

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is classified into two key subtypes, classical and basal, with basal PDAC predicting worse survival. Using in vitro drug assays, genetic manipulation experiments, and in vivo drug studies in human patient-derived xenografts (PDXs) of PDAC, we found that basal PDACs were uniquely sensitive to transcriptional inhibition by targeting cyclin-dependent kinase 7 (CDK7) and CDK9, and this sensitivity was recapitulated in the basal subtype of breast cancer. We showed in cell lines, PDXs, and publicly available patient datasets that basal PDAC was characterized by inactivation of the integrated stress response (ISR), which leads to a higher rate of global mRNA translation. Moreover, we identified the histone deacetylase sirtuin 6 (SIRT6) as a critical regulator of a constitutively active ISR. Using expression analysis, polysome sequencing, immunofluorescence, and cycloheximide chase experiments, we found that SIRT6 regulated protein stability by binding activating transcription factor 4 (ATF4) in nuclear speckles and protecting it from proteasomal degradation. In human PDAC cell lines and organoids as well as in murine PDAC genetically engineered mouse models where SIRT6 was deleted or down-regulated, we demonstrated that SIRT6 loss both defined the basal PDAC subtype and led to reduced ATF4 protein stability and a nonfunctional ISR, causing a marked vulnerability to CDK7 and CDK9 inhibitors. Thus, we have uncovered an important mechanism regulating a stress-induced transcriptional program that may be exploited with targeted therapies in particularly aggressive PDAC.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is an extremely lethal disease with a 5-year survival of 11% and is likely to become the second leading cause of cancer-related deaths within a decade (1). PDAC pathogenesis is characterized by a gradual progression through increasingly dysplastic precursor lesions toward invasive and lastly metastatic PDAC (2). This progression is paired with the early acquisition of oncogenic KRAS (Kirsten rat sarcoma virus) mutations and the subsequent loss of tumor suppressor genes CDKN2A (p16INK4a) (cyclin-dependent kinase inhibitor 2A), TP53 (transformation-related protein 53), and components of the transforming growth factor β (TGFβ) pathway such as SMAD4 (SMAD family member 4), TGFβR1 (transforming growth factor β receptor 1), and TGFβR2, each of which is mutated in more than 50% of patients. The creation of genetically engineered mouse models (GEMMs) with different combinations of these genes has faithfully recapitulated the disease and validated their importance in the initiation and progression of PDAC (3, 4). The various genetic analyses of PDAC have led to the same conclusion that PDACs are genetically very similar—both between patients and between primary and metastatic tumors within the same patient (5, 6). However, clinical experience suggests that pancreatic cancer is much more diverse, given the heterogeneity of responses to chemotherapy and corresponding range of survivals observed.
To understand differences in clinical behavior, transcriptional signatures have been examined from resected tumors, biopsies, patient-derived xenografts (PDXs), organoids, and cell lines (7, 8). The consensus across several studies is that there are two predominant subtypes of PDAC: classical [includes immunogenic, aberrantly differentiated endocrine exocrine (ADEX), classical A, and classical B] and basal [alternatively named and overlaps with quasi-mesenchymal (QM), squamous, basal A, and basal B] and that the basal subtype confers a poorer overall prognosis (7–11). Basal PDAC tends to be poorly differentiated with some exhibiting squamous features, whereas the classical subtype is well differentiated and maintains epithelial characteristics. Basal PDAC is defined by a complete loss of endodermal identity [low HNF4A (hepatocyte nuclear factor 4α) and GATA6 (GATA binding protein 6)]; acquired expression programs characteristic of squamous tumors, including TGF-β signaling, hypoxia response, metabolic reprogramming, and epithelial to mesenchymal transition (EMT); a reduced dependence on oncogenic KRAS for growth (8); higher frequency for inactivation of TP53, CDKN2A (11), and chromatin modifiers, including MLL2 (lysine methyltransferase 2D), MLL3 (lysine methyltransferase 2C), and KDM6A (lysine demethylase 6A); MYC (MYC protooncogene, bHLH transcription factor) amplification (6, 12); and MYC pathway activation (8).
The mechanism by which the highly aggressive basal PDAC subtype is established remains poorly understood, although recent analysis of the PDAC epigenome suggests that epigenetic dysregulation may be involved (13). An unbiased characterization of the PDAC epigenome can predict PDAC subtype and prognosis, which has not been possible with genetics alone (13, 14). Sirtuin 6 (SIRT6) is a NAD+ (nicotinamide adenine dinucleotide)–dependent histone deacetylase that acts as a repressor of MYC-driven transcription and has been implicated in cellular stress resistance, genomic stability, aging, and energy homeostasis (15, 16). It acts as a strong tumor suppressor in both human and murine PDAC, where SIRT6 inactivation cooperates with oncogenic KRAS to drive a more aggressive and highly metastatic form of disease (17, 18).
We showed here that basal PDAC cells are highly sensitive to transcriptional CDK inhibition. We demonstrated that SIRT6 defines the classical subtype and regulates activating transcription factor 4 (ATF4), the master regulator of the integrated stress response (ISR). Inhibition of transcriptional CDKs elevates cellular stress catastrophically in basal PDAC cells, whereas this stress can instead be resolved in classical PDAC cells. The vulnerability of basal PDAC cells to CDK inhibition lies in their inability to activate ATF4 to mitigate cellular stress in the absence of SIRT6.
RESULTS
Low SIRT6 expression correlates with basal state
To establish whether SIRT6 expression correlates with PDAC subtype, we first analyzed data generated from a previous large-scale integrated genomic analysis of human PDACs (8), which were classified into classical and basal PDAC subtypes (Fig. 1A). Because stromal contamination in human tumors may confound such transcriptional analyses, we also looked at a panel of 12 human PDAC cell lines classified as either classical or basal PDAC by previous groups. All classical PDAC cell lines expressed higher amounts of SIRT6 than any basal PDAC cell line (Fig. 1B). Moreover, ectopic expression of wild-type (WT) SIRT6 but not a catalytically inactive mutant (HY) form of SIRT6 reduced RNA and protein expression of the ΔN isoform of TP63 (tumor protein 63), a key biomarker of the basal subtype, in two independent basal PDAC lines (fig. S1, A to C).
Fig. 1. THZ1 specifically induces apoptosis in basal but not classical PDAC.
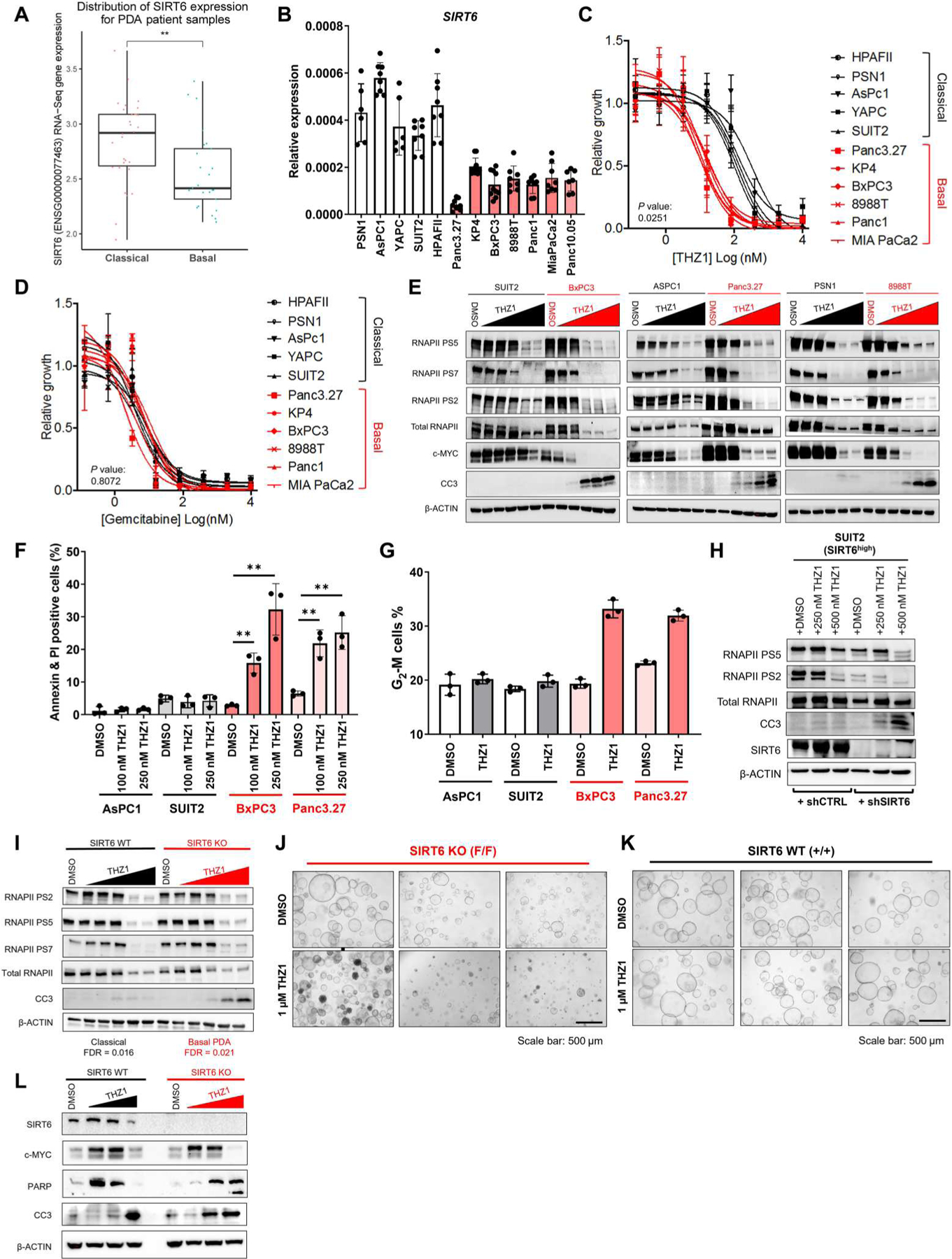
(A) SIRT6 expression from RNA-seq data of human PDAC samples classified as belonging to either basal or classical subtypes. (B) qRT-PCR for SIRT6 in a panel of human PDAC cell lines. (C and D) Proliferation curves for basal PDAC (red) compared with classical (black) PDAC cell lines treated with increasing doses of THZ1 (C) and gemcitabine (D). Significant difference in median inhibitory concentration (IC50) values for basal versus classical indicated by P ≤ 0.05 (two-tailed unpaired Student’s t test). (E) Western blot for RNA polymerase II (RNAPII) phosphorylation of serine-5 (PS5), serine-7 (PS7), serine-2 (PS2), MYC, cleaved caspase-3 (CC3), and β-ACTIN in basal (red) compared with classical (black) PDAC cell lines treated with increasing doses of THZ1 (20 nM to 5 μM) for 16 hours. (F) Quantification of annexin + propidium iodide–positive cells in classical (gray) versus basal (red) PDAC lines after THZ1 treatment. (G) Quantification of G2-M cell populations in classical (gray) versus basal (red) PDAC lines after THZ1 treatment. (H) Western blots for PS5, PS2, RNAPII, CC3, SIRT6, and β-ACTIN in classical cell line expressing either short hairpins targeting SIRT6 (shSIRT6) or control hairpins (shCTRL), treated with increasing doses of THZ1 (250 and 500 nM). (I) Western blots for PS2, PS5, PS7, RNAPII, CC3, and β-ACTIN in 2D cell lines derived from GEMM pancreatic tumors with (+/+) and without (F/F) SIRT6 expression treated with increasing concentrations of THZ1 for 16 hours. SIRT6 WT and SIRT6 KO cell lines were subtyped as classical and basal, respectively, by RNA-seq with significance defined as FDR < 0.05. (J and K) Bright-field imaging of organoids derived from GEMM pancreatic tumors with (+/+) and without (F/F) SIRT6 expression treated with either DMSO or 1 μM THZ1 for 24 hours. All bright-field images have a scale bar of 500 μm. (L) Western blots of SIRT6, c-MYC, poly(ADP-ribose) polymerase (PARP), CC3, and β-ACTIN in organoids derived from GEMM pancreatic tumors with and without SIRT6 expression treated with incremental doses of THZ1 (500 nM to 2.5 μM) for 16 hours. Error bars represent ±SEM between technical duplicates. Data are representative of at least three independent experiments. *P ≤ 0.05; **P ≤ 0.01 (two-tailed unpaired Student’s t test).
