

Abstract
Members of the human gut microbiome enzymatically process many bioactive molecules in the gastrointestinal (GI) tract. Most gut bacterial modifications characterized to date are hydrolytic or reductive in nature. Here we report that abundant human gut bacteria from the phylum Bacteroidetes perform conjugative modifications by selectively sulfonating steroidal metabolites. While sulfonation is a ubiquitous biochemical modification, this activity had not yet been characterized in gut microbes. Using genetic and biochemical approaches, we identify a widespread biosynthetic gene cluster that encodes both a sulfotransferase (BtSULT, BT0416) and enzymes that synthesize the sulfonate donor adenosine 3’-phosphate-5’-phosphosulfate (PAPS), including an APS kinase (CysC, BT0413) and an ATP sulfurylase (CysD and CysN, BT0414-BT0415). BtSULT selectively sulfonates steroidal metabolites with a flat A/B ring fusion, including cholesterol. Germ-free mice monocolonized with Bacteroides thetaiotaomicron ΔBT0416 exhibited reduced gastrointestinal levels of cholesterol sulfate (Ch-S) compared to B. thetaiotaomicron wild type-colonized mice. The presence of BtSULT and BtSULT homologs in bacteria inhibited leukocyte migration in vitro and in vivo, and abundances of cluster genes were significantly reduced in patients with inflammatory bowel diseases. Together, these data provide a mechanism by which gut bacteria sulfonate steroidal metabolites and suggest that these compounds can modulate immune cell trafficking in the host.
Introduction
Human gut microorganisms chemically modify a plethora of endobiotics and xenobiotics that enter the gut, often altering the biological functions of these molecules1. Most of the transformations elucidated to date are either hydrolytic, cleaving chemical bonds and breaking down larger molecules, or reductive, transferring electrons into the substrate2,3. In contrast, host enzymes generally utilize conjugative and oxidative chemistry to modify molecules4.
One of the keystone conjugative transformations in host metabolism is sulfonation, or the enzymatic conversion of alcohols to sulfate esters. Organisms deploy sulfonation to detoxify compounds including drugs4 and hormones5,6 and generate signaling molecules7. Sulfotransferases (SULTs) catalyze the transfer of a sulfonate group from cofactor adenosine 3’-phosphate-5’-phosphosulfate (PAPS) to an acceptor group of a substrate (Fig. 1a).
Fig. 1 |. The human gut bacterium B. thetaiotaomicron VPI-5482 (B. theta) transforms cholesterol (Ch) to cholesterol sulfate (Ch-S) in monocolonized GF mice.

a, Sulfotransferase (SULT) enzymes utilize the cofactor 3’-phosphoadenosine-5’-phosphosulfate (PAPS) to catalyze the sulfonation of a substrate such as cholesterol (Ch), resulting in the formation of a sulfate ester product (cholesterol-sulfate, Ch-S). The sulfate group is highlighted with red text. SULTs have been characterized in mammals and certain environmental and pathogenic prokaryotes but have not yet been characterized in human-associated commensal bacteria.
b, Design of monocolonization mouse experiment. GF B6 mice were either colonized with B. theta or were left in the GF state. Following confirmation of bacterial colonization, mice were fed a high-fat diet for 4 weeks. Ileum, liver, and cecal contents were then collected for UPLC-MS analysis and gene expression analyses.
c, Levels of Ch-S in cecal contents were higher in B. theta-colonized mice than in GF mice (n=8 mice per group, two-tailed Welch’s t test).
d, No significant differences in the expression of the host gene Sult2b1b were observed in the distal ileum or liver of B. theta-monocolonized mice compared to GF mice as measured by quantitative PCR (n=8 mice per group, two-tailed Welch’s t test, n.s. = not significant).
e, B. theta converted Ch to Ch-S in culture. B. theta was incubated with Ch (100 μM) for 48 h and Ch-S production was quantified by UPLC-MS. (n=3 biological replicates). All data are presented as mean ± SEM.
The mammalian genome encodes more than 10 SULTs5,7 that sulfonate compounds including bile acids (BAs), steroids, phenols, and neurotransmitters5. SULTs have also been identified in select pathogenic and environmental prokaryotes, including mycobacteria8, Escherichia coli9, and Cyanobacteria10, and these enzymes sulfonate glycolipids8, carbohydrates10,11, and alcohols12,13. However, SULTs have not yet been characterized in human-associated commensal microbes.
Sulfonation transforms nonpolar compounds into hydrophilic metabolites that are more readily excreted14,15. Many sulfonated products elicit biological functions, including cholesterol 3-sulfate (Ch-S) (Fig. 1a), which regulates serine protease activity and membrane functions16–19. Recently, Ch-S was reported to regulate immune responses by inhibiting T-cell receptor signaling20 and blocking leukocyte migration21. Cholesterol (Ch) is among the most abundant metabolites in the human gut; on average, the intestine is exposed to 1 to 2.4 g of Ch per day22–25. Whether gut microbes convert Ch to Ch-S and thereby modulate host immune function, however, was unknown.
Here, we report that Bacteroides thetaiotaomicron, a prominent gut microbe, sulfonates steroid-like molecules. Through bioinformatic searches, we identified a SULT-containing gene cluster that is widespread among gut Bacteroidetes, abundant and prevalent across human gut-microbiome profiles, and responsible for sulfonation of metabolites in vitro. We then show that the B. thetaiotaomicron SULT (BtSULT) displays substrate preference based on steroidal structure and is required for sulfonation by B. thetaiotaomicron in vivo. Bacterial supernatants from Ch-S-producing bacteria inhibited the migration of leukocytes in vitro, and colonization of mice with BtSULT-containing compared to BtSULT-deficient bacteria inhibited migration of T cells to mesenteric lymph nodes (MLNs). In addition, the abundance of SULT cluster genes were reduced in patients with inflammatory bowel diseases (IBD). Together, these findings broaden the transformations known to be carried out by gut microbes and suggest that bacterial SULTs produce molecules that affect host immune cell trafficking.
Results
B. thetaiotaomicron produces Ch-S in vivo
We serendipitously observed significantly higher levels of Ch-S in cecal contents of germ-free (GF) mice monocolonized with B. thetaiotaomicron and fed a high-fat diet compared to GF control animals (Fig. 1b–c). Ch-S can be produced by the host enzyme SULT2b1b6. We quantified Sult2b1b gene expression levels in the distal ileum and liver and observed no significant differences between monocolonized and GF mice (Fig. 1d). These results raised the question of whether B. thetaiotaomicron itself possesses sulfotransferase activity and contributed to the increased Ch-S levels in monocolonized animals. To test whether B. thetaiotaomicron produces Ch-S, we incubated B. thetaiotaomicron with Ch and monitored Ch-S production by ultra-high performance liquid chromatography-mass spectrometry (UPLC-MS). We detected increasing concentrations of Ch-S over 48 hours in cultures containing Ch, while no Ch-S was detected in bacteria-only or media plus Ch controls (Fig. 1e). These results indicate that B. thetaiotaomicron produces Ch-S from Ch in vitro and suggest that B. thetaiotaomicron contributed to the increased gastrointestinal levels of Ch-S in vivo.
Identification of a SULT gene cluster in B. thetaiotaomicron
Bacteria possess different pathways to scavenge sulfur from their environment for growth and metabolism26,27. Sulfonation is part of assimilatory sulfur metabolism (Extended Data Fig. 1)26,28,29. The cysteine biosynthesis (cys) genes in this pathway are highly conserved across multiple domains of life and allow microbes to respire anaerobically by using inorganic sulfate as a terminal electron acceptor (Extended Data Fig. 1). This pathway also supplies the cofactors needed for sulfonation29. BLASTP searches30,31 using cys genes from E. coli K1226,27 as references revealed a localized gene cluster containing homologs for adenosine triphosphate (ATP) sulfurylase subunit 1 (CysN, BT0415, E value = 9e-157), ATP sulfurylase subunit 2 (CysD, BT0414, E value = 1e-158), and APS kinase (CysC, BT0413, E value = 2e-58), followed by a hypothetical protein belonging to the sulfotransferase family (BT0416) in the genome of B. thetaiotaomicron VPI-5482 (Fig. 2a). Notably, BLASTP searches using the AprBA gene from Desulfovibrio desulfuricans DSM 642 (G449DRAFT_1613) did not reveal homologs in B. thetaiotaomicron, suggesting that this organism does not contain a complete dissimilatory sulfur metabolism pathway (Supplementary Table 2a, Extended Data Fig. 1). Based on these findings, we hypothesized that B. thetaiotaomicron catalyzes the conversion of Ch to Ch-S via the biosynthetic gene cluster BT0413-BT0416, that BT0416 encodes a SULT, and that BT0413-BT0415 encode CysC, CysD, and CysN for PAPS biosynthesis.
Fig. 2 |. A biosynthetic gene cluster in human gut bacteria converts Ch to Ch-S.
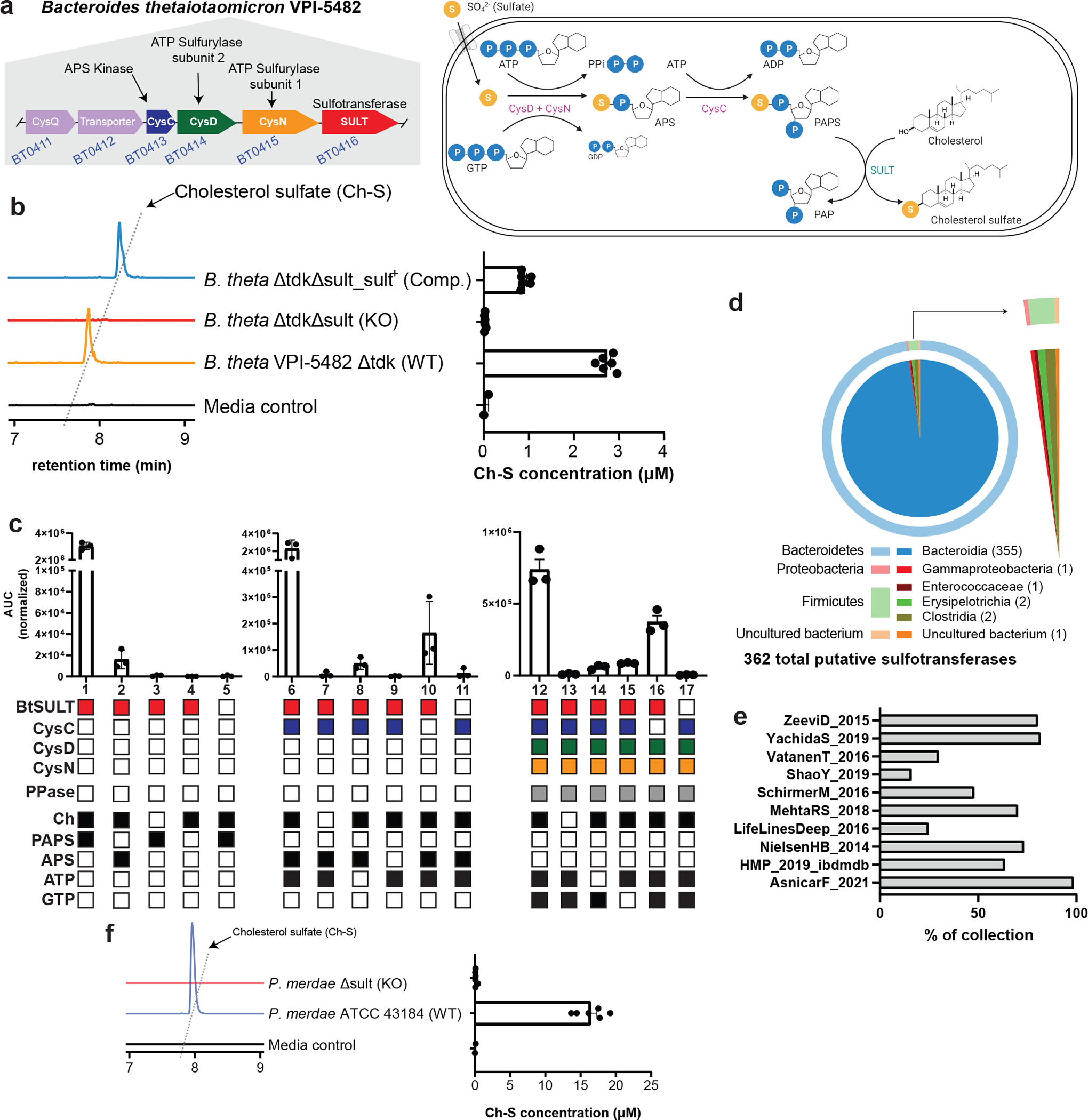
a, Biosynthetic gene cluster containing cys genes as well as a putative sulfotransferase in B. theta. After transport of sulfate into the bacterial cell, ATP sulfurylase (CysD&N) catalyzes the conversion of ATP and sulfate to APS. APS kinase (CysC) 3’-phosphorylates APS to generate PAPS. Finally, SULT (BT0416) catalyzes the transformation of Ch to Ch-S.
b, Representative extracted ion chromatogram (EIC) traces (left) and quantified production of Ch-S (right) by B. theta WT, Δsult, and Δsult_sult+ show sult-dependent Ch-S production. Data are presented as mean ± S.E.M (n=6 from two pooled experiments).
c, In vitro reconstruction of the BtSULT pathway using purified enzymes with precursor molecule production coupled to BtSULT activity. Conditions 1–5 demonstrate that BtSULT utilizes PAPS as a sulfonate source (1), can use APS under non-ideal conditions (2), and BtSULT, Ch, and a cofactor are required for sulfonation (3–5). Conditions 6–11 demonstrate that CysC catalyzes the conversion of APS and ATP to PAPS (6) and ATP, APS, and CysC are each necessary to catalyze PAPS formation (8, 9, and 10). Conditions 12–17 represent the complete biosynthetic pathway, showing that CysD&N, CysC, GTP, and ATP can form PAPS (12). Filled boxes indicate the presence BtSULT (red), CysC (blue), CysD (green), CysN (orange), pyrophosphatase (grey), and small molecules (black) in a given reaction. Data are presented as mean ± S.E.M (n=3 replicates per reaction).
d, Illustration of 362 total putative SULTs (60% identity across sequenced bacterial genomes) retrieved from the NCBI database. The outer ring displays the relative distribution at the phylum level. The inner circle shows class level distribution. The number of bacteria for each genus has been indicated next to the genus name.
e, Metagenomic prevalence of the BT0416 gene in representative samples from the 10 largest collections shows BT0416 presence at varying levels across collections, with greater than 50% occurrence in more than half the available collections.
f, Representative EIC traces (left) and quantified Ch-S production (right) by P. merdae WT and Δsult show sult-dependent Ch-S production. Data are presented as mean ± S.E.M (n = 6 from two pooled experiments).
In vitro characterization of the BtSULT gene cluster
To characterize the putative SULT, we constructed a knockout strain using counterselectable allelic exchange (B. thetaiotaomicron Δsult)32 and assessed Ch-S production (Extended Data Fig. 2a). We observed no production of Ch-S by B. thetaiotaomicron Δsult, confirming that BT0416 is responsible for Ch-S production in vitro (Fig. 2b). Complementation of the deletion mutant with BT0416 restored the ability to produce Ch-S (Fig. 2b). Neither deletion of BT0416 nor complementation of the deletion strain affected the growth of B. thetaiotaomicron (Extended Data Fig. 2b, 2c). Together, these data indicate that BT0416 is a bacterial sulfotransferase and is required for sulfonation of Ch in B. thetaiotaomicron.
BT0412 is homologous to sodium/sulfate symporters and likely transports inorganic sulfate into the cell where it can be used by CysD, CysN, and CysC to generate the PAPS cofactor. Genetic deletion of BT0412 hindered bacterial growth (Extended Data Fig. 2d–h), but this mutant still produced Ch-S (Extended Data Fig. 2i). These results are consistent with previous findings, as Bacteroides species can harvest sulfate from small organic molecules such as cysteine, which can then be used to synthesize PAPS33. Supplementation of media with additional organic sulfur sources was necessary for growth of a second B. thetaiotaomicron Δ0413–0415 strain, which exhibited a significant growth defect when compared to B. thetaiotaomicron wild-type (Extended Data Fig. 2j, e–h) and was unable to produce Ch-S in culture (Extended Data Fig. 2i). Chemical complementation of lysed B. thetaiotaomicron Δ0413–0415 with PAPS rescued Ch-S production (Extended Data Fig. 2k). These data indicate that BT0413-BT0415 are required to support sulfonation and robust growth in B. thetaiotaomicron while BT0412 contributes to but is not required for sulfonation.
To further characterize the proteins encoded by BT0413-BT0416, we heterologously expressed and purified these four enzymes. We then performed a series of assays to confirm the function of each enzyme in sulfonation (Fig. 2c). Due to the similar chemical properties and masses of pathway cofactors, chromatographic and/or spectrometric separation was not possible; therefore, PAPS biosynthesis activities (CysD, CysN, and CysC) were assessed by coupling PAPS production to production of Ch-S by BtSULT.
