

Abstract
Huntington’s disease arises from a toxic gain of function in the huntingtin (HTT) gene. As a result, many HTT-lowering therapies are being pursued in clinical studies, including those that reduce HTT RNA and protein expression in the liver. To investigate potential impacts, we characterized molecular, cellular, and metabolic impacts of chronic HTT lowering in mouse hepatocytes. Lifelong hepatocyte HTT loss is associated with multiple physiological changes, including increased circulating bile acids, cholesterol and urea, hypoglycemia, and impaired adhesion. HTT loss causes a clear shift in the normal zonal patterns of liver gene expression, such that pericentral gene expression is reduced. These alterations in liver zonation in livers lacking HTT are observed at the transcriptional, histological and plasma metabolite level. We have extended these phenotypes physiologically with a metabolic challenge of acetaminophen, for which the HTT loss results in toxicity resistance. Our data reveal an unexpected role for HTT in regulating hepatic zonation, and we find that loss of HTT in hepatocytes mimics the phenotypes caused by impaired hepatic β-catenin function.
Keywords: Huntington’s disease, Huntingtin, Knock-out, B-catenin, Liver zonation
Graphical Abstract
Introduction
Huntington’s disease (HD) is a fatal autosomal dominant neurodegenerative disease arising from the expansion of a glutamine-coding CAG tract near the 5’ end of huntingtin (HTT) (MacDonald et al, 1993). Progressive changes in mood, cognition and characteristic movement symptoms have been mapped in people carrying HD mutations. Unfortunately, no disease modifying treatments have been approved (Ross et al, 2014). Recently a major focus in the HD field has been huntingtin mRNA and protein (HTT and HTT) lowering therapies, which have advanced to human clinical studies with a diverse range of Huntingtin lowering agents (Tabrizi et al, 2019). Notably, multiple companies are investigating small molecule splice modulator drugs that result in degradation of Htt’s mRNA via nonsense mediated decay - treatment with these drugs results in robust reductions of Htt in the liver, suggesting that better understanding the impact of HTT lowering on hepatic physiology is well justified (Bhattacharyya et al, 2021a; Keller et al, 2022a).
HTT is highly conserved and widely expressed (Saudou & Humbert, 2016). Population studies reveal that loss of function mutations in HTT are much rarer than expected by chance, suggesting Htt is poorly tolerant of these mutations (e.g. gnomAD - observed/expected pLoF in HTT = 0.12, pLI = 1) (Karczewski et al, 2020). Indeed, Htt knockout in mice results in early embryonic lethality (Duyao et al, 1995; Nasir et al, 1995; Zeitlin et al, 1995) and hypomorphic alleles that express less Htt than normal in humans cause a profound neurodevelopmental disorder whose symptoms are distinct from the HD (Lopes et al, 2016; Rodan et al, 2016). HTT has been associated with many cellular pathways and roles including regulating autophagy, transcription, vesicular trafficking, cellular polarity, adhesion and others (Saudou & Humbert, 2016).
To better understand normal HTT functions, and how these might predict the risks for ongoing HTT lowering studies in HD patients, we generated mice that lack Htt expression in hepatocytes. Mice lacking Htt in hepatocytes are viable and appear healthy, which enabled us to conduct studies on the impact of HTT loss in vivo. We observe many molecular, transcriptional, and physiological changes in mice which lack Htt expression in hepatocytes. Most notably, we observe a shift in the normal zonated pattern of gene expression in the liver, consistent with a loss of pericentral hepatocyte identity.
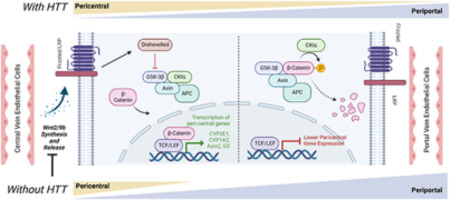
Hepatocyte identity and gene expression varies across several axes, including a gradient of regionalized gene expression changes between the pericentral and periportal vein regions (Russell & Monga, 2017). The acquisition and maintenance of pericentral gene expression in hepatocytes is under the positive control of β-catenin signaling, which in turn is activated by the paracrine release of specific Wnt species from liver endothelial cells (ECs) (Hu et al, 2022). In short, we observe transcriptional, histological and physiological evidence of reduced pericentral hepatocytes in the livers of HttLKO/LKO mice, suggesting a periportalization of the liver after HTT loss, very similar to what has been observed in mice with directly impaired hepatic β-catenin signaling (Tan et al, 2006; Yang et al, 2014; Preziosi et al, 2017; Hu et al, 2022).
Results
Generation and validation of hepatocyte-specific Htt knockout (HttLKO/LKO)
To generate mice lacking hepatic Htt, we crossed a previously described allele in which exon-1 of the Htt is flanked by loxP sites (Htttm2Szi, hereafter Httfl/fl) (Dragatsis et al, 2000) to B6.Cg-Speer6ps1Tg(Alb-cre)21Mgn/J, yielding an allele we refer to as hepatocyte-specific Htt knockout mice (HttLKO). HttLKO/LKO mice were born in expected Mendelian ratios when crossing to homozygosity (Table S1), breed with normal fecundity as homozygotes (mean = 6.3, s.d.= 2.4 pups per litter) (NAGASAWA et al, 1973) and are indistinguishable from littermates in terms of gross appearance and behavior. Htt transcripts and protein are markedly reduced in the livers of HttLKO/LKO mice (Fig. 1A–B), while HTT expression is spared in a survey of other peripheral tissues based on western blot analysis (Fig. 1B–C). Complete HTT loss in bulk liver assessments is not predicted (or seen, Fig. 1A–C), as our Cre line drives HTT knockout selectively in hepatocytes, but not the other cell types that comprise approximately 20% of liver cells (Poisson et al, 2017). These data suggest we successfully generated viable constitutive HttLKO/LKO mice, which lack both Htt and HTT in hepatocytes, but not in other organs examined.
Fig 1: Huntingtin transcript, protein levels, and body weight of hepatocyte-specific Huntingtin knockout mice (HttLKO/LKO).

(A) RNAseq reveals Htt mRNA is reduced in HttLKO/LKO mice compared to wildtype littermates (N = 5/genotype, p < 0.0001). (B-C) Western blot reveals that liver HTT is reduced in HttLKO/LKO mice compared to wildtype littermates (N = 4/genotype, t(3.8) = 4.9; p = 0.035), while brain, heart, and kidney are unchanged (Brain, t(5.2) = 2.2; p = 0.33; Heart, t(3.7) = 2.1; p = 0.43; Kidney, t(5.8) = t1.1; p = 1). Bonferroni correction was used to adjust for multiple comparisons. (D) HttLKO/LKO mice weigh less than wildtype littermates (9.5% reduction; N = 11 WT, 20 LKO, p < 0.0001). * indicates significant statistical difference.
Physiological and Adhesion Phenotypes
We generated large cohorts of female Htt+/+ and HttLKO/LKO mice for metabolic analyses. Throughout their lives, HttLKO/LKO mice weigh subtly less than Htt+/+ mice (Fig. 1D, linear mixed effects model genotype effect F(1,30) = 28.9, p < 0.001). There are no corresponding differences in terminal organ weights (liver, perigonadal white fat, interscapular brown adipose tissue, spleen), when considered either as raw weights or normalized to body weights (Table S2). Plasma chemistry measurements in 6-month-old HttLKO/LKO mice (Table S3) reveal increased circulating blood urea nitrogen (BUN; Fig. 2A; 29% increase; N = 11/genotype, t-test t(14.8) = 3.8; p = 0.002) and total cholesterol (Fig. 2B; 35% increase; N = 11/genotype, t-test t(12.4) = 3.6; p = 0.003), while no differences were observed in the levels of liver enzymes alanine aminotransferase (Fig. 2C, N = 11/genotype, t-test t(9.25) = −0.2; p = 0.88) or aspartate aminotransferase (Fig. 2D, N = 11/genotype, t-test t(9.9) = −0.21; p = 0.84), indicating that these changes occur in the absence of gross liver damage in aged HttLKO/LKO mice. We also found that HttLKO/LKO mice have increased level of bile acids when tested at 2 months (Fig. 2E, 189% increase; N = 163 (Htt+/+) and 34 (HttLKO/LKO), t-test t(40.5) = 2.6; p = 0.01). Collectively, these data reveal a pattern of altered systemic urea, cholesterol and bile acid metabolism in HttLKO/LKO mice.
