

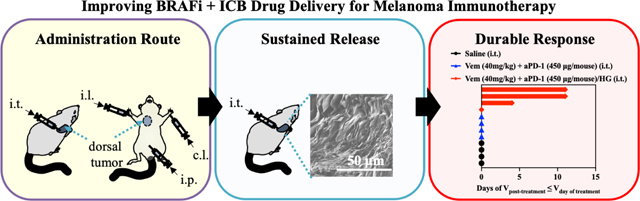
Abstract
The wide prevalence of BRAF mutations in diagnosed melanomas drove the clinical advancement of BRAF inhibitors in combination with immune checkpoint blockade for treatment of advanced disease. However, deficits in therapeutic potencies and safety profiles motivate the development of more effective strategies that improve the combination therapy’s therapeutic index. Herein, we demonstrate the benefits of a locoregional chemoimmunotherapy delivery system, a novel thermosensitive hydrogel comprised of gelatin and Pluronic® F127 components already widely used in humans in both commercial and clinical products, for the co-delivery of a small molecule BRAF inhibitor with immune checkpoint blockade antibody for the treatment of BRAF-mutated melanoma. In vivo evaluation of administration route and immune checkpoint target effects revealed intratumoral administration of antagonistic programmed cell death protein 1 antibody (aPD-1) lead to potent antitumor therapy in combination with BRAF inhibitor vemurafenib. The thermosensitive F127-g-Gelatin hydrogel that was evaluated in multiple murine models of BRAF-mutated melanoma that facilitated prolonged local drug release within the tumor (>1 week) substantially improved local immunomodulation, tumor control, rates of tumor response, and animal survival. Thermosensitive F127-g-Gelatin hydrogels thus improve upon the clinical benefits of vemurafenib and aPD-1 in a locoregional chemo-immunotherapy approach for the treatment of BRAF-mutated melanoma.
Keywords: BRAF inhibitor, immune checkpoint blockade, thermosensitive hydrogel, sustained release, drug delivery system
Graphical Abstract
1. Introduction
With rapidly increasing rates of melanoma over recent decades, BRAF has been identified as a highly attractive therapeutic target since half of melanoma cases are BRAF-mutated [1–4]. BRAF is a signaling molecule in the mitogen-activated protein kinase (MAPK) pathway, which governs the growth, differentiation, and apoptosis of cells by associating with epidermal growth factor (EGF), Kirsten rat sarcoma viral oncogene homologue (KRAS), mitogen-activated protein kinase kinase (MEK), extracellular signal-regulated kinase (ERK), phosphoinositide 3-kinase (PI3K), and mechanistic target of rapamycin (mTOR) [4]. While a wild-type BRAF monomer induces the low activity of its downstream MEK, activation of its upstream RAS promotes the homo- or heterodimerization of BRAF, which elevates MEK activity [5]. However, a mutation in an ATP-binding pocket of BRAF leads to the consecutive unchecked hyperactivation of the RAS/RAF/MEK/ERK pathway, which promotes the growth, differentiation, and survival of cancer cells [5,6]. Vemurafenib (Vem), Dabrafenib, and Encorafenib approved by U.S. Food and Drug Administration (FDA) as BRAF inhibitors (BRAFi) to treat metastatic melanoma in clinic, have shown high therapeutic potencies. Nevertheless, most patients develop acquired resistance to BRAFi, which is attributed to secondary mutations in other pathways, dysregulated metabolism, and BRAFV600E overexpression [7–10]. Numerous combination therapies with various inhibitors blocking upstream and downstream of BRAF have been explored, such as EGFR inhibitors (Cetuximab, Panitumumab), IGF1R inhibitors (Ganitumab), KRAS inhibitors (Dacomitinib), and MEK inhibitors (Trametinib, Binimetinib, Cobimetinib) [1–6]. To date, Dabrafenib plus Trametinib, Vem plus Cobimetinib, and Encorafenib plus Binimetinib have received FDA-approval [11].
It is now established that the therapeutic effects of BRAFi originate not only from its direct effects on the RAS/RAF/MEK/ERK pathway in cancer cells [1–11], but also from the host immune response. Therapeutic BRAFi increases CD8+ T [12–16] and natural killer (NK) cells in tumors [15] as well as decreases the level of myeloid-derived suppressor cells (MDSCs) [13] and regulatory T cells (Tregs) [13]. However, programmed cell death ligand (PD-L) 1, an immune checkpoint whose ligand is programmed cell death (PD) 1, is up-regulated on cancer cells in response to BRAFi [14,15]. Furthermore, NKG2D ligand that mediates the cytotoxicity of NK and T cells is downregulated in tumors [15], which can compensate for the CD8+ T and NK cell-mediated antitumor immune response induced by BRAFi. Considering the blockbuster clinical success of agonist antibodies of immune checkpoints, so called immune checkpoint blockade (ICB) therapies, that invigorate the antitumor immune response to result in durable cures, it is now established that the combination of targeted therapies including BRAFi with ICB can elicit more potent disease control [14,17–21].
Clinically, Vem is administered orally or systemically [22], but systemic toxicity including liver injury and kidney failure is a known side effect [22–24]. To mitigate these toxicity risks, the potential of topical administration of Vem in a form of ointments has been explored, demonstrating enhanced antitumor effects and improved liver, kidney, and lung biosafety compared to oral and systemic intraperitoneal (i.p.) administration [22]. Our group also recently reported the potential for locoregional administration of ICB antibodies to improve tumor control relative to conventional systemic administration by promoting the immune response in tumor microenvironment and secondary lymphoid tissues [25]. Furthermore, locoregional sustained delivery of immune modulating agents including ICB antibodies with hydrogels and scaffolds can improve the antitumor therapeutic index with durable immune response [25–32].
Herein, we report a locoregional sustained drug delivery system (DDS) for Vem and antagonistic PD-1 monoclonal antibody (aPD-1) combination therapy for the treatment of BRAF-mutated melanoma. Various drug combinations and routes of administrations were explored, revealing intratumoral (i.t.) administration of VEM in combination with aPD-1 to result in the most potent control of an in vivo murine model of BRAF-mutated melanomas. The potential of this newly developed sustained delivery system, thermosensitive hydrogels (F127-g-Gelatin) comprised of gelatin and Pluronic® F127 [32], on therapeutic benefits on Vem and aPD-1 combination therapy was investigated. The delivery system achieved sustained drug release that was controlled by hydrogel degradation. As a result of sustained delivery, i.t. administration of Vem and aPD-1 within the hydrogel system not only reduced the drug dose and number of administrations required to achieve therapeutic benefit, but also improved the therapeutic index with the enhanced memory CD8+ T cell response in the tumor compared to the free drugs. This demonstrates the potential for the F127-g-Gelatin sustained drug delivery system in potentiating locoregional chemoimmunotherapy for BRAF-mutated melanoma.
2. Materials & Methods
2.1. In vitro cytotoxicity test of Vem
103 D4M, B16F10, or NIH3T3 cells in DMEM (90 μL, Gibco™) containing fetal bovine serum (10%, Gibco™) and Antibiotic-Antimycotic (1X, Gibco™) were seeded in the 96-well cell culture plates (Falcon™). After overnight incubation in 37°C CO2 incubator, cells were treated with Vem (10 μL) at different concentrations. After incubating for 2 days at 37°C in a CO2 incubator, AlamarBlue™ cell viability reagent (5 μL, Invitrogen™) was added to culture suspensions. After 1 h incubation at 37°C in a CO2 incubator, fluorescence (ex: 560 nm, em: 590 nm) was measured by Synergy H4 microplate reader (BioTek).
2.2. Animals
All animal procedures were approved by Georgia Tech’s IACUC. Animals were acquired from Jackson Laboratories.
2.3. Optimization of D4M cell numbers for establishing D4M tumor models
0.5×105, 1×105, or 5×105 D4M cells in saline (30 μL) were subcutaneously inoculated to the right dorsal skin of the C57Bl/6 (8–10 weeks). The volume of the D4M tumors was determined by an ellipsoid calculation, with dimensions measured by caliper. Animal survival is depicted with Kaplan-Meier plots.
2.4. Administration- and ICBs-dependent Vem-mediated tumor therapy.
5×105 D4M cells in saline (30 μL) were subcutaneously inoculated to the right dorsal skin of the C57Bl/6 (8–10 weeks) on day 0. Free Vem was formulated in 20% DMSO containing hydroxypropyl methylcelluose (HPMC, 0.05 mg μL−1). Vem (10 mg kg−1, 100 μL) was treated i.p. every day from day 7 to day 16. aPD-1, aCTLA-4, or aPD-1/aCTLA-4 (each 150 μg mouse−1, 30 μL) were administered intraperitoneally (i.p.), intratumorally (i.t.), intradermally to tissue ipsilateral (i.l.) to the tumor, and intradermally to tissue contralateral (c.l.) to the tumor on day 8, 11, and 14. For tumor re-challenge studies, 5×105 D4M cells in saline (30 μL) were subcutaneously inoculated to the left dorsal skin of the mice survived after tumor inoculation and Vem and ICBs therapy. Tumor volumes were determined by an ellipsoid calculation, which dimensions were measured with caliper. Animal survival is depicted with Kaplan-Meier plots.
2.5. Synthesis of F127-g-Gelatin polymer
F127-g-Gelatin polymer was synthesized by our previously reported procedure [32]. In brief, 4-nitrophenyl chloroformate (3.2 g, Sigma Aldrich) dissolved in 50 mL DCM was added to Pluronic® F127 (20 g, Sigma Aldrich) dissolved in dichloromethane (50 mL, Sigma Aldrich), followed by vigorous stirring for overnight. Cold diethyl ether (2750 mL, Sigma Aldrich) was used to precipitate the resultant 4-nitrophenyl chloroformate functionalized F127. After vacuum filtration, the 4-nitrophenyl chloroformate functionalized F127 dissolved in 33.3 % (v/v) ethanol (150 mL, Sigma Aldrich) was reacted with gelatin type A (10 g, Sigma Aldrich, 300g bloom) dissolved in deionized water (1 L) containing triethylamine (15 mL, Sigma Aldrich) for overnight. The final product, F127-g-Gelatin, was yielded with dialysis (Spectrum Industries, MWCO 100 KDa) against deionized water for 1.5 days and then lyophilization for 3 days.
2.6. Characterization of F127-g-Gelatin polymer
Bruker Advance 400 MHz FT-NMR 1H nuclear magnetic resonance spectroscopy (1H NMR) was used to confirm the chemical compositions of F127-g-Gelatin polymer. Hitachi SU-8230 at accelerating voltage 1 kV and 10 μA emission current was used to take scanning electron microscope (SEM) images to observe microstructure of dry F127-g-Gelatin hydrogel that had prepared by incubating 4.5 wt.% hydrogel in 37 °C water bath for 1 h, rapid-frozen in liquid nitrogen, and lyophilized for 3 days. Synergy H4 microplate reader was used to determine the critical micellar concentrations (CMC) of the polymer by measuring the ratiometric emission (em) fluorescence (373 nm/383 nm) of pyrene (final concentrations=0.6 μM) at 336 nm excitation (ex) in the difference concentrations of polymer solutions.
