reads overlapping with multiple viral genes, only the first 70bp of each read were aligned and counted. The resulting counts matrix of viral alignment was appended to the host gene counts from step 1.
The Seurat (v4.0.4) (55) pipeline was used for downstream quality control and analysis. Cellranger-filtered data was read into Seurat. Host genes were removed if identified in fewer than 10 cells while viral gene intergenic regions were removed if found in no cells. Cells were removed if they expressed < 50 genes or viral regions, < 5000 UMIs, > 5 % total UMIs from mitochondria, or > 70000 UMIs. The filtered data was normalized by dividing gene counts by total counts per cell and multiplied by 10,000 followed by natural-log transformation. The top 2,000 most variable genes were scaled with total UMI and percentage mitochondria regressed out. These were used to calculate principal components (PCs) with the top 20 used to perform Uniform Manifold Approximation and Projection (UMAP) and determining the k-nearest neighbors and clustering.
Clusters were identified by over-representation analysis and examining the top enriched markers. Canonical cell type markers were plotting in UMAP space and using dot plots. Heatmaps, dot plots, bar plots, scatter plots, and violin plots were generated using Seurat and ggplot2 R packages (v3.4.0) (56). Groups of cells in heatmaps were randomly subset to the size of the smallest group.
Flow Cytometry
3T12 and J774 cells were infected with MHV68 LANAβlac or MHV68.H.GFP at an MOI of 1 PFU/cell and harvested at the indicated time points followed by staining for flow cytometric analysis. Samples treated with the recombinant murine IL-4 were given 10% cDMEM with 10ng/mL of IL-4 eighteen hours prior to infection and replaced with fresh media containing IL-4 immediately following infection. Cell suspensions were stained with LIVE/DEAD fixable Near-IR Dead Cell stain kit (Invitrogen, Cat. No. L10119) at a 1:1000 dilution in BSS wash (111mM Dextrose, 2mM KH2PO4, 10mM Na2HPO4, 25.8 CaCl2•2H2O, 2.7mM KCl, 137mM NaCl, 19.7mM MgCl•6H2O, 16.6mM MgSO4). Cells were then stained for CCF2 or protein detection. Cells for CCF2 detection were stained with CCF2-AM (Thermo Fisher, Cat. No. K1023) at a 1:10 dilution following the manufacturer’s protocol. To detect proteins, cells were stained with a Fc receptor block (Fc Shield, Tonbo Biosciences, Cat. No. 70–0161), a rabbit antibody against the MHV68 ORF4 protein (vRCA, 1:400 dilution) (gift of the Virgin Lab, Washington University St. Louis (35, 51)) labeled with a Zenon R-phycoerythrin antirabbit IgG reagent (Invitrogen, Cat. No. Z25307), per manufacturer’s recommendations, and a AF647-conjugated anti-mouse phospho-histone H2AX antibody (1:800 dilution) (clone JBW30). Samples were fixed in 1% paraformaldehyde prior to analysis using the Bio-Rad ZE5/YETI Cell Analyzer or an Agilent Novocyte Penteon flow cytometer. All flow cytometry experiments included unstained, single-stain and full minus one controls to define background fluorescence, fluorescent signal spread and compensation.
Light microscopy
At the indicated time points, 6-well plates of 3T12 or J774 cells were imaged on a Nikon Eclipse Ti2 inverted light microscope equipped with Iris 15 camera (Photometris) and use of NIS elements software. Plates were mounted on a stage heated to 37°C. A 10x phase objective was used to capture brightfield images. Immediately following image acquisition, samples were harvested and processed for flow cytometric analysis.
Statistical Analysis and Software
Data analysis, graphing and statistical analyses were performed using GraphPad Prism (version 9.4.0; GraphPad Software, San Diego, California USA, www.graphpad.com). Flow cytometry data were analyzed using FlowJo (version 10.8.1. Ashland, OR: Becton, Dickinson and Company; 2022). scRNA-seq data processing and analysis were done as described above. Statistical significance was tested by unpaired, nonparametric, Mann-Whitney t test or 1-way ANOVA for comparing replicate values from experiments. RNA-Seq data have been deposited to NCBI GEO and will be made publicly available upon manuscript acceptance.