Basal and classical PDACs have unique therapeutic vulnerabilities
SIRT6 acts as a potent co-repressor for the MYC oncogene. SIRT6low/basal human PDAC cell lines and SIRT6 knockout (SIRT6 KO) cell lines derived from PDAC GEMMs demonstrated elevated amounts of chromatin-bound MYC and a dependency on MYC expression for growth and survival (17). Studies have shown that the CDK7 inhibitor THZ1 can selectively target super-enhancer (SE)–regulated genes such as MYC in some cancers. Unlike most kinase inhibitors, THZ1 binds to an allosteric site on CDK7, CDK12, and CDK13 rather than its adenosine triphosphate (ATP)–binding pocket, thereby affording high specificity with few off-target effects at clinically relevant doses (19). It should be noted that inhibition of CDK12 and CDK13 occurred at slightly higher concentrations when compared with CDK7 (20). THZ1 has recently been shown to markedly inhibit tumor cell growth in multiple cancer types (13, 19, 21–24). In these studies, THZ1 was found to achieve its antiproliferative effect through selective inhibition of target genes driven by SEs, which normally function to amplify lineage-specific transcription factors in these cancers (25). SEs are areas within the genome typically marked by H3K27ac (histone H3 acetylated at lysine-27), H3K9ac (histone H3 acetylated at lysine-9), and H3K4me1 (histone H3 monomethylated at lysine-4) and occupied by paused RNA polymerase II (RNAPII) (26, 27). CDK7 functions as a known regulator of RNAPII-mediated initiation, pause establishment, and elongation through the direct phosphorylation of the RNAPII C-terminal domain (CTD) (19). Thus, it has been suggested that CDK7 inhibition by THZ1 leads to reduced occupancy of RNAPII within SEs (28).
To evaluate the potential efficacy of THZ1 in basal PDAC, we treated a panel of seven human basal PDAC cell lines with THZ1 and compared their sensitivity with five human classical PDAC lines (Fig. 1C). SIRT6low/basal PDAC lines were highly sensitive to THZ1, whereas SIRT6high/classical PDAC lines were not as sensitive. By contrast, both basal and classical PDAC lines were equally sensitive to gemcitabine (Fig. 1D), the standard of care treatment for pancreatic cancer. Moreover, THZ1 sensitivity was inversely correlated with SIRT6 expression across a broad panel of 22 PDAC lines (fig. S1D). Treatment with THZ1 resulted in a similar reduction in phosphorylation of both substrates of CDK7 within the RNAPII CTD, referred to as phospho–serine-5 (PS5) and PS7, as well as the transition to productive elongation marker PS2, in both basal and classical PDAC lines (Fig. 1E). MYC protein was also greatly reduced in both cell subtypes. However, activation of the apoptotic marker cleaved caspase-3 was observed as early as 16 hours after treatment with THZ1 at 500 nM in basal PDAC lines, whereas no activation of cleaved caspase-3 was seen in classical PDAC lines even at doses 10-fold higher. Consistent with these results, annexin V staining was markedly increased after 24 hours of treatment with 100 nM THZ1 in basal but not classical PDAC cell lines (Fig. 1F and fig. S1E). Propidium iodide staining revealed an accumulation of basal PDAC cells in G2-M phase after 24 hours of treatment with 100 nM THZ1, suggesting a G2-M arrest, as previously observed in neuroblastoma (13). No effects on cell cycle were observed in classical PDAC cell lines (Fig. 1G and fig. S1F). Moreover, inhibition of SIRT6 using short hairpin RNA (shRNA) sensitized SIRT6high/classical PDAC to THZ1 treatment (Fig. 1H) without changing expression of CDK proteins (fig. S1G). Last, both two-dimensional (2D) PDAC lines (Fig. 1I) and 3D organoids (Fig. 1, J to L) derived from our SIRT6 KO GEMM demonstrated marked sensitivity to THZ1, similar to that observed in our human basal PDAC cell lines. Thus, reduced amounts of SIRT6 correlate with the basal PDAC subtype and can predict THZ1 sensitivity in PDAC.
Inhibition of CDK7 but not MYC is both necessary and sufficient for THZ1-induced apoptosis in basal PDAC
We next sought to define the specificity of CDK7 inhibition by THZ1 and determine whether CDK7 inhibition was necessary or sufficient for the induction of apoptosis in basal PDAC cells. We evaluated the efficacy of THZ1-R, a structural analog of THZ1 that cannot covalently bind to CDK7 (19). As expected, neither basal nor classical PDAC cell lines were sensitive to THZ1-R (fig. S2, A and B). We then tested whether ectopic expression of a mutant form of CDK7, CDK7 C312S, which cannot covalently bind THZ1 (19), could prevent THZ1 from inducing apoptosis in basal PDAC cells and found that expression of CDK7 C312S but not WT CDK7 prevented both cleaved caspase-3 activation and MYC suppression in two independent basal PDAC lines (fig. S2, C and D). Last, to specifically assess whether CDK7 inhibition is sufficient for the induction of apoptosis in basal PDAC cells, we used CRISPR-Cas9–expressing lentivirus to knock out CDK7 in two basal and two classical PDAC lines. Treatment with two independent single-guide RNAs (sgRNAs) against CDK7, but not a nontargeting control sgRNA, inhibited cell growth and induced apoptosis in basal but not classical PDAC cell lines (fig. S2, E to H). Thus, inhibition of CDK7 is both necessary and sufficient for the efficacy of THZ1 in SIRT6low/basal PDAC. Enforced ectopic expression of MYC in three independent basal PDAC lines using a doxycycline-inducible promoter restored MYC protein but could not rescue the induction of THZ1-mediated apoptosis (fig. S2, I and J). We previously showed that SIRT6 inactivation promoted PDAC progression and metastasis through up-regulation of LIN28b; however, LIN28b and let-7 targets remained constant upon treatment with THZ1 (fig. S2K). Thus, THZ1-induced apoptosis in basal PDAC is not due to transcriptional inhibition of MYC or LIN28b.
SE-regulated genes are not preferentially down-regulated by THZ1 in basal PDAC
The efficacy of THZ1 at inducing apoptosis in cancer may be through the selective inhibition of SE-regulated genes (including MYC). MYC was potently and rapidly down-regulated in PDAC cells after treatment with THZ1, but this did not appear to be unique to basal PDAC cell lines, because we also saw a reduction in MYC expression in classical cell lines (Fig. 1E). We therefore performed a combined RNA sequencing (RNA-seq) and H3K27ac chromatin immunoprecipitation–sequencing (ChIP-seq) analysis of basal and classical PDAC cells after THZ1 treatment to determine whether SE-regulated genes were selectively targeted. More than 2000 genes were down-regulated in both BxPC3 and Panc3.27 (basal) cells by THZ1, and, of these, 1437 were commonly down-regulated. However, only 87 genes were down-regulated in SUIT2 (classical) cells after 6 hours of THZ1 treatment (Fig. 2A), and most SE-regulated genes were not down-regulated by THZ1 in all three cell lines tested (Fig. 2B). The MYC SE itself was not more decorated by H3K27ac in basal PDAC compared with classical PDAC (Fig. 2, C and D). Instead, both gene ontology (GO) and gene set enrichment analysis (GSEA) demonstrated that genes related to transcription were potently and specifically repressed in basal PDAC cells after THZ1 exposure (Fig. 2E). To determine whether the effects of THZ1 and flavopiridol (an ATP-competitive CDK9 inhibitor) on transcription differed in basal compared with classical PDAC, we subjected classical and basal cells to increasing doses of each compound (19, 28, 29). Nuclei were isolated and nascent transcripts were extended by the addition of radiolabeled cytidine triphosphate in a nuclear walk-on reaction (30, 31) followed by denaturing gel analysis of the labeled transcripts. The signals from transcripts generated by paused Pol II (30 to 70 nucleotides) were quantified. THZ1 treatment, which interferes with pausing, led to nearly identical fold reductions of paused transcripts in both cell types, whereas blocking productive elongation with flavopiridol showed the same result (Fig. 2F). Thus, both basal and classical cells responded similarly to THZ1 and flavopiridol. Together, our results confirmed a specific dependence on transcription in basal PDAC; however, whether this specificity was due to the inhibition of a specific gene program remained unclear.
Fig. 2. THZ1 inhibits a broad transcriptional program in basal but not classical PDAC.
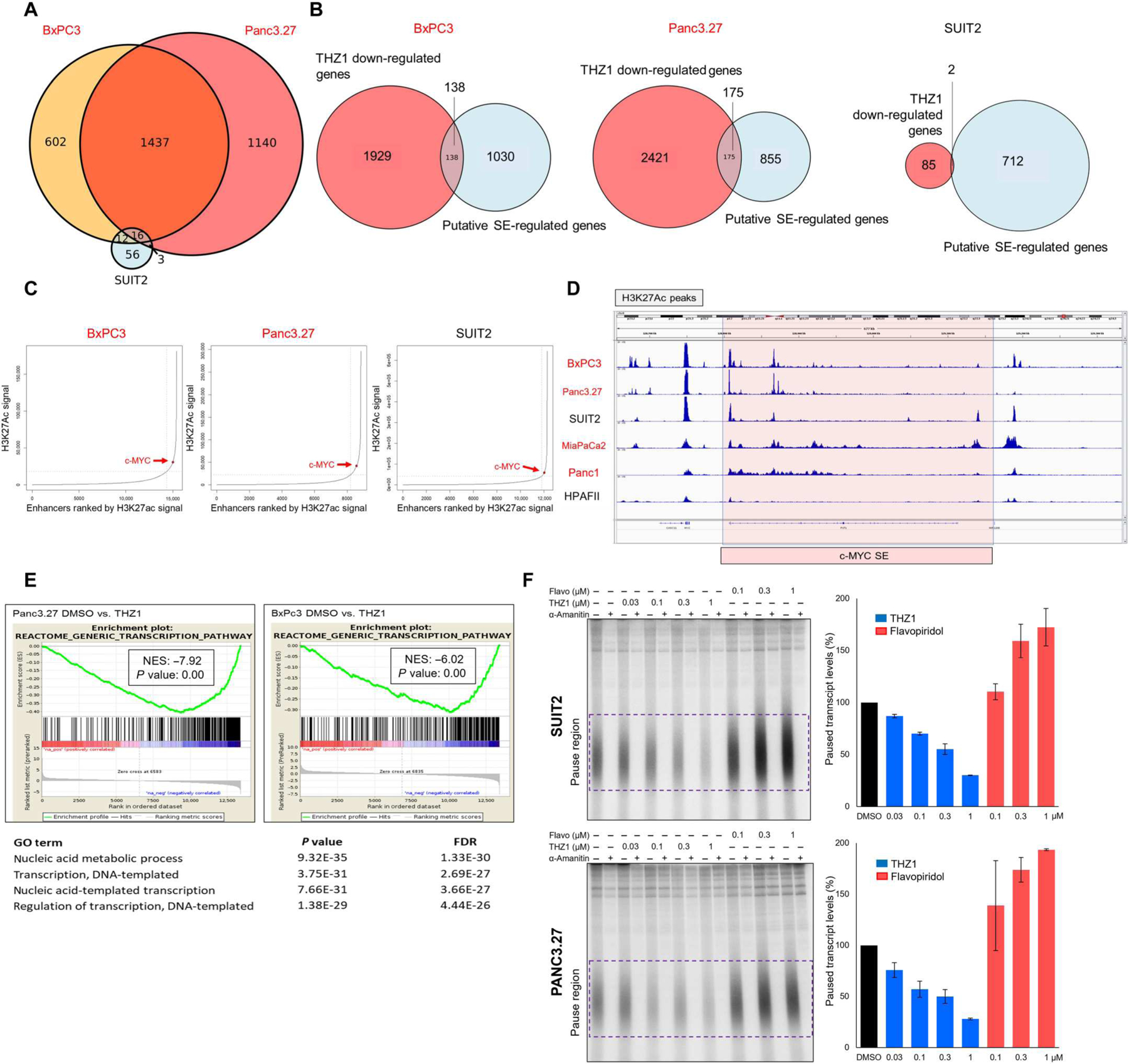
(A) Venn diagram showing overlap of down-regulated genes between basal lines BxPC3 and Panc3.27 and the classical PDAC line SUIT2 after THZ1 treatment. (B) Venn diagram showing overlap between down-regulated genes from RNA-seq experiment and putative super-enhancer (SE)–regulated genes in the indicated PDAC cell lines. (C) Enhancer regions plotted in order of increasing input-normalized H3K27ac ChIP-seq signal, comparing basal (red) and classical (black) lines. SEs are defined as being to the right of the curve inflection point, indicated by a dashed vertical line, and c-MYC is highlighted by a red point. (D) H3K27ac peaks at a putative c-MYC enhancer in PDAC, comparing basal (red) and classical (black) lines. (E) Gene set enrichment analyses (GSEAs) of differentially expressed genes and significant gene ontology (GO) pathways suppressed in basal PDAC lines after THZ1 treatment. (F) Nuclear walk on assays of SUIT2 and Panc3.27 cells treated with the indicated amounts of THZ1 and flavopiridol. Autoradiographs of denaturing gels of transcripts generated (left) and quantification of the paused transcripts contained within the purple box (right) are shown. Error bars represent SD from two biological replicates.
