

Abstract
Mu-opioid receptor (μOR) agonists such as fentanyl have long been used for pain management, but are considered a major public health concern owing to their adverse side effects, including lethal overdose1. Here, in an effort to design safer therapeutic agents, we report an approach targeting a conserved sodium ion-binding site2 found in μOR3 and many other class A G-protein-coupled receptors with bitopic fentanyl derivatives that are functionalized via a linker with a positively charged guanidino group. Cryo-electron microscopy structures of the most potent bitopic ligands in complex with μOR highlight the key interactions between the guanidine of the ligands and the key Asp2.50 residue in the Na+ site. Two bitopics (C5 and C6 guano) maintain nanomolar potency and high efficacy at Gi subtypes and show strongly reduced arrestin recruitment–one (C6 guano) also shows the lowest Gz efficacy among the panel of μOR agonists, including partial and biased morphinan and fentanyl analogues. In mice, C6 guano displayed μOR-dependent antinociception with attenuated adverse effects, supporting the μOR sodium ion-binding site as a potential target for the design of safer analgesics. In general, our study suggests that bitopic ligands that engage the sodium ion-binding pocket in class A G-protein-coupled receptors can be designed to control their efficacy and functional selectivity profiles for Gi, Go and Gz subtypes and arrestins, thus modulating their in vivo pharmacology.
Opioid receptors, members of the class A family of G-protein-coupled receptors4 (GPCRs), are key molecular targets in pain management. The challenge remains to engage specific subtypes of opioid receptor and to trigger a specific functional response that can produce in vivo analgesia without side effects such as tolerance, respiratory depression and addiction5. Some approaches to mitigate these adverse reactions6 include the development of biased μOR agonists7–11, peripherally restricted agonists12, ligands targeting μOR splice variants13, opioids with mixed actions at other subtypes10,14,15 and compounds that bind to μOR only in acidic environments16. Most of these past approaches have targeted the orthosteric site, although positive allosteric modulators have recently been reported at μORs17. Here we aimed to target a distinct Na+-binding allosteric site, which is highly conserved in a vast majority of family A GPCRs2 and is detected crystallographically in high-resolution inactive state structures2,18 and biochemically in at least 26 diverse family A GPCRs.
Allosteric modulation of Gi and Go GPCR subtypes by sodium was first observed in the 1970s at the μOR19. The presence of physiological NaCl concentrations was found to shift the receptor towards the inactive state, thus reducing agonist binding2,20,21. As recent structural studies reveal, the pocket undergoes marked conformational changes upon receptor activation and is critical for the modulation of signalling in family A GPCRs22. It has also been shown that the Na+-binding site residues serve as major ‘efficacy switches’18 that can bias the GPCR signalling to either Gi protein or β-arrestin-2 pathways. The polar cavity with the Na+ site has also been characterized by molecular dynamics in inactive μOR21,23, whereas the high-resolution structure of μOR bound to the agonist Bu72 (Protein Data Bank (PDB): 5C1M) revealed activation-related conformational changes in the cavity that prevent sodium binding but retain several water molecules3 (Fig. 1a).
Fig. 1 |. Targeting the Na+ site with fentanyl-based bitopic ligands and characterization of lead compounds in binding, G-protein and arrestin signalling assays.
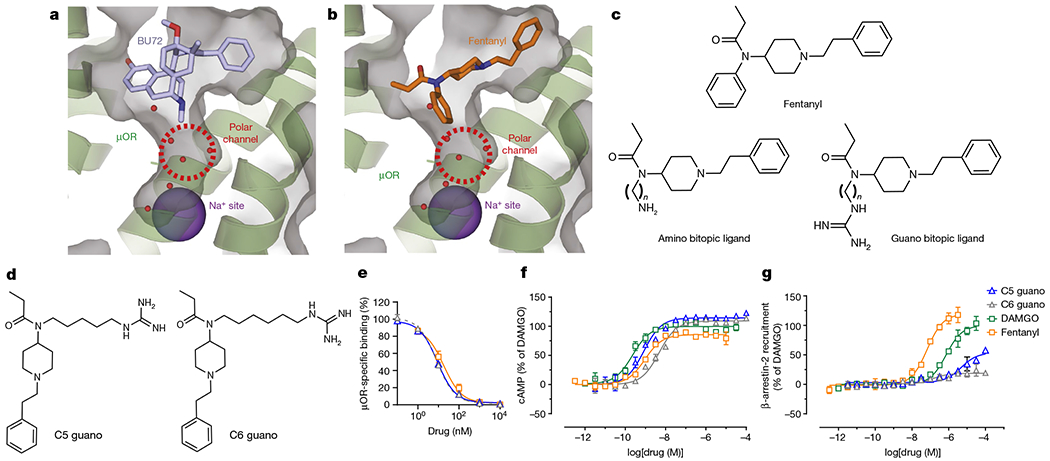
a, Structure of Bu72 bound to μOR, showing the orthosteric site and the unoccupied Na+ site. The μOR structure is shown in green ribbon as well as transparent grey surface. b, Docking of fentanyl in μOR, showing the orthosteric site and the unoccupied Na+ site. c, Chemical structures of fentanyl and designed bitopic ligands. d, Chemical structures of the lead fentanyl guano bitopic ligands. e, Binding affinities at the μOR. Lead bitopic ligands (guano fentanyls) were characterized in binding assays in CHO cells expressing μOR using [125I]IBNtxA as radioligand. C5 guano and C6 guano had similar affinity to fentanyl. Data are mean ± s.e.m. (n = 3 experiments each done in triplicate). f,g, cAMP inhibition (f) and Tango assay for β-arrestin-2 recruitment on μOR (g) with the bitopic ligands. The guano bitopic derivatives show high G-protein agonism with poor recruitment of β-arrestin-2; C5 guano was the most active relative to DAMGO. Data are mean ± s.e.m. (n = 3 experiments each done in duplicate). Extended Data Table 1 shows values for all panels and data for all bitopic ligands.
Moreover, this structure revealed a water-filled polar channel linking the orthosteric pocket with the Na+ site, raising the possibility of developing ligands that interact directly with the Na+ site. The high conservation and key functional role of this allosteric pocket suggest that it is suitable as a target for allosteric modulators and bitopic ligands with unique properties for some family A GPCRs, including the μOR. To address this hypothesis, we used a structure-based approach and designed a series of bitopic ligands on the fentanyl scaffold. These bitopic ligands target both the orthosteric pocket of the μOR and extend into the polar channel towards the Na+ site24 (Fig. 1b,c). The binding pose of two fentanyl bitopic ligands was confirmed by structure determination using cryo-electron microscopy (cryo-EM). Functional characterization of these ligands revealed that the extension of a positively charged guanidine group into the Na+ site resulted in a near complete loss of arrestin recruitment, and, surprisingly, substantial changes in the relative potency and efficacy for the six Gi, Go and Gz subtypes. Finally, the lead bitopic ligand, C6 guano, showed antinociception with reduced side effects compared with classical μOR agonists such as morphine or the lethal agonist fentanyl. Owing to the high conservation of the Na+−binding pocket, similar bitopic ligand designs may be suitable for effective control of functional selectivity in other class A GPCRs.
Bitopic μOR ligands based on the fentanyl scaffold
Fentanyl is a synthetic μOR-selective agonist that is 100-fold more potent as an analgesic than morphine11,25. Unlike morphine, fentanyl also is highly efficacious for inducing arrestin translocation and is a full agonist for Gi-mediated signalling, whereas morphine is a partial agonist. Fentanyl also acts rapidly and is commonly used to manage postoperative and severe cancer-related pain, and is known to bind specifically to the orthosteric site of μOR–but in the USA, its use has led to around 60,000 deaths due to overdose26.
Here we predicted the binding poses of fentanyl on the basis of the crystal structure of the active state μOR with the N-terminus truncated at residue M653 (Fig. 1b), and computationally designed and synthesized a small library of fentanyl derivatives designed to extend through the polar channel below the orthosteric pocket to the Na+-binding pocket (Extended Data Fig.1). Although alternative models for fentanyl binding have been suggested27,28, the docking model used here has been validated by cryo-EM with the fentanyl analogue lofentanil29, as well as by cryo-EM complexes with the designed bitopic ligand reported here. The models predicted the distance between the amide nitrogen of fentanyl and the carboxy group of the D1142.50 residue of the allosteric Na+-binding site to be approximately 13 Å (superscripts indicate Ballesteros–Weinstein numbering for GPCRs30). In order to engage the allosteric Na+-binding site, we replaced the classical fentanyl benzene ring with an aliphatic chain linker (Cn, where n = 3, 5, 6, 7, 9 or 11) connected to a positively charged ‘warhead’, an amine or guanidine (guano) group (Fig. 1c). The bitopic compound with a C7 linker (C7 guano) was predicted to be optimal for the formation of a direct salt bridge between the amino warhead and D1142.50 carboxylic acid, and the C6 linker (C6 guano) was predicted to be optimal for the interaction for the guanidine warhead. The syntheses of amino and guano bitopic ligands31 on the fentanyl scaffold are presented in Supplementary Fig. 1.
Functional assessment of fentanyl-based scaffold
We first evaluated the binding affinities of fentanyl bitopic ligands on μOR using a competitive radioligand assay with labelled 3′-iodobenzoyl-6β-naltrexamide13 ([125I]IBNtxA) (Fig. 1e and Extended Data Table 1). The binding data clearly indicate a difference between guano- and amino-fentanyl bitopic ligands. The amino-fentanyl compounds showed reduced affinity towards μOR, suggesting that the amino warhead does not allow efficient interaction with the polar and negatively charged residues predicted to line the sodium binding pocket (D1142.50, N1503.35, N3287.45, S3297.46 and S1543.39). Furthermore, the extension of the aliphatic linker did not seem to substantially affect the binding of such molecules, with (inhibition constant) Ki values of 281 and 369 nM for C9 amino and C7 amino ligands, respectively.
By contrast, all guanidino derivatives displayed moderate to high affinity towards μOR with Ki values ranging from 590 nM for the C11-guano derivative to 4.6 nM for C5 guano and 4.1 nM for C6 guano, which represents a slight improvement over fentanyl binding (Ki = 9 nM). The guanidine moiety appears to provide a better fit and additional hydrogen bond donors and/or acceptors, potentially enhancing interaction in the polar cavity of μOR, with Ki values differing by up to 1,000-fold compared with their amino counterparts. Structure–activity relationship analysis showed that whereas the optimal affinity was obtained with C5 guano and C6 guano, C3 guano was too short to reach the Na+ pocket and showed reduced binding affinity (Ki = 77 nM). Further extension of the bitopic linker beyond C6 had a negative effect on the affinity. C7 guano showed increased Ki values, probably owing to steric clash because of inadequate space in the polar pocket for the extended linker.
To assess the functional properties of the bitopic ligands on μOR, we used the Tango assay for β-arrestin-2 recruitment and cAMP assays for Gi protein activation (Fig. 1f,g and Extended Data Table 1). As expected from their low affinity, the amino fentanyls displayed only weak stimulation of the Gi pathway, with half-maximal effective concentration (EC50) values all above 293 nM and no measurable β-arrestin-2 recruitment at the highest tested drug concentrations of 10 μM for the majority of analogues. Among guanidino bitopic ligands, the highest affinity C5 guano ligand demonstrated full agonistic activity in cAMP assays (maximum G-protein activation (Emax) = 114%) with subnanomolar potency (EC50 = 0.8 nM), in the same range as the standard full agonist DAMGO (EC50 = 0.9 nM). Following the same trend as affinity, the potency of guanidine derivatives was inversely correlated with the aliphatic linker chain length from C6 guano (EC50 = 4.7 nM) to C11 guano (EC50 = 1017 nM).
