

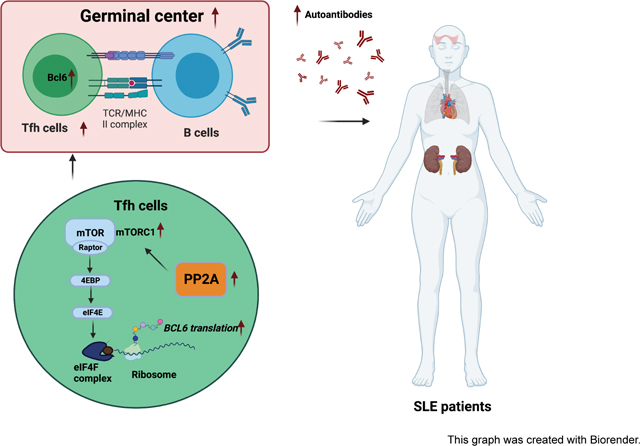
Abstract
Follicular helper T (Tfh) cells are important for generating humoral immune responses by helping B cells form germinal centers (GCs) and the production of high-affinity antibodies. However, aberrant Tfh cell expansion also contributes to the generation of self-reactive autoantibodies and promotes autoantibody-mediated autoimmune diseases such as systemic lupus erythematosus (SLE). Protein phosphatase 2A catalytic subunit alpha isoform (PP2A Cα) expression levels are elevated in peripheral T cells of SLE patients and positively correlate with autoantibody titers and disease activity. Here, we demonstrate a critical role of PP2A in Tfh differentiation by using T cell restricted PP2A Cα deficient mice. We observed impaired Tfh differentiation and GC response in two different classical Tfh induction models. Mechanistic studies revealed that downregulation of protein translation of the Tfh lineage transcription factor BCL6 in PP2A deficient T cells. Importantly, we found that PP2A deficiency by either gene knockout or chemical inhibition alleviated lupus severity in mice. Lastly, we confirmed a positive correlation between PP2A Cα and BCL6 protein levels in human CD4+ T cells from patients with SLE. In summary, our study revealed a critical role of PP2A in regulating Tfh cells and suggests it is a potential therapeutic target for lupus.
Keywords: PP2A, Tfh, SLE, mTOR
Graphical Abstract
1. Introduction
T follicular helper (Tfh) cells constitute a specialized subset of effector CD4+ T cells, which help B cells undergo class switching, as well as antibody affinity maturation in germinal centers during humoral immune responses [1]. Tfh cells are defined by high expression of surface markers CXCR5, ICOS and PD1, which facilitate the migration of Tfh cells toward B cell follicles and ensure their interaction with B cells [2–4]. The transcription factor (TF) BCL6 is the lineage-defining factor for Tfh cells and is critical for Tfh differentiation and maintenance [5, 6]. As a transcriptional repressor, BCL6 can directly bind and suppress non-Tfh-associated genes like Tbx21, Gata3, and Rora. It can also indirectly upregulate expression of Tfh-associated genes such as Cxcr5 and Pdcd1 via repression of transcription repressors like BLIMP1 [6, 7].
Dysregulated Tfh cells can disturb GC reactions and generate autoreactive antibodies with the potential to induce autoimmune diseases. For example, increased circulating Tfh (cTfh) cells in peripheral blood have often been found in patients with SLE, a typical autoimmune disease mediated by autoantibodies [8, 9]. Tfh cell numbers also increase in lupus animal models, in parallel to disease development and progression. Furthermore, genetic deficiency of genes such as P2rx7 [10], Def6, Swap70 [11] and Srsf1 [12] can increase Tfh cell numbers and lead to spontaneous or aggravated lupus in mice. In contrast, inhibiting the development of Tfh in mice can prevent the progression of SLE [13, 14]. Therefore, targeting Tfh cells may be a feasible approach to control autoimmune diseases like SLE.
Phosphatase PP2A is one of the major Ser/Thr phosphatases in eukaryotes, which is critical for many cellular functions including cell survival, proliferation, activation, and differentiation. PP2A is composed of three subunits, a structural A, a regulatory B, and a catalytic C subunit. The PP2A C subunit has two isoforms (PP2A Cα and PP2A Cβ), and the expression of PP2A Cα is ten times higher than PP2A Cβ [15, 16]. It has been reported that PP2A Cα protein expression levels are elevated in peripheral T cells of SLE patients and positively correlate with disease activities [8]. PP2A has been reported to be essential in Th17 cell differentiation and requisite for the regulatory function of Treg cells [17, 18]. However, evidence from these studies is not sufficient to establish a solid link between PP2A and SLE. For example, despite the overwhelming evidence that IL17 contributes to lupus pathology, IL17A deficiency in lupus prone MRL/lpr mice or IL17A neutralization in NZB/NZW mice does not affect the course of nephritis [19]. Furthermore, the function of PP2A in Treg cells would be predicted to promote immune tolerance rather than autoimmunity [18]. Lastly, it has been suggested that PP2A expression promotes SLE by diminishing IL2 production, but low dose recombinant human IL2 treatment in lupus could not restore the aberrant Tfh expansion to normal levels [20]. In addition, other autoimmune diseases like rheumatoid arthritis (RA), in which there is lower IL2 levels but equal expression of PP2A compared to healthy controls, have conflicting data regarding Tfh cell frequencies [8, 21]. Given the critical role of Tfh cells in SLE, the fundamental question of whether PP2A affects the differentiation and function of Tfh cells and contributes to the production of autoreactive antibodies in SLE remains an important question.
In our present study, we demonstrate that PP2A is essential for Tfh differentiation. We further show that PP2A deficiency translationally downregulated BCL6 expression via the mTORC1–4EBP-eIF4E axis. Additionally, both PP2A T cell deficiency and PP2A inhibition significantly protected mice from lupus with fewer autoantibodies in serum and milder injuries in kidney. Finally, we confirmed a positive correlation between PP2A Cα and BCL6 protein expression in CD4+ T cells of lupus patients. Overall, our study suggests that PP2A is a potential therapeutic target for the treatment of SLE.
2. Materials and methods
2.1. Mice and cell lines
Ppp2ca-floxed mice were provided by X.G. B6 mice and mice with Cre recombinase driven by the distal promoter of the gene encoding the kinase Lck were bought from the Jackson Laboratory. Lupus prone MRL/MpJ-Faslpr (MRL/lpr) mice and their control strain MRL/MpJ mice were purchased from Shanghai laboratory animal center of the Chinese Academy of Sciences (CAS) and bred at the Zhejiang University laboratory animal center under specific pathogen-free conditions. All control mice were littermate controls. The experimental protocols were approved by the review committee of Zhejiang University School of Medicine. All animal experiments comply with the national research council’s guide for the care and use of laboratory animals. Jurkat cell line for in vitro studies, Platinum-E (Plat-E) cell line for retroviral packaging, 293FT cell line for lentivirus packaging were purchased from American Type Culture Collection (ATCC).
2.2. Mice immunization and treatment
Mice were age and sex-matched within each experiment (6–10-week-old) and were bred and maintained in the animal facility of Zhejiang University School of Medicine (Hangzhou, China).
Both male and female mice were used in SRBC and NP-KLH (4-hydroxy-3-nitrophenyl-Keyhole Limpet Hemocyanin, Biosearchtech) immunization model. For SRBC immunization, mice were immunized with 2×108 SRBCs (Baiji biotech, China) intraperitoneally (i.p.) and sacrificed on day 7 or as indicated in the manuscript. For antigen-specific antibody immunization, mice were immunized with 100 μg NP-KLH precipitated with alum plus 1 μg LPS on day 0 or 100 μg NP-KLH in an equal amount of incomplete Freund’s adjuvant (Thermo Fisher Scientific) on day 14 for the boost dose. Spleens were cut into two halves, with one half used for flow cytometry, and the other half for imaging studies.
For the chromatin-immunized lupus model, female C57BL/6 mice at 8-week-old were subcutaneously (s.c.) immunized with chromatin extracted from splenocytes as reported [22]. In brief, nuclear chromatin samples were prepared from splenocytes stimulated with 5 μg/mL ConA (Sigma) for 72 hours. Mice were immunized with 200 μg of chromatin emulsified complete Freund’s adjuvant (CFA; Invitrogen) and boosted with 100 μg of chromatin in incomplete Freund’s adjuvant (IFA; Invitrogen) on day 14 and day 28 after the first immunization. Mice immunized with adjuvants emulsified PBS were used as controls.
For the SLE treatment model, MRL/lpr and MRL/MpJ mice were treated with PP2A inhibitor CAN (0.5 mg/kg of body weight) or PBS i.p. once every 2 days from week 8 to week 16. Bodyweight was recorded once per week. Blood was collected at week 20. Spleens were processed for flow cytometry. Left kidneys were processed for H&E and PAS staining from paraffin-embedded tissue sections. Right kidneys were processed to frozen sections for immunofluorescence staining.
2.3. Human tonsils and blood samples
All human work has been carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki). Human tonsils from patients with adenotonsillar hypertrophy causing sleep disordered breathing or obstructive sleep apnea were obtained from the Division of Pediatric Otolaryngology, Children’s National Hospital, Washington, DC, USA. Tonsil cells were mechanically disrupted to make a single cell suspension one day after tonsillectomy. Cells were then washed with PBS and banked with 90% fetal bovine serum (FBS, VWR) and 10% DMSO. Banked tonsil cells were thawed with thaw medium (RPMI with 10% of FBS, 0.1mg/ml DNase1 (Roche), and 10mM HEPES) in a 37°C water bath for 2–3 mins. Cells were diluted by incremental addition of a 1:1 volume of media to 8ml, then centrifuged at 1600rpm for 5min. Cells were then resuspended in 300 μL of thaw media, incubated at room temp for 5 min, then washed with media without DNase1, taken up 10ml, and filtered in a 100mm strainer before spinning down for culture. Tonsillar cells were then co-cultured with or without PP2A inhibitor CAN for 16 hours before flow cytometry assessment.
For samples in Figure 7, 10 female patients who met the requirement of the revised criteria of the American College of Rheumatology for SLE were enrolled. Age-matched healthy females were recruited as controls. Disease activity scores were evaluated based on the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI): no activity (SLEDAI = 0–4), mild activity (SLEDAI = 5 to 10), moderate activity (SLEDAI = 11 to 14), and high activity (SLEDAI ≥ 15). 10 patients with SLEDAI less or equal to 4 were enrolled in the cohort. Patient information is listed in Supplemental Table 2. PBMCs were extracted with lymphocytes separation medium (CEDARLAN,CL5020) and assayed with flow cytometry or sorted for CD4+ T cells for western blotting.