In order to prepare TRITC-labeled aPD-1 (aPD-1-TRITC), aPD-1 (4.1 mg, Bioxcell, clone RMP1–14) in DPBS without calcium and magnesium (PBS(−/−)) (500 μL, Gibco™) was reacted with TRITC (100 μg, Thermofisher Science) in dimethyl sulfoxide (DMSO, 100 μL, Sigma Aldrich) for 2 h, followed by purification with Zeba desalting column (MWCO 7 KDa, Thermofisher Science) four times. Synergy H4 microplate reader was used to observe self-quenching of aPD-1-TRITC in F127-g-Gelatin by quantifying the fluorescence of TRITC in aPD-1-TRITC (ex: 547 nm, em: 579 nm) in the different concentrations of F127-g-Gelatin solutions.
2.7. . In vitro residence stability of the hydrogel and drug release test with the hydrogel
In vitro residence stability of F127-g-Gelatin hydrogel was evaluated by measuring the weight of F127-g-Gelatin hydrogel hydrated by DPBS with calcium and magnesium (PBS (+/+)) (Gibco™) in the same volume of PBS (+/+) over time. In detail, 4.5 wt.% F127-g-Gelatin hydrogel in PBS (+/+) (300 μL) without or with Vem (0.7 mg mL−1 in 4.5 wt.% F127-g-Gelatin hydrogel) or aPD-1-TRITC (0.6 mg mL−1 in 4.5 wt.% F127-g-Gelatin hydrogel) was added in the 1.5 mL e-tube in 37 °C water incubator. After 30 min incubation, PBS (+/+) (300 μL) with or without 2.5 U mL−1 MMP-9 (collagenase IV, Gibco™) was added. At predetermined time intervals, supernatants were taken. The remaining mass of hydrated hydrogel were measured and then fresh PBS (+/+) (300 μL) with or without 2.5 U mL−1 MMP-9 was added.
Drug release testing was performed by measuring the contents of Vem or aPD-1-TRITC in the supernatants sampled from F127-g-Gelatin hydrogel incubated with PBS (+/+) over time. In detail, 4.5 wt.% F127-g-Gelatin hydrogel in PBS (+/+) (300 μL) with Vem (0.7 mg mL−1 in 4.5 wt.% F127-g-Gelatin hydrogel) or aPD-1-TRITC (0.6 mg mL−1 in 4.5 wt.% F127-g-Gelatin hydrogel) was added in the 1.5 mL e-tube in 37 °C water incubator. After 30 min incubation, PBS (+/+) (300 μL) with or without 2.5 U mL−1 MMP-9 was added. At predetermined time intervals, supernatants were taken and then replaced with fresh PBS (+/+) (300 μL) with or without 2.5 U mL−1 MMP-9. Agilent 1260 Infinity II High Performance Liquid Chromatography (HPLC) system (G7111B, G7129A, G7130A, G7114A) equipped with Agilent Poroshell 120 EC-C18 column (4.6 mm X 100, 2.7 μm) was employed to evaluate the contents of Vem in the supernatants in the condition of gradient elution of acetonitrile and deionized water (% acetonitrile=30–90) at flow rate 1 mL min−1 at 25 °C. Synergy H4 microplate reader was utilized to quantify the fluorescence of TRITC in aPD-1-TRITC (ex: 547 nm, em: 579 nm).
2.8. In vivo residence stability of the hydrogel
4.5 wt.% F127-g-Gelatin hydrogel in saline (30 μL) was subcutaneously administered to the right dorsal skin of the C57Bl/6 (8–10 weeks). The volume of hydrogel was determined by an ellipsoid calculation, which dimensions were measured with caliper.
2.9. In vivo biodistribution of aPD-1 with the hydrogel
To prepare AF647-labeled aPD-1 (aPD-1-AF647), aPD-1 (15.65 mg) in PBS (−/−) (1.23 mL) was reacted with AlexaFluor™647 NHS Ester (AF647-NHS, Invitrogen™) in DMSO (30 μL of 10 mM) for 2 h, followed by purification with Zeba desalting column (MWCO 7 KDa, Thermofisher Science) 5 times.
5×105 D4M cells in saline (30 μL) were subcutaneously inoculated to the right dorsal skin of the C57Bl/6 (8–10 weeks) on day 0. Free aPD-1-AF647, Free Vem+aPD-1-AF647, 4.5 wt.% F127-g-Gelatin hydrogel containing aPD-1-AF647 or 4.5 wt.% F127-g-Gelatin hydrogel containing Vem and aPD-1-AF647 in 30 μL were i.t. administered to the D4M tumor-bearing mice on day 7. The dose of Vem and aPD-1-AF647 is 10 mg kg−1 and 100 μg mouse−1, respectively. Mice were sacrificed on day 8, 11, and 14. FastPrep-24 (MP Biomedicals) was utilized to homogenize tumors, tumor draining lymph nodes (dLNs), non draining lymph nodes (ndLNs), spleens, livers, kidney, lungs, and blood harvested in 1.4 mm zirconium bead filled tubes (OPS Diagnostics). Synergy H4 microplate reader was used to record the fluorescence of aPD-1-AF647 (ex: 650 nm, em: 670 nm) in each tissue. Different concentrations of aPD-1-AF647 were mixed with homogenized tissues harvested from tumor-, drug-, and hydrogel-free mice to establish the standard curves.
2.10. In vivo tumor therapy and immune profiles with the hydrogel containing Vem and aPD-1
5×105 D4M cells in saline (30 μL) were subcutaneously inoculated to the right dorsal skin of the C57Bl/6 (8–10 weeks) on day 0. Vem (10, 20, 40 mg kg−1) and aPD-1 (300 or 450 μg mouse−1) in 30 μL saline or in, 30 μL of 4.5 wt.% F127-g-Gelatin hydrogel (30 μL) was i.t. administered on day 7. The volume of tumors was determined by an ellipsoid calculation, which dimensions were measured with caliper. Animal survival is depicted with Kaplan-Meier plots. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels in serum were measured from blood collected from facial vein on day 14 by using alanine aminotransferase (ALT) activity colorimetry/fluorometry (Biovision) and aspartate aminotransferase (AST) activity colorimetric assay kits (Biovision).
For immune profiles, another set of mice with same schedule of tumor inoculations and treatment was sacrificed to harvest tumors, dLNs, and spleens on day 14. 70 μm strainer (Corning) and ACK lysis buffer (Lonza) were used to harvest splenocytes. 70 μm strainer (Corning) and collagenase D (Roche, 1 mg mL−1) were used to harvest lymphocytes and tumoral immune cells. To prevent non-specific interactions between cells and antibodies, cells were incubated with 2.4G2 on ice for 5 min. To stain live cells, Zombie Aqua Fixable viability dye (Biolegend) were treated to cells, followed by incubation at room temperature (RT) for 30 min. The resultant cells were stained with antibody mixtures except Foxp3 on ice for 30 min. After the cells were fixed and permeabilized with Foxp3 Fixation/Permeabilization working solution (eBioscience™ Foxp3/Transcription Factor Staining Buffer Set, Invitrogen™) on ice for 60 min, Foxp3 and Tcf1 staining was proceeded on ice for 75 min. Cells were profiled with LSR Fortessa flow cytometry (BD Biosciences) and then analyzed with FlowJo (FlowJo LLC).
The SM1 tumor model was used as an additional model of BRAF-mutated melanoma. SM1 cells were kindly donated from Dr. Antoni Ribas at the University of California Los Angeles. To passage and amplify the SM1 cells, SM1 cells (1×106 cells) in 30 uL were subcutaneously inoculate to the right dorsal skin of NOD scid gamma (NSG) mouse (8 weeks). When size of a tumor reached to 5–10 mm, SM1 cancer cells were harvested by using 70 μm strainer after the tumors were excised from the sacrificed mouse, and then dispersed in 50 μL saline. To establish the SM1 tumor model, 30 uL of the cell solutions above were subcutaneously inoculated to the right dorsal skin of the C57Bl/6 (8–10 weeks) on day 0. Vem (40 mg kg−1) and aPD-1 (450 μg mouse−1) with 4.5 wt.% F127-g-Gelatin hydrogel (30 μL) was i.t. administered when tumor size reaches to 5–10 mm. As a control group, one time administration of free Vem (40 mg kg−1) formulated in 20% DMSO containing hydroxypropyl methylcelluose (HPMC, 0.05 mg μL−1, 100 μL) i.t. and aPD-1 (450 μg mouse−1, 30 μL) was treated i.t. Tumor volumes and survival were measured as same with the above.
2.11. Statistics
Plotting graphs and statistical analysis were performed with Prism software (Graphpad). In vitro and in vivo data are reported with a formulation of mean ± standard deviation (SD) and mean ± standard error of mean (SEM), respectively. While two-tailed paired Student t-test was used for statistical comparisons of two groups, two-way ANOVA or one-way ANOVA with Tukey post-hoc hypothesis were employed for statistical comparisons of multiple groups; ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
3. Results & Discussion
3.1. Administration route- and ICB-dependent Vem-mediated tumor therapy effects on D4M melanomas
D4M is a metastatic murine melanoma cell line bearing the BRAFV600E mutation, which can be passaged in vitro and then used for in vivo disease modeling in either immune-deficient or syngeneic mice [33]. Consistent with its BRAF status, D4M cells treated in in vitro culture were Vem sensitive (Fig. S1A), whereas negligible effects of Vem were found for BRAF wild-type melanoma cells (B16F10, Fig. S1B) and normal (non-malignant) fibroblasts (NIH3T3, Fig. S1C).
To identify effective administration routes and combinations for Vem (10 mg kg−1, administered i.p. a total of 10 times) with ICB, aCTLA-4, aPD-1, or aCTLA-4 and aPD-1 in combination (each 150 μg mouse−1, administered a total of 3 times) were administered i.p., i.t., i.l., or c.l. (Fig. 1–3, Fig. S2, Table S1–S3) into mice bearing melanomas injected in the right dorsal skin formed from 5×105 D4M cells. aCTLA-4 was found to significantly improve the therapeutic effects of Vem with respect to tumor growth and/or survival regardless of its administration route (Fig. 1, Fig. S2A, Table S1), though this effect was least pronounced i.l., which only prolonged delay in tumor growth post-treatment in a greater number of animals (Fig. S2A). Most notably, administration of aCTLA-4 i.p. and i.t. in Vem treated (i.p.) animals led to 20% complete tumor regression of D4M tumors. Likewise, aPD-1 significantly promoted the antitumor effects of Vem, regardless of its administration route (Fig. 2, Fig. S2B, Table S2). However, the therapeutic effects of the combined Vem and aPD-1 (Fig. 2) were significantly better than the combined Vem and aCTLA-4 (Fig. 1) in terms of tumor growth, survival, and complete response (CR), the latter being defined herein as complete tumor regression. In particular, administration of aPD-1 i.t. allowed for 60% CR of tumors in Vem treated (i.p.) animals, which was more efficient than other administration routes of aPD-1 as well as aCTLA-4. However, the combination of Vem with aCTLA-4/aPD-1 failed to improve efficacy over Vem alone (Fig. 3, Fig. S2C, Table S3) or in combination with either aCTLA-4 (Fig. 1) or aPD-1 (Fig 2) individually, which can be explained by blockade of multiple immune checkpoints in combination not always resulting in synergies due to compensatory upregulation of the other immune checkpoint pathways [34,35] and tumor-specific sensitivities to ICB [36,37]. Despite different rates of response and therapeutic efficacy, mice that had experienced CR to the combined Vem and aCTLA-4, the combined Vem and aPD-1, and the combined Vem and aCTLA-4/aPD-1 exhibited 100% resistance to the re-challenge of D4M tumors inoculated in the left dorsal skin (Fig. 4) contralateral to the regressed tumor completely treated with the combined Vem and ICB therapy (Fig. 1–3). These results indicate that when regression occurs with combined Vem and ICB therapy, this response is associated with long-term systemic immunological memory. Results in this model imply that i.t. administration of aPD-1 synergizes optimally with BRAFi anticancer therapy in the treatment of BRAF-mutated melanoma.