Of note, the bitopic guanidine-containing derivatives with the strongest affinity and potency in cAMP assays did not recruit β-arrestin-2 efficiently. For example, C5 guano in Tango assays had EC50 = 4,710 nM and Emax = 54%, whereas for DAMGO, EC50 = 723 nM and Emax = 100%, and for fentanyl, EC50 = 66 nM and Emax = 119 %. For C6 and C7 derivatives in Tango assays, Emax was less than 20% relative to DAMGO, and in the cAMP assay, Emax was 109% and 107% relative to DAMGO, respectively. Thus, the designed extensions of the fentanyl scaffold into the polar pocket below the orthosteric site markedly reduced the β-arrestin-2 recruitment capacity of fentanyl11, which is considered one of the strongest β-arrestin-2 recruiting opiates.
Structural validation of bitopic ligand design
Given the pharmacological data showing that C5 guano and C6 guano are the most potent bitopic ligands with respect to cAMP inhibition with reduced β-arrestin-2 activity, we used cryo-EM to determine the structures of the μOR–i1 complex bound to C5 guano and C6 guano at 3.2 Å and 3.3 Å global resolution, respectively (Fig. 2, Extended Data Figs. 2 and 3 and Extended Data Table 2). The bitopic ligand-bound μOR structures reveal the expected binding pose of the ligands, with their fentanyl scaffolds overlapping with the fentanyl model in the orthosteric pocket and the guanidine extension protruding towards the sodium site (Fig. 2b,c,f,g). The conformation of the bitopic ligand-bound receptor is very similar to the high-resolution structure of BU72-bound active μOR (PDB: 5C1M) with all-atom root-mean-squared deviation (r.m.s.d.) being 0.957 Å for C5 guano and 1.189 Å for C6 guano, for residues lining the orthosteric pocket (Extended Data Fig. 4). Unlike C5 guano and C6 guano, BU72 does not extend into the polar pocket and the all-atom r.m.s.d. of the residues predicted to form the Na+-binding pocket and polar channel is 0.884 Å for C5 guano and 1.056 Å for C6 guano (Extended Data Fig. 4b,d).
Fig. 2 |. Structures of bitopic ligands bound to μOR.
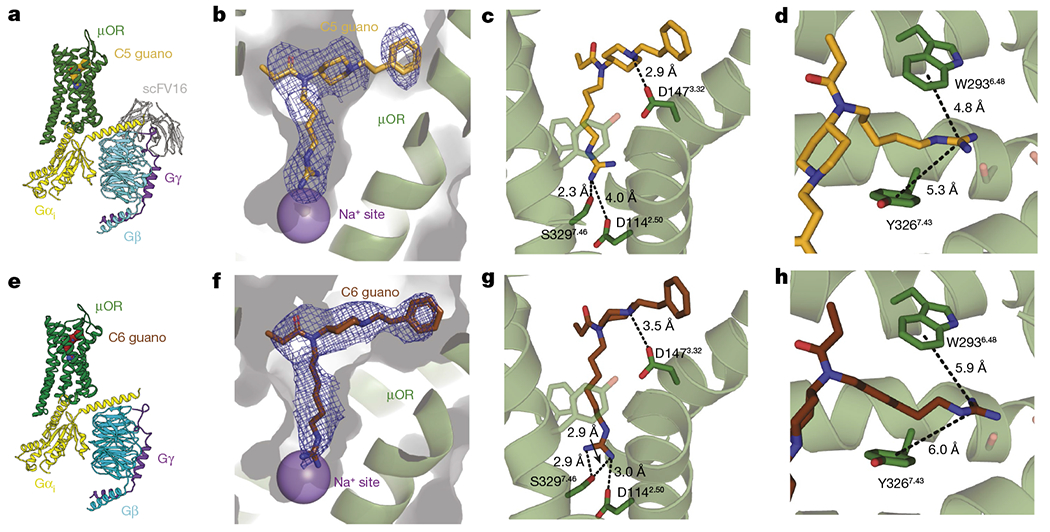
a,e, Cryo-EM structure of C5 guano (a) and C6 guano (e) bound to μOR–Gi–scFv16 complex coloured by subunit. b,f, View of the C5 guano (b) and C6 guano (f) cryo-EM density. The cryo-EM maps are contoured at 5.0σ. The functional groups of the bitopic ligands target site 2 above the Na+-binding site, as expected. c,g, Salt-bridge interactions between C5 guano (c) or C6 guano (g) and residues in the μOR pocket. d,h, Cation–π interactions between C5 guano (d) or C6 guano (h) and residues in the μOR pocket.
In both structures, we observe direct interaction between the orthosteric residue D1473.32 and the piperidine ring nitrogen atom, which is consistent with the docked fentanyl model (Fig. 2c,g). This confirms that C5 and C6 guano bitopic ligands maintain the same specific interaction to the orthosteric pocket as fentanyl.
The structure also reveals that the shorter C5 linker affords only limited interaction (distance 4.0 Å) between the basic guanidine warhead of C5 guano and the major anchor of the Na+ binding site, the acidic D1142.50 side chain (Fig. 2c). Nevertheless, it is possible that an interaction between the warhead and the aspartate is dynamically mediated by water molecules that are not resolved in the 3.2 Å resolution cryo-EM map. The lack of direct contact with D1142.50 can be compensated by a strong hydrogen-bonded local interaction of the guanidine warhead with S3297.46, which is another key residue of the Na+-binding site (Fig. 2c). The structure of C6 guano-bound μOR has subtle localized differences compared with C5 guano-bound μOR. In the C6 guano-bound structure, the distance between D1142.50 and the guanidine warhead was 3 Å, enabling the formation of a direct salt bridge. Besides interacting with the polar residues of the Na+ binding site, the positive charge of the guanidine warhead might also form weak cation–π interactions with the aromatic residues located at the polar channel32 (Fig. 2d,h), further stabilizing the binding pose of guano bitopic ligands with appropriate aliphatic linker sizes.
Molecular dynamics of the μOR complexes
The results presented above demonstrate that the unique binding interactions observed for C5 guano and C6 guano bitopic ligands with μOR (Fig. 2) lead to signalling profiles that are very distinct from their orthosteric templates. To obtain further conformational insights into the ligand–receptor interactions, we analysed the dynamics of the direct and water-mediated interactions of C5 guano and C6 guano and other derivatives in the active state μOR. We performed 10 independent molecular dynamics runs of approximately 1-μs duration for each complex in a lipid bilayer membrane (Extended Data Fig. 5). The initial structure for the C5 guano molecular dynamics simulation was derived from the corresponding cryo-EM structure, whereas the initial C6 guano complex was obtained by docking of the ligand into the structure of μOR defined by the C5 guano complex. The molecular dynamics trajectories were used to calculate (1) the distance between the ligand piperidine nitrogen atom and carboxylate oxygens of D1473.32, (2) the distance between guano nitrogen atoms of the ligand and carboxylate oxygens of D1142.50, and (3) the number of waters bridging the guanidine group and carboxylate oxygens of D1142.50 (Extended Data Fig. 5f,g). The conformations of the fentanyl core in the orthosteric pocket were stable for all ligands, with more than 96% of molecular dynamics frames maintaining either direct salt bridge (more than 85% of frames, defined as an N–O distance of less than 3.5 Å) or water-mediated interactions between piperidine nitrogen and carboxylate oxygens of D1473.32. The interactions between guanidine and D1142.50 were also maintained in at least 90% of the frames, however, with different ratios of direct salt bridges and water-mediated interactions. For C5 guano, 33% of the trajectory frames had 1 or 2 water molecules bridging guanidine group and D1142.50, whereas 57% of the frames showed direct salt-bridge interactions between C5 guano and D1142.50, with a relatively rapid exchange between these interaction modes. For C6 guano, this direct salt-bridge bonding to D1142.50 increased to 71% of all frames, with an additional 23% of indirect interactions, in line with a more optimal distance match predicted for the C6 linker. Our observation of a vast majority (more than 90%) of the molecular dynamics conformations maintaining either a water-mediated or a direct interaction between C5 guano and D1142.50 of the Na+-binding pocket of μOR suggests the importance of these interactions for the ligand affinity and functional properties.
Intrinsic efficacy compared with classical agonists
We next pharmacologically assessed our guano bitopic ligands using bioluminescence resonance energy transfer (BRET)-based assays for measuring arrestin recruitment and G-protein heterotrimer dissociation (TRUPATH), including at both arrestin isoforms (β-arrestin-1 and β-arrestin-2) and at all Gα subtypes (Gi1, Gi2, Gi3, GoA, GoB and Gz), through which μOR signals25,33. Specifically, we compared their signalling profiles with those of several other μOR ligands, including chemically and functionally diverse endogenous and exogenous agonists (Fig. 3, Extended Data Fig. 6 and Extended Data Tables 3 and 4). For brevity, we focus here on our two most potent compounds, C5 guano and C6 guano, in comparison with selected other ligands with pharmacological reference to the synthetic peptide agonist DAMGO.
Fig. 3 |. Profiling of C5 guano, C6 guano and μOR using TRUPATH Gαβγ biosensors and β-arrestin-1 and β-arrestin-2 efficacy.
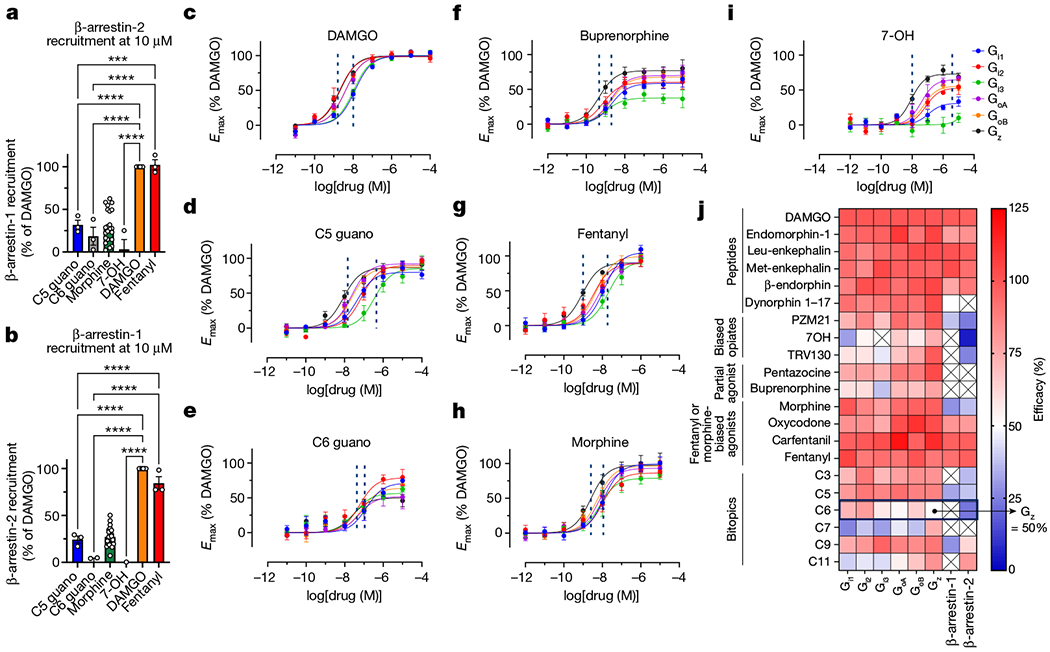
a,b, BRET assays for β-arrestin-2 (a) and β-arrestin-1 (b) recruitment in the presence of 10 μM C5 guano, C6 guano, morphine, fentanyl, buprenorphine, 7-OH and DAMGO (all relative to DAMGO). a, C5 guano and C6 guano showed significantly reduced β-arrestin-2 recruitment compared with DAMGO. Minimum, median and maximum values–C5 guano: 23.9%, 29.0%, 42,5%; C6 guano: 2.7%, 14.6%, 38.4%; morphine: −6.0%, 25.8%, 62.6%; 7-OH:−13.1%, −2.4%, 25.2%; DAMGO: 100%, 100%, 100%; fentanyl: 93.5%, 99.7%, 113.8%. b, Similarly, C5 guano and C6 guano showed significantly reduced β-arrestin-1 recruitment compared with DAMGO. Minimum, median and median values–C5 guano: 17.0%, 26.6%, 29.4%; C6 guano: −7.9%, 4.2%, 4.8%; morphine: 7.2%, 25.6%, 50.2%; 7-OH: −8.0%, −1.4%, 0.3%; DAMGO: 100%, 100%, 100%; fentanyl: 77.4%, 77.4%, 98.4. Data are mean ± s.e.m. (n = three experiments each done in duplicate). Primary statistics for a,b are provided in Supplementary Table 2. c–i, Gα-subtype selectivity using TRUPATH on μOR for DAMGO (c), C5 guano (d), C6 guano (e), buprenorphine (f), fentanyl (g), morphine (h) and 7-OH (i). C6 guano showed distinct potencies and efficacies for all six Gα-protein subtypes (Gi1, Gi2, Gi3, GoA, GoB and Gz) with the lowest efficacy for the Gz subtype. j, Efficacy heat map for opioid peptides, biased agonists, partial agonists, morphine or fentanyl template agonists and bitopic ligands using TRUPATH and β-arrestin-1 and β-arrestin-2 activity. Raw curves and values for all compounds are presented in Extended Data Fig. 6 and Extended Data Tables 2 and 3. Data are mean ± s.e.m. (n = three experiments each done in duplicate).