2.4. Flow cytometry
For mice experiments, single-cell suspension of splenic or lymph node cells was incubated with purified anti-mouse CD16/32 antibody (BioLegend) for 5 min to block Fc receptors and then stained with indicated antibodies in staining buffer (PBS supplemented with 1% FBS and 5 mM EDTA). For intracytokine detection in mice, PMA (50 ng/ml, Beyotime), ionomycin (500 ng/ml) with GolgipPlug (BD) were used for stimulation for 4 h. For nuclear factor staining, cells were stained using Bioscience Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific) according to the manufacturer’s protocol. For intracellular phosphorylated antibodies detection, cells were fixed with 4% paraformaldehyde and permeabilized with ice cold MeOH.
The following antibodies were used in mice experiments: anti-mouse CXCR5 (REA215) was from Miltenyi. Anti-mouse BCL6 (K112–91), anti-mouse CD95 (JoL), anti-mouse PD1 (J43) and anti- Foxp3 mouse (FJK-16s) were all from BD Biosciences. Anti-mouse GL7 (GL7), anti-mouse IgG (X56), anti-mouse CD138 (281–2), anti-mouse CD19 (1D3/CD19), anti-mouse SLAM (TC15–12F12.2), anti-mouse biotin IgD (11.26C.2a), anti-mouse biotin Gr-1 (RB6–8C5), anti-mouse biotin IgM (RMM-1), anti-mouse CD44 (IM7), anti-mouse ICOS (C389.4A), anti-mouse CD4 (RM4–5), anti-mouse TCRβ (H57–597) and Streptavidin were all from BioLegend. Anti-p-4EBP(T37/46) (236B4), anti-p-S6(S235/236) (D52.2.2E) were from CST. Samples were acquired on a LSR Fortessa (BD) or NovoCyte (ACEA), and data were analyzed using FlowJo vX.10.0.7 software.
For tonsil cells, two million tonsil cells were cultured at 1×107 cells/ml in 96 well plates with the indicated amount of PP2A inhibitor Cantharidin (Sigma) in the culture medium for 16 hours. For intracytokine detection, GolgiSTOP (monensin, BD), Golgiplus (BFA, BD), PMA (50ng/ml), and Ionomycin (1000 ng/ml) were added into the culture medium and stimulated for 2.5 h. Cells were first stained with live dead dye for 15 min at RT, washed twice, and then incubated with monocyte blocker (BioLegend) for 5 min. Antibodies for chemokine receptors (anti-CCR7 for 10 min, anti-CXCR5 for 5 min) were sequentially added at RT. Antibody mix containing the rest of the surface antibodies and brilliant stain buffer plus (BD) was then added directly to the cells and incubated for 30 min at RT in the dark. Surface-stained cells were washed and fixed with Cytofix Fixation Buffer (BD) at RT for 20 min, washed with permeabilization buffer (eBioscience) twice, then intracytokine antibody mix was added for 30 min. Nuclear transcription factor (TF) staining was processed the same as in mice experiments. Stained cells were collected using spectral flow cytometry (Aurora, Cytek) within 24 hours.
Live/dead Blue and anti-human CD45 (HI30) were purchased from Thermo. Anti-human CCR7 (G043H7), antihuman CD3 (SK7), anti-human CD19 (HIB19), anti-human PD1 (EH12.2H7), anti-human ICOS (C398.4A), anti-human CD14 (63D3), anti-human FOXP3 (206D), anti-human Tbet (4B10), anti-human IFNγ (B27) and anti-human TNFa (MAb 11) were all from BioLegend; anti-human GATA3 (L50-823), anti-human CXCR5 (RF8B2), anti-human CD45RA (5H9), anti-human CD56 (NCAM16.2), anti-human CD8 (SK1), anti-human BCL6 (K112–91) and anti-human IL21 (3A3-N2.1) were all from BD; anti-human CD4 (SK3) was from Cytek.
2.5. Cell sorting
Mice CD4+ T cells were enriched by negative selection with a CD4+ isolation kit (Stem Cell). Human PBMC and mice CD4+ T cell single-cell suspensions were stained with indicated antibodies for 30min at 4°C, and then washed twice. Stained cells were then sorted with FACS Aria II BD. Anti-mouse CD44 (IM7), anti-mouse CD4 (RM4–5), anti-mouse CD25 (PC61) and anti-mouse CD62L (MEL-14) were used for naive CD4+ T cell sorting from mice. And anti-human CD25 (BC96), anti-human CD4 (RPA-T4) anti-human CD45RA (HI100), and anti-human CCR7 (GO43H7) were used for CD4+T cell sorting from human PBMC (BioLegend).
2.6. T cell in vitro stimulation
Jurkat and naïve CD4+ T cells were activated with plate bounded anti-CD3 and CD28 (anti-mouse CD3, Invitrogen, anti-mouse CD28 and anti-human CD3/28, BioLegend, 2.5 μg/ml each) for the indicated time. Other than anti-CD3 and CD28, conditions for Th0 and Tfh-like cells: Th0 (anti-IL4 10 μg/ml, anti-IFNγ 10 μg/m, BioLegend); Tfh-like (anti-IL4 10 μg/ml, anti-IFNγ 10 μg/ml, anti-IL12 10 μg/ml, anti-TGFβ 10 μg/ml, BioLegend; IL6 100 ng/ml and IL21 50 ng/ml, PeproTech).
For the T cell coculture regents, rapamycin was purchased from Cell Signaling Technology. 4EGI-1 and Cycloheximide (CHX) were purchased from MedChemExpress. Cantharidin (CAN), okadaic acid (OA), and MG132 were purchased from Selleckchem. Sphingomyelinase (SMase) was purchased from Merck.
2.7. Western blotting and M7GTP pull-down assay
Cells were lysed in 2×SDS loading buffer (100 mM Tris-HCl, 4% SDS, 20% glycerol, 2% 2-mercaptoethanol, and 0.05% bromophenol blue). The samples were then boiled and loaded on 10% or 12% SDS-PAGE gels and transferred to nitrocellulose membranes (Pall). The membranes were blocked for 1 h in blocking buffer (5% skimmed milk and 0.1%Tween20 in TBS) at room temperature. After incubation with primary and secondary antibodies, immunoblotting was detected with ECL blotting reagents (Thermo Fisher).
For the M7GTP pull-down assay, cells were homogenized in Western and IP buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, Beyotime) for 30 mins and flipped in 1.5 ml Eppendorf tubes for 30 min at 4°C and then centrifuged at 12,000g for 5 min at 4°C to remove cellular debris. Proteins were immunoprecipitated by incubation of lysates with M7GTP Sepharose Beads (Jena Bioscience) overnight at 4°C. The presence of immunocomplex proteins was determined by Western Blotting.
The following antibodies were used: anti-BCL6 (561520) and anti-PP2A Cα (610556) was from BD Biosciences. Anti-p-4EBP1(Thr37/46) (2855), anti-p-4EBP1(Thr70) (9455), anti-pS6K (Ser389) (9234), anti-eIF4E (9742), anti-eIF4G (2489) and anti-S6K (9202) were all from CST. Anti-eIF2α (ab169528), anti-mTOR (ab32028), anti-4EBP (ab30521) and anti-pS6K (ab2418) were all from Abcam.
2.8. Real-time PCR analysis
RNA was extracted using a total RNA extraction reagent (Vazyme Biotech). Complementary DNA was synthesized using HiScript® II Reverse Transcriptase (Vazyme Biotech) according to the manufacturer’s instructions. qPCR was performed using SYBR Green (Vazyme Biotech) on a CFX96 Touch Real-Time PCR (Bio-Rad). All samples were individually normalized to GAPDH in humans and Gapdh in mice.
The following primers were used:
Human BCL6: forward, 5’-TCA CCA ACA CCA AGG TGC AA-3’; reverse,5’-TGT AAT CGC CCT TAT CGT CGT-3’.
Human GAPDH: forward, 5’-ACA ACT TTG GTA TCG TGG AAG G-3’; reverse, 5’-GCC ATC ACG CCA CAG TTT C-3’.
Human GATA3: forward, 5’-GCC CCT CAT TAA GCC CAA G-3’; reverse, 5’-TTG TGG TGG TCT GAC AGT TCG-3’.
Mice Bcl6: forward, 5’-CCG GCA CGC TAG TGA TGT T-3’; reverse, 5’-TGT CTT ATG GGC TCT AAA CTG CT-3’.
Mice Gapdh: forward, 5’-AGG TCG GTG TGA ACG GAT TTG-3’; reverse, 5’-TGT AGA CCA TGT AGT TGA GGT CA-3’.
2.9. ELISA
For affinity maturation, high-affinity and low-affinity NP-specific antibodies were measured by ELISA with 10 mg/mL NP7-BSA or NP23-BSA (Bioresearch Technologies) as the respective coating reagent. Histone-specific antibodies were detected using plates coated with histone (50 μg/ml) (Sigma). dsDNA-specific antibodies were detected using plates coated with dsDNA (20 μg/ml) (Sigma). A peroxidase-labeled Fc-specific anti-mouse IgG or IgM detection antibody was used as the secondary antibody (SouthernBiotech).
2.10. Urine protein assay
Urine protein levels from the Chromatin induced mice were evaluated every two weeks. Urine protein levels from MRL/lpr mice were evaluated at week 19. Urine was collected 3 consecutive days at 8 am and 3 pm and pooled for one test. The urine protein concentration was analyzed by Pierce™ BCA Protein Assay Kit (Thermo scientific, 23225).
2.11. Vectors and transductions
Plat-E cells were cells for retroviral packaging and transfected with PMX-retrovirus using PEI transfection method. 293FT cells were cells for lentiviral packaging and co-transfected with the lentiviral core vectors psPAX2 and pMD2.G at a ratio of 4:3:1 using the calcium phosphate transfection method. All virus-containing supernatants were collected 48–72h after transfection. Supernatants were filtered through a 0.45 µm PES membrane before use. Naive CD4+T cells were stimulated with anti-CD3/28 for 24h, and then spun infected with PMX-HA-Ppp2ca or control viral supernatant in the presence of polybrene (8 μg/ml) and 10 mM HEPES at 32°C with a speed of 2500 rpm for 90 mins. Jurkat cells were spun infected with lentivirus encoding human HA-PPP2CA, FLAG-BCL6 or control viral supernatant in the presence of polybrene (8 μg/ml) and 10 mM HEPES at 32°C with a speed of 2500 rpm for 90 mins. The shRNAs targeting PPP2CA were constructed in lentivirus (Sigma). List as shPPP2CA-shRNA1: 5’-TGG AAC TTG ACG ATA CTC TAA-3’, shPPP2CA-shRNA2: 5’-CCC ATG TTG TTC TTT GTT ATT-3’. Control shRNA: 5’-GTT CTC CGA ACG TGT CAC GT-3’. Forty-eight hours after infection, the cells were selected by culture with 2 mg/mL puromycin (Sigma) or blasticidin (Invitrogen).