Fig. 1. Administration routes-dependency of combined Vem and aCTLA-4 therpay on D4M models.
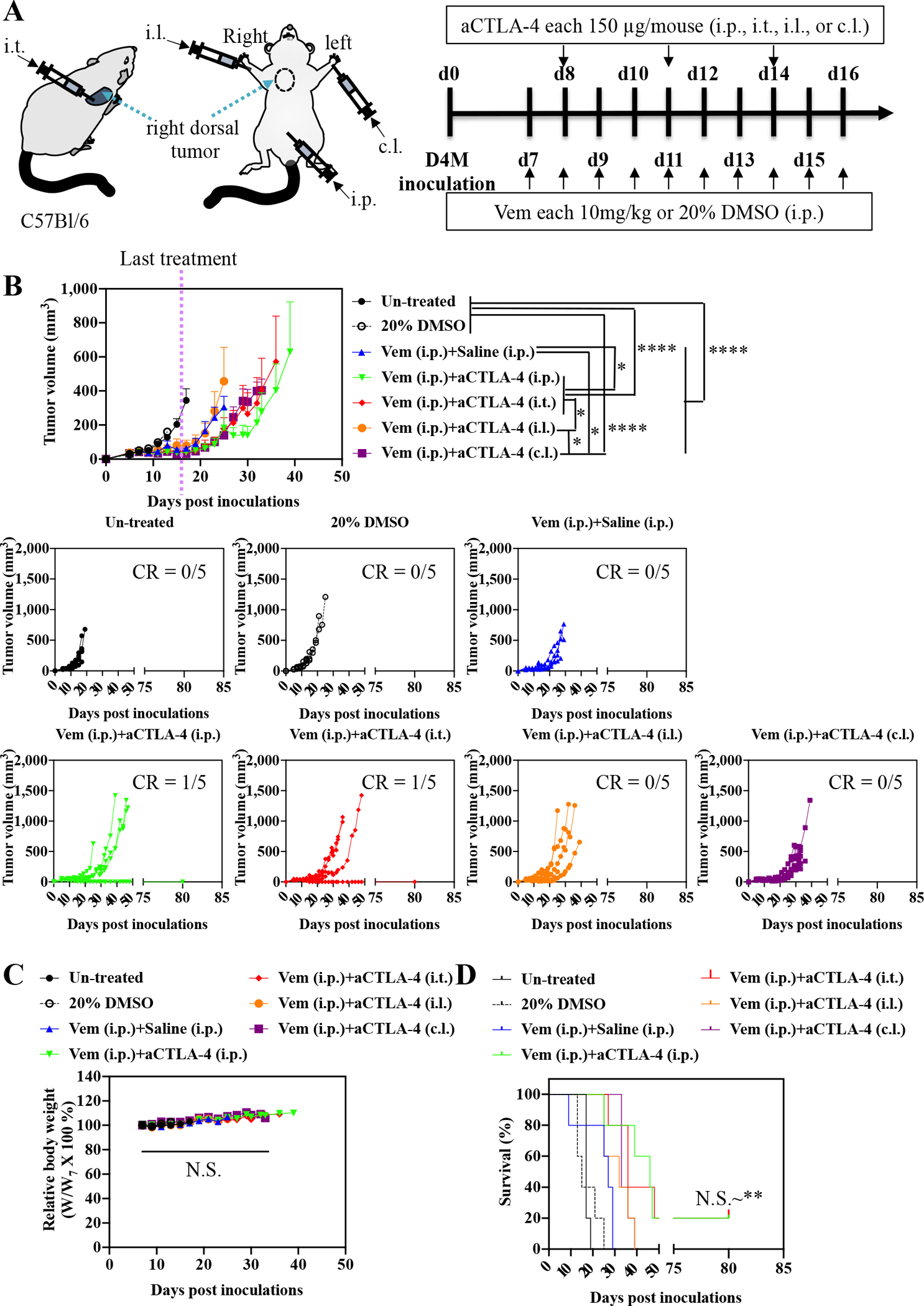
(A) Scheme for administration routes and treatment schedule. 5×105 D4M cells in 30 μL saline were subcutaneously inoculated in C57Bl/6 on day 0. Vem (10 mg/kg, 100 μL) was treated i.p. every day from day 7 to day 16. aCTLA-4 (150 μg mouse−1, 30 μL) was administered i.p., i.t., i.l. or c.l. 3 times every 3 days from day 8. (B) Average and individual tumor volumes (n=5). (C) Weight changes after treatment (n=5). (D) Kaplan–Meier survival curves (n=5). Data are presented as mean±SEM. Two-way ANOVA using Tukey post-hoc statistical hypothesis was employed for (B) and (C). Log-rank using Mentel-Cox statistical hypothesis was used for survival (D). ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05. N.S. means “Non Significant”. Statistical values for (D) are listed in Table S1.
Fig. 3. Antagonistic effects of combined aCTLA-4 and aPD-1 in Vem-mediated therapy on D4M models.
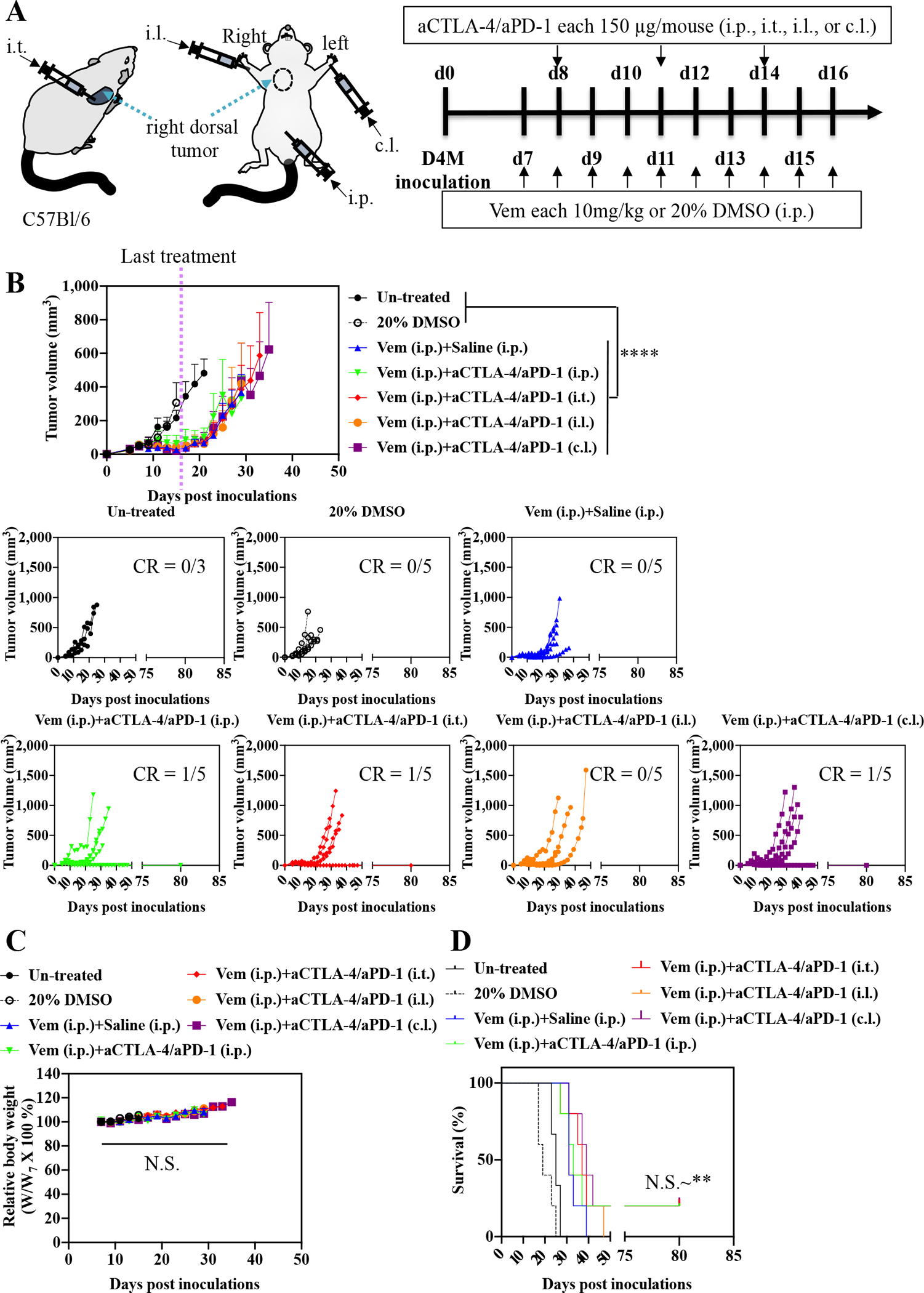
(A) Scheme for administration routes and treatment schedule. 5×105 D4M cells in 30 μL saline were subcutaneously inoculated in C57Bl/6 on day 0. Vem (10 mg/kg, 100 μL) was treated i.p. every day from day 7 to day 16. Mixture of aCTLA-4 and aPD-1 (each 150 μg mouse−1, total 30 μL) was administered i.p., i.t., i.l. or c.l. 3 times every 3 days from day 8. (B) Average and individual tumor volumes (n=5). (C) Weight changes after treatment (n=5). (D) Kaplan–Meier survival curves (n=5). Data are presented as mean±SEM. Two-way ANOVA using Tukey post-hoc statistical hypothesis was employed for (B) and (C). Log-rank using Mentel-Cox statistical hypothesis was used for survival (D). ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05. N.S. means “Non Significant”. Statistical values for (D) are listed in Table S3.
Fig. 2. Administration routes-dependency of combined Vem and aPD-1 therpay on D4M models.