Previous efforts to develop opioid drugs with reduced side effects have been based on the hypothesis that β-arrestin-2 mediates deleterious responses such as respiratory depression7,11,34. The β-arrestin-2 recruitment profile of bitopic ligands was similar to the Tango assay. C5 guano (Emax = 38%) and C6 guano (Emax = 22%) showed progressively reduced β-arrestin-2 efficacy compared with fentanyl (Emax = 97%) but similar to morphine (Emax = 27%) (Fig. 3a). The G-protein-biased compounds, such as PZM21, 7-OH and TRV130, weakly recruited β-arrestin-2 (Emax = 24%, no response and 26%, respectively). Of note, these G-protein-biased compounds, for which we observed no or reduced β-arrestin-2 recruitment over the tested concentration range, have shown adverse effects (including respiratory depression) in certain preclinical animal models35–37 and in human clinical trials38, suggesting that low or even no β-arrestin-2-independent signalling is insufficient for at least certain unwanted physiological responses6,9,39.
We also examined β-arrestin-1 recruitment. C5 guano showed 32% efficacy in β-arrestin-1 recruitment assays–compared with 92% for fentanyl and 27% for morphine–whereas other bitopic ligands such as C6 guano showed no measurable β-arrestin-1 efficacy (Fig. 3b).
It has been proposed recently that low intrinsic G-protein efficacy, rather than abolished β-arrestin-2 activity, more favourably results in reduced side effects35. This suggestion was made on the basis of BRET-based measurements of heterotrimeric G-protein dissociation, which displays lower amplification and sensitivity to receptor reserve than conventional functional assays, and thus more accurately reflects measurements of ligand intrinsic efficacy33. As μOR signals through all Gi, Go and Gz family members, and this previous study only investigated one Gi2 subtype, we sought to more thoroughly characterize the concept with both existing and our novel ligands using the TRUPATH platform. The ligands differed in the efficacy and order of potency for specific Gi, Go and Gz subtypes, as well as in the range of potencies from the most- to least-sensitive G-protein isoform (dashed lines in Fig. 3c–i indicate the range of potencies; Extended Data Fig. 6, Extended Data Tables 3 and 4 and Supplementary Fig. 2). For DAMGO, the potency order is (Gz ≈ Gi2 > GoA ≈ GoB > Gi3 ≈ Gi1) with the EC50 ranging from 2.1 nM for Gz to 12.4 nM for Gi1. Fentanyl had a similar potency order (Gz > Gi2 > GoA ≈ GoB > Gi1 > Gi3), whereas the EC50 range was greater, 0.9 nM for Gz to 19 nM for Gi3. Morphine had a lower efficacy than DAMGO at all Gi and Go subtypes, with the lowest efficacy at Gi3 (79%). The distribution of EC50 values for morphine was similar to that for DAMGO (Gz > GoB ≈ GoA ≈ Gi3 > Gi2 ≈ Gi1), ranging from 2.4 nM for Gz to 11 nM for Gi1.
Similarly diverse profiles were observed with most exogenous opioids, whereas endogenous opioids were full or nearly full agonists at all transducers tested (an exception being Dyn1-17 at the arrestins). For C5 guano and C6 guano, we observed similarly unique signalling profiles, including between these compounds, despite their chemically similar scaffolds. For example, C5 guano displayed a wide range of potencies (lowest and highest EC50: Gi3, 350 nM and Gz, 8.8 nM) but a narrow range of efficacies (lowest and highest Emax: Gi1, 80% and GoA, 92%), whereas C6 guano displayed a narrow range of potencies (lowest and highest EC50: Gi1, 133 nM and Gz, 25n M) but a wide range of efficacies (lowest and highest Emax: Gz, 50% and Gi2, 79%), with overall lower efficacy. Of note, we observed that C6 guano displayed a lower efficacy at Gz both relative to other G proteins and in terms of efficacy relative to that of all other agonists (Fig. 3j). Notably, this includes the partial agonists buprenorphine and pentazocine (Emax, Gz = 78% and 103%) as well as biased agonists such as 7-OH, TRV130 and PZM21 (Emax, Gz = 73%, 98% and 98% respectively).
In summary, C6 guano shows marginal decreases in G-protein potency and lower intrinsic efficacy relative to classical μOR agonists such as DAMGO, fentanyl, morphine and oxycodone, and had the lowest efficacy for Gz. C6 guano showed diminished arrestin recruitment relative to DAMGO, other opioid peptides, fentanyl and oxycodone, but similar arrestin recruitment to morphine.
Selectivity across species and other GPCRs
Although there is high species homology between human μOR and mouse μOR, given that the lead bitopic ligand was to be profiled in mice, we screened C5 guano and C6 guano and several other μOR agonists for binding and Gi1 and β-arrestin-2 activity (Supplementary Table 1). The binding affinity for C6 guano at both human and mouse μOR was similar–that is, 1.2 and 1.08 nM, respectively (Supplementary Table 1a). Differences in Gi1 potency and efficacy across drugs across species were marginal. The EC50 of C6 guano at Gi1 was 132 nM for human μOR and 39 nM for mouse μOR, and the Emax was 71% and 79%, respectively. Crucially, C6 guano retains partial agonism and poor β-arrestin-2 recruitment at mouse μOR (Supplementary Table 1b).
C5 guano and C6 guano were counter-screened across around 45 targets using competitive radioligand binding assays through the Psychoactive Drug Screening Program at the National Institute of Mental Health40. In this assay platform, the affinity at μOR was 6.1 and 2.7 nM for C5 guano and C6 guano, respectively, and there was very little off-target binding (Supplementary Table 1c). The highest off-target affinities were found at the κ-opioid receptor (κOR) (224 and 259 nM for C5 and C6 guano bitopic ligands, respectively) and at the δ-opioid receptor (δOR) (1,530 and 312 nM, respectively), showing around 100-fold selectivity for C6 guano. The nearest non-opioid target for C5 guano was the histamine H1 receptor, over which it exhibited 45-fold selectivity; C6 guano exhibited more 400-fold selectivity over H1 and the α1A adrenoreceptor. When screened in BRET-based Gi1 signalling assays at κOR and δOR, both C5 and C6 guano showed reasonable selectivity for μOR (Extended Data Table 1 and Supplementary Figs. 2 and 3a–c).
In vivo pharmacology of C6 guano
Given the distinct differences in Gα-subtype signalling displayed by C6 guano compared with the other agonists, we further examined the pharmacology of this analogue in vivo using mouse models. The antinociceptive effect of C6 guano was evaluated in vivo in mice using a standard 55 °C warm water tail-withdrawal assay10,15, with the compound administered supraspinally (intracerebroventricular (ICV)), since it showed no systemic activity when administered intraperitoneally, probably because the two positive charges result in suboptimal blood–brain barrier penetration. The median antinociceptive dose (ED50) by ICV (and 95% confidence interval) for C6 guano was 18.77 nmol (5.49–55.54 nmol) (Fig. 4a), slightly higher than the ED50 of morphine, 6.6 nmol (4.4–8.43 nmol) (Supplementary Fig. 4a). A ceiling effect of antinociception was observed with C6 guano with increasing dosage from 30 to 300 nmol. C6 guano antinociception was found to be significantly attenuated in μOR-knockout mice (Fig. 4b). Before further analysis, we tested its stability in brain homogenates and carried out brain exposure studies. C6 guano showed high metabolic stability (Supplementary Fig. 4b). When assessed in pharmacokinetics assays at a 100 nmol ICV dose, C6 guano showed decent exposure at all tested time points. At the peak antinociceptive time point of 20 min, C6 guano showed 450 fold higher brain exposure than the ligand’s agonistic potency at mouse μOR (Supplementary Fig. 4c). These results suggest that C6 guano is stable and has desirable μOR occupancy for in vivo activity. C6 guano was further characterized in detail for potential adverse effects. Locomotor effects were evaluated using the Comprehensive Lab Animal Monitoring System (CLAMS) assay10,25. Ambulations induced by 300 nmol C6 guano did not differ significantly from vehicle, in contrast to morphine, which showed hyperlocomotion at 100 and 300 nmol doses (5× and 15× of the ED50, respectively) (Fig. 4c). We also measured breath rates in the same CLAMS assay at 100 and 300 nmol doses. Treatment with C6 guano resulted in an increase, whereas morphine caused a decrease in breath rates with a 100 nmol ICV dose (Fig. 4d). Increased breath rates were also observed in μOR-knockout mice, suggesting that they were an off-target effect, independent of μOR. The mechanism underlying this increased breath rate remains unknown. An ideal μOR ligand would not have effects on respiration. However, in parallel testing to distinguish our ligand from a biased μOR agonist, we tested the respiratory effects of 7-hydroxymitragynine (7-OH). This ligand has μOR-dependent antinociceptive actions and retains hyperlocomotion as well as conditioned place preference (CPP) in mice41. Subcutaneous injection of 4.5 mg kg−1 7-OH (15× the antinociceptive ED50) showed respiratory depression similar to that caused by morphine (30 mg kg−1 by subcutaneous injection; 15× the antinociceptive ED50) (Supplementary Fig. 4d). The results are in agreement with results of oral administration of 7-OH36. Notably, other biased ligands9,35,42 have also been shown to retain respiratory depression or provide marginal benefits over morphine.
Fig. 4 |. C6 guano exhibits μOR-mediated antinociception without CPP, CPA or hyperlocomotor effects.