BtSULT produced Ch-S in the presence of Ch and PAPS (Fig. 2c-1), but when Ch (Fig. 2c-3), PAPS (Fig. 2c-4), or BtSULT (Fig. 2c-5) was removed, no Ch-S formed. These data show that BtSULT encodes a sulfotransferase that utilizes PAPS to sulfonate Ch and produce Ch-S. In addition, APS, the sulfonated PAPS precursor, can act as a cofactor for BtSULT, although this reaction is less efficient (Fig. 2c-2). The ability of a bacterial SULT to use alternative cofactors is precedented9; however, APS has not been previously identified as a sulfonate source.
Moving up the biosynthetic pathway, BtSULT and CysC generated Ch-S from Ch, APS, and ATP (Fig. 2c-6). When substrate (Fig. 2c-7) or APS (Fig. 2c-9) was removed, no product formed. These data indicate that CysC is an APS kinase that converts APS to PAPS. When APS was present without ATP (Fig. 2c-8) or CysC (Fig. 2c-10), Ch-S was formed at lower levels, again suggesting that APS can act as a sulfonate donor when PAPS is not present.
Finally, we considered APS biosynthesis. BtSULT, CysC, CysD, and CysN together with sulfate, ATP, and GTP catalyzed the conversion of Ch to Ch-S (Fig. 2c-12). Removal of substrate (Fig. 2c-13) or BtSULT (Fig. 2c-16) ablated Ch-S production. The absence of ATP or GTP (Fig. 2c-14–15) reduced Ch-S production. These data complete the in vitro reconstitution of cholesterol sulfonation and show that CysD, CysN, and CysC process sulfate and ATP into the PAPS cofactor, which serves as the primary sulfonate donor for sulfonation.
Using overlapping end-point PCR34, we confirmed that BT0411-BT0416 are cotranscribed in a single polycistronic mRNA and are therefore part of an operon (Extended Data Fig. 2l). Addition of Ch did not induce BT0416 expression as determined by qPCR (Extended Data Fig. 2m), suggesting that these genes are constitutively expressed in B. thetaiotaomicron. Together with the findings that deletion of the cys genes BT0413-BT0415 impairs B. thetaiotaomicron growth (Extended Data Fig. 2e–h), these results suggest that PAPS biosynthesis contributes to viability and that BtSULT is co-transcribed with these genes but is not required for growth.
BLASTP searches using known bacterial sulfolipid transporter genes35 did not reveal a potential Ch-S transporter. We hypothesized that Ch-S is released into the extracellular environment when microbes lyse upon cell death. To investigate this hypothesis, we quantified Ch-S levels in washed cell pellets compared to supernatants during early and late stationary phase (48h and 168h, respectively). At 48h, the majority of Ch-S was contained within the pellet; while Ch-S levels were higher in supernatant at 168h, consistent with release of Ch-S following cell death and lysis (Extended Data Fig. 2n,o). Prior research has shown that a substantial portion of the human gut microbiome consists of damaged or lysed cells at any given time36, suggesting that cell death is a plausible mechanism for Ch-S release into the gut lumen.
BT0416 is widespread in gut Bacteroidetes species
We next sought to uncover additional gut bacteria possessing sulfonation activity. Using BT0416 as a query sequence, we conducted BLASTP searches against bacterial genomes in the NCBI non-redundant protein database37,38, and identified 362 bacterial genomes containing SULT homologs. Most of these bacteria belonged to the phylum Bacteroidetes, one of the two predominant phyla in the human gut microbiome39. We also found BT0416 homologs in Firmicutes, Proteobacteria, and one uncultured bacterium (Fig. 2d, Supplementary Table 3).
To assess the prevalence of BT0416 in human microbiota, we queried the curatedMetagnomicData program40 for BT0416 homologs. We observed homologs in 77 of 78 collections within the curated data and a composite of the BtSULT gene cluster (BT0413-BT0416) at high levels in more than half of the collections (Extended Data Fig. 3a, Supplementary Table 4). Additionally, BtSULT homologs were identified in most samples in more than half of the collections (Fig. 2e). These observations suggest that BtSULT homologs are widely distributed in human microbiomes.
Next, we incubated a BtSULT homolog-containing Bacteroidetes species, Parabacteroides merdae ATCC 43184 (P. merdae), with Ch. Like B. thetaiotaomicron, P. merdae also produced Ch-S in vitro (Fig. 2f). We then generated a deletion strain (P. merdaeΔPARMER_01922 or P. merdae Δsult, Extended Data Fig. 2p). This strain did not produce Ch-S in vitro (Fig. 2f), and genetic deletion of PARMER_01922 did not affect bacterial growth (Extended Data Fig. 2q). Additional screening using Bacteroidetes strains and representative members of other gut phyla revealed that all sulfonating strains contained homologs of BtSULT and the PAPS biosynthesis genes BT0413-BT0415, while non-sulfonating strains lacked a homologous sulfotransferase (Extended Data Fig. 3b, Supplementary Table 2b,c). These data suggest that PARMER_01922 and BtSULT homologs in other gut organisms encode functional sulfotransferases and provide further evidence that both BtSULT and PAPS cofactor biosynthesis are necessary for sulfonation.
BtSULT sulfonates metabolites with a flat A/B ring system
Given that mammalian SULTs sulfonate diverse substrates and the human gut contains a range of cholesterol-like molecules5–7, we investigated whether BtSULT possessed promiscuous sulfotransferase activity. We evaluated the activities of B. thetaiotaomicron cultures and purified BtSULT against a panel of molecules, including (1) predominant human and rodent BAs, which possess a pendant D-ring side chain, stereochemical variations, and substitutions at the 3, 6, 7, and 12 positions of the steroidal core; (2) steroids with no D-ring side chain (i.e., estradiol and dehydroepiandrosterone (DHEA)); (3) steroid-like compounds with D ring side chain modifications (i.e., ergosterol, cholesterol, β-sitosterol, pregnenolone, and lanosterol); and (4) non-steroidal compounds containing 6-membered carbocycles (i.e., cyclohexanol and calcifediol) (Fig. 3a).
Fig. 3 |. Characterization of selective SULT activity in human gut bacteria.

a, Library of substrates tested for conversion by BtSULT. Positive substrates (red) that were sulfonated include compounds with a flat 5α trans-fused steroidal ring system and compounds with a flat 5-ene A/B ring fusion. Negative substrates (blue) that were not sulfonated include compounds with bent, 5β cis-fused ring systems.
b, In vitro incubation of substrates (30 μM) with either B. theta (48 hours) or purified BtSULT (0.5 hours and 4 hours) resulted in conversion of flat A/B ring substrates to their sulfonated forms (n=3 biological replicates). Normalized intensities are presented (area under the curve of the extracted ion chromatogram (EIC)) and normalized to the internal standard glycocholic acid (GCA). Data are presented as mean ± S.E.M.
c, Three dimensional structural representations of the different A/B ring conformations tested against BtSULT. Substrate structures accepted include flat trans- and 5-ene compounds, while structures either not sulfonated or sulfonated less efficiently include bent cis-fused structures. Red text highlights favored substrate configuration.
d, Three dimensional structural representations of the different LCA isomers, including bent (LCA and isoLCA) and flat (alloLCA and isoalloLCA) ring conformations, as well as the 5-ene flat structure of cholesterol. Red text highlights favored substrate configuration.
BtSULT displayed a relatively narrow substrate scope, preferentially sulfonating substrates containing a flat A/B ring system found in 5α trans-fused (e.g., isoallolithocholic acid (isoalloLCA), allolithocholic acid (alloLCA)) or 5-ene molecules (e.g., DHEA, ergosterol, cholesterol, pregnenolone, β-sitosterol, lanosterol, and calcifediol) (Fig. 3b,c). BtSULT did not sulfonate compounds with a bent A/B ring system found in 5β cis-fused molecules (e.g., cholic acid (CA), chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), ursodeoxycholic acid (UDCA)) (Fig. 3b,d). These results indicate that BtSULT does not sulfonate most BAs, an abundant class of steroid-like molecules found in high concentrations in the human gut41. BtSULT did sulfonate isoalloLCA, a bacterial BA metabolite with a 5α trans-fused A/B ring that was recently found to enhance the differentiation of naive T cells into regulatory T (Treg) cells42. We found that isoalloLCA-3-sulfate did not induce Treg differentiation in vitro, suggesting that bacterial sulfonation can alter biological activities (Extended Data Fig. 4a). BtSULT did not sulfonate cyclohexanol (Fig. 3b), suggesting that additional degrees of substrate recognition provided by a steroid-like backbone may be required for sulfonation by BtSULT.
One notable exception that we observed was the sulfonation of isolithocholic acid (isoLCA), which has a 5β-cis A/B ring system. We recently found that certain Bacteroidetes species, including B. thetaiotaomicron and P. merdae, convert isoLCA to isoalloLCA using a four-gene cluster43 (Extended Data Fig. 4b,c). We hypothesized that B. thetaiotaomicron is converting isoLCA to isoalloLCA and then producing isoalloLCA-3-sulfate. However, isoLCA-3-sulfate and isoalloLCA-3-sulfate are isomers and could not be separated chromatographically. To determine the identity of the sulfonated product, we cultured P. merdae wild-type and P. merdae ΔPARMER_04016–18, the isoalloLCA cluster deletion mutant, with isoLCA and monitored sulfonation by UPLC-MS (Extended Data Fig. 4d). While P. merdae wild-type produced both isoalloLCA and the sulfonated LCA isomer, P. merdae ΔPARMER_04016–18 did not produce either isoalloLCA or the sulfonated peak. These data show that the production of the sulfonated LCA isomer depends on the presence of the isoalloLCA gene cluster in P. merdae and suggest that BtSULT sulfonates isoalloLCA but not isoLCA in culture. While we did observe production of isoLCA-3-sulfate by purified BtSULT, the results are unlikely to be relevant in live bacterial cells, as we observed low turnover of isoLCA compared to isoalloLCA at 0.5 hours by the purified enzyme (Fig. 3b).
BtSULT produces Ch-S in vivo
To examine whether BtSULT is contributing to Ch-S production in vivo, we quantified the levels of Ch-S in mice monocolonized with B. thetaiotaomicron wild-type, B. thetaiotaomicron Δsult or B. thetaiotaomicron Δsult_sult+ (Fig. 4a). Bacterial colonization levels were comparable for all strains (Fig. 4b). We observed significantly higher levels of Ch-S in feces and cecal contents (Fig. 4c) of B. thetaiotaomicron wild-type-colonized mice compared to B. thetaiotaomicron Δsult-colonized mice, and knockout-colonized and GF mice possessed similar levels of Ch-S (Fig. 4c). B. thetaiotaomicron Δsult_sult+-monocolonized mice also possessed higher levels of Ch-S compared to both GF and knockout-colonized mice, although the levels were lower than in wild-type-colonized mice. We also quantified Ch-S levels in GF and conventional (SPF) mice; however, the levels were not comparable, likely due to increased Sult2B1b expression in GF livers (Extended Data Fig. 5). We observed that levels of β-sitosterol sulfate were increased in wild-type compared to knockout-colonized mice (Extended Data Fig. 4e). β-sitosterol is a plant steroid present in both mouse chow and human food, suggesting that BtSULT can also sulfonate dietary metabolites in vivo. These data provide evidence that BtSULT produces Ch-S in vivo and suggest that this enzyme may contribute to the production of other sulfonated steroid-like metabolites.
Fig. 4 |. Ch-S is produced by BtSULT in vivo.
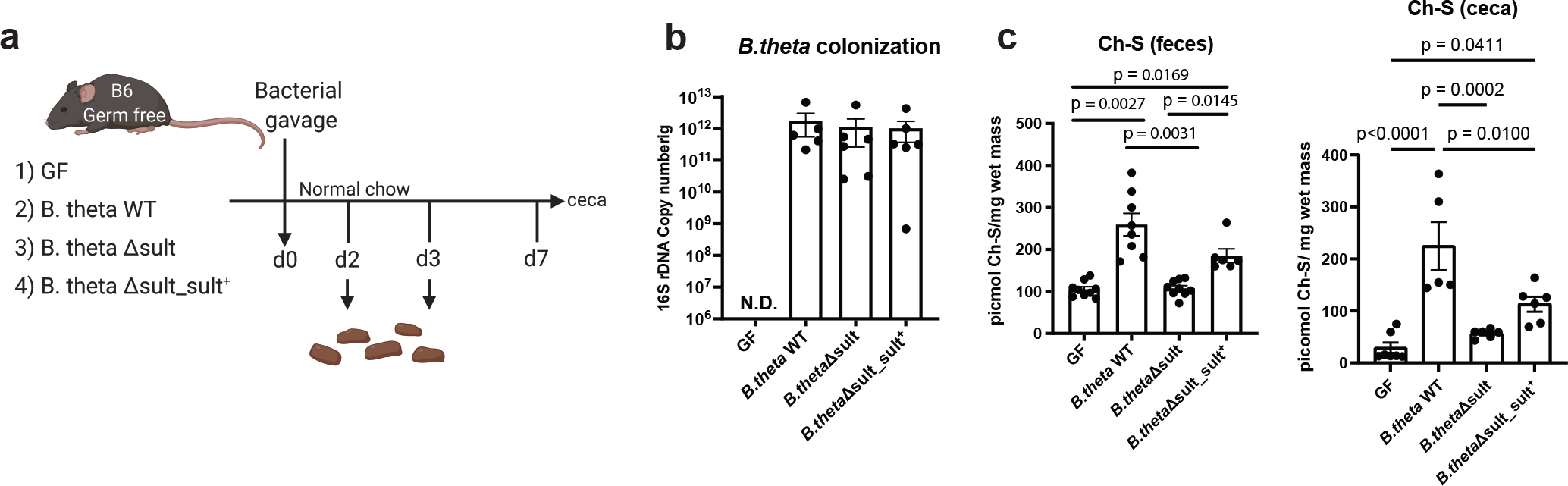
a, Design of BtSULT monocolonization experiment. GF B6 mice were colonized with B. theta WT, Δsult, Δsult_sult+ or were left in the GF state and fed a normal chow diet for 7 days. Fecal samples were collected on day 2 for bacterial colonization check. Ch-S levels were analyzed from day 3 fecal materials and cecal samples upon sacrifice on day 7 using UPLC-MS.
b, No significant differences in colonization efficiency were observed among B. theta strains (GF and B.theta WT groups, n=5, B.theta sult KO and B.theta sult KO complemented groups, n=6, results pooled from two separate experiments, data are presented as mean ± SEM, one-way ANOVA with Dunnett’s multiple comparison test. There were no detectable copy numbers from the GF group.).
c, Left, Ch-S levels in fecal samples (day 3) from mice colonized with B. theta WT or B. theta Δsult_sult+ were significantly higher than Ch-S levels in GF mice or B. theta KO colonized mice (GF, n=9; B.theta WT, n=8, B.theta sult KO, n=9, B.theta sult KO complement, n=6, results pooled from three separate experiments, data are presented as mean ± SEM, one-way ANOVA followed by Dunnett’s multiple comparison test). Right, Ch-S levels in cecal contents of mice monocolonized with B. theta WT were significantly higher than Ch-S levels in GF or B. theta KO-colonized mice (GF, n=7, B.theta WT, n=5, B.theta sult KO and B.theta sult KO complemented groups, n=6, results pooled from three separate experiments, data are presented as mean ± SEM, one-way ANOVA followed by Tukey’s multiple comparisons).
Many of the SULT substrates, including cholesterol, exhibit low water solubility, raising questions about how accessible these compounds are to gut bacterial enzymes. Phospholipids are present in the lumen and increase the solubility of Ch44. To investigate whether increasing the concentration of phospholipids could raise Ch-S production levels, we incubated B. thetaiotaomicron with Ch in rich media (BHI+), defined media, or nutrient-supplemented defined media with or without lecithin45. Contrary to expectation, lecithin addition did not enhance Ch-S production in defined media and reduced Ch-S production in rich and enhanced defined media (Extended Data Fig. 6a). Bacterial growth was not affected by lecithin (Extended Data Fig. 6b). Moreover, we observed micromolar (~picomol/mg) concentrations of Ch in cecal contents and feces from mice and in human feces (Extended Data Fig. 6c–e). These data suggest that Ch is accessible to gut bacteria in the lower GI tract and that low Ch solubility is not limiting bacterial Ch-S production.
Sulfonation outcompetes desulfation in vitro and ex vivo
Gut bacteria encode sulfatases (SULFs) that catalyze desulfation2,27,46. We therefore investigated whether SULF activity counteracts SULT activity. Previous work indicated that Ch-S was not a substrate for the steroid sulfatase in Peptococcus niger H446. However, recent work identified SULFs in a variety of Bacteroides species, including B. thetaiotaomicron2. To test whether the B. thetaiotaomicron SULF is active against Ch-S, we incubated B. thetaiotaomicron Δsult whole cells and lysate with Ch-S and monitored metabolite levels over 8 hours (Extended Data Fig. 7a). We observed a substantial decrease in Ch-S levels within 1 hour in lysate but not in whole cells. Together with our SULT results, these data indicate that B. thetaiotaomicron possesses intracellular SULT and SULF activities and suggest that SULF is active on intracellular Ch-S, but extracellular Ch-S is not imported and subject to desulfation. Based on these data, we propose a model for Bacteroides Ch-S production. BtSULT converts intracellular Ch and imported sulfate to Ch-S. While intracellular desulfation to Ch occurs, sulfonation and Ch-S production are favored. As bacteria die and lyse, Ch-S is released into the gut lumen (Extended Data Fig. 7b).