Fig 2: Hepatocyte-specific Htt knockout mice have reduced circulating glucose, elevated urea, total cholesterol and bile acids, but normal liver enzymes, glucose disposal, insulin sensitivity and gluconeogenesis.

(A) At 6-months of age, HttLKO/LKO mice have increased circulating blood urea nitrogen (BUN), (B) increased circulating total cholesterol, (C) normal ALT and (D) AST I increased circulating bile acids and (F) reduced fasting glucose. HttLKO/LKO mice display normal glucose clearance (G) when challenged with bolus of glucose in a glucose tolerance test, display normal hypoglycemia and recovery (H) when challenged with an insulin tolerance test, and no change in time required to convert pyruvate and clear the resulting glucose (I) in a pyruvate tolerance test. Star symbol (*) indicates significant statistical difference.
Given the liver’s key role in systemic glucose metabolism, and HTT’s proposed controversial role in regulating organismal and cellular metabolism (Brustovetsky, 2016), we investigated systemic glucose metabolism in HttLKO/LKO mice 6- and 13-months of age. At both ages, we observe significant 18-hour fasting hypoglycemia in HttLKO/LKO compared to Htt+/+ mice (Fig. 2F, ages pooled for plotting, 27% reduction; N = 66 wildtype, 22 HttLKO/LKO, t-test t(42.9) = 5.4; p < 0.0001). To probe changes in glucose disposal we conducted glucose tolerance tests (GTT) and insulin tolerance tests (ITT), finding no effect of genotype in plasma glucose levels during these challenges after normalization of baseline hypoglycemia (6-month data shown in Fig. 2G–I; GTT: N = 10/genotype/age, ANOVA, F(1,37) = 1.1, p = 0.3; Fig. 2H ITT: N = 10/genotype, ANOVA, F(1,37) = 7.26, p = 0.88). Hepatic gluconeogenesis, as measured by the pyruvate tolerance test (PTT) was also unchanged in HttLKO/LKO mice compared to wildtype mice at 6- and 13-months of age (Fig. 2I; N = 10/genotype, ANOVA, F(1,37) = 1.26, p = 0.27). Together, these results suggest that while loss of HTT is associated with mild hypoglycemia, systemic glucose metabolism and gluconeogenesis are normal in HttLKO/LKO mice.
During portal vein perfusions for the isolation of purified primary hepatocytes from HttLKO/LKO mice, we observed abnormal blistering of Glisson’s capsule, which surrounds the liver, due to the rapid collapse of the matrix of underlying hepatocytes (Fig. 3A). This blistering of the liver capsule is observed in each HttLKO/LKO mouse (N > 200 mice). To test whether expanded polyglutamine HTT (mHTT) leads to similar adhesion changes, we examined liver blistering in HttQ111/Q111 mice, which express only mHTT, and observed no blistering (N = 20 mice). We also examined two different lines of mice with reduced HTT levels - first HttLKO/+ mice that express 50% wildtype HTT levels (N = 5 mice) and HttQ175/Q175 (N=6), which are known to express only ~25% of HTT levels (Southwell et al, 2016). Neither line expressing low HTT levels produced any blistering, suggesting that HTT reduction to between 0% and 25% of wildtype levels is required for this phenotype to occur, and that it is solely caused by HTT loss, not mHTT expression.
Fig 3: Hepatocyte-specific Htt knockout mice display impaired cellular adhesion.

(A) WT liver (left) during perfusion shows normal intact liver and Glisson’s capsule while HttLKO/LKO mice show swelling or ‘blistering’ of the Glisson’s capsule as it pulls away from the underlying hepatocyte matrix (right, arrowhead). (B) List of mice with modified Htt alleles and corresponding percent of mice that display blistering.
Transcriptional Signature of HttLKO/LKO Livers Reveals Fate Loss, Altered Zonation and Inflammation
To establish the transcriptional consequences of chronic HTT lowering in vivo, we conducted RNA sequencing (RNASeq) from livers of 10-month-old HttLKO/LKO mice. We observe 730 genes that are significantly up-regulated and 389 genes that are downregulated in the livers of 10-month-old HttLKO/LKO mice (log2 fold-change (FC) cutoff of +/−0.5, adjusted p-value cutoff of 0.05; Fig. 4A). On manual inspection of a volcano plot (Fig. 4A), a number of the most down-regulated genes were observed to be required for core hepatocyte functions. To more formally test this observation, we utilized the TissueEnrich algorithm (Jain & Tuteja, 2018), which uses publicly available cross-tissue gene expression data to determine whether a gene list has a higher than expected number of tissuerestricted genes. Interestingly, amongst 389 robustly down-regulated genes in HttLKO/LKO livers, 81 are liver-restricted genes, a result which is highly unlikely to occur by chance (Fig. 4B; log2(FC) = 5.4, p = 2.8e-36). Of the 730 up-regulated genes, only 6 are liver-specific, not corresponding to an enrichment in liver-specific genes given a list of this size (Fig. 1B; log2(FC) = 0.25, p = 0.99), and a larger number are enriched for expression in other tissue types, including thymus and cortex (Fig. 1B; log2(FC) = 5.2, p = 1.6e-17; log2(FC) = 2.6, p = 4.7e-07). Consistent with these gene-level changes, down-regulated genes HttLKO/LKO livers were enriched in gene-ontology pathways involving canonical hepatic functions (e.g. long-chain fatty acid metabolic processes - GO:0001676), while up-regulated pathways are less clearly related to hepatocyte functions, but notably feature a number of pro-inflammatory pathways, including “T cell activation - GO:0042110” and “cellular response to type I interferon - GO:0071357)” (Fig. 4C,D).
Fig 4: Transcriptional analyses of livers from HttLKO/LKO mice reveals down-regulation of hepatocytes-specific genes, and up-regulation of non-hepatic genes.

(A) A volcano plot of 16,852 genes highlights 389 downregulated (Log2 FC < −0.5) and 730 upregulated (Log2 FC > 0.5) genes with adjusted p < 0.05. (B) TissueEnrich shows that downregulated genes are significantly overrepresented in genes annotated for expression in liver, while upregulated genes are significantly overrepresented in non-liver tissue genes. (C) The top 20 enriched gene ontology pathways of biological processes (p < 0.01) from down-regulated genes (Log2(FC) < −0.5 and adjusted p < 0.05) are canonically liver related pathways while (D) the top 20 enriched gene ontology pathways of biological processes (p < 0.01) from up-regulated genes (Log2(FC) > 0.5 and adjusted p < 0.05) are not hepatocyte specific pathways.