Basal PDACs are sensitive to inhibitors of transcriptional CDKs
To further elucidate the mechanism of action of THZ1 in basal PDAC, we extended our evaluation of the efficacy of CDK transcriptional inhibitors to include specific inhibitors for CDK4/6, CDK9, CDK7, and CDK12/13 (20) in our panel of PDAC cell lines. Basal PDAC cell lines were 5- to 10-fold more sensitive to both CDK9 (flavopiridol) (Fig. 3, A and B) and CDK7 (YKL-5-124) (Fig. 3, C and D) inhibition than classical PDAC, whereas basal and classical PDAC were equally sensitive to the CDK4/6 cell cycle inhibitor palbociclib (Fig. 3E). Basal PDAC exhibited only minimal sensitivity to inhibition of CDK12/13 (THZ531) (fig. S3A). Treatment with CDK7, CDK9, and CDK12/13 inhibitors led to reductions in RNAPII phosphorylation marks in both basal and classical lines as well as specific induction of apoptosis as measured by cleaved caspase-3 only in basal PDAC lines. MYC protein was reduced similarly between basal and classical PDAC lines (Fig. 3, B and D, and fig. S3B). To determine whether basal PDAC was more sensitive to transcriptional inhibitors in general, we assessed the efficacy of bromodomain inhibitors, a class of non–CDK-targeting transcriptional inhibitors. Our panel of PDAC cell lines showed equivalent sensitivity to bromodomain inhibition with JQ1, I-BET151, and I-BET762 (fig. S3C). These data indicate that the differential sensitivity between PDAC subtypes is selective for inhibitors of transcriptional CDKs. To further investigate this, we used CRISPR-Cas9–expressing lentivirus to knock out CDK9, CDK12, or CDK13 in two basal and two classical PDAC lines. Treatment with two independent sgRNAs against CDK9, but not a nontargeting control sgRNA, inhibited cell growth significantly more in basal PDAC cell lines (P = 0.0025 and 0.00013) compared with classical (P = 0.118 and 0.036) (fig. S3, D to F). KO of CDK12 only minimally reduced growth in basal lines compared with classical, whereas CDK13 KO induced no change in growth in both basal and classical PDAC cell lines (fig. S3, G to L). Together, these data support that basal PDAC is selectively more vulnerable to loss of CDK7 and CDK9 function rather than loss of CDK12 and CDK13 function. Considering the structural and functional differences between CDK7 and CDK9, our data demonstrate a multifaceted dependency on CDK-regulated transcription in basal PDAC that is not due to a specific CDK7-regulated pathway or gene program. Instead, our results uncovered a therapeutic vulnerability specifically in the basal but not the classical PDAC subtype and prompted us to further explore the mechanistic basis for this “transcriptional CDK addiction.”
Fig. 3. Basal PDAC lines are differentially sensitive to inhibitors of transcriptional CDKs.
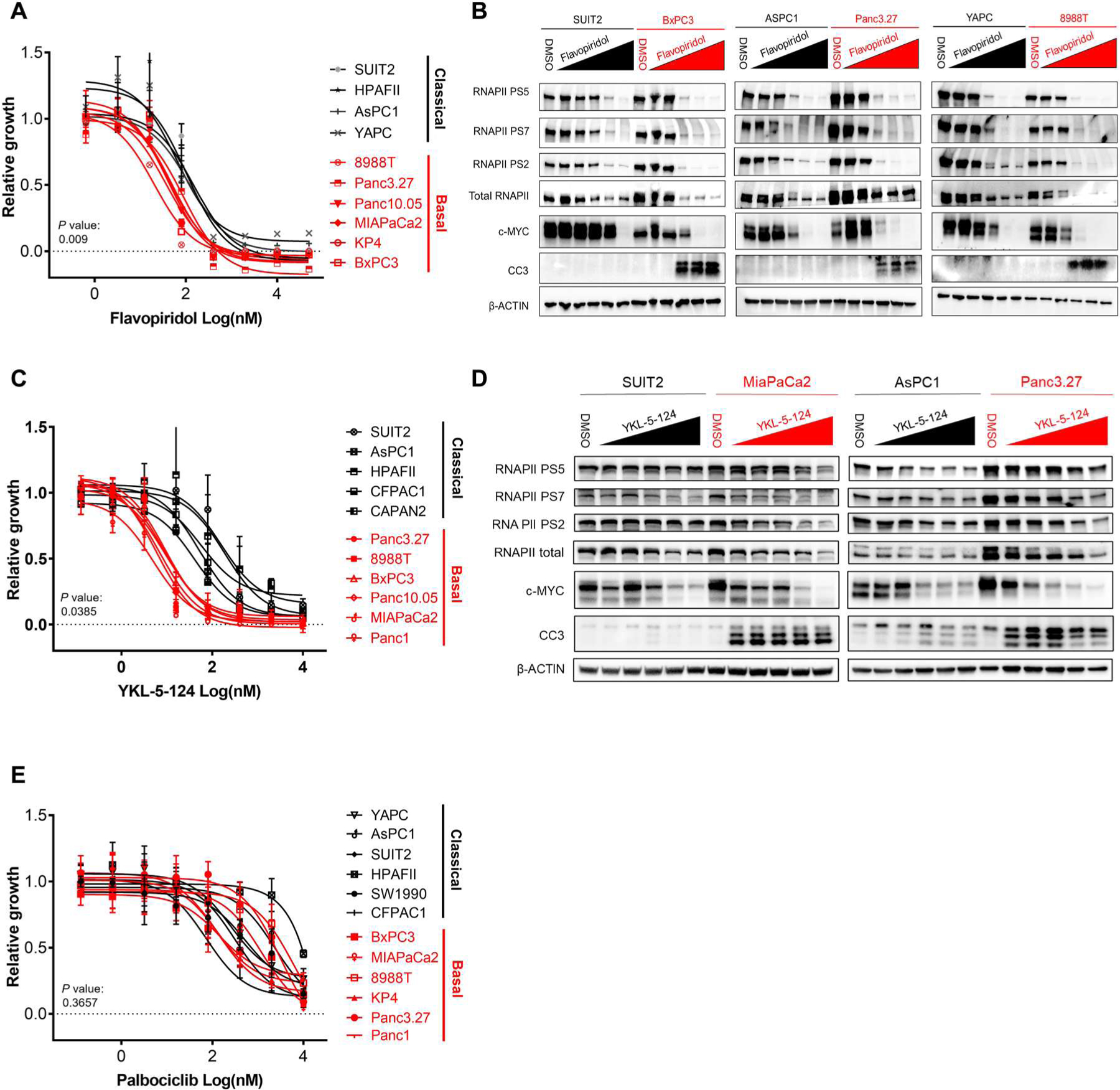
(A) Proliferation curves for basal PDAC (red) compared with classical (black) PDAC cell lines treated with increasing doses of flavopiridol. (B) Western blots for RNAPII, PS5, PS7, PS2, MYC, CC3, and β-ACTIN in basal (red) compared with classical (black) PDAC cell lines treated with increasing doses of flavopiridol (20 nM to 5 μM). (C) Proliferation curves for basal PDAC (red) compared with classical (black) PDAC cell lines treated with increasing doses of YKL-5–124. (D) Western blots for RNAPII, PS5, PS7, PS2, MYC, CC3, and β-ACTIN in basal (red) compared with classical (black) PDAC cell lines treated with increasing doses of YKL-5–124 (20 nM to 5 μM). (E) Proliferation curves for basal PDAC (red) compared with classical (black) PDAC cell lines treated with increasing doses of palbociclib. Proliferation curves were performed in duplicate, and data are represented as mean ± SEM among three independent experiments. Significant differences in IC50 values for basal versus classical indicated by P ≤ 0.05 (two-tailed unpaired Student’s t test with Welch’s correction for those with unequal variances).
SIRT6 regulates the ISR in PDAC
We next examined whether mRNA translation rates were different between basal and classical PDAC. Global de novo protein synthesis was markedly different as measured by puromycin incorporation (Fig. 4A) (32). We observed that basal PDAC cells had increased translation compared with classical PDAC cells. Furthermore, treatment with THZ1 markedly inhibited translation in basal PDAC but had no impact in classical PDAC (Fig. 4B). Last, basal cells were more sensitive to the translation inhibitor puromycin and homoharringtonine, confirming their greater reliance on mRNA translation (fig. S4A).
Fig. 4. SIRT6 regulates the ISR in PDAC.
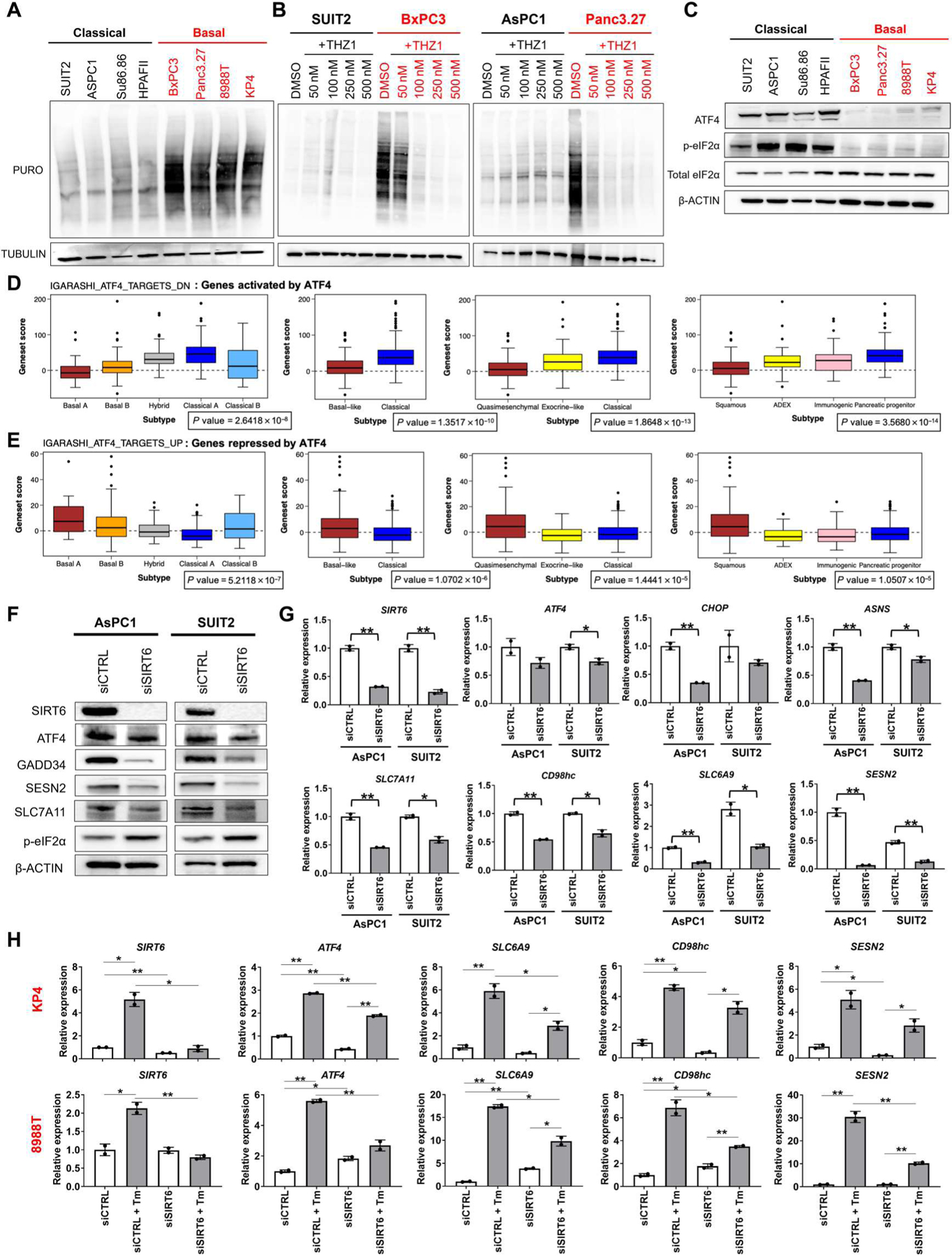
(A) Western blots for puromycin incorporation and TUBULIN in classical (black) and basal (red) PDAC lines. (B) Western blots for puromycin incorporation and TUBULIN in two classical (black) and two basal (red) PDAC lines treated with incremental doses of THZ1. (C) Western blots for ISR markers ATF4, phosphorylated eIF2α (p-eIF2α), total eIF2α, and β-ACTIN in basal (red) and classical (black) PDAC lines. (D and E) Bioinformatic analysis of publicly available data comparing expression of ATF4 target genes according to four published PDAC transcriptional subtyping classifications. Wilcoxon rank sum test was performed comparing basal versus classical (or subtype equivalent) to give an FDR-adjusted P value. (F) Western blots for SIRT6; ISR markers ATF4, GADD34, and p-eIF2α; ATF4 target genes SESN2, ASNS, SLC7A11; and β-ACTIN in classical lines treated with nonspecific and SIRT6-specific small interfering RNAs (siRNAs). (G) qRT-PCRs for SIRT6, ISR markers, and ATF4 target genes in classical lines treated with nonspecific and SIRT6-specific siRNAs. (H) qRT-PCRs for SIRT6, ATF4, and ATF4 target genes in two basal lines treated with 10 μM tunicamycin (Tm) for 5 hours in the presence of nonspecific and SIRT6-specific siRNAs. Error bars represent ±SEM between technical duplicates. Data are representative of at least two independent experiments. *P ≤ 0.05; **P ≤ 0.01 (two-tailed unpaired Student’s t test).