To investigate the interplay between SULT and SULF activity in complex microbial communities, equal concentrations of Ch and Ch-S were added to human fecal slurries (Fc and Fe) and the resultant mixtures were cultured for 7 days (Extended Data Fig. 7c–d). We observed increasing concentrations of Ch-S in samples from both donors over time, indicating that both microbial communities favored Ch-S production over desulfation. These data demonstrate that SULT activity is present in human microbiomes and sulfonation can be favored over desulfation in complex human gut microbial communities.
Bacterial Ch-S inhibits leukocyte migration in vitro
We next investigated whether bacterial production of Ch-S could affect host cellular responses. While Ch-S was reported to inhibit leukocyte migration and contribute to an immunosuppressive environment in the murine eye, whether bacterial Ch-S production affects immune cell migration had not been reported47. To investigate this question, we utilized a transwell chemotaxis assay. Specifically, we quantified the migration of CD90.2+ immune cells in response to the chemokine CCL21. Compared to a vehicle, CCL21 treatment led to increased migration of leukocytes, while leukocyte migration was not affected by Ch (Fig. 5a). Consistent with previous results47, Ch-S treatment decreased leukocyte migration in a dose-dependent manner (Fig. 5a) suggesting that Ch-S may negatively influence immune cell migration.
Fig. 5 |. BtSULT modulates immune cell trafficking and is negatively correlated with IBD in patients.
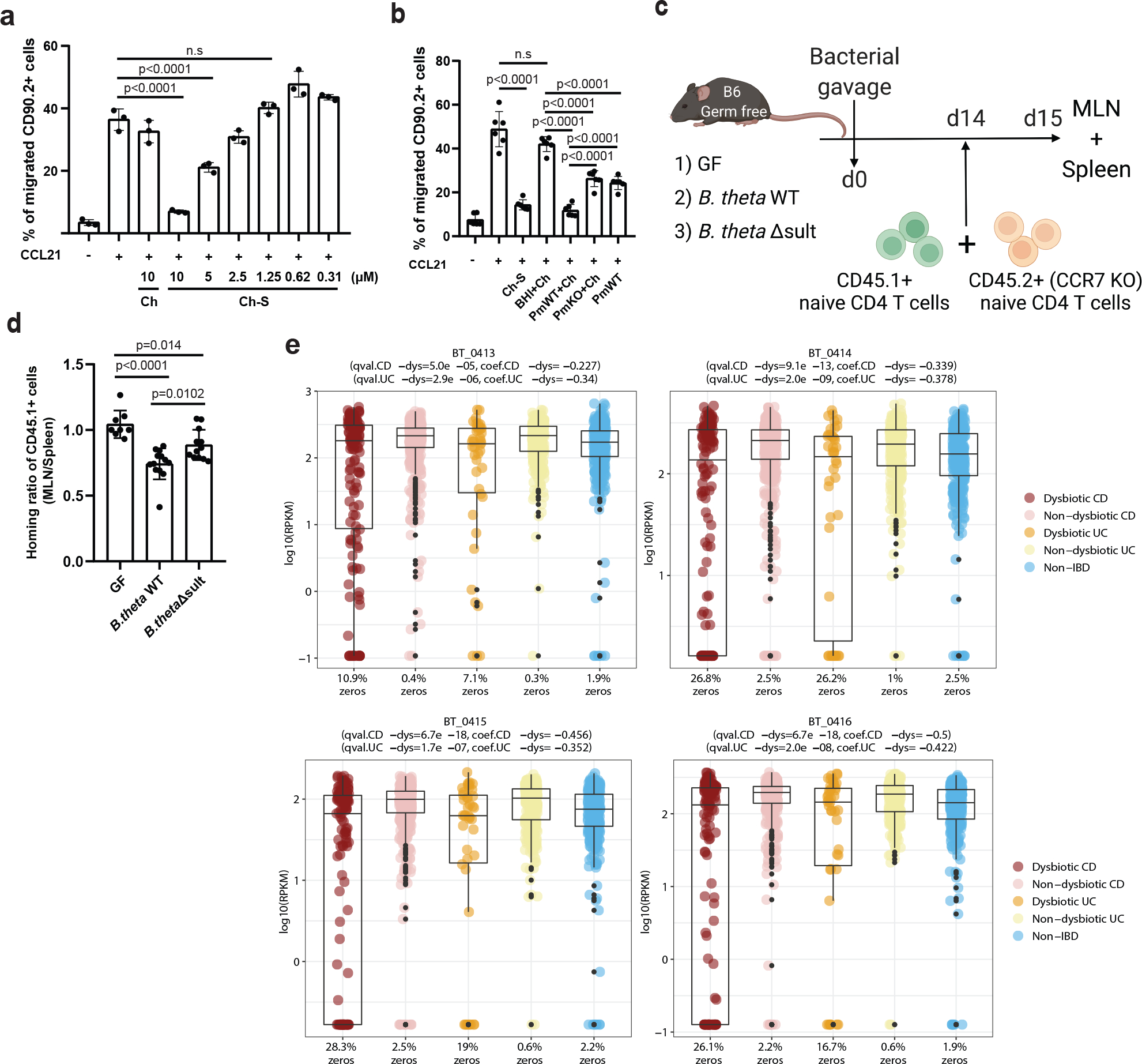
a, Ch-S treatment reduced CCL21-induced leukocyte migration. Splenocytes were treated with vehicle, Ch, or Ch-S at indicated concentrations. The percentage of migrated leukocytes (CD90.2+) in the lower versus the upper chamber was evaluated after 6 hours. (n=3 biological replicates per condition, data are presented as mean ± S.D, one-way ANOVA followed by Tukey’s multiple comparison test, n.s.=not significant).
b, Transwell chemotaxis assay was performed similarly to a) cells were incubated with the following supernatants; BHI+Ch (BHI media+Ch), PmWT+Ch (P. merdae wild-type, cultured with Ch), PmKO+Ch (P. merdae KO cultured with Ch), and PmWT (P. merdae wild-type without Ch) (n=6 biological replicates per condition, data pooled from two independent experiments, presented as mean ± S.D., one-way ANOVA followed by Tukey’s multiple comparison test, n.s=not significant).
c, In the T cell transfer experiment, recipient mice were GF or monocolonized with B. theta WT or B. theta Δsult. CFSE-labeled naïve CD4 T-cells (CD45.1+ wild-type and CD45.2+ CCR7-deficient) were mixed and co-transferred into recipient mice. After 24h, cells were isolated from the spleens and MLNs of recipients, stained with antibodies against CD45, CD4, CD25, CD44, and CD62L.
d, Migration of donor cell lymphocytes to MLNs, presented as the normalized ratio of migrated cells-to-MLN divided by spleen, was significantly impaired with B. theta WT (n=13) compared to GF (n=8) or B. thetaΔsult (n=12). Data pooled from three independent experiments, presented as mean ± S.D., with one-way ANOVA followed by Tukey’s multiple comparison test).
e, The normalized abundances of APS kinase (BT0413), ATP sulfurylase (BT0414, BT0415), and sulfotransferase (BT0416) homologs were significantly depleted (FDR q-values < 0.05) in the dysbiotic states of both CD (50 patients) and UC (30 patients) subjects from the HMP2 cohort compared to their individual baselines (26 patients). The percentage of zeros is shown on the x axis. For the box plots, the center line indicates the median (second quartile) and the box limits indicate first and third quartiles of the data. The points outside of box plot whiskers are outliers. Statistical analysis was performed using a linear mixed model and its coefficient and significance (FDR-adjusted P values) are shown (Supplementary Table 6).
We then investigated whether bacterially produced Ch-S could suppress leukocyte migration. Immune cells were incubated with supernatants from P. merdae wild-type or P. merdae Δsult cultures cultured in the presence of Ch. We verified Ch-S levels by UPLC-MS in the P. merdae wild-type and P. merdae Δsult cultures (Supplementary Fig. 1). The supernatants of P. merdae wild-type + Ch, but not P. merdae Δsult + Ch, cultures significantly reduced the percentage of the leukocytes that migrated to the lower chamber compared to supernatants of P. merdae wild-type only control (Fig. 5b). Neither Ch-S nor bacterial supernatant affected immune cell viability (Extended Data Fig. 8a–b). Together, these data indicate that bacterially produced Ch-S inhibits leukocyte migration in vitro.
Presence of BtSULT inhibits leukocyte migration in vivo
Naïve CD4 T cells exhibit a high-level expression of C-C chemokine receptor type 7 (CCR7) and migrate to secondary lymph nodes such as MLNs in response to CCL2148. We investigated whether colonization of GF mice with Ch-S producing bacteria inhibits T cell migration in vivo in a CCL21-CCR7 dependent manner. First, we confirmed that supernatants from P. merdae wild-type reduced CCL21-induced CD4 T cell migration in vitro (Extended Data Fig. 8c). Next, we adoptively transferred naïve CD4 T cells from CD45.1+ wild-type and CD45.2+ CCR7-deficient mice into GF, B. thetaiotaomicron WT-colonized, and B. thetaiotaomicron Δsult-colonized mice and assessed migration to MLNs versus spleens after 24h (Fig. 5c,d). Compared to GF mice or those colonized with B. thetaiotaomicron Δsult, mice with B. thetaiotaomicron WT exhibited reduced ratios of donor WT CD4 T-cells (CD45.1+) in MLNs (Fig. 5d). In contrast, CCR7-deficient CD4 T cell (CD45.2+) ratios were similar regardless of cohort (Extended Data Fig. 8d). These results demonstrate that the presence of BtSULT-containing microbes is sufficient to suppress the migration of CD4 T-cells to MLN in vivo via a yet-to-be elucidated mechanism. Together with the in vitro findings, these data indicate that bacterial enzymes that produce Ch-S likely affect immune cell trafficking and responses.
At this time, the mechanism by which BtSULT affects host immune cell migration is not clear and may be indirect. Although we observed higher levels of Ch-S in intestinal contents and feces in mice colonized with BtSULT-containing compared to BtSULT-deficient bacteria (Fig. 4c, Extended Data Fig. 9a,b), we did not observe significant differences in Ch-S levels in plasma (Extended Data Fig. 9c). We observed a significant increase in host Sult2b1b expression in the ileum of GF mice compared to monocolonized mice (Extended Data Fig. 9d–f); however, we still observed significantly higher Ch-S levels in WT colonized mice in spite of potential reduced host contribution (Extended Data Fig. 9a,b). We observed no significant differences in host steroid sulfatase expression in the liver, ileum or colon (Extended Data Fig. 9g–i). It is possible that the constraint of harvesting tissues at a specific timepoint combined with ad libitum feeding and host Ch-S production masked the effects of bacterial Ch-S production on Ch-S levels outside the gut. Alternatively, it is possible that luminal Ch-S production could affect host immune response without reaching systemic circulation through pathways such as dendritic cell sampling of gut contents49,50 or transport of bacterial metabolites to local immune cells through bacterial outer membrane vesicles51,52. These hypotheses have not been tested, however, and thus the mechanisms by which metabolites produced by bacteria in the gut lumen affect host immune response warrants further exploration.
BtSULT cluster and Ch-S abundant in human feces
Chemokine-driven immune cell migration is critical to controlling inflammatory responses in many tissues, including the gut mucosa53. Given the finding that bacterial production of Ch-S affects leukocyte migration, we investigated whether the abundances of BtSULT cluster genes were altered in IBD patients. We utilized 54metagenomic profiles of stool samples from subjects with Crohn’s disease, ulcerative colitis, and non-IBD controls from the HMP2 IBDMDB cohort54. Through targeted functional profiling of HMP2 metagenomes, we revealed that the abundances of all four genes from the BtSULT pathway cluster were significantly depleted in the dysbiotic state of both CD and UC patients compared to their non-dysbiotic baselines (Fig. 5e), even when controlling for phylum-level taxonomic changes (Extended Data Fig. 10a). The mean amount of fecal Ch-S was not different between CD and UC patients and non-IBD controls (Extended Data Fig. 10b,c). Variations in host production of Ch-S could contribute to a normalization in Ch-S levels detected in feces. Moreover, bacterial production of Ch-S in the intestinal lumen may exert local immunological effects. Importantly, Ch-S was abundant in human feces (mean values of 21.0 μM, 26.6 μM, and 23.0 μM in UC, CD, and non-IBD individuals, respectively) and exhibited substantial interpersonal variations (ranges of 2–116 μM, 2–170 μM, and 3–115 μM in UC, CD, and non-IBD individuals, respectively). Additionally, we found a significant, positive correlation between BtSULT homolog abundance and fecal Ch-S concentration across the entirety of the HMP2 cohort (Extended Data Fig. 10d). Taken together with our findings that human fecal communities convert Ch to Ch-S in varying amounts, it is likely that both bacterial and mammalian enzymes contribute Ch-S production in the human host.
Discussion
Here, we used genome mining to identify a candidate bacterial SULT in the abundant gut commensal B. thetaiotaomicron. Through heterologous expression and genetic deletion of the BT0416 gene in two Bacteroidetes strains, we identified an operon required for both generation of the PAPS cofactor and sulfonation of steroid-like molecules55
Sulfonation is known to affect the solubility of compounds, their biological activities, and their passage across the gut epithelium into circulation56–58. Indeed, we found that bacterially produced Ch-S affected the migration of leukocytes. Additionally, we found that sulfonation of isoalloLCA inhibits its activity to induce Treg differentiation42,43. Thus, the levels and substrate specificities of sulfonating bacteria in the gut could modulate immune responses in the host through the generation of sulfonated metabolites.
Further analyses revealed that BT0416 is both abundant in human gut microbes, and prevalent in the guts of broad adult populations. Interestingly, we also found the abundances of BtSULT cluster genes are significantly lower in the feces of patients with dysbiotic CD and UC compared to their non-dysbiotic controls. We also found that the presence of BtSULT limits immune cell migration in vitro and in vivo, an effect that has been shown to suppress immune response47. Further studies, however, are needed to determine whether gut bacterial production of Ch-S could induce anti-inflammatory effects in the host.
Whether bacterial conversion of Ch to Ch-S has other effects on the host is not yet known. Ch is one of the most abundant metabolites in the human gut, and this compound has been linked to human diseases, including cardiovascular disease, neurodegenerative diseases, autoimmune diseases59, and cancers60. Future work may reveal additional ways in which bacterial sulfonation of Ch affects host health and disease. Additionally, we found that BtSULT sulfonates a number of bioactive metabolites, including DHEA, an abundant circulating steroid and androgen and estrogen precursor61, calcifediol, a form of vitamin D62, and pregnenolone, a neurosteroid63,64. In an example of how bacterial sulfonation could affect the biological functions of metabolites, pregnenolone is an endocannabinoid63,64, while pregnenolone-sulfate is a negative allosteric modulator of the γ-aminobutyric acid alpha (GABAA) receptor65 and a positive allosteric modulator of the N-methyl-D-aspartate receptor (NMDA) receptor66.
Our efforts to uncover the origins of Ch-S production in colonized mice has revealed a previously uncharacterized group of gut bacterial enzymes. Studying the BtSULT cluster and its corresponding products provides opportunities to discover bioactive molecules and to understand the biosynthetic mechanisms governing steroid metabolism. These studies will facilitate the rational prediction of SULT substrate structures and act as a first step toward understanding the effects of bacterial sulfonation on host health and disease.
Methods
Statement on Ethical Compliance
All experiments performed in this study comply with ethical regulations. All mouse experiments were performed under full compliance with IACUC approved protocol (IS00001676) and guidelines of Harvard Medical School. Human fecal samples were collected under the Harvard Medical School IRB# 19–1887. The HMP2 samples were collected and analyzed as previously published54 and was reviewed by the Institutional Review Boards at each sampling site: overall Partners Data Coordination (IRB #2013P002215); MGH Adult cohort (IRB #2004P001067); MGH Paediatrics (IRB #2014P001115); Emory (IRB #IRB00071468); Cincinnati Children’s Hospital Medical Center (2013– 7586); and Cedars-Sinai Medical Center (3358/CR00011696). Bacterial samples were stored and experiments were performed following protocols approved by the Harvard Medical School Committee on Microbiological Safety (#21–097).
Mice
C57BL/6J mice were purchased from Jackson laboratory and were housed in an individually ventilated cage system (Tecniplast) at 20–22 °C and 40–55% humidity and under a 12 h–12 h light–dark cycle at the specific-pathogen-free New Research Building facility of Harvard Medical School. Germ-free C57BL/6NCrl mice were maintained in gnotobiotic isolators as appropriate. All experiments were conducted on sex-matched 5–10 week old mice. Mice were colonized with designated bacteria by oral gavage with bacterial culture in the biosafety station and kept in Isocages for the duration of the experiment. Control powder meal (Teklad Global 19% protein extruded diet, #2019) was autoclaved and provided to mice during the experiment. Feces were collected 2 or 3 days post-colonization for colonization check or fecal Ch-S quantification, and cecal materials were harvested on the last day for Ch-S quantification by UPLC-MS. All experiments involving mice were performed using IACUC approved protocols under either Brigham and Women’s Hospital Center for Comparative Medicine (monocolonization vs GF experiment) or the Institutional Animal Care and Use Committee at Harvard Medical School (B. thetaiotaomicron Δsult and WT colonization experiment).