Concerned that these proinflammatory signals might result from immune cell infiltration, thereby changing the balance of cell types present in our bulk liver RNASeq analysis, we scored five axes of potential hepatic pathology, including immune cell infiltration, hepatocyte swelling, fibrosis, lipid accumulation and necrosis, finding no alterations in HttLKO/LKO livers compared to Htt+/+ (Table S4). We also profiled 42 inflammatory markers in the plasma of Htt+/+ and HttLKO/LKO mice at 6- and at 13-months and find that 25 of 42 analytes provided results that were above the lower limit of detection and present in a robust number of animals (Table S5). Only very modest changes were observed in three circulating inflammatory markers, including an increase of tissue inhibitors of matrix metalloproteinases (TIMP-1; 2-way ANOVA, Genotype effect F(1, 21 )=5.3; p = 0.03) and modestly reduced levels of Eotaxin-1 in HttLKO/LKO mice at both ages (2-way ANOVA, Genotype effect F(1, 21)=6.2; p = 0.021). These results suggest that a frank increase in immune cell infiltration is unlikely to explain the pro-inflammatory gene expression we observe, with only limited changes observed in circulating inflammatory modulators.
Huntingtin has been proposed to play a number of important roles in regulating transcription, in part via interactions with transcription factors (TFs) (Saudou & Humbert, 2016), so we examined which TF target genes are enriched in differentially expressed genes (DEGs) in HttLKO/LKO livers. Using EnrichR to study enrichments amongst the ChIP Enrichment Analysis (ChEA 2022) dataset, we observe that genes which are up-regulated in HttLKO/LKO livers are enriched amongst genes annotated as PRC2 complex member SUZ12-target genes (62 of 1019 genes, odds ratio = 1.77, adjusted p = 0.02) (Lachmann et al, 2010; Kuleshov et al, 2016; Xie et al, 2021). In contrast, down-regulated DEGs in HttLKO/LKO livers are enriched in genes regulated by canonical hepatic transcription factors, for example LXR (81 of1578 genes, odds ratio = 3.2, adjusted p < 1.3e-13). Individual genes aberrantly up-regulated in HttLKO/LKO livers include neurotransmitter receptors (e.g. Adora1, Gria3, adj p = 0.015 and 0.003; Fig. 5A) and a number of cell fate-determining transcription factor genes not normally expressed in adult hepatocytes, including Runx2 and Pax5 (adjusted p = 0.024 and 0.03; Fig. 5A). These data are consistent with a model in which loss of HTT in hepatocytes drives aberrant derepression of important cell identity and fate-determining genes, including a notable upregulation of PRC2-target genes.
Fig 5: Transcriptional analyses of livers from HttLKO/LKO mice reveals down-regulation of hepatocytes-specific genes, and up-regulation of non-hepatic genes.

(A) Down-regulated DEGs in HttLKO/LKO livers are enriched with genes regulated by canonical hepatic transcription factors, including LXR, while up-regulated DEGs are enriched with genes regulated by the PRC2 complex SUZ12-target genes. (B) Neural genes Gria3 and Adora1 are upregulated in HttLKO/LKO livers, as are developmental transcription factor genes Pax5 and Runx2. Star symbol (*) indicates significant statistical difference.
Liver Zonation Changes Suggest Impaired β-Catenin Signaling
Increased circulating urea in HttLKO/LKO mice (Fig. 2) suggests potential dysfunction of one of two major urea handling pathways–namely the urea cycle, which occurs in periportal (PP) hepatocytes, or glutamine synthesis which is strictly localized to pericentral (PC) hepatocytes (Halpern et al, 2017). Expression of urea cycle genes in HttLKO/LKO livers is overall unchanged (e.g. Ass1; Fig. 6A; t = −0.3, p = 0.89), while levels of glutamine synthetase trend lower (Glul; Fig. 6A, log2FC = −0.71, t = −2.66, p = 0.1). In the adult mouse liver, Glul expression is restricted to a narrow band of PC hepatocytes (Halpern et al, 2017), reflecting the general zonation of hepatocyte functions along the PC/PP gradient. Cadherin proteins are also differentially expressed in these zones: PC regions express N-cadherin while PP regions express E-cadherin (Doi et al, 2007). At the RNA level, we observe no change in Cdh2 (N-cadherin; Fig. 7C) expression, while we see increases in Cdh1 (E-cadherin; Fig. 7C; log2FC = 1.4, t = 7.5, adjusted p-value = 0.001). This was reflected histologically, where we observe a qualitatively altered expression pattern of E-cadherin immunoreactivity, but no change in N-cadherin immunoreactivity in the livers of 6-month-old HttLKO/LKO mice (Fig. 7A). We also see a robust reduction in other PC enriched proteins, including Cyp1a2 and Cyp2e1, both histologically (Cyp1a2, 24% reduction, t(17.9) = 10.29, p = 6.22e-9; Cyp2e1, 31% reduction, t(8.6) = 6.2, p = 0.0002) and transcriptionally (Cyp1a2, t = −3.6, p = 0.04; Cyp2e1, t = −4.1, p = 0.02; Fig 7A–C). These findings are consistent with altered zonation in HttLKO/LKO livers contributing to the observed gene expression alterations and metabolic findings, including impaired urea clearance.
Fig. 6: Hepatic transcriptional analyses from HttLKO/LKO mice reveal down-regulation of pericentral genes.

(A) Argininosuccinate Synthase 1 (Ass1) expression is unchanged while glutamine synthase (Glul) is modestly decreased. (B) The transcripts of specific Wnt genes are downregulated, Wnt2 and Wnt9b. (C) LKO DEGs (Log2(FC) < −0.5 or Log2(FC) > 0.5 and p < 0.05) that are annotated as pericentral genes are near-exclusively negative, while (D) DEGs that are manually annotated as periportal do not follow a predominant pattern. Star symbol (*) indicates significant statistical difference.
Fig 7: The livers of mice with huntingtin-deficient hepatocytes have altered zonation.

(A) Expression patterns of pericentral markers CYP1A2 and CYP2E1, but not CDH2, show reduced expression from pericentral zone into the periportal zone, while periportal marker CDH1 shows a qualitatively increased expression near the pericentral zones, yet non-significant alteration in total expression. Quantified IHC (B) and RNA sequencing (C) shows reduced expression of pericentral proteins in HttLKO/LKO mice (Cyp1a2, Cyp2e1,) and corresponding transcripts (RNAseq), while periportal marker CHD1 shows unchanged IHC (p = 0.17) but increased transcript levels p = 0.001). Scale bar = 350 um (CYP1A2, CYP2E1) or 100 um (CDH1, CDH2).* indicates significant statistical difference.
To further probe the altered hepatocyte zonation in HttLKO/LKO livers, we divided our list of differentially expressed genes into those up- and down-regulated in HttLKO/LKO livers and compared those lists to a published consensus list of genes enriched in pericentral or periportal hepatocytes (Ghallab et al, 2019). Amongst the 34 DEGs (p < 0.05) in HttLKO/LKO livers annotated as “periportal”, we observe no clear trend in directionality of gene expression changes (Fig. 6D, 12 genes up-regulated and 22 genes down-regulated in HttLKO/LKO livers). In striking contrast, amongst the 95 DEGs in HttLKO/LKO livers annotated as “pericentral”, virtually all are down-regulated (Fig. 6C, 5 genes up-regulated and 90 genes down-regulated in HttLKO/LKO livers, p < 0.05). Pericentral hepatocyte gene expression in development and adulthood is activated and maintained by Wnt/β-catenin signaling (Russell & Monga, 2017). Specific Wnts that are expressed in liver endothelial cells in the pericentral region, including Wnt2 and Wnt9b, control gene expression in hepatocytes at this region (Wang et al, 2015; Hu et al, 2022). Expression of Wnt2 and Wnt9b is downregulated in HttLKO/LKO livers (Fig. 6B; Wnt2: Log2(FC) = −1.2, t = −3.7, p = 0.037; Wnt9b: Log2(FC) = −1.29, t = −5.5, p = 0.006), consistent with the pericentral phenotype observed in HttLKO/LKO, suggesting a non-cell autonomous link between hepatocyte Htt expression and endothelial Wnt expression. Thus, convergent histological and transcriptional data suggest a loss of pericentral hepatocyte identity in HttLKO/LKO livers, which is consistent with impaired β-catenin signaling after Htt loss.