A variety of inputs regulate global translation in both healthy and cancer cells, many of which converge upon the ISR. Activation of the ISR occurs when eukaryotic translation initiation factor 2 (eIF2α) is phosphorylated at serine-51. EIF2α phosphorylation causes transient but marked attenuation of global protein synthesis, with the exception of select transcription factors, such as ATF4. Serving as an integration point for the ISR, ATF4 helps to orchestrate the cell’s response to the perturbing stress by activating transcription of genes necessary for either stress reduction or apoptosis. In turn, the ISR can be rapidly deactivated through the dephosphorylation of eIF2α by a complex of growth arrest and DNA damage–inducible protein (GADD34) and protein phosphatase 1 (PP1). Thus, the ISR is a reversible mechanism through which cells can markedly reduce global translation to survive a wide variety of stresses.
We interrogated the ISR in a panel of basal and classical PDAC cell lines. Virtually all classical PDAC lines had high baseline activation of the ISR, as measured by high ATF4 expression and phosphorylation of eIF2α, which correlated with low overall translation rates. Conversely, all basal PDAC lines had low baseline activation of the ISR coupled with relatively high translation rates (Fig. 4, A and C). We further sought to validate these findings using publicly available data from human PDAC tumors. Given that ATF4 is principally translationally regulated, and publicly available ATF4 expression data show no differential expression between subtypes in PDAC, we opted to examine expression of ATF4 target genes within the publicly available datasets as a proxy for ISR activation. We found that genes activated by ATF4 were significantly up-regulated (P < 2.7 × 10−8) in classical PDAC compared with basal PDAC according to four published PDAC transcriptional subtyping classifications. The inverse was also found to be true; genes repressed by ATF4 were significantly up-regulated (P < 1.5 × 10−5) in basal PDAC compared with classical PDAC by the same classifications (Fig. 4, D and E). We next sought to understand how classical lines maintained activation of the ISR, whereas it remained silent in basal lines. As noted, SIRT6 has been implicated, albeit indirectly, in distinct cellular responses to other stressors such as hypoxia and oxidative stress (33). SIRT6 has also been shown to bind ATF4, which prompted us to investigate its role in regulating ATF4 and/or function in PDAC cells (34). We found that in classical PDAC cells, loss of SIRT6 led to a decrease in overall ATF4 protein abundance (Fig. 4F and fig. S4B) and a decrease in amount of ATF4 bound to chromatin (fig. S4C). In addition, loss of SIRT6 in classical PDAC cells reduced expression of ATF4 target gene transcripts and proteins, including GADD34 (protein phosphatase 1 regulatory subunit 15A), CHOP (DNA damage–inducible transcript 3), ASNS (asparagine synthetase), Sestrin 2 (SESN2), and the amino acid transporters SLC7A11 (solute carrier family 7 member 11), SLC6A9 (solute carrier family 6 member 9), and CD98hc (CD98 heavy chain) (Fig. 4, F and G). SIRT6 KO cells from our PDAC GEMMs also demonstrated the same effect (fig. S4, D and E). These targets suggested that SIRT6 regulates ATF4 target genes specifically involved in amino acid deprivation and not oxidative stress or autophagy (fig. S4F). Conversely, ectopic expression of WT SIRT6, but not the catalytic mutant SIRT6 HY, in basal PDAC cells robustly increased ATF4 target gene expression, suggesting that SIRT6 deacetylase activity was necessary for its regulation of ATF4-mediated transcription (fig. S4G). The essential role for SIRT6 in increasing ATF4, and as an adaptive response, became apparent in basal cells that are prevented from increasing SIRT6 in response to stress. In both KP4 and 8988T basal cells, tunicamycin treatment of the siCTRL causes an increase in expression of SIRT6, ATF4, and ATF4 target genes. Tunicamycin is a known inducer of ATF4 (35), which in our basal model also increased SIRT6, thereby enabling ATF4 induction. Knockdown of SIRT6 renders basal PDAC cells incapable of fully inducing ATF4 and ATF4 target genes in response to tunicamycin [which elevates endoplasmic reticulum (ER) stress by inhibiting N-linked glycosylation] (36). In addition, the already low amounts of ATF4 in basal cells are further reduced by knockdown of residual SIRT6 (Fig. 4H). Together, these data validate the necessity of SIRT6 for complete induction of ATF4.
SIRT6 regulates ATF4 stability
To investigate the principal mechanism by which SIRT6 regulates ATF4, we performed a time course of ATF4 expression dynamics by measuring mRNA and protein abundance upon SIRT6 knockdown. We found that ATF4 protein was reduced after 48 hours of SIRT6 knockdown, but changes in ATF4 mRNA did not occur until 72 hours (Fig. 5, A and B). Because ATF4 is predominantly translationally regulated and our data showed that ATF4 protein changes occur before mRNA changes with SIRT6 knockdown, we decided to investigate whether SIRT6 regulates ATF4 protein translation. To do this, we performed polysome profiling on classical cells with and without SIRT6 knockdown. Polysome profiling uses cycloheximide to block protein synthesis and freeze the ribosomes on the mRNA transcripts, enabling assessment of a transcript bound at different stages of ribosome assembly and translation. A sucrose gradient and ultracentrifugation are used to separate the monosomes from polysomes, and abundance is measured by absorption. We found no change in global translation, as indicated by similar polysome traces between control and SIRT6 knockdown even with reduction in SIRT6 mRNA abundance verified by quantitative reverse transcription polymerase chain reaction (qRT-PCR) (fig. S5, A and B). ATF4 mRNA abundance showed no difference in amount of transcript bound to ribosomes in each fraction (fig. S5C) as well as no change in amount of cumulative ATF4 mRNA in monosomes or polysomes (fig. S5D) with SIRT6 knockdown as measured by qRT-PCR. Last, SIRT6 knockdown and ATF4 protein reduction were verified in our classical cells through paired Western blots (fig. S5E). Therefore, we concluded from these data that ATF4 protein translation is not substantially affected by SIRT6.
Fig. 5. SIRT6 regulates stability of ATF4 protein.
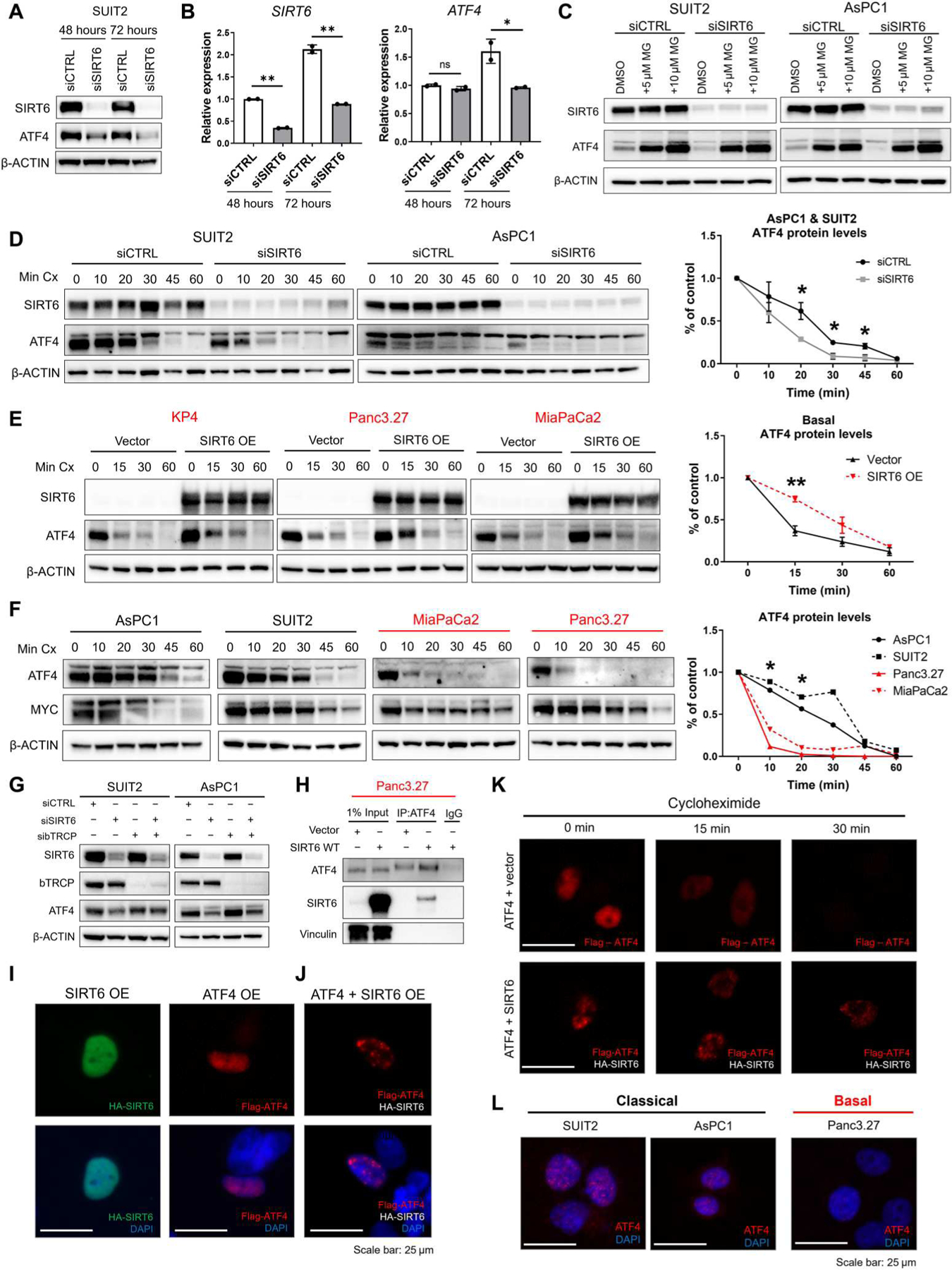
(A and B) Western blots (A) for SIRT6, ATF4, and β-ACTIN and qRT-PCR (B) for SIRT6 and ATF4 mRNA abundance in SUIT2 cells transfected with nonspecific or SIRT6-specific siRNAs for 48 or 72 hours. (C) Western blots for SIRT6, ATF4, and β-ACTIN in classical PDAC cell lines transfected with nonspecific or SIRT6-specific siRNAs and treated with 5 or 10 μM MG-132 (MG) for 6 hours. (D) Western blots for SIRT6, ATF4, and β-ACTIN and quantification of relative ATF4 abundance in classical PDAC cell lines transfected with nonspecific or SIRT6-specific siRNAs and treated with cycloheximide (Cx) (150 μg/ml) for 10, 20, 30, 45, or 60 min. (E) Western blots for SIRT6, ATF4, and β-ACTIN and quantification of relative ATF4 abundance in basal PDAC cell lines transfected with empty vector, SIRT6 WT overexpression, or SIRT6 HY overexpression plasmids and treated with cycloheximide (150 μg/ml) for 15, 30, or 60 min. (F) Western blots for ATF4, MYC, and β-ACTIN and quantification of relative ATF4 abundance in classical and basal PDAC cell lines treated with cycloheximide (150 μg/ml) for 10, 20, 30, 45, or 60 min. Significance indicates basal versus classical comparison. (G) Western blots for SIRT6, β-TRCP, ATF4, and β-ACTIN in classical lines treated with nonspecific (siCTRL), SIRT6-specific (siSIRT6), and β-TRCP–specific (sibTRCP) siRNAs. (H) Immunoprecipitation of ATF4 followed by Western blotting for ATF4, SIRT6, and vinculin in Panc3.27 cells transfected with empty vector or SIRT6 overexpression plasmids. (I) Immunofluorescence for single transfection of SIRT6 overexpression (OE) with a hemagglutinin (HA) tag (green) or ATF4 overexpression with a FLAG tag (red) in Panc3.27 cells. (J) Immunofluorescence for cotransfection of SIRT6 and ATF4 overexpression in Panc3.27 cells stained for FLAG-ATF4 (red). (K) Immunofluorescence for cotransfection of ATF4 plus empty vector overexpression and ATF4 plus SIRT6 overexpression in Panc3.27 cells with a cycloheximide treatment of 0, 15, and 30 min stained for FLAG-ATF4 (red). (L) Immunofluorescence for endogenous ATF4 (red) in two classical and one basal cell line. All immunofluorescence images have a scale bar of 25 μm. Data are representative of at least two independent experiments. *P ≤ 0.05 and **P ≤ 0.01 (two-tailed unpaired Student’s t test).