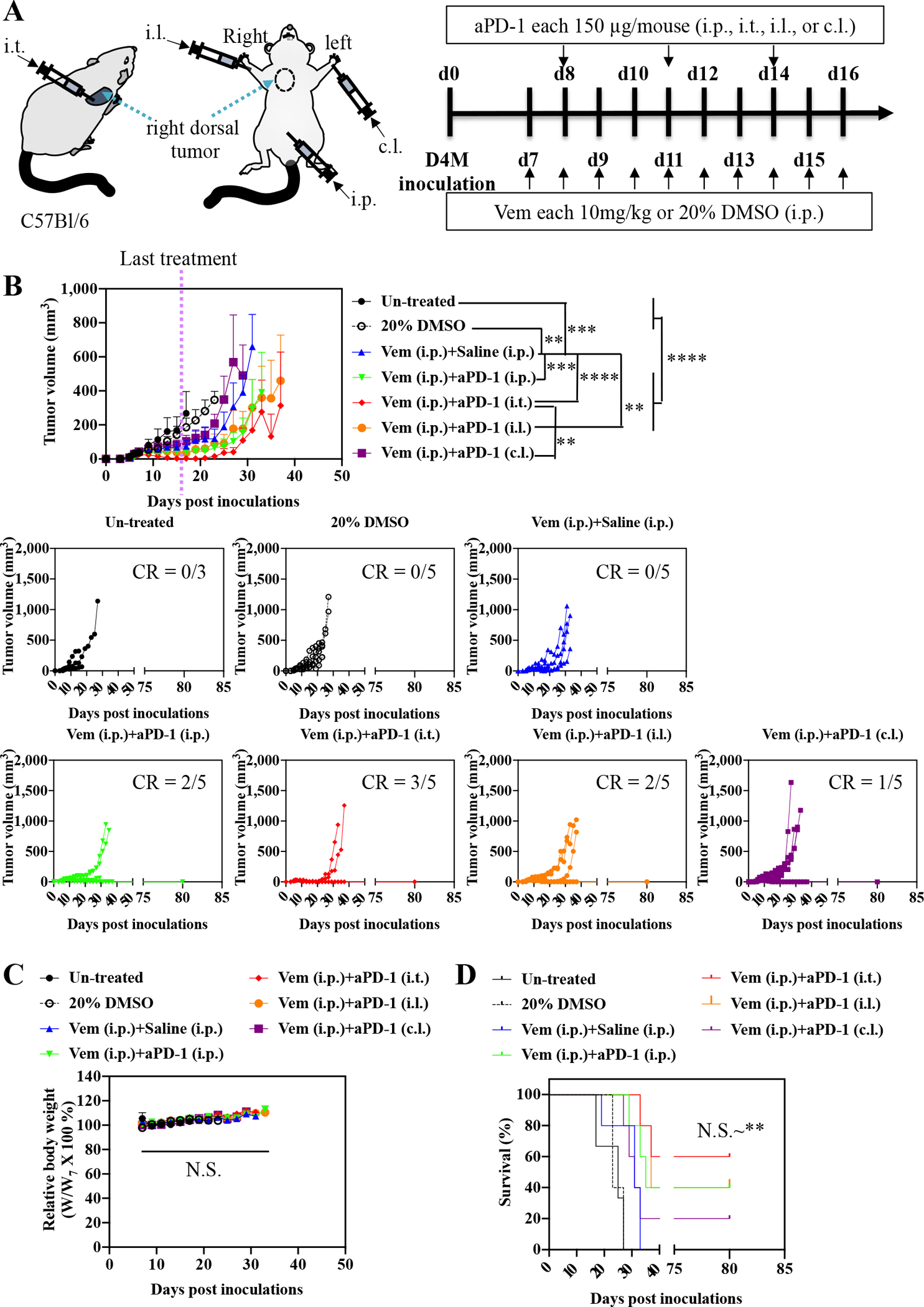
(A) Scheme for administration routes and treatment schedule. 5×105 D4M cells in 30 μL saline were subcutaneously inoculated in C57Bl/6 on day 0. Vem (10 mg/kg, 100 μL) was treated i.p. every day from day 7 to day 16. aPD-1(150 μg mouse−1, 30 μL) was administered i.p., i.t., i.l. or c.l. 3 times every 3 days from day 8. (B) Average and individual tumor volumes (n=5). (C) Weight changes after treatment (n=5). (D) Kaplan–Meier survival curves (n=5). Data are presented as mean±SEM. Two-way ANOVA using Tukey post-hoc statistical hypothesis was employed for (B) and (C). Log-rank using Mentel-Cox statistical hypothesis was used for survival (D). ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05. N.S. means “Non Significant”. Statistical values for (D) are listed in Table S2.
Fig. 4. Re-challenge of tumor on the mice that had experienced CR after combined Vem and ICBs therapy in Fig 1–3.
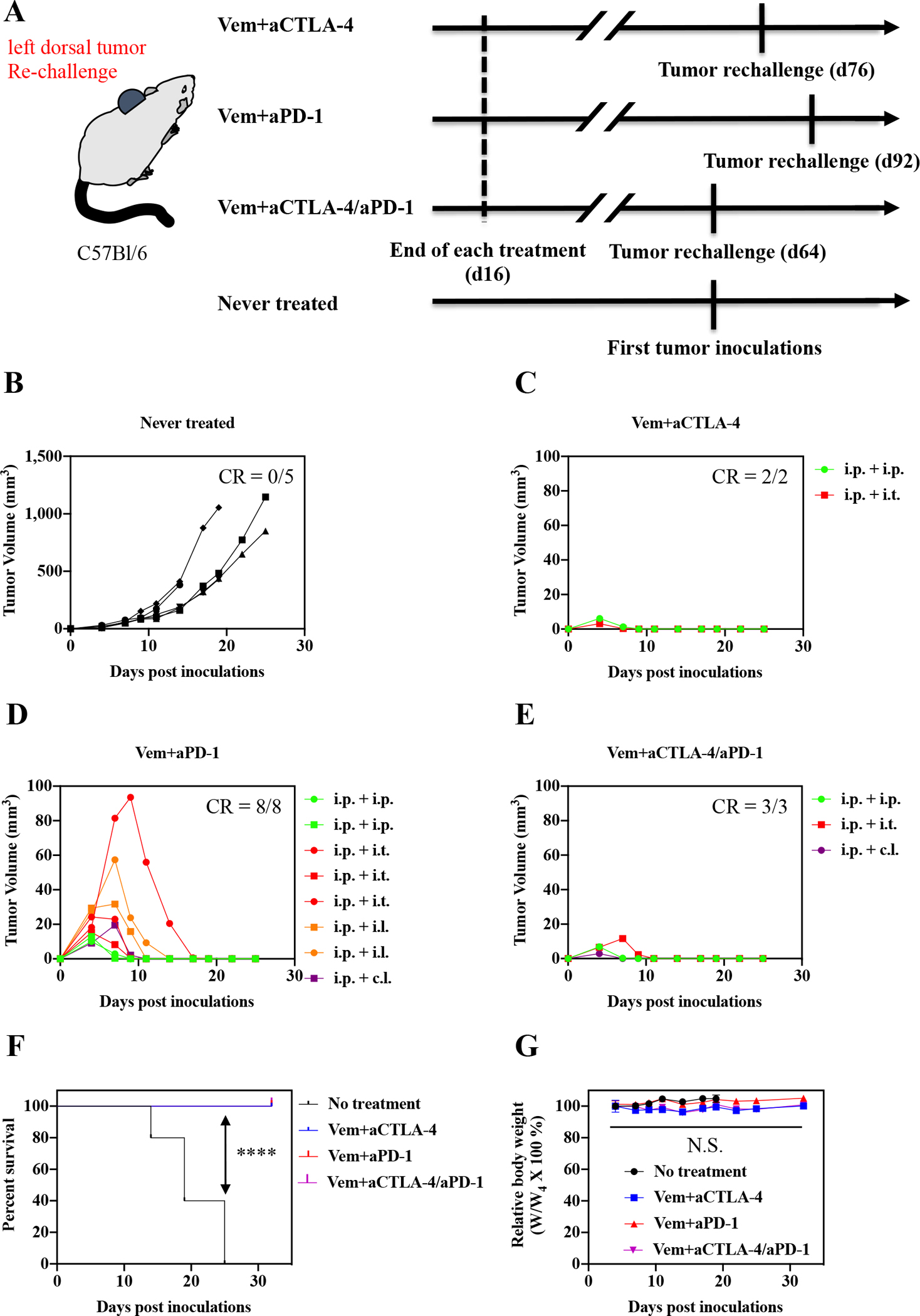
(A) Scheme for tumor re-challenge and treatment schedules. 5×105 D4M cells in 30 μL saline were subcutaneously inoculated to the left dorsal of the mice survived from combined Vem and ICBs therapy. (B) Individual tumor volumes of the mice that had never experienced tumor inoculations and combined Vem and ICBs therapy. (C-E) Individual re-challenged tumor volumes of the mice that survived after having experienced the tumor inoculations on right dorsal and (C) combined Vem (i.p.) and aCTLA-4 (i.p. or i.t.) therapy, (D) combined Vem (i.p.) and aPD-1 (i.p., i.t.,i.l., or c.l.) therapy, or (E) combined Vem (i.p.) and aCTLA-4/aPD-1 (i.p., i.t., or c.l.) therapy. (F) Kaplan–Meier survival curves after re-challenge. (G) Weight changes tumor after re-challenge. Log-rank using Mentel-Cox statistical hypothesis was used for statistical analysis of (F). ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05. Two-way ANOVA using Tukey post-hoc statistical hypothesis was employed for (G) presented with mean±SEM.. N.S. means “Non Significant”.
3.2. Sustained release of Vem and aPD-1 with F127-g-Gelatin thermosensitive hydrogel
Based on the improved synergies of Vem and aPD-1 therapy when administered i.t. in mediating tumor control (Fig. 1–4) and the potential of sustained DDSs to potentiate the effects of immunotherapy [25–32], we hypothesized that a sustained release hydrogel would promote the antitumor effects of i.t. administered Vem and aPD-1. Our group recently reported a thermosensitive hydrogel (F127-g-Gelatin) comprised of biocompatible and renal clearable Pluronic® F127 and biocompatible and biodegradable gelatin [32]. As F127-g-Gelatin hydrogels not only contain hydrophobic polypropylene oxide blocks capable of loading hydrophobic drugs, but also interact with protein drugs [32], the F127-g-Gelatin formulation was expected to enable the efficient loading and sustained release of Vem and aPD-1, which can lead to durable antitumor chemoimmunotherapy on BRAF-mutated melanomas.
F127-g-Gelatin was synthesized via two-step reaction. Briefly, 4-nitrophenyl chloroformate facilitated the conversion of hydroxyl groups of Pluronic® F127 to nitrophenyl carbonates, a good leaving group. Amine groups of gelatins were reacted with the nitrophenyl carbonates to afford F127-g-Gelatin polymer (Fig. S3). 1H NMR confirmed the successful synthesis of F127-g-Gelatin polymer that is comprised of 61.6 ± 3.1 % F127 and 38.4 % gelatin in mass ratio (Fig. S4). The F127-g-Gelatin polymer behaved as a thermosensitive hydrogel with a phase-transition from solution to hydrogel at 4.0–7.0 wt.% aqueous solution when its temperature was increased to 37°C [32]. Scanning electron microscopy (SEM) images of 4.5 wt.% hydrogel sampled by incubation at 37°C, rapid-freeze with liquid nitrogen, and lyophilization (Fig. 5A) showed the sheet-like structures capable of entrapping small molecule and biomolecular solutes to facilitate their release via diffusion.
Fig. 5. In vitro residence stability and drug release behavior of F127-g-Gelatin hydrogels and in vivo biodistribution of aPD-1 released from F127-g-Gelatin thermosensitive hydrogels.
