

Stenotrophomonas maltophilia codes for a tetracycline-modifying FAD-dependent monooxygenase that shares an overall fold with the tetracycline destructase TetX and possesses a unique active site within this enzyme family.
Keywords: FAD-dependent monooxygenases, tetracycline, antibiotic resistance
Abstract
The resistance of the emerging human pathogen Stenotrophomonas maltophilia to tetracycline antibiotics mainly depends on multidrug efflux pumps and ribosomal protection enzymes. However, the genomes of several strains of this Gram-negative bacterium code for a FAD-dependent monooxygenase (SmTetX) homologous to tetracycline destructases. This protein was recombinantly produced and its structure and function were investigated. Activity assays using SmTetX showed its ability to modify oxytetracycline with a catalytic rate comparable to those of other destructases. SmTetX shares its fold with the tetracycline destructase TetX from Bacteroides thetaiotaomicron; however, its active site possesses an aromatic region that is unique in this enzyme family. A docking study confirmed tetracycline and its analogues to be the preferred binders amongst various classes of antibiotics.
1. Introduction
Antibiotic resistance has become a global health problem of continually increasing magnitude (Lewis, 2020 ▸; Murray et al., 2022 ▸). Stenotrophomonas maltophilia is an opportunistic multidrug-resistant pathogen (Brooke, 2012 ▸) which is responsible for an increasing number of infections (Brooke, 2014 ▸; Chang et al., 2015 ▸). This Gram-negative bacterium attacks immunocompromised hosts in hospital environments, particularly patients affected by cystic fibrosis, lung cancer or chronic obstructive pulmonary disease (Esposito et al., 2017 ▸). Apart from respiratory tract infections, S. maltophilia can also be associated with bacteremia, urinary tract infections, meningitis, endocarditis, osteomyelitis and biliary sepsis. However, the efficacy of antibiotic therapy is often limited due to the low susceptibility of this pathogen to a broad spectrum of drugs, including tetracycline antibiotics (Sánchez, 2015 ▸; Gajdács & Urbán, 2019 ▸).
The molecular structure of tetracycline antibiotics comprises a linear fusion of four rings with various functional groups attached (Chopra & Roberts, 2001 ▸). These molecules are used in the treatment of a broad spectrum of bacterial infections by binding to the 30S subunit of the ribosome, which inhibits protein synthesis (Jenner et al., 2013 ▸; Brodersen et al., 2000 ▸). Tetracycline antibiotics have been widely used against both Gram-positive and Gram-negative bacteria in medicine, and also in the food industry to protect plants, aquacultures and animal growth.
The excessive use of antibiotics has contributed to the development of various mechanisms of drug resistance in bacteria. In the case of tetracycline antibiotics, the leading resistance mechanisms are the overexpression of chromosomally encoded multidrug efflux pumps (Zhao et al., 2018 ▸), ribosomal protection proteins and enzymatic modification of antibiotic molecules (Nguyen et al., 2014 ▸; Grossman, 2016 ▸; He et al., 2015 ▸). Multidrug efflux pumps are essential for the strong resistance of S. maltophilia to tetracycline antibiotics. There is also some evidence that S. maltophilia may have a tetracycline-specific efflux pump (He et al., 2015 ▸). The first two mechanisms, efflux pumps and ribosomal protection, have been suppressed by the design of new semi-synthetic variants of tetracycline antibiotics (for example tigecycline; Yaghoubi et al., 2022 ▸) using a targeted selection of functional groups bound to the tetracycline ring (Sum et al., 1994 ▸). However, the third mechanism, enzymatic modification, which was discovered relatively recently (Yang et al., 2004 ▸; Moore et al., 2005 ▸), still represents a serious threat. The enzymes of the tetracycline destructase family (EC 1.14.13.231; Cheng et al., 2022 ▸; Fang et al., 2020 ▸; Markley & Wencewicz, 2018 ▸) are capable of covalent modification of the tetracycline scaffold, which causes degradation to inactive and unstable compounds, for example 11a-hydroxy-oxytetracycline (Yang et al., 2004 ▸): see the reaction scheme in Fig. 1 ▸. The structural and functional properties of such enzymes from other bacteria have been elucidated by determining crystal structures, for example of TetX from Bacteroides thetaiotaomicron (PDB entries 2y6r and 4a6n; Volkers et al., 2011 ▸, 2013 ▸), TetX2 from B. thetaiotaomicron (PDB entry 3p9u; Walkiewicz et al., 2011 ▸), TetX4 from Escherichia coli (PDB entry 7epw; Cheng et al., 2021 ▸), TetX6 from Chryseobacterium oncorhynchi (PDB entries 8er0 and 8er1; Kumar et al., 2023 ▸), TetX7 from Pseudomonas aeruginosa (PDB entry 6wg9; Gasparrini et al., 2020 ▸), Tet(56) from Legionella longbeachae (PDB entry 5tum; Park et al., 2017 ▸) and Tet(50), Tet(51) and Tet(55) from an uncultured bacterium (PDB entries 5tue, 5tuk and 5tul; Park et al., 2017 ▸). The tetracycline destructases are classified as class A FAD-dependent monooxygenases (FDOs), which cover numerous proteins with diverse purposes and functions that primarily catalyze the hydroxylation of aromatic substrates. Their characteristic structural feature is the presence of a FAD-binding domain that tightly binds the FAD prosthetic group. Their catalytic function is dependent on NADH or NADPH co-factors.
Figure 1.

Simplified reaction scheme of the degradation of oxytetracycline catalyzed by the tetracycline destructase TetX. The four individual rings of the molecule are labelled in green. The C atoms susceptible to modification by tetracycline destructases are labelled with their numbers.
To the best of our knowledge, the degradation of tetracycline antibiotics by S. maltophilia has not yet been studied. Antibiotic-modifying enzymes for β-lactams and aminoglycosides have been reported in S. maltophilia, but not for tetracyclines (Gil-Gil et al., 2020 ▸). However, several strains of S. maltophilia code for a FAD-dependent monooxygenase (NCBI Reference Sequence WP_049406473; O’Leary et al., 2016 ▸) that shares 28% sequence identity with the tetracycline destructase TetX. We carried out a biophysical and structural analysis of the recombinant form of this enzyme to elucidate whether it could contribute to the resistance of S. maltophilia to tetracycline antibiotics. The nucleotide sequence coding for the enzyme has been found in clinical isolates from India, Australia, Europe (Esposito et al., 2017 ▸; Lira et al., 2017 ▸) and the USA (Pak et al., 2015 ▸; Roach et al., 2015 ▸). Our results show its close structural and functional relationship to the tetracycline destructase enzyme family.
2. Materials and methods
2.1. Bioinformatic analysis and target selection
A search for nucleotide sequences coding for proteins similar to tetracycline destructases was performed on the NCBI database (NCBI Resource Coordinators, 2016 ▸) using Protein BLAST (Boratyn et al., 2013 ▸). The amino-acid sequence of TetX from B. thetaiotaomicron (Volkers et al., 2011 ▸; PDB entry 2y6r) was used as a search template. The sequence of SmTetX, a putative FAD-dependent monooxygenase from S. maltophilia strain AB550 (Permala et al., 2018 ▸) with NCBI Reference Sequence WP_049406473 (O’Leary et al., 2016 ▸; Arita et al., 2021 ▸) included in GenBank entry CP028899 (Glady-Croue et al., 2018 ▸), shares 28% sequence identity and 41% sequence similarity with TetX according to EMBOSS Needle (Rice et al., 2000 ▸).
2.2. Recombinant expression
The target amino-acid sequence (NCBI Reference Sequence WP_049406473) was back-translated and codon usage was optimized for expression in E. coli with OPTIMIZER (Puigbò et al., 2007 ▸). A plasmid encoding the SmTetX protein including a 6×His tag at the N-terminus of the translated protein was synthesized and cloned into the pET-28a(+)-TEV expression vector via NdeI and BamHI restriction sites by GenScript, USA. The cleavage site for Tobacco etch virus (TEV) protease was placed between the His tag and the target SmTetX sequence (Supplementary Fig. S1).
The plasmid was transformed into competent E. coli strain Lemo21 (DE3) cells (New England Biolabs) using the heat-shock method. Cell precultures in LB medium were incubated in a shaker at 32°C and 180 rev min−1 overnight. The medium was supplemented with 50 µg ml−1 kanamycin and 25 µg ml−1 chloramphenicol. 10 ml preculture was added to 1 l Power Broth medium (Molecular Dimensions, catalogue No. MD12-106) and the cell culture was incubated at 30°C and 150 rev min−1 until the OD600 reached ∼0.5. Expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at 20°C for 16 h. The cells were harvested after centrifugation of the culture at 4°C and 5000g for 30 min.
2.3. Purification and characterization
The cell pellet was homogenized in lysis buffer consisting of 50 mM Tris–HCl, 500 mM NaCl, 30 mM imidazole pH 8, protease-inhibitor cocktail (Sigma–Aldrich, catalogue No, P8849) and DNase I (Sigma–Aldrich, catalogue No. D4263). The cells were gently lysed by sonication (Qsonica Q700). After centrifugation at 40 000g for 30 min and filtration using a Puradisc 25 mm PTFE 1.0 µm (Whatman), the clarified lysate was loaded onto an equilibrated 5 ml HisTrap FF column (GE Healthcare) for Ni–NTA affinity chromatography. The column was then washed with 50 mM Tris–HCl, 500 mM NaCl, 30 mM imidazole pH 8 and the protein was eluted using the same buffer with a step gradient to 170 mM imidazole using an ÄKTApurifier FPLC system (GE Healthcare/Amersham Biosciences) at 10°C.
