

Abstract
Epstein–Barr virus (EBV) is an oncogenic herpesvirus associated with several cancers of lymphocytic and epithelial origin1–3. EBV encodes EBNA1, which binds to a cluster of 20 copies of an 18-base-pair palindromic sequence in the EBV genome4–6. EBNA1 also associates with host chromosomes at non-sequence-specific sites7, thereby enabling viral persistence. Here we show that the sequence-specific DNA-binding domain of EBNA1 binds to a cluster of tandemly repeated copies of an EBV-like, 18-base-pair imperfect palindromic sequence encompassing a region of about 21 kilobases at human chromosome 11q23. In situ visualization of the repetitive EBNA1-binding site reveals aberrant structures on mitotic chromosomes characteristic of inherently fragile DNA. We demonstrate that increasing levels of EBNA1 binding trigger dosedependent breakage at 11q23, producing a fusogenic centromere-containing fragment and an acentric distal fragment, with both mis-segregated into micronuclei in the next cell cycles. In cells latently infected with EBV, elevating EBNA1 abundance by as little as twofold was sufficient to trigger breakage at 11q23. Examination of whole-genome sequencing of EBV-associated nasopharyngeal carcinomas revealed that structural variants are highly enriched on chromosome 11. Presence of EBV is also shown to be associated with an enrichment of chromosome 11 rearrangements across 2,439 tumours from 38 cancer types. Our results identify a previously unappreciated link between EBV and genomic instability, wherein EBNA1-induced breakage at 11q23 triggers acquisition of structural variations in chromosome 11.
EBV is an oncogenic herpesvirus detected in various cancers of lymphocytic and epithelial origin1–3. Long-term latent infection in the form of extrachromosomal viral episomes has been widely used to define EBV-associated tumours. Persistence of the EBV genome in the host nucleus is mediated by the viral protein EBNA1, which is known to be expressed in all forms of EBV latency in proliferating cells8. The carboxy-terminal DNA-binding domain of EBNA1 binds a specific 18-base-pair (bp) palindromic sequence tandemly repeated 20 times at the EBV origin of replication4–6. EBNA1 also associates with the human genome through non-sequence-specific binding, thereby physically attaching the EBV genome to host chromosomes7. Several studies have shown that EBNA1 is important for EBV-mediated immortalization of B cells9,10. However, the degree to which EBNA1 directly contributes to tumorigenesis or is simply required for EBV maintenance remains controversial11. Several non-sequence-specific EBNA1-binding sites in the human genome undergo EBNA1-mediated expression changes linked to increased cellular survival12–15. EBNA1 has also been proposed to promote tumorigenesis by inhibiting p53, disrupting promyelocytic leukaemia bodies and inducing oxidative stress11.
To better understand the role of EBNA1 in cellular transformation, we expressed a Flag-tagged allele of full-length EBNA1 in non-EBV-infected human cells (Extended Data Fig. 1a,b). In sharp contrast to the large number of globally distributed EBNA1-binding sites in EBV-infected nuclei12–15, EBNA1 was highly enriched at two punctate foci in the nuclei of non-EBV-infected primary retinal pigment epithelium (RPE) cells as well as tumour-derived DLD1 cells and HeLa cells (Extended Data Fig. 1c). Up to four distinct foci were also observed in pseudo-tetraploid U2OS osteosarcoma cells. Foci formation was specific to the human genome as only diffuse nuclear signal was seen in mouse cells. Focal enrichment of sequence-specific DNA-binding proteins such as EBNA1 probably labels repetitive loci containing an enrichment of the recognition sequence, much like localization of a complex of nuclease-dead Cas9 (dCas9) and single guide RNA (sgRNA) at a repetitive locus containing hundreds of clustered copies of the sgRNA target sequence16,17 (Extended Data Fig. 1d).
As non-EBV-infected cells are not expected to possess the 18-bp palindromic EBNA1 recognition sequence from the EBV genome, we next reasoned that focal enrichment of EBNA1 was mediated by sequence-specific binding to a cluster of EBV-like 18-bp palindromic sequences in the endogenous human genome. We expressed truncation alleles of EBNA1 that harboured or lacked its sequence-specific DNA-binding domain18 (Fig. 1a,b). As predicted, whereas localization of EBNA1(ΔGAGR) lacking the unrelated glycine-alanine and glycine-arginine repeat domains was indistinguishable from that of full-length EBNA1, deletion of the DNA-binding domain (EBNA1(ΔDBD)) abolished appearance of the single pair of punctate foci, producing only diffuse nuclear signal (Fig. 1c,d). Thus, our data strongly suggest that there is a cluster of EBV-like 18-bp palindromic sequences in the endogenous human genome (Fig. 1e).