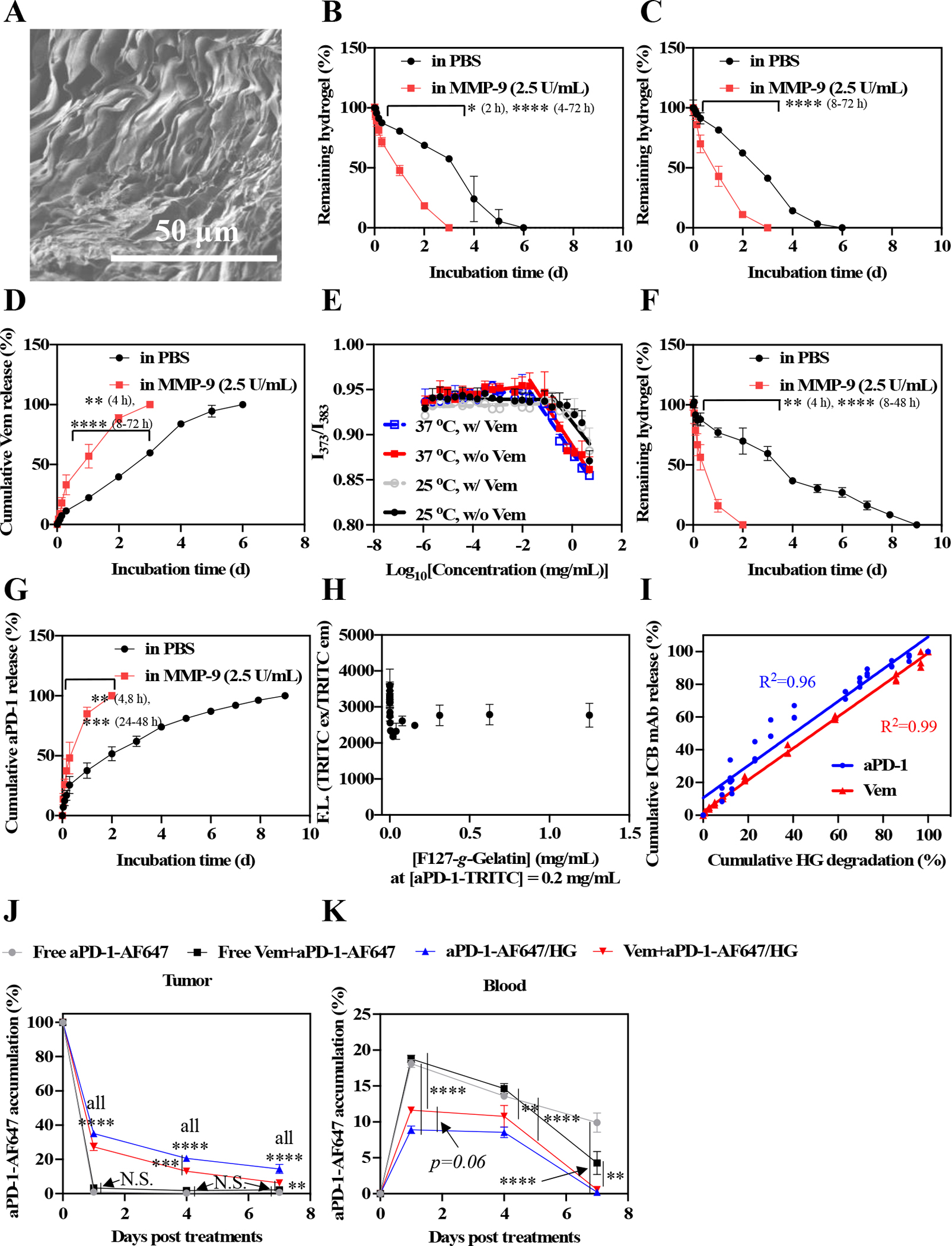
(A) Dry SEM image of dry 4.5 wt.% F127-g-Gelatin hydrogel. (B) In vitro residence stability of 4.5 wt.% bare F127-g-Gelatin hydrogel (n=3–4). (C) In vitro residence stability of 4.5 wt.% F127-g-Gelatin hydrogel (300 μL) containing Vem (20 mg mL−1 in the F127-g-Gelatin hydrogel) (n=4). (D) In vitro Vem release behaviors from 4.5 wt.% F127-g-Gelatin hydrogel (300 μL) containing Vem (20 mg mL−1 in the F127-g-Gelatin hydrogel) (n=4). (E) CMC measurement of F127-g-Gelatin at 25 °C and 37 °C with (w/) or without (w/o) Vem. CMC is the intersection of two distinctive linear lines determined by ratiometric fluorescence (373 nm/383 nm) of pyrenes. (F) In vitro residence stability of 4.5 wt.% F127-g-Gelatin hydrogel (300 μL) containing aPD-1-TRITC (6.67 mg mL−1 in the F127-g-Gelatin hydrogel) (n=4). (G) In vitro Vem release behaviors from 4.5 wt.% F127-g-Gelatin hydrogel (300 μL) containing aPD-1-TRITC (6.67 mg mL−1 in the F127-g-Gelatin hydrogel) (n=4). (H) F127-g-Gelatin concentration-dependent fluorescence of aPD-1-TRITC (n=3). (I) Correlation graph between hydrogel degradation and Vem/aPD-1-TRITC release (n=3–4). (J,K) In vivo local and systemic biodistribution of aPD-1 released from F127-g-Gelatin thermosensitive hydrogels. Free aPD-1-AF647, free aPD-1-AF647 in the presence of Vem, aPD-1-AF647 with 4.5 wt.% F127-g-Gelatin hydrogel, aPD-1-AF647 with 4.5 wt.% F127-g-Gelatin hydrogel containing Vem (Vem and aPD-1 dose equivalent to 10 mg kg−1 and 100 μg mouse−1, respectively) (n=4) were administered into the tumor established with D4M 5×105 cells in 30 μL saline on day 0. Mice were sacrificed on day 1, 4, and 7. Biodistribution of aPD-1 in (J) tumor, and (K) blood. Data are presented as (A-I) mean±SD or (J,K) mean±SEM. Two-way ANOVA using Tukey post-hoc statistical hypothesis was employed for (B-D,F,G,J,K). ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05. N.S. means “Non Significant”.
The residence stability of the F127-g-Gelatin hydrogel and drug release profiles were investigated. Bare F127-g-Gelatin hydrogels were gradually degraded and disrupted over the course of 6 days when incubated at 37°C (Fig. 5B). High residence stability of F127-g-Gelatin hydrogel without toxicity was also observed in vivo as well (Fig. S5). Due to the enzymatic degradation of gelatin components in F127-g-Gelatin, co-incubation with matrix metalloproteinase-9 (MMP-9) that is overexpressed by various melanomas [32, 38–40] was found to accelerate the degradation of F127-g-Gelatin hydrogels (Fig. 5B). Furthermore, the Vem load negligibly affected the residence stability of F127-g-Gelatin hydrogels (Fig. 5B,C). Reverse-phase gradient HPLC was employed to quantify Vem released from F127-g-Gelatin hydrogels (Fig. S6). Vem was released from F127-g-Gelatin hydrogel in phosphate-buffered saline (PBS) for 6 d in a sustained manner, while MMP-9 accelerated the release of Vem from F127-g-Gelatin hydrogel (Fig. 5D). The critical micellar concentrations (CMCs) of F127-g-Gelatin polymer were detected at 25 °C and 37 °C (Fig. 5E, Table S4), indicating the presence of hydrophobic interactions potentially between Vem and F127-g-Gelatin polymer. However, pyrene-assisted CMC of F127-g-Gelatin was not changed by Vem (Fig. 5E), correlating with the insignificant effects of Vem on residence stability of F127-g-Gelatin hydrogel (Fig. 5B,C). TRITC labeled aPD-1 (aPD-1-TRITC) was used to investigate the effects of aPD-1 on the residence stability and the aPD-1 release profiles. Like Vem loaded hydrogels, the residence stability and drug release behaviors of aPD-1-TRITC loaded F127-g-Gelatin hydrogels were altered by coincubation with MMP-9 (Fig. 5F,G). However, aPD-1-TRITC loaded F127-g-Gelatin hydrogels demonstrated significantly prolonged residence stability compared to bare and Vem loaded hydrogels (Fig. 5F), which affected aPD-1 release behaviors (Fig. 5G). Interestingly, fluorescence of aPD-1-TRITC sharply dropped and was then restored with increasing F127-g-Gelatin concentration (Fig. 5H). This phenomenon is attributed to the self-quenching effects of fluorophores that are aggregated or clustered to form dark dimers in the nanoparticles and polymers [41], implying the interactions of aPD-1 and F127-g-Gelatin polymers. Consistent with the expected interactions between drugs and F127-g-Gelatin polymer, drug release correlated with hydrogel degradation rate (Fig. 5I). In summary, F127-g-Gelatin hydrogel not only allowed the load of Vem and aPD-1, but also facilitated sustained drug release that was hydrogel controlled.
3.3. In vivo biodistribution of aPD-1 with the hydrogel on D4M tumor models
To explore hydrogel effects on drug biodistribution, 4.5 wt.% F127-g-Gelatin hydrogels loaded with AlexaFluor™647 labeled aPD-1 (aPD-1-AF647) at 100 μg mouse−1 were injected into day 7 D4M melanomas and fluorescence quantified in the tumor as well as systemic tissues over time (Fig. 5J,K, Fig. S7). F127-g-Gelatin hydrogels resulted in enhanced and sustained retention of aPD-1 in the tumor (Fig. 5J) even 7 d after the administration, effects accompanied by reduced systemic exposure of aPD-1 in the spleen (Fig. S7A), Liver (Fig. S7B), kidney (Fig. S7C), lung (Fig. S7D), and blood (Fig. 5K), compared to i.t. bolus delivery of aPD-1-AF647. Interestingly, Vem significantly reduced tumor retention of aPD-1 (Fig. 5J), which may be ascribed to the improved perfusion of tumors associated with BRAFi and thus clearance of released drug [42]. Nevertheless, F127-g-Gelatin hydrogels imparted the significantly higher accumulation of aPD-1 in the tumor with significantly lower systemic exposure than bolus delivery in the absence and presence of Vem, which raised an expectation of durable antitumor chemo-immunotherapy.
3.4. In vivo tumor therapy and immune profiles of the hydrogel containing Vem and aPD-1 on D4M tumor models
The therapeutic potential of F127-g-Gelatin hydrogels to potentiate the combination of Vem and aPD-1 was assessed in the D4M tumor model (Fig. 6). In contrast with the combination of daily i.p. administration of Vem (each 10 mg kg−1, total 10 times = total 100 mg kg−1) with i.t. aPD-1 every 3 days (each 150 μg mouse−1, total 3 times = total 450 μg mouse−1) (Fig. 2), a single i.t. administration (Fig. 6A) of free Vem (20 mg kg−1) and free aPD-1 (300 μg mouse−1) failed to achieve tumor control (Fig. 6B). However, a single i.t. administration of Vem (20 mg kg−1) and aPD-1 (300 μg mouse−1) formulated within the F127-g-Gelatin hydrogel achieved more potent tumor control with 60% partial response (PR), defined as a 30% decrease in tumor size after treatment, compared to treatment with saline (0% PR), unloaded (drug-free) hydrogel (HG) (0% PR), and bolus delivery of the drugs in combination (0% PR) (Fig. 6C). In addition, the hydrogel formulations negligibly affected levels of alanine aminotransferase(ALT)/aspartate aminotransferase(AST) (Fig. 6D) and body weight (Fig. 6E), supportive of F127-g-Gelatin’s biocompatibility. Furthermore, the enhanced antitumor effects with F127-g-Gelatin hydrogels allowed significantly prolonged animal survival, compared to bolus delivery (Fig. 6F, Table S5). These results demonstrate the utility of hydrogel-mediated sustained release to impart therapeutic benefit without the need for repeated dosing, yet this represents just one approach among many conceivable strategies for combination therapies enabled by this drug delivery system. Further optimization studies to explore best combinational dose for each drug, decoupling of drugs (a single injection of drugs vs. separate injection of each drug), and drug formulation (Vem in hydrogel + free aPD-1 vs. Free Vem + aPD-1 in hydrogel vs. Vem+aPD-1 in hydrogel, Vem in hydrogel + aPD-1 in hydrogel) would pave the way for improving clinical benefits of Vem and aPD-1 therapy in the treatment of BRAF-mutated tumors.