The buffer was exchanged to 50 mM Tris–HCl, 150 mM NaCl pH 8, 0.5 mM dithiothreitol (DTT), 1 mM ethylenediaminetetraacetic acid using a 3 kDa cutoff Nanosep centrifugal device (Pall Corporation); the final protein concentration was 1 mg ml−1. TEV cleavage was performed at 4°C for 16 h using TEV protease at a concentration of 0.05 mg ml−1; TEV protease production and the enzymatic reaction were carried out according to a standard protocol (Tropea et al., 2009 ▸). Tag-free SmTetX was separated on a 5 ml HisTrap FF column (GE Healthcare).
Size-exclusion chromatography was performed using a Superdex 75 Increase 10/300 GL column (GE Healthcare) equilibrated with buffer consisting of 25 mM Tris–HCl, 150 mM NaCl pH 8. Sample purity was checked by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Supplementary Fig. S2); the gel was made using the TGX FastCast Acrylamide Kit 12% (Bio-Rad) and a Colour Prestained Protein Standard, Broad Range marker (New England Biolabs) and was stained with InstantBlue (Expedeon). The protein sample was characterized by nanoscale differential scanning fluorimetry (NanoDSF) carried out in a Prometheus NT.48 (NanoTemper) and isoelectric focusing (IEF; Novex pH 3–10 IEF, 5% polyacrylamide gel, ThermoFisher Scientific; analysis was performed using the protocol recommended by the manufacturer). The UV–Vis spectrum of the protein (Supplementary Fig. S3) at a concentration of 3 mg ml−1 was measured in 100 mM [tris(hydroxymethyl)methylamino]propanesulfonic acid (TAPS) pH 8.5 in a quartz cuvette (optical path 10 mm) using a Specord 50 Plus spectrophotometer (Analytik Jena).
2.4. Structural mass spectrometry
For this analysis, SmTetX was alkylated by iodoacetamide at a final concentration of 30 mM. After 30 min incubation in the dark, trypsin was added to a concentration of 0.1 µg ml−1 and the reaction mixture was incubated overnight at 37°C.
Samples were analysed using a liquid-chromatography system (Agilent 1200 series, Agilent Technologies) connected to a timsToF Pro PASEF mass spectrometer equipped with a CaptiveSpray ion source (Bruker Daltonics) operated in positive data-dependent mode. 5 µl peptide mixture was injected using an autosampler onto a C18 trap column (UHPLC Fully Porous Polar C18 2.1 mm ID, Phenomenex). After 5 min of trapping at a flow rate of 20 µl min−1, peptides were separated on a C18 column (Luna Omega 3 µm Polar C18 100 Å, 150 × 0.3 mm, Phenomenex) using a linear 35 min water–acetonitrile gradient from 5 to 35%(v/v) acetonitrile at a flow rate of 4 µl min−1. Both the trap and analytical columns were heated to 50°C. Parameters from the PASEF method for standard proteomics were used for the timsTOF Pro settings.
The raw data were processed using the PeaksStudio 10.0 software (Bioinformatics Solutions, Canada). The search parameters were set up as follows: enzyme − trypsin (specific), carbamidomethylation and oxidation of methionine as a variable modification. Data were searched against the SmTetX sequence (Supplementary Fig. S4).
2.5. Small-angle X-ray scattering
After size-exclusion chromatography (described above), the protein in the most concentrated fraction (4.4 mg ml−1) was transferred to buffer composed of 25 mM bis-Tris, 150 mM NaCl pH 6.5 via overnight dialysis using Slide-A-Lyzer MINI Dialysis Devices, 3.5K MWCO (Thermo Scientific). Protein samples were centrifuged at 15 000 rev min−1 for 15 min prior to small-angle X-ray scattering (SAXS) measurements. Two samples were analysed: protein with reducing agent (30 mM DTT) and a control sample of protein without any additive. Data were collected at the Centre of Molecular Structure (BIOCEV), Czech Republic using a MetalJet C2+ X-ray source with a gallium anode (Excillum), SAXSpoint 2.0 (Anton Paar) and an EIGER R 1M detector (Dectris). Scattering images were processed in Aares, custom-made software for the in-house source developed by J. Stránský. Further analysis and processing was performed using the ATSAS software package (Manalastas-Cantos et al., 2021 ▸). Parameters and statistics relating to the measurements are listed in Supplementary Table S1. The scattering images are available at https://doi.org/10.5281/zenodo.7348780. The processed data including ab initio shape determination have been deposited in the SASBDB database under accession codes SASDPW7 and SASDPV7 for sample with and without reducing agent, respectively (Supplementary Fig. S5).
2.6. Activity assay
A spectrophotometric assay was performed in 96-well plates (BRAND, Wertheim, Germany) at 25°C in triplicate using a similar setup as reported previously (Forsberg et al., 2015 ▸; Gasparrini et al., 2020 ▸; Moore et al., 2005 ▸; Rudra et al., 2018 ▸; Yang et al., 2004 ▸). Enzymatic reactions of the substrates (0.5 mM oxytetracycline, Sigma–Aldrich, catalogue No O5875; 0.5 mM NADPH, Sigma–Aldrich, catalogue No. 10621692001) were carried out with 0.1 µM SmTetX in 100 mM TAPS buffer pH 8.5 in a total volume of 100 µl. Changes in the optical density were monitored using a Spark microplate reader (Tecan) in the UV–Vis spectrum with 1 min intervals.
2.7. Crystallization, diffraction data collection and processing
The optimal protein buffer for crystallization was selected according to screening using the nanoscale differential scanning fluorimetry method conducted on a Prometheus NT.48 (NanoTemper). The protein sample was transferred to buffer consisting of 20 mM bis-Tris, 50 mM NaCl pH 6.5 and concentrated to 10 mg ml−1 using a 3 kDa cutoff Nanosep centrifugal device (Pall Corporation). When searching for the optimal crystallization condition, we used several commercial crystallization screens, including our acidic screen (Fejfarová et al., 2016 ▸). 96-well crystallization plates were set up by a Gryphon crystallization robot (Art Robbins) using the sitting-drop vapour-diffusion method and were stored and monitored in an RI1000 protein crystallization imager (Formulatrix) at a temperature of 20°C; the protein:reservoir ratios were 2:1, 1:1 and 1:2 in a 0.3 µl drop. The initial crystallization hits from the MORPHEUS screen (Molecular Dimensions; Gorrec, 2009 ▸) were further optimized in hanging drops using the microseeding method and Additive Screen (Hampton Research). The final condition consisted of 12%(w/v) PEG 8000, 24%(v/v) ethylene glycol, 60 mM sodium nitrate, 60 mM disodium hydrogen phosphate, 60 mM ammonium sulfate, 100 mM MES–imidazole pH 6.5, 4% acetone; the protein:reservoir ratio was 2:1 in a 1.5 µl drop.
The crystals were harvested in LithoLoops (Molecular Dimensions) and vitrified in liquid nitrogen without cryoprotection owing to the presence of ethylene glycol at a sufficient concentration in the crystallization conditions. Diffraction data were collected on beamline 14.1 of the BESSY II synchrotron-radiation source (Helmholtz Zentrum Berlin, Germany; Mueller et al., 2015 ▸) using a mini-kappa goniometer and a PILATUS 6M detector (Dectris) under the control of MXCuBE (Oscarsson et al., 2019 ▸). The data set was processed in XDSGUI (Kabsch, 2010 ▸) and initially scaled in AIMLESS from the CCP4 suite (Evans & Murshudov, 2013 ▸; Agirre et al., 2023 ▸). A limited range of diffraction images (2400 images, corresponding to 240° of the total rotational angle) were processed due to an increase in R meas per image in the final stage of data collection. The diffraction data exhibited high anisotropy: the suggested diffraction limit according to the criterion of I/σ(I) being higher than 1.5 varied from 2.69 to 1.96 Å for different directions in reciprocal space, as reported in AIMLESS. After anisotropy correction with STARANISO (Tickle et al., 2018 ▸), the phase problem was solved with a combination of MoRDa (Vagin & Lebedev, 2015 ▸; Krissinel et al., 2018 ▸) and Phaser (McCoy et al., 2007 ▸) at 2.4 Å resolution. The crystal structure of AbsH3 (Clinger et al., 2021 ▸; PDB entry 6n04) was used as a template; its FAD-binding domain and substrate-binding domain were placed individually into the unit cell. The structure model was refined with REFMAC5 (Kovalevskiy et al., 2018 ▸) using restraints for FAD from AceDRG (Long et al., 2017 ▸) and manually edited as in Švecová et al. (2021 ▸); harmonic restraints were applied to several water molecules to avoid clashes. Manual modifications and real-space refinement were carried out in Coot (Emsley et al., 2010 ▸). The high-resolution diffraction limit (1.95 Å) was determined by the paired refinement protocol with PAIREF (Karplus & Diederichs, 2012 ▸; Malý et al., 2020 ▸, 2021 ▸). Regions in the model that were difficult to interpret due to a lack of signal were resolved using a combination of polder maps (Liebschner et al., 2017 ▸), composite omit maps (Terwilliger et al., 2008 ▸) and feature-enhanced maps (Afonine et al., 2015 ▸) from the Phenix package (Liebschner et al., 2019 ▸). The final structure was refined using all reflections and was validated with Coot, MolProbity (Williams et al., 2018 ▸) and the wwPDB validation service (Berman et al., 2003 ▸). Data-collection, processing and refinement statistics are shown in Table 1 ▸. The diffraction images are available from the Structural Biology Data Grid (https://data.sbgrid.org/) at https://doi.org/10.15785/SBGRID/956. The coordinates and structure-factor amplitudes were deposited in the PDB with accession code 8aq8. The presented structure alignments and calculations of root-mean-square deviation (r.m.s.d.) were carried out in PyMOL 2.5 (Schrödinger). The crystal structure and its similarity to other protein structures were investigated with ProFunc (Laskowski et al., 2005 ▸), PISA (Krissinel & Henrick, 2007 ▸), STRIDE (Heinig & Frishman, 2004 ▸), PDBsum (Laskowski et al., 2018 ▸), PDBeFold (Krissinel & Henrick, 2004 ▸), VAST (Madej et al., 2014 ▸) and DALI (Holm, 2020 ▸).