Chemicals
Authentic standards were acquired for Ch-S (Avanti Polar Lipids #700016), DHEA-S (Cayman chemical 15873), pregnenolone-sulfate (Sigma P162), and estradiol-sulfate (Sigma E9505). Additional standards were synthesized and authenticated as noted below.
Synthetic procedures
Sulfonated standards were prepared for calcifediol, ergosterol, β-sitosterol, isoLCA, alloLCA, isoalloLCA according to a previously reported protocol67. Briefly, all anhydrous reactions were run under a positive pressure of argon or nitrogen. Silica gel column chromatography was performed using 60 Å silica gel (230−400 mesh). NMR spectra recorded in the listed solvents and the residual solvent peak or TMS was used as the internal reference. For calcifediol, ergosterol, β-sitosterol, alloLCA, and isoalloLCA, synthesis was carried out as per reported procedure67. To a 0.1 M solution of the alcoholic precursor (1.0 equiv.) in pyridine, SO3:pyridine (2.5 equiv., except for calcifediol where 1.3 equiv. was used) was added, and the resulting solution was stirred at rt for 18 h. The reaction mixture was concentrated using a rotary evaporator. The resulting slurry was resuspended in 5 mL of 10:1 dichloromethane:methanol and 5 mL of a saturated solution of sodium bicarbonate was added. The biphasic solution was concentrated using a rotary evaporator. The resulting crude compound was then purified by silica gel chromatography (80% dichloromethane/20% methanol) to provide the sulfonated compounds.
Ergosterol-3S.
White solid (0.14 g, 23%); 1H NMR (400 MHz, DMSO-d6) δ 5.48 (s, 1H), 5.32 (s, 1H), 5.26–5.14 (m, 2H), 3.95–3.89 (m, 1H), 2.53 (s, 1H), 2.17 (t, J = 12.8 Hz, 1H), 2.03–1.17 (m, 18H), 0.99 (d, J = 6.4 Hz, 3H), 0.88–0.85 (m, 6H), 0.81–0.78 (m, 6H), 0.57 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 140.82, 140.43, 135.79, 131.89, 119.59, 116.57, 75.53, 55.49, 54.33, 45.99, 42.83, 42.49, 40.33, 38.83, 38.26, 37.04, 32.92, 29.56, 28.37, 23.02, 21.41, 20.98, 20.21, 19.91, 17.77, 16.32, 12.82, 12.25; HRMS (m/z): [M - H]- calcd. for C28H43O4S, 475.2888; found, 475.2912. Supplementary Figures 2–3.
β-sitosterol-3S.
White solid (94.0 mg, 16%); 1H NMR (400 MHz, DMSO-d6) δ 5.26 (s, 1H), 3.86–3.78 (m, 1H), 2.35 (dd, J = 13.6, 4.4 Hz, 1H), 2.12 (t, J = 12.0 Hz, 1H), 1.95–1.75 (m, 5H), 1.63–0.73 (m, 36H), 0.63 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 150.42, 141.18, 56.59, 55.90, 50.02, 45.61, 42.32, 37.36, 36.54, 35.95, 33.82, 31.90, 31.85, 29.25, 29.18, 28.26, 25.94, 24.32, 23.09, 23.07, 21.96, 21.04, 20.17, 19.50, 19.40, 19.08, 12.24, 12.14, 12.12,; HRMS (m/z): [M - H]- calcd. for C29H49O4S, 493.3357; found, 493.3376. Supplementary Figures 4–5.
alloLCA-3S.
White solid (12.0 mg, 50%); TLC Rf = 0.2 (20% methanol/ 80% dichloromethane); 1H NMR (400 MHz, DMSO-d6) δ 11.89 (br s, 1H), 4.25 (s, 1H), 2.22–2.14 (m, 1H), 2.08–2.01 (m, 1H), 1.90 (d, J = 11.6 Hz, 1H), 1.76 (d, J = 13.2 Hz, 2H), 1.68–0.98 (m, 21H), 0.85 (d, J = 6.0 Hz, 4H), 0.71–0.60 (m, 7H); 13C NMR (100 MHz, DMSO-d6) δ 175.91, 71.53, 71.47, 56.55, 55.93, 54.44, 42.62, 35.60, 35.50, 35.32, 33.83, 32.89, 32.18, 31.55, 31.27, 28.57, 28.05, 26.87, 24.23, 20.82, 18.60, 12.39, 12.36, 11.73; HRMS (m/z): [M - H]- calcd. for C24H39O6S, 455.2473; found, 455.2483. Supplementary Figures 6–7.
isoalloLCA-3S.
White solid (16.0 mg, 16%); TLC Rf = 0.15 (20% methanol/ 80% dichloromethane); 1H NMR (400 MHz, DMSO-d6) δ 11.91 (br s, 1H), 3.95–3.87 (m, 1H), 2.22–2.02 (m, 2H), 1.90–0.81(m, 28H), 0.73 (s, 3H), 0.62–0.57 (m, 4H); 13C NMR (100 MHz, DMSO-d6) δ 175.76, 75.52, 56.37, 55.99, 54.10, 44.87, 42.63, 39.90, 37.06, 35.66, 35.47, 35.40, 35.30, 32.11, 31.46, 31.23, 29.04, 28.79, 28.07, 24.26, 21.22, 18.59, 12.45, 12.44, 12.37, 12.34; HRMS (m/z): [M - H]- calcd. for C24H39O6S, 455.2473; found, 455.2499. Supplementary Figures 8–9.
Calcifediol-3S.
White solid (10.0 mg, 28%); TLC Rf = 0.06 (20% methanol/ 80% dichloromethane); 1H NMR (400 MHz, 1:1 CDCl3:DMSO-d6) δ 5.90 (d, J = 11.2 Hz, 1H), 5.68 (d, J = 11.6 Hz, 1H), 4.70 (d, J = 1.6 Hz, 1H), 4.46 (d, J = 2.4 Hz, 1H), 4.24–4.18 (m, 1H), 2.49 (dd, J = 12.0, 7.6 Hz, 1H), 2.40 (dd, J = 13.2, 4.0 Hz, 1H), 2.14–2.07 (m, 2H), 1.88–1.45 (m, 8H), 1.37–0.85 (m, 26H), 0.75–0.68 (m, 1H), 0.68–0.64 (m, 3H), 0.21 (s, 3H); 13C NMR (100 MHz, 1:1 CDCl3:DMSO-d6) δ 144.45, 141.08, 134.27, 121.62, 117.03, 111.35, 76.136, 76.06, 69.80, 56.06, 55.71, 45.15, 43.49, 42.55, 40.01, 35.87, 35.55, 32.34, 31.43, 28.23, 27.74, 27.60, 27.47, 26.95, 22.81, 21.47, 20.16, 17.86, 17.72, 10.86; HRMS (m/z): [M - H]- calcd. for C27H43O5S, 479.2837; found, 479.2862. Supplementary Figures 10–11.
For isoLCA, synthesis was carried out as per reported procedure67. To a suspension of isolithocholic acid (20.0 mg, 0.025 M) in pyridine at room temperature (rt) was added pyridine sulfur trioxide (170 mg, 21.4 equiv.) slowly. The resulting solution was stirred at rt for 30 min, and then quenched with 2 mL 1 N sodium hydroxide solution. The reaction mixture was further diluted with 1 mL ethyl acetate. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 × 2 mL). The combined aqueous layers were concentrated under reduced pressure, and the crude compound was then purified by silica gel chromatography (40% methanol/60% dichloromethane) to provide the target compound 3S-isolithocholic acid (15.1 mg, 60%) as a yellow powder.
3S-isoLCA.
1H NMR (400 MHz, CDCl3): δ 2.36–2.16 (m, 2H), 2.03–1.75 (m, 8H), 1.70–1.58 (m, 2H), 1.56–1.53 (m, 2H), 1.48–1.42 (m, 6H), 1.35–1.29 (m, 4H), 1.24–1.08 (m, 6H), 0.98–0.96 (m, 6H), 0.71 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 213.53, 75.63, 56.44, 56.07, 42.50, 40.09, 39.86, 39.02, 36.87, 35.68, 35.36, 34.39, 31.14, 31.05, 30.02, 27.80, 26.24, 25.95, 25.32, 23.83, 22.89, 20.80, 17.35, 11.06. HRMS (m/z): [M] calcd for C24H40O6S, 456.2546, found 456.3145. Supplementary Figures 12–13.
Putative sulfonated derivatives of lanosterol were detected and assigned based on their masses (505 m/z [M-H]) are quantified as areas under the curve.
Microbial strains
All strains used in this study are listed in Supplementary Table 1b. Culturing of human gut bacteria was performed in an anaerobic chamber (Coy Laboratory Products) with a gas mixture of 5% hydrogen and 20% carbon dioxide (balance nitrogen) unless otherwise stated. Individual strains were plated from glycerol stocks onto brain heart infusion agar (Bacto) supplemented with 5 mg/L hemin, 2.5 uL/L Vitamin K, 500 mg/L cysteine HCl (BHI+), and grown for 2–3 days. Colonies were then inoculated into 3 mL of BHI+ liquid media in Falcon™ Round-Bottom polystyrene tubes and grown for 48 hours at 37 °C to provide starter cultures, which were diluted 1:100 in triplicate into 5 mL fresh BHI+ media containing 100 μM of the corresponding substrate or vehicle control. Cultures were grown at 37 °C for time course experiments. An aliquot of culture (0.5 mL) was harvested and used for sulfonated metabolite quantification. All experiments were performed in triplicate and repeated twice unless otherwise stated.
Culturing B. thetaiotaomicron with phospholipid additive
Starter cultures of B. thetaiotaomicron in BHI+ media were diluted at 1:100 into BHI+ media, defined media for Bacteroides,68 or defined media for Bacteroides supplemented with ATCC trace mineral solution (1%), ATCC trace vitamin solution (1%), D(−)arabinose (20% w/v), D-(+) cellulobiose (1g L−1), D-(+) maltose (1g L−1), D-(−)-fructose (1g L−1), and casamino acids (10% w/v). All media configurations were assessed in the presence and absence of lecithin (10 mg/mL) as a lipid additive. All cultures contained substrate cholesterol (30 μM). The cultures were assessed for growth and Ch-S production was evaluated after 7 days.
Culturing of SPF mouse cecal samples
The cecal contents of 5 SPF mice were resuspended in Cullen–Haiser Gut (CHG) media (which consists of brain heart infusion media (Bacto BHI, BD) supplemented with 1% BBL vitamin K1-hemin solution (BD), 1% trace minerals solution (ATCC), 1% trace vitamins solution (ATCC), 5% fetal bovine serum (FBS) (Hyclone), 1 g l−1 cellubiose, 1 g l−1 maltose and 1 g l−1 fructose) media to a concentration of 10 mg/mL and then incubated at 37°C under anaerobic conditions for 2 days. The starter culture was then diluted 1:100 in fresh BHI+ media with 15 μM cholesterol sulfate and 15 μM cholesterol. Samples were collected at 0, 3 and 7 days for downstream quantification.
Culturing of human fecal samples
Human fecal samples were obtained from two individuals (Donors Fc and Fe) under HMS IRB Protocol # IRB19–1887 from the Alm Lab at the Massachusetts Institute of Technology. A sample of ~150 mg was removed via sterile blade from the frozen sample, massed, and resuspended in CHG media at a final concentration of 10 mg/mL. The slurry was divided into 3 cultures of equal volume and an initial time point taken. The culture was then supplemented with 15 μM cholesterol sulfate and 15 μM cholesterol. Time points were collected at 0, 3, and 7 days for downstream quantification.
Determination of Ch-S export in B. thetaiotaomicron
B. thetaiotaomicron was cultured in BHI+ media in the presence of Ch (30 μM) for 48 and 168 hours. Samples were taken by removing 1 mL of homogeneous culture from the culture tube. The 1 mL sample was split into 2 volumes of 500 μL, with one volume retained for analysis. The second fraction was centrifuged for 2 minutes at 7,000 × g, and the media supernatant retained for analysis. The cell pellet was washed in sterile PBS by gentle pipetting, centrifuged at 7,000 × g, and the supernatant was then decanted. The final pellet was resuspended in 500 μL of PBS and retained for analysis. Whole culture, pellet, and media supernatant were extracted in the same manner as bacterial samples for product quantification. Data were reported as pellet and supernatant %Ch-S compared to paired whole culture.
qRT-PCR for host sulfotransferase gene expression
Frozen mouse distal ilea and livers were obtained from a monocolonization experiment with B. thetaiotaomicron VPI-5482 Δtdk69. Briefly, age- and weight-matched male germ-free C57BL/6 mice were maintained in gnotobiotic isolators at the Massachusetts Host-Microbiome Center for three weeks and then transferred to pre-sterilized CLAMS cages at Brigham and Women’s Hospital (BWH) Metabolic Core facility for one week. Mice were fed a high-fat diet (Research Diets D12492) for 4 weeks. Total RNA was extracted using RNeasy Mini Kit (Qiagen) with on-column DNase treatment using RNase-Free DNase Set (Qiagen) followed by a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems) (n=8). The resultant cDNA was diluted 10X and analyzed by qRT–PCR using LightCycler 480 SYBR Green I Master (Roche). Reactions were performed in a 384- well format using a QuantStudio 7 Pro Real-Time PCR (Life Technologies) at Harvard Medical School’s ICCB-Longwood Screening Facility. The 2−ΔΔCT method70 was used to calculate relative changes in gene expression and all results were normalized to the mouse ribosomal protein L32 mRNA (Supplementary Table 1a).
Additional qPCR analysis was done for later monocolonized experiments in a similar manner. Briefly, germ free (n=6) C57BL/6 mice and groups monocolonized with B. thetaiotaomicron WT (n=6) or Δsult (n=6) were fed a normal chow diet for 7 days post colonization (Fig. 4a) prior to sacrifice. At sacrifice, liver, ileum, and colon tissues were harvested and RNA and cDNA prepared as described above. The expression levels of mSult2b1b and mSTS were then acquired, normalized to the mouse ribosomal protein L32 mRNA (primers in Supplementary Table 1a) and calculated using the 2−ΔΔCT method70.
BT0416 qPCR
B. thetaiotaomicron starter culture was diluted at a ratio of 1:40 into fresh BHI+ media. Cholesterol (30 μM) or ethanol (0.06% v/v) was added and the cultures were incubated at 37°C. Time points were collected as 1 mL volumes, with the exception 4 mL was collected at 1 hour, and centrifuged to pellet before being stored at −80°C. Pellets were removed and mRNA was extracted using the Qiagen AllPrep Bacterial DNA/RNA/Protein kit using the standard protocol. Extracted mRNA was then reverse transcribed using a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). The resulting cDNA was diluted and analyzed by qRT-PCR using LightCycler 480 SYBER Green I Master (Roche). Reactions were performed in a 384-well plate at the ICCB-Longwood Screening Center at Harvard Medical School in a Thermo Fischer QuantStudio 7 Pro instrument.
Determination of operon structure BT0411–0416
B. thetaiotaomicron was cultured in BHI+ media with cholesterol (30 μM) for 8 hours at 37°C. Cells were harvested from 1 mL of culture, and extraction of both mRNA and gDNA was performed using an AllPrep Bacterial Kit (Qiagen). The mRNA was prepared in the same manner as for qPCR (see above). The resulting cDNA was used for downstream PCR analysis. End-point overlapping PCR34 was performed on paired gDNA and cDNA using Q5 High Fidelity Polymerase (NEB) with primers coordinated to amplify the regions between BT0411 and BT0412, BT0412 and BT0413, etc (Supplementary Table 1a). Samples were assessed via agarose gel in biological triplicate with representative paired samples shown (Extended Data Fig. 2l).
Construction of B. thetaiotaomicron and P. merdae knockouts
Mutants were created in the B. thetaiotaomicron VPI-5482 and P. merdae ATCC 43184 (P. merdae) backgrounds. The BtΔ0416 and PmΔ01922 strains were constructed using previously described counterselectable allelic exchange methods32,71. Briefly, ~1 kb fragments upstream and downstream of the BT0416 and PARMER01922 genes were cloned and fused using primer pairs (BT0416KO UF/UR and DF/DR; PARMER_01922KO UF/UR and DF/DR) and ligated into the suicide vector pExchange-tdk and pLGB13, respectively. The resulting vectors were transformed into Escherichia coli S17–1 λ pir and then conjugated into B. thetaiotaomicron and P. merdae. Single-crossover integrants were selected on BHI-blood agar plates containing 200 μg/mL gentamicin and 25 μg/mL erythromycin for B. thetaiotaomicron, and 5 μg/ml erythromycin and 200 μg/mL gentamicin for P. merdae, cultured in BHI+ medium overnight, and then plated onto BHI-blood agar plates containing 200 μg/ml 5-fluoro-2-deoxyuridine (FUdR) for B. thetaiotaomicron and 100 ng/ml anhydrotetracycline (aTC) for P. merdae. Candidate BT0416 and PARMER01922 deletions were screened by PCR using the diagnostic primers listed in Supplementary Table 1a and confirmed by DNA sequencing to identify isolates that had lost the gene.