Fig. 6. Antitumor effects and tumoral immune profiles of combined Vem and aPD-1 with F127-g-Gelatin hydrogel on D4M models.
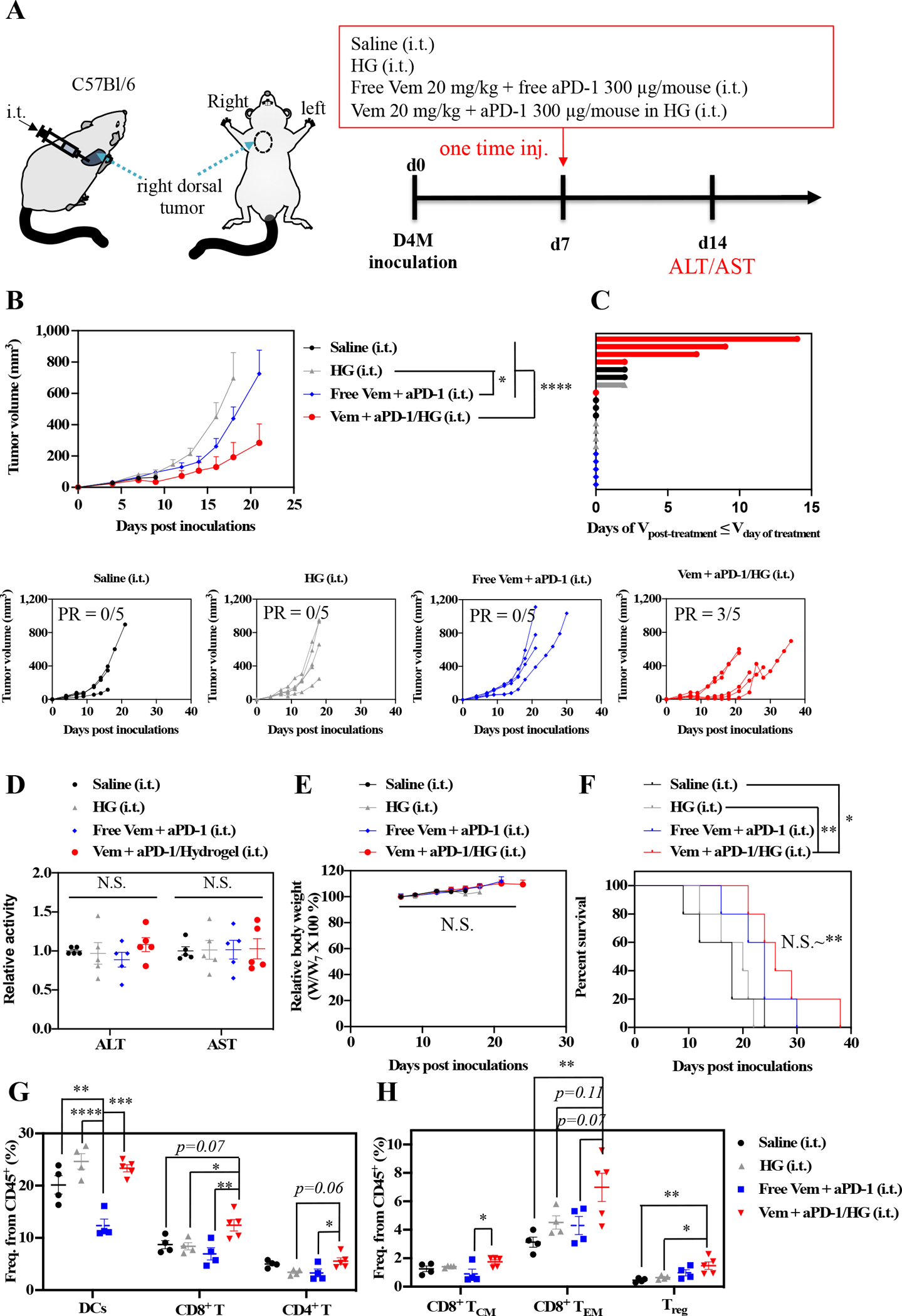
(A) Scheme for administration routes and treatment schedule. 5×105 D4M cells in 30 μL saline were subcutaneously inoculated in C57Bl/6 on day 0. Mixture of Vem (20 mg kg−1) and aPD-1 (300 μg mouse−1) in saline (30 μL) or F127-g-Gelatin hydrogel (30 μL) was administered i.t. on day 7. (B) Average and individual tumor volumes (n=5). (C) Swimmer plot to present therapeutic response, which depicts how long that mouse’s tumor growth curve remained flat or lower than its initial volume at time of treatment (n=5). (D) ALT/AST results from blood serum (n=5). (E) Weight changes after treatment (n=5). (F) Kaplan–Meier survival curves (n=5). (G,H) Profiles of DCs and T cells in tumor on day 14 (n=4–5). Frequency of (G) CD45+CD11c+ (DCs), CD45+CD3+CD8+ (CD8+ T) cells, CD45+CD3+CD4+ (CD4+ T), (H) CD45+CD3+CD8+CD62L+CD44+ (central memory CD8+ T cells, CD8+ TCM), CD45+CD3+CD8+CD62L−CD44+ (Effector memory CD8+ T cells, CD8+ TEM), and CD45+CD3+CD4+Foxp3+ (Treg) cells. Data are presented as mean±SEM. Data are presented as mean±SEM. Two-way ANOVA using Tukey post-hoc statistical hypothesis was employed for (B) and (E). One-way ANOVA using Tukey post-hoc statistical hypothesis was employed for (D), (G), and (H). Log-rank using Mentel-Cox statistical hypothesis was used for survival (F). ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05. N.S. means “Non Significant”. Statistical values for (E) are listed in Table S5.
Immune profiles day 14 post-treatment were assessed (Fig. S8, Table S6) revealing negligible effects of combined Vem and aPD-1 therapy on T lymphocyte responses in lymphoid tissues, but a reduced DC frequency in the spleen (Fig. S9). However, delivery in the F127-g-Gelatin hydrogels significantly increased the infiltration of CD8+ T cells into treated tumors compared to treatment with saline, unloaded HG, and bolus delivery of Vem and aPD-1 in combination (Fig. 6G). Intratumoral frequencies of CD8+ TCM and CD8+ TEM were also significantly increased by treatment of Vem and aPD-1 in combination when formulated in the HG but not in their free form (Fig. 6H). On the other hand, only subtle increases in CD4+ T (Fig. 6G) and Treg (Fig. 6H) cell frequencies were found in tumors treated with Vem and aPD-1 in combination formulated in the HG. The combination therapy of Vem with aPD-1 formulated within the F127-g-Gelatin hydrogel that results in improved tumor control and animal survival is associated with expansion of memory CD8+ T cells within the tumor.
3.5. Dose-dependent in vivo tumor therapy with the hydrogel containing Vem and aPD-1
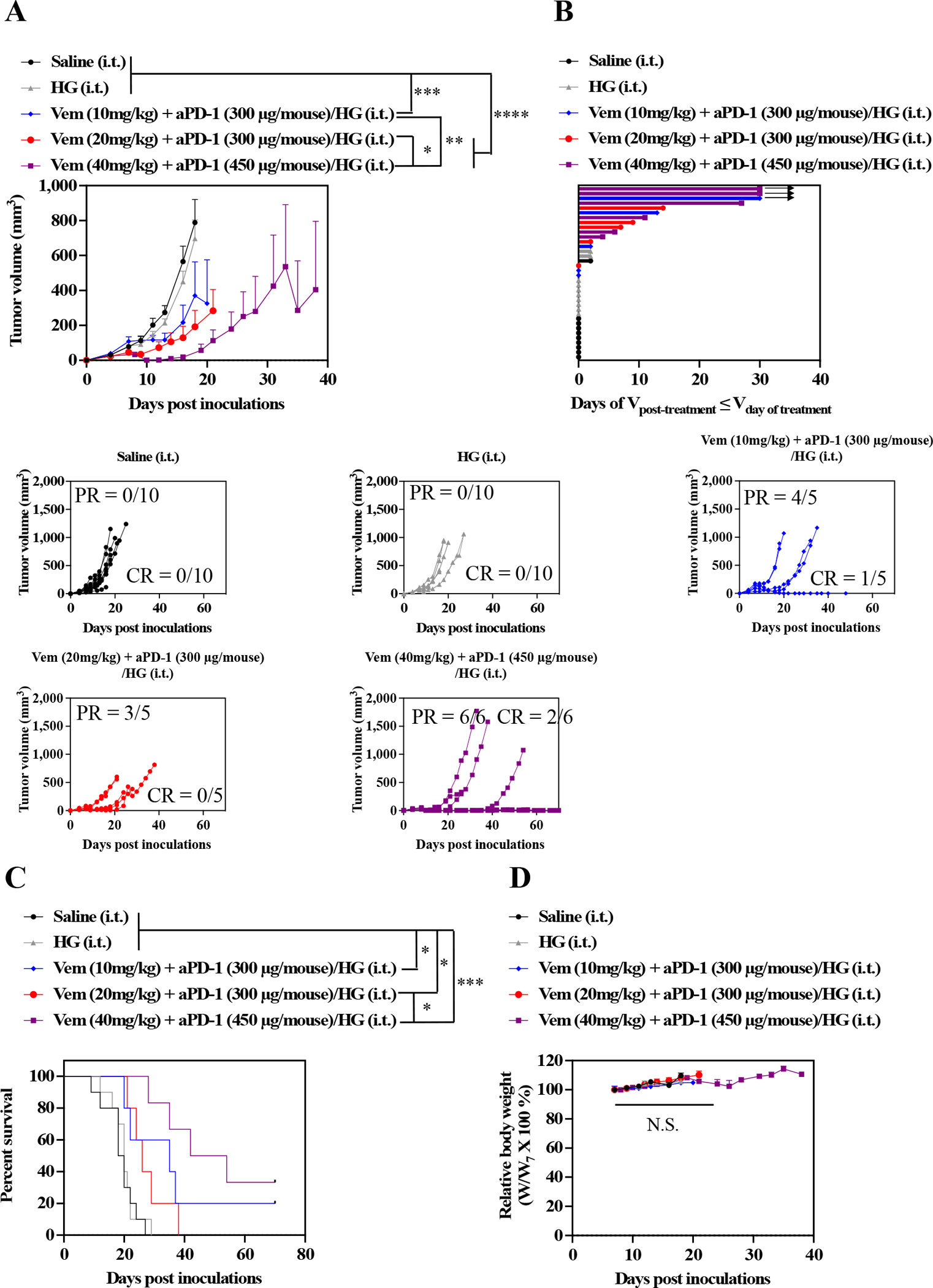
Dose-dependent anticancer therapeutic effects of combined Vem (10, 20, or 40 mg kg−1) and aPD-1 (300, or 450 μg mouse−1) formulated in F127-g-Gelatin hydrogels were investigated (Fig. 7, Table S7). As expected, increasing the Vem dose also achieved higher rates of tumor regression (Fig. 7A) and PR/CR (80% PR/20 % CR in Vem 10 mg kg−1 + aPD-1 300 μg mouse−1, 60% PR/0 % CR in Vem 20 mg kg−1 + aPD-1 300 μg mouse−1, and 100% PR/33 % CR in Vem 40 mg kg−1 + aPD-1 450 μg mouse−1) (Fig. 7B), which resulted in significantly prolonged survival of the mice (Fig. 7C, Table S7) with negligible body weight changes (Fig. 7D).