Table 1. Data-collection and merging statistics (after anisotropy correction) and structure-refinement parameters for SmTetX.
Values in parentheses are for the highest resolution shell.
| PDB entry | 8aq8 |
| Data collection | |
| X-ray source | BL14.1, BESSY II |
| Wavelength (Å) | 0.9180 |
| Detector | PILATUS 6M |
| Temperature (K) | 100 |
| Crystal-to-detector distance (mm) | 424 |
| No. of oscillation images | 2400 |
| Exposure time per image (s) | 0.1 |
| Oscillation width (°) | 0.1 |
| Data processing | |
| Space group | P21 |
| a, b, c (Å) | 52.9, 160.5, 95.6 |
| α, β, γ (°) | 90, 95.9, 90 |
| Resolution (Å) | 48.21–1.95 (2.01–1.95) |
| No. of reflections | 467117 (25410) |
| No. of unique reflections | 98291 (4915) |
| Multiplicity | 4.8 (5.2) |
| Completeness (spherical) (%) | 85.3 (48.5) |
| Completeness (ellipsoidal) (%) | 96.3 (92.9) |
| R meas | 0.156 (1.702) |
| R p.i.m. | 0.070 (0.729) |
| Mean I/σ(I) | 8.6 (1.1) |
| CC1/2 | 0.997 (0.378) |
| CC* | 0.999 (0.741) |
| Mosaicity (°) | 0.2 |
| Solvent content (%) | 52.6 |
| Refinement | |
| R work | 0.2002 (0.3185) |
| R free (5% reflections) | 0.2410 (0.3289) |
| R all | 0.2022 (0.3175) |
| CCwork | 0.964 (0.592) |
| CCfree | 0.938 (0.617) |
| Mean ADP (Å2) | 28.9 |
| No. of protein chains in asymmetric unit | 4 |
| No. of atoms | 11859 |
| No. of water molecules | 1111 |
| Ligands | 4 × FAD, 6 × ethylene glycol, 2 × polyethylene glycol, 3 ×
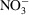 , 2 ×
, 2 ×
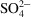 , 4 × Cl−
, 4 × Cl−
|
| R.m.s.d. from ideal bond lengths (Å) | 0.009 |
| R.m.s.d. from ideal angles (°) | 1.498 |
| Ramachandran favoured (%) | 97 |
| Ramachandran outliers (%) | 0 |
| Rama-Z score (Sobolev et al., 2020 ▸) | −1.3 ± 0.2 |
We also attempted to solve the structure of SmTetX in complex with a tetracycline antibiotic using soaking and co-crystallization, without success.
2.8. Molecular docking
The initial geometries of the studied antibiotics were obtained from ChEMBL (Mendez et al., 2019 ▸) and PubChem (Kim et al., 2021 ▸) in SDF format. When necessary, the structures were converted to 3D coordinates and H atoms were added with Open Babel version 3.1.1 (O’Boyle et al., 2011 ▸). The geometry of the ligands was further optimized with ORCA version 5.0.3 (Neese et al., 2020 ▸) at the dispersion-corrected DFT (RI-TPSS-D3/SVP) level of theory in two steps. Firstly, hydrogen positions were optimized while heavy-atom coordinates were kept fixed, followed by full all-atom geometry optimization. For the ligand–protein docking, the structures were converted to PDBQT format (partial atomic charges and atomic types assigned) using the prepare_receptor4 and prepare_ligand4 Python scripts from MGLTools 1.5.7 (Sanner, 1999 ▸) for the protein and ligands, respectively. Docking was performed with AutoDock Vina version 1.2.3 (Eberhardt et al., 2021 ▸; Trott & Olson, 2010 ▸) using a 40 × 40 × 40 Å box centred around the C2′ atom of FAD with focus on the re site. For each ligand, a set of predicted most stable complex geometries was obtained from the results of 20 independent AutoDock Vina runs. The poses were visualized in PyMOL 2.5 (Schrödinger). The flexible side-chain docking into the re site included protein residues Thr47, Leu48, Asp49, His51, Lys102, Glu104, Phe181, Ser201, Phe203, Phe212, Gln214, Tyr224, Val301 and Trp354 found in the vicinity of the antibiotic ligands.
3. Results
3.1. Recombinant form of SmTetX
The FAD-dependent monooxygenase from S. maltophilia (NCBI Reference Sequence WP_049406473) was recombinantly expressed in E. coli cells (Supplementary Fig. S2). The protein consists of 364 amino-acid residues and a FAD prosthetic group that causes the characteristic yellow colour of the sample. The UV–Vis spectrum of SmTetX shows a dominant oxidized state of FAD, but the reduced state is also partially present (Supplementary Fig. S3). The estimated melting temperature of SmTetX, as determined using NanoDSF, is ∼48°C and its isoelectric point is 5.4, as determined using IEF. The structure of the SmTetX monomer based on the crystal structure (described below) fits both SAXS data sets well with a χ2, as calculated by CRYSOL, of close to 1 (Supplementary Table S1 and Supplementary Fig. S5). Thus, SmTetX is a monomer in solution. Mass-spectrometric analysis of SmTetX peptide fragments shows a basically complete coverage of the construct sequence and the absence of disulfide bridges, as all of the cysteine residues in the protein were observed to be alkylated (Supplementary Fig. S4).
3.2. Activity against oxytetracycline
SmTetX was tested for activity against oxytetracycline (OTC). The analysed substrates, OTC and nicotinamide adenine dinucleotide phosphate (NADPH), exhibit absorption peaks at 368 and 340 nm, respectively. Thus, the activity against OTC was monitored similarly to previous studies (Forsberg et al., 2015 ▸; Gasparrini et al., 2020 ▸; Moore et al., 2005 ▸; Rudra et al., 2018 ▸; Yang et al., 2004 ▸) using the decrease in absorption at 400 nm. SmTetX causes a decrease in the absorbance at 400 nm in the absorption spectrum of OTC that indicates enzymatic activity (Figs. 2 ▸ b and 2 ▸ h). The changes in the OTC+enzyme spectra appear in the presence, and even in the absence, of the NADPH co-factor (Figs. 2 ▸ a and 2 ▸ g). The estimated turnover numbers for OTC modification by the enzyme, based on the absorbance change at 400 nm during the first 2 min, for OTC and for OTC with NADPH are k cat ≃ 2.6 ± 0.7 and k cat ≃ 2.0 ± 0.7 s−1, respectively. The estimated turnover numbers for NADPH oxidation by the enzyme, based on the absorbance change at 340 nm during the first 2 min, for NADPH and for NADPH with OTC are k cat ≃ 1.6 ± 0.8 and k cat ≃ 2.1 ± 0.7 s−1, respectively. Both reactions appear to be saturated after several minutes as no further decrease in absorbance can be observed. The protein is stable under the reaction conditions used, as was confirmed by NanoDSF; therefore, the loss of activity cannot be attributed to enzyme degradation. The enzyme also oxidizes NADPH in the absence of a tetracycline antibiotic (Figs. 2 ▸ c and 2 ▸ i). This reaction also achieves saturation in several minutes.
Figure 2.

SmTetX causes changes in the UV–Vis spectrum of oxytetracycline (OTC) and NADPH. The initial concentrations of the reagents, which are listed in each panel, were [OTC] = 0.5 mM, [NADPH] = 0.5 mM and [enzyme] = 0.1 µM. The individual enzymatic assays were performed in parallel in 100 mM TAPS pH 8.5. (a–f) Absorbance scans are plotted at 1 min intervals over a time course of 10 min, represented by a rainbow colour gradient [from red through yellow to blue as shown in (a)]. Background absorbance of the buffer was subtracted. (g–l) Monitoring of the decrease in absorbance of OTC (400 nm) and NADPH (340 nm); standard errors of the mean from measurements in triplicate are shown.