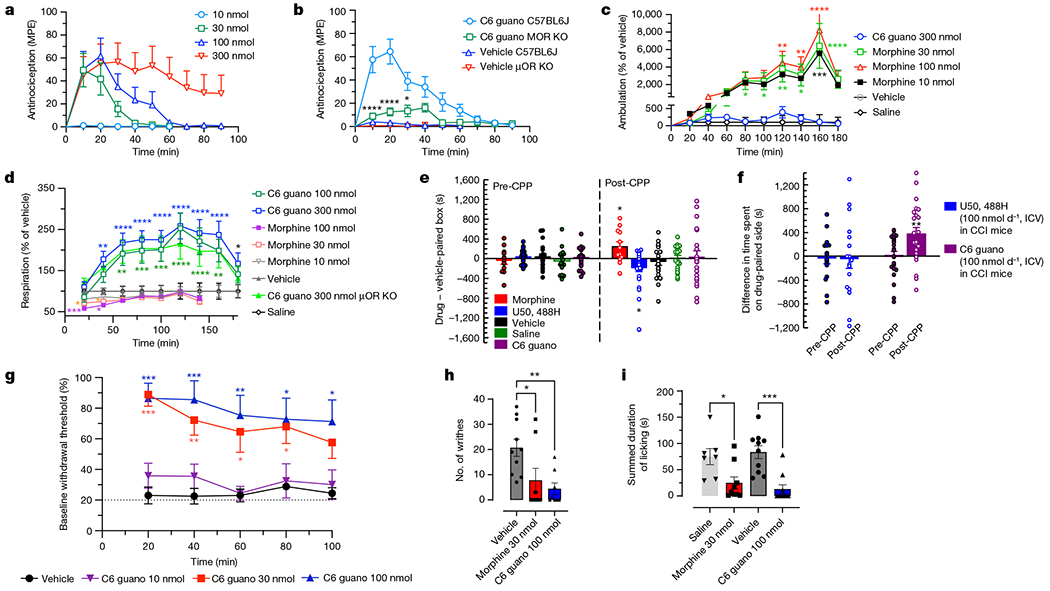
a, Antinociceptive time course: C57BL/6J mice were administered C6 guano by ICV injection and antinociception was measured using the 55 °C tail-withdrawal assay. Data are shown as mean percentage antinociception (MPE) ± s.e.m. An ED50 (with 95% confidence interval) of 18.77 nmol (5.49–55.54 nmol) was calculated for C6 guano. b, Antinociception by 100 nmol ICV C6 guano was attenuated in μOR-knockout (KO) mice compared with wild-type (C57BL/6J) or vehicle-treated (veh) mice. Data are MPE ± s.e.m. c,d, Locomotor effects and respiratory depression. C57BL/6J mice were administered either saline, vehicle, morphine (10, 30 or 100 nmol ICV), C6 guano (100 or 300 nmol ICV) and ambulation (c) or number of breaths (d) was measured every minute and averaged in 20-min bins. Data are presented as percentage of vehicle response ± s.e.m. c, Morphine treatment resulted in hyperlocomotion, whereas the C6 guano (300 nmol ICV) effect was not significantly different from ICV vehicle at any time point. d, Morphine did not result in any significant decrease in respiration rate at 10 nmol, but caused respiratory depression at 30 nmol and 100 nmol doses. C6 guano caused increased respiration rate at 100 nmol and 300 nmol doses compared with vehicle. There were no significant differences between wild-type and μOR-knockout mice treated with 300 nmol ICV C6 guano. e, C6 guano (100 nmol ICV) treatment did not result in CPP or CPA changes relative to vehicle, whereas morphine (30 nmol ICV) resulted in CPP and U50,488H (100 nmol ICV) resulted in CPA. Minimum, median and maximum values–U50,488H pre-conditioning (pre-CPP): −207 s, 52 s, 344 s; U50,488H post-conditioning (post-CPP): −1,436 s, −109 s, 178 s; morphine pre-CPP: −542 s, −23 s, 413 s; morphine post-CPP: −299 s, 154 s, 819 s; C6 guano pre-CPP: −364 s, 43 s, 499 s; C6 guano post-CPP: −878 s, −42 s, 1,164 s; saline pre-CPP: −416 s, −93 s, 595 s; saline post-CPP:−411 s, 129 s, 438 s; vehicle pre-CPP: −377 s, 43 s, 573 s; vehicle post-CPP: −865 s, −75 s, 533 s. f, Comparison of C6 guano and U50,488H in an operant model of antinociception using CCI–CPP. A schematic representation of the CCI–CPP model protocol is presented in the Supplementary Methods. Points represent the difference in time spent on the drug-paired side. Data are mean ± s.e.m. U50,488H treatment in this model did not result in CPP or CPA, whereas C6 guano treatment resulted in CPP. Minimum, median and maximum values: U50,488H pre-CPP: −771 s, −52 s, 699 s; U50,488H post-CPP: −1,170 s, −67 s, 1,293 s; C6 guano pre-CPP: −767 s, 101 s, 438 s; C6 guano post-CPP: −566 s, 388 s, 1,397 s. g, Dose- and time-dependent anti-allodynic activity of C6 guano in a CCI model of neuropathic pain. Mechanical allodynia produced by sciatic nerve ligation was reduced between 20 and 100 min after treatment with 30 nmol or 100 nmol ICV C6 guano but not by 10 nmol ICV guano or vehicle. Data are mean ± s.e.m. h, Antinociception by C6 guano in the mouse acetic acid writhing test. Treatment with 100 nmol ICV C6 guano or 30 nmol ICV morphine showed significant antinociception compared with vehicle or saline. Data plotted are number of writhes counted over 15 min after administration of compound. Minimum, median and maximum values: vehicle: 7, 19.5, 37; morphine: 0, 0, 32; C6 guano: 0, 0.5, 17. i, Evaluation of C6 guano for antinociceptive effects in the formalin-induced inflammation assay in mouse. Treatment with 100 nmol ICV C6 guano or 30 nmol ICV morphine showed significant antinociception compared with vehicle or saline. Minimum, median and maximum values: vehicle: 34 s, 93.5 s, 151 s; morphine: 1 s, 7 s, 95 s; C6 guano: 0 s, 0 s, 78 s; saline: 29 s, 74 s, 150 s. n values and primary statistics for all panels are provided in Supplementary Table 2.
Next, we tested C6 guano for reward or aversive behaviour in a CPP paradigm in mouse. ICV treatment with 100 nmol C6 guano did not induce CPP or conditioned place aversion (CPA); by contrast, morphine at an equianalgesic dose resulted in CPP and treatment with the κOR agonist U50,488H resulted in CPA (Fig. 4e).
For validation of antinociceptive effects beyond a thermal model, we tested C6 guano in models of operant pain, neuropathic pain (the chronic constriction injury (CCI) model), visceral pain (acetic acid writhing) and inflammatory pain (the formalin assay). In an operant pain model that subjected mice exposed to CCI to place conditioning in the CPP assay43, 100 nmol ICV U50,488H did not result in CPP or CPA, consistent with previous reports with systemic U50,488H43. By contrast, place conditioning with 100 nmol ICV C6 guano resulted in CPP, suggesting that it can blunt the aversive emotions associated with neuropathic pain (Fig. 4f). Extending this finding, treatment with C6 guano dose-dependently ameliorated mechanical allodynia displayed by mice exposed to CCI, with near-maximal efficacy and a longer time course against this model of neuropathic pain than observed with the thermal pain assays (Fig. 4g). Moreover, mice given 100 nmol ICV C6 guano demonstrated antinociception equivalent to that of 30 nmol ICV morphine in the writhing assay (Fig. 4h) and in the formalin assay (Fig. 4i).
Overall, our in vivo tests suggest that C6 guano produced antinociception against diverse pain types but did not show typical μOR-mediated adverse effects.
Discussion
Functional selectivity has been proposed as a path to more precise pharmacology, to enable the separation of therapeutic efficacy from side effects in several receptor families, including opioid9, angiotensin44, adrenergic, dopamine, serotonin and other receptors45.
Here we used bitopic extensions of the fentanyl scaffold to design functionally selective ligands for μOR, a typical family A GPCR. Our results demonstrate that guanidine extension of the ligand (C5 guano and C6 guano) can engage the conserved D2.50 side chain of the sodium site, either directly via a salt bridge or indirectly via water. Binding of the guanidine moiety in the sodium pocket can markedly reduce or even abolish β-arrestin-1 and β-arrestin-2 recruitment compared with the parent ligand fentanyl, while maintaining G-protein partial agonism in less amplified BRET-based assays with limited receptor reserve. We also observed a distinct Gi Go and Gz activity profile of C6 guano, which engages the D2.50 residue directly, compared with C5 guano, which engages the D2.50 site more indirectly via water. Notably, there was a selective decrease in efficacy in Gz with C6 guano compared with the other tested compounds (Fig. 3j), and a narrower spread of Gα-subtype potency compared with C5 guano (Fig. 3e) and partial agonists such as pentazocine and buprenorphine, full agonists such as DAMGO, fentanyl, carfentanil and other G-protein-biased μOR partial agonists, morphinans and opioid peptides. It should be noted that replacement of the n-aniline ring with small aliphatic groups46 has been shown to result in abolished β-arrestin recruitment; however, this also reduces G-protein efficacy (by 60–70%) and potency (80–1,000-fold) compared with the parent fentanyl. By contrast, our C5 and C6 bitopic ligands maintained fentanyl-like high affinity as well as cAMP potency and efficacy. It is possible that the guanidine functional group that engages D2.50 can differentially affect the conformation and/or dynamics of the helices involved in receptor activation. Thus, the bulky guanidine fitting the sodium pocket may be able to block the well-characterized inward shift of transmembrane helix (TM)7 previously associated with arrestin signalling, while simultaneously allowing the outward shift of TM6 that opens the G-protein-binding site44,47.
It is currently believed that the analgesia, respiratory depression and physical dependence mediated by the μOR are all dependent on G-protein pathways39,48 and not on β–arrestin-2 as previously proposed34. μOR is coupled to six distinct G-protein isoforms, and the possibility that functional selectivity for some of these isoforms can be used to separate analgesia from adverse effects remains largely unexplored. As our results suggest, the bitopic design that extends orthosteric ligands such as fentanyl into the conserved sodium pocket may provide a platform for the development of such Gα-subtype-biased ligands.
Several reports have suggested that Gi, Go and Gz subtypes may have different roles in the physiological responses to μOR agonists. For example, in studies using antisense oligonucleotides to reduce the expression of individual Gi, Go and Gz subtypes, DAMGO elicits activation of Gi2, Gz and Gq, whereas endomorphin-1 involves the activation of Gi1, Gi3 and Gz to produce supraspinal analgesia, suggesting that different Gα subunits mediate analgesia in response to these two μOR ligands49–51. The role of Gz in opioid-induced antinociception and adverse effects is less well understood. Gz-knockout animals have shown little or no change in supraspinal analgesic effects of morphine52, yet they exhibit a marked increase in analgesic tolerance53,54 and a decrease in lethality54. Studies in Go-knockout mice suggest that supraspinal analgesia is primarily mediated by one or both Go subtypes. Homozygous Go-knockout mice are not viable; however, supraspinal analgesia in heterozygous Go-knockout mice is diminished, even though spinal analgesic actions of morphine remain intact55. Supraspinal analgesia of morphine is intact in mice deficient in Gi2 or Gi355. To our knowledge, there are no reported studies in Gi1-knockout mice, possibly owing to lethality in homozygotes. Of note, studies using ICV injection of antisense oligonucleotides to suppress Gi, Go and Gz subtype expression have shown that all Gi, Go and Gz antisense oligonucleotides suppress the analgesia produced by heroin, suggesting all subtypes contribute to analgesia51. Thus, the physiological roles of Gα-subtypes are incompletely understood and compounds with distinct Gi, Go and Gz signalling profiles may enable us to assess the roles of specific subtypes in the therapeutic and adverse effects of μOR agonists. To this end, C6 guano could be a valuable tool and provide a platform to probe the pharmacology of Gz coupled to μOR. When evaluated in animal models, C6 guano exhibited supraspinal analgesia mediated by μOR in a range of rodent pain models while showing attenuated abuse potential and aversion. It is possible that reduced Gz efficacy and/or lower efficacy at all Gα-subtypes and/or other opioid targets is responsible for this distinct in vivo profile of C6 guano. Unexpectedly, C6 guano caused an increase in respiration rate via an unknown mechanism independent of μOR activation.