2.12. Polysome analysis
Jurkat cells that transfected with eithter shScramble or shPPP2CA were used in polysome assay. Polysome was prepared as described previously [23]. Briefly, Jurkat cells were seeded in T75 at 1^107 cells per dish and cultured overnight. Cells were then treated with 100 ug/ml CHX for 5 min before lysis, washed in ice-cold PBS (without calcium) plus 100 ug/ml CHX, and then lysed in polysome lysis buffer (15 mM HEPES-KOH (pH 7.4), 7.5 mM MgCl2, 100 mM KCl, 2 mM DTT, 1.0% Triton X-100, 100 ug/ml CHX, and one tablet of EDTA-free protease inhibitors (Roche) per 25 ml). Lysates were normalized by protein content using Bradford reagent (Bio-rad) and either layered onto 11 ml 10–50% sucrose density gradients (15 mM HEPES-KOH, 7.5 mM MgCl2, 100 mM KCl, 2 mM DTT, 100 mg/ml CHX, 20 U/ml SuperaseIn, 10–50% RNase-free sucrose) or adjusted to 0.5% SDS and reserved for total RNA isolation. Gradients were centrifuged in an SW-41Tirotor at 32,000 r.p.m. at 4°C for 2 h, and then sampled using a Labconco Auto Densi-Flow Gradient Fractionator connected to an Isco Tris pump with constant monitoring of optical density (OD) at 254 nM. PolyA1 synthetic luciferase mRNA (5 ng, Promega) was added to each fraction for normalization. RNA was extracted from diluted fractions using RNA extraction reagent (Vazyme Biotech). Complementary DNA was synthesized using HiScript® II Reverse Transcriptase (Vazyme Biotech). Transcript abundance was determined by real-time PCR. Measurements were then normalized to luciferase abundance and plotted as percent detected. Oligonucleotides used for qPCR firefly luciferase:
forward: 5’-ATC CGG AAG CGA CCA ACG CC-3’, reverse: 5’-GTC GGG AAG ACC TGC CAC GC-3’.
2.13. Bulk RNAseq and Gene Set Enrichment Analysis (GESA)
Naive CD4+ T cells from WT and cKO mice with three replicates were stimulated by plate bounded anti-CD3 and anti-CD28 for 1 hour. mRNA was extracted using RNAeasy Plus Mini kit (Qiagen). mRNA profiles of in vitro stimulated WT and cKO naive CD4+ T cells were generated by deep sequencing of three biological replicates by Novaseq. fastp software (https://github.com/OpenGene/fastp) was used to remove the reads that contained adaptor contamination, low quality bases and undetermined bases with default parameter. Then sequence quality was also verified using fastp. We used HISAT2 (https://ccb.jhu.edu/software/hisat2) to map reads to the reference genome of Mus musculus GRCm38. The mapped reads of each sample were assembled using StringTie (https://ccb.jhu.edu/software/stringtie) with default parameters. Then, all transcriptomes from all samples were merged to reconstruct a comprehensive transcriptome using gffcompare (https://github.com/gpertea/gffcompare/). Differential gene lists between stimulated WT and cKO naïve CD4+ T cells were created by edgeR and an absolute fold change of 1.3 and FDR less than 0.05 were set as cut-off for significantly changed genes. GSEA (4.1.0) was used to interpret gene expression data. DESeq2 package from GenePattern was first used to normalize the raw counts and generate compatible input files for GESA (4.1.0). All gene counts were used in the GSEA analysis. C7.immunesigbd.v7.4.symbols was selected as the gene sets database. Each analysis was arranged 1000 times to determine the p-value. P < 0.05 and false discovery rate (FDR) < 0.25 were considered to have significant differences.
2.14. Imaging
For confocal microscopy, frozen sections of mice kidneys or spleens were fixed with cold acetone and stained by direct immunofluorescence (Alexa Flour 594 anti-mouse IgD (Biolegend,11–26c.2a), Alexa Flour 488 anti-mouse CD4 (Biolegend, RM4–5)) and FITC anti-mouse IgG (Biolegend, Poly4060). Images were captured with Nikon A1R.
Mouse kidneys were harvested, fixed in 10% buffered formalin, and embedded in paraffin. Sections (4 μm) were stained with H&E and PAS. Pathological changes in the kidneys were semi-quantitatively scored on a scale of 0–3 (0 = no changes; 3 = severe cell proliferation/ infiltration and crescent formation) as described previously [24].
2.15. Ingenuity Pathway Analysis
Pathway enrichment by Ingenuity Pathway Analysis (IPA, Invitrogen) of 269 proteins identified from literature where Jurkat T cells were treated with SMase (0.5 units/mL) or vehicle (50% glycerol in PBS) for 1h and then lysed for protein immunoprecipitation with a PP2A Cα-specific antibody [18].
2.16. GO and KEGG analysis of phosphorylation mass spectrometry data
The raw datasets were derived from a dataset from a previous study in our lab [25]. GO and KEGG enrichment analysis was performed with R (v3.0.0), visualization was generated with the ggplot2 package, and the GO and KEGG terms with the adjusted p value of less than 0.01 were considered significantly enriched.
2.17. PP2A phosphatase activity assay
PP2A phosphatase activity was assessed using the PP2A Phosphatase Activity Assay Kit (GMS50042.3; Laier biotech) according to the manufacturer’s instructions. Briefly, cells were treated with cell lysis buffer and incubated with GENMED regents sequentially (all regents were provided by the kit). PP2A phosphatase activities from samples were measured by colorimetry and phosphate concentrations were calculated from a standard curve created using serial dilutions of a standard phosphate solution.
2.18. Statistics
Results are shown as the means ± SEM or means ± S.D. indicated in the figure legend. Statistical analyses were performed in GraphPad Prism (V8). Statistical significance was determined by a t-test (2-tailed) for 2 groups. Pearson correlation analysis was used in correlation analysis. P values of less than 0.05 were considered significant if not specified.
2.19. Study approval
All animal procedures were approved by the Institutional Animal Care and Use Committee of Zhejiang University, School of Medicine (protocol number ZJU202100122). The human PBMC samples used in this study were obtained from the Department of Rheumatology, Sir Run Run Shaw Hospital, Zhejiang University, School of Medicine (protocol number 20210319100608647). The tonsils were obtained under a protocol approved by the Institutional Review Board (IRB) at Children’s National Hospital (IRB protocol number 00009806). For samples from lupus patients, written informed consent was received before participation. For tonsil samples, written informed consent was obtained from parents/guardians of all enrolled participants, and assent was obtained from minor participants over 7 years of age. Human tonsil samples were deidentified.
3. Results
3.1. PP2A is required for Tfh differentiation and GC response
To evaluate the role of PP2A in Tfh cells, we first compared the protein levels of PP2A Cα in Tfh (CXCR5+PD1hiCD44+CD4+) and non-Tfh (CXCR5−1PD1lowCD44+CD4+) cells from mouse spleens and found that Tfh cells expressed more abundant PP2A Cα than non-Tfh cells (Fig. 1A–1B)
Figure 1. Conditional PP2A Cα loss in peripheral T cells impaired Tfh differentiation and GC formation.

(A) The sorting strategy of Tfh and non-Tfh cells from non-immunized C57/B6 mice. (B) The protein expression of BCL6 and PP2A Cα from Tfh and non-Tfh cells by western blotting. (C) Schematic diagram of experimental design. PP2A WT and cKO mice were immunized with SRBCs and analyzed on day 7 (n = 8–14 mice per genotype). (D) Representative flow cytometric plots of CXCR5+PD1hi, CXCR5+SLAMlo, CXCR5+BCL6hi populations among CD4+CD44+ T cells from spleens of the indicated genotype. (E) Frequencies and cell numbers of populations in D. (F) Representative flow cytometric staining of IL21 expression gated on CXCR5+PD1+CD44+CD4+ T cells from spleens of the indicated genotype. (G) Statistical analysis of mean fluorescence intensities (MFI) of IL21 in F. (H) Representative flow cytometric plots of GL7+Fas+ GC B cells for the indicated genotype. (I) Frequencies and cell numbers of populations in H. (J) Representative flow cytometric plots of light zone (LZ) GC B cells (CD86+) and dark zone (DZ) GC B cells (CXCR4+) for the indicated genotype. (K) Statistical analysis of LZ/DZ ratios in I. (L) Representative tissue sections from the spleen by immunofluorescence staining (CD4: green, IgD: red). (M) Statistical analysis of L was quantified by the average intensity of CD4 staining in GC areas from each genotype and calculated by Image J (n = 5 mice from each genotype, one section per mouse). Data are representative of two independent experiments and analyzed by two-sided student’s t-test. Error bars of Means ± SEM. *p<0.05; **p<0.01; ***p<0.001.
We next used Ppp2ca T-cell conditional knockout mice (Ppp2cafl/fl disLck-Cre, PP2A cKO) to answer if PP2A plays a role in Tfh cell differentiation. PP2A cKO mice were previously described and shown to exhibit approximately 50% reduction of PP2A phosphatase activity. PP2A WT (Ppp2cafl/+ disLck-Cre or Ppp2ca+/+ disLck-Cre) and cKO were immunized with sheep red blood cells (SRBC, Fig. 1C) and evaluated at day 7 after immunization. Tfh cell generation was significantly reduced in PP2A cKO mice compared with PP2A WT mice, as indicated by the decreased number and frequency of CXCR5+PD-1hi, CXCR5+SLAM− and CXCR5+BCL6hi Tfh cells (gated on CD4+CD44+) (Fig. 1D–1E). Moreover, the secretion of IL21 within the CXCR5+PD-1hi Tfh population was reduced compared with WT mice, suggesting that loss of PP2A not only constrained the differentiation of Tfh cells but also led to functional impairment of differentiated Tfh cells (Fig. 1F–1G). Accordingly, GC B cell frequencies were strongly reduced in PP2A cKO mice (Fig. 1H–1I), along with decreased ratios of the light zone (LZ) to dark zone (DZ) GC B cells (Fig. 1J–1K). Loss of PP2A also led to a marked decline of T cell density in the GC area (Fig. 1L-M), consistent with impaired GC formation due to fewer GC-entranced Tfh cells.