mTOR (mammalian target of rapamycin) is known to activate ATF4 through a mechanism distinct from its canonical induction by the ISR, which prompted us to examine this in the context of SIRT6 (35). We performed SIRT6 knockdown in two classical PDAC cell lines and assessed mTOR pathway components. Loss of SIRT6 resulted in clear ATF4 reduction but no change in expression or phosphorylation of mTOR and its pathway components (fig. S5F). Therefore, we concluded that the mTOR pathway is not integral to SIRT6 regulation of ATF4.
To determine whether SIRT6 regulated ATF4 protein degradation, we treated classical cells with a proteosome inhibitor (MG132) and observed accumulation of ATF4 protein to be comparable regardless of SIRT6 status (Fig. 5C), indicating that the proteosome is involved in SIRT6 regulation of ATF4. In addition, treating classical cells with cycloheximide to block protein synthesis caused ATF4 protein abundance to decline at a significantly faster rate (P < 0.05) with SIRT6 knockdown (Fig. 5D). Conversely, restoration of SIRT6 in basal cells and treating with cycloheximide caused ATF4 protein abundance to decline at a slower rate as compared with the empty vector control (Fig. 5E), confirming that SIRT6 stabilizes ATF4 protein.
To determine whether ATF4 instability is a general feature of basal PDAC given its low SIRT6 status, we treated four basal and classical PDAC cell lines with cycloheximide to observe ATF4 protein stability at baseline. We found that ATF4 protein degraded at a significantly faster rate (P < 0.05) in the basal cell lines than in the classical cell lines (Fig. 5F), validating that SIRT6 protein expression dictates ATF4 protein stability. Moreover, we sought to determine whether ATF4 was targeted for degradation by β-TRCP (β-transducin repeat-containing E3 ubiquitin protein ligase), the receptor component of the E3 ubiquitin ligase previously shown to bind and target ATF4 for proteosome degradation (37). We knocked down β-TRCP in the absence of SIRT6 and achieved a partial rescue of ATF4 protein abundance (Fig. 5G). Therefore, we concluded that SIRT6 control of ATF4 protein stability is protective against β-TRCP–mediated proteasomal degradation and explains the differential ATF4 protein amounts in basal and classical PDAC.
To further investigate how SIRT6 stabilizes ATF4, we performed coimmunoprecipitation to assess direct binding of SIRT6 and ATF4. ATF4 was pulled down in either a basal cell line expressing SIRT6 or an empty vector control. SIRT6 was shown to specifically bind to ATF4 with no nonspecific binding in the empty vector or isotope control (Fig. 5H). Next, immunofluorescence was performed to determine whether ATF4 localization changed upon stabilization by SIRT6. Individual overexpression of only SIRT6 or only ATF4 in a basal line showed diffuse localization of each protein throughout the nucleus (Fig. 5I). However, when SIRT6 and ATF4 were both overexpressed in the same cell, we saw a marked change in ATF4 localization to a distinct speckling pattern within the nucleus (Fig. 5J). To identify whether this speckling pattern was protective against degradation of ATF4, we overexpressed ATF4 plus an empty vector control or ATF4 plus SIRT6 in the same basal line and treated with cycloheximide. As expected, ATF4 staining showed rapid degradation with cycloheximide treatment in the absence of SIRT6. Conversely, in the presence of SIRT6, we saw stabilization of ATF4 and maintenance of the speckling pattern throughout cycloheximide treatment (Fig. 5K).
Previous work has identified that stabilization of ATF4 occurs when ATF4 is localized to nuclear speckles (38). To determine whether SIRT6 induced ATF4 localization to nuclear speckles in PDAC, we performed the same overexpression experiment as in Fig. 5K and stained for nuclear speckles with the SC35 antibody. In the absence of SIRT6, there was no colocalization of ATF4 and the SC35 nuclear speckles marker; however, the addition of SIRT6 induced colocalization of ATF4 and SC35 as well as maintained this colocalization during cycloheximide treatment (fig. S5G). Moreover, SIRT6 colocalized to these speckles with ATF4 while maintaining localization in other regions of the nucleus (fig. S5H). Last, endogenous staining of ATF4 in SIRT6high/classical lines showed a similar speckling pattern to the ATF4 and SIRT6 overexpression in a SIRT6low line (Fig. 5L). For comparison, the SIRT6low/basal line showed no endogenous ATF4 staining (Fig. 5L). Together, these data indicated that SIRT6 stabilizes ATF4 through binding and directing ATF4 to nuclear speckles, which protects ATF4 from degradation by the proteosome. This protection of ATF4 by SIRT6 thereby enables transcriptional activation of ATF4 target genes and control of the ISR.
Inability to induce ISR confers sensitivity to transcriptional inhibition
To determine whether ISR activation could protect basal cells from transcriptional CDK inhibition, we altered GADD34 expression in both basal and classical PDAC cell lines. Knockdown of GADD34 was used to induce activation of the ISR by keeping eIF2a phosphorylated. GADD34 knockdown both increased p-eIF2α between DMSO (dimethyl sulfoxide) siCTRL and DMSO siGADD34 and significantly reduced sensitivity to THZ1 (P < 0.04), as shown by loss of cleaved caspase-3 activation and reduced annexin/propidium iodide staining in two independent basal PDAC cell lines (Fig. 6A and fig. S6A). Conversely, preventing activation of the ISR in classical PDAC cells by overexpression of GADD34 resulted in both reduced p-eIF2α and significantly higher amounts of apoptosis in response to THZ1 (P < 0.03) (Fig. 6B and fig. S6B). Similarly, knockdown of ATF4 sensitized classical PDAC cells to THZ1 (Fig. 6C). Thus, our results suggest that the inability to launch the ISR in basal PDAC cells renders them sensitive to transcriptional inhibition, whereas a constitutively active ISR in classical PDAC lines confers resistance.
Fig. 6. The ISR confers sensitivity or resistance to transcriptional inhibitors.
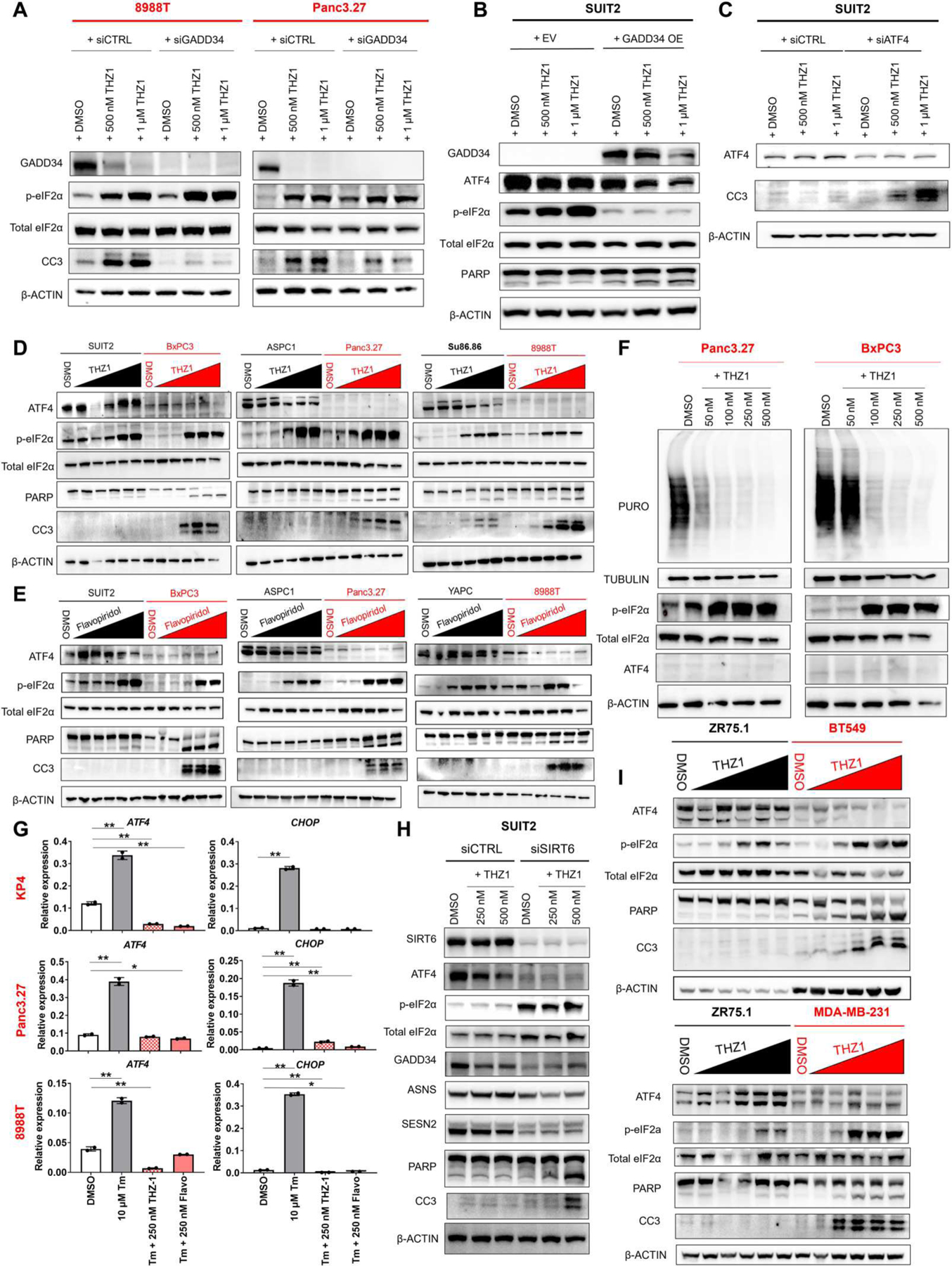
(A) Western blots for GADD34, p-eIF2α, total eIF2α, CC3, and β-ACTIN in two basal PDAC lines transfected with control or GADD34-specific siRNAs and treated with increasing doses of THZ1. (B) Western blots for GADD34, ATF4, p-eIF2α, total eIF2α, PARP, and β-ACTIN in a classical PDAC line transduced with either an empty vector or GADD34 overexpression and treated with increasing doses of THZ1. (C) Western blots for ATF4, CC3, and β-ACTIN in a classical PDAC line transfected with either control or ATF4-specific siRNAs and treated with increasing doses of THZ1. (D) Western blots for ATF4, p-eIF2α, total eIF2α, PARP, CC3, and β-ACTIN in basal (red) compared with classical (black) PDAC cell lines treated with increasing doses of THZ1 (20 nM to 5 μM). (E) Western blots for ATF4, p-eIF2α, total eIF2α, PARP, CC3, and β-ACTIN in basal (red) compared with classical (black) PDAC cell lines treated with increasing doses of flavopiridol (20 nM to 5 μM). (F) Puromycin incorporation and Western blotting for TUBULIN, p-eIF2α, total eIF2α, ATF4, and β-ACTIN in basal PDAC cell lines treated with increasing doses of THZ1. (G) qRT-PCRs of ATF4 and CHOP in basal lines treated with tunicamycin only, tunicamycin with THZ1, and tunicamycin with flavopiridol. Total treatment time for tunicamycin was 5 hours, and total treatment time for THZ1 and flavopiridol was 16 hours. (H) Western blots for SIRT6, ATF4, p-eIF2α, total eIF2α, GADD34, ASNS, SESN2, PARP, CC3, and β-ACTIN in SUIT2 PDAC cells transfected with nonspecific or SIRT6-specific siRNAs and treated with 250 or 500 nM THZ1 for 20 hours. (I) Western blots for ATF4, p-eIF2α, total eIF2α, PARP, CC3, and β-ACTIN in basal BT549 and MDA-MB-231 (red) compared with luminal ZR75.1 (black) breast cancer cell lines treated with increasing doses of THZ1 (20 nM to 5 μM). Error bars represent ±SEM between technical duplicates. Data are representative of at least two independent experiments. *P ≤ 0.05; **P ≤ 0.01 (two-tailed unpaired Student’s t test).