Mutants for ΔBT0412 were prepared in a similar manner to Δsult utilizing the primers specified in Supplementary Table 1a. For the ΔBT0413–5 mutants, initial efforts failed at secondary selection with FUdR; however, we were able to rescue and isolate viable mutants by restreaking colonies directly from the Gentamycin+Erythromycin plates onto plates of BHI+ agar with 10% horse blood, 200 μg/mL FudR, 0.5% w/v cysteine, 0.1% w/v methionine, and 1% trace metals (ATCC). These plates were incubated for 4 days at 37°C under anaerobic conditions before colonies were selected and cultured in BHI+ media supplemented with 0.5% w/v cysteine, 0.1% w/v methionine, and 1% trace metals (ATCC). Cultures were identified by PCR and sequence confirmed. Positive mutants were then screened for the sulfonation phenotype.
For B. thetaiotaomicron complementation strain construction, a previously described method based on the mobilizable Bacteroides element NBU2 was used72. Briefly, BT0416 was PCR-amplified, cloned into the constitutive expression vector pNBU2_erm_us1311, which contains the 300 bp region upstream of BT1311 (σ70), and transformed into E. coli S17 λ pir73,74. The resulting vector was then conjugated into the B. thetaiotaomicron recipient strain (BtΔ0416). Integrants were selected from BHI+ with 10% horse blood agar plate containing 200 μg/mL gentamicin and 25 μg/mL erythromycin. Candidate B. thetaiotaomicron complementation strain (BtΔ0416_0416+) was confirmed via PCR and sequencing.
Cloning and expression of candidate sult and cys genes
The amino acid sequences for B. thetaiotaomicron SULT and Cys proteins were obtained from the IMG database. Codon optimized sequences were generated and purchased from Twist Biosciences, cloned into either pET28a (cysC and cysN) or pET21a (sult and cysD) expression vectors with a TEV cleavable, C-terminal 6-histidine affinity tag under an isopropyl β-d-1-thiogalactopyranoside (IPTG)-inducible operon. Later, cloning of the sult gene was performed to generate an N-terminal 6-His-SUMO-tagged sult gene in the pTD68 vector for soluble expression of SULT. The sult gene was inserted into the pTD68 vector using PCR amplification (SULT_Fwd and SULT_Rvs, Supplementary Table 1a) and a standard cut-and-paste cloning protocol. Successful cloning was verified by commercial Sanger sequencing.
The expression and purifications of CysN, CysD, and CysC were carried out according to the same protocol. The relevant plasmid was transformed into E. coli BL21 pLysS cells under antibiotic selection. One liter Luria Broth cultures were inoculated with a 1:100 dilution of overnight starter culture and antibiotics and grown at 37 °C to an optical density (OD600) of 0.5–0.6 before induction with 1 mM of IPTG. The cultures were then grown overnight at 18 °C before being harvested by centrifugation at 8,980 x g and the pellets stored at −80 °C. The pellets were thawed and resuspended in a lysis buffer (50 mM Sodium Phosphate Buffer/ 500 mM NaCl/ 30 mM imidazole, pH = 7.2). Cells were lysed by sonication and the lysate clarified by centrifugation at 48,380 x g. The clarified lysate was filtered and loaded onto a 1 mL HisTrap HP (Cytiva) column via an FPLC system. The bound protein was eluted on a linear imidazole gradient from 30 to 500 mM. The protein-containing fractions were determined based on absorbance at 280 nm and their contents confirmed by SDS-PAGE (Supplementary Fig. 14a, b, and d). Select fractions were combined and dialyzed into storage buffer (50 mM sodium phosphate/ 500 mM NaCl pH 7.2). The dialyzed protein was concentrated, quantified, snap frozen with liquid nitrogen, and stored at −80 °C. Quantification was accomplished by SDS-PAGE gel analysis, compared to a bovine serum albumin (BSA) standard curve (Supplementary. Fig. 14c and e, Supplementary Table 5) with ImageJ or Bradford assay.
The growth and expression of the B. thetaiotaomicron SULT protein was carried out in BL21 (DE3) cells. One liter Luria Broth cultures were inoculated with a 1:100 dilution of overnight starter culture under antibiotic selection and grown at 37 °C to an OD600 of 0.6 before induction with 1 mM of IPTG. The induced cultures were grown at 37 °C for an additional 6 hours before harvesting and storage in 2 L aliquots. Pellets were later thawed in a lysis buffer (30 mM HEPES/ 500 mM NaCl/ 30 mM imidazole/ 2 mM DTT, pH 7) and purified in a similar manner to the Cys proteins. The eluted protein fractions were pooled, concentrated, supplemented with recombinant ULP1 protease, and dialyzed overnight into cleavage buffer (30 mM HEPES/ 150 mM NaCl/ 2 mM DTT, pH 7). The dialyzed protein was reloaded on the HisTrap column by syringe and the column washed with lysis buffer. The flowthrough and wash were pooled, concentrated, quantified, and immediately used in assays without freezing or storage (Note: Loss of activity was observed for SULT upon freeze thaw across a variety of conditions and purifications).
Purified enzyme assays
Reactions were coupled to BtSULT conversion due to similar chemical properties and masses of ATP and PAPS (506.996 and 506.986) and adenosine 5’-phosphosulfate (APS), adenosine 5’-diphosphate (ADP), and adenosine 3’,5’-diphosphate (PAP) (427.020, 427.029, and 427.029). All assays were performed using freshly purified BtSULT. In the substrate conversion assays, a concentration of 30 μM of indicated substrate was incubated with 1 μM SULT and 100 μM PAPS (Sigma-Aldrich) in 50 mM sodium phosphate / 150 mM NaCl, pH 7.2 for 0.5 and 4 hours at 37 °C. The coupling assay was performed at 37 °C in a buffer of 50 mM Tris/10 mM Na2SO4/ 5 mM MgCl2, pH 7. For each component (when present), the concentration was held constant at the following: 100 μM PAPS (Sigma), 100 μM APS (Cayman Chemical), 5 mM ATP (Sigma), 5 mM GTP (Sigma), 50 μM cholesterol (Sigma), 0.5 U pyrophosphatase (New England Biosciences), 5 μM CysC, 2.5 μM CysD, 2.5 μM CysN, and 1 μM BtSULT. Time points were taken at 4 hours and overnight. All samples were prepared in triplicate and time points were taken as 250 μL volumes and snap frozen in liquid nitrogen to halt the reaction.
Bacterial lysis assay for SULF activity
B. thetaiotaomicron Δsult was cultured anaerobically in 45 mL BHI+ media at 37°C for 48 hours and then harvested by centrifugation at 4,100 x g for 20 minutes at 4°C. The pellet was then resuspended in lysis buffer (50 mM Potassium Phosphate/50 mM NaCl/2 mM DTT, pH=7). Three volumes of resuspended cells were transferred to Eppendorf tubes for unlysed controls. The remaining cell suspension was lysed via sonication. The raw lysate was separated into aliquots. All aliquots, both unlysed and lysed, were diluted 1:1 with lysis buffer + 80 μM Ch-S ([Ch-S]final=40μM). Reactions were incubated at 37°C under anaerobic atmosphere. Time points were collected at 1h, 4h, and 8h by taking 250 μL aliquots and snap frozen for later extraction and analysis.
Bacterial lysis assay for SULT activity
B. thetaiotaomicron ΔBT0413–5 was cultured anaerobically and lysed in a similar manner as described for the SULF lysis assay. Equal volumes of lysate and whole cell were assessed for SULT activity by incubation with Ch (30 μM) and chemical PAPS (100 μM) at 37°C for 4 hours. The reaction was snap frozen in liquid nitrogen for later extraction and analysis.
UPLC-MS analyses
Sample preparation for bacterial and pure protein samples
Samples were acidified to a pH of 1 with HCl (Sigma) with GCA (Steraloids) as the internal standard and extracted twice with ethyl acetate (Sigma). The organic phase was collected and dried down using a turbovap (Biotage). Dried extracts were resuspended in 75% HPLC-grade methanol in dH2O and analyzed by UPLC-MS (Agilent Technologies 1290 Infinity II UPLC system coupled online to an Agilent Technologies 6120 Quadrupole LC/MS spectrometry in negative electrospray mode) using a published method69,75 with modifications outlined as follows. Extracted molecule solutions were injected onto a Phenomenex 1.7 μm, C18 100 Å, 100 × 21 mm LC column with a flow rate of 0.350 mL/min using 0.05% formic acid in water as mobile phase A and acetone as mobile phase B. The following gradient was applied: 0–1 min: 25–60% B, 1–5 min: 60–70% B, 5–6 min: 70–100% B, 6–7 min: 100% B isocratic, 7–8 min: 100–25% B, 8–10 min: 25% isocratic. Extracted ion chromatograms and areas under the curve were generated using Agilent ChemStation C.01.07 SR3. Exported data were analyzed in Microsoft Excel and GraphPad Prism V9.
Sample preparation for mouse tissue quantification
Compounds were extracted from mouse cecal samples and quantified by UPLC-MS as previously reported69. GCA was used as the internal standard for mouse samples. The limits of detection of individual bile acids in tissues (in picomol/mg wet mass) are as follows: GCA, 0.8; Ch-S, 0.1.
Extraction of Ch and Ch-S from plasma
A volume of 100 uL of plasma was aliquoted and supplemented with the internal standards 25-hydroxy-cholesterol-3-sulfate and d7-cholesterol (10 μM each). The plasma was then diluted with equal volumes of water and ammonium acetate (0.5 M stock solution) before incubating for 1 hour at room temperature. Samples were then extracted twice with ethyl acetate (Sigma). The organic phase was collected and dried down using a turbovap (Biotage) before being resuspended in 100 μL methanol. Prepared samples were then analyzed for Ch-S levels by the previously described UPLC-MS methods for biological samples. Ch quantification was carried out by analysis of the extract via GC-MS (Agilent Technologies 7890B GC System coupled to a 5977B Electron Ionization Mass Spec detector). Briefly, 1 μL of sample was injected into the inlet held at 320°C, 11.65 psi on a split stream at a 5:1 ratio. The sample was run on an Agilent J&W HP-5MS Ultra Inert Column (Model 19091S-433 UI) held at 120°C for 1 minute, ramping to 308°C at 50°C/min, then increasing to 315°C over the following minute. The column was held at 315°C for 4 minutes before returning to 120°C. The detector was set to selected ion monitoring for m/z 386 and 393. Samples were normalized to internal standard and quantified against a standard curve. Data were collected by Agilent GCMS MassHunter B.07.005.2479. Extracted ion chromatograms and areas under the curve were generated using Agilent MassHunter Qualitative Analysis B.007. Exported data were analyzed in Microsoft Excel and GraphPad Prism V9.
Identification of Ch-S from HMP2 cohort.
LC–MS data were acquired using C18-negative mode LC–MS methods as previously54. Peaks of unknown ID were confirmed using an authentic standard run alongside with the quality control reference stool pool generated in the HMP2 study. Ch-S identity was confirmed by matching m/z in negative mode and retention time, and subsequently verified using LC-MS/MS (Supplementary Fig. 15). Concentration was determined by comparison of a reference sample to an authentic standard curve (see UPLC-MS methods). Extracted ion chromatograms were generated using QualBrowser (Xcalibur v.4.1.31.9; Thermo Fisher Scientific). The commercial standard used was cholesterol-3-sulfate, sodium salt (Avanti Polar Lipids, 700016). Ch-S peak in HMP2: QI9100 (Supplementary Fig. 15).
In vitro CD4+ T Cell Culture
Naive CD4+ T cells were purified from the dissociated spleens and lymph nodes of C57BL/6N mice by fluorescence-associated cell sorting (FACS) (CD4+CD62LhiCD44lo 572). Purified cells were cultured under TH0 condition stimulated with anti-CD3e (5 μg/mL, clone 145– 2C11, eBioscience), anti-CD28 (5 μg/mL, clone 37.51, eBioscience), and human IL-2 (100 U/mL, Peprotech). Bile acids were added on day 0. IsoalloLCA dissolved in DMSO was resuspended in culture media and sonicated before being added to the culture. Cells were harvested at 72 hours followed by staining with anti-CD4 (RM4–5, eBioscience) or anti-Foxp3 (FJK-16s, 580 eBioscience) antibodies, and analyzed with LSR II flow cytometer (BD).
Preparation of bacterial supernatant for transwell chemotaxis assays
Bacterially produced Ch-S was obtained by culturing P. merdae ATCC 43184 (WT) and P. merdae Δsult in the presence of 500 μM Ch for 168 hours alongside media controls (n = 3) and P. merdae WT with no additional Ch. A 1.25 mL sample was removed from each culture, acidified to pH 1 with HCl (Sigma), and extracted twice with ethyl acetate (Sigma). The organic phase was collected and evaporated to dryness using a turbovap (Biotage). Dried extracts were resuspended in 100 μL of HPLC-grade methanol (Sigma) and quantified by running 1:10 dilutions on the UPLC-MS (Agilent Technologies 1290 Infinity II UPLC system coupled online to an Agilent Technologies 6120 Quadrupole LC/MS spectrometry in negative electrospray mode) alongside a standard curve according to the above method. Quantified samples were used for immune cell migration assays (see below).
Transwell chemotaxis assay
Murine splenocytes (1 × 107 cells/ml) of spleen were isolated from C57BL/6 mice as follows. Mash the spleen through 70 μm cell strainer into the 60mm dish. Collect suspended cells and spin down at 500xg for 5 min. For RBC removal, incubate the pellet with 1ml ACK lysis buffer (ThermoFisher Scientific, Cat. A1049201) for 1–2min. After washing with PBS, Isolated splenocytes were pre-incubated at 37 °C for 1 h in ex-vivo20 medium (Lonza, Cat. 04–448Q), with the indicated concentration of Ch and Ch-S or with bacterial cultured supernatants (1 × 106 cells per condition). After pre-incubation, treated cells (1 × 106) were plated onto the upper chamber of Transwells (Corning, Cat. 3421). CCL21 (200ng/ml, R&D Systems, Cat. 457–6C-025/CF) was added to the bottom chamber. After incubation at 37 °C for 6 h, cells migrated to the bottom chamber were collected and stained with the following antibodies; CD45-APC/Cy7 (1:300, BD pharmingen), CD90.2-PE/Cy7 (1:300, Biolegend), CD4-APC (1:300, Biolegend) and CD8-eFluor450 (1:300, ebioscience). The percentage of the migrated leukocytes was calculated by dividing the number of cells in the bottom chamber by the number of total immune cells added to the upper chamber and then multiplying the result by 100. To determine cell viability, cells were also stained with Live/Dead fixable dye Aqua (ThermoFisher). All flow cytometry analyses were performed on an LSR II flow cytometer (BD) with BD FACS DIVA software (BD) and data were analyzed with FlowJo software (TreeStar). Gating strategy and representative data are shown as Supplementary Figures 16a and 17a,b
Lymph node homing assay
CD4 T cells were enriched using the naïve CD4 T cell isolation kit (Milteni Biotec, Cat. 130–104-453) from mononuclear cells isolated from the spleens and lymph nodes of congenically marked, wild-type (CD45.1) and CCR7-deficient (CD45.2) mice. A mixture of CD45.1-naïve CD4 T cells (3×106) and CD45.2-naïve CD4 T cells (1.5×106) were labeled with carboxyfluorescein succinimidyl ester (CFSE) and transferred into GF, B. thetaiotaomicron WT-monocolonized, or BtSULT Δsult-monocolonized B6 mice by retro-orbital injection. After 24h, recipient mice were sacrificed, and immune cells were isolated from the spleens and MLNs and stained with the following antibodies: CD45-Pacific blue (1:300, Biolegned), CD4-BV605 (1:300, Biolegend), CD25-PE (1:200, ebioscience), CD44-APC (1:300, ebiosience), CD62L-PE/Cy7 (1:300, Biolegend), CD45.1-PerCP/Cy5.5 (1:200, ebioscience) and CD45.2-APC/Fire750 (1:200, Biolegend). The ratio of donor CD4 T cells in either MLNs or spleens was first determined by dividing the number of CFSE+ CD4+ T cells (donor cells) by that of CD45+ cells (recipient cells) in each tissue. We then determine the relative migration of donor cells into the MLNs over the spleens of recipient mice by calculating the normalized ratio. Gating strategy and representative data are shown as Supplementary Figures 16b and 17c.
Bioinformatic analyses
BLASTP searches for cys genes in B. thetaiotaomicron
BLASTP searches were performed against the B. thetaiotaomicron reference genome (Bacteroides thetaiotaomicron VPI-5482) in the Integrated Microbial Genomes, the US Department of Energy’s Joint Genome Institute (IMG JGI)30,31 (May 4, 2021) using the amino acid sequences for E. coli K12 CysC (NP_417230.1), CysD (NP_417232.1), and CysN (NP_417231.1) as query sequences (cutoff E-value of 10−5 for all BLASTP searches).
BLASTP searches for dissimilatory sulfur metabolism genes
Protein query sequences of sulfur adenylyltransferase (Sat), APS reductase (Apr), and dissimilatory sulfite reductase (Dsr) from a known sulfate reducer Desulfovibrio desulfuricans DSM 642 (G449DRAFT_1613) were aligned against the Bacteroides thetaiotaomicron reference genome (Bacteroides thetaiotaomicron VPI-5482) in the IMG JGI30,31 database(March 6, 2022) using BLASTP with a cutoff E-value of 10-5. No homologs were found in the B. thetaiotaomicron genome. Results are shown in Supplementary Table 2a.