In summary, we have demonstrated that functional selectivity of the fentanyl scaffold can be controlled by bitopic extension of the ligand into the μOR sodium pocket. This is consistent with reports in which the highly conserved allosteric pocket has a central role in the functional mechanism of family A GPCRs2,56. Previously, major changes in receptor basal activity18,57–59, efficacy2,56–59 and functional selectivity switches2,18,56 that can bias GPCR signalling towards either Gi-protein or β-arrestin-2 pathways were observed for point mutations in the Na+-binding pocket for a variety of family A receptors. Our study reveals that such functional modulation can be achieved with bitopic ligands specifically designed to interact with the sodium pocket. This design was validated by cryo-EM structural studies and detailed analysis of the distinct signalling profiles at μOR of the bitopic ligands. Moreover, our bitopic ligands were able to modulate functional selectivity both between G protein and β-arrestin-2, as well as between G-protein subtypes, resulting in a new class of antinociceptive agents with attenuated adverse opioid effects. Exceptional conservation of the sodium pocket across GPCR structures2 suggests that such bitopic ligands could be designed for many other family A GPCRs. Although the functional effects would differ between different receptors, the rational design of such bitopic ligands could provide highly versatile pharmacological probes for exploring the multidimensional signalling landscape of GPCRs.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-022-05588-y.
Extended Data
Extended Data Fig. 1 |. Docking of a fentanyl based bitopic targeting the Na+ binding site.
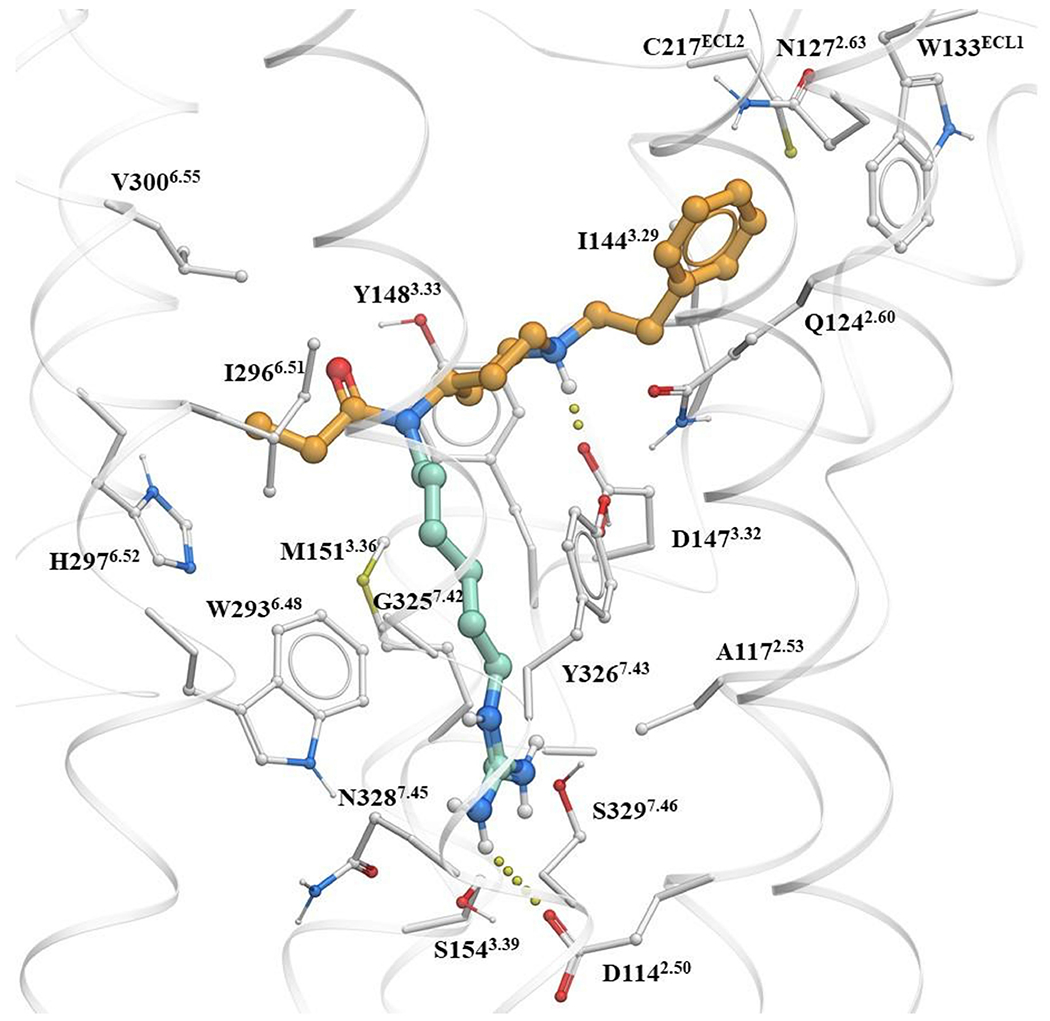
Molecular docking of a fentanyl based bitopic ligand shows that the functional head group can target the Na+ pocket.
Extended Data Fig. 2 |. Cryo-EM data processing work-flows.

Representative micrographs, 2D classes, 3D classes and data processing procedures for (A) C5-guano and (B) C6-guano bound μOR-Gi-scFv16 complex.
Extended Data Fig. 3 |. Global and local resolutions for cryo-EM maps.
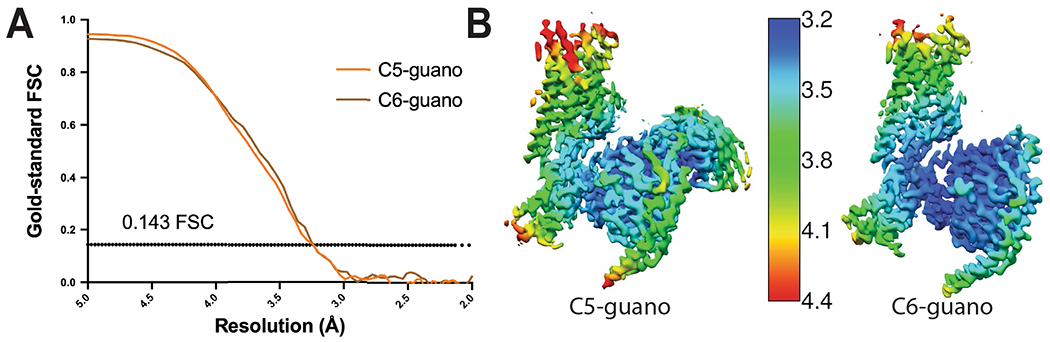
(A) Gold-standard FSC curves for C5-guano and C6-guano bound μOR-Gi structures. Overall resolution is 3.2 Å for C5-guano bound μOR–Gi-scFv16 and 3.3 Å for C6-guano bound μOR–Gi using the gold Standard FSC = 0.143 criterion. (B) Local resolution map of C5 guano and C6 guano bound μOR–Gi structures. (C) Data collection, refinement, and model statistic of two structures. Extended Data Table 2. Cryo-EM data collection, refinement and validation statistics.
Extended Data Fig. 4 |. Comparison of bitopic structures to BU72 structure.
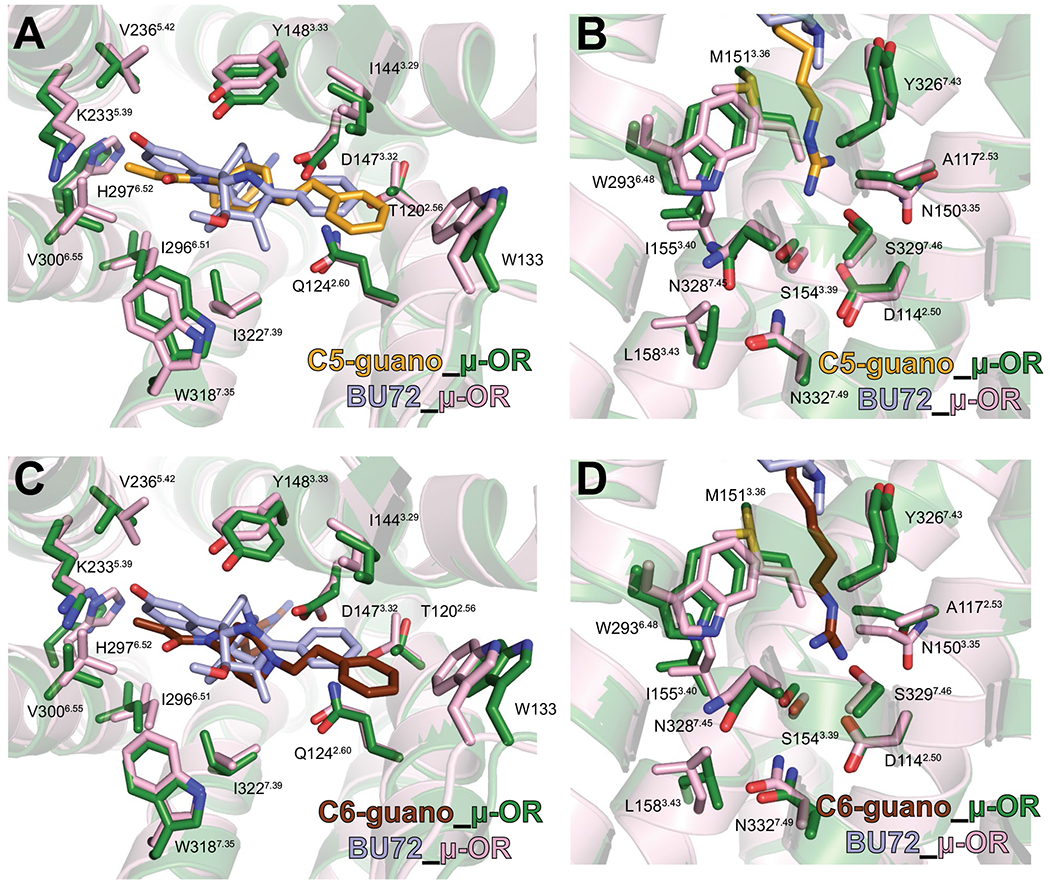
A, C, Side chains of μOR orthosteric pocket residues are shown for the C5-guano (A) and C6-guano (C) bound μOR–Gi complex (green) in comparison with the BU72 bound μOR (PDB code 5C1M; pink). The orthosteric pocket residues of μOR in complex with bitopic ligands and BU72 show nearly identical conformations. B, D, Side chains of μOR site-2 and Na+ site residues are shown for the C5 guano (B) and C6 guano (D) bound μOR–Gi complex (green) in comparison with the BU72 bound μOR (PDB code 5C1M; pink). The site-2 and Na+ site residues of μOR in complex with bitopic ligands and BU72 show nearly identical conformations.
Extended Data Fig. 5 |. Analysis of dynamics of direct and water mediated interactions of bitopic ligands.