Follicular regulatory T cells (Tfr) are a subset of regulatory T cells which negatively regulate GC reactions. In contrast to the findings in Tfh cells, there were no differences in either frequencies of Treg cells (FOXP3+CD4+) or Tfr cells (FOXP3+CXCR5+PD1+CD4+) between PP2A WT and cKO mice but the ratios of Tfh/Tfr were decreased significantly (S1A-1C).
3.2. PP2A cKO mice produce less T cell-dependent antibodies
To further evaluate the impact of PP2A on GC activity, we immunized PP2A WT and cKO mice with NP-KLH (Fig. 2A). As what was found in the SRBC-induced model, impaired Tfh and GC B cell differentiation, as well as decreased LZ/DZ ratios and IL21 levels were observed in PP2A cKO mice 14 days after immunization (Fig. 2B–2C, and S1D-1I). Consistently, serum concentrations of both high- and low-affinity antibodies (IgG1 and IgM) specific for NP7 and NP23 were significantly reduced in PP2A cKO with respect to WT mice (Fig. 2D), indicating broad impairment of antibody production in PP2A cKO mice. To assess the memory responses, mice were immune boosted on day 14 and analyzed on day 21. Not only were GC B cell percentages and numbers decreased, but also the numbers and the frequencies of NP-specific IgG1+ memory B cells were reduced (Fig. 2E–2G). Similarly, high- or low-affinity NP-specific IgG1 and IgM antibodies were decreased in PP2A cKO mice (Fig. 2H). These data demonstrate that PP2A deficiency in T cells reduces the efficiency of T cell-dependent antibody production.
Figure 2. Conditional PP2A Cα loss in peripheral T cells attenuates antibody response.

(A) Schematic diagram of experimental design. Mice were immunized with NP-KLH in Alum and analyzed on day 14 for primary response (n = 8–14 mice per genotype) or boost with NP-KLH in IFA on day 14 and then analyzed on day 21 for secondary response (10–12 mice per genotype). (B-D) Primary response: (B) Representative flow cytometric profiling of GC B cells for the indicated genotype. (C) Frequencies and cell numbers of GC B cells. (D) Serum anti-NP7 and anti-NP23 specific IgM or IgG1 antibodies from the primary response of NP-KLH immunized mice by ELISA. (E-H) Secondary response: (E) Representative flow cytometric profiling of NP+IgG1+ memory B cells for the indicated genotype. Dump channel including IgM, IgD, CD138 and Gr-1. (F) Statistical analysis of populations in E. (G) Statistical analysis of GC B cells (GL7+FAS+ of B220+ B cells). (H) Serum anti-NP7 and anti-NP23 specific IgM or IgG1 antibodies from the secondary response of NP-KLH immunized mice by ELISA. Data are representative of two independent experiments and analyzed by two-sided student’s t-test. Error bars of Means ± SEM. *p<0.05; **p<0.01; ***p<0.001.
3.3. Aberrant BCL6 protein expression in PP2A cKO T cells
To investigate how PP2A regulates Tfh differentiation, we evaluated the dynamics of Tfh cell generation after SRBC immunization in PP2A WT and cKO mice (Fig. S2A). PP2A cKO mice had similar frequencies of pre-Tfh (CXCR5+BCL6lowCD4+CD44+) cells, but significantly decreased GC-Tfh (CXCR5+BCL6hi CD4+CD44+) frequencies on day 2 (Fig. S2B); in contrast, on day 7, pre-Tfh frequencies were significantly reduced as well as GC-Tfh cells (Fig. S2C). Of note, the expression of BCL6 within GC-Tfh (CXCR5+PD1hi) cells was also significantly reduced in PP2A cKO mice (Fig. 3A). Similar results were found in the NP-KLH-induced model on day 14 (Fig. 3B). These data indicated that CXCR5 induction during the early stage of Tfh cell priming is intact in the context of PP2A deficiency. Thus, PP2A may affect CXCR5 and Tfh differentiation by regulating the critical transcription factor BCL6.
Figure 3. PP2A controls BCL6 protein expression but not transcription.

(A) Representative flow cytometric profiling of BCL6 expression in Tfh (CXCR5+PD1hiCD44+CD4+) cells after SRBC immunization for the indicated genotype. Statistical analysis of BCL6 MFI is shown on the right panel. (B) Representative flow cytometric profiling of BCL6 expression in Tfh (CXCR5+PD1hiCD44+CD4+) cells after NP-KLH immunization for the indicated genotype. Statistical analysis of BCL6 MFI is shown on the right panel. (C) Naive CD4+ T cells were cultured with plate bounded anti-CD3 with or without anti-CD28 for 24 hours. The expression of BCL6 and PP2A Cα was analyzed by western blotting. (D) Naive CD4+ T cells from PP2A WT or cKO mice were cultured with plate bounded anti-CD3/28 for 6 hours. The expression of BCL6 and PP2A Cα was analyzed by western blotting. (E) Naive CD4+ T cells from PP2A WT or cKO mice were cultured under anti-CD3/28 stimulation for the indicated time. The expression of Bcl6 mRNA was measured by qPCR. (F) Bulk RNAseq was performed with PP2A WT and cKO CD4+ naïve T cells (three biological replicates per genotype) after stimulating with plate bounded anti-CD3/28 antibodies for 1 hour. GSEA analysis used all normalized gene counts as input from PP2A WT and cKO CD4+ naïve T cells showed enrichment of Tfh-cell signature gene set (ES: 0.35, NES 1.67, p-val 0.000, FDR 0.061). This gene set was derived from a comparison between CXCR5+BCL6+Tfh cells and CXCR5−BCL6−CD4+ T cells. Figure (A-E) are representative of two independent experiments and analyzed by two-sided student’s t-test; NS, not significant. Error bars of Means ± SEM. *p<0.05; **p<0.01; ***p<0.001.
The early dysregulation of BCL6 protein expression in PP2A cKO cells suggested that PP2A directly affects the induction of BCL6 expression after T-cell activation. To investigate this, we probed BCL6 expression in primary naive CD4+ T cells and Jurkat cells in vitro before and after TCR stimulation. The results showed that anti-CD3 stimulation alone induced BCL6 expression and additional anti-CD28 treatment could amplify BCL6 expression (Fig. 3C and S3A). We found that the expression of BCL6 peaked at 6 hours after stimulation in primary naive CD4+ T cells and 24 hours in Jurkat cells (Fig. S3B-S3C). Thus, these two time points were used in the following assays of each corresponding cell type. Remarkably, PP2A cKO CD4+ T cells exhibited much lower BCL6 protein levels compared to the WT controls (Fig. 3D and S3B). Consistently, the expression of BCL6 was decreased in Jurkat cells after either knockdown of PP2A Cα by shRNA, or inhibition of PP2A by Cantharidin (CAN) or Okadaic acid (OA) (Fig. S3D-S3F), while increased in Jurkat cells when treated with PP2A activator Sphingomyelinase (SMase) (Fig. S3G). SMase increases endogenous ceramide content and constrains the inhibitory effects of SET on PP2A complex [34]. CAN of 5 uM reduced PP2A activity to one-third in treated Jurkat cells (Fig. S3H).
To determine how PP2A impairs BCL6 expression, we first compared Bcl6 mRNA levels in PP2A WT and cKO naïve CD4+ T cells at different time points following TCR stimulation. Bcl6 mRNA expression peaked at 3 hours after stimulation and then declined with time (Fig. 3E). To our surprise, no difference was observed between WT and cKO CD4+ T cells (Fig. 3E). Similar results were observed in Jurkat cells treated with either PP2A inhibitor CAN or shPPP2CA, except for a mild reduction at 3h (Fig. S3I). RNAseq analysis was performed with anti-CD3 plus anti-CD28 treated primary naïve CD4+ T cells. Again, Bcl6 was not among the differentially expressed genes (Supplemental Table 1). However, CXCR5+BCL6+ Tfh signature genes were enriched in activated WT CD4+ T cells but not in PP2A cKO cells (Fig. 3F). IRF4 and DNMT1 have been suggested to regulate BCL6 and be regulated by PP2A [26–28]. Again, no difference was observed in their expression levels between PP2A WT and cKO, in either Th0 or Tfh-like culture conditions (Fig. S3J). IL2 which has been considered as an inhibitory stimulator for BCL6 expression is negatively regulated by PP2A [8, 29]. We thus tested if the changes of BCL6 in PP2A deficiency were due to effects from IL2. However, IL2 neutralization failed to rescue BCL6 expression in PP2A cKO cells (Fig. S3K). In sum, these data suggest the dysregulation of BCL6 protein expression in PP2A deficient lymphocytes upon TCR stimulation, while this effect is independent of Bcl6 gene transcriptional regulation.
3.4. Translational downregulation of BCL6 protein levels via disruption of eIF4F complex formation in PP2A cKO T cells
We then investigated whether the reduced BCL6 protein in activated PP2A deficient CD4+ T cells was due to aberrant protein translation and/or degradation. Of note, BCL6 contains three PEST domains (proline, glutamic acid, serine, and threonine), which are associated with rapid protein degradation. Notably, in FLAG-BCL6 overexpressed Jurkat cells (transduced by lentivirus), whereas FLAG and BCL6 protein levels were strikingly reduced by either PP2A inhibitor or shRNA knockdown with or without TCR stimulation, their mRNA transcripts were not affected (Fig. 4A–4C). Since the lentivirus initiates FLAG-BCL6 transcription under an independent promoter, this again indicated that the reduced BCL6 protein in PP2A deficient cells was irrelevant to transcriptional regulation and may occur post-transcriptionally. We next evaluated the protein degradation of BCL6 protein by treating cells with the protein synthesis inhibitor cycloheximide (CHX). BCL6 protein was rapidly degraded following CHX treatment. However, the rates of BCL6 degradation in CAN treated or untreated cells were similar (Fig. S4A-S4B). Accordingly, inhibiting proteasome-mediated protein degradation in CAN treated activated Jurkat cells or primary PP2A cKO T cells by MG132 failed to increase BCL6 protein expression to the levels of MG132 treated control cells (Fig. S4C-S4D). These results demonstrated that the attenuation of BCL6 protein in PP2A cKO T cells is not likely due to protein degradation.
Figure 4. Protein synthesis of BCL6 is decreased in PP2A Cα deficiency.