BLASTP searches for cys and btSult homologs in sulfonating bacterial strains
Protein query sequences of BT0413 (CysC), BT0414 (CysD), BT0415 (CysN), and BT0416 (BtSult) were aligned against the bacteria selected for activity screening (Extended Data Figure 3b) in the NCBI non-redundant protein database37,38 and IMG JGI30,31 database using BLASTP with a cutoff E-value of 10−5 (March 6, 2022). Top hit homologs for each bacterium were recorded with both E-value and percent identity (Supplementary Table 2b) along with the gene locus tag (Supplementary Table 2c).
BLASTP searches for sulfolipid transporters in B. thetaiotaomicron
Protein query sequences of known membrane transporters MmpL8, MmpL1, MmpL2, and Sap from Mycobacterium were aligned against the Bacteroides thetaiotaomicron reference genome (Bacteroides thetaiotaomicron VPI-5482) in the IMG JGI30,31 database with a cutoff E-value of 10-5. No homologs were identified in the B. thetaiotaomicron genome in any search (December 14, 2021; March 11, 2022).
Bioinformatic identification of putative sulfotransferases in human gut bacterial species
The distribution of the sulfotransferase (SULT) was examined by using the BT0416 sequence as the query for a BLASTP search in the NCBI non-redundant protein database37,38 (August 22, 2021) using 60% identification as the cut-off (Supplementary Table 3).
Biosynthetic gene cluster profiling in human microbiome metagenomic datasets
We identified the homologs of CysC (BT0413), CysD (BT0414), CysN (BT0415) and SULT (BT0416) by detecting their UniRef90 annotations (i.e. protein sequences with >90% amino acid identity and >80% coverage) in Uniref76 (release 2019_01). We queried these UniRef90 gene families to curatedMetagenomicData v3.0.140, and extracted the quantitative gene families which have been uniformly processed with HUMAnN377. Then, we assessed the prevalence of the homologs of each cys gene (BT0413-BT0416) among the stool samples for each study in curatedMetagenomicData.
Statistical analysis of biosynthetic genes in human IBD cohorts
Microbial Data Overview
We used a publicly available IBD metagenomics dataset for determining the differential abundance (DA) of the biosynthetic genes in disease/dysbiotic conditions, specifically the IBDMDB study within the integrated Human Microbiome Project (HMP2 or iHMP)54. HMP2 is a longitudinal cohort containing 132 participants with CD (n = 67), UC (n = 38), and non-IBD controls (n = 27) followed for up to one year. We downloaded the HMP2 metagenomic sequencing reads and metabolomic profiles from the Inflammatory Bowel Disease Multi’omics Databases (http://ibdmdb.org) as of July 2020. These reads included 1,298 metagenomes samples and 546 MBX samples from 106 subjects (CD, n = 50; UC, n = 30; and non-IBD, n = 26). The metagenomic sequencing reads had been previously quality controlled by the KneadData workflow. Microbiome-associated IBD activity was previously defined in this cohort by comparing gut microbial composition of IBD patients to non-IBD controls54.
Identifying Differentially Abundant Microbial Genes
We used ShortBRED78 to accurately profile the abundance of genes involved in the sulfonation biosynthetic pathway in metagenomes from the HMP254 dataset. In order to construct ShortBRED markers for these gene families, we first gathered homologs of CysC, CysD, CysN, SULT by performing BLASTP79 searches against the NCBI nr protein database80 using APS kinase (BT0413), ATP sulfurylase (BT0414, BT0415), and sulfotransferase (BT0416) sequences from B. thetaiotaomicron VPI 5432 as queries. Database sequences were selected for marker construction if they exhibited >80% query coverage, >60% alignment identity. These sequences were combined with UniRef90 (June 2020 version) as a comprehensive background for marker identification using ShortBRED-identify under default settings. We then applied the resulting marker sequences to profile the abundances of CysC, CysD, CysN, SULT from HMP2 metagenomes using ShortBRED-quantify (under default settings). Abundances are reported in RPKM units (read mapped per kilobase of coding sequence per million sample reads).
We tested the abundances of CysC, CysD, CysN, and SULT homologs for differential abundance over dysbiosis state within diagnosis and dysbiosis states (diagnosis:dysbiosis). The normalized gene abundance was log-transformed with zero values smoothed by half of the smallest non-zero measurement on a per-feature basis. We used a per-feature mixed-effects linear model within the MaAsLin 2 package (http://huttenhower.sph.harvard.edu/maaslin) as below:
That is, the transformed abundance of each gene was modelled as a function of diagnosis (a categorical variable with non-IBD as the reference state) and dysbiosis state as a nested binary variable (per-diagnosis non-dysbiotic samples serving as the reference state), adjusting for consent age as a continuous covariate, antibiotics as a binary covariate. A per-subject random effect was used to control for repeated measures. Nominal P values were adjusted for multiple hypothesis testing using the Benjamini-Hochberg method.
Identifying differentially abundant metabolites
HMP2 subjects were compared between “active” (dysbiotic) and “inactive” (non-dysbiotic) states within individual time courses as described previously54. Prior to statistical model fitting, gut metabolome profiles of HMP2 subjects were 1) median-normalized to reduce technical sample-to-sample variation; 2) prevalence-filtered to remove low-confidence features (requiring > 30% non-zero values); and 3) log-transformed for variance-stabilization (replacing zero values with half the smallest non-zero measurement on a per-feature basis).
Differential abundance (DA)
status over disease activity (dysbiosis) was determined within the longitudinally sampled HMP2 cohort by evaluating the following linear mixed-effects model for each metabolite using R’s nlme package:
Diagnosis was coded as a categorical variable (CD, UC, non-IBD control) with non-IBD control as the reference state.
Dysbiosis within diagnosis (diagnosis:dysbiosis) was used to determine DA status, with per-diagnosis non-dysbiotic samples serving as the reference state. Age at study consent was included as a continuous covariate and per-sample antibiotics exposure as a binary covariate. A per-subject random effect was included to compensate for repeated sampling and to “absorb” potential confounders that were invariant over subjects (e.g., recruitment site).
Model coefficients of the diagnosis:dysbiosis (HMP2) terms was interpreted as DA effect sizes, and their associated two-tailed p-values were used to determine statistical significance (Wald’s test). Where applicable, simultaneously derived p-values were adjusted for multiple hypothesis testing using the Benjamini-Hochberg FDR method (Supp. Table 7; Extended Data Fig. 10 b,c).
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and the Supplementary Information. The HMP2 IBD 16S and RNA-seq data are available through NCBI under BioProject ID PRJNA675599 and GEO accession number GSE179740, respectively, and the metabolomics workbench study ST000923. The curatedMetagenomicData are available online (https://waldronlab.io/curatedMetagenomicData/, with individual accession IDs listed in Supplementary Table 9). Comparison of Ch-S standard as part of the HMP2 analysis is published in supplemental data (Supplementary Figure 15).
Code availability
The software packages used in this study are free and open source. MaAsLin2 is available via http://huttenhower.sph.harvard.edu/maaslin as source code and installable packages. The R package limma is available online (https://www.bioconductor.org/packages/release/bioc/html/limma.html). The curatedMetagenomicData40 package is available online (https://waldronlab.io/curatedMetagenomicData/). Analysis scripts employing these packages are available from the authors upon request.
Statistics and Reproducibility
No statistics were used to determine sample sizes. Mouse sample sizes were determined by available mice controlling for sex (male) and age (5–10) weeks. No animals were excluded from any experimental data set. Individual cohort sizes are listed with figures. All animal data is the result of pooled samples from two or more independent experiments (noted in figure legends). Human donors Fc and Fe were male, between the ages of 30 and 35, and had healthy body mass indices. No other information is known. The population for HMP2 samples were recorded in initial publication54. Mice in each experiment were randomly separated into cohorts, with the exception of CCR7-KO strain mice for lymph node homing assay. Data collection and analysis were not performed blinded to experimental conditions. Data distribution was assumed to be normal but this was not formally tested.
Extended Data
Extended Data Fig. 1 |. Overview of the assimilatory and dissimilatory sulfur metabolism pathways in bacteria.

Depiction of the assimilatory (blue) and dissimilatory (red) pathways for sulfur metabolism in bacteria showing the key molecules and enzymes involved. While the assimilatory pathway activates sulfur for subsequent incorporation in amino acids, cofactors, and other metabolites, the dissimilatory pathway utilizes sulfur as an electron sink in order to turn over cofactors, rid the cell of excess electrons, and produce hydrogen sulfide. All enzymes are listed by their common name.26,28,29,55
Extended Data Fig. 2 |. Generation of mutant strains, growth, and gene profiles in B. theta and P. merdae strains.

a, PCR verification of counter-selected deletion and complemented clones of the BT0416 gene in B. theta VPI-5482 Δtdk (WT). Lane 1: B. theta Δtdk (WT), lane 2: B. theta ΔtdkΔsult_sult+ (complemented strain), lane 3: B. theta ΔtdkΔsult (KO), and lane 4: NEB 1 kb DNA ladder. PCR primers, BT0416_F/ BT0416_R. Data presented represent individual PCR reactions.
b, Growth profiles of B. theta cultured in BHI+ media displayed no difference between B. theta WT, KO, and Δsult_sult+. Culture turbidity was measured as optical density at 600 nm (OD600). Values are mean ± SEM, n=3 biological replicates.
c, Growth profiles of B. theta VPI-5482 Δtdk and VPI-5482 Δsult show no difference when incubated with either Ch (30 μM) or Ch-S (30 μM) in BHI+ media. Culture turbidity was measured as optical density at 600 nm (OD600). Values are mean ± SEM, n=3 biological replicates.
d, PCR of counter-selected colonies for the deletion of BT0412 in B. theta VPI-5482 Δtdk. Lane L: NEB 1kb DNA ladder, Lanes 1–4,5: negative cultures, Lane 4: B. theta ΔBT0412 (KO). Lane 6: Wild type culture amplification. PCR primers: BT0412_UF2, BT0412_DR2. Data presented represent individual PCR reactions.
e, Growth curves plotting OD600 of B. theta WT, ΔBT0412, and ΔBT0413–5 cultures in BHI+ media with 30 μM Ch, data shown are mean ± SEM, n = 3 biological replicates.
f-h, OD600 measures of WT, ΔBT0412, and ΔBT0413–5 cultures at i) 4h, j), 8h, and k) 24h, showing significant defects in the growth of both knockout strains. Data are shown as mean ± SEM with one-way ANOVA multiple comparisons to WT with Dunnett’s correction, n = 3 biological replicates.
I, Representative extracted ion chromatogram (EIC) UPLC-MS traces (left) and quantified production (right) show that B. theta and B. theta ΔBT0412 produces Ch-S when incubated with Ch (100 μM) over 72 hours while B. theta ΔBT0413–5 does not produce Ch-S under the same conditions. Data are presented as mean ± SEM, n=3 biological replicates.
j, PCR of counter-selected colonies for the deletion of BT0413–5 in B. theta VPI-5482 Δtdk. Lane L: NEB 1kb DNA ladder, Lanes 1: Wild type culture amplification, Lanes 2–12,14,16: negative cultures, Lanes 13,15: B. theta ΔBT0413–5 (KO). PCR primers: BT0413–5_UF2, BT0413–5_DR2. Data presented represent individual PCR reactions.
k, B. theta ΔBT0413–5 culture lysate produced Ch-S when chemically complemented with PAPS (100 μM). No Ch-S production was detected in the absence of PAPS. Data are shown as mean ± SEM, n = 3 replicates.
l, End-point overlapping PCR of genes in BtSULT cluster with gDNA (lanes 2, 4, 6, 8, 10, and 12) and paired cDNA (lanes 3, 5, 7, 9, 11, and 13) derived from extracted mRNA. Representative samples (n=3) run on 0.8% agarose gel with NEB 1kb ladder (L).
m, As measured by RT-qPCR, the expression of BT0416 in B. thetaiotaomicron was not affected by the addition of cholesterol at 1, 6, and 8 hours post Ch addition (n = 3 biological replicates per group, data are normalized to 16S rRNA and shown as mean ± SEM with Welch’s t-test with 2-tailed p-value).
n-o, Cultures of B. theta in BHI+ media with Ch (30 μM) were harvested at 48 and 168 hours and Ch-S levels were assessed in whole culture, media supernatant, and washed pellet. The data are reported as percentage of Ch-S in pellet and supernatant compared to paired whole cell culture. n = 3 biological replicates, mean ± SEM with Welch’s t-test with two-tailed correction.
p, PCR verification of counter-selected clones for deletion of the PARMER_01922 gene in P. merdae ATCC 43184 (WT). Lane 1: P. merdae (WT), lane 2: P. merdae Δsult (KO), and lane 4: NEB 1 kb DNA ladder. PCR primers, PARMER01922_F/ PARMER01922_R.
q, Growth profiles of P. merdae cultured in BHI+ media displayed no difference between P. merdae WT and Δsult. Culture turbidity was measured as optical density at 600 nm (OD600). Values are mean ± SEM, n=3 biological replicates. Data presented represent individual PCR reactions.
Extended Data Fig. 3 |. Metagenomic prevalence of BT0413-BT0416.
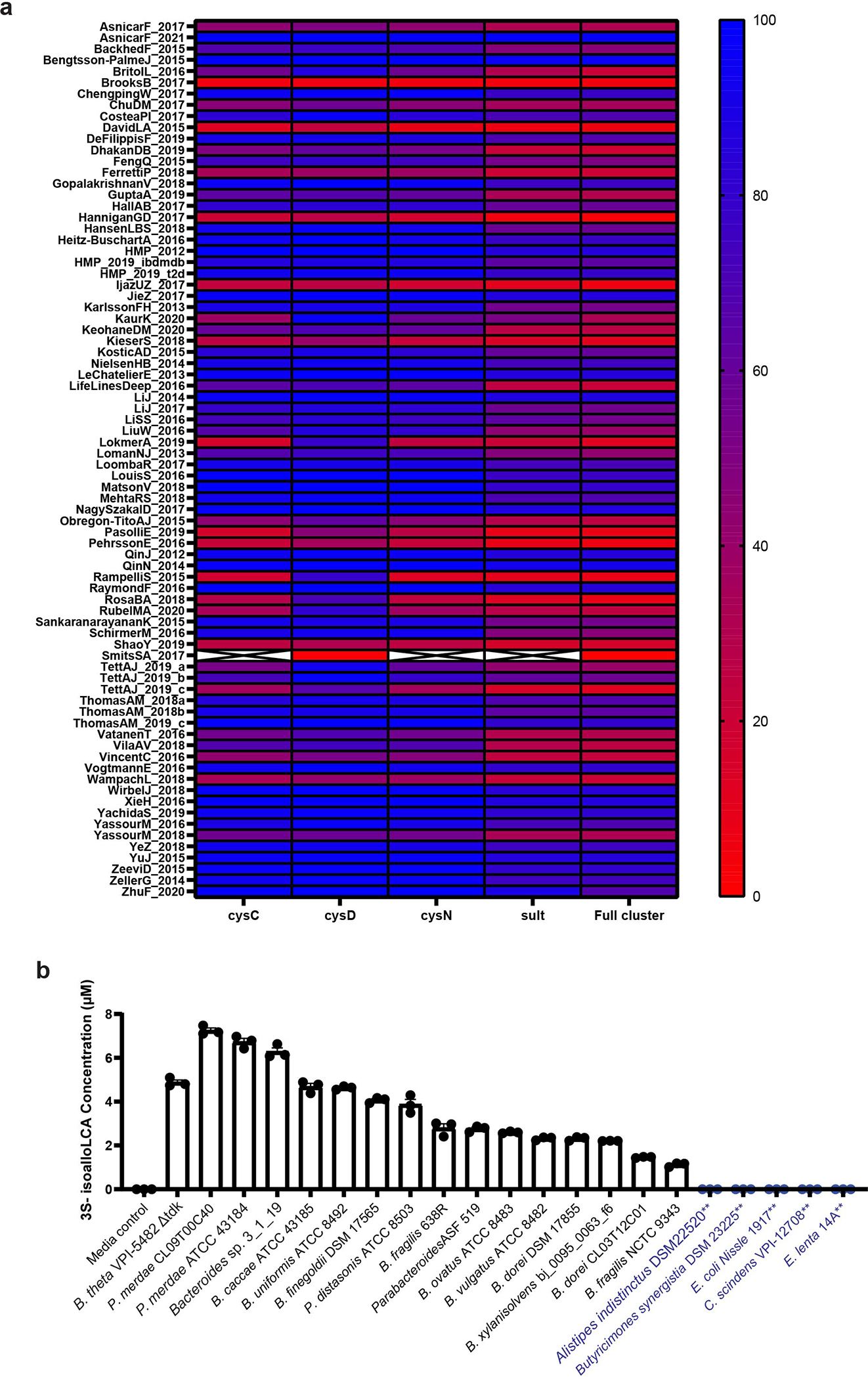
a, Metagenomic prevalence of genes across the collections available in the curatedMetagenomicData program. Each row of the heat map represents a different collection, with each column representing an individual gene. The full cluster annotation is the percent of samples in each collection that possess all 4 genes (BT0413-BT0416).
b, IsoalloLCA-3-sulfate was produced by species from the Bacteroidetes phylum containing a homolog for BtSULT (black text) and was not produced by Bacteroides species (Alistipes indistinctus DSM 22520, Butyricimonas synergistica DSM 23225) and other bacteria (E. coli Nissle 1917, C. scindens VPI 12708, and E. lenta 14A) that lack a BT0416 homolog (blue text) (n=3 biological replicates per group, data are mean ± SEM).