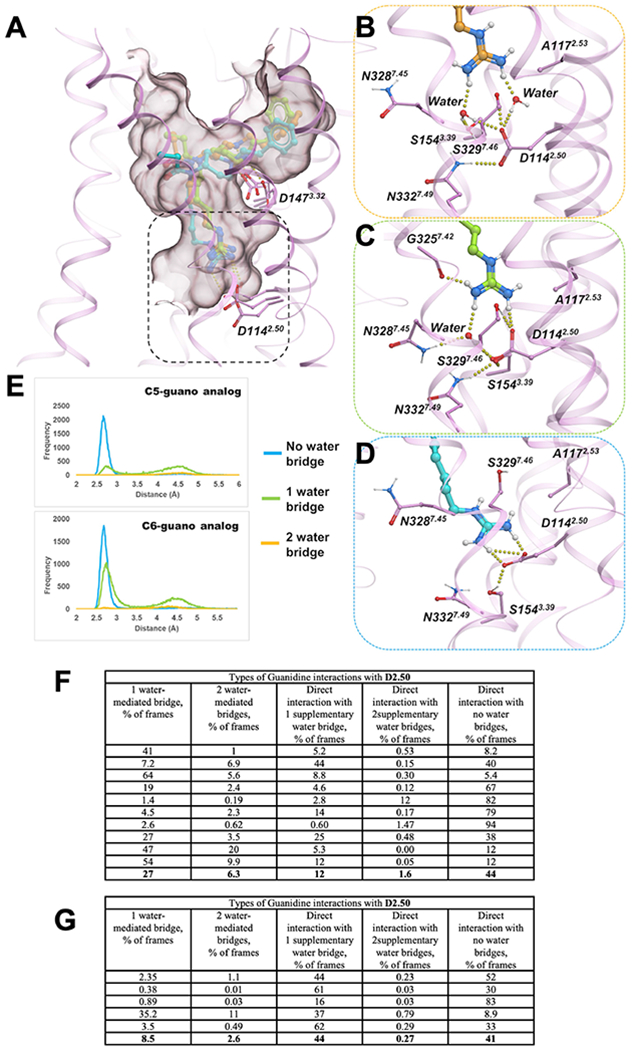
A) Overlay of three examples of C5 guano conformations bound to active state MOR (pink cartoon/sticks) during MD simulations (B) Detailed view of the interactions between guano moiety of C5 guano (orange sticks) and D1142.50 mediated by two water molecule (C) Direct salt bridge interactions between C5 guano (light green sticks) and D1162.50 supplemented by an additional water-mediated hydrogen bond. (D) direct salt bridge interactions between C5 guano (cyan sticks) and D1142.50 (E) Probability densities of distances between guano nitrogen atoms and D1142.50 carboxylate oxygens. Each chart plots probability density for frames with two bridging waters (orange), one bridging water (green), and no bridging waters (cyan). (F) Categorization and relative proportion of D2.50 and D3.32 mediated interactions in 10 independent C5 guano-μOR MD trajectories for 1 μs each. Among the cumulative frames from the 10 μs MD runs, close to 1/3rd of the frames-maintained guano-D2.50 interactions exclusively through water-mediated hydrogen bonds, while ~57% frames formed direct salt- bridges with or without supplementary water mediated interactions. Therefore, close to 90% of the frames maintained D2.50-guano interactions. The piperidine-D3.32 interactions were observed to be even more stable, with over 96% of the frames indicating direct salt bridge or water-mediated hydrogen bonds. (G) Categorization and relative proportion of D2.50 and D3.32 mediated interactions in 5 independent C6-μOR trajectories for 1 μs each. Overall, the number of direct interactions with D2.50 increased from 57% to 85% (compared to C5), perhaps resulting from the increase in linker length by a carbon atom that decreases the overall distances to D2.50 residue.
Extended Data Fig. 6 |. Profiling of chemically and pharmacologically distinct μOR agonists using TRUPATH, arrestin signaling.
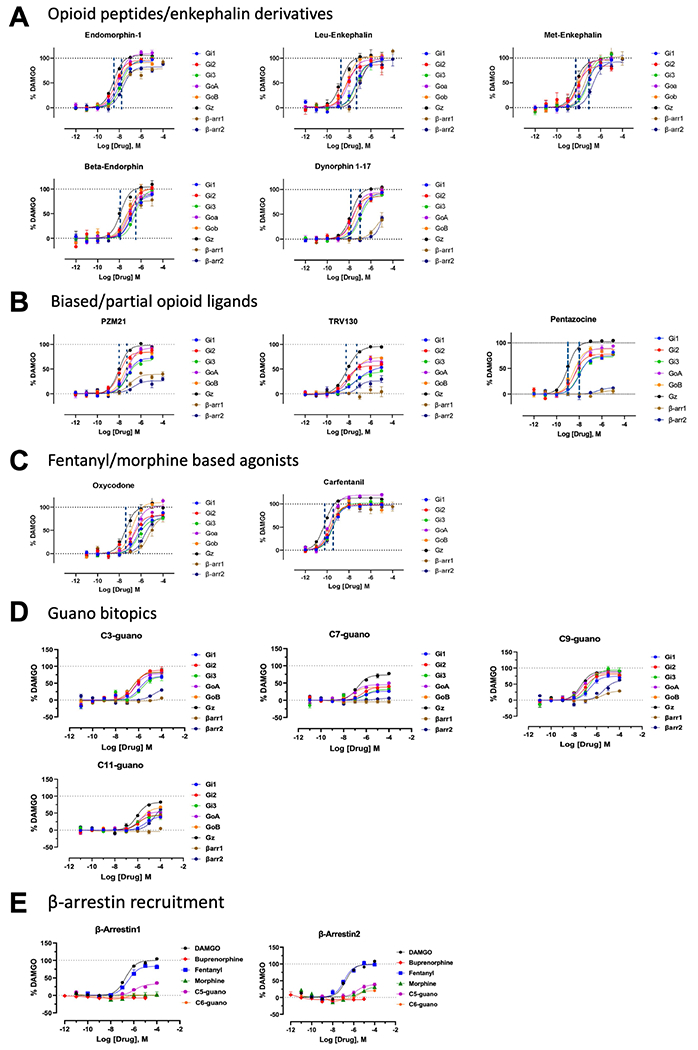
A) Peptides: Endomorphin-1, Leu-enkephalin, Met-enkephalin, Beta- endorphin and Dynorphin A (1-17). Dynorphin A (1-17) showed reduced arrestin recruitment while other peptides retained robust arrestin recruitment among peptides tested. B) Opioid biased agonists and partials: PZM21, TRV130, Gα- subtype selectivity and arrestin recruitment on μOR. PZM21, 7-OH and TRV130 showed <50% efficacy for arrestin1/2. Highest efficacy for all three biased agonists was seen at the Gz-subtype. μOR partial agonist pentazocine was a full agonist at the Gz subtype. C) Oxycodone and Carfentanil, Gα- subtype selectivity and arrestin recruitment on μOR. Carfentanil showed near maximal efficacy at all Gα- subtypes and arrestin1/2. Oxycodone was a full agonist at Gz and showed >50% efficacy at β-arrestin2. D) Fentanyl guano bitopics show differential G-protein and arrestin efficacy with increased chain length.
Extended Data Table 1 |.
Summary of binding affinities, cAMP and arrestin recruitment values of fentanyl amino and guano bitopics on μOR
| Binding a | cAMPb | β-arrestin2b | |||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Ligands | Ki ± SEM (nM) | EC50 (nM) | pEC50 ± SEM | Emax ± SEM (%) | EC50 (nM) | pEC50 ± SEM | Emax ± SEM (%) |
| C3 amino | > 1000 | 12300 | 4.9 ± 0.2 | 142 ± 20 | n.d. | n.d. | 8.2 ± 1.1 |
| C3 guano | 77 ± 3.0 | 164 | 6.8 ± 0.1 | 107 ± 2.3 | n.d. | n.d. | 6.9 ± 2.4 |
| C5 amino | 1179 ± 13 | 469 | 6.3 ± 0.2 | 90 ± 5.9 | n.d. | n.d. | n.d. |
| C5 guano | 4.6 ± 0.7 | 0.8 | 9.1 ± 0.1 | 104 ± 3.1 | 4710 | 5.3 ± 0.1 | 54 ± 3.2 |
| C6 amino | 1033 ± 15 | 844 | 6.1 ± 0.1 | 126 ± 4.7 | n.d. | n.d. | 20 ± 2.3 |
| C6 guano | 4.1 ± 0.3 | 4.7 | 8.3 ± 0.1 | 137 ± 2.1 | n.d. | n.d. | 19 ± 1.4 |
| C7 amino | 369 ± 7.0 | 436 | 6.4 ± 0.1 | 109 ± 3.9 | n.d. | n.d. | n.d. |
| C7 guano | 41 ± 8.0 | 62 | 7.2 ± 0.1 | 109 ± 2.4 | n.d. | n.d. | 13 ± 1.4 |
| C9 amino | 281 ± 2.2 | 360 | 6.4 ± 0.1 | 106 ± 4.6 | 66720 | 4.2 ± 0.2 | 56 ± 9.4 |
| C9 guano | 58 ± 8.0 | 21.3 | 7.7 ± 0.1 | 128 ± 3.2 | 792 | 5.1 ± 0.1 | 120 ± 6.6 |
| C11amino | 589 ± 13 | 293.2 | 6.5 ± 0.1 | 105 ± 3.6 | n.d. | n.d. | n.d |
| C11 guano | 165 ± 4 | 1020 | 6.0 ± 0.1 | 118 ± 5.2 | n.d. | n.d. | n.d. |
| Fentanyl | 9.1 ± 3.1 | 0.90 | 9.0 ± 0.1 | 86 ± 1.8 | 66.2 | 7.2 ± 0.1 | 119 ± 2.7 |
| DAMGO | n.d. | 0.40 | 9.4 ± 0.1 | 99 ± 4 | 723 | 6.1 ± 0.1 | 100 ± 2.8 |
Ki binding competition studies were performed with the indicated compound against 125I-IBNtxA (0.1 nM) in membranes from CHO cells stably expressing the μOR cloned mouse opioid receptor. Results are presented as Ki ± SEM (nM) from three independent experiments performed in triplicate.
Potency and efficacy data were obtained using agonist induced inhibition measured by cyclic AMP (cAMP) and Tango assay for β-arrestin 2 recruitment, respectively, from three independent experiments performed in triplicate. Efficacy is represented as EC50 (nM) and percent maximal stimulation (Emax) relative to standard agonist DAMGO. “n.d.” Denotes not determined. Efficacy<20%.
Extended Data Table 2 |.
Cryo-EM data collection, refinement and validation statistics
| C5-guano-μOR-Gi-scFvl6 (EMDB-26314) (PDB 7U2L) |
C6-guano-μOR-Gi (EMDB-26313) (PDB 7U2K) |
|
|---|---|---|
| Data collection and processing | ||
|
| ||
| Magnification | 130,000 | 130,000 |
| Voltage (kV) | 300 | 300 |
| Electron exposure (e–/Å2) | 67 | 68.5 |
| Defocus range (μm) | 0.7-2 | 0.7-2 |
| Pixel size (Å) | 1.06 | 0.8676 |
| Symmetry imposed | C1 | C1 |
| Initial particle images (no.) | 2,037,265 | 994,998 |
| Final particle images (no.) | 164,647 | 232,146 |
| Map resolution (Å) | 3.2 | 3.3 |
| FSC threshold | 0.143 | 0.143 |
| Map resolution range (Å) | 3.2-4.9 | 3.2-4.6 |
| Refinement | ||
| Initial model used (PDB code) | 6DDE | 6DDE |
| Model resolution (Å) | 3.2 | 3.2 |
| FSC threshold | 0.143 | 0.143 |
| Model resolution range (Å) | 2.9-3.4 | 2.8-3.4 |
| Map sharpening B factor (Å2) | −161.76 | −103.46 |
| Model composition | ||
| Non-hydrogen atoms | 8739 | 6759 |
| Protein residues | 1122 | 889 |
| Ligands | 1 | 1 |
| B factors (Å2) | ||
| Protein | 60.76 | 92.70 |
| Ligand | 78.28 | 125.85 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.004 | 0.009 |
| Bond angles (°) | 0.566 | 0.768 |
| Validation | ||
| MolProbity score | 1.72 | 1.78 |
| Clashscore | 8.03 | 7.07 |
| Poor rotamers (%) | 0 | 0 |
| Ramachandran plot | ||
| Favored (%) | 95.93 | 94.18 |
| Allowed (%) | 4.07 | 5.82 |
| Disallowed (%) | 0 | 0 |
Statistics of the cryo-EM maps and coordinates.
Extended Data Table 3 |.