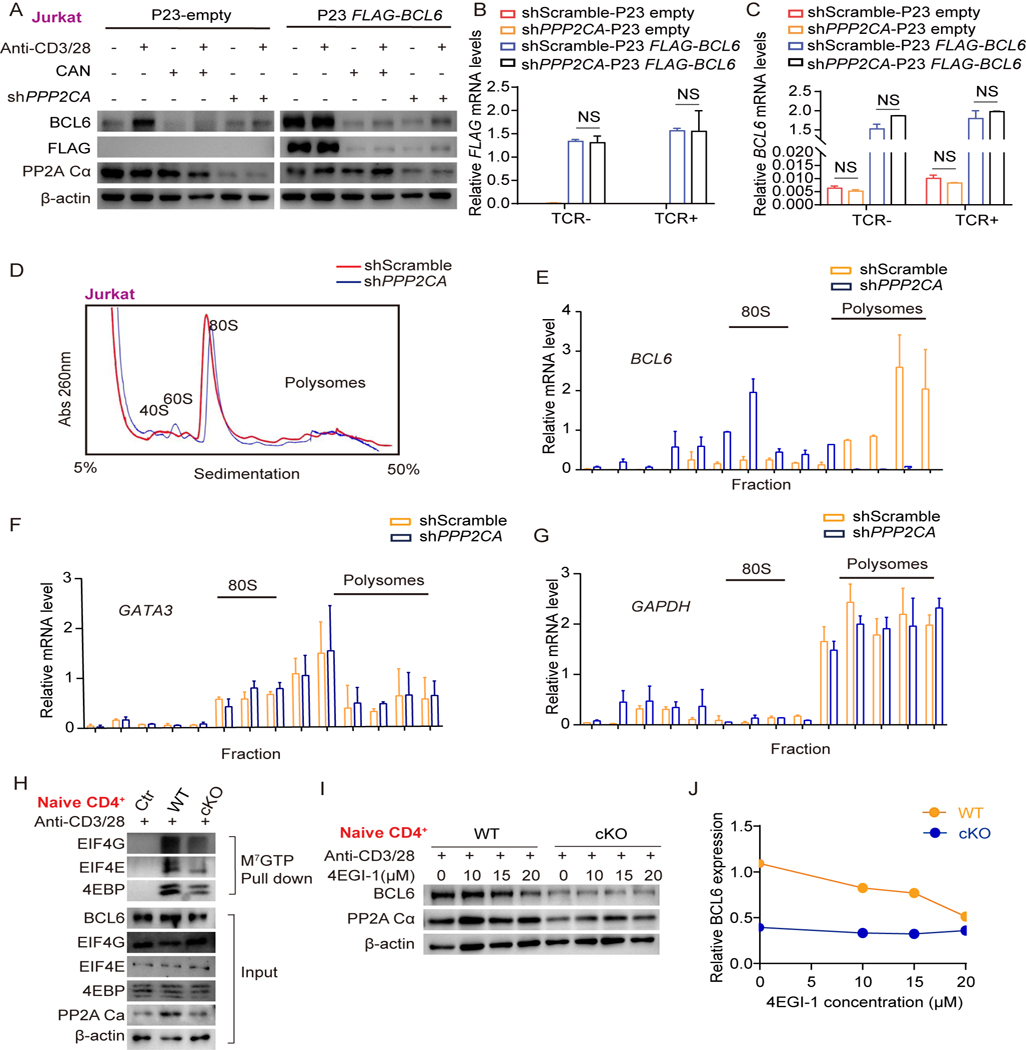
(A) P23-FLAG-BCL6 or P23-empty was transfected in shPPP2CA or shScramble Jurkat cells. Cells were either stimulated or unstimulated with the plate bounded anti-CD3/28 with or without PP2A inhibitor CAN (5μM) for 24 hours. The expression of BCL6, FLAG, and PP2A Cα was analyzed by western blotting. (B and C) Expression of BCL6 and FLAG mRNA was measured by qPCR. (D) Polysome profiles of Jurkat cells which were transfected with shScramble or shPPP2CA and cultured with plate bounded anti-CD3/28 for 24 hours (Abs260, absorbance of light at 260 nm). (E-G) BCL6, GATA3, and GAPDH mRNA abundance in different fractions were quantified by qPCR. (H) Assembly of the eIF4F complex was evaluated by M7GTP immunoprecipitation using extracts from PP2A WT and cKO naive CD4+ T cells which were stimulated with anti-CD3/28 for 6 hours. (I) PP2A WT and cKO naive CD4+ T cells were treated with vehicle control or the indicated doses of 4EGI-1 and stimulated with anti-CD3/28 for 6 hours. The expression of BCL6 and PP2A Cα was analyzed by western blotting. (J) Quantification of BCL6 signal intensity by densitometry over the time course of 4EGI-1 treatment. The levels of β-actin were set as controls. Data are representative of two independent experiments and are analyzed by two-sided student’s t-test; NS, not significant. Error bars of Mean ± S.D. *p<0.05; **p<0.01; ***p<0.001.
When we analyzed Ser/Thr phosphorylation profiling data from PP2A WT and cKO thymocytes [30], we noticed an enrichment of changes in cellular processes including mRNA binding, processing, and RNA transportation (Fig. S5A). To assess BCL6 translational efficiency, polyribosome fractionation analysis by sucrose density gradient centrifugation was performed to track the distribution and quantification of ribosome binding BCL6 mRNAs using extracts from activated shPPP2CA-treated or shScramble-treated Jurkat cells. Polysome profile showed comparable absorbance in gradient fractions corresponding to the monosomes and polysomes, suggesting intact global translation in PP2A deficient Jurkat cells (Fig. 4D). BCL6 mRNAs were then detected by RT-PCR in each fraction. A clear differential distribution was observed: BCL6 mRNA was distributed mainly in heavy polysomes in shScramble-treated control Jurkat cells but switched to light polysomes in shPPP2CA-treated cells, indicating inefficient initiation of translation in PP2A deficiency (Fig. 4E). Of note, the polysome distribution of GATA3 and GAPDH mRNA was largely superimposable even in shPPP2CA-treated cells, suggesting that translational regulation by PP2A is selective (Fig. 4F–4G).
Eukaryotic translation initiation relies on the binding of the eIF4F complex, which consists of eIF4E, eIF4G, and eIF4A, to the cap presented at the 5’end of all mRNAs [31]. The eIF4E binding protein 4EBP competes with eIF4G for eIF4E binding and prevents the formation of the eIF4F complex, which has been reported to be involved in BCL6 de novo synthesis [11]. Thus, we conducted a m7G cap pull-down assay to evaluate assembly of translational active eIF4F (eIF4E-eIF4G-mRNA) complex in both activated CD4+ T cells and Jurkat cells. We found that the enrichment of cap binding proteins eIF4G and eIF4E was reduced in PP2A cKO T cells (Fig. 4H) or Jurkat cells treated with CAN or shRNA (Fig. S5B-S5C). 4EGI-1 as a small-molecule inhibitor that mimicked the function of 4EBP, can bind to eIF4E and interfere with its interaction with eIF4G. We observed that 4EGI-1 treatment decreased the BCL6 expression in WT CD4+ T cells and Jurkat cells to a level equivalent to PP2A cKO or knockdown cells, while it didn’t further decrease the expression of BCL6 protein in PP2A cKO T cells and shPPP2CA treated Jurkat cells (Fig. 4I–4J, S5D-S5E). Protein translation initiation could also be regulated by the phosphorylation of initiation factor eIF2α [32]. But PP2A deficiency didn’t change the phosphorylation status of eIF2α (Fig. S5F). Thus, PP2A deficiency selectively disrupts cap-dependent translation initiation of BCL6 by interfering with eIF4F complex formation.
3.5. PP2A regulates BCL6 translation via modulating mTORC1 mediated 4EBP phosphorylation
Phosphorylated 4EBP mediated by mTORC1 affects its binding to eIF4E. Hypophosphorylated 4EBP isoforms interact strongly with eIF4E, whereas hyperphosphorylated isoforms do not [33]. The Ingenuity Pathway Analysis (IPA) of the published data from PP2A immunoprecipitation of PP2A activator SMase treated Jurkat cells, revealed the association of PP2A Ca with the mTOR pathway components (Fig. 5A). Therefore, the disrupted eIF4F complex in PP2A deficiency may result from aberrant mTOR-4EBP phosphorylation upon TCR stimulation (Fig. 5B). As expected, our results showed that p-S6K (Ser371) and p-4EBP (Thr37/46) was attenuated in PP2A inhibited Jurkat cells (Fig. 5C). The decrease of phosphorylated S6K is also seen in PP2A cKO naïve CD4+ T cells upon anti-CD3 and anti-CD28 stimulation (Fig. S6A). The downregulated phosphorylation of S6K and/or 4EBP could extend to 24 hours in PP2A deficient Jurkat cells and 6 hours in primary CD4 cKO T cells after stimulation, when the expression of BCL6 reaches the peak (Fig. 5D–5E and S6B). Similar trend was also identified by mTORC1 effectors p-S6K (Thr389) and p-4EBP (Thr70) in PP2A deficient jurkat cells or p-S6K (Thr389) in cKO CD4+ T cells (Fig. S6C-D). To further verify that the decreased BCL6 protein in PP2A deficiency was affected by reduced mTORC1 activity, we used mTORC1 inhibitor rapamycin (Rapa) to treat PP2A deficient Jurkat or naïve cKO CD4+ T cells. A reduction of BCL6 protein expression was observed in Jurkat and WT CD4+ T cells, while the changes in PP2A deficient T cells were subtle (Fig. 5F–5G, S6E). Consistently, the expression of BCL6 was increased along with mTORC1 effectors (p-S6K (Ser371) and p-4EBP (Thr37/46)) when we overexpressed PP2A Ca in Jurkat cells (Fig. 5H). Surprisingly, a discrepant phosphorylation trend between S6 and S6K was observed. The phosphorylation of S6 (Ser235/236) was increased as reported in literature [18], either in Jurkat cells or PP2A deficient primary CD4+ T cells, in contrast to the phosphorylation of 4EBP, which was decreased as observed by our western blotting data (Fig. S6G-J). S6 can be phosphorylated on Ser235/236 by RSK and others, as well as mTORC1 [35] (Fig. 5B). We previously reported increased pERK in cKO CD4+ T cells [36] (Fig. S6D in the cited paper), an upstream activator of RSK, supporting a likely effect of RSK on residue Ser235/236. These data support that insufficient mTORC1 activation contributes to 4EBP hypophosphorylation and disrupts eIF4F complex formation for BCL6 translation in PP2A deficient T cells upon TCR stimulation.
Figure 5. The decrease of BCL6 protein in PP2A deficient T cells is mTOR dependent.