Extended Data Fig. 4 |. Sulfonation of isoalloLCA alters its biochemical properties.
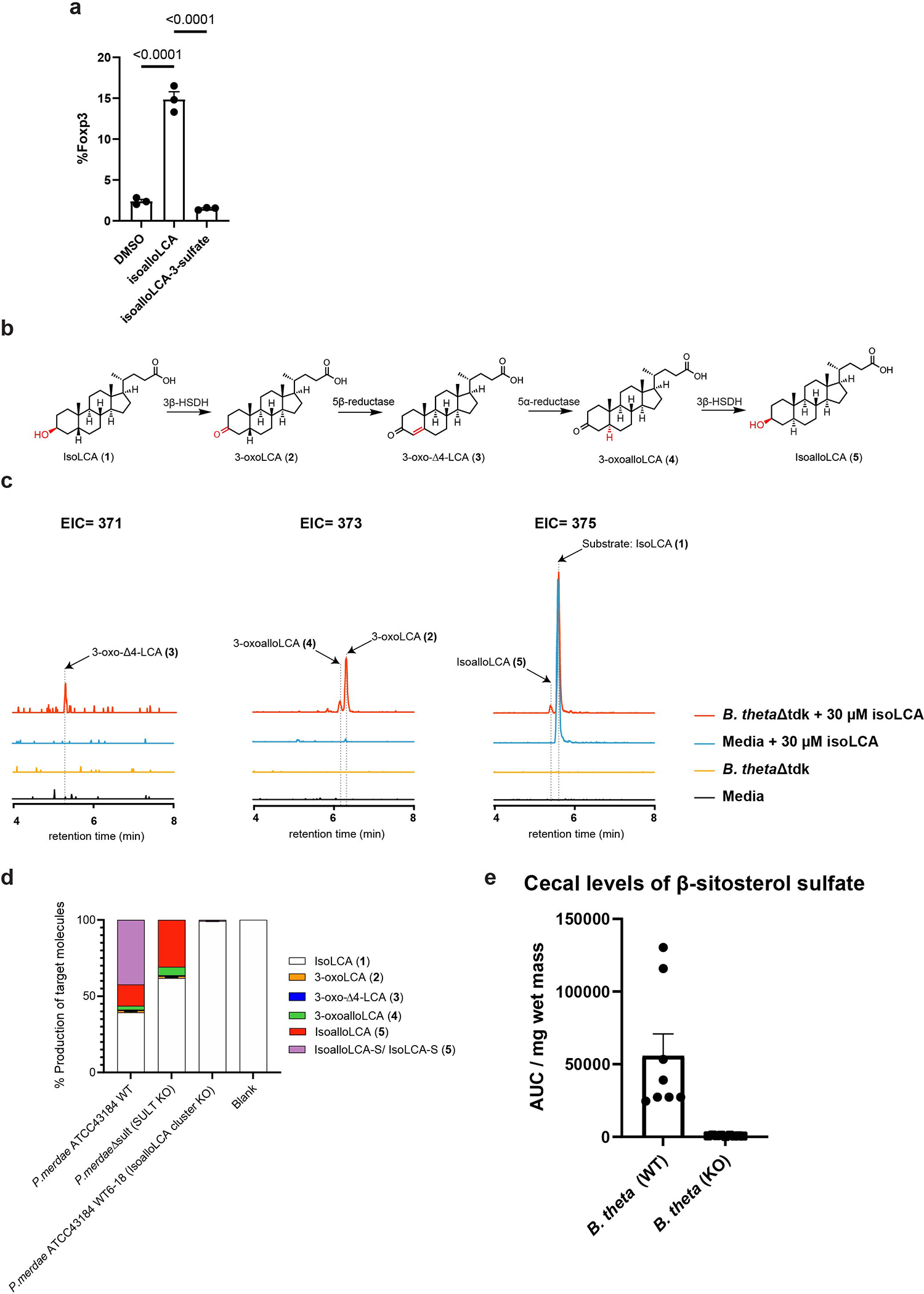
a, Flow cytometry analysis and quantification of native CD4+ T-cells cultured under TH0 conditions (anit-CD3, anti-CD28, and IL-2) conditions in the presence of DMSO, isoalloLCA, or isoalloLCA 3-sulfate for 72 hours are shown. Cells were stained with FOXP3 as a marker for Treg cells (n = 3 biological replicates per group, data are shown as the mean ± SEM, one-way ANOVA followed by Tukey’s multiple comparison test).
b, Previously elucidated biosynthetic pathway in Bacteroidetes species for the conversion of isoLCA (1) to isoalloLCA (5). The site of biotransformation is highlighted in red for each step.
c, Extracted ion chromatograms (EICs) showing that B. theta Δtdk (WT) cultures incubated with isoLCA produced isoalloLCA (5) as well as the intermediates 3-oxoLCA (2), 3-oxo-Δ4-LCA (3), and 3-oxoalloLCA (4), supporting the hypothesis that subsequent actions of a 3β-hydroxysteroid dehydrogenase (HSDH), 5β-reductase, a 5α-reductase, and a 3β-HSDH are responsible for the conversion of isoLCA to isoalloLCA by B. theta.
d, The composition of bile acid metabolites was determined for cultures of P. merdae ATCC 43184, P. merdae Δsult, and P. merdae ΔPARMER_04016–18 incubated for 48 hours with isoLCA (100 μM). We observed a complete loss of the sulfonated compound in P. merdae Δsult culture, while the P. merdae ΔPARMER_04016–18 culture exhibited a loss of isoLCA-derived intermediates and a marked decrease in production of sulfonated compound compared to the WT culture. The composition of LCA derived molecules was determined by UPLC-MS (n = 3 biological replicates).
e, β-sitosterol-3-sulfate levels in the cecal contents of mice monocolonized with B. theta WT were elevated when compared to levels in B. theta KO colonized mice (B. theta WT n = 8, B. theta KO n = 9, results pooled from three separate experiments, data are presented as mean ± SEM).
Extended Data Fig. 5 |. Comparison of cholesterol metabolite and gene expression levels between GF and SPF mice.

a-c, Cholesterol sulfate levels were quantified in the a) ceca, b) feces, and c) plasma of GF (n=5) and SPF (n=5) and found to be higher in GF mice across these tissues. Data are shown as mean ± SEM with Welch’s two-tailed t-test.
d-f, Cholesterol levels were quantified in the d) ceca, e) feces, and f) plasma of the same mice by GCMS and found to not be significantly different. Data are shown as mean ± SEM with Welch’s two-tailed t-test.
g-i, Expression of host Sult2B1b was measured by quantitative PCR in the g) liver, h) ileum, and i) colon of GF mice (n=5) compared to SPF mice (n=5) and found to have significant differences in both the liver and colon, data are normalized to L32 and shown as mean ± SEM with Welch’s two-tailed t-test.
j-l, Expression of host steroid sulfatase (sts) was measured by quantitative PCR in the j) liver, k) ileum, and l) colon of GF mice (n=5) compared to SPF mice (n=5) and found to be significantly different in the ileum, data are normalized to L32 and shown as mean ± SEM with Welch’s two-tailed t-test..
Extended Data Fig. 6 |. Cholesterol availability in vitro and in vivo.

a, B. thetaiotaomicron WT cultured in different types of media (BHI+, defined media, and “Enhanced” defined media) with and without lecithin (10 mg/L) exhibited lower levels of Ch-S production compared to BHI+ media. Data are shown as mean ± SEM with one-way ANOVA with Dunnett’s correction, comparing to BHI+ production, n = 3 biological replicates.
b, The OD600 of cultures from a) was taken after 48 hour of incubation. No significant differences in growth levels were observed between pairs of cultures. Data are shown as mean ± SEM with Welch’s two tailed t-test comparing between pairs of cultures, n = 3 biological replicates.
c, Levels of Ch quantified by GCMS in cecal contents of germ-free (n=5) and SPF (n=5) mice. Data are presented as mean ± SEM with Welch’s two-tailed t-test.
d, Levels of Ch quantified by GCMS in fecal pellets of germ-free (n=5) and SPF (n=5) mice. Data are presented as mean ± SEM with Welch’s two-tailed t-test.
e, Concentration of Ch found in human feces (n = 2 biological samples) in technical triplicate. Data are shown as mean ± SEM.
Extended Data Fig. 7 |. Sulfonation versus desulfation by gut bacteria.

a, Ch-S levels were reduced when this metabolite was incubated with B. thetaiotaomicron lysate but not when incubated with whole cells. Data are shown as mean ± SEM with Welch’s t-test with two tails between each time point, n = 3 biological replicates.
b, Proposed model of Ch-S production and release by B. thetaiotaomicron over cell lifecycle. During lag and exponential phase, Ch-S is produced within the cell and exists in equilibrium with Ch. This equilibrium is controlled by both BtSULT and bacterial SULF, with BtSULT favoring Ch-S production. During late stationary phase, cell death and lysis leads to the release of ChS into extracellular environment.
c,d, Slurries of fecal samples from two healthy donors,I) Fe and (d) Fc, were independently cultured for 7 days following addition of Ch and Ch-S (15 μM each) to test the relative activity of SULT vs. SULF in an ex vivo microbiome. Levels of Ch-S were significantly increased in both cultures after 7 days. Data are shown as mean ± SEM with one-way ANOVA followed by Tukey’s multiple comparisons test, n = 3 replicate cultures per donor.
Extended Data Fig. 8|. BtSULT suppressed T cell migration in vitro and in vivo.
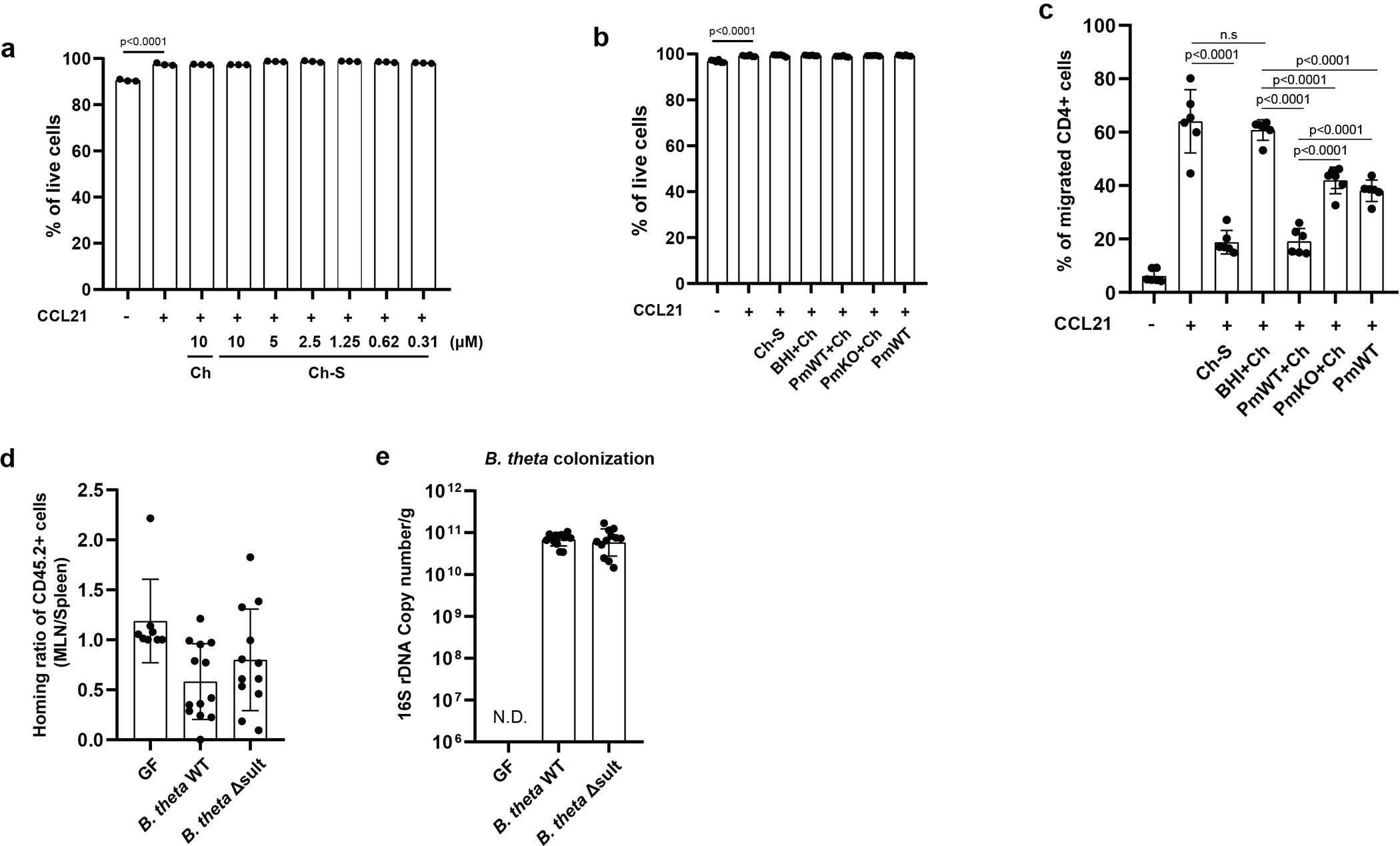
a, Ch-S treatment did not affect leukocyte viability (data correspond to Fig. 4d). Live cell percentage was measured by live/dead dye staining (right) (n=3 biological replicates per condition, data are presented as mean ± S.D, one-way ANOVA followed by Tukey’s multiple comparison test).
b, Bacterial supernatant treatment did not affect leukocyte viability (data correspond to Fig. 4e). Cells were incubated with the following supernatants: BHI+Ch (BHI media + Ch), PmWT+Ch (P. merdae wild-type, cultured with Ch), PmKO+Ch (P. merdae KO cultured with Ch), and PmWT (P. merdae wild-type without Ch) (n=6 biological replicates per condition, data pooled from two independent experiments, data are presented as mean ± S.D., one-way ANOVA followed by Tukey’s multiple comparison test n.s = not significant).
c, Ch-S produced by P. merdae suppressed T cell migration following treatment with CCL21 (200ng/mL). The migrated T cells were identified using CD45, CD90.2 and CD4 antibodies (n=6 biological replicates per condition, data pooled from two independent experiments, data are presented as mean ± S.D., one-way ANOVA followed by Tukey’s multiple comparison test, n.s = not significant).
d, The migration of CCR7-deficient CD45.2+ T cells to MLNs is comparable regardless of recipient mice (data correspond to Fig. 4g). Cells isolated from spleen and MLNs of recipient mice post-24 hours. Results are presented as the normalized ratio of the migrated cells to MLNs divided by the migrated cells to spleens (GF, n=8; B. thetaiotaomicron WT, n=13; B. thetaiotaomicron sult KO, n=12; data pooled from three independent experiments, data are presented as mean ± S.D., one-way ANOVA followed by Tukey’s multiple comparison test).
e, Comparable colonization of B. thetaiotaomicron strains. qPCR analyses for detecting the B. thetaiotaomicron 16S rRNA gene in fecal samples (GF, n=8; B. thetaiotaomicron WT, n=13; B. thetaiotaomicron Δsult, n=12; data pooled from three independent experiments, N.D=not detected).
Extended Data Fig. 9 |. Ch-S levels in mouse intestinal contents and blood.

a, Ch-S levels in cecal contents of mice colonized with B. thetaiotaomicron WT were significantly higher than those in GF mice or B. thetaiotaomicron KO colonized mice (GF, n=6; B. theta WT, n=6; B. theta KO, n=6), data are presented as mean ± SEM, one-way ANOVA followed by Tukey’s multiple comparisons.
b, Ch-S levels in fecal contents of mice colonized with B. thetaiotaomicron WT were significantly higher than those in GF mice or B. thetaiotaomicron KO colonized mice (GF, n=6; B. theta WT, n=6; B. theta KO, n=6, data are presented as mean ± SEM, one-way ANOVA followed by Tukey’s multiple comparisons).
c, Ch-S levels in the plasma of mice colonized with B. thetaiotaomicron WT were not significantly higher than those in GF mice or B. thetaiotaomicron KO colonized mice (GF, n=6; B. theta WT, n=6; B. theta KO, n=6, data are presented as mean ± SEM, one-way ANOVA followed by Tukey’s multiple comparisons).
d, No significant differences in the expression of the host gene sult2b1b were observed in the liver of B. theta WT or Δsult-colonized mice compared to GF mice as measured by quantitative PCR (n=6 mice per group), data are normalized to L32 and shown as mean ± SEM with one-way ANOVA followed by Tukey’s multiple comparisons, (n.s. = not significant).
e, Expression of host gene sult2b1b was found to be significantly upregulated in the ileum of GF mice compared to mice colonized with B. theta WT or Δsult as measured by quantitative PCR (n=6 mice per group), data are normalized to L32 and shown as mean ± SEM with one-way ANOVA followed by Tukey’s multiple comparisons.
f, No significant differences in the expression of the host gene sult2b1b were observed in the colon of B. theta WT or Δsult-colonized mice compared to GF mice as measured by quantitative PCR (n=6 mice per group), data are normalized to L32 and shown as mean ± SEM with one-way ANOVA followed by Tukey’s multiple comparisons, (n.s. = not significant).
g-i, No significant differences in the expression of the host gene steryl sulfatase (sts) were observed in the g) liver, h) ileum, or i) colon of B. theta WT or Δsult-colonized mice compared to GF mice as measured by quantitative PCR (n=6 mice per group), data are normalized to L32 and shown as mean ± SEM with one-way ANOVA followed by Tukey’s multiple comparisons, (n.s. = not significant).