Acetaminophen Challenge
Mice with impaired hepatic β-catenin signaling experience “periportalization” of the liver, similar to our observations in HttLKO/LKO livers. This change results in differential sensitivity to a range of challenges, including iron overload (Preziosi et al, 2017), ethanol intoxication (Liu et al, 2012) and acetaminophen (APAP) overdose (Apte et al, 2009). In the liver, APAP is metabolized by CYP2E1 and CYP1A2 into N-acetyl-p-benzoquinone imine (NAPQI), which is in turn toxic to hepatocytes (Raucy et al, 1989). Both CYPs are β-catenin target genes expressed pericentrally, rendering mice with hepatocyte-specific deletion of β-catenin resistant to APAP toxicity via lower levels of these enzymes (Yang et al, 2014). Consistent with our hypothesis, we observe that HttLKO/LKO mice have markedly reduced circulating liver enzymes (79.6% reduction of ALT, t(13.7) = 3.4, p = 0.004; 80% reduction of AST, t(12.7) = 3.6, p = 0.003) after a 20-hour APAP challenge (Fig. 8). This confirms that HttLKO/LKO mice share physiological sensitivities to those of mice with impaired β-catenin signaling and further supports liver periportalization in mice lacking HTT in hepatocytes.
Fig 8: HttLKO/LKO mice are resistant to a toxic acetaminophen challenge.

Circulating levels of hepatic enzymes ALT and AST were quantified after a 20-hour acetaminophen challenge. This reveals a markedly lower level in HttLKO/LKO mice compared to wildtypes (79.6% reduction of; 80% reduction of AST; Htt+/+ N = 9, HttLKO/LKO N = 7). * indicates significant statistical difference.
Discussion
Despite more than 30 years of intensive studying following the identification of the causal gene in Huntington’s disease, there remains much to learn about HTT function–especially outside of the central nervous system. We developed and characterized mice in which Htt is excised from hepatocytes, leading to important pathophysiological alterations owing to observed transcriptional changes. Our work examining the consequences of HTT knock-out in hepatocytes expands the understanding of wildtype HTT functions, as well as describing an unexpected role in cellular fate and liver zonation.
A primary motivation for this study is to better understand loss of function liabilities in on-going HTT-lowering trials in HD patients (Tabrizi et al, 2019). One approach to HTT lowering in clinical studies in HD patients involves small-molecule RNA splicing modulators which induce nonsense mediated decay of HTT transcript, reducing both wildtype and mutant HTT protein (Bhattacharyya et al, 2021b; Keller et al, 2022b). This approach is expected to reduce HTT levels in central and peripheral tissues, notably including the liver, which is exposed to high levels of oral drugs (Almazroo et al, 2017). Our data suggest that these trials should include monitoring for changes seen in HttLKO/LKO mice including: hypoglycemia, hypercholesterolemia, uremia and increased circulating bile acids (Fig. 2). Our acetaminophen challenge results provide functional evidence that HTT loss in hepatocytes impacts clinically-relevant hepatic physiology, including first pass metabolism of drugs by Cyp enzymes (Fig. 7). More broadly, hepatic dysregulation of β-catenin signaling and consequential alterations in zonation are associated with important physiological consequences. Missense mutations in Wnt co-receptor LRP6 leads to early coronary artery disease (CAD) in human mutation carriers (Mani et al, 2007) and knockin mice, who develop hepatosteatosis, even on regular chow diets (Go et al, 2014). Many supportive mouse studies have established that impaired β-catenin signaling leads to liver periportalization which is associated with increased vulnerability to ethanol intoxication (Liu et al, 2012), iron overload (Preziosi et al, 2017) and resistance to acetaminophen challenge (Yang et al, 2014). While expression of several periportal genes is not altered in the absence of hepatic HTT, it is likely that their distribution across the liver lobule may be altered and thus the observation of periportalization of the liver is in fact true and functional. We have recently shown that loss of Wnt2 and Wnt9b together from the endothelial cells did not alter the overall expression of periportal genes like Ass1, but instead led to their appearance in pericentral zone hepatocytes (Hu et al, 2022). This is planned for future studies using single cell spatial transcriptomics.
Decreased Wnt-β-catenin signaling in the HttLKO/LKO mice is also supported by additional observations. Notably, mice with β-catenin knockout in hepatocytes show increased basal bile acid levels similar to HttLKO/LKO (Behari et al, 2010). These mice also exhibit a basal compensatory increase in farnesoid X receptor (FXR) activation, leading to decreased bile acid synthesis from cholesterol and higher basal cholesterol levels, again similar to HttLKO/LKO mice. Finally, mice with hepatic β-catenin knockout showed higher basal NF-κB activation and increased baseline inflammation (Nejak‐Bowen et al, 2013). Indeed, HttLKO/LKO mice also display higher T cell signature in our RNA-Seq analysis and modest changes in circulating pro-inflammatory modulators, although the exact mechanism of this observation requires further investigation.
While only a limited number of studies have investigated the role of wildtype HTT on metabolism, a broader literature exists on the impact of polyglutamine expanded mHTT expression on cellular and organismal metabolism. Urea cycle dysfunction has been described in both HD patients and HD model mice, which is improved in mouse models via low-protein diet, leading to improvements in HD-relevant symptoms in parallel to improved urea cycle function (Chiang et al, 2007). These urea cycle deficiencies were proposed to be rooted in reduced transcription of key urea cycle genes, including argininosuccinate lyase and argininosuccinate synthetase by the C/EBPα transcription factor. Subsequently, markedly increased urea was observed in the brains of both HD transgenic model sheep and HD patients (Patassini et al, 2015; Handley et al, 2017). While there is no evidence of clinically-impactful hepatic dysfunction in HD patients, an elegant series of studies probed hepatic mitochondrial function in HD mutation carriers using ingestion of 13C-labeled methionine, which is catabolized in hepatic mitochondria to 13CO2 that can be monitored in the breath (Stuwe et al, 2013; Hoffmann et al, 2014). These studies revealed early, progressive reductions in 13CO2 release in HD mutation carriers, consistent with reduced number or function of hepatic mitochondria.
Amongst many other cellular pathways, HTT plays roles in transcriptional regulation. HTT can bind and regulate many transcription factors and transcriptional regulatory proteins, including key members of the basal transcriptional machinery (Steffan et al, 2000; Zhai et al, 2005; Seong et al, 2009; Gao et al, 2019) as well as regulate cytoplasmic retention of transcriptional regulators including repressor element 1-silencing (REST) (Zuccato et al, 2003; Shimojo, 2008). Consistent with HTT’s roles in transcriptional regulation, we observe that loss of Htt is associated with a wide range of transcriptional changes in the livers of HttLKO/LKO mice (Fig. 4). Several notable observations emerge from our transcriptional data. First, expression of pericentral genes are markedly reduced, while periportal genes show no clear direction of change (Fig. 6C–D). Second, genes in core pathways associated with liver function, which are the targets of hepatic transcription factors such as LXR, are down regulated (Fig. 5). Third, non-hepatocyte genes, and PRC2-complex-target genes, are up-regulated (Fig. 5). These data provide additional confirmation about the periportalization of the liver, and suggest that an up-regulation of cell-type inappropriate genes is a molecular phenotype associated with Htt loss.