(A) Pathway enrichment by Ingenuity Pathway Analysis (IPA) of published 269 proteins immunoprecipitated with a PP2A Cα-specific antibody from PP2A activator SMase treated Jurkat cells. (B) Schematic of phosphorylation sites on S6K, S6 and 4EBP that were evaluated in our study. (C and D) Western blotting of phosphorylated and total 4EBP and S6K in Jurkat cells which were treated with CAN or vehicle and stimulated with anti-CD3/28 antibodies for the indicated time. (E) Western blotting of total BCL6, phosphorylated and total 4EBP and S6K levels in Jurkat cells which were transfected with shPPP2CA or shScrambleand and stimulated with anti-CD3/28 antibodies for 24 hours. (F) The phosphorylated and total levels of 4EBP and S6K in shPPP2CA or shScramble transfected Jurkat cells were treated with or without Rapamycin (Rapa) and stimulated with anti-CD3/28 for 24 hours. (G) Quantification of BCL6 signal intensity by densitometry over the time course of Rapa treatment. (H) P23-HA-PPP2CA or P23-empty was transfected in Jurkat cells. Cells were either stimulated or unstimulated with plate bounded anti-CD3/28 antibodies for 24 hours. The protein expression of BCL6, HA, PP2A Cα, phosphorylated and total 4EBP and S6K were analyzed by western blotting. Phosphrylation levels were analyzed by calculating the greyscale ratios of phosphorylated protein to their total protein. Relative Bcl6 protein levels were normalized by greyscale ratios of Bcl6 protein to their β-actin controls. Faster migrating form of p-4EBP proteins was indicated by arraow. Data are representative of at least two independent experiments.
3.6. Ppp2ca knockout or PP2A inhibitor treatment alleviates disease severity in lupus mice
Given the essential role of PP2A in Tfh cell differentiation, we further delineated the role of PP2A in the murine model of lupus. We first used a chromatin-immunized lupus mouse model [22, 37, 38] (Fig. S7A). After 12 weeks post immunization, WT mice displayed clear hallmarks of lupus presentation, including proteinuria and high titers of autoantibodies (IgG of anti-histone and anti-dsDNA antibodies), while these levels were remarkably lower in PP2A cKO mice (Fig. S7B-S7D). Hematoxylin and eosin (H&E) and Periodic-Acid-Schiff (PAS) staining of kidney also showed decreased lymphocyte infiltration and lipogranulomas formation along with less IgG detected in the glomeruli of PP2A cKO mice (Fig. S7E-S7G). As expected, lower Tfh (gated as CXCR5+PD-1hi, CXCR5+ICOShi, or CXCR5+BCL6hi) cell numbers and frequencies were also observed in PP2A cKO mice spleen (Fig. S8A-S8B), as well as the reduced cell numbers and frequencies of GC B cells and plasma B cells (Fig. S8C-S8F). Thus, these data supported that PP2A deficiency protects lupus development by modulating Tfh cells.
We then explored the therapeutic potential of PP2A inhibitors in lupus treatment. MRL/lpr mice spontaneously develop hyperactive neutrophils and lupus-associated autoantibodies by 4–5 months of age. We treated female MRL/lpr mice and control MRL/MpJ mice with i.p. PP2A inhibitor CAN (0.5 mg/kg/2 days) or PBS at 8-week-old (Fig. 6A). The inhibition effect of CAN on PP2A was verified in B6 mice by phosphatase assay (Fig. S9A). These mice were observed for 20 weeks after the initiation of treatment. Strikingly, CAN treatment significantly improved MRL/lpr survival at 20 weeks (Fig. 6B). The dynamic change of the body weight in each group also reflected the attenuated disease severity in CAN treatment MRL/lpr mice (Fig. 6C). Consistently, proteinuria levels (Fig. 6D) and the serum antibody of IgG and IgM isotypes of anti-dsDNA titers (Fig. 6E-6F) were decreased in CAN treated MRL/lpr mice, along with the reduced infiltration of inflammatory cells and crescent formation in the kidney (Fig. 6G-6I). Lipogranulomas and immune complex deposition were also fewer in the glomeruli of CAN-treated MRL/lpr mice (Fig. 6J).
Figure 6. PP2A inhibitor CAN protects mice from SLE.
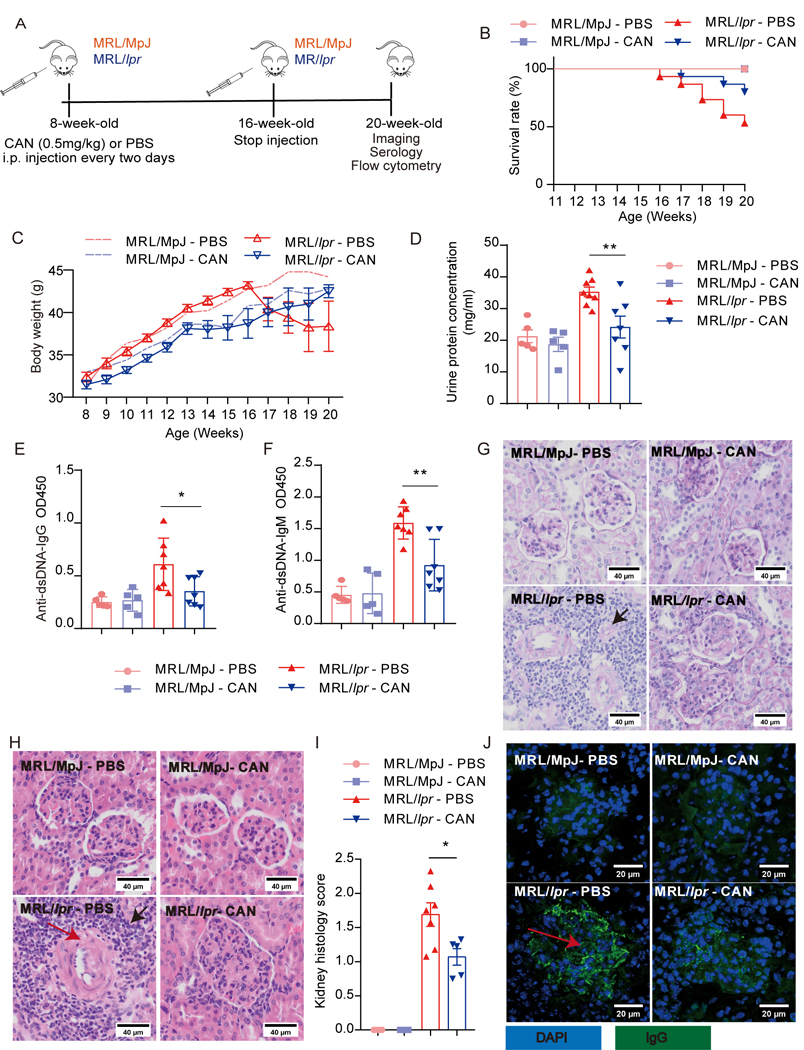
(A) Schematic diagram of experimental design. PP2A inhibitor CAN (0.5 mg/kg) or PBS was administered i.p. every two days from 8-week-old to 16-week-old and then analyzed at 20-week-old (MRL/lpr-CAN: n = 15; MRL/lpr-PBS: n = 15; MRL/MpJ-CAN: n = 5; MRL/MpJ-PBS: n = 5). (B) The survival rate of the MRL/lpr mice treated with CAN or PBS. MRL/MpJ mice are control mice with the same gene background as MRL/lpr mice. (C) The bodyweight of mice was evaluated at the indicated age. (D) The urine protein concentration of mice in each group at week 19. (E and F) Anti-dsDNA IgG and IgM antibodies in the serum of the mice were detected by ELISA. (G) Representative PAS staining from kidney sections with the portion of affected glomeruli. Original magnification, ×40. Bars represent 40 µm. (H) Representative HE staining of the glomerular areas from the kidneys. Black arrow, infiltration of numerous lymphocytes; red arrow, crescent. Original magnification, ×40. Bars represent 40 µm. (I) Histology scores of H&E staining from kidney sections. (J) Immunofluorescence staining of IgG deposition in the glomerulus. Red arrow, immune complex precipitation. Original magnification, ×40. Bars represent 20 µm. Data are representative of two independent experiments and analyzed by two-sided student’s t-test. Error bars of Means ± SEM. *p<0.05; **p<0.01; ***p<0.001.
Thus, these data demonstrate the potential therapeutic efficacy of PP2A inhibition in a lupus model.
3.7. Positive correlation between the expression of PP2A Cα and BCL6 in CD4+ T cells from lupus patients
To establish translational evidence of the connection between SLE, PP2A, and BCL6 in human patients, we analyzed peripheral cTfh cells from 10 female SLE patients and 10 age and gender matched healthy controls (HC). The cTfh (CXCR5+PD-1+) cells were gated on CD4+CD25−CD45RA− cell population (Fig. 7A). The frequency of these cTfh cells had a trend of subtle increase in the SLE patients (Fig. 7B). Then we measured the expression of PP2A Cα and BCL6 levels in lupus and HC CD4+ T cells by western blotting. The result showed that the expression of both PP2A Cα subunit and BCL6 were elevated in SLE patients and displayed a positive correlation (Fig. 7C-7F). To obtain functional evidence of this regulation, we treated human both HC and SLE patient CD4+ T cells with CAN, and a dose-dependent suppression of BCL6 expression was observed (Fig. 7G). We also cultured human tonsil cells with the PP2A inhibitor CAN. The frequencies of CXCR5hiPD-1hi and BCL6hiICOShi were decreased significantly by CAN in a dose-dependent manner, as well as the expression of BCL6 (Fig. 7H-7K). Of note, the frequency of Treg cells gated with CD4+FOXP3+ was comparable, and the expression of GATA3 and T-bet didn’t change (Fig. 7J-7K). Cytokine production was evaluated ex vivo upon phorbol 12-myristate 13-acetate (PMA) and ionomycin stimulation. Again, only the production of IL21 was decreased in CXCR5hiCD4+ T cells (Fig. 7L), while no obvious differences were found in IFNγ and TNFα production. This indicated other T helper types were less affected (Fig. 7M). Thus, these results support that the PP2A-BCL6 regulation axis is also valid in human CD4+ T cells and suggest PP2A inhibitor as a potential drug for treating SLE.
Figure 7. PP2A Cα expression positively correlates with BCL6 protein in CD4+ T cells of lupus patients.