Extended Data Fig. 10 |. BT0413-BT0416 homologs show significant differential abundance after adjusting for variation in underlying taxonomic abundance.

a, Accounting for underlying variation in the taxonomic abundance of SULT possessing bacteria (Bacteroidetes), with phyla abundances as additional covariates to normalize the abundance of genes. BT0413-BT0416 homologues were profiled from HMP2 metagenomes (n = 1,595 samples from 130 subjects; linear mixed-effects model coefficient for dysbiosis within diagnosis, FDR-adjusted p-values < 0.05). The percentage of zeros in each condition are added as x-axis tick labels. Boxplot ‘boxes’ indicate the first, second (median), and third quartiles of the data. Whiskers indicate the inner fences of the data. Statistical analysis was performed using a linear mixed-effect model and its coefficient and significance, FDR-adjusted p-values, are shown.
b,c, Ch-S was not significantly depleted in patients with CD or UC either when patients were separated into dysbiotic and non-dysbiotic states (b) or patients were not separated based on dysbiotic state (c) relative to non-IBD control samples in the HMP2 cohort (n = 47, n = 169, n = 12, n = 110 and n = 122 for individuals with dysbiotic CD, with non-dysbiotic CD, with dysbiotic UC, with non-dysbiotic UC and without IBD, respectively). The percentage of zeros is shown on the x axis. For the box plots, the center line indicates the median (second quartile) and the box limits indicate first and third quartiles of the data. The points outside of box plot whiskers are outliers. Statistical analysis was performed using a linear mixed model and its coefficient and significance (FDR-adjusted P values) are shown (Supplementary Table 7).
d, A significant, positive relationship was observed between the abundance of BtSULT homologs and Ch-S concentrations in fecal samples from the HMP2 cohort based on linear regression analysis (n=400 participants). Regression fit is shown as a solid line (red) with dashed lines (red) indicating the confidence intervals at 99%. Statistics are shown in Supplementary Table 8.
Supplementary Material
Acknowledgments
We thank members of the Devlin, Huh, and Clardy labs (Harvard Medical School-HMS) for helpful discussions. We thank the HMS ICCB-Longwood Screening Facility at Harvard Medical School for their expertise and instrument support. GF versus B. thetaiotaomicron-monocolonized mouse experiments in Figure 1 were performed with generous support from the Massachusetts Host-Microbiome Center, supported by grant P30DK034854. We thank Laurie E. Comstock (HMS, University of Chicago) for the pLGB13 plasmid (Addgene plasmid #126618) and Leonor García-Bayona (University of Chicago) for her technical support. We thank Steve Blacklow (HMS) for the use of the pTD68 plasmid and accompanying ULP1 protease. We are grateful to Tom Rapoport (HMS) for the use of the AKTA explorer FPLC system (Amersham Biosciences). We thank E. Alm and T. Nguyen (Center for Microbiome Informatics and Therapeutics, Massachusetts Institute of Technology) for providing human feces. We are grateful to the human patients who participated in the HMP2 studies. This work was supported by National Institutes of Health grants MIRA R35 GM128618 (A.S.D.), R01 DK110559 (J.R.H. and A.S.D.), U54DE023798 (C.H.), and R24DK110499 (C.H.), and a Harvard Medical School Dean’s Innovation Grant in the Basic and Social Sciences (A.S.D. and J.R.H.), a John and Virginia Kaneb Fellowship (A.S.D.), a Harvard Medical School Christopher Walsh Fellowship (L.Y.) and a Wellington Postdoctoral Fellowship (L.Y.). The computations in this paper were run in part on the FASRC Cannon cluster supported by the FAS Division of Science Research Computing Group at Harvard University. Figure panels in Fig 1b, 2a, 4a, 5c, Extended Data Fig. 1 and Extended Data Fig. 7b were created using BioRender.
Footnotes
Competing interests
A.S.D. is a consultant for Takeda Pharmaceuticals and Axial Therapeutics. J.R.H. is a consultant for CJ Research Center, LLC. C.H. is on the scientific advisory boards of Seres Therapeutics, Empress Therapeutics, and ZOE Nutrition. The remaining authors declare no competing interests.
References
- 1.Liou CS et al. A Metabolic Pathway for Activation of Dietary Glucosinolates by a Human Gut Symbiont. Cell 180, 717–728.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ervin SM et al. Structural Insights into Endobiotic Reactivation by Human Gut Microbiome-Encoded Sulfatases. Biochemistry-us 59, 3939–3950 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Koppel N, Rekdal VM & Balskus EP Chemical transformation of xenobiotics by the human gut microbiota. Science 356, 2770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jancova P, Anzenbacher P & Anzenbacherova E PHASE II DRUG METABOLIZING ENZYMES. Biomed Pap 154, 103–116 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Lindsay J, Wang LL, Li Y & Zhou SF Structure, function and polymorphism of human cytosolic sulfotransferases. Curr Drug Metab 9, 99–105 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Javitt NB, Lee YC, Shimizu C, Fuda H & Strott CA Cholesterol and Hydroxycholesterol Sulfotransferases: Identification, Distinction from Dehydroepiandrosterone Sulfotransferase, and Differential Tissue Expression. Endocrinology 142, 2978–2984 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Strott CA Sulfonation and Molecular Action. Endocr Rev 23, 703–732 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Mougous JD, Green RE, Williams SJ, Brenner SE & Bertozzi CR Sulfotransferases and Sulfatases in Mycobacteria. Chem Biol 9, 767–776 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Malojčić G et al. A structural and biochemical basis for PAPS-independent sulfuryl transfer by aryl sulfotransferase from uropathogenic Escherichia coli. Proc National Acad Sci 105, 19217–19222 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maeda K, Okuda Y, Enomoto G, Watanabe S & Ikeuchi M Biosynthesis of a sulfated exopolysaccharide, synechan, and bloom formation in the model cyanobacterium Synechocystis sp. strain PCC 6803. Elife 10, e66538 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehrhardt DW et al. In vitro sulfotransferase activity of NodH, a nodulation protein of Rhizobium meliloti required for host-specific nodulation. J Bacteriol 177, 6237–6245 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gu L et al. Polyketide Decarboxylative Chain Termination Preceded by O-Sulfonation in Curacin A Biosynthesis. J Am Chem Soc 131, 16033–16035 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malojčić G et al. A structural and biochemical basis for PAPS-independent sulfuryl transfer by aryl sulfotransferase from uropathogenic Escherichia coli. Proc National Acad Sci 105, 19217–19222 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jancova P, Anzenbacher P & Anzenbacherova E PHASE II DRUG METABOLIZING ENZYMES. Biomed Pap 154, 103–116 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Lindsay J, Wang LL, Li Y & Zhou SF Structure, function and polymorphism of human cytosolic sulfotransferases. Curr Drug Metab 9, 99–105 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Strott CA & Higashi Y Cholesterol sulfate in human physiology what’s it all about? J Lipid Res 44, 1268–1278 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Iwamori M, Iwamori Y & Ito N Regulation of the Activities of Thrombin and Plasmin by Cholesterol Sulfate as a Physiological Inhibitor in Human Plasma. J Biochem 125, 594–601 (1999). [DOI] [PubMed] [Google Scholar]
- 18.Cheetham JJ, Epand RM, Andrews M & Flanagan TD Cholesterol sulfate inhibits the fusion of Sendai virus to biological and model membranes. J Biol Chem 265, 12404–12409 (1990). [PubMed] [Google Scholar]
- 19.Williams ML, Hughes-Fulford M & Elias PM Inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity and sterol synthesis by cholesterol sulfate in cultured fibroblasts. Biochimica Et Biophysica Acta Bba - Mol Cell Res 845, 349–357 (1985). [DOI] [PubMed] [Google Scholar]
- 20.Wang F, Beck-García K, Zorzin C, Schamel WWA & Davis MM Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat Immunol 17, 844–850 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakurai T et al. Cholesterol sulfate is a DOCK2 inhibitor that mediates tissue-specific immune evasion in the eye. Sci Signal 11, 4874 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Xu Z, McClure ST & Appel LJ Dietary Cholesterol Intake and Sources among U.S Adults: Results from National Health and Nutrition Examination Surveys (NHANES), 2001–2014. Nutrients 10, 771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennion LJ & Grundy SM Effects of obesity and caloric intake on biliary lipid metabolism in man. J Clin Invest 56, 996–1011 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kenny DJ et al. Cholesterol Metabolism by Uncultured Human Gut Bacteria Influences Host Cholesterol Level. Cell Host Microbe 28, 245–257.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stephen D,T & John M,D. Sterol absorption by the small intestine. Curr Opin Lipidol 14, (2003). [DOI] [PubMed] [Google Scholar]
- 26.Kredich NM Biosynthesis of Cysteine. Ecosal Plus 3, (2008). [DOI] [PubMed] [Google Scholar]
- 27.Carbonero F, Benefiel AC, Alizadeh-Ghamsari AH & Gaskins HR Microbial pathways in colonic sulfur metabolism and links with health and disease. Front Physiol 3, 448 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rückert C Sulfate reduction in microorganisms—recent advances and biotechnological applications. Curr Opin Microbiol 33, 140–146 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Kushkevych I et al. Recent Advances in Metabolic Pathways of Sulfate Reduction in Intestinal Bacteria. Cells 9, 698 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen I-MA et al. The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic Acids Res 49, 939 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukherjee S et al. Genomes OnLine Database (GOLD) v.8: overview and updates. Nucleic Acids Res 49, 983 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koropatkin NM, Martens EC, Gordon JI & Smith TJ Starch Catabolism by a Prominent Human Gut Symbiont Is Directed by the Recognition of Amylose Helices. Structure 16, 1105–1115 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macy JM & Probst I The Biology of Gastrointestinal Bacteroides. Annu Rev Microbiol 33, 561–594 (1979). [DOI] [PubMed] [Google Scholar]
- 34.Adams AND et al. A Novel Family of RNA-Binding Proteins Regulate Polysaccharide Metabolism in Bacteroides thetaiotaomicron. J Bacteriol 203, e00217–21 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Domenech P, Reed MB & Barry CE Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect Immun 73, 3492–501 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maurice CF, Haiser HJ & Turnbaugh PJ Xenobiotics Shape the Physiology and Gene Expression of the Active Human Gut Microbiome. Cell 152, 39–50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Altschul SF et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF et al. Protein database searches using compositionally adjusted substitution matrices. Febs J 272, 5101–5109 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arumugam M et al. Enterotypes of the human gut microbiome. Nature 473, 174–180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pasolli E et al. Accessible, curated metagenomic data through ExperimentHub. Nat Methods 14, 1023–1024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamilton JP et al. Human cecal bile acids: concentration and spectrum. Am J Physiol-gastr L 293, G256–G263 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Hang S et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li W et al. A bacterial bile acid metabolite modulates Treg activity through the nuclear hormone receptor NR4A1. Cell Host Microbe (2021) doi: 10.1016/j.chom.2021.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson B,A, Lewis, et al. The Lipid Bilayer. in Molecular Biology of the Cell vol. 4 (Garland Science New York, 2002). [Google Scholar]
- 45.Gérard P et al. Bacteroides sp. strain D8, the first cholesterol-reducing bacterium isolated from human feces. Appl Environ Microb 73, 5742–9 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eldere JV, Parmentier G, Asselberghs S & Eyssen H Partial characterization of the steroidsulfatases in Peptococcus niger H4. Appl Environ Microb 57, 69–76 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakurai T et al. Cholesterol sulfate is a DOCK2 inhibitor that mediates tissue-specific immune evasion in the eye. Sci Signal 11, 4874 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Bjorkdahl O et al. Characterization of CC-chemokine receptor 7 expression on murine T cells in lymphoid tissues. Immunology 110, 170–179 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zegarra-Ruiz DF et al. Thymic development of gut-microbiota-specific T cells. Nature 594, 413–417 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Diehl GE et al. Microbiota Restrict Trafficking of Bacteria to Mesenteric Lymph Nodes by CX3CR1hi Cells. Nature 494, 116–120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hickey CA et al. Colitogenic Bacteroides thetaiotaomicron Antigens Access Host Immune Cells in a Sulfatase-Dependent Manner via Outer Membrane Vesicles. Cell Host Microbe 17, 672–680 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bittel M et al. Visualizing transfer of microbial biomolecules by outer membrane vesicles in microbe-host-communication in vivo. J Extracell Vesicles 10, e12159 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McNamee EN et al. Chemokine receptor CCR7 regulates the intestinal TH1/TH17/Treg balance during Crohnˈs-like murine ileitis. J Leukocyte Biol 97, 1011–1022 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lloyd-Price J et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peck SC et al. A glycyl radical enzyme enables hydrogen sulfide production by the human intestinal bacterium Bilophila wadsworthia. Proc National Acad Sci 116, 201815661 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chaudhari SN et al. Bariatric surgery reveals a gut-restricted TGR5 agonist with anti-diabetic effects. Nat Chem Biol 17, 20–29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alnouti Y Bile Acid Sulfation: A Pathway of Bile Acid Elimination and Detoxification. Toxicol Sci 108, 225–246 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Sato H et al. Novel Potent and Selective Bile Acid Derivatives as TGR5 Agonists: Biological Screening, Structure−Activity Relationships, and Molecular Modeling Studies. J Med Chem 51, 1831–1841 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Wu S, Li W, Han J, Sun Q & Qureshi AA Hypercholesterolemia and Risk of Incident Psoriasis and Psoriatic Arthritis in US Women. Arthritis Rheumatol 66, 304–310 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ding X, Zhang W, Li S & Yang H The role of cholesterol metabolism in cancer. Am J Cancer Res 9, 219–227 (2019). [PMC free article] [PubMed] [Google Scholar]
- 61.Traish AM, Kang HP, Saad F & Guay AT Dehydroepiandrosterone (DHEA)—A Precursor Steroid or an Active Hormone in Human Physiology (CME). J Sex Medicine 8, 2960–2982 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Nomenclature of Vitamin D. Eur J Biochem 124, 223–227 (1982). [PubMed] [Google Scholar]
- 63.Busquets-Garcia A et al. Pregnenolone blocks cannabinoid-induced acute psychotic-like states in mice. Mol Psychiatr 22, 1594–1603 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vallée M et al. Pregnenolone Can Protect the Brain from Cannabis Intoxication. Science 343, 94–98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Majewska MD, Mienville J-M & Vicini S Neurosteroid pregnenolone sulfate antagonizes electrophysiological responses to GABA in neurons. Neurosci Lett 90, 279–284 (1988). [DOI] [PubMed] [Google Scholar]
- 66.Wu FS, Gibbs TT & Farb DH Pregnenolone sulfate: a positive allosteric modulator at the N-methyl-D-aspartate receptor. Mol Pharmacol 40, 333–6 (1991). [PubMed] [Google Scholar]
- 67.Adhikari AA et al. A Gut-Restricted Lithocholic Acid Analog as an Inhibitor of Gut Bacterial Bile Salt Hydrolases. Acs Chem Biol 16, 1401–1412 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bacic MK & Smith CJ Laboratory Maintenance and Cultivation of Bacteroides Species. Curr Protoc Microbiol 9, 13C.1.1–13C.1.21 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yao L et al. A selective gut bacterial bile salt hydrolase alters host metabolism. Elife 7, e37182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Livak KJ & Schmittgen TD Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔC T Method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 71.García-Bayona L & Comstock LE Streamlined Genetic Manipulation of Diverse Bacteroides and Parabacteroides Isolates from the Human Gut Microbiota. Mbio 10, e01762–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang J, Shoemaker NB, Wang G-R & Salyers AA Characterization of a Bacteroides Mobilizable Transposon, NBU2, Which Carries a Functional Lincomycin Resistance Gene. J Bacteriol 182, 3559–3571 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cullen TW et al. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science 347, 170–175 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Degnan PH, Barry NA, Mok KC, Taga ME & Goodman AL Human Gut Microbes Use Multiple Transporters to Distinguish Vitamin B12 Analogs and Compete in the Gut. Cell Host Microbe 15, 47–57 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Swann JR et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc National Acad Sci 108, 4523–4530 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Suzek BE, Huang H, McGarvey P, Mazumder R & Wu CH UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23, 1282–1288 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Beghini F et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 10, e65088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaminski J et al. High-Specificity Targeted Functional Profiling in Microbial Communities with ShortBRED. Plos Comput Biol 11, e1004557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Camacho C et al. BLAST+: architecture and applications. Bmc Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.O’Leary NA et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44, D733–D745 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and the Supplementary Information. The HMP2 IBD 16S and RNA-seq data are available through NCBI under BioProject ID PRJNA675599 and GEO accession number GSE179740, respectively, and the metabolomics workbench study ST000923. The curatedMetagenomicData are available online (https://waldronlab.io/curatedMetagenomicData/, with individual accession IDs listed in Supplementary Table 9). Comparison of Ch-S standard as part of the HMP2 analysis is published in supplemental data (Supplementary Figure 15).