3.3. Crystal structure
Crystals of SmTetX provided severely anisotropic diffraction data. Nevertheless, the structure of the protein could be solved using molecular replacement and refined at 1.95 Å resolution. Anisotropy correction of the diffraction data (Tickle et al., 2018 ▸) and the paired refinement protocol (Karplus & Diederichs, 2012 ▸; Malý et al., 2020 ▸) proved to be beneficial for structure solution. The asymmetric unit consists of four SmTetX molecules with pairwise r.m.s.d.s on Cα atoms below 0.34 Å; the substrate-binding site remains structurally unchanged across the individual protein chains. There are two pairs of identical covalent dimers cross-linked via two disulfide bridges, i.e. chains A and B are linked via Cys172(A)–Cys172(B) and Cys281(A)–Cys281(B) disulfide bridges. The enzyme shares its fold with the class A FAD-dependent monooxygenases (Paul et al., 2021 ▸; Toplak et al., 2021 ▸). The overall structure of the monomer is divided into a smaller substrate-binding domain (residues Glu75–Ser99 and Pro176–Tyr274) and a larger FAD-binding domain (Met1–His74, Lys100–Leu175 and Ser275–Pro323), which are stabilized by a long C-terminal α-helix (Asp324–Arg364) (Figs. 3 ▸ a and 3 ▸ c).
Figure 3.

Structural analysis of SmTetX. The substrate-binding domain is coloured red, the FAD-binding domain green and the C-terminal helix blue. C atoms of FAD are shown in grey in stick representation and a chloride anion is shown as a yellow sphere. (a, b) Overall fold represented in secondary structure (a) and surface (b) views along the access tunnel to the re site. (c) Schematic diagram of the SmTetX domain structure; the numbers correspond to the native amino-acid sequence. (d) The binding of FAD in combined stick and secondary-structure representation: a view from the flavin si site. The 2mF o − DF c electron density is contoured in blue at the 1σ level. C atoms of the residues belonging to the GXGXXG, DG and GD binding motifs are coloured yellow. (e) Interactions of FAD with the protein. Hydrogen bonds are shown in orange with distances in Å. The graphics were prepared in PyMOL 2.5 (Schrödinger) and LigPlot+ (Laskowski & Swindells, 2011 ▸).
The substrate-binding domain is composed of a seven-stranded parallel β-sheet and four α-helices that are exposed to solvent. The FAD-binding domain consists of a three-stranded antiparallel β-sheet, a five-stranded parallel β-sheet, a two-stranded antiparallel β-sheet, six α-helices and one 310-helix as analysed by STRIDE (Heinig & Frishman, 2004 ▸). Part of the Rossmann fold (βαβ motif) of the FAD-binding domain is located at the N-terminus and possesses the GXGXXG sequence motif (residues Gly11–Gly16) characteristic for binding the adenine moiety of FAD (Dym & Eisenberg, 2001 ▸; Kleiger & Eisenberg, 2002 ▸). The FAD adenine moiety is also bound by the previously described Asp159-Gly160 (DG) motif and the diphosphate moiety by the Gly292-Asp293 (GD) motif, similarly to other class A FDOs (Huijbers et al., 2014 ▸; Paul et al., 2021 ▸). The whole FAD molecule is well localized in electron density and is bound via 11 hydrogen bridges to Ala15, Glu34, Arg107, Leu129, Asp293 and Val305 (Figs. 3 ▸ d and 3 ▸ e).
The isoalloxazine moiety of FAD is accessible to solvent through two large cavities that are located between the substrate-binding and FAD-binding domains (Fig. 3 ▸ b). The binding sites in these cavities are further named according to their position with respect to the flavin ring as the re site (substrate-binding site) and the si site (NADPH-binding site). FAD is observed in the IN conformation in our structure, i.e. FAD is elongated and its isoalloxazine moiety reaches into the re site. A large portion of the FAD molecule is accessible to solvent and participates in the formation of the si site.
The re site consists of the main chain of the loop Pro300–Gly302 and the side chains of a large nonpolar aromatic region (Phe181, Phe203, Phe205, Phe212, Tyr224 and Trp354), a polar region (Ser201, Gln214 and Cys222) and Asp49 (Fig. 4 ▸ a). A chloride anion was modelled in the re site near the FAD isoalloxazine moiety according to the signal strength in the 2mF o − DF c map, the typical distances to adjacent atoms and their chemical properties. It is bound at the pyrimidine moiety of isoalloxazine and coordinated by the main-chain N atoms of the Pro300–Val303 loop. Several residues at the protein termini (Met1-Gln2, Gly358–Gly364) and the re site (Gly93–Asp101) could not be modelled due to a lack of electron density.
Figure 4.

The active site of SmTetX is located at the re site. (a) Composition of the active site in combined stick and secondary-structure representation. C atoms of the substrate-binding domain are coloured red, those of the FAD-binding domain green and those of the C-terminal helix blue. The chloride anion is shown as a yellow sphere. (b, c) Alignment of SmTetX with the TetX–chlortetracycline (CTC) complex (Volkers et al., 2011 ▸; PDB entry 2y6r) in yellow shown in stick representation. The r.m.s.d. was 3.74 Å using 284 Cα atoms of chains A. The graphics were prepared in PyMOL 2.5 (Schrödinger).
3.4. Docking of antibiotic representatives
In order to inspect the interactions of the protein with likely ligands, in silico ligand docking of selected tetracycline molecules (Supplementary Fig. S6) and other antibiotic representatives into the binding re site were carried out (Table 2 ▸). As these ligands can appear in multiple tautomeric forms, we took the keto (C3)–amino (C4) tautomerism into account. However, we also tested the enolate (C3)–ammonium (C4) tautomerism of tetracycline (Aleksandrov et al., 2007 ▸) and obtained similar results both in terms of relative energies in the unbound solvated state at the RI-TPSS-D3/TZVP/CPCM level of theory as well as in the predicted binding affinity to the studied protein. The molecule of anhydrotetracycline was docked to act as a potential substrate (SmTetX in FAD-IN conformation), similarly to as observed in the complex with TetX6 (Kumar et al., 2023 ▸); nevertheless, it should be noted that anhydrotetracycline inhibits Tet(50) when bound in the re site with the FAD-OUT conformation (Park et al., 2017 ▸). The chloride anion present in the re site in our crystal structure was taken into account in the calculations.
Table 2. The lowest calculated binding energies of selected antibiotics in the binding re site determined by molecular docking.
Tetracycline representatives are labelled with an asterisk.
| Antibiotic molecule | Affinity (kcal mol−1) |
|---|---|
| *Anhydrotetracycline | −9.08 |
| Ampicillin | −8.81 |
| *Oxytetracycline | −8.70 |
| *Tetracycline | −8.65 |
| Rifampin | −8.58 |
| Streptomycin | −8.41 |
| *Chlortetracycline | −8.17 |
| Levocetirizine | −8.03 |
| Amoxicillin | −7.83 |
| Amikacin | −7.79 |
| Erythromycin | −7.69 |
| Mitomycin | −7.67 |
| Chloramphenicol | −7.62 |
| Paromomycin | −7.48 |
| Rifamycin SV | −7.39 |
| Lincomycin | −7.38 |
| Abyssomicin C | −7.38 |
| Kanamycin | −7.38 |
| Clarithromycin | −6.95 |
| Gentamicin | −6.83 |
| Metronidazole | −5.31 |
4. Discussion
SmTetX shows activity against oxytetracycline (OTC; Fig. 2 ▸), which would classify the enzyme as a member of the tetracycline destructase family (Cheng et al., 2022 ▸; Markley & Wencewicz, 2018 ▸). Our estimated turnover number of SmTetX towards OTC as a substrate (2.6 ± 0.7 s−1) is comparable to that of the tetracycline destructase TetX2 (1.3 ± 0.1 s−1; Yang et al., 2004 ▸) and that of TetX4 towards tigecycline (2.03 ± 0.03 s−1; Cheng et al., 2021 ▸). However, all of the described tetracycline destructases strictly require NADPH for their catalytic activity. SmTetX oxidizes the NADPH co-factor in the absence of OTC (Figs. 2 ▸ c and 2 ▸ i). This is consistent within the family as NADPH reduces FAD prior to the formation of C4a-hydroperoxyflavin, which is needed for electrophilic attack on a tetracycline substrate. Remarkably, the changes in the OTC absorption spectra for SmTetX occurred even in the absence of NADPH (Figs. 2 ▸ a and 2 ▸ g). The reactions reach saturation before substrate exhaustion. Both observations can be explained by the partial presence of the reduced state of FAD in SmTetX as indicated by the UV–Vis absorption spectrum (Supplementary Fig. S3).
The overall fold of SmTetX is very similar to those of the tetracycline destructases TetX (Volkers et al., 2011 ▸) and TetX2 (Walkiewicz et al., 2011 ▸) from B. thetaiotaomicron, TetX4 from E. coli (Cheng et al., 2021 ▸) and TetX7 from P. aeruginosa (Gasparrini et al., 2020 ▸).