(A-F) Healthy controls were age and gender-matched with SLE patients: (A) Human cTfh gating strategy. (B) Statistical analysis of cTfh frequencies in SLE and HC cohorts. (C) The expression levels of BCL6 and PP2A Cα from CD4+ T cells of healthy controls and SLE patients were analyzed by western blotting. (D and E) Quantification of BCL6 and PP2A Cα signal intensity by densitometry. The levels of β-actin were set as control. Each dot represents one individual. (F) Correlation analysis from relative PP2A Cα and BCL6 levels from SLE and HC participants. (G) Western blotting of BCL6 in human naive CD4+ T cells (from HC or SLE subjects) which were treated with CAN and cultured with plate bounded anti-CD3/28 antibodies for 24 hours. (H-M) Tonsillar cells were co-cultured with the indicated concentration of PP2A inhibitor CAN for 16 hours before flow cytometry assessment. Flow cytometric analysis of CXCR5+PD1hi (H), BCL6hiICOShi (I) GC-Tfh cells, and Treg cells (J) under the indicated condition. (K) Representative flow cytometric profiling of BCL6, GATA3, and T-bet expression gated on CD4+ T cells for the indicated condition. (L) Representative flow cytometric profiling of CXCR5+IL21+ in CD4+ T cells for the indicated condition. (M) Representative flow cytometric profiling of IFNγ and TNFα expression of CD4+ T cells under the indicated condition. Data are representative of two independent experiments with similar results and analyzed by two-sided student’s t-test; Error bars of Means ± SEM. Pearson correlation was applied in (F). *p<0.05; **p<0.01; ***p<0.001, ****p<0.0001.
4. Discussion
In the present study, we revealed the positive regulatory role of PP2A in B cell-mediated antibody response through promoting Tfh differentiation. We also uncovered the underlying mechanisms and demonstrated that PP2A regulates BCL6 expression at the level of protein translation via mTORC1–4EBP-eIF4E axis in CD4+ T cells, providing an underlying mechanism. Furthermore, we found that a T cell specific PP2A knockout impairs Tfh generation and reduces disease severity in the inducible SLE mouse model.
Previous studies have provided evidence that PP2A protein and catalytic activity are increased in patients with SLE [8]. Here we confirmed a significantly positive correlation between BCL6 and PP2A Cα levels in SLE patients. In addition, we found that human tonsil cells cultured with PP2A inhibitor resulted in reduced Tfh cell numbers. This experimental evidence, together with the genetic evidence from the PP2A cKO mice, helps clarify the regulatory role of PP2A in SLE. We further demonstrated that treatment with the PP2A inhibitor CAN increased the survival rate in MRL/lpr lupus-prone mice. Interestingly, PP2A inhibitor CAN as a traditional Chinese medicine has been used in treating various tumors (including hepatocarcinoma, lung cancer, and colon cancer) [39], and the PP2A activator FLY720 has also been used in treating autoimmune diseases such as Multiple Sclerosis [40]. Our discovery thus highlights the translational potential for PP2A inhibitors as a therapeutic candidate for clinical SLE management. However, due to the heterogeneity of SLE, PP2A inhibitors should be prioritized in treating lupus patients with high cTfh frequencies. Also, given the broad spectrum of protein phosphatases functions and off-target effects such as PP1 inhibition by common PP2A inhibitors [41–43], the concentration window for treatment and the safety of using PP2A inhibitors needs to be evaluated very carefully. Further studies, for example, titrating a more reasonable CAN dose in vivo or developing PP2A precise inhibitors, will be important aspects to investigate in the future.
As previously described, PP2A is a protein with multiple functions in different cell types. In T cells, several different regulatory functions and mechanisms have been reported to explain how overactivated PP2A regulates SLE in human patients. For example, PP2A could regulate CD3ζ and FcRγ transcription through dephosphorylation of EIF-1 to enhance early CD3/TCR-mediated signaling events in human SLE patient T cells [44]. It has also been reported that PP2A can suppress the production of IL2 in human patients with SLE by regulating pCREB expression and pCREB binding to the IL2 promoter [8]. In addition, PP2A controls the expression of DNMT1 through the MEK/ERK signaling pathway and influences the expression of methylation-sensitive genes like CD70, and ITGAL in human SLE T cells [28]. However, these mechanisms were not obvious in PP2A cKO T cells.
Previous studies showed that PP2A activation can decrease mTORC1 activity and promote Treg suppressive activity [18]. An increase of p-S6 (Ser235/236) in PP2A deficient T cells was also observed in our experiments [18], suggesting a possible role of PP2A in downregulating mTORC1 activity. However, when we studied more carefully, we found that the activations of the two major mTORC1 downstream substrates, 4EBP and S6K, were downregulated upon PP2A deficiency. Since S6 can be phosphorylated by both RSK and S6K [35, 45], the discrepancy between the phosphorylation status of S6K and S6 could reflect an effect from RSK or an additional regulation effect of S6 by PP2A. In our previous study, we have observed increased pERK, an upstream activator of RSK, in PP2A deficient cells, which supports a possible role of RSK (Fig. S6D in our published work) [17]. These data fit to our understanding towards the broader regulatory roles of phosphatases and suggested a more complicated regulatory function of PP2A in T cells besides the presently described function of regulating BCL6 and Tfh differentiation.
A recent study has indicated that the mTORC1–4EBP-eIF4E axis regulates Tfh differentiation via BCL6 translation [46]. The eIF4F complex regulates the translation of all mRNAs, however, it changes in its levels and/or activity which can selectively alter the translation of a subset of “eIF4E-sensitive” mRNAs without impacting the global protein synthesis. In agreement with these findings, PP2A deficiency disrupted the eIF4F complex in the same way [33]. We further found that disruptive eIF4F complex due to 4EBP hypophosphorylation in PP2A deficiency T cells specifically affects the translation of BCL6 without causing global protein expression changes. A previous study showed that an inhibitor of mTOR diminishes the capacity of eIF4E to bind 5’ terminal oligopyrimidine (TOP) motifs and TOP-like mRNA much more than other mRNAs [23]. Of note, S6K could cooperately work with 4EBP in selective translational control of 5’TOP mRNA independent of S6 [47] the Bcl6 gene contains the TOP-like motif. Furthermore, it has been known that mTORC1 activity is increased in SLE T cells including increased 4EBP phosphorylation while mTORC2 is reduced compared with T cells from healthy donors, which is consistent with our conclusion [48]. In all, our research thus suggested a new pathway that PP2A regulates Tfh cells in both mice and humans.
A limitation of our study is the lack of revealing a dephosphorylation substrate of PP2A that contributes to stabilizing BCL6 protein translation. However, this is a common obstacle in phosphatase studies due to the nature of broad specificity and the transient interactions between the enzymes and substrates. The complicated regulation pattern we found in this study may also reflect the behavior of a phosphatase, which may generate accumulated effects from multiple substrates [30] by affecting more than one pathway. Other mechanisms other than those we reported here are possible. Moreover, the final outcome of PP2A deficiency for a pathway appears to be determined by the cumulative impact of each micro-modulation. In addition to the complex of multiple substrates, PP2A exerts different functions through distinct substrate specific regulatory subunits (PP2A B subunit) [49]. Among the total 15 regulatory subunits, 14 of them are highly expressed in Jurkat cells (data not shown). The B subunit specifically mediating the mTORC1–4EBP-eIF4E-BCL6 axis is currently unknown. Moreover, in this study, we only tested the effects of PP2A inhibitors in a lupus model. Last, in this study, we only tested the effects of PP2A inhibitors in a lupus model. Other autoimmune diseases presented with aberrant Tfh expansion and abnormal autoantibody production may also benefit from modulating PP2A activity, which needs to be further explored.
5. Conclusion
In summary, we showed that PP2A plays an important role in producing autoantibodies in lupus via promoting the differentiation of Tfh cells. PP2A is a potential therapeutic target for lupus.
Supplementary Material
Highlights:
PP2A is requisite for the development of Tfh cells and normal GC function
PP2A deficiency led to the down regulation of BCL6 translation
PP2A T cell deficiency or PP2A inhibition significantly protected mice from lupus
Positive correlation of PP2A Ca and BCL6 protein levels in human lupus CD4+ T cells
Acknowledgments
We thank Dr. Liwei Lv for providing chromatin-induced lupus mouse model technical support; Shuangshuang Liu, Yanwei Li, Jiajia Wang, and Yingying Huang from the core facilities (Zhejiang University School of Medicine) for technical assistance in histology and FACS analysis. Yang Yao for technical assistance. The Department of Rheumatology in Sir Run Run Shaw Hospital for helping with healthy and lupus participants recruitment. This work was supported by National Natural Science Foundation of China Grants 31900628 (to Q.X.); U21A20199 and 31930038 (to L.L); in part by the Intramural Research Program of NIAID, NIH (Q.X., K.M. and P.L.S.);Innovative research team of high-level local universities in Shanghai SHSMU-ZLCX 20211600 (to L.L); China postdoctoral science foundation 2022M720087 (to K.X.Z), Medical Health Science and Technology Project of Zhejiang Provincial Health Commission Grant 2020KY585 (to Q.X.); and the Educational Commission of Zhejiang Province Grant Y202043492 (to Q.X.).
Footnotes
Author statement
Yu Jiang: Conceptualization; Methodology; Investigation; Data Curation; Writing - Original Draft preparation, Reviewing & Editing. Xuexiao Jin: Investigation; Writing - Reviewing & Editing. Zhexu Chi: Investigation; Writing - Review & Editing. Yadan Bai: Investigation. Kalpana Manthiram: Resources; Writing – Editing Original Draft. Pamela Mudd: Resources. Kaixiang Zhu: Funding acquisition. Yongmei Han: Resources. Lie Wang: Scientific inputs; Methodology. Pamela L. Schwartzberg: Resources; Scientific inputs; Writing - Review & Editing. Xiang Gao: Resources. Linrong Lu: Conceptualization; Methodology; Writing - Original Draft preparation, Reviewing & Editing; Supervision; Funding acquisition. Qin Xu: Conceptualization; Methodology; Writing - Original Draft preparation, Reviewing & Editing; Investigation; Supervision; Funding acquisition.
Data deposition
The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, GSE number is GSE208185.