We have previously observed that expression of mHTT in the striatum of HttQ111/+ mice is associated with an unexpected scrambling of the gene expression profiles across a wide range of cell types: in every cell type, cell-type-appropriate genes are down regulated while inappropriate genes are upregulated (Malaiya et al, 2021). Other groups had previously observed alterations in tissue-specific gene expression in HD tissues (Achour et al, 2015; Langfelder et al, 2016; Gras et al, 2017), suggesting that there may be a convergence of gain- and loss-of-function phenotypes for mHTT, in that both appear to be driving a loss of cell fate commitment/homeostasis. While β-catenin-mediated transcriptional regulation is a widespread pathway, the β-catenin protein itself lacks both a transactivation domain and DNA binding domain, and is expressed across many different cell types and developmental stages (Söderholm & Cantù, 2021). This conundrum has led to the suggestion that β-catenin’s role in transcriptional regulation plays a highly tissue- and developmental stage-dependent role in crafting transcription via dynamic binding to partner proteins–first in the cytoplasm and then in the nucleus–to carry out cell- and developmental-stage selective tuning of gene expression.
Previous work has demonstrated functional interactions between HTT and β-catenin, where HTT levels have been shown to be regulated via direct interactions with multiple members of the destruction complex (Godin et al, 2010). This interaction has been proposed to aberrantly stabilize β-catenin, leading to enhanced mHTT toxicity in neurons, which was subsequently confirmed in a Drosophila model of HD, in which reduced Wingless/Wnt signaling was protective and overexpression of Armadillo/β-catenin was deleterious (Dupont et al, 2012). More recently, investigation into neurodevelopmental functions discovered that HTT plays a role in RAB11-mediated N-cadherin trafficking during neuronal polarization (Barnat et al, 2017), and that HTT co-localizes with junctional complex proteins, including β-catenin, in the ventricular zone during neurogenesis in human embryos (Barnat et al, 2020). This latter study suggests that mHTT expression in the developing brain is associated with perturbations in β-catenin-mediated functions, consistent with our finding that loss of wildtype HTT perturbs liver zonation.
A limitation of our work in extending to HTT lowering agents in clinical trials in HD patients is that our mouse lacks HTT expression both during development and through adulthood, as the Cre driver we have used is actie as early asas early as E10.5 (Weisend et al, 2009). This is very different from a human HD patient exposed to HTT lowering in the liver after being treated with agents that are being tested in adult humans. We are conducting ongoing investigations, which focus on HTT knockout and knockdown in adult mice, to determine the extent to which the phenotypes we describe here are developmental in origin.
In sum, our findings implicate HTT in the regulation of hepatocyte cell fate, liver zonation and liver physiology via regulation of β-catenin signaling and identify an intriguing axis of HTT-Wnt-β-catenin crosstalk requiring further mechanistic work. Our studies also have important and unexpected implications for HTT-lowering treatments that reach the liver.
Methods
Mice
We generated a hepatocyte-specific C57BL/6 Htt knockout mouse (HttLKO) by first back-crossing Htttm2Szi (Httfl/fl (Dragatsis et al, 2000)) to a C57BL/6 background using speed congenics (Charles River) and confirmed to be 99.9% B6 based on a 384 SNP panel and then crossing to B6.Cg-Tg(Alb-cre)21Mgn/J (JAX stock 003574) (Postic et al, 1999). All HttLKO , Httfl/fl and Htt+/+ mice were bred at Western Washington University (WWU) and housed in cages of 3–6 mice with access to food and water ad libitum unless otherwise mentioned. Vivarium lights were on a 12-hour light/dark cycle. The WWU institutional animal care and use committee approved the generation of the HttLKO mice and all procedures under protocols 16–006 and 14–006, and 23–70.
Immunoblotting
Total HTT levels were quantified via western blotting. Tissues were homogenized in tubes with 1.5-mm zirconium oxide beads for 2 minutes at 4,000 rpm using a benchtop homogenizer (Beadblaster, Benchmark) in RIPA buffer (150 mM NaCl, 25 mM Tris-HCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing Halt protease and phosphatase inhibitors (Thermo Fisher Scientific). Concentration of protein lysates was determined by BCA assay (Pierce) according to the manufacturer’s protocol.
Equal amounts of reduced protein were loaded into 3–8% Tris-Acetate Mini Gels (Thermo Fisher Scientific) and separated electrophoretically by molecular weight. Protein was transferred at 4℃ for 17 – 20 hours to a PVDF membrane. Membranes were stained with a Revert total protein stain (LiCor) according to manufacturer’s protocol and imaged for 2 minutes at 700 nm on an Odyssey Fc (LiCor) to quantify the total protein in each lane for downstream normalization. Total protein dye was removed and membranes were blocked (Odyssey blocking buffer, LiCor) for 1 hour at room temperature and probed overnight at 4℃ with a monoclonal rabbit anti-HTT antibody (Abcam: EPR5526; Ab_109115) diluted 1:1,000 in blocking buffer plus 0.2% tween, followed by a 1 hour room temperature incubation in goat anti-rabbit IgG IRDye 680RD-conjugated secondary antibody (LiCor:
926–68071; Ab_10956166) diluted 1:15,000 in blocking buffer plus 0.2% tween and 0.01% SDS. Membranes were imaged for 2 minutes at 700 nm on an Odyssey Fc and quantified in Image Studio (LiCor). HTT protein was quantified by normalizing the HTT band signal to the total protein signal in each lane.
RNA Sequencing
Liver tissue for RNA sequencing (RNAseq) was harvested from female Htt+/+ and HttLKO/LKO mice (N = 6 per genotype) sacrificed at 289 (SD = 13.7) days old. RNA was extracted (RNeasy Lipid Tissue Mini Kit, Qiagen) and confirmed for RIN > 7 (Agilent 2100 Bioanalyzer). cDNA libraries were constructed at Azenta (South Plainfield, NJ) using the Illumina TruSeq RNA Sample Prep Kit using and sequenced on a HiSeq 2000 (2 × 150bp) to a read depth of 7.6 × 10^7 +/− 9.9×10^6. Sequence reads were trimmed to remove adapter sequences and nucleotides with poor quality using Trimmomatic v.0.36. Reads were aligned to the Mus musculus GRCm38 reference genome assembly (ENSEMBL) using STAR aligner v.2.5.2b. Unique gene hit counts were calculated by using featureCounts from the Subread package v.1.5.2 (Supplemental Data File 1). Differential gene expression was conducted using the edgeR (Robinson et al, 2010), with voom (Law et al, 2014) and pathway enrichment was assessed using enrichR (Chen et al, 2013; Kuleshov et al, 2016).
Metabolic Challenges
For the pyruvate tolerance test (PTT), mice were fasted at the beginning of their dark phase (6 pm) for 18 hours and IP injected with pyruvate (2,000 mg/kg) at 12 pm on Monday. Blood glucose from tail prick was measured (Contour next EZ glucometer, Bayer) at 0, 15, 30, 60, 90, 120, and 150 minutes post injection. For the glucose tolerance test (GTT) mice were fasted for 6 hours at 8 am on Wednesday and IP injected with glucose (2000 mg/kg) at 2 pm. Blood glucose was measured as above at 0, 15, 30, 60, 90, and 120 minutes post injection. For the insulin tolerance test (ITT) mice were fasted for 6 hours on Friday at 8 am before IP insulin injection (0.75 U/kg; Henry Schein Animal Health). Blood glucose was measured as above at 0, 15, 30, 60, 90, 120, 150, and 180 minutes post injection.