The crystal structure contains SmTetX in the form of covalent dimers, which are most likely to be artefacts of crystallization. Other experimental methods (SEC and SAXS; Supplementary Fig. S5) indicate a monomeric state of the protein in solution, and mass spectrometry confirms the absence of disulfide bridges. The SmTetX monomer organization observed in our structure is very close to that of similar enzymes. Thus, the covalent links between the SmTetX chains very likely do not affect the structure of the SmTetX monomer itself. We assume that SmTetX acts as a monomer in solution and not as a dimer as found in our crystal structure.
The binding of FAD to SmTetX is conserved within the family of class A FMOs. In our case, we only observe the IN conformation of FAD; however, the size of the si site would also allow the accommodation of the bent OUT conformation that was observed in the crystal structure of Tet(50) (Park et al., 2017 ▸).
Despite numerous co-crystallization and soaking trials, our attempts to experimentally determine complexes of SmTetX with tetracycline antibiotics or co-factors failed. Therefore, we identified putative binding sites according to the structures of similar complexes. We hypothesize that the si site is the binding site for NADPH as in the case of 4-hydroxybenzoate hydroxylase, which is the most thoroughly studied class A FMO (Crozier-Reabe & Moran, 2012 ▸; Eppink et al., 1998 ▸, 1999 ▸). On the basis of structures of tetracycline destructases in complex with tetracyclines (Cheng et al., 2022 ▸; Park et al., 2017 ▸; Volkers et al., 2011 ▸, 2013 ▸), we hypothesize that the substrate-binding site is located in the re site (Fig. 4 ▸ a).
The destructases TetX and Tet(50) and the putative reductase AbsH3 from the abyssomicin biosynthesis pathway (used in molecular replacement; Clinger et al., 2021 ▸; PDB entry 6n04) are the most similar enzymes in terms of sequence, overall structure and structure of the active site. Structure-based sequence alignment (Fig. 5 ▸) reveals many similarities in the substrate-binding site as well as some unique features of SmTetX. The most conserved parts are regions Thr47–Leu50 (SmTetX numbering), which is placed next to the pyrimidine moiety of isoalloxazine, and Pro300–Gly302, which is located above the re side of the isoalloxazine ring.
Figure 5.

Multiple structure-based sequence alignment of SmTetX with the tetracycline destructases TetX from B. thetaiotaomicron (PDB entry 2y6r; Volkers et al., 2011 ▸) and Tet(50) (PDB entry 5tue; Park et al., 2017 ▸), and the putative reductase AbsH3 from Streptomyces sp. (PDB entry 6n04; Clinger et al., 2021 ▸). Regions significant for the re site of the enzyme are shown. The key residues in the binding pocket of SmTetX are highlighted by a black background (and shown in Fig. 4 ▸). Identity, similarity and dissimilarity of the key amino acids are marked with green, orange and red backgrounds, respectively. *The sequence and structure of this region are the same for TetX, TetX2, TetX4 and TetX7 (Cheng et al., 2022 ▸).
Further discussion is based on a structural comparison of SmTetX with TetX, the most similar tetracycline destructase with a thoroughly described structure and function. Several important residues are identical in SmTetX and TetX, i.e. Asp49, His51, Phe212, Pro300 and Gly302 (SmTetX numbering). The side chains of Phe205 and Phe351 in SmTetX correspond to their phenylalanine analogues in TetX, but they originate from different secondary-structure elements (Figs. 4 ▸ b and 5 ▸ c). The aromatic residues Phe203 and Trp354 in SmTetX replace Met203 and Met375 in TetX to conserve the nonpolar character of these sites. The major difference in SmTetX are residues Phe181 and Tyr224, which expose their aromatic side chains to the re site and thus change the nature of the active site, in contrast to Asn190 and Gln192 in TetX, which participate in substrate binding (PDB entry 2y6r). Moreover, Cys222 and Ser201 in SmTetX replace His234 and Arg213, respectively, in TetX. Overall, of the 15 residues forming the substrate-binding pocket of SmTetX, five are identical, four have a similar character and six have a significantly different character. The differences raise questions about the actual binding of tetracycline antibiotics, i.e. whether the re site allows the accommodation of such ligands and whether their binding modes are compatible with the known tetracycline modification sites reported for tetracycline destructases.
Due to the lack of a structure of SmTetX in complex with a ligand, we attempted to investigate the binding of tetracycline representatives (tetracycline, oxytetracycline, anhydrotetracycline and chlortetracycline) by the use of computational ligand docking. The calculations could be influenced by the unmodelled region at the entrance to the re site (Gly93–Asp101) in the SmTetX crystal structure (Fig. 3 ▸ a). However, the amino-acid sequence and length of this region are similar to those of TetX, where this loop is not directly involved in ligand binding, for example in the TetX–chlortetracycline complex (PDB entry 2y6r). Therefore, we assume that the missing residues do not affect the docking results for molecules of the size of tetracycline antibiotics and smaller.
In the docking study, tetracycline representatives were among the highest affinity binders, for example anhydrotetracycline (–9.1 kcal mol−1) and OTC (–8.7 kcal mol−1) (Table 2 ▸ and Supplementary Fig. S6). The side chains of Phe181 and Tyr224 of SmTetX, representing the largest difference in the active site with respect to TetX, modify the bottom of the binding cavity and lead to a small shift of the docked ligands compared, for example, with the TetX–chlortetracycline complex (Figs. 4 ▸ b and 4 ▸ c). Nevertheless, the calculated poses of OTC place its C atoms that are susceptible to modification by tetracycline destructases, i.e. C1, C2, C3 and C11a, in the vicinity of the reactive C4a of FAD (Supplementary Figs. S6 and S7). Recently, a structure of Tet(X6) in complex with anhydrotetracycline bound in a substrate-like orientation was published (PDB entry 8er0; Kumar et al., 2023 ▸). In comparison with our docking of this ligand, the positions of the A and D rings (Fig. 1 ▸) are swapped.
Apart from tetracycline antibiotics, ampicillin, rifampin and streptomycin were also shown to be high-affinity binders. Interestingly, the class A FAD-dependent monooxygenases include the rifamycin-inactivating enzyme Rox from Streptomyces venezuelae (PDB entries 5vqb and 6brd; Koteva et al., 2018 ▸) that is capable of rifampin inactivation. In comparison with the tetracycline destructases, the Rox structure contains a larger active site and two additional consecutive α-helices.
Gln214 of SmTetX lining the outer edge of the re site is found in alternative conformations in the structure, suggesting that it possesses flexibility that might be involved in ligand binding. Interestingly, in the flexible side-chain docking calculations, a twist of the glutamine side chain (Supplementary Fig. S7) was indicated to a conformation analogous to Asn226 in the TetX–chlortetracycline complex (PDB entry 2y6r; Figs. 4 ▸ b and 4 ▸ c). These docking results show two possible binding modes of OTC: both are compatible with enzymatic modification and they differ in the location of the A and D rings of OTC.
Therefore, the re site of SmTetX is capable of accommodating tetracycline antibiotics and the suggested poses would enable electrophilic attack of hydroperoxyflavin on the tetracycline ring.
5. Conclusion
Several clinical isolates of S. maltophilia contain a reported FAD-dependent monooxygenase similar to the tetracycline destructase TetX from B. thetaiotaomicron. We have shown that this enzyme is capable of activity towards oxytetracycline and that its structure is very similar to that of TetX. SmTetX has a unique structural feature in the active site within this enzyme family: a large aromatic region formed by Phe181, Phe203, Phe205, Phe212, Tyr224 and Trp354. According to our in silico binding analysis, despite the remarkable differences in the active site, tetracycline representatives are among the highest affinity binders of the tested antibiotics and the aromatic region probably participates in tetracycline binding. Furthermore, the rate of oxytetracycline degradation is comparable to the rates shown by other destructases. Hence, these results provide evidence that S. maltophilia codes for an enzyme that is capable of degrading tetracycline antibiotics, even if such a mechanism is not essential for its resistance to these antibiotics.
6. Related literature
The following references are cited in the supporting information for this article: Islam et al. (2013 ▸) and Schwinn et al. (2020 ▸).
Supplementary Material
PDB reference: SmTetX, 8aq8
SASBDB reference: SmTetX, without reducing agent, SASDPV7
SASBDB reference: with reducing agent, SASDPV7
Supplementary Figures and Table. DOI: 10.1107/S2053230X23005381/va5051sup1.pdf
Raw diffraction images for PDB entry 8aq8.: https://doi.org/10.15785/SBGRID/956
Raw scattering images for SASBDB entries SASDPV7 and SASDPW7.: https://doi.org/10.5281/zenodo.7348780
Acknowledgments
We would like to thank all of the staff at the Centre of Molecular Structure at BIOCEV and also acknowledge all of the staff of beamline 14.1 at the BESSY II synchrotron (Helmholtz-Zentrum Berlin, Berlin, Germany). We acknowledge CMS-BIOCEV (biophysical techniques, crystallization of proteins and nucleic acids, diffraction techniques, structural mass spectrometry) of CIISB, Instruct-CZ Centre, supported by MEYS CR (LM2015043 and LM2018127) and CZ.02.1.01/0.0/0.0/18_046/0015974.