Acknowledgments:
We thank Dr. Brian Russo’s lab, especially Jenna Vickery for the use and training of their Nikon Eclipse Ti2 inverted light microscope, and Christine Childs, Kristina Terrell and Dmitry Baturin of the University of Colorado Cancer Center Flow Cytometry Shared Resource for training and assistance with flow cytometry. This study was supported by National Institutes of Health grant R01 AI157201 awarded to LvD and ETC, with additional support by the National Institutes of Health Cancer Center Support Grant (P30CA046934), including assistance from the Bioinformatics and Biostatistics Shared Resource (RRID: SCR_021983), Flow Cytometry Shared Resource (RRID: SCR_022035), and Genomics Shared Resource (RRID: SCR_021984).
References:
- 1.Longnecker R, Neipel F. 2007. Introduction to the human gamma-herpesviruses. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis, Cambridge. [PubMed] [Google Scholar]
- 2.Mariggio G, Koch S, Schulz TF. 2017. Kaposi sarcoma herpesvirus pathogenesis. Philos Trans R Soc Lond B Biol Sci 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Young LS, Yap LF, Murray PG. 2016. Epstein-Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer 16:789–802. [DOI] [PubMed] [Google Scholar]
- 4.Virgin HWt, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol 71:5894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Tibbetts SA, Krug LT. 2021. Conquering the Host: Determinants of Pathogenesis Learned from Murine Gammaherpesvirus 68. Annu Rev Virol 8:349–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cieniewicz B, Santana AL, Minkah N, Krug LT. 2016. Interplay of Murine Gammaherpesvirus 68 with NF-kappaB Signaling of the Host. Front Microbiol 7:1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flano E, Husain SM, Sample JT, Woodland DL, Blackman MA. 2000. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J Immunol 165:1074–81. [DOI] [PubMed] [Google Scholar]
- 8.Flano E, Kim IJ, Moore J, Woodland DL, Blackman MA. 2003. Differential gammaherpesvirus distribution in distinct anatomical locations and cell subsets during persistent infection in mice. J Immunol 170:3828–34. [DOI] [PubMed] [Google Scholar]
- 9.Sunil-Chandra NP, Efstathiou S, Nash AA. 1992. Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. J Gen Virol 73 (Pt 12):3275–9. [DOI] [PubMed] [Google Scholar]
- 10.Babcock GJ, Decker LL, Volk M, Thorley-Lawson DA. 1998. EBV persistence in memory B cells in vivo. Immunity 9:395–404. [DOI] [PubMed] [Google Scholar]
- 11.Aalam F, Nabiee R, Castano JR, Totonchy J. 2020. Analysis of KSHV B lymphocyte lineage tropism in human tonsil reveals efficient infection of CD138+ plasma cells. PLoS Pathog 16:e1008968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frederico B, Milho R, May JS, Gillet L, Stevenson PG. 2012. Myeloid infection links epithelial and B cell tropisms of Murid Herpesvirus-4. PLoS Pathog 8:e1002935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weck KE, Kim SS, Virgin HI, Speck SH. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J Virol 73:3273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawler C, Milho R, May JS, Stevenson PG. 2015. Rhadinovirus host entry by cooperative infection. PLoS Pathog 11:e1004761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosa GT, Gillet L, Smith CM, de Lima BD, Stevenson PG. 2007. IgG fc receptors provide an alternative infection route for murine gamma-herpesvirus-68. PLoS One 2:e560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tarakanova VL, Leung-Pineda V, Hwang S, Yang CW, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, Virgin HWt. 2007. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1:275–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan CSE, Lawler C, Stevenson PG. 2017. CD8+ T cell evasion mandates CD4+ T cell control of chronic gamma-herpesvirus infection. PLoS Pathog 13:e1006311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frederico B, Chao B, May JS, Belz GT, Stevenson PG. 2014. A murid gammaherpesviruses exploits normal splenic immune communication routes for systemic spread. Cell Host Microbe 15:457–70. [DOI] [PubMed] [Google Scholar]
- 19.Frederico B, Chao B, Lawler C, May JS, Stevenson PG. 2015. Subcapsular sinus macrophages limit acute gammaherpesvirus dissemination. J Gen Virol 96:2314–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suarez AL, van Dyk LF. 2008. Endothelial cells support persistent gammaherpesvirus 68 infection. PLoS Pathog 4:e1000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sunil-Chandra NP, Efstathiou S, Nash AA. 1993. Interactions of murine gammaherpesvirus 68 with B and T cell lines. Virology 193:825–33. [DOI] [PubMed] [Google Scholar]