To test this hypothesis, we treated a panel of three classical and three basal PDAC cell lines with increasing doses of either THZ1 or flavopiridol and monitored for the expression of ISR markers ATF4 and p-eIF2α as well as the induction of apoptosis (Fig. 6, D and E). All six cell lines were able to induce phosphorylation of eIF2α, whereas only the basal lines suppressed global mRNA translation upon drug treatment (Figs. 4B and 6F). However, induction of the ISR remained incomplete in the SIRT6low/basal PDAC lines because none was able to express ATF4 (Fig. 6, D to F). In contrast, tunicamycin was able to induce the expression of both ATF4 and its target gene CHOP in all basal PDAC lines tested (Figs. 4, I and J, and 6G), a response not seen when these cells were treated with either THZ1 or flavopiridol (Fig. 6G). Moreover, silencing of SIRT6 in SIRT6high/classical PDAC reduced baseline expression of ATF4 to basal PDAC amounts. These cells behaved similarly to basal PDAC cells in the presence of THZ1, where p-eIF2α was increased, but ATF4 and many ATF4 target genes were not able to be induced, thus sensitizing classical lines to THZ1 treatment (Fig. 6H).
Last, we asked whether the key biological differences in stress response between the more aggressive, mesenchymal-like basal subtype and the less aggressive, epithelial-like classical subtype may extrapolate to other cancers with similar subtype classifications. The basal subtype of PDAC was classified as such because of overlap in transcriptional signatures with basal tumors of the bladder and breast (9). In addition, basal breast cancers, including triple-negative breast cancers, were recently shown to be sensitive to THZ1 when compared with the ER+ luminal subtype (24). We found that, similar to basal PDAC, basal breast cancer cell lines (BT549 and MDA-MB-231) failed to activate a complete ISR program and were more sensitive to THZ1 than their luminal counterparts (Fig. 6I). Thus, although the upstream signaling of the ISR remained intact in basal PDAC and basal breast cancer cell lines, they were unable to activate downstream regulators in the presence of transcriptional inhibitors such as THZ1 or flavopiridol, rendering them vulnerable to this class of compounds.
To show that the ISR confers differential sensitivity to transcriptional inhibition in vivo, we implanted SIRT6 KO and SIRT6 WT GEMM-derived PDAC organoids subcutaneously in immunocompetent syngeneic C57BL/6 mice and treated them daily with flavopiridol for 3 weeks. Flavopiridol inhibited the growth of the SIRT6 KO but not the SIRT6 WT organoid-derived tumors (Fig. 7A). The histology of the implanted PDAC organoid tumors was independently verified by a pathologist to be indistinguishable from the original GEMM tumors for both SIRT6 KO and SIRT6 WT models (Fig. 7B).
Fig. 7. SIRT6 expression correlates with basal PDAC status and flavopiridol sensitivity in vivo.
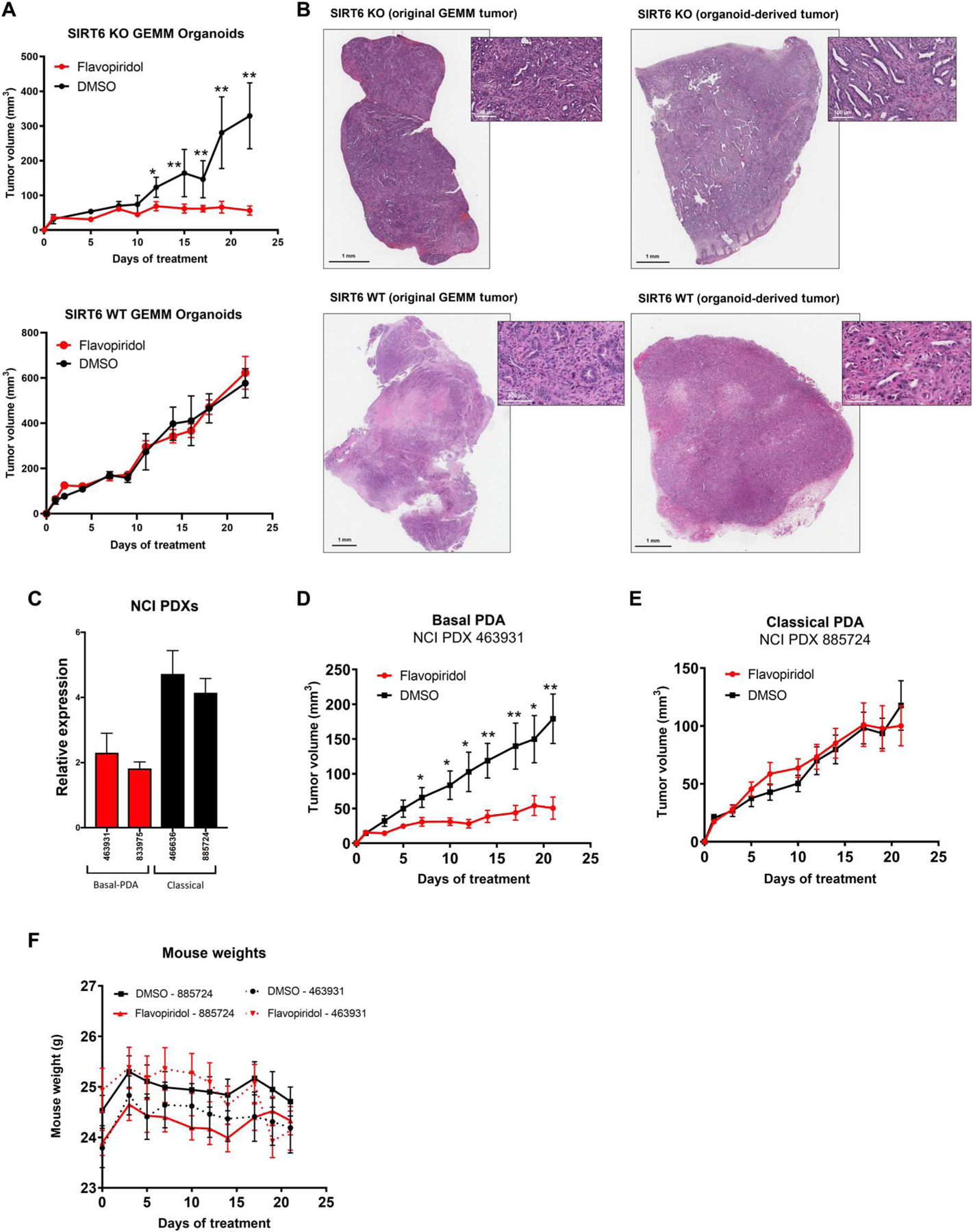
(A) Tumor volume of organoid xenografts derived from SIRT6 KO and SIRT6 WT mice after treatment with once-daily flavopiridol (5 mg/kg) (red) compared with DMSO control (black) for 21 days (DMSO n = 3, flavopiridol n = 3). (B) Hematoxylin and eosin staining showing comparison between original tumor and organoid-derived tumor from the same source for SIRT6 KO and SIRT6 WT. (C) PDAC subtype–specific expression of SIRT6 in patient-derived xenografts (PDXs) from the NCI. (D and E) Tumor volume of NCI PDX 463931 (basal) (D) and NCI PDX 885724 (classical) (E) xenografts after treatment with once-daily flavopiridol (5 mg/kg) (red) compared with DMSO control (black) for 21 days (DMSO n = 10, flavopiridol n = 10). (F) Whole-body weights of mice implanted with NCI PDX 885724 (classical) and NCI PDX 463931 (basal) xenografts after treatment with once-daily flavopiridol (5 mg/kg) (red) compared with DMSO control (black) for 21 days. *P ≤ 0.05 and **P ≤ 0.01 (two-tailed unpaired Student’s t test).
We next used two different PDX models, subtyped as classical and basal, respectively, on the basis of publicly available RNA-seq data from the NCI (National Cancer Institute) patient-derived model repository (PDMR) and confirmed the designations based on abundance of SIRT6 expression (Fig. 7C). The models were treated with flavopiridol for 3 weeks, and growth of basal PDXs was inhibited (Fig. 7D). In comparison, classical PDXs were not sensitive (Fig. 7E). Flavopiridol also did not cause substantial toxicity, as assessed by maintenance of animal weight throughout treatment (Fig. 7F). Together, these results suggested that transcriptional inhibition of the basal subtype of PDAC, defined by SIRT6 expression, is a specific and potent treatment strategy for this deadly disease.
DISCUSSION
Here, we have found that SIRT6, a NAD+-dependent histone deacetylase, can regulate ATF4 stability in response to cellular stresses and is highly expressed in the classical PDAC subtype, whereas it is lost in the basal PDAC subtype. As a result, basal PDAC has a suppressed ISR at baseline, whereas classical PDAC has an elevated ISR at baseline. We further found that SIRT6 is required for ISR activation through its regulation of ATF4 stability, which enables the classical PDAC subtype to survive stressors such as inhibition of transcriptional CDKs. Normally, basal PDAC cells would induce SIRT6 in response to an applied stressor, with a subsequent increase in ATF4 and an adaptive response to that stress. However, when the applied stress is transcriptional CDK inhibition, SIRT6 cannot be induced and is therefore not present to stabilize ATF4, which prevents the ISR from becoming active. This reduced or inactive ISR prevents the cell from adequately responding to the stress of transcriptional CDK inhibition, which drives the basal PDAC cells toward cell death. Genetic manipulation of key regulators of the ISR, including ATF4 and GADD34, was sufficient to alter sensitivity to these inhibitors of transcriptional CDKs in both subtypes.
PDAC regulation of ATF4 through stability and degradation has not been studied as in-depth as regulation of ATF4 through translational control. However, some insightful work has found that ATF4 has a targeted E3 ligase receptor (β-TRCP), which directly interacts with and guides ATF4 to the proteasome for degradation. This interaction is mediated by ATF4 phosphorylation at serine-219, which is within a similar motif as other β-TRCP substrates (37). We found that β-TRCP is important for ATF4 degradation by the proteasome in PDAC, and loss of β-TRCP in the absence of SIRT6 enables a partial rescue of ATF4 protein abundance. However, because this rescue is partial and not complete, β-TRCP may not be the only E3 ligase receptor component that binds ATF4. Considering that loss of SIRT6 enables ATF4 to escape the protection of nuclear speckles, this would increase ATF4 abundance outside of the speckles and increase potential to bind with other E3 ligase receptor components. In addition, it has been identified that epigenetic modifiers such as histone acetyltransferases are able to stabilize ATF4 through mechanisms independent of their catalytic activity. The histone acetyltransferase p300 can stabilize and localize ATF4 to nuclear speckles, thereby increasing its transcriptional activity (38). Our data identify that this model of epigenetic modifiers, such as p300 and SIRT6, stabilizing and localizing ATF4 to nuclear speckles holds true in PDAC. This model illustrates how protein stability encompasses another layer of posttranslational regulation of the ATF4 signaling pathway. Further work will be needed to determine whether the catalytic activity of SIRT6 is necessary for its ability to stabilize ATF4.