Fig. 7. Dose-dependent antitumor effects of combined Vem and aPD-1 with F127-g-Gelatin hydrogel on D4M models.
(A) Average and individual tumor volumes (n=5–10). (B) Swimmer plot to present therapeutic response, which depicts how long that mouse’s tumor growth curve remained flat or lower than its initial volume at time of treatment (n=5 or 10). Arrow indicates complete responder, which continues to have low tumor volume for remainder of measurement days. (C) Kaplan–Meier survival curves (n=5–10). (D) Weight changes after treatment (n=5–10). Data are presented as mean±SEM. Two-way ANOVA using Tukey post-hoc statistical hypothesis was employed for (A) and (D). Log-rank using Mentel-Cox statistical hypothesis was used for survival (C). ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05. N.S. means “Non Significant”. Statistical values for (C) are listed in Table S7.
3.6. In vivo tumor therapy with the hydrogel containing Vem and aPD-1 on SM1 tumor models
The therapeutic benefit of F127-g-Gelatin hydrogels on combined Vem and aPD-1 chemoimmunotherapy was further demonstrated in the SM1 model of BRAF mutated melanoma (Fig. 8). Like the D4M model, this melanoma line was established from the inducible BRAFv600E mouse model and is moderately sensitive to Vem due to the CD-KN2A gene deletion and BRAF and MITF gene amplification in addition to the BRAFV600E mutation [43,44]. SM1 cells were passaged in NSG mice, harvested and subcutaneously inoculated to the right dorsal skin of C57Bl/6 animals. Single i.t. administration of Vem (40 mg kg−1) and aPD-1 (450 μg mouse−1) in combination when formulated within the F127-g-Gelatin hydrogel led to prolonged tumor control than bolus delivery of free drug without any bodyweight changes (Fig. 8A–C, Fig. S10, Table S8), with a PR of 50 % and CR of 25 %. These results (Fig 2, Fig. 6–8, Fig. S2B, Fig. S10) fortify the F127-g-Gelatin thermosensitive hydrogel as improving the therapeutic index, achieving dose sparing, and reducing number of injections required to treat BRAF-mutated tumors using BRAFi and aPD-1 in combination.
Fig. 8. Antitumor effects of combined Vem and aPD-1 with F127-g-Gelatin hydrogel on SM1 models.
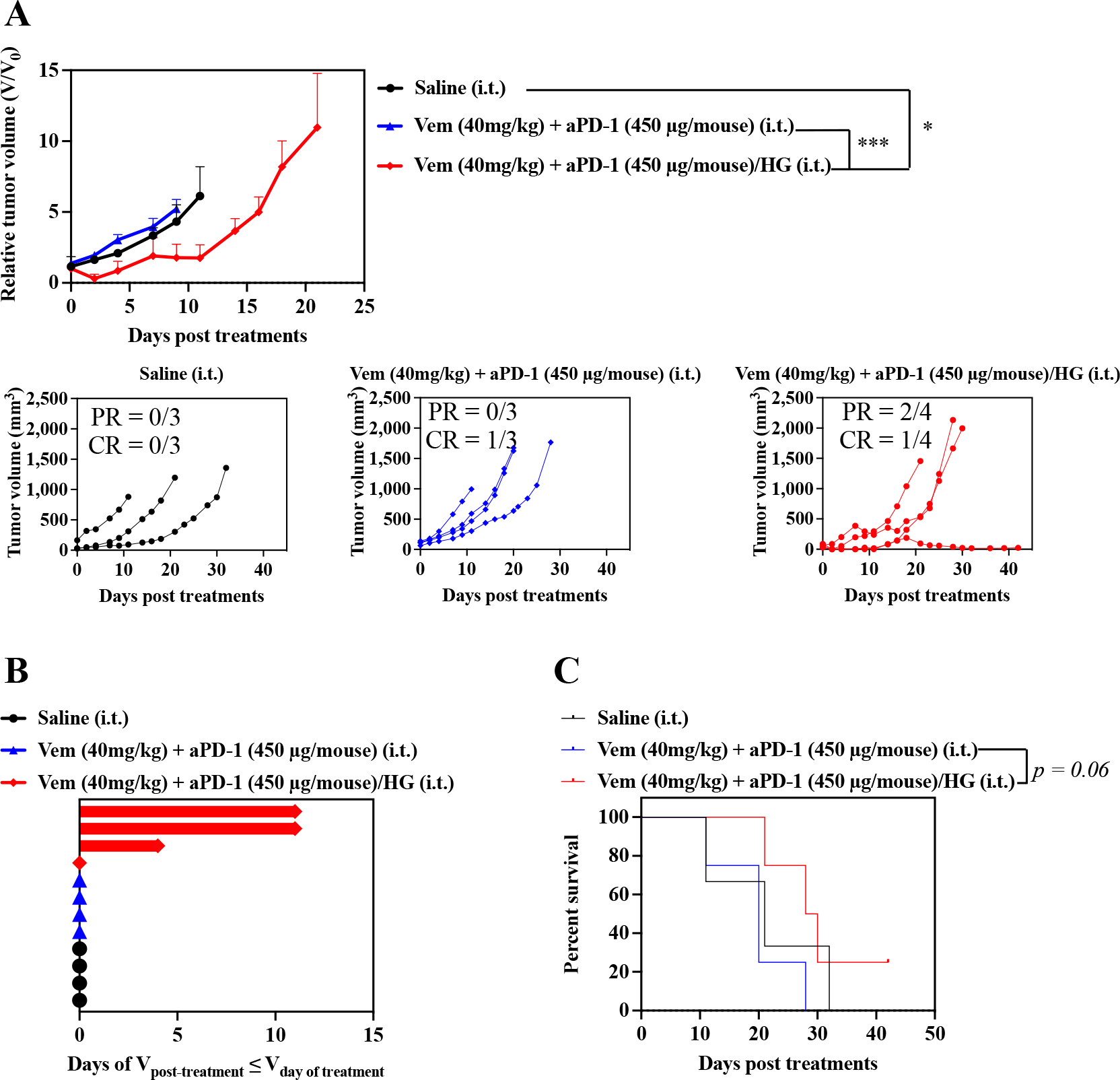
SM1 cells were passaged in NSG mice. After harvested and resuspended in 160 μL saline, 30 μL of SM1 cell solutions were subcutaneously inoculated in C57Bl/6. Treatments were started when SM1 tumor size reached to 5–10 mm. Mice treated with saline, or combined single i.t. administrations of Vem (40 mg kg−1) and aPD-1 (450 μg mouse−1) were employed as control groups. F127-g-Gelatin hydrogels containing combined Vem (40 mg kg−1) and aPD-1 (450 μg mouse−1) were i.t. administered one time. (A) Relative average and individual tumor volumes (n=3–4). (B) Swimmer plot to present therapeutic response, which depicts how long that mouse’s tumor growth curve remained flat or lower than its initial volume at time of treatment (n=3–4). (C) Kaplan–Meier survival curves (n=3–4). Data are presented as mean±SEM. Twoway ANOVA using Tukey post-hoc statistical hypothesis was employed for (A). Log-rank using Mentel-Cox statistical hypothesis was used for survival (C). ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05. Statistical values for (C) are listed in Table S8.
4. Conclusion
In summary, therapeutic synergies of Vem with ICB for the treatment of BRAF-mutated melanoma were achieved by targeting aPD-1 and focusing drug actions to directly within the tumor microenvironment through locoregional delivery. The benefits of this sustained release formulation were substantial with respect to achieving tumor control, increasing the rate of response, immunomodulation, and prolonging animal survival. Our findings, fortified in multiple murine tumor models, demonstrate the potential of F127-g-Gelatin hydrogels for sustained locoregional chemoimmunotherapy drug delivery in the treatment of BRAF-mutated cancer.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health (NIH) grants R01CA207619 (S.N.T.), R01CA247484 (S.N.T.), and S10OD016264, and Department of Defense Grant CA150523 (S.N.T.). M.P.M. was supported by a National Science Foundation Graduate Research Fellowship.
Footnotes
Declaration of competing interest
J.K. and S.N.T. are inventors on a submitted patent related to the technology described in this manuscript.