- 22.Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HI. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J Virol 70:6775–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nealy MS, Coleman CB, Li H, Tibbetts SA. 2010. Use of a virus-encoded enzymatic marker reveals that a stable fraction of memory B cells expresses latency-associated nuclear antigen throughout chronic gammaherpesvirus infection. J Virol 84:7523–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diebel KW, Oko LM, Medina EM, Niemeyer BF, Warren CJ, Claypool DJ, Tibbetts SA, Cool CD, Clambey ET, van Dyk LF. 2015. Gammaherpesvirus small noncoding RNAs are bifunctional elements that regulate infection and contribute to virulence in vivo. mBio 6:e01670–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng BY, Zhi J, Santana A, Khan S, Salinas E, Forrest JC, Zheng Y, Jaggi S, Leatherwood J, Krug LT. 2012. Tiled microarray identification of novel viral transcript structures and distinct transcriptional profiles during two modes of productive murine gammaherpesvirus 68 infection. J Virol 86:4340–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forrest JC, Speck SH. 2008. Establishment of B-cell lines latently infected with reactivation-competent murine gammaherpesvirus 68 provides evidence for viral alteration of a DNA damage-signaling cascade. J Virol 82:7688–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berger JN, Sanford B, Kimball AK, Oko LM, Kaspar RE, Niemeyer BF, Jones KL, Clambey ET, van Dyk LF. 2020. Redefining De Novo Gammaherpesvirus Infection Through High-Dimensional, Single-Cell Analysis of Virus and Host. bioRxiv doi: 10.1101/2020.08.11.203117:2020.08.11.203117. [DOI] [Google Scholar]
- 28.Wang G, Zarek C, Chang T, Tao L, Lowe A, Reese TA. 2021. Th2 Cytokine Modulates Herpesvirus Reactivation in a Cell Type Specific Manner. J Virol doi: 10.1128/JVI.0194620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reese TA, Wakeman BS, Choi HS, Hufford MM, Huang SC, Zhang X, Buck MD, Jezewski A, Kambal A, Liu CY, Goel G, Murray PJ, Xavier RJ, Kaplan MH, Renne R, Speck SH, Artyomov MN, Pearce EJ, Virgin HW. 2014. Helminth infection reactivates latent gamma-herpesvirus via cytokine competition at a viral promoter. Science 345:5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulkarni A, Anderson AG, Merullo DP, Konopka G. 2019. Beyond bulk: a review of single cell transcriptomics methodologies and applications. Curr Opin Biotechnol 58:129136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Skike ND, Minkah NK, Hogan CH, Wu G, Benziger PT, Oldenburg DG, Kara M, Kim-Holzapfel DM, White DW, Tibbetts SA, French JB, Krug LT. 2018. Viral FGARAT ORF75A promotes early events in lytic infection and gammaherpesvirus pathogenesis in mice. PLoS Pathog 14:e1006843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Z, Schwartz S, Wagner L, Miller W. 2000. A greedy algorithm for aligning DNA sequences. J Comput Biol 7:203–14. [DOI] [PubMed] [Google Scholar]
- 33.Lalezari JP, Stagg RJ, Jaffe HS, Hitchcock MJ, Drew WL. 1996. A preclinical and clinical overview of the nucleotide-based antiviral agent cidofovir (HPMPC). Adv Exp Med Biol 394:105–15. [DOI] [PubMed] [Google Scholar]
- 34.Andrei G, Topalis D, De Schutter T, Snoeck R. 2015. Insights into the mechanism of action of cidofovir and other acyclic nucleoside phosphonates against polyoma- and papillomaviruses and non-viral induced neoplasia. Antiviral Res 114:21–46. [DOI] [PubMed] [Google Scholar]
- 35.Kapadia SB, Molina H, van Berkel V, Speck SH, Virgin HWt. 1999. Murine gammaherpesvirus 68 encodes a functional regulator of complement activation. J Virol 73:7658–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riggs JB, Medina EM, Perrenoud LJ, Bonilla DL, Clambey ET, van Dyk LF, Berg LJ. 2021. Optimized Detection of Acute MHV68 Infection With a Reporter System Identifies La1rge Peritoneal Macrophages as a Dominant Target of Primary Infection. Front Microbiol 12:656979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez-Guzman D, Rickabaugh T, Wu TT, Brown H, Cole S, Song MJ, Tong L, Sun R. 2003. Transcription program of murine gammaherpesvirus 68. J Virol 77:10488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rose TM, Bruce AG, Barcy S, Fitzgibbon M, Matsumoto LR, Ikoma M, Casper C, Orem J, Phipps W. 2018. Quantitative RNAseq analysis of Ugandan KS tumors reveals KSHV gene expression dominated by transcription from the LTd downstream latency promoter. PLoS Pathog 14:e1007441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramaswami R, Tagawa T, Mahesh G, Serquina A, Koparde V, Lurain K, Dremel S, Li X, Mungale A, Beran A, Ohler ZW, Bassel L, Warner A, Mangusan R, Widell A, Ekwede I, Krug LT, Uldrick TS, Yarchoan R, Ziegelbauer JM. 2022. Transcriptional analysis identifies overlapping and tissue-distinct profiles between Kaposi sarcoma tumors of the skin and gastrointestinal tract. bioRxiv doi: 10.1101/2022.03.18.484923:2022.03.18.484923. [DOI] [Google Scholar]