Fig. 1 |. Sequence-specific DNA-binding domain mediates enrichment of EBNA1 at a single genomic locus in the endogenous human genome.
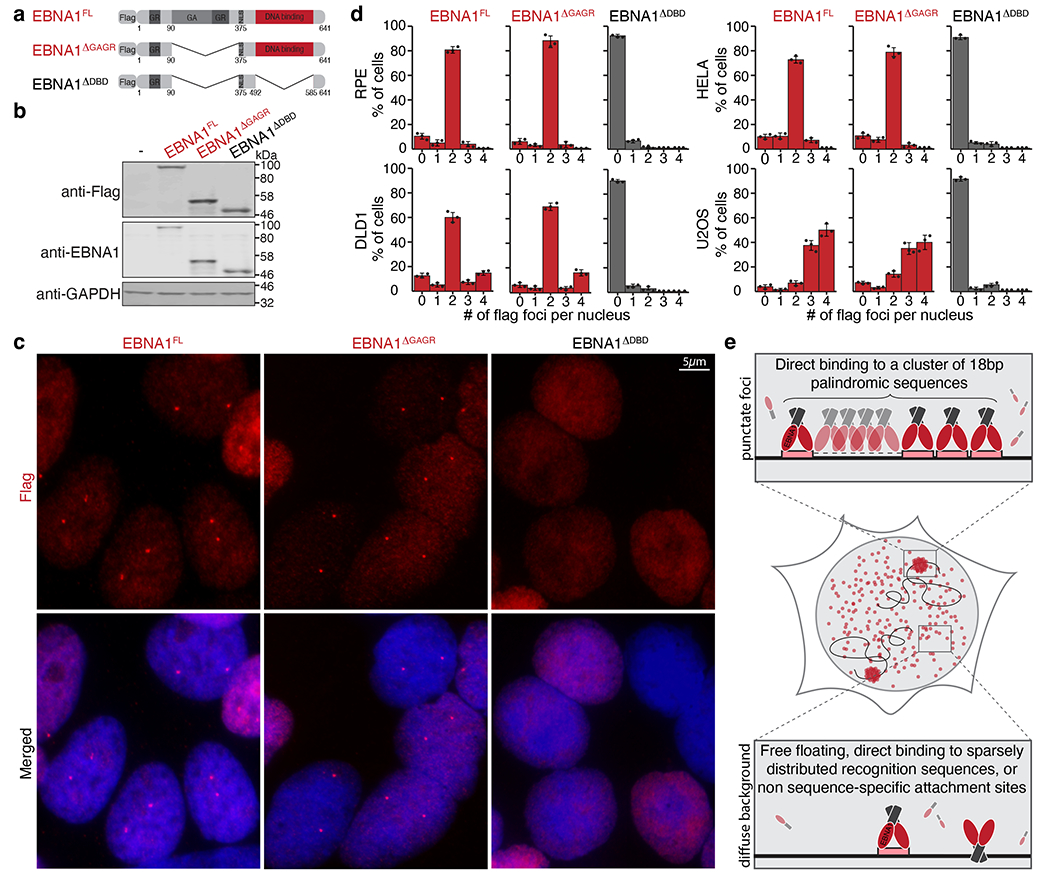
a, Schematic representation of the Flag-tagged alleles of EBNA1. GR, glycine– arginine-rich region; GA, glycine–alanine repeat region; NLS, nuclear localization signal. b, Representative immunoblots of cells expressing the indicated Flag-tagged alleles of EBNA1. For gel source data, see Supplementary Fig. 1. c, Representative anti-Flag (red) immunofluorescence micrographs showing localization of the alleles in DLD1 cells. Scale bar, 5 μm. d, Quantification of localization of the alleles of EBNA1 in the indicated cell lines either at day 1 following transient expression in RPE, HeLa and U2OS cells, or at day 2 following stable transduction in DLD1 cells. A total of 908 RPE cells, 902 DLD1 cells, 901 HeLa cells and 893 U2OS cells were counted. Data are presented as mean values Å} s.e.m. from three independent experiments. e, Schematic representation of the hypothesis that EBNA1 foci formation is mediated by sequence-specific enrichment through the DNA-binding domain (red) at a recognition sequence (pink) clustered at a repetitive site in the human genome.