These tests began at either 160 (+/− 2) or 366 (+/− 6) days of age. Htt+/+ mice with high bile acids (>20 umol/L) were excluded from metabolic testing as this correlates to high likelihood of portosytemic shunts (Cudalbu et al, 2013). All mice received an intravenous tail vein injection of 150 uL sterile saline 3 months prior to testing. Htt+/+ mice were imported from Jackson Labs (Bar Harbor, Maine) 3.5 months prior to testing.
For the acetaminophen challenge, 94 +/− 7 day old mice were IP injected with 300 mg/kg APAP (Sigma) freshly dissolved in PBS at 3 pm and were sacrificed 20 hours later. Cardiac blood was processed and measured for ALT and AST as described below.
Blistering Assay
To probe mice for adhesion deficits, mice were perfused via the hepatic portal vein (HPV) with Hanks’ buffered saline solution (Gibco) without calcium and magnesium with EGTA (0.5 mM). Upon cannulation of the HPV, the inferior vena cava (IVC) was cut and perfusion began immediately at ~10 ml/min for 5 minutes. Intermittent clamping (5 seconds, twice per minute) of the IVC was done to ensure buffer completely filled all liver lobes. Care was taken to ensure that IVC clamping would not burst the Glisson’s capsule in livers that displayed blistering. HttQ175/Q175 and HttQ111/Q111 mice were imported from Jackson Labs 2 weeks prior to testing.
Histology
Left lateral and caudate liver lobes were collected from 6 month (165+/− 9) male mice and drop-fixed in 10% neutral buffered formalin (BBC biochemical) for 72 hr. After fixation, livers were stored in PBS + 0.02% sodium azide until they were paraffin embedded, cut into 5-μm sections, and mounted on glass slides.
For immunohistochemistry, sections were deparaffinized and rehydrated to distilled water as described previously (Bragg et al, 2017) [67]. Sections underwent heat-mediated antigen retrieval in a pressure cooker for 20 min in pH = 6 sodium citrate buffer (CYP2E1, CYP1A2). After cooling for 30 minutes, samples were placed in 3% H2O2 for 10 min to quench endogenous peroxide activity. After washing with PBS, slides were blocked for 10 min with Super Block (ScyTek). The primary antibodies were incubated at the following concentrations in PBS: CYP2E1 (Sigma, HPA009128, 1:100), CYP1A2 (Santa Cruz Biotechnology, sc53241, 1:100) N-cadherin (Abcam, ab18203, 1:100), E-cadherin (Cell Signaling, 14472, 1:100). Samples were washed with PBS three times and incubated with the appropriate biotinylated secondary antibody (Vector Laboratories) diluted 1:250 (CYP2E1, CYP1A2) for 15 min at room temperature in antibody diluent or 1:500 (N-cadherin, E-cadherin) for 30 minutes. Samples were washed with PBS three times and sensitized with the Vectastain ABC kit (Vector Laboratories, PK-6101). Following three washes with PBS, color was developed with DAB Peroxidase Substrate Kit (Vector Laboratories, SK-4100), followed by quenching in distilled water. Slides were counterstained with hematoxylin (Thermo Scientific, 7211), dehydrated in xylenes, and coverslips applied with Cytoseal XYL (Thermo Scientific, 8312–4).
Slides were imaged on an Olympus BX51 using Neurolucida software. Quantification was completed in FIJI/ImageJ (Schindelin et al, 2012) by thresholding to select image areas positive for immunostaining. Experimenters were blind to genotype for antibody application, image acquisition, and quantification.
For gross pathology, H&E stained sections were scored by a veterinary pathologist (Phoenix central laboratories, Mukilteo, WA) blinded to genotype for immune cell infiltration, hepatocyte swelling, fibrosis, lipid accumulation and necrosis. Each category was scored on a 4-point scale (0 = absent, 1 = minimal to mild, 2 = moderate, 3 = severe).
Plasma profiling
Clinical chemistry plasma analysis for 12 analytes (Table S3) was completed using an Atellica Clinical Chemistry Analyzer (Siemens) at Phoenix Central Labs (Mukilteo, WA). Analysis for 42 inflammatory markers (Table S5) was completed using a rodent multianalyte panel (Rodent MAP 4.0) based on the Luminex platform at Ampersand Biosciences (Lake Clear, NY).
Plasma was collected from mice following a lethal injection of sodium pentobarbital (Fatal Plus, Henry Schein) at 176 days (SD = 2.2 days) or 387 days (SD = 2.1 days). For all analytes (except bile acids), blood was collected via cardiac puncture. Plasma for bile acids was collected via submandibular vein from live mice at 62+/−11 days. Plasma was flash frozen following collection in heparinized microtainers (BD, cat. no. 365965) and purification by centrifugation at 1,300 × g for 10 minutes, followed by 2,500 × g for 15 min.
Statistical analysis
Statistics were processed in R (Team, 2022). For RNAseq statistics, see RNAseq section. For all other data, comparisons with two groups used Welch’s t-tests to account for unequal variance between groups. To account for multiple comparisons, we adjusted alpha levels using a Bonferroni adjustment. For factorial tests, we used ANVOA with Tukey’s tests for post hoc analyses. For a summary of tests and group sizes, see Table S6. Data presented in Figs 1,2,5–8 used boxplots - horizontal lines indicate 25th, 50th and 75th percentile, while the vertical whiskers indicate the range of data. Data falling outside 1.5 times the interquartile range are graphed as isolated points but were not excluded from statistical analysis. Graphics were produced using ggplot2 (Wickham, 2016) and Illustrator (Adobe) and BioRender.com.
Supplementary Material
Acknowledgments
Funding provided by CHDI Foundation grant to JBC, NINDS grant NS124728 to SZ, NIDDK grants R01DK062277 and R01DK103775 to SPM, and diagnostic and technical assistance for liver histology was provided by Pittsburgh Liver Research Center’s Clinical Biospecimen Repository and Processing Core which is funded by NIDDK grant P30DK120531 (PI: SPM).
Conflict of Interest Statement
SK, DH, TFV are full time employees of CHDI Foundation. JBC has consulting income from Cajal Neuroscience and Guidepoint. SPM is on the scientific advisory board or a consultant for Antlera, Surrozen, Alnylam and Vicero Inc. in addition to non-related research funding from Alnylam and Fog Pharmaceuticals.