Funding Statement
This work was supported by the Ministry of Education, Youth and Sports CR, projects CAAS (grant No. CZ.02.1.01/0.0/0.0/16_019/0000778 to the Faculty of Nuclear Sciences and Physical Engineering, Czech Technical University in Prague), ELIBIO (grant No. CZ.02.1.01/0.0/0.0/15_003/0000447 to the Institute of Biotechnology AS CR) and BIOCEV (grant No. CZ.1.05/1.1.00/02.0109 to the Institute of Biotechnology AS CR), from the ERDF fund, by institutional support from the Institute of Biotechnology AS CR (RVO 86652036) and by the Grant Agency of the Czech Technical University in Prague (grant No. SGS22/114/OHK4/2T/14).
References
- Afonine, P. V., Moriarty, N. W., Mustyakimov, M., Sobolev, O. V., Terwilliger, T. C., Turk, D., Urzhumtsev, A. & Adams, P. D. (2015). Acta Cryst. D71, 646–666. [DOI] [PMC free article] [PubMed]
- Agirre, J., Atanasova, M., Bagdonas, H., Ballard, C. B., Baslé, A., Beilsten-Edmands, J., Borges, R. J., Brown, D. G., Burgos-Mármol, J. J., Berrisford, J. M., Bond, P. S., Caballero, I., Catapano, L., Chojnowski, G., Cook, A. G., Cowtan, K. D., Croll, T. I., Debreczeni, J. É., Devenish, N. E., Dodson, E. J., Drevon, T. R., Emsley, P., Evans, G., Evans, P. R., Fando, M., Foadi, J., Fuentes-Montero, L., Garman, E. F., Gerstel, M., Gildea, R. J., Hatti, K., Hekkelman, M. L., Heuser, P., Hoh, S. W., Hough, M. A., Jenkins, H. T., Jiménez, E., Joosten, R. P., Keegan, R. M., Keep, N., Krissinel, E. B., Kolenko, P., Kovalevskiy, O., Lamzin, V. S., Lawson, D. M., Lebedev, A. A., Leslie, A. G. W., Lohkamp, B., Long, F., Malý, M., McCoy, A. J., McNicholas, S. J., Medina, A., Millán, C., Murray, J. W., Murshudov, G. N., Nicholls, R. A., Noble, M. E. M., Oeffner, R., Pannu, N. S., Parkhurst, J. M., Pearce, N., Pereira, J., Perrakis, A., Powell, H. R., Read, R. J., Rigden, D. J., Rochira, W., Sammito, M., Sánchez Rodríguez, F., Sheldrick, G. M., Shelley, K. L., Simkovic, F., Simpkin, A. J., Skubak, P., Sobolev, E., Steiner, R. A., Stevenson, K., Tews, I., Thomas, J. M. H., Thorn, A., Valls, J. T., Uski, V., Usón, I., Vagin, A., Velankar, S., Vollmar, M., Walden, H., Waterman, D., Wilson, K. S., Winn, M. D., Winter, G., Wojdyr, M. & Yamashita, K. (2023). Acta Cryst. D79, 449–461.
- Aleksandrov, A., Proft, J., Hinrichs, W. & Simonson, T. (2007). ChemBioChem, 8, 675–685. [DOI] [PubMed]
- Arita, M., Karsch-Mizrachi, I. & Cochrane, G. (2021). Nucleic Acids Res. 49, D121–D124. [DOI] [PMC free article] [PubMed]
- Berman, H., Henrick, K. & Nakamura, H. (2003). Nat. Struct. Mol. Biol. 10, 980. [DOI] [PubMed]
- Boratyn, G. M., Camacho, C., Cooper, P. S., Coulouris, G., Fong, A., Ma, N., Madden, T. L., Matten, W. T., McGinnis, S. D., Merezhuk, Y., Raytselis, Y., Sayers, E. W., Tao, T., Ye, J. & Zaretskaya, I. (2013). Nucleic Acids Res. 41, W29–W33. [DOI] [PMC free article] [PubMed]
- Brodersen, D. E., Clemons, W. M., Carter, A. P., Morgan-Warren, R. J., Wimberly, B. T. & Ramakrishnan, V. (2000). Cell, 103, 1143–1154. [DOI] [PubMed]
- Brooke, J. S. (2012). Clin. Microbiol. Rev. 25, 2–41. [DOI] [PMC free article] [PubMed]
- Brooke, J. S. (2014). Expert Rev. Anti Infect. Ther. 12, 1–4. [DOI] [PubMed]
- Chang, Y.-T., Lin, C.-Y., Chen, Y.-H. & Hsueh, P.-R. (2015). Front. Microbiol. 6, 893. [DOI] [PMC free article] [PubMed]
- Cheng, Q., Cheung, Y., Liu, C., Chan, E. W. C., Wong, K. Y., Zhang, R. & Chen, S. (2022). Commun. Biol. 5, 522. [DOI] [PMC free article] [PubMed]
- Cheng, Q., Cheung, Y., Liu, C., Xiao, Q., Sun, B., Zhou, J., Chan, E. W. C., Zhang, R. & Chen, S. (2021). BMC Biol. 19, 262. [DOI] [PMC free article] [PubMed]
- Chopra, I. & Roberts, M. (2001). Microbiol. Mol. Biol. Rev. 65, 232–260. [DOI] [PMC free article] [PubMed]
- Clinger, J. A., Wang, X., Cai, W., Zhu, Y., Miller, M. D., Zhan, C., Van Lanen, S. G., Thorson, J. S. & Phillips, G. N. (2021). Proteins, 89, 132–137. [DOI] [PMC free article] [PubMed]
- Crozier-Reabe, K. & Moran, G. (2012). Int. J. Mol. Sci. 13, 15601–15639. [DOI] [PMC free article] [PubMed]
- Dym, O. & Eisenberg, D. (2001). Protein Sci. 10, 1712–1728. [DOI] [PMC free article] [PubMed]
- Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. (2021). J. Chem. Inf. Model. 61, 3891–3898. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Eppink, M. H. M., Bunthof, C., Schreuder, H. A. & van Berkel, W. J. H. (1999). FEBS Lett. 443, 251–255. [DOI] [PubMed]
- Eppink, M. H. M., Schreuder, H. A. & van Berkel, W. J. H. (1998). Eur. J. Biochem. 253, 194–201. [DOI] [PubMed]
- Esposito, A., Pompilio, A., Bettua, C., Crocetta, V., Giacobazzi, E., Fiscarelli, E., Jousson, O. & Di Bonaventura, G. (2017). Front. Microbiol. 8, 1590. [DOI] [PMC free article] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- Fang, L., Chen, C., Cui, C., Li, X., Zhang, Y., Liao, X., Sun, J. & Liu, Y. (2020). BioEssays, 42, 2000014. [DOI] [PubMed]
- Fejfarová, K., Kádek, A., Mrázek, H., Hausner, J., Tretyachenko, V., Koval’, T., Man, P., Hašek, J. & Dohnálek, J. (2016). Acta Cryst. F72, 24–28. [DOI] [PMC free article] [PubMed]
- Forsberg, K. J., Patel, S., Wencewicz, T. A. & Dantas, G. (2015). Chem. Biol. 22, 888–897. [DOI] [PMC free article] [PubMed]
- Gajdács, M. & Urbán, E. (2019). Health Serv. Res. Manag. Epidemiol. 6, 233339281987077. [DOI] [PMC free article] [PubMed]
- Gasparrini, A. J., Markley, J. L., Kumar, H., Wang, B., Fang, L., Irum, S., Symister, C. T., Wallace, M., Burnham, C. D., Andleeb, S., Tolia, N. H., Wencewicz, T. A. & Dantas, G. (2020). Commun. Biol. 3, 241. [DOI] [PMC free article] [PubMed]
- Gil-Gil, T., Martínez, J. L. & Blanco, P. (2020). Expert Rev. Anti Infect. Ther. 18, 335–347. [DOI] [PubMed]
- Glady-Croue, J., Niu, X.-Z., Ramsay, J. P., Watkin, E., Murphy, R. J. T. & Croue, J.-P. (2018). Sci. Total Environ. 626, 1005–1011. [DOI] [PubMed]
- Gorrec, F. (2009). J. Appl. Cryst. 42, 1035–1042. [DOI] [PMC free article] [PubMed]
- Grossman, T. H. (2016). Cold Spring Harb. Perspect. Med. 6, a025387. [DOI] [PMC free article] [PubMed]
- He, T., Shen, J., Schwarz, S., Wu, C. & Wang, Y. (2015). J. Antimicrob. Chemother. 70, 1031–1036. [DOI] [PubMed]
- Heinig, M. & Frishman, D. (2004). Nucleic Acids Res. 32, W500–W502. [DOI] [PMC free article] [PubMed]
- Holm, L. (2020). Protein Sci. 29, 128–140. [DOI] [PMC free article] [PubMed]
- Huijbers, M. M. E., Montersino, S., Westphal, A. H., Tischler, D. & van Berkel, W. J. H. (2014). Arch. Biochem. Biophys. 544, 2–17. [DOI] [PubMed]
- Islam, M., Honma, M., Nakabayashi, T., Kinjo, M. & Ohta, N. (2013). Int. J. Mol. Sci. 14, 1952–1963. [DOI] [PMC free article] [PubMed]
- Jenner, L., Starosta, A. L., Terry, D. S., Mikolajka, A., Filonava, L., Yusupov, M., Blanchard, S. C., Wilson, D. N. & Yusupova, G. (2013). Proc. Natl Acad. Sci. USA, 110, 3812–3816. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Karplus, P. A. & Diederichs, K. (2012). Science, 336, 1030–1033. [DOI] [PMC free article] [PubMed]
- Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., Li, Q., Shoemaker, B. A., Thiessen, P. A., Yu, B., Zaslavsky, L., Zhang, J. & Bolton, E. E. (2021). Nucleic Acids Res. 49, D1388–D1395. [DOI] [PMC free article] [PubMed]
- Kleiger, G. & Eisenberg, D. (2002). J. Mol. Biol. 323, 69–76. [DOI] [PubMed]
- Koteva, K., Cox, G., Kelso, J. K., Surette, M. D., Zubyk, H. L., Ejim, L., Stogios, P., Savchenko, A., Sørensen, D. & Wright, G. D. (2018). Cell Chem. Biol. 25, 403–412. [DOI] [PubMed]
- Kovalevskiy, O., Nicholls, R. A., Long, F., Carlon, A. & Murshudov, G. N. (2018). Acta Cryst. D74, 215–227. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Krissinel, E., Uski, V., Lebedev, A., Winn, M. & Ballard, C. (2018). Acta Cryst. D74, 143–151. [DOI] [PMC free article] [PubMed]
- Kumar, H., Williford, E. E., Blake, K. S., Virgin-Downey, B., Dantas, G., Wencewicz, T. A. & Tolia, N. H. (2023). Commun. Biol. 6, 423. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A., Jabłońska, J., Pravda, L., Vařeková, R. S. & Thornton, J. M. (2018). Protein Sci. 27, 129–134. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A. & Swindells, M. B. (2011). J. Chem. Inf. Model. 51, 2778–2786. [DOI] [PubMed]
- Laskowski, R. A., Watson, J. D. & Thornton, J. M. (2005). Nucleic Acids Res. 33, W89–W93. [DOI] [PMC free article] [PubMed]
- Lewis, K. (2020). Cell, 181, 29–45. [DOI] [PubMed]
- Liebschner, D., Afonine, P. V., Baker, M. L., Bunkóczi, G., Chen, V. B., Croll, T. I., Hintze, B., Hung, L.-W., Jain, S., McCoy, A. J., Moriarty, N. W., Oeffner, R. D., Poon, B. K., Prisant, M. G., Read, R. J., Richardson, J. S., Richardson, D. C., Sammito, M. D., Sobolev, O. V., Stockwell, D. H., Terwilliger, T. C., Urzhumtsev, A. G., Videau, L. L., Williams, C. J. & Adams, P. D. (2019). Acta Cryst. D75, 861–877.