We next searched the previously reported 903 EBNA1-binding sites identified from genome-wide chromatin immunoprecipitation sequencing12 in cells latently infected with EBV. We found that one particular binding site appears as a cluster at chromosome 11q23. Indeed, when we examined the corresponding 30-bp chromatin immunoprecipitation sequencing binding motif, we recognized that seven nucleotides (5′-GGGTAAC-3′) of the binding motif resemble half of the 18-bp palindromic EBNA1 recognition sequence (5′-GGGTAGCATATGCTACCC-3′) from the EBV genome5. Remarkably, using human genome assembly 38 (GRCh38), we found that 11q23 contains a cluster of regularly interspersed variants of the EBV 18-bp palindromic sequence with a central non-palindromic 4-bp core (Fig. 2a–f). Indeed, genome-wide search for the human 18-bp imperfect palindrome revealed only 1 cluster in the human genome located at 11q23 with an 188-fold enrichment compared to baseline copies across the genome (P value: 3.4 Å~ 10−5; Extended Data Fig. 2a). When we examined available long-read (about 25 kb)-sequenced genomes of two individuals of Ashkenazi descent and two individuals of Chinese descent, we found a difference of about 2.6-fold in repeat copy number between the two groups (Extended Data Fig. 2b,c), indicative of genetic polymorphism in the human population. Interspecies alignment revealed that this repetitive site is evolutionarily conserved among the great apes (Extended Data Fig. 3a,b).
Fig. 2 |. A cluster of regularly interspersed copies of an 18-bp imperfect palindromic sequence at 11q23 mediates sequence-specific enrichment of EBNA1.
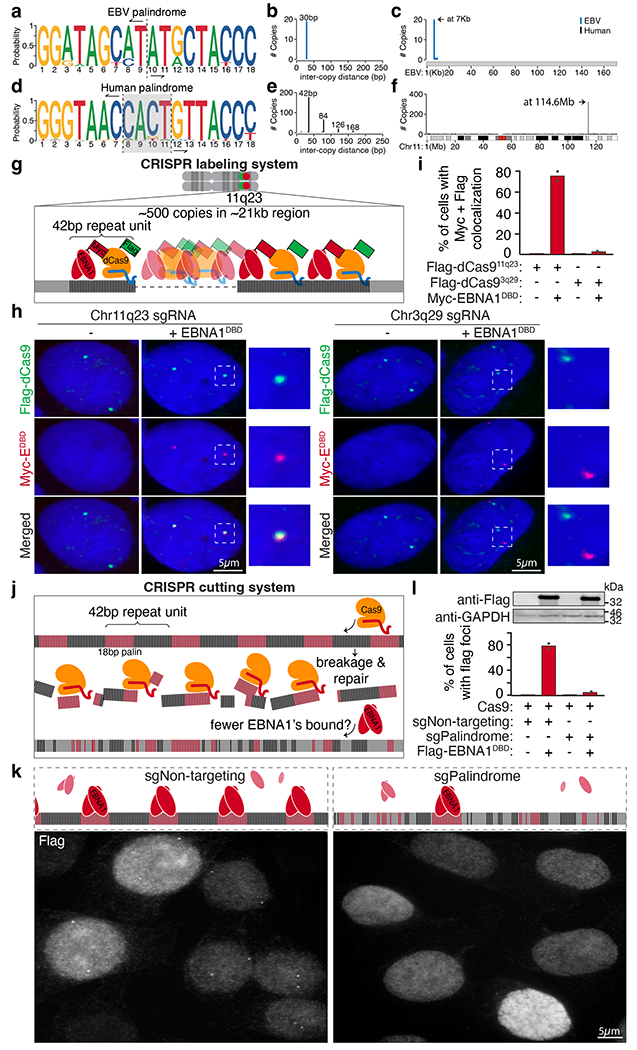
a–c, Consensus motif logo (a), inter-copy distance (b) and position (c) of 18-bp palindromic repeats in the EBV genome. d–f, Consensus motif logo (d), inter-copy distance (e) and position (f) of 18-bp imperfect palindromic repeats with up to 6 variant and 2 asymmetric nucleotides on human chromosome 11. Vertical dashed lines in the consensus motif logos represent the point of symmetry of the palindromes. The grey shaded area highlights the 4-bp non-palindromic core of the human palindrome. g, Schematic representation of the CRISPR labelling system. h, Representative anti-Flag (green) and anti-MYC (red) immunofluorescence micrographs showing localization of Flag–dCas9 directed by the indicated sgRNAs labelling either 11q23 or 3q29 with and without expression of MYC–EBNA1(DBD). Scale bars, 5 μm. i, Quantification of staining in h. A total of 95 cells were counted for Flag–dCas9 labelling 11q23. A total of 110 cells were counted for Flag–dCas9 labelling 3q29. Data are presented as bars representing mean values from two independent experiments. j, Schematic representation of the CRISPR cutting system. k, Representative anti-Flag immunofluorescence micrographs showing localization of Flag–EBNA1(DBD) in cells treated with either non-targeting sgRNA (sgNon-targeting) or palindrome-targeting sgRNA (sgPalindrome). Scale bar, 5 μm. For gel source data, see Supplementary Fig. 1. l, Quantification of staining in k. A total of 410 cells were counted for Cas9-mediated cutting with non-targeting sgRNA. A total of 630 cells were counted for Cas9-mediated cutting with palindrome-targeting sgRNA. Data are presented as bars representing mean values from two independent experiments.