- 40.Tarakanova VL, Molleston JM, Goodwin M, Virgin HWt. 2010. MHV68 complement regulatory protein facilitates MHV68 replication in primary macrophages in a complement independent manner. Virology 396:323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mounce BC, Tsan FC, Droit L, Kohler S, Reitsma JM, Cirillo LA, Tarakanova VL. 2011. Gammaherpesvirus gene expression and DNA synthesis are facilitated by viral protein kinase and histone variant H2AX. Virology 420:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodwin MM, Canny S, Steed A, Virgin HW. 2010. Murine gammaherpesvirus 68 has evolved gamma interferon and stat1-repressible promoters for the lytic switch gene 50. J Virol 84:3711–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goodwin MM, Molleston JM, Canny S, Abou El Hassan M, Willert EK, Bremner R, Virgin HW. 2010. Histone deacetylases and the nuclear receptor corepressor regulate lyticlatent switch gene 50 in murine gammaherpesvirus 68-infected macrophages. J Virol 84:12039–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lange PT, Schorl C, Sahoo D, Tarakanova VL. 2018. Liver X Receptors Suppress Activity of Cholesterol and Fatty Acid Synthesis Pathways To Oppose Gammaherpesvirus Replication. mBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aurubin CA, Knaack DA, Sahoo D, Tarakanova VL. 2021. Low-Density Lipoprotein Receptor Suppresses the Endogenous Cholesterol Synthesis Pathway To Oppose Gammaherpesvirus Replication in Primary Macrophages. J Virol 95:e0064921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steed A, Buch T, Waisman A, Virgin HWt. 2007. Gamma interferon blocks gammaherpesvirus reactivation from latency in a cell type-specific manner. J Virol 81:6134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zarek CM, Dende C, Coronado J, Pendse M, Dryden P, Hooper LV, Reese TA. 2022. Coinfection with Intestinal Parasite Expands Resident Macrophages and Impairs Control of Chronic Herpesvirus Infection. bioRxiv doi: 10.1101/2022.10.05.510926:2022.10.05.510926. [DOI] [Google Scholar]
- 48.Freeman ML, Lanzer KG, Cookenham T, Peters B, Sidney J, Wu TT, Sun R, Woodland DL, Sette A, Blackman MA. 2010. Two kinetic patterns of epitope-specific CD8 T-cell responses following murine gammaherpesvirus 68 infection. J Virol 84:2881–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gredmark-Russ S, Cheung EJ, Isaacson MK, Ploegh HL, Grotenbreg GM. 2008. The CD8 T-cell response against murine gammaherpesvirus 68 is directed toward a broad repertoire of epitopes from both early and late antigens. J Virol 82:12205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freeman ML, Burkum CE, Cookenham T, Roberts AD, Lanzer KG, Huston GE, Jensen MK, Sidney J, Peters B, Kohlmeier JE, Woodland DL, van Dyk LF, Sette A, Blackman MA. 2014. CD4 T cells specific for a latency-associated gamma-herpesvirus epitope are polyfunctional and cytotoxic. J Immunol 193:5827–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oko LM, Kimball AK, Kaspar RE, Knox AN, Coleman CB, Rochford R, Chang T, Alderete B, van Dyk LF, Clambey ET. 2019. Multidimensional analysis of Gammaherpesvirus RNA expression reveals unexpected heterogeneity of gene expression. PLoS Pathog 15:e1007849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coomes SM, Farmen S, Wilke CA, Laouar Y, Moore BB. 2011. Severe gammaherpesvirus-induced pneumonitis and fibrosis in syngeneic bone marrow transplant mice is related to effects of transforming growth factor-beta. Am J Pathol 179:2382–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH. 2017. Massively parallel digital transcriptional profiling of single cells. Nat Commun 8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R. 2021. Integrated analysis of multimodal single-cell data. Cell 184:3573–3587 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wickham H. 2016. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York. [Google Scholar]