- Liebschner, D., Afonine, P. V., Moriarty, N. W., Poon, B. K., Sobolev, O. V., Terwilliger, T. C. & Adams, P. D. (2017). Acta Cryst. D73, 148–157. [DOI] [PMC free article] [PubMed]
- Lira, F., Berg, G. & Martínez, J. L. (2017). Front. Microbiol. 8, 2190. [DOI] [PMC free article] [PubMed]
- Long, F., Nicholls, R. A., Emsley, P., Gražulis, S., Merkys, A., Vaitkus, A. & Murshudov, G. N. (2017). Acta Cryst. D73, 112–122. [DOI] [PMC free article] [PubMed]
- Madej, T., Lanczycki, C. J., Zhang, D., Thiessen, P. A., Geer, R. C., Marchler-Bauer, A. & Bryant, S. H. (2014). Nucleic Acids Res. 42, D297–D303. [DOI] [PMC free article] [PubMed]
- Malý, M., Diederichs, K., Dohnálek, J. & Kolenko, P. (2020). IUCrJ, 7, 681–692. [DOI] [PMC free article] [PubMed]
- Malý, M., Diederichs, K., Dohnálek, J. & Kolenko, P. (2021). Acta Cryst. F77, 226–229. [DOI] [PMC free article] [PubMed]
- Manalastas-Cantos, K., Konarev, P. V., Hajizadeh, N. R., Kikhney, A. G., Petoukhov, M. V., Molodenskiy, D. S., Panjkovich, A., Mertens, H. D. T., Gruzinov, A., Borges, C., Jeffries, C. M., Svergun, D. I. & Franke, D. (2021). J. Appl. Cryst. 54, 343–355. [DOI] [PMC free article] [PubMed]
- Markley, J. L. & Wencewicz, T. A. (2018). Front. Microbiol. 9, 1058. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Mendez, D., Gaulton, A., Bento, A. P., Chambers, J., De Veij, M., Félix, E., Magariños, M. P., Mosquera, J. F., Mutowo, P., Nowotka, M., Gordillo-Marañón, M., Hunter, F., Junco, L., Mugumbate, G., Rodriguez-Lopez, M., Atkinson, F., Bosc, N., Radoux, C. J., Segura-Cabrera, A., Hersey, A. & Leach, A. R. (2019). Nucleic Acids Res. 47, D930–D940. [DOI] [PMC free article] [PubMed]
- Moore, I. F., Hughes, D. W. & Wright, G. D. (2005). Biochemistry, 44, 11829–11835. [DOI] [PubMed]
- Mueller, U., Förster, R., Hellmig, M., Huschmann, F. U., Kastner, A., Malecki, P., Pühringer, S., Röwer, M., Sparta, K., Steffien, M., Ühlein, M., Wilk, P. & Weiss, M. S. (2015). Eur. Phys. J. Plus, 130, 141.
- Murray, C. J., Ikuta, K. S., Sharara, F., Swetschinski, L., Robles Aguilar, G., Gray, A., Han, C., Bisignano, C., Rao, P., Wool, E., Johnson, S. C., Browne, A. J., Chipeta, M. G., Fell, F., Hackett, S., Haines-Woodhouse, G., Kashef Hamadani, B. H., Kumaran, E. A. P., McManigal, B., Achalapong, S., Agarwal, R., Akech, S., Albertson, S., Amuasi, J., Andrews, J., Aravkin, A., Ashley, E., Babin, F., Bailey, F., Baker, S., Basnyat, B., Bekker, A., Bender, R., Berkley, J. A., Bethou, A., Bielicki, J., Boonkasidecha, S., Bukosia, J., Carvalheiro, C., Castañeda-Orjuela, C., Chansamouth, V., Chaurasia, S., Chiurchiù, S., Chowdhury, F., Clotaire Donatien, R., Cook, A. J., Cooper, B., Cressey, T. R., Criollo-Mora, E., Cunningham, M., Darboe, S., Day, N. P. J., De Luca, M., Dokova, K., Dramowski, A., Dunachie, S. J., Duong Bich, T., Eckmanns, T., Eibach, D., Emami, A., Feasey, N., Fisher-Pearson, N., Forrest, K., Garcia, C., Garrett, D., Gastmeier, P., Giref, A. Z., Greer, R. C., Gupta, V., Haller, S., Haselbeck, A., Hay, S. I., Holm, M., Hopkins, S., Hsia, Y., Iregbu, K. C., Jacobs, J., Jarovsky, D., Javanmardi, F., Jenney, A. W. J., Khorana, M., Khusuwan, S., Kissoon, N., Kobeissi, E., Kostyanev, T., Krapp, F., Krumkamp, R., Kumar, A., Kyu, H. H., Lim, C., Lim, K., Limmathurotsakul, D., Loftus, M. J., Lunn, M., Ma, J., Manoharan, A., Marks, F., May, J., Mayxay, M., Mturi, N., Munera-Huertas, T., Musicha, P., Musila, L. A., Mussi-Pinhata, M. M., Naidu, R. N., Nakamura, T., Nanavati, R., Nangia, S., Newton, P., Ngoun, C., Novotney, A., Nwakanma, D., Obiero, C. W., Ochoa, T. J., Olivas-Martinez, A., Olliaro, P., Ooko, E., Ortiz-Brizuela, E., Ounchanum, P., Pak, G. D., Paredes, J. L., Peleg, A. Y., Perrone, C., Phe, T., Phommasone, K., Plakkal, N., Ponce-de-Leon, A., Raad, M., Ramdin, T., Rattanavong, S., Riddell, A., Roberts, T., Robotham, J. V., Roca, A., Rosenthal, V. D., Rudd, K. E., Russell, N., Sader, H. S., Saengchan, W., Schnall, J., Scott, J. A. G., Seekaew, S., Sharland, M., Shivamallappa, M., Sifuentes-Osornio, J., Simpson, A. J., Steenkeste, N., Stewardson, A. J., Stoeva, T., Tasak, N., Thaiprakong, A., Thwaites, G., Tigoi, C., Turner, C., Turner, P., van Doorn, H. R., Velaphi, S., Vongpradith, A., Vongsouvath, M., Vu, H., Walsh, T., Walson, J. L., Waner, S., Wangrangsimakul, T., Wannapinij, P., Wozniak, T., Young Sharma, T. E. M. W., Yu, K. C., Zheng, P., Sartorius, B., Lopez, A. D., Stergachis, A., Moore, C., Dolecek, C. & Naghavi, M. (2022). Lancet, 399, 629–655.
- NCBI Resource Coordinators (2016). Nucleic Acids Res. 44, D7–D19.