Using a CRISPR labelling system (Fig. 2g and Extended Data Fig. 4a), we validated that MYC–EBNA1(DBD) foci colocalized with Flag-tagged dCas9 foci targeting a portion of the 42-bp repeat sequence at 11q23, but not with Flag–dCas9 foci targeting a separate repetitive site at 3q29 (Fig. 2h–l and Extended Data Fig. 4c,d; ref. 16). We next examined whether a CRISPR cutting system to eliminate the 18-bp palindromic sequence would abolish formation of EBNA1 foci (Fig. 2j and Extended Data Fig. 4b). Whereas transient treatment with Cas9 and non-targeting sgRNA did not reduce the percentage of cells producing EBNA1 foci relative to the parental cells (Fig. 1d), treatment with Cas9 and palindrome-targeting sgRNA sharply reduced the frequency of cells with foci (Fig. 2k,l and Extended Data Fig. 4e,f), consistent with most cells having lost most or all of the 18-bp palindromic sequences. Together, these CRISPR-based efforts demonstrate that the sequence-specific EBNA1 foci in the endogenous human genome are precisely positioned at the cluster of regularly interspersed copies of 18-bp imperfect palindromic sequences at 11q23. This is consistent with previous findings that mutations at the central 4-bp core of the 18-bp palindrome were well tolerated for sequence-specific binding by EBNA1 (ref. 19).
Repetitive sequences present an intrinsic challenge to genome stability20,21. In the presence of additional replication stress, repetitive DNA is known to fail faithful replication, producing aberrant structures before mitosis and/or gaps on mitotic chromosomes, termed fragile sites22,23, that correlate with recurrent breakpoints in cancer24–26. Consequently, breakage at fragile sites has long been thought to enable deletion of tumour suppressor genes, amplification of oncogenes and/or production of fusion oncogenes in which rearrangements are initiated by breakage at the fragile site. In contrast to twin foci on duplicated sister chromatids in mitosis, fragile DNA is known to appear as a single dot indicating loss or failure to replicate and decatenate or as several irregular dots that represent defective condensation of stalled replication intermediates27. Using a fluorescently labelled BAC probe against a 290-kb region located 4 Mb distal to the EBNA1-binding site (Fig. 3a), we determined that aberrant structures indicative of replication or condensation errors at 11q23 were formed on about 40% of mitotic chromosome 11, a frequency increasing to about 50–60% after replication stress induction by aphidicolin (Fig. 3c,e). Indeed, use of oligonucleotide-based fluorescence in situ hybridization (oligo-FISH) with a fluorescently labelled 15-bp oligonucleotide against the repeat unit (Fig. 3b) revealed similar appearance and frequencies of replication-stress-enhanced aberrant structures (Fig. 3d,f) formed at the repetitive EBNA1-binding site containing the 18-bp imperfect palindromic sequences. Furthermore, these aberrant structures were consistent with those formed at the 3q29 repetitive site16 (Extended Data Fig. 5) as well as those previously reported at fragile telomeres28 (detected with a fluorescently tagged oligonucleotide against telomeric repeats).
Fig. 3 |. Repetitive DNA containing the 18-bp imperfect palindromes forms aberrant structures characteristic of fragile DNA and undergoes breakage and micronucleation after induced EBNA1 expression.