Potency table for drugs profiled in Extended Data Fig. 6
| Potency | ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Compounds | Gi1 EC50 nM (pEC50 ± SEM) |
Gi2 EC50 nM (pEC50 ± SEM) |
Gi3 EC50 nM (pEC50 ± SEM) |
G0a EC50 nM (pEC50 ± SEM) |
G0b EC50 nM (pEC50 ± SEM) |
Gz EC50 nM (pEC50 ± SEM) |
β-arrl EC50 nM (pEC50 ± SEM) |
β-arr2 EC50 nM (pEC50 ± SEM) |
| Opioid Peptides | ||||||||
| DAMGO | 18 (7.7 ± 0.1) | 5 (8.2±0.1) | 15 (7.8±0.1) | 5 (8.3±0.1) | 2.5 (8.6±0.1) | 5.6 (9.2±0.1) | 189 (6.7±0.1) | 171 (6.8±0.1) |
| Endomorphin-1 | 9.9 (8.0 ± 0.1) | 4.3 (8.4 ± 0.1) | 16 (7.8 ± 0.1) | 8.0 (8.1 ± 0.1) | 6.0 (8.2 ± 0.1) | 3.0 (8.5 ± 0.1) | 11 (7.9 ± 0.1) | 21 (7.7 ± 0.1) |
| Leu-Enkephalin | 39 (7.4 ± 0.1) | 5.1 (8.3 ± 0.2) | 47 (7.3 ± 0.1) | 8.6 (8.1 ± 0.1) | 6.7 (8.2 ± 0.1) | 1.8 (8.8 ± 0.1) | 98 (7.0 ± 0.2) | 90 (7.1 ± 0.2) |
| Met-Enkephalin | 54 (7.3 ± 0.1) | 10 (8.00 ± 0.3) | 79 (7.1 ± 0.1) | 12 (7.9 ± 0.1) | 8.6 (8.1 ± 0.1) | 5.3 (8.3 ± 0.1) | 39 (7.4 ± 0.2) | 193 (6.7 ± 0.2) |
| Beta-Enkephalin | 128 (6.9 ± 0.1) | 93 (7.0 ± 0.2) | 293 (6.5 ± 0.1) | 62 (7.2 ± 0.1) | 55 (7.3 ± 0.2) | 13 (7.9 ± 0.1) | 28 (7.6 ± 0.2) | 120 (6.9 ± 0.1) |
| Dynorphin 1-17 | 88 (7.1 ± 0.1) | 18 (7.7 ± 0.1) | 115 (6.7 ± 0.1) | 25 (7.6 ± 0.1) | 43 (7.4 ± 0.1) | 167 (7.8 ± 0.1) | nd | nd |
|
| ||||||||
| Biased Opiates | ||||||||
| PZM21 | 53 (7.3 ± 0.07) | 10 (8.0 ± 0.1) | 55 (7.3 ± 0.1) | 52 (7.3 ± 0.1) | 28 (7.6 ± 0.1) | 9.8 (8.0 ± 0.1) | 104 (7.0 ± 0.2) | 199 (6.7 ± 0.4) |
| 7-OH | 111 (6.9 ± 0.3) | 76 (7.1 ± 0.2) | nd | 39 (7.4 ± 0.2) | 55 (7.3 ± 0.2) | 8.6 (8.1 ± 0.1) | nd | nd |
| TRV130 | 51 (7.3 ± 0.1) | 5.9 (8.2 ± 0.2) | 26 (7.6 ± 0.2) | 12 (7.9 ± 0.2) | 18 (7.7 ± 0.1) | 6.4 (8.2 ± 0.1) | nd | 73 (7.1 ± 0.4) |
|
| ||||||||
| Partial agonists | ||||||||
| Pentazocine | 10 (8.0 ± 0.1) | 4.3 (8.4 ± 0.1) | 8.6 (8.1 ± 0.1) | 5.1 (8.3 ± 0.1) | 3.3 (8.5 ± 0.1) | 1.0 (9.0 ± 0.1) | nd | nd |
| Buprenorphine | 2.1 (8.6 ± 0.2) | 0.7 (9.1 ± 0.2) | 1.0 (9.0 ± 0.3) | 1.8 (8.7 ± 0.2) | 2.0 (8.9 ± 0.3) | 0.4 (9.3 ± 1.6) | nd | nd |
|
| ||||||||
| Prototypic agonists | ||||||||
| Oxycodone | 486 (6.3 ± 0.1) | 278 (6.6 ± 0.2) | 565 (6.3 ± 0.1) | 286 (6.5 ± 0.2) | 146 (6.8 ± 0.1) | 39 (7.4 ± 0.2) | 5300 (5.3 ±0.2) | 2061 (5.7 ± 0.1) |
| Carfentanil | 0.30 (9.5 ± 0.1) | 0.21 (9.6 ± 0.1) | 0.43 (9.4 ± 0.1) | 0.21 (9.7 ± 0.1) | 0.22 (9.8 ± 0.1) | 0.14 (10.2 ± 0.1) | 0.20 (9.7 ±0.2) | 0.32 (9.5 ± 0.2) |
| Fentanyl | 12.7 (7.9 ± 0.1) | 3.4 (8.4 ± 0.2) | 19.4 (7.7 ± 0.2) | 6.8 (8.2 ± 0.1) | 4.9 (8.3 ± 0.1) | 0.9 (9.0 ± 0.1) | 10.5 (8.0 ± 0.1) | 7.8 (8.1 ± O.q) |
| Morphine | 11.9 (8.0 ± 0.1) | 10.4 (8.0 ± 0.2) | 7.2 (8.1 ± 0.2) | 6.9 (8.2 ± 0.1) | 5.5 (8.3 ± 0.1) | 2.4 (8.6 ± 0.1) | 205 (6.7 ± 0.1) | 118 (6.9 ± 0.2) |
|
| ||||||||
| Bitopics | ||||||||
| C3-guano | 1234 (5.9 ± 0.2) | 562 (6.2 ± 0.2) | 1811 (5.7 ± 0.2) | 505 (6.3 ± 0.2) | 338 (6.5 ± 0.1) | 305 (6.5 ± 0.2) | n.d. | 23571(4.6 ± 0.5) |
| C5-guano | 453 (7.3 ± 0.1) | 79 (7.1 ± 0.1) | 350 (6.5 ± 0.2) | 23 (7.6 ± 0.1) | 24 (7.6 ± 0.2) | 8.8 (8.0 ± 0.1) | 1210 (5.9 ± 0.2) | 1760(5.7 ± 0.3) |
| C6-guano | 132 (6.9 ± 0.2) | 58 (7.2 ± 0.2) | 33 (7.5 ± 0.3) | 54 (57.3± 0.3) | 72 (7.1 ± 0.2) | 25 (7.6 ± 0.3) | n.d. | 5191(5.3±0.6) |
| C7-guano | 531 (6.3 ± 0.2) | 450 (6.3 ± 0.2) | 833 (6.1 ± 0.4) | 81 (7.1 ± 0.2) | 63 (7.2 ± 0.2) | 161 (6.8 ± 0.1) | n.d. | n.d. |
| C9-guano | 93.1 (7.0 ± 0.1) | 60 (7.2 ± 0.2) | 181 (6.7 ± 0.1) | 27 (7.6 ± 0.1) | 43 (7.6 ± 0.1) | 27 (7.6 ± 0.1) | 3215 (5.5 ± 0.2) | 4089 (5.4 ± 0.2) |
| C11-guano | 4745 (5.3 ± 0.1) | 1311 (5.9 ± 0.3) | 1065 (6.0 ± 0.3) | 1242 (5.9 ± 0.1) | 2038 (5.7 ± 0.1) | 861 (6.1 ± 0.1) | n.d. | 30885 (4.5 ± 0.2) |
Potency [EC50 nM (pEC50 ± SEM)] are reported as estimates from simultaneous curve fitting of all biological replicates and include standard error.
Extended Data Table 4 |.
Efficacy table for drugs profiled in Extended Data Fig. 6
| Efficacy | ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Compounds | Gi1 Emax % ± SEM |
Gi2 Emax % ± SEM |
Gi3 Emax % ± SEM |
G0a Emax % ± SEM |
G0b Emax % ± SEM |
Gz Emax % ± SEM |
β-arrl Emax % ± SEM |
β-arr2 Emax % ± SEM |
| Opioid Peptides | ||||||||
| DAMGO | 99 ± 2 | 98 ± 3 | 98 ± 2 | 99 ± 2 | 100 ± 2 | 100 ± 2 | 100 ± 3.1 | 99± 2.8 |
| Endomorphin-1 | 93 ± 1.6 | 95 ± 1.7 | 96 ± 1.4 | 109 ± 2.7 | 91 ± 2.6 | 105 ± 1.1 | 78 ± 3.1 | 82 ± 2.5 |
| Leu-Enkephalin | 92 ± 5.5 | 85 ± 7.2 | 93 ± 4.7 | 96 ± 3.8 | 102 ± 4.5 | 103 ± 5.2 | 100 ± 6.2 | 97 ± 6.8 |
| Met-Enkephalin | 92 ± 4.6 | 84 ± 6.6 | 103 ± 5.6 | 97 ± 3.7 | 96 ± 3.1 | 96 ± 3.1 | 102 ± 4.9 | 92 ± 4.5 |
| Beta-Enkephalin | 87 ± 3.6 | 102 ± 6.1 | 99 ± 2.8 | 91 ± 3.2 | 99 ± 5.9 | 104 ± 4.5 | 76 ± 4.8 | 90 ± 5.3 |
| Dynorphin 1–17 | 92 ± 2.3 | 86 ± 3.8 | 90 ± 2.4 | 94 ± 4.4 | 92 ± 3.0 | 103 ± 1.4 | 50 ± 15 | 42±1.03 |
|
| ||||||||
| Biased Opiates | ||||||||
| PZM21 | 72 ± 2.0 | 84 ± 3.0 | 68 ± 1.9 | 92 ± 4.0 | 84 ± 3.1 | 98 ± 1.6 | 38 ± 3.3 | 24 ± 3.7 |
| 7-OH | 31 ± 4.9 | 53 ± 5.1 | nd | 65 ± 4.9 | 56 ± 3.5 | 73 ± 2.8 | nd | nd |
| TRV130 | 58 ± 3.8 | 58 ± 4.2 | 45 ± 3.3 | 67 ± 5.3 | 76 ± 4.4 | 98 ± 2.2 | nd | 26 ± 5.9 |
|
| ||||||||
| Partial agonists | ||||||||
| Pentazocine | 75 ± 1.8 | 78 ± 3.8 | 73 ± 2.3 | 89 ± 4.0 | 89 ± 3.2 | 103 ± 1.5 | 7±4.51 | 13±4.04 |
| Buprenorphine | 59 ± 3.6 | 60 ± 3.7 | 38 ± 4.0 | 70 ± 4.3 | 68 ± 3.3 | 77 ± 3.5 | nd | nd |
|
| ||||||||
| Prototypic agonists | ||||||||
| Oxycodone | 82 ± 3.2 | 83 ± 5.7 | 75 ± 3.8 | 102 ± 5.2 | 110 ± 3.4 | 102 ± 4.9 | 81 ± 7.9 | 80 ± 3.9 |
| Carfentanil | 97 ± 1.8 | 99 ± 2.2 | 102 ± 1.8 | 119 ± 3.8 | 96 ± 4.1 | 112 ± 1.7 | 96 ± 7.0 | 97 ± 6.7 |
| Fentanyl | 105 ± 4.8 | 89 ± 6.2 | 92 ± 6.5 | 91 ± 4.3 | 100 ± 4.2 | 90 ± 3.4 | 92 ± 2.2 | 97 ± 3.4 |
| Morphine | 99 ± 4.1 | 86 ± 5.5 | 79 ± 6.2 | 93 ± 3.9 | 96 ± 3.2 | 98 ± 4.6 | 29 ± 2.0 | 37 ± 4.2 |
|
| ||||||||
| Bitopics | ||||||||
| C3-guano | 71 ± 6.3 | 90 ± 6.5 | 70 ± 8.5 | 78 ± 5.1 | 83 ± 3.6 | 82 ± 5.3 | n.d. | 37 ± 11 |
| C5-guano | 80 ± 3.6 | 90 ± 3.7 | 85 ± 5.5 | 92 ± 2.3 | 87 ± 3.7 | 86 ± 3.2 | 32 ± 3.0 | 38 ± 5.3 |
| C6-guano | 71 ± 7.7 | 79 ± 7.2 | 56 ± 6.1 | 52 ± 7.0 | 64 ± 4.1 | 50 ± 5.3 | n.d. | 23±7.6 |
| C7-guano | 26 ± 1.8 | 38 ± 2.6 | 31 ± 5.2 | 45 ± 2.7 | 38 ± 2.6 | 73 ± 2.8 | n.d. | n.d. |
| C9-guano | 83 ± 3.4 | 96 ± 5.5 | 98 ± 4.6 | 95 ± 4.0 | 96 ± 2.8 | 99 ± 4.2 | 29 ± 2.7 | 61 ± 7.0 |
| C11-guano | 41 ± 2.5 | 45 ± 3.7 | 43 ± 5.2 | 54 ± 3.4 | 67 ± 3.2 | 82 ± 3.2 | n.d. | 80 ± 12 |
Efficacy (Emax% ± SEM) are reported as estimates from simultaneous curve fitting of all biological replicates and include standard error.