- Neese, F., Wennmohs, F., Becker, U. & Riplinger, C. (2020). J. Chem. Phys. 152, 224108. [DOI] [PubMed]
- Nguyen, F., Starosta, A. L., Arenz, S., Sohmen, D., Dönhöfer, A. & Wilson, D. N. (2014). Biol. Chem. 395, 559–575. [DOI] [PubMed]
- O’Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T. & Hutchison, G. R. (2011). J. Cheminform, 3, 33. [DOI] [PMC free article] [PubMed]
- O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., Rajput, B., Robbertse, B., Smith-White, B., Ako-Adjei, D., Astashyn, A., Badretdin, A., Bao, Y., Blinkova, O., Brover, V., Chetvernin, V., Choi, J., Cox, E., Ermolaeva, O., Farrell, C. M., Goldfarb, T., Gupta, T., Haft, D., Hatcher, E., Hlavina, W., Joardar, V. S., Kodali, V. K., Li, W., Maglott, D., Masterson, P., McGarvey, K. M., Murphy, M. R., O’Neill, K., Pujar, S., Rangwala, S. H., Rausch, D., Riddick, L. D., Schoch, C., Shkeda, A., Storz, S. S., Sun, H., Thibaud-Nissen, F., Tolstoy, I., Tully, R. E., Vatsan, A. R., Wallin, C., Webb, D., Wu, W., Landrum, M. J., Kimchi, A., Tatusova, T., DiCuccio, M., Kitts, P., Murphy, T. D. & Pruitt, K. D. (2016). Nucleic Acids Res. 44, D733–D745. [DOI] [PMC free article] [PubMed]
- Oscarsson, M., Beteva, A., Flot, D., Gordon, E., Guijarro, M., Leonard, G., McSweeney, S., Monaco, S., Mueller-Dieckmann, C., Nanao, M., Nurizzo, D., Popov, A., von Stetten, D., Svensson, O., Rey-Bakaikoa, V., Chado, I., Chavas, L., Gadea, L., Gourhant, P., Isabet, T., Legrand, P., Savko, M., Sirigu, S., Shepard, W., Thompson, A., Mueller, U., Nan, J., Eguiraun, M., Bolmsten, F., Nardella, A., Milàn-Otero, A., Thunnissen, M., Hellmig, M., Kastner, A., Schmuckermaier, L., Gerlach, M., Feiler, C., Weiss, M. S., Bowler, M. W., Gobbo, A., Papp, G., Sinoir, J., McCarthy, A., Karpics, I., Nikolova, M., Bourenkov, G., Schneider, T., Andreu, J., Cuní, G., Juanhuix, J., Boer, R., Fogh, R., Keller, P., Flensburg, C., Paciorek, W., Vonrhein, C., Bricogne, G. & de Sanctis, D. (2019). J. Synchrotron Rad. 26, 393–405. [DOI] [PMC free article] [PubMed]
- Pak, T. R., Altman, D. R., Attie, O., Sebra, R., Hamula, C. L., Lewis, M., Deikus, G., Newman, L. C., Fang, G., Hand, J., Patel, G., Wallach, F., Schadt, E. E., Huprikar, S., van Bakel, H., Kasarskis, A. & Bashir, A. (2015). Antimicrob. Agents Chemother. 59, 7117–7120. [DOI] [PMC free article] [PubMed]
- Park, J., Gasparrini, A. J., Reck, M. R., Symister, C. T., Elliott, J. L., Vogel, J. P., Wencewicz, T. A., Dantas, G. & Tolia, N. H. (2017). Nat. Chem. Biol. 13, 730–736. [DOI] [PMC free article] [PubMed]
- Paul, C. E., Eggerichs, D., Westphal, A. H., Tischler, D. & van Berkel, W. J. H. (2021). Biotechnol. Adv. 51, 107712. [DOI] [PubMed]
- Permala, R. R., Glady-Croue, J., Watkin, E. L. J., Ramsay, J. P. & Croue, J.-P. (2018). Microbiol. Resour. Announc. 7, e00914-18. [DOI] [PMC free article] [PubMed]
- Puigbò, P., Guzmán, E., Romeu, A. & Garcia-Vallvé, S. (2007). Nucleic Acids Res. 35, W126–W131. [DOI] [PMC free article] [PubMed]
- Rice, P., Longden, I. & Bleasby, A. (2000). Trends Genet. 16, 276–277. [DOI] [PubMed]
- Roach, D. J., Burton, J. N., Lee, C., Stackhouse, B., Butler-Wu, S. M., Cookson, B. T., Shendure, J. & Salipante, S. J. (2015). PLoS Genet. 11, e1005413. [DOI] [PMC free article] [PubMed]
- Rudra, P., Hurst-Hess, K., Lappierre, P. & Ghosh, P. (2018). Antimicrob. Agents Chemother. 62, e00119-18. [DOI] [PMC free article] [PubMed]
- Sánchez, M. B. (2015). Front. Microbiol. 6, 658. [DOI] [PMC free article] [PubMed]
- Sanner, M. F. (1999). J. Mol. Graph. Model. 17, 57–61. [PubMed]
- Schwinn, K., Ferré, N. & Huix-Rotllant, M. (2020). Phys. Chem. Chem. Phys. 22, 12447–12455. [DOI] [PubMed]
- Sobolev, O. V., Afonine, P. V., Moriarty, N. W., Hekkelman, M. L., Joosten, R. P., Perrakis, A. & Adams, P. D. (2020). Structure, 28, 1249–1258. [DOI] [PMC free article] [PubMed]
- Sum, P.-E., Lee, V. J., Testa, R. T., Hlavka, J. J., Ellestad, G. A., Bloom, J. D., Gluzman, Y. & Tally, F. P. (1994). J. Med. Chem. 37, 184–188. [DOI] [PubMed]
- Švecová, L., Østergaard, L. H., Skálová, T., Schnorr, K. M., Koval’, T., Kolenko, P., Stránský, J., Sedlák, D., Dušková, J., Trundová, M., Hašek, J. & Dohnálek, J. (2021). Acta Cryst. D77, 755–775. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C., Grosse-Kunstleve, R. W., Afonine, P. V., Moriarty, N. W., Adams, P. D., Read, R. J., Zwart, P. H. & Hung, L.-W. (2008). Acta Cryst. D64, 515–524. [DOI] [PMC free article] [PubMed]
- Tickle, I. J., Flensburg, C., Keller, P., Paciorek, W., Sharff, A., Vonrhein, C. & Bricogne, G. (2018). STARANISO. Cambridge: Global Phasing Ltd.
- Toplak, M., Matthews, A. & Teufel, R. (2021). Arch. Biochem. Biophys. 698, 108732. [DOI] [PubMed]
- Tropea, J. E., Cherry, S. & Waugh, D. S. (2009). Methods Mol. Biol. 498, 297–307. [DOI] [PubMed]
- Trott, O. & Olson, A. J. (2010). J. Comput. Chem. 31, 455–461. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Lebedev, A. (2015). Acta Cryst. A71, s19.
- Volkers, G., Damas, J. M., Palm, G. J., Panjikar, S., Soares, C. M. & Hinrichs, W. (2013). Acta Cryst. D69, 1758–1767. [DOI] [PubMed]
- Volkers, G., Palm, G. J., Weiss, M. S., Wright, G. D. & Hinrichs, W. (2011). FEBS Lett. 585, 1061–1066. [DOI] [PubMed]
- Walkiewicz, K., Davlieva, M., Wu, G. & Shamoo, Y. (2011). Proteins, 79, 2335–2340. [DOI] [PMC free article] [PubMed]
- Williams, C. J., Headd, J. J., Moriarty, N. W., Prisant, M. G., Videau, L. L., Deis, L. N., Verma, V., Keedy, D. A., Hintze, B. J., Chen, V. B., Jain, S., Lewis, S. M., Arendall, W. B., Snoeyink, J., Adams, P. D., Lovell, S. C., Richardson, J. S. & Richardson, D. C. (2018). Protein Sci. 27, 293–315. [DOI] [PMC free article] [PubMed]
- Yaghoubi, S., Zekiy, A. O., Krutova, M., Gholami, M., Kouhsari, E., Sholeh, M., Ghafouri, Z. & Maleki, F. (2022). Eur. J. Clin. Microbiol. Infect. Dis. 41, 1003–1022. [DOI] [PMC free article] [PubMed]
- Yang, W., Moore, I. F., Koteva, K. P., Bareich, D. C., Hughes, D. W. & Wright, G. D. (2004). J. Biol. Chem. 279, 52346–52352. [DOI] [PubMed]
- Zhao, J., Liu, Y., Liu, Y., Wang, D., Ni, W., Wang, R., Liu, Y. & Zhang, B. (2018). Front. Microbiol. 9, 549. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: SmTetX, 8aq8
SASBDB reference: SmTetX, without reducing agent, SASDPV7
SASBDB reference: with reducing agent, SASDPV7
Supplementary Figures and Table. DOI: 10.1107/S2053230X23005381/va5051sup1.pdf
Raw diffraction images for PDB entry 8aq8.: https://doi.org/10.15785/SBGRID/956
Raw scattering images for SASBDB entries SASDPV7 and SASDPW7.: https://doi.org/10.5281/zenodo.7348780