Competing interests
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity, 2011;34:947–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chen X, Ma W, Zhang T, Wu L, Qi H. Phenotypic Tfh development promoted by CXCR5-controlled re-localization and IL-6 from radiation-resistant cells. Protein & cell, 2015;6:825–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H. PD-1 Controls Follicular T Helper Cell Positioning and Function. Immunity, 2018;49:264–74 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W. et al. T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature, 2015;517:214–8. [DOI] [PubMed] [Google Scholar]
- [5].Robert J ACP, DiToro Daniel, Yusuf Isharat, Eto Danelle,, Burton Barnett ALD, Craft Joe, Crotty Shane. Bcl6 and Blimp-1 Are Reciprocal and Antagonistic Regulators of T Follicular Helper Cell Differentiation. science, 2009;325. [DOI] [PMC free article] [PubMed]
- [6].Hatzi K, Nance JP, Kroenke MA, Bothwell M, Haddad EK, Melnick A. et al. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. The Journal of experimental medicine, 2015;212:539–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Choi J, Crotty S. Bcl6-Mediated Transcriptional Regulation of Follicular Helper T cells (TFH). Trends Immunol, 2021;42:336–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Katsiari CG, Kyttaris VC, Juang YT, Tsokos GC Protein phosphatase 2A is a negative regulator of IL-2 production in patients with systemic lupus erythematosus. The Journal of clinical investigation, 2005;115:3193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Choi JY, Ho JH, Pasoto SG, Bunin V, Kim ST, Carrasco S. et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis & rheumatology, 2015;67:988–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Faliti CE, Gualtierotti R, Rottoli E, Gerosa M, Perruzza L, Romagnani A. et al. P2X7 receptor restrains pathogenic Tfh cell generation in systemic lupus erythematosus. The Journal of experimental medicine, 2019;216:317–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yi W, Gupta S, Ricker E, Manni M, Jessberger R, Chinenov Y. et al. The mTORC1–4E-BP-eIF4E axis controls de novo Bcl6 protein synthesis in T cells and systemic autoimmunity. Nature communications, 2017;8:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Katsuyama T, Li H, Comte D, Tsokos GC, Moulton VR Splicing factor SRSF1 controls T cell hyperactivity and systemic autoimmunity. The Journal of clinical investigation, 2019;129:5411–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gao X, Song Y, Du P, Yang S, Cui H, Lu S. et al. Administration of a microRNA-21 inhibitor improves the lupus-like phenotype in MRL/lpr mice by repressing Tfh cell-mediated autoimmune responses. Int Immunopharmacol, 2022;106:108578. [DOI] [PubMed] [Google Scholar]
- [14].Duan X, Shen C, Zhang X, Wu L, Chen J, Ma B. et al. Toll-like receptor 7 agonist imiquimod prevents the progression of SLE in MRL/lpr mice via inhibiting the differentiation of T follicular helper cells. Int Immunopharmacol, 2020;80:106239. [DOI] [PubMed] [Google Scholar]
- [15].Ruvolo PP The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA clinical, 2016;6:87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhou J, Pham HT, Walter G. The formation and activity of PP2A holoenzymes do not depend on the isoform of the catalytic subunit. J Biol Chem, 2003;278:8617–22. [DOI] [PubMed] [Google Scholar]
- [17].Xu Q, Jin X, Zheng M, Rohila D, Fu G, Wen Z. et al. Phosphatase PP2A is essential for TH17 differentiation. Proc Natl Acad Sci U S A, 2019;116:982–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Apostolidis SA, Rodriguez-Rodriguez N, Suarez-Fueyo A, Dioufa N, Ozcan E, Crispin JC et al. Phosphatase PP2A is requisite for the function of regulatory T cells. Nature immunology, 2016;17:556–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schmidt T, Paust HJ, Krebs CF, Turner JE, Kaffke A, Bennstein SB et al. Function of the Th17/interleukin-17A immune response in murine lupus nephritis. Arthritis & rheumatology, 2015;67:475–87. [DOI] [PubMed] [Google Scholar]
- [20].He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J. et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med, 2016;22:991–3. [DOI] [PubMed] [Google Scholar]
- [21].Grasshoff H, Comduhr S, Monne LR, Muller A, Lamprecht P, Riemekasten G. et al. Low-Dose IL-2 Therapy in Autoimmune and Rheumatic Diseases. Front Immunol, 2021;12:648408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ma K, Li J, Wang X, Lin X, Du W, Yang X. et al. TLR4(+)CXCR4(+) plasma cells drive nephritis development in systemic lupus erythematosus. Ann Rheum Dis, 2018;77:1498–506. [DOI] [PubMed] [Google Scholar]
- [23].Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM A unifying model for mTORC1-mediated regulation of mRNA translation. Nature, 2012;485:109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jin X, Xu Q, Pu C, Zhu K, Lu C, Jiang Y. et al. Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cellular & molecular immunology, 2021;18:1896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mingzhu Zheng DL, Zhao Zhishan, Shytikov Dmytro, Xu Qin, Jin Xuexiao, Liang Jingjing,, Jun Lou SW, Wang Lie, Hu Hu, Zhou Yiting, Gao Xiang, and Lu Linrong. Protein phosphatase 2A has an essential role in promoting thymocyte survival during selection. PNAS, 2019. [DOI] [PMC free article] [PubMed]
- [26].Jiao J, Lv Z, Zhang P, Wang Y, Yuan M, Yu X. et al. AID assists DNMT1 to attenuate BCL6 expression through DNA methylation in diffuse large B-cell lymphoma cell lines. Neoplasia, 2020;22:142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yu HR, Kim YJ, Lee HR KSHV vIRF4 enhances BCL6 transcription via downregulation of IRF4 expression. Biochem Biophys Res Commun, 2018;496:1128–33. [DOI] [PubMed] [Google Scholar]
- [28].Sunahori K, Nagpal K, Hedrich CM, Mizui M, Fitzgerald LM, Tsokos GC The catalytic subunit of protein phosphatase 2A (PP2Ac) promotes DNA hypomethylation by suppressing the phosphorylated mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK)/phosphorylated ERK/DNMT1 protein pathway in T-cells from controls and systemic lupus erythematosus patients. J Biol Chem, 2013;288:21936–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liao W, Spolski R, Li P, Du N, West EE, Ren M. et al. Opposing actions of IL-2 and IL-21 on Th9 differentiation correlate with their differential regulation of BCL6 expression. Proc Natl Acad Sci U S A, 2014;111:3508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zheng M, Li D, Zhao Z, Shytikov D, Xu Q, Jin X. et al. Protein phosphatase 2A has an essential role in promoting thymocyte survival during selection. Proc Natl Acad Sci U S A, 2019;116:12422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hinnebusch AG The Scanning Mechanism of Eukaryotic Translation Initiation. Annual Review of Biochemistry, 2014;83:779–812. [DOI] [PubMed] [Google Scholar]
- [32].Kim SM, Yoon SY, Choi JE, Park JS, Choi JM, Nguyen T. et al. Activation of eukaryotic initiation factor-2 alpha-kinases in okadaic acid-treated neurons. Neuroscience, 2010;169:1831–9. [DOI] [PubMed] [Google Scholar]
- [33].Roux PP, Topisirovic I. Regulation of mRNA translation by signaling pathways. Cold Spring Harbor perspectives in biology, 2012;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tian HP, Qiu TZ, Zhao J, Li LX, Guo J. Sphingomyelinase-induced ceramide production stimulate calcium-independent JNK and PP2A activation following cerebral ischemia. Brain Inj, 2009;23:1073–80. [DOI] [PubMed] [Google Scholar]
- [35].Salmond RJ, Emery J, Okkenhaug K, Zamoyska R. MAPK, phosphatidylinositol 3-kinase, and mammalian target of rapamycin pathways converge at the level of ribosomal protein S6 phosphorylation to control metabolic signaling in CD8 T cells. J Immunol, 2009;183:7388–97. [DOI] [PubMed] [Google Scholar]
- [36].Qin Xu XJ, Zheng Mingzhu, Rohila Deepak, Fu Guotong, Wen Zhuoyu, Lou Jun,, Songquan RS, Wang Lie, HuHu, Gao Xiang, and Lu Linrong. Phosphatase PP2A is essential for TH17 differentiation. PNAS, 2019:982–7. [DOI] [PMC free article] [PubMed]
- [37].Hong LI Y.-y. Z., SUN Ya-nan, Xi-yi HUANG, JIA Yong-feng, LI Duan. Induction of systemic lupus erythematosus syndrome in BALB/c mice by immunization with active chromatin. Acta Pharmacol Sin, 2004;25:807–11. [PubMed] [Google Scholar]
- [38].Wen Z, Xu L, Xu W, Xiong S. Retinoic Acid Receptor-Related Orphan Nuclear Receptor gammat Licenses the Differentiation and Function of a Unique Subset of Follicular Helper T Cells in Response to Immunogenic Self-DNA in Systemic Lupus Erythematosus. Arthritis & rheumatology, 2021;73:1489–500. [DOI] [PubMed] [Google Scholar]
- [39].Mazhar S, Taylor SE, Sangodkar J, Narla G. Targeting PP2A in cancer: Combination therapies. Biochim Biophys Acta Mol Cell Res, 2019;1866:51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Khan MM, Kalim UU, Khan MH, Lahesmaa R. PP2A and Its Inhibitors in Helper T-Cell Differentiation and Autoimmunity. Front Immunol, 2021;12:786857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang G, Dong J, Deng L. Overview of Cantharidin and its Analogues. Curr Med Chem, 2018;25:2034–44. [DOI] [PubMed] [Google Scholar]
- [42].Honkanen RE Cantharidin, another natural toxin that inhibits the activity of serine/threonine protein phosphatases types 1 and 2A. FEBS Lett, 1993;330:283–6. [DOI] [PubMed] [Google Scholar]
- [43].Fernandez JJ, Candenas ML, Souto ML, Trujillo MM & Norte M. Okadaic Acid, Useful Tool for Studying Cellular Processes. Curr Med Chem, 2002;9:229–62. [DOI] [PubMed] [Google Scholar]
- [44].Juang YT, Wang Y, Jiang G, Peng HB, Ergin S, Finnell M. et al. PP2A dephosphorylates Elf-1 and determines the expression of CD3zeta and FcRgamma in human systemic lupus erythematosus T cells. J Immunol, 2008;181:3658–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Salmond RJ, Brownlie RJ, Meyuhas O, Zamoyska R. Mechanistic Target of Rapamycin Complex 1/S6 Kinase 1 Signals Influence T Cell Activation Independently of Ribosomal Protein S6 Phosphorylation. J Immunol, 2015;195:4615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yi W, Gupta S, Ricker E, Manni M, Jessberger R, Chinenov Y. et al. The mTORC1–4E-BP-eIF4E axis controls de novo Bcl6 protein synthesis in T cells and systemic autoimmunity. Nature Communications, 2017;8:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].D. e. a. Barth-Baus. S6 phosphorylation-independent pathways regulate translation of 5′-terminal oligopyrimidine tract-containing mRNAs in differentiating hematopoietic cells. Nucleic Acids Res, 2002;30:1919–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kato H, Perl A. Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4-CD8-double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. J Immunol, 2014;192:4134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochimica et biophysica acta, 2009;1795:1–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.