Reprogramming of protein synthesis can collaborate with epigenetic and metabolic programs to determine cell state. Global translation is frequently dysregulated in cancer, and it remains an open question whether translation drives or merely reflects cellular identity. The ISR and control of global translation through phosphorylated eIF2α may have an evolutionarily conserved role in cell plasticity in response to stress (39). Studies of stem cells in murine models and human cancers have linked p-eIF2α and the ISR to cancer cell plasticity and maintenance of stem cell populations (40, 41). Protein synthesis rates may dictate cell lineage in epithelial populations as well. For example, progenitor basal cells exhibit higher amounts of protein synthesis compared with luminal cells within the murine prostate. This observation is maintained in the context of prostate cancer (42). Similarly, in transdifferentiated (EMT) breast cancer cells, overproduction of extracellular matrix components leads to ER stress and activation of the PERK (eukaryotic translation initiation factor 2 α kinase 3)/eIF2α axis, which is paralleled by invasion and metastasis (43). Breast cancer cells can also increase plasticity in response to mTOR inhibitors and chemotherapeutics that induce translational activation of stemness factors NANOG (Nanog homeobox), SNAIL (snail family transcriptional repressor 1), and NODAL (nodal growth differentiation factor). These effects are overcome with drugs that antagonize the translational reprogramming caused by p-eIF2α, suggesting that the ISR drives breast cancer plasticity (44). In the epidermis, an eIF2B5-mediated translational program leads to loss of progenitor self-renewal, which limits tumor initiation and growth (45). Collectively, these observations suggest a multifaceted role for the ISR in cancer cell phenotypic switching.
Our study is limited by several factors. First, we recognize that patient tumors can be heterogeneous in their subtypes, but the more we understand about the individual subtypes, the better we can develop targeted therapies that can convert cell state, constrain plasticity, and kill the resulting tumor population. In addition, we recognize that knockdown of SIRT6 in classical PDAC does not reduce ATF4 abundance as low as that seen in basal PDAC. Considering the complexity of ATF4 regulation, other mechanisms in addition to SIRT6 may control ATF4 abundance in PDAC. Specifically, there could be additional modes of regulation keeping ATF4 low in basal PDAC other than lack of SIRT6. Although we observe marked sensitivity of basal PDAC to inhibitors of transcriptional CDKs in mouse models, this may not always predict response rates in human patients. However, we have aimed to make our preclinical studies as relevant as possible by implanting murine PDAC organoids into immunocompetent syngeneic C57BL/6 mice to preserve the effects of the immune system on treatment. In addition, we treated PDX models in immunocompromised mice to account for differential responses between human and murine tumors.
Last, examining the ability for subtype switching through epigenetic adaptation and inhibition of key drivers of the basal/classical state would be an important future extension of this study to see how this could alter therapeutic response and potential acquired resistance to therapy over time. An understanding of this regulation may allow us to potentially convert classical tumors to basal tumors to reduce intratumoral heterogeneity and constrain plasticity before applying subtype-specific therapies like transcriptional CDK inhibition.
Treatment of cancer patients with a highly selective and potent oral CDK7 inhibitor has already shown promising results in a phase 1 clinical trial across multiple tumor types (46). Clinical activity was most notable in pancreatic cancer, where stable disease was noted in 38% of patients. Most adverse events were low grade and reversible. These results have led to expansion of this trial in PDAC (46). Our study provides an understanding of the biology that determines sensitivity to CDK7 inhibitors, which will aid further clinical development of these inhibitors in PDAC. Investigations of resistance mechanisms and combination therapies with transcriptional CDK inhibitors are important next steps in providing successful therapeutic options to patients with PDAC.
MATERIALS AND METHODS
Study design
The goals of this study were to uncover a functional relationship between PDAC subtypes and SIRT6 expression as well as define the mechanism of subtype-specific sensitivity to inhibition of transcriptional CDKs in PDAC. This objective was accomplished by (i) characterizing SIRT6 regulation of the ISR as a defining feature of PDAC subtypes, (ii) dissecting the mechanism by which SIRT6 controls ATF4 and thereby influences cellular responses to stress in PDAC, (iii) identifying the role of the ISR in sensitivity and resistance to inhibitors of transcriptional CDKs, and (iv) conducting a series of in vitro and in vivo studies to delineate the therapeutic potential of transcriptional CDK inhibitors in basal PDAC. For all experiments, our sample size was determined on the basis of past experience and published literature. We used the maximum number of mice available for a given experiment based on the following criteria: the number of mice available within the correct age range per strain and tumor availability after implantation of human PDX tissue or organoids. For all studies, mice were randomly assigned to each treatment group. A blinded pathologist performed all histological analyses for murine studies. The number of replicates is specified in each figure legend.
Organoid xenograft
All mouse procedures were conducted in accordance with the Fred Hutchinson Cancer Center (FHCC) Institutional Animal Care and Use Committee (IACUC) guidelines and the ARRIVE guidelines. C57BL/6J (BL/6J) mice (000664, JAX) were purchased from the Jackson Laboratory. Injections of organoid cultures for the generation of subcutaneously grafted organoid tumors were conducted as described previously (47). Typically, 1 × 106 to 2 × 106 cells in a 100-μl suspension of 50% Matrigel (Corning) in phosphate-buffered saline (PBS) were injected into each flank of the mouse. When average tumor diameters of 3 to 5 mm3 were reached, mice were treated with a once daily intraperitoneal injection of vehicle or flavopiridol (5 mg/kg; S1230, Selleck) diluted with 5% dextrose in water solution (600063, Bound Tree Medical) for 21 days. Caliper measurements of tumors and body weights were recorded three times per week.
Patient-derived xenograft
PDX models were acquired from the NCI PDMR: 8333975-119-r (lot no. CD1183), 463931-005-r (lot no. LS2265), 466636-057-r (lot no. MD0903), and 885724-159-r (lot no. AM1179). Vials of cryopreserved PDX tissue fragments were revived and subsequently repassaged as fresh whole tissue into both NSG (NOD scid gamma) and nude mice (Foxn1nu). Freshly repassaged tissue was then used for implantation of study cohorts of nude mice (002019, JAX). All work in mice was approved by IACUC (FHCC IACUC protocol 50935-200016). Every PDX model tested negative for human pathogens (IDEXX h-IMPACT panel). An aseptic standardized procedure was used uniformly: ~1- to 2-mm3 PDX tissue chunks (37°C) were rinsed in RPMI, suspended in Matrigel (Corning), and implanted subcutaneously into the right flanks of 6- to 8-week-old female mice (see strain notes above) under isoflurane anesthesia. Preemptive analgesia was provided using buprenorphine SR (0.05 mg/kg). Post-surgically, tumor growth and body condition were monitored, and tumors were measured with electronic calipers. When average tumor diameters of 3 to 5 mm3 were reached, mice were treated with a once daily intraperitoneal injection of vehicle or flavopiridol (5 mg/kg; S1230, Selleck) diluted with 5% dextrose in water solution (600063, Bound Tree Medical) for 21 days. Caliper measurements of tumors and body weights were recorded three times per week.
Statistical analyses
Statistical significance was determined by specific tests and presented as means ± SEM as indicated in the figure legends. Experimental raw values were depicted when possible or normalized to internal controls. When comparing data from two groups, paired or unpaired Student’s two-tailed t test was used to determine significance, which was set at a P value of <0.05. Statistical analyses were performed using GraphPad Prism and R or Python packages. RNA-seq data were analyzed using Python package HTSeq and Bioconductor packages edgeR and goseq. ChIP-seq data were analyzed using Python packages MACS2 and ROSE. Both RNA-seq and ChIP-seq data used R package VennDiagram, and false discovery rates (FDRs) were calculated using the Benjamini-Hochberg method and cut off at 5%. Pearson’s correlation coefficient and corresponding P value were used to measure the extent of correlation between SIRT6 expression and THZ1 sensitivity. Additional information can be found in Supplementary Materials and Methods.
Supplementary Material
Acknowledgments:
We thank R. Eisenman as well as past and present members of the Kugel laboratory for helpful discussions and specifically L. Boila for helpful insight with the flow cytometry experiments and N. Yamamoto for help compiling statistical information.
Funding:
This work was supported in part by NIH grants 5R37CA241472 (to S.K.), R01CA223483 (to S.R.H.), GM126908 (to D.H.P.), GM135362 (to A.C.H.), and R37CA230617 (to A.C.H.); a V scholar award (V218-004) from the V Foundation for Cancer Research (to S.K.); a Swim across America Pancreas Cancer Development Research Award (to S.K.); Chromosome Metabolism (to N.K.); Cancer training grant 5T32CA009657 (to N.K.); American Cancer Society Postdoctoral Fellowship (133107-PF-20-012-01) (to N.K.); and a Walter Benjamin fellowship from the German Research Foundation (to S. Dobersch). This research was supported by the Genomics and Bioinformatics Core and by Elizabeth Cromwell from the Preclinical Modeling Core in Shared Resource of the Fred Hutch/University of Washington Cancer Consortium (P30 CA015704).
Footnotes
Supplementary Materials
This PDF file includes:
Other Supplementary Material for this manuscript includes the following: MDAR Reproducibility Checklist
Competing interests: S. Dobersch owns stocks from QIAGEN and Merck. All other authors declare no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. The RNA-seq and ChIP-seq data generated in this study are publicly available in GEO at the National Center for Biotechnology Information under the accession number GSE181606.