References
- Achour M, Gras SL, Keime C, Parmentier F, Lejeune F-X, Boutillier A-L, Néri C, Davidson I, Merienne K (2015) Neuronal identity genes regulated by super-enhancers are preferentially down-regulated in the striatum of Huntington’s disease mice. Hum Mol Genet 24: 3481–3496. doi: 10.1093/hmg/ddv099. [DOI] [PubMed] [Google Scholar]
- Almazroo OA, Miah MK, Venkataramanan R (2017) Drug Metabolism in the Liver. Clin Liver Dis 21: 1–20. doi: 10.1016/j.cld.2016.08.001. [DOI] [PubMed] [Google Scholar]
- Apte U, Singh S, Zeng G, Cieply B, Virji MA, Wu T, Monga SPS (2009) Beta-Catenin Activation Promotes Liver Regeneration after Acetaminophen-Induced Injury. Am J Pathology 175: 1056–1065. doi: 10.2353/ajpath.2009.080976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnat M, Capizzi M, Aparicio E, Boluda S, Wennagel D, Kacher R, Kassem R, Lenoir S, Agasse F, Braz BY et al. (2020) Huntington’s disease alters human neurodevelopment. Science: eaax3338. doi: 10.1126/science.aax3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnat M, Friec JL, Benstaali C, Humbert S (2017) Huntingtin-Mediated Multipolar-Bipolar Transition of Newborn Cortical Neurons Is Critical for Their Postnatal Neuronal Morphology. Neuron 93: 99–114. doi: 10.1016/j.neuron.2016.11.035. [DOI] [PubMed] [Google Scholar]
- Behari J, Yeh T-H, Krauland L, Otruba W, Cieply B, Hauth B, Apte U, Wu T, Evans R, Monga SPS (2010) Liver-Specific β-Catenin Knockout Mice Exhibit Defective Bile Acid and Cholesterol Homeostasis and Increased Susceptibility to Diet-Induced Steatohepatitis. Am J Pathology 176: 744–753. doi: 10.2353/ajpath.2010.090667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, Trotta CR, Narasimhan J, Wiedinger KJ, Li W, Effenberger KA, Woll MG, Jani MB, Risher N, Yeh S et al. (2021a) Small molecule splicing modifiers with systemic HTT-lowering activity. Nat Commun 12: 7299. doi: 10.1038/s41467-021-27157-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, Trotta CR, Narasimhan J, Wiedinger KJ, Li W, Effenberger KA, Woll MG, Jani MB, Risher N, Yeh S et al. (2021b) Small molecule splicing modifiers with systemic HTT-lowering activity. Nat Commun 12: 7299. doi: 10.1038/s41467-021-27157-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragg RM, Coffey SR, Weston RM, Ament SA, Cantle JP, Minnig S, Funk CC, Shuttleworth DD, Woods EL, Sullivan BR et al. (2017) Motivational, proteostatic and transcriptional deficits precede synapse loss, gliosis and neurodegeneration in the B6.HttQ111/+ model of Huntington’s disease. Sci Rep-uk 7: 41570. doi: 10.1038/srep41570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N (2016) Mutant Huntingtin and Elusive Defects in Oxidative Metabolism and Mitochondrial Calcium Handling. Mol Neurobiol 53: 2944–2953. doi: 10.1007/s12035-015-9188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform 14: 128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang M-C, Chen H-M, Lee Y-H, Chang H-H, Wu Y-C, Soong B-W, Chen C-M, Wu Y-R, Liu C-S, Niu D-M et al. (2007) Dysregulation of C/EBPα by mutant Huntingtin causes the urea cycle deficiency in Huntington’s disease. Hum Mol Genet 16: 483–498. doi: 10.1093/hmg/ddl481. [DOI] [PubMed] [Google Scholar]
- Cudalbu C, McLin VA, Lei H, Duarte JMN, Rougemont A-L, Oldani G, Terraz S, Toso C, Gruetter R (2013) The C57BL/6J Mouse Exhibits Sporadic Congenital Portosystemic Shunts. Plos One 8: e69782. doi: 10.1371/journal.pone.0069782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi Y, Tamura S, Nammo T, Fukui K, Kiso S, Nagafuchi A (2007) Development of complementary expression patterns of E‐ and N‐cadherin in the mouse liver. Hepatol Res 37: 230–237. doi: 10.1111/j.1872-034x.2007.00028.x. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S (2000) Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet 26: 300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- Dupont P, Besson M-T, Devaux J, Liévens J-C (2012) Reducing canonical Wingless/Wnt signaling pathway confers protection against mutant Huntingtin toxicity in Drosophila. Neurobiol Dis 47: 237–247. doi: 10.1016/j.nbd.2012.04.007. [DOI] [PubMed] [Google Scholar]
- Duyao M, Auerbach A, Ryan A, Persichetti F, Barnes G, McNeil S, Ge P, Vonsattel J, Gusella J, Joyner A et al. (1995) Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 269: 407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- Gao R, Chakraborty A, Geater C, Pradhan S, Gordon KL, Snowden J, Yuan S, Dickey AS, Choudhary S, Ashizawa T et al. (2019) Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. Elife 8: e42988. doi: 10.7554/elife.42988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghallab A, Myllys M, Holland CH, Zaza A, Murad W, Hassan R, Ahmed YA, Abbas T, Abdelrahim EA, Schneider KM et al. (2019) Influence of Liver Fibrosis on Lobular Zonation. Cells 8: 1556. doi: 10.3390/cells8121556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go G, Srivastava R, Hernandez-Ono A, Gang G, Smith SB, Booth CJ, Ginsberg HN, Mani A (2014) The Combined Hyperlipidemia Caused by Impaired Wnt-LRP6 Signaling Is Reversed by Wnt3a Rescue. Cell Metab 19: 209–220. doi: 10.1016/j.cmet.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin JD, Poizat G, Hickey MA, Maschat F, Humbert S (2010) Mutant huntingtin‐impaired degradation of β-catenin causes neurotoxicity in Huntington’s disease. Embo J 29: 2433–2445. doi: 10.1038/emboj.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras SL, Keime C, Anthony A, Lotz C, Longprez LD, Brouillet E, Cassel J-C, Boutillier A-L, Merienne K (2017) Altered enhancer transcription underlies Huntington’s disease striatal transcriptional signature. Sci Rep-uk 7: 42875. doi: 10.1038/srep42875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern KB, Shenhav R, Matcovitch-Natan O, Tóth B, Lemze D, Golan M, Massasa EE, Baydatch S, Landen S, Moor AE et al. (2017) Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542: 352. doi: 10.1038/nature21065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handley RR, Reid SJ, Brauning R, Maclean P, Mears ER, Fourie I, Patassini S, Cooper GJS, Rudiger SR, McLaughlan CJ et al. (2017) Brain urea increase is an early Huntington’s disease pathogenic event observed in a prodromal transgenic sheep model and HD cases. Proc National Acad Sci 114: E11293–E11302. doi: 10.1073/pnas.1711243115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann R, Stüwe SH, Goetze O, Banasch M, Klotz P, Lukas C, Tegenthoff M, Beste C, Orth M, Saft C (2014) Progressive hepatic mitochondrial dysfunction in premanifest Huntington’s disease. Movement Disord 29: 831–834. doi: 10.1002/mds.25862. [DOI] [PubMed] [Google Scholar]
- Hu S, Liu S, Bian Y, Poddar M, Singh S, Cao C, McGaughey J, Bell A, Blazer LL, Adams JJ et al. (2022) Single-cell spatial transcriptomics reveals a dynamic control of metabolic zonation and liver regeneration by endothelial cell Wnt2 and Wnt9b. Cell Reports Medicine 3: 100754. doi: 10.1016/j.xcrm.2022.100754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Tuteja G (2018) TissueEnrich: Tissue-specific gene enrichment analysis. Bioinformatics 35: 1966–1967. doi: 10.1093/bioinformatics/bty890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581: 434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller CG, Shin Y, Monteys AM, Renaud N, Beibel M, Teider N, Peters T, Faller T, St-Cyr S, Knehr J et al. (2022a) An orally available, brain penetrant, small molecule lowers huntingtin levels by enhancing pseudoexon inclusion. Nat Commun 13: 1150. doi: 10.1038/s41467-022-28653-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller CG, Shin Y, Monteys AM, Renaud N, Beibel M, Teider N, Peters T, Faller T, St-Cyr S, Knehr J et al. (2022b) An orally available, brain penetrant, small molecule lowers huntingtin levels by enhancing pseudoexon inclusion. Nat Commun 13: 1150. doi: 10.1038/s41467-022-28653-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A et al. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma’ayan A (2010) ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26: 2438–2444. doi: 10.1093/bioinformatics/btq466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu X-H, Ramos EM, El-Zein K, Zhao Y et al. (2016) Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci 19: 623–633. doi: 10.1038/nn.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK (2014) voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15: R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Yeh T, Singh VP, Shiva S, Krauland L, Li H, Zhang P, Kharbanda K, Ritov V, Monga SPS et al. (2012) β-Catenin is essential for ethanol metabolism and protection against alcohol‐mediated liver steatosis in mice. Hepatology 55: 931–940. doi: 10.1002/hep.24766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes F, Barbosa M, Ameur A, Soares G, Sá J de, Dias AI, Oliveira G, Cabral P, Temudo T, Calado E et al. (2016) Identification of novel genetic causes of Rett syndrome-like phenotypes. Journal of medical genetics 53: 190–199. doi: 10.1136/jmedgenet-2015-103568. [DOI] [PubMed] [Google Scholar]
- MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N et al. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72: 971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Malaiya S, Cortes-Gutierrez M, Herb BR, Coffey SR, Legg SRW, Cantle JP, Colantuoni C, Carroll JB, Ament SA (2021) Single-Nucleus RNA-Seq Reveals Dysregulation of Striatal Cell Identity Due to Huntington’s Disease Mutations. J Neurosci 41: 5534–5552. doi: 10.1523/jneurosci.2074-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani A, Radhakrishnan J, Wang H, Mani A, Mani M-A, Nelson-Williams C, Carew KS, Mane S, Najmabadi H, Wu D et al. (2007) LRP6 Mutation in a Family with Early Coronary Disease and Metabolic Risk Factors. Science 315: 1278–1282. doi: 10.1126/science.1136370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAGASAWA H, MIYAMOTO M, FUJIMOTO M (1973) Reproductivity in Inbred Strains of Mice and Project for Their Efficient Production. Exp Anim Tokyo 22: 119–126. doi: 10.1538/expanim1957.22.2_119. [DOI] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR (1995) Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 81: 811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- Nejak‐Bowen K, Kikuchi A, Monga SPS (2013) Beta‐catenin–NF‐κB interactions in murine hepatocytes: A complex to die for. Hepatology 57: 763–774. doi: 10.1002/hep.26042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patassini S, Begley P, Reid SJ, Xu J, Church SJ, Curtis M, Dragunow M, Waldvogel HJ, Unwin RD, Snell RG et al. (2015) Identification of elevated urea as a severe, ubiquitous metabolic defect in the brain of patients with Huntington’s disease. Biochemical and biophysical research communications 468: 161–166. doi: 10.1016/j.bbrc.2015.10.140. [DOI] [PubMed] [Google Scholar]
- Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, Rautou P-E (2017) Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol 66: 212–227. doi: 10.1016/j.jhep.2016.07.009. [DOI] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA (1999) Dual Roles for Glucokinase in Glucose Homeostasis as Determined by Liver and Pancreatic β Cell-specific Gene Knock-outs Using Cre Recombinase*. J Biol Chem 274: 305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- Preziosi ME, Singh S, Valore EV, Jung G, Popovic B, Poddar M, Nagarajan S, Ganz T, Monga SP (2017) Mice lacking liver-specific β-catenin develop steatohepatitis and fibrosis after iron overload. J Hepatol 67: 360–369. doi: 10.1016/j.jhep.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucy JL, Lasker JM, Lieber CS, Black M (1989) Acetaminophen activation by human liver cytochromes P450IIE1 and P450IA2. Arch Biochem Biophys 271: 270–283. doi: 10.1016/0003-9861(89)90278-6. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodan LH, Cohen J, Fatemi A, Gillis T, Lucente D, Gusella J, Picker JD (2016) A novel neurodevelopmental disorder associated with compound heterozygous variants in the huntingtin gene. Eur J Hum Genet 24: 1826–1827. doi: 10.1038/ejhg.2016.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross C, Aylward E, Wild E (2014) Huntington disease: natural history, biomarkers and prospects for therapeutics. [DOI] [PubMed] [Google Scholar]
- Russell JO, Monga SS (2017) Wnt/β-Catenin Signaling in Liver Development, Homeostasis, and Pathobiology. Annu Rev Pathology Mech Dis 13: 1–28. doi: 10.1146/annurev-pathol-020117-044010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F, Humbert S (2016) The Biology of Huntingtin. Neuron 89: 910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong IS, Woda JM, Song J-J, Lloret A, Abeyrathne PD, Woo CJ, Gregory G, Lee J-M, Wheeler VC, Walz T et al. (2009) Huntingtin facilitates polycomb repressive complex 2. Hum Mol Genet 19: 573–583. doi: 10.1093/hmg/ddp524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojo M (2008) Huntingtin Regulates RE1-silencing Transcription Factor/Neuron-restrictive Silencer Factor (REST/NRSF) Nuclear Trafficking Indirectly through a Complex with REST/NRSF-interacting LIM Domain Protein (RILP) and Dynactin p150Glued *. J Biol Chem 283: 34880–34886. doi: 10.1074/jbc.m804183200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderholm S, Cantù C (2021) The WNT/β‐catenin dependent transcription: A tissue‐specific business. Wires Mech Dis 13: e1511. doi: 10.1002/wsbm.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwell AL, Smith-Dijak A, Kay C, Sepers M, Villanueva EB, Parsons MP, Xie Y, Anderson L, Felczak B, Waltl S et al. (2016) An enhanced Q175 knock-in mouse model of Huntington disease with higher mutant huntingtin levels and accelerated disease phenotypes. Hum Mol Genet 25: 3654–3675. doi: 10.1093/hmg/ddw212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu Y-Z, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM (2000) The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc National Acad Sci 97: 6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuwe S, Goetze O, Lukas C, Klotz P, Hoffmann R, Banasch M, Orth M, Schmidt W, Gold R, Saft C (2013) Hepatic mitochondrial dysfunction in manifest and premanifest Huntington disease. Neurology 80: 743–746. doi: 10.1212/wnl.0b013e318282514e. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Ghosh R, Leavitt BR (2019) Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron 101: 801–819. doi: 10.1016/j.neuron.2019.01.039. [DOI] [PubMed] [Google Scholar]
- Tan X, Behari J, Cieply B, Michalopoulos GK, Monga SPS (2006) Conditional Deletion of β-Catenin Reveals Its Role in Liver Growth and Regeneration. Gastroenterology 131: 1561–1572. doi: 10.1053/j.gastro.2006.08.042. [DOI] [PubMed] [Google Scholar]
- Team RC (2022) R: A language and environment for statistical computing. R Foundation for Statistical Computing http://www.R-project.org/ (2016).
- Wang B, Zhao L, Fish M, Logan CY, Nusse R (2015) Self-renewing diploid Axin2+ cells fuel homeostatic renewal of the liver. Nature 524: 180–185. doi: 10.1038/nature14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisend CM, Kundert JA, Suvorova ES, Prigge JR, Schmidt EE (2009) Cre activity in fetal albCre mouse hepatocytes: Utility for developmental studies. Genesis 47: 789–792. doi: 10.1002/dvg.20568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016) ggplot2: Elegant Graphics for Data Analysis (New York: Springer-Verlag; ). [Google Scholar]
- Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM et al. (2021) Gene Set Knowledge Discovery with Enrichr. Curr Protoc 1: e90. doi: 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mowry LE, Nejak‐Bowen KN, Okabe H, Diegel CR, Lang RA, Williams BO, Monga SP (2014) Beta-catenin signaling in murine liver zonation and regeneration: A Wnt‐Wnt situation! Hepatology 60: 964–976. doi: 10.1002/hep.27082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin S, Liu J-P, Chapman DL, Papaioannou VE, Efstratiadis A (1995) Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet 11: 155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- Zhai W, Jeong H, Cui L, Krainc D, Tjian R (2005) In Vitro Analysis of Huntingtin-Mediated Transcriptional Repression Reveals Multiple Transcription Factor Targets. Cell 123: 1241–1253. doi: 10.1016/j.cell.2005.10.030. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T et al. (2003) Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet 35: 76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.