Supplementary Material
Acknowledgements
This work was supported by an American Heart Association Postdoctoral Fellowship (H.W.), NIH grants R33045884 (S.M.), R01DA042888 and R01DA007242 (Y.X.P.), R37DA036246 (B.K.K. and G.S.), R33DA038858 and P01DA035764 (V.K.), and R21DA048650 and R00DA038725 (R.A.-H.). B.K.K. and G.S. are additionally supported by the Mathers Foundation and R.A.-H. is supported through the Brain and Behavior Research Foundation. The State of Florida, Executive Office of the Governor’s Office of Tourism, Trade, and Economic Development provides funding to J.P.M. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748 to MSKCC. Cryo-EM data collection was performed at the Stanford-SLAC Cryo-EM Facilities, supported by Stanford University, SLAC and the National Institutes of Health S10 Instrumentation Programs. The authors thank E. Montabana and C. Zhang for their support with cryo-EM data collection; and Stanford University and the Stanford Research Computing Center for providing computational resources and support that contributed to these research results. Cryo-EM data processing for this project was performed on the Sherlock cluster. The authors acknowledge the Center for Advanced Research Computing (CARC) at the University of Southern California for providing computing resources that have contributed to the research results reported in this study. Receptor binding profiles were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program (NIMH PDSP), contract no. HHSN-271-2018-00023-C. B.L.R. is director of NIMH PDSP at the University of North Carolina at Chapel Hill and J.D. is project officer of NIMH PDSP at NIMH, Bethesda.
Footnotes
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Competing interests S.M. and Y.X.P. are founders of Sparian Biosciences. B.K.K. is a founder of and consultant for ConfometRx. G.S. is a cofounder of Deep Apple. All other authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-022-05588-y.
Data availability
The cryo-EM maps and corresponding coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-26314 (C5 guano–μOR–Gi–scFv16) and EMD-26313 (C6 guano–μOR–Gi) and the Protein Data Bank (PDB) under accession codes 7U2L (C5 guano–μOR–Gi–scFv16) and 7U2K (C6 guano–μOR–Gi). The authors declare that all the data supporting the findings of this study are available within the article, extended data and supplementary information files. All compounds can be made available on reasonable requests from the authors. Source data are provided with this paper.
References
- 1.DeWeerdt S. Tracing the US opioid crisis to its roots. Nature 573, S10–S12 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Zarzycka B, Zaidi SA, Roth BL & Katritch V Harnessing ion-binding sites for GPCR pharmacology. Pharmacol. Rev 71, 571–595 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang W. et al. Structural insights into μ-opioid receptor activation. Nature 524, 315–321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hilger D, Masureel M & Kobilka BK Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol 25, 4–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasternak GW & Pan Y-X Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev 65, 1257–317 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varga BR, Streicher JM & Majumdar S Strategies towards safer opioid analgesics—a review of old and upcoming targets. Br. J. Pharmacol 10.1111/bph.15760 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manglik A et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeWire SM et al. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphines. J. Pharmacol. Exp. Ther 344, 708–717 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Faouzi A, Varga BR & Majumdar S Biased opioid ligands. Molecules 25, 4257 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uprety R et al. Controlling opioid receptor functional selectivity by targeting distinct subpockets of the orthosteric site. eLife 10, e56519 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmid CL et al. Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 171, 1165–1175.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eans SO et al. Parallel synthesis of hexahydrodiimidazodiazepines heterocyclic peptidomimetics and their in vitro and in vivo activities at μ (MOR), δ (DOR), and κ (KOR) opioid receptors. J. Med. Chem 58, 4905–4917 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Majumdar S et al. Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proc. Natl Acad. Sci. USA 108, 19778–19783 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiguchi N et al. BU10038 as a safe opioid analgesic with fewer side-effects after systemic and intrathecal administration in primates. Br. J. Anaesth 122, e146–e156 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Váradi A et al. Mitragynine/corynantheidine pseudoindoxyls as opioid analgesics with mu agonism and delta antagonism, which do not recruit β-arrestin-2. J. Med. Chem 59, 8381–8397 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massaly N, Temp J, Machelska H & Stein C Uncovering the analgesic effects of a PH-dependent mu-opioid receptor agonist using a model of nonevoked ongoing pain. Pain 161, 2798–2804 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Kandasamy R et al. Positive allosteric modulation of the mu-opioid receptor produces analgesia with reduced side effects. Proc. Natl Acad. Sci. USA 118, e2000017118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fenalti G et al. Molecular control of δ-opioid receptor signalling. Nature 506, 191–196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pert CB, Pasternak G & Snyder SH Opiate agonists and antagonists discriminated by receptor binding in brain. Science 182, 1359–1361 (1973). [DOI] [PubMed] [Google Scholar]
- 20.Hu X et al. Kinetic and thermodynamic insights into sodium ion translocation through the μ-opioid receptor from molecular dynamics and machine learning analysis. PLoS Comput. Biol 15, e1006689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shang Y et al. Mechanistic insights into the allosteric modulation of opioid receptors by sodium ions. Biochemistry 53, 5140–5149 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu W et al. Structural basis for allosteric regulation of GPCRS by sodium ions. Science 337, 232–236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Selvam B, Shamsi Z & Shukla D Universality of the sodium ion binding mechanism in class A G-protein-coupled receptors. Angew. Chem. Int. Ed. Engl 57, 3048–3053 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Manglik A et al. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakraborty S et al. A novel mitragynine analog with low-efficacy mu opioid receptor agonism displays antinociception with attenuated adverse effects. J. Med. Chem 64, 13873–13892 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Overdose Death Rates. National Institute on Drug Abuse https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates (accessed 23 September 2019).
- 27.Lipiński PFJ, Jarończyk M, Dobrowolski JC & Sadlej J Molecular dynamics of fentanyl bound to μ-opioid receptor. J. Mol. Model 25, 144 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Subramanian G, Paterlini MG, Portoghese PS & Ferguson DM Molecular docking reveals a novel binding site model for fentanyl at the mu-opioid receptor. J. Med. Chem 43, 381–391 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Qu Q et al. Structural insights into distinct signaling profiles of the μOR activated by diverse agonists. Nat. Chem. Biol 10.1038/s41589-022-01208-y (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ballasteros JA & Weinstein H Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Meth. Neurosci 25, 366–428 (1995). [Google Scholar]
- 31.Dardonville C et al. Synthesis and pharmacological studies of new hybrid derivatives of fentanyl active at the μ-opioid receptor and I2-imidazoline binding sites. Bioorg. Med. Chem 14, 6570–6580 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Gallivan JP & Dougherty DA Cation-π interactions in structural biology. Proc. Natl Acad. Sci. USA 96, 9459–9464 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olsen RHJ et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol 16, 841–849 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raehal KM, Walker JKL & Bohn LM Morphine side effects in β-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther 314, 1195–1201 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Gillis A et al. Low intrinsic efficacy for g protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal 13, 31 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Hill R, Kruegel AC, Javitch JA, Lane JR & Canals M The respiratory depressant effects of mitragynine are limited by its conversion to 7-OH mitragynine. Br. J. Pharmacol 179, 3875–3885 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He L et al. Pharmacological and genetic manipulations at the μ-opioid receptor reveal arrestin-3 engagement limits analgesic tolerance and does not exacerbate respiratory depression in mice. Neuropsychopharmacology 46, 2241–2249 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anesthetic and Analgesic Drug Products Advisory Committee. Oliceridine FDA Advisory Committee Briefing Document (FDA, 2018). [Google Scholar]
- 39.Kliewer A et al. Phosphorylation-deficient G-protein-biased μ-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun 10, 367 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Besnard J et al. Automated design of ligands to polypharmacological profiles. Nature 492, 215–220 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chakraborty S et al. Oxidative metabolism as a modulator of kratom’s biological actions. J. Med. Chem 64, 16553–16572 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill R et al. The novel μ-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br. J. Pharmacol 175, 2653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson LL et al. Characterization of CM-398, a novel selective sigma-2 receptor ligand, as a potential therapeutic for neuropathic pain. Molecules 27, 3617 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wingler L et al. Angiotensin analogs with divergent bias stabilize distinct receptor conformations. Cell 176, 468–478.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wootten D, Christopoulos A, Marti-Solano M, Babu MM & Sexton PM Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol 19, 638–653 (2018). [DOI] [PubMed] [Google Scholar]
- 46.de Waal PW et al. Molecular mechanisms of fentanyl mediated β-arrestin biased signaling. PLoS Comput. Biol 16, e1007394 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu JJ, Horst R, Katritch V, Stevens RC, & Wuthrich K Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kliewer A et al. Morphine-induced respiratory depression is independent of β-arrestin2 signalling. Br. J. Pharmacol 177, 2923–2931 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Standifer KM, Rossi GC & Pasternak GW Differential blockade of opioid analgesia by antisense oligodeoxynucleotides directed against various G protein alpha subunits. Mol. Pharmacol 50, 293–298 (1996). [PubMed] [Google Scholar]
- 50.Sánchez-Blázquez P, Rodríguez-Díaz M, DeAntonio I & Garzón J Endomorphin-1 and endomorphin-2 show differences in their activation of μ opioid receptor-regulated G proteins in supraspinal antinociception in mice. J. Pharmacol. Exp. Ther 291, 12–18 (1999). [PubMed] [Google Scholar]
- 51.Sánchez-Blázquez P, Gómez-Serranillos P & Garzón J Agonists determine the pattern of G-protein activation in μ-opioid receptor-mediated supraspinal analgesia. Brain Res. Bull 54, 229–235 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Yang J et al. Loss of signaling through the G protein, Gz, results in abnormal platelet activation and altered responses to psychoactive drugs. Proc. Natl Acad. Sci. USA 97, 9984–9989 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hendry IA et al. Hypertolerance to morphine in G(Zα)-deficient mice. Brain Res. 870, 10–19 (2000). [DOI] [PubMed] [Google Scholar]
- 54.Leck KJ et al. Deletion of guanine nucleotide binding protein αz subunit in mice induces a gene dose dependent tolerance to morphine. Neuropharmacology 46, 836–846 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Lamberts JT, Jutkiewicz EM, Mortensen RM & Traynor JR Mu-opioid receptor coupling to Gαo plays an important role in opioid antinociception. Neuropsychopharmacology 36, 2041–2053 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Katritch V et al. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci 39, 233–244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schöppe J et al. Crystal structures of the human neurokinin 1 receptor in complex with clinically used antagonists. Nat. Commun 10, 17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Massink A et al. Sodium ion binding pocket mutations and adenosine A2A receptor function. Mol. Pharmacol 87, 305–313 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Capaldi S et al. Allosteric sodium binding cavity in GPR3: a novel player in modulation of aβ production. Sci. Rep 8, 11102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM maps and corresponding coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-26314 (C5 guano–μOR–Gi–scFv16) and EMD-26313 (C6 guano–μOR–Gi) and the Protein Data Bank (PDB) under accession codes 7U2L (C5 guano–μOR–Gi–scFv16) and 7U2K (C6 guano–μOR–Gi). The authors declare that all the data supporting the findings of this study are available within the article, extended data and supplementary information files. All compounds can be made available on reasonable requests from the authors. Source data are provided with this paper.