CRediT authorship contribution statement
Jihoon Kim: Conceptualization, Methodology, Investigation, Formal analysis, Visualization, Writing - Original draft, Review & Editing. Paul A. Archer: Investigation, Formal analysis, Writing - Review & Editing. Margaret P. Manspeaker: Investigation, Formal analysis. Alexa R. C. Avecilla: Investigation, Formal analysis. Brian P. Pollack: Conceptualization, Writing – Review & Editing. Susan N. Thomas: Conceptualization, Supervision, Project administration, Funding acquisition, Writing – Review & Editing.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.biomaterials.xxxx.xxxxxx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P, Vemurafenib: the first drug approved for BRAF-mutant cancer, Nat. Rev. Drug Discov. 11 (2012) 873–886, 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- [2].Salama AKS, Flaherty KT, BRAF in Melanoma: Current Strategies and Future Directions, Clin. Cancer Res. 19 (2013) 4326–4334, 10.1158/1078-0432.CCR-13-0779 [DOI] [PubMed] [Google Scholar]
- [3].Vultur A, Villanueva J, Herlyn M, Targeting BRAF in Advanced Melanoma: A First Step toward Manageable Disease, Clin. Cancer Res. 17 (2011) 1658–1663, 10.1158/1078-0432.CCR-10-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Morkel M, Riemer P, Bläker H, Sers C, Similar but different: distinct roles for KRAS and BRAF oncogenes in colorectal cancer development and therapy resistance, Oncotarget 6 (2015) 20785–20800, 10.18632/oncotarget.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Poulikakos PI, Rosen N, Mutant BRAF Melanomas—Dependence and Resistance, Cancer Cell 19 (2011) 11–15, 10.1016/j.ccr.2011.01.008 [DOI] [PubMed] [Google Scholar]
- [6].Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung W, Bravo BJ, Giannetti AM, Peck A, Masselot A, Morales T, Smith D, Brandhuber BJ, Hymowitz SG, Malek S, Structure of the BRAF-MEK Complex Reveals a Kinase Activity Independent Role for BRAF in MAPK Signaling, Cancer cell 26 (2014) 402–413, 10.1016/j.ccr.2014.07.007. [DOI] [PubMed] [Google Scholar]
- [7].Fedorenko IV, Paraiso KHT, Smalley KSM, Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma, Biochem. Pharmacol. 82 (2011) 201–209, 10.1016/j.bcp.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD, Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance, Nature 494 (2013) 251–255, 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Luebker SA, Koepsell SA , Diverse Mechanisms of BRAF Inhibitor Resistance in Melanoma Identified in Clinical and Preclinical Studies, Front. Oncol. 9 (2019) 268, 10.3389/fonc.2019.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Monsma DJ, Cherba DM, Eugster EE, Dylewski DL, Davidson PT, Peterson CA, Borgman AS, Winn ME, Dykema KJ, Webb CP, MacKeigan JP, Duesbery NS, Nickoloff BJ, Monks NR, Melanoma patient derived xenografts acquire distinct Vemurafenib resistance mechanisms, Am. J. Cancer Res. 5 (2015) 1507–1518. [PMC free article] [PubMed] [Google Scholar]
- [11].Yu C, Liu X, Yang J, Zhang M, Jin H, Ma X, Shi H, Combination of Immunotherapy With Targeted Therapy: Theory and Practice in Metastatic Melanoma, Front. Immunol. 10 (2019) 990, 10.3389/fimmu.2019.00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, Lo JA, Hodi FS, Freeman GJ, Bosenberg MW, McMahon M, Flaherty KT, Fisher DE, Sharpe AH, Wargo JA, Response to BRAF Inhibition in Melanoma Is Enhanced When Combined with Immune Checkpoint Blockade, Cancer Immunol. Res. 2 (2014) 643–654, 10.1158/2326-6066.CIR-13-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Steinberg SM, Zhang P, Malik BT, Boni A, Shabaneh TB, Byrne KT, Mullins DW, Brinckerhoff CE, Ernstoff MS, Bosenberg MW, Turk MJ, BRAF Inhibition Alleviates Immune Suppression in Murine Autochthonous Melanoma, Cancer Immunol. Res. 2 (2014) 1044–1050, 10.1158/2326-6066.CIR-14-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, Peng W, Sullivan RJ, Lawrence DP, Hodi FS, Overwijk WW, Lizee G, Murphy GF, Hwu P, Flaherty KT, Fisher DE, Wargo JA, BRAF Inhibition Is Associated with Enhanced Melanoma Antigen Expression and a More Favorable Tumor Microenvironment in Patients with Metastatic Melanoma, Clin. Cancer Res. 19 (2013) 1225–1231, 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bellmann L, Cappellano G, Schachtl-Riess JF, Prokopi A, Seretis A, Ortner D, Tripp CH, Brinckerhoff CE, Mullins DW, Stoitzner P, A TLR7 agonist strengthens T and NK cell function during BRAF-targeted therapy in a preclinical melanoma model, Int. J. Cancer 146 (2020) 1409–1420, 10.1002/ijc.32777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Peiffer L, Farahpour F, Sriram A, Spassova I, Hoffmann D, Kubat L, Stoitzner P, Gambichler T, Sucker A, Ugurel S, Schadendorf D, Becker JC, BRAF and MEK inhibition in melanoma patients enables reprogramming of tumor infiltrating lymphocytes, Cancer Immunol. Immunother. 70 (2021) 1635–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Romero D, Time for adjuvant vemurafenib?, Nat. Rev. Clin. Oncol. 15 (2018) 265, 10.1038/nrclinonc.2018.39. [DOI] [PubMed] [Google Scholar]
- [18].Menaa F, Latest Approved Therapies for Metastatic Melanoma: What Comes Next?, J. Skin Cancer (2013) 735282, 10.1155/2013/735282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J, Hepatotoxicity with combination of vemurafenib and ipilimumab, N. Engl. J. Med. 368 (2013) 1365–1366, 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- [20].Sullivan RJ, Hamid O, Gonzalez R, Infante JR, Patel MR, Hodi FS, Lewis KD, Tawbi HA, Hernandez G, Wongchenko MJ, Chang Y, Roberts L, Ballinger M, Yan Y, Cha E, Hwu P, Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients, Nat. Med. 25 (2019) 929–935. [DOI] [PubMed] [Google Scholar]
- [21].Borch TH, Harbst K, Rana AH, Andersen R, Martinenaite E, Kongsted P, Pedersen M, Nielsen M, Kjeldsen JW, Kverneland AH, Lauss M, Hölmich LR, Hendel H, Met Ö, Jönsson G, Donia M, Svane IM, ImmunoTher J. Cancer 9 (2021) e002703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zou L, Ding W, Zhang Y, Cheng S, Li F, Ruan R, Wei P, Qiu B, Peptide-modified vemurafenib-loaded liposomes for targeted inhibition of melanoma via the skin, Biomaterials 182 (2018) 1–12, 10.1016/j.biomaterials.2018.08.013. [DOI] [PubMed] [Google Scholar]
- [23].Spengler EK, Kleiner DE, Fontana RJ, Vemurafenib-Induced Granulomatous Hepatitis, Hepatology 65 (2017) 745–748, 10.1002/hep.28692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Launay-Vacher V, Zimner-Rapuch S, Poulalhon N, Fraisse T, Garrigue V, Gosselin M, Amet S, Janus N, Deray G, Acute Renal Failure Associated With the New BRAF Inhibitor Vemurafenib, Cancer 120 (2014) 2158–2163, 10.1002/cncr.28709. [DOI] [PubMed] [Google Scholar]
- [25].Francis DM, Manspeaker MP, Schudel A, Sestito LF, O’Melia MJ, Kissick HT, Pollack BP, Waller EK, Thomas SN, Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy, Sci. Transl. Med. 12 (2020) eaay3775, 10.1126/scitranslmed.aay3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tam HH, Melo MB, Kang M, Pelet JM, Ruda VM, Foley MH, hu JK, Kumari S, Crampton J, Baldeon AD, Sanders RW, Moore JP, Crotty S, Langer R, Anderson DG, Chakraborty AK, Irvine DJ, Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination, Proc. Natl. Acad. Sci. U. S. A. 113 (2016) E6639–E6648, 10.1073/pnas.1606050113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Boopathy AV, Mandal A, Kulp DW, Menis S, Bennett NR, Watkins HC, Wang W, Martin JT, Thai NT, He Y, Schief WR, Hammond PT, Irvine DJ, Enhancing humoral immunity via sustained-release implantable microneedle patch vaccination, Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 16473–16478, 10.1073/pnas.1902179116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kim NW, Kim S-Y, Lee JE, Yin Y, Lee JH, Lim SY, Kim ES, Duong HTT, Kim HK, Kim S, Kim J-E, Lee DS, Kim J, Lee MS, Lim YT, Jeong JH, Enhanced Cancer Vaccination by In Situ Nanomicelle-Generating Dissolving Microneedles, ACS Nano 12 (2018) 9702–9713, 10.1021/acsnano.8b04146. [DOI] [PubMed] [Google Scholar]
- [29].Yu S, Wang C, Yu J, Wang J, Lu Y, Zhang Y, Zhang X, Hu Q, Sun W, He C, Chen X, Gu Z, Injectable Bioresponsive Gel Depot for Enhanced Immune Checkpoint Blockade, Adv. Mater. 30 (2018) 1801527, 10.1002/adma.201801527. [DOI] [PubMed] [Google Scholar]
- [30].Chung CK, Fransen MF, van der Maaden K, Campos Y, Garcia-Couce J, Kralisch D, Chan A, Ossendorp F, Cruz LJ, Thermosensitive hydrogels as sustained drug delivery system for CTLA-4 checkpoint blocking antibodies, J. Control. Release 323 (2020) 1–11, 10.1016/j.jconrel.2020.03.050. [DOI] [PubMed] [Google Scholar]
- [31].Kim J, Francis DM, Thomas SN, In Situ Crosslinked Hydrogel Depot for Sustained Antibody Release Improves Immune Checkpoint Blockade Cancer Immunotherapy, Nanomaterials 11 (2021) 471, 10.3390/nano11020471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kim J, Francis DM, Sestito LF, Archer PA, Manspeaker MP, O’Melia MJ, Thomas SN, Thermosensitive hydrogel releasing nitric oxide donor and anti-CTLA-4 micelles for anti-tumor immunotherapy, Nat. Comm. 13, (2022) 1479, 10.1038/s41467-022-29121-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hooijkaas A, Gadiot J, Morrow M, Stewart R, Schumacher T, Blank CU, Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma, Oncoimmunol. 1 (5) (2012) 609–617, 10.4161/onci.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang R-Y, Francois A, McGray AR, Miliotto A, Odunsi K, Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer, Oncoimmunol. 6 (1) (2017) e1249561, 10.1080/2162402X.2016.1249561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wei SC, Anang N-AAS, Sharma R, Andrews MC, Reuben A, Levine JH, Cogdill AP, Mancuso JJ, Wargo JA, Pe’er D, Allison JP, Combination anti–CTLA-4 plus anti–PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies, Proc. Natl. Acad. Sci. U. S. A. 116 (45) (2019) 22699–22709, 10.1073/pnas.1821218116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Duperret EK, Wise MC, Trautz A, Villarreal DO, Ferraro B, Walters J, Yan J, Khan A, Masteller E, Humeau L, Weiner DB, Synergy of Immune Checkpoint Blockade with a Novel Synthetic Consensus DNA Vaccine Targeting TERT, Mol. Ther. 26 (2) (2018) 435–445, 10.1016/j.ymthe.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pardoll DM, The blockade of immune checkpoints in cancer immunotherapy, Nat. Rev. Cancer 12 (4) (2016) 252–264, 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Su K, Wang C, Recent advances in the use of gelatin in biomedical research, Biotechnol. Lett. 37 (2015) 2139–2145, 10.1007/s10529-015-1907-0. [DOI] [PubMed] [Google Scholar]
- [39].Huang S-C, Ho C-T, Lin-Shiau S-Y, Lin J-K, Carnosol inhibits the invasion of B16/F10 mouse melanoma cells by suppressing metalloproteinase-9 through down-regulating nuclear factor-kappaB and c-Jun, Biochem. Pharmacol. 69 (2005) 221–232, 10.1016/j.bcp.2004.09.019. [DOI] [PubMed] [Google Scholar]
- [40].Lugowska I, Kowalska M, Fuksiewicz M, Kotowicz B, Mierzejewska E, Kosela-Paterczyk H, Szamotulska K, Rutkowski P, Serum markers in early-stage and locally advanced melanoma, Tumor Biol. 36 (2015) 8277–8285, 10.1007/s13277-015-3564-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Swiecicki J-M, Thiebaut F, Di Pisa M, Gourdin-Bertin S, Tailhades J, Mansuy C, Burlina F, Chwetzoff S, Trugnan G, Chassaing G, Lavielle S, How to unveil self-quenched fluorophores and subsequently map the subcellular distribution of exogenous peptides, Sci. Rep. 6 (2016) 20237, 10.1038/srep20237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Comunanza V, Cora D, Orso F, Consonni FM, Middonti E, Di Nicolantonio F, Buzdin A, Sica A, Medico E, Sangiolo D, Taverna D, Bussolino F, VEGF blockade enhances the antitumor effect of BRAFV600E inhibition, EMBO Mol. Med. 9 (2017) 219–237, 10.15252/emmm.201505774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hu-Lieskovan S, Mok S, Moreno BH, Tsoi J, Robert L, Goedert L, Pinheiro EM, Koya RC, Graeber TG, Comin-Anduix B, Ribas A, Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma, Sci. Transl. Med. 7 (2015) 279ra41, 10.1126/scitranslmed.aaa4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, Minasyan A, Graham NA, Graeber TG, Chodon T, Ribas A, BRAF Inhibitor Vemurafenib Improves the Antitumor Activity of Adoptive Cell Immunotherapy, Cancer Res. 72 (2012) 3928–3937, 10.1158/0008-5472.CAN-11-2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.