REFERENCES AND NOTES
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM, Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 74, 2913–2921 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Ryan DP, Hong TS, Bardeesy N, Pancreatic adenocarcinoma. N. Engl. J. Med 371, 1039–1049 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA, Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA, Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Brar G, Blais EM, Joseph Bender R, Brody JR, Sohal D, Madhavan S, Picozzi VJ, Hendifar AE, Chung VM, Halverson D, Mikhail S, Matrisian LM, Rahib L, Petricoin E, Pishvaian MJ, Multi-omic molecular comparison of primary versus metastatic pancreatic tumours. Br. J. Cancer 121, 264–270 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayashi A, Fan J, Chen R, Ho Y-J, Makohon-Moore AP, Lecomte N, Zhong Y, Hong J, Huang J, Sakamoto H, Attiyeh MA, Kohutek ZA, Zhang L, Boumiza A, Kappagantula R, Baez P, Bai J, Lisi M, Chadalavada K, Melchor JP, Wong W, Nanjangud GJ, Basturk O, O’Reilly EM, Klimstra DS, Hruban RH, Wood LD, Overholtzer M, Iacobuzio-Donahue CA, A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma. Nat. Cancer 1, 59–74 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, Feiler HS, Ko AH, Olshen AB, Danenberg KL, Tempero MA, Spellman PT, Hanahan D, Gray JW, Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med 17, 500–503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, Nourse C, Murtaugh LC, Harliwong I, Idrisoglu S, Manning S, Nourbakhsh E, Wani S, Fink L, Holmes O, Chin V, Anderson MJ, Kazakoff S, Leonard C, Newell F, Waddell N, Wood S, Xu Q, Wilson PJ, Cloonan N, Kassahn KS, Taylor D, Quek K, Robertson A, Pantano L, Mincarelli L, Sanchez LN, Evers L, Wu J, Pinese M, Cowley MJ, Jones MD, Colvin EK, Nagrial AM, Humphrey ES, Chantrill LA, Mawson A, Humphris J, Chou A, Pajic M, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Lovell JA, Merrett ND, Toon CW, Epari K, Nguyen NQ, Barbour A, Zeps N, Moran-Jones K, Jamieson NB, Graham JS, Duthie F, Oien K, Hair J, Grutzmann R, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Rusev B, Capelli P, Salvia R, Tortora G, Mukhopadhyay D, Petersen GM; Australian Pancreatic Cancer Genome Initiative, Munzy DM, Fisher WE, Karim SA, Eshleman JR, Hruban RH, Pilarsky C, Morton JP, Sansom OJ, Scarpa A, Musgrove EA, Bailey UMH, Hofmann O, Sutherland RL, Wheeler DA, Gill AJ, Gibbs RA, Pearson JV, Waddell N, Biankin AV, Grimmond SM, Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, Smyla JK, Anderson JM, Kim HJ, Bentrem DJ, Talamonti MS, Iacobuzio-Donahue CA, Hollingsworth MA, Yeh JJ, Virtual micro-dissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet 47, 1168–1178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puleo F, Nicolle R, Blum Y, Cros J, Marisa L, Demetter P, Quertinmont E, Svrcek M, Elarouci N, Iovanna J, Franchimont D, Verset L, Galdon MG, Devière J, de Reyniès A, Laurent-Puig P, Van Laethem JL, Bachet J-B, Maréchal R, Stratification of pancreatic ductal adenocarcinomas based on tumor and microenvironment features. Gastroenterology 155, 1999–2013.e3 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O’Kane GM, Connor AA, Denroche RE, Grant RC, McLeod J, Wilson JM, Jang GH, Zhang A, Liang SB, Borgida A, Chadwick D, Kalimuthu S, Lungu I, Bartlett JMS, Krzyzanowski PM, Sandhu V, Tiriac H, Froeling FEM, Karasinska JM, Topham JT, Renouf DJ, Schaeffer DF, Jones SJM, Marra MA, Laskin J, Chetty R, Stein LD, Zogopoulos G, Haibe-Kains B, Campbell PJ, Tuveson DA, Knox JJ, Fischer SE, Gallinger S, Notta F, Author correction: Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet 52, 463 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Campagna D, Cope L, Lakkur SS, Henderson C, Laheru D, Iacobuzio-Donahue CA, Gene expression profiles associated with advanced pancreatic cancer. Int. J. Clin. Exp. Pathol 1, 32–43 (2008). [PMC free article] [PubMed] [Google Scholar]
- 13.Lomberk G, Blum Y, Nicolle R, Nair A, Gaonkar KS, Marisa L, Mathison A, Sun Z, Yan H, Elarouci N, Armenoult L, Ayadi M, Ordog T, Lee J-H, Oliver G, Klee E, Moutardier V, Gayet O, Bian B, Duconseil P, Gilabert M, Bigonnet M, Garcia S, Turrini O, Delpero JR, Giovannini M, Grandval P, Gasmi M, Secq V, De Reynies A, Dusetti N, Iovanna J, Urrutia R, Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun 9, 1978 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaferia GR, Balestrieri C, Prosperini E, Nicoli P, Spaggiari P, Zerbi A, Natoli G, Dissection of transcriptional and cis-regulatory control of differentiation in human pancreatic cancer. EMBO J 35, 595–617 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosch-Presegué L, Vaquero A, Sirtuins in stress response: Guardians of the genome. Oncogene 33, 3764–3775 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Kugel S, Mostoslavsky R, Chromatin and beyond: The multitasking roles for SIRT6. Trends Biochem. Sci 39, 72–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kugel S, Sebastian C, Fitamant J, Ross KN, Saha SK, Jain E, Gladden A, Arora KS, Kato Y, Rivera MN, Ramaswamy S, Sadreyev RI, Goren A, Deshpande V, Bardeesy N, Mostoslavsky R, SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell 165, 1401–1415 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sebastian C, Zwaans BMM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D, Cosentino C, Greenson JK, MacDonald AI, McGlynn L, Maxwell F, Edwards J, Giacosa S, Guccione E, Weissleder R, Bernstein BE, Regev A, Shiels PG, Lombard DB, Mostoslavsky R, The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 151, 1185–1199 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, Jenkins CE, Hannett NM, McMillin D, Sanda T, Sim T, Kim ND, Look T, Mitsiades CS, Weng AP, Brown JR, Benes CH, Marto JA, Young RA, Gray NS, Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, Ficarro SB, Elkins JM, Liang Y, Hannett NM, Manz T, Hao M, Bartkowiak B, Greenleaf AL, Marto JA, Geyer M, Bullock AN, Young RA, Gray NS, Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol 12, 876–884 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, Zhang L, Li M, Meng W, Ponnuswami A, Sun W, Ma J, Hulleman E, Swigut T, Wysocka J, Tang Y, Monje M, Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 31, 635–652.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, Zhang J, Shimamura T, Capelletti M, Reibel JB, Cavanaugh JD, Gao P, Liu Y, Michaelsen SR, Poulsen HS, Aref AR, Barbie DA, Bradner JE, George RE, Gray NS, Young RA, Wong K-K, Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 26, 909–922 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z, Peng H, Wang X, Yin X, Ma P, Jing Y, Cai M-C, Liu J, Zhang M, Zhang S, Shi K, Gao W-Q, Di W, Zhuang G, Preclinical efficacy and molecular mechanism of targeting CDK7-dependent transcriptional addiction in ovarian cancer. Mol. Cancer Ther 16, 1739–1750 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, Yuzugullu H, Von T, Li H, Lin Z, Stover DG, Lim E, Wang ZC, Iglehart JD, Young RA, Gray NS, Zhao JJ, CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 163, 174–186 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinz S, Romanoski CE, Benner C, Glass CK, The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol 16, 144–154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA, Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sur I, Taipale J, The role of enhancers in cancer. Nat. Rev. Cancer 16, 483–493 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Nilson KA, Guo J, Turek ME, Brogie JE, Delaney E, Luse DS, Price DH, THZ1 reveals roles for Cdk7 in co-transcriptional capping and pausing. Mol. Cell 59, 576–587 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chao SH, Price DH, Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem 276, 31793–31799 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Nilson KA, Lawson CK, Mullen NJ, Ball CB, Spector BM, Meier JL, Price DH, Oxidative stress rapidly stabilizes promoter-proximal paused Pol II across the human genome. Nucleic Acids Res 45, 11088–11105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ball CB, Nilson KA, Price DH, Use of the nuclear walk-on methodology to determine sites of RNA polymerase II initiation and pausing and quantify nascent RNAs in cells. Methods 159–160, 165–176 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jana S, Deo R, Hough RP, Liu Y, Horn JL, Wright JL, Lam H-M, Webster KR, Chiang GG, Sonenberg N, Hsieh AC, mRNA translation is a therapeutic vulnerability necessary for bladder epithelial transformation. JCI Insight 6, e144920 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Z, Huang Y, Zhu L, Yang K, Liang K, Tan J, Yu B, SIRT6 promotes angiogenesis and hemorrhage of carotid plaque via regulating HIF-1α and reactive oxygen species. Cell Death Dis 12, 77 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang N, Yang X, Yuan F, Zhang L, Wang Y, Wang L, Mao Z, Luo J, Zhang H, Zhu WG, Zhao Y, Increased amino acid uptake supports autophagy-deficient cell survival upon glutamine deprivation. Cell Rep 23, 3006–3020 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Park Y, Reyna-Neyra A, Philippe L, Thoreen CC, mTORC1 balances cellular amino acid supply with demand for protein synthesis through post-transcriptional control of ATF4. Cell Rep 19, 1083–1090 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D, An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Lassot I, Segeral E, Berlioz-Torrent C, Durand H, Groussin L, Hai T, Benarous R, Margottin-Goguet F, ATF4 degradation relies on a phosphorylation-dependent interaction with the SCFβTrCP ubiquitin ligase. Mol. Cell. Biol 21, 2192–2202 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lassot I, Estrabaud E, Emiliani S, Benkirane M, Benarous R, Margottin-Goguet F, p300 modulates ATF4 stability and transcriptional activity independently of its acetyltransferase domain. J. Biol. Chem 280, 41537–41545 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Garcia-Jimenez C, Goding CR, Starvation and pseudo-starvation as drivers of cancer metastasis through translation reprogramming. Cell Metab 29, 254–267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friend K, Brooks HA, Propson NE, Thomson JA, Kimble J, Embryonic stem cell growth factors regulate eIF2α phosphorylation. PLOS ONE 10, e0139076 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Galen P, Mbong N, Kreso A, Schoof EM, Wagenblast E, Ng SWK, Krivdova G, Jin L, Nakauchi H, Dick JE, Integrated stress response activity marks stem cells in normal hematopoiesis and leukemia. Cell Rep 25, 1109–1117.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Hsieh AC, Nguyen HG, Wen L, Edlind MP, Carroll PR, Kim W, Ruggero D, Cell type-specific abundance of 4EBP1 primes prostate cancer sensitivity or resistance to PI3K pathway inhibitors. Sci. Signal 8, ra116 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng YX, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JH, Proia TA, Jin DX, Reinhardt F, Ploegh HL, Wang Q, Gupta PB, Epithelial-to-mesenchymal transition activates PERK–eIF2α and sensitizes cells to endoplasmic reticulum stress. Cancer Discov 4, 702–715 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Jewer M, Lee L, Leibovitch M, Zhang G, Liu J, Findlay SD, Vincent KM, Tandoc K, Dieters-Castator D, Quail DF, Dutta I, Coatham M, Xu Z, Puri A, Guan BJ, Hatzoglou M, Brumwell A, Uniacke J, Patsis C, Koromilas A, Schueler J, Siegers GM, Topisirovic I, Postovit L-M, Translational control of breast cancer plasticity. Nat. Commun 11, 2498 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai EY, Kufeld MN, Schuster S, Arora S, Larkin M, Germanos AA, Hsieh AC, Beronja S, Selective translation of cell fate regulators mediates tolerance to broad oncogenic stress. Cell Stem Cell 27, 270–283.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma M, Bashir B, Hamilton E, Juric D, Papadopoulos K, Richardson D, Shapiro G, Hodgson G, Ke N, D’Ippolito A, Henry S, Zhu L, Rosario M, Jolin H, Roth D, Klimek V, Madigan C, Kelly M, 518MO Tolerability and preliminary clinical activity of SY-5609, a highly potent and selective oral CDK7 inhibitor, in patients with advanced solid tumors. Ann. Oncol 32, S587–S588 (2021). [Google Scholar]
- 47.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, Tuveson DA, IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov 9, 282–301 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, Gracanin A, Oni T, Yu KH, van Boxtel R, Huch M, Rivera KD, Wilson JP, Feigin ME, Ohlund D, Handly-Santana A, Ardito-Abraham CM, Ludwig M, Elyada E, Alagesan B, Biffi G, Yordanov GN, Delcuze B, Creighton B, Wright K, Park Y, Morsink FH, Molenaar IQ, Borel Rinkes IH, Cuppen E, Hao Y, Jin Y, Nijman IJ, Iacobuzio-Donahue C, Leach SD, Pappin DJ, Hammell M, Klimstra DS, Basturk O, Hruban RH, Offerhaus GJ, Vries RG, Clevers H, Tuveson DA, Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanjana NE, Shalem O, Zhang F, Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F, Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trapnell C, Pachter L, Salzberg SL, TopHat: Discovering splice junctions with RNA-seq. Bioinformatics 25, 1105–1111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anders S, Pyl PT, Huber W, HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson MD, McCarthy DJ, Smyth GK, edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen H, Boutros PC, VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12, 35 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Young MD, Wakefield MJ, Smyth GK, Oshlack A, Gene Ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol 11, R14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP, Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langmead B, Trapnell C, Pop M, Salzberg SL, Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA, Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA, Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G, GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T, deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Kane GM, Grunwald BT, Jang G-H, Masoomian M, Picardo S, Grant RC, Denroche RE, Zhang A, Wang Y, Lam B, Krzyzanowski PM, Lungu IM, Bartlett JMS, Peralta M, Vyas F, Khokha R, Biagi J, Chadwick D, Ramotar S, Hutchinson S, Dodd A, Wilson JM, Notta F, Zogopoulos G, Gallinger S, Knox JJ, Fischer SE, GATA6 expression distinguishes classical and basal-like subtypes in advanced pancreatic cancer. Clin. Cancer Res 26, 4901–4910 (2020). [DOI] [PubMed] [Google Scholar]
- 65.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, Loney F, May B, Milacic M, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Weiser J, Wu G, Stein L, Hermjakob H, D’Eustachio P, The reactome pathway knowledgebase. Nucleic Acids Res 48, D498–D503 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Igarashi T, Izumi H, Uchiumi T, Nishio K, Arao T, Tanabe M, Uramoto H, Sugio K, Yasumoto K, Sasaguri Y, Wang KY, Otsuji Y, Kohno K, Clock and ATF4 transcription system regulates drug resistance in human cancer cell lines. Oncogene 26, 4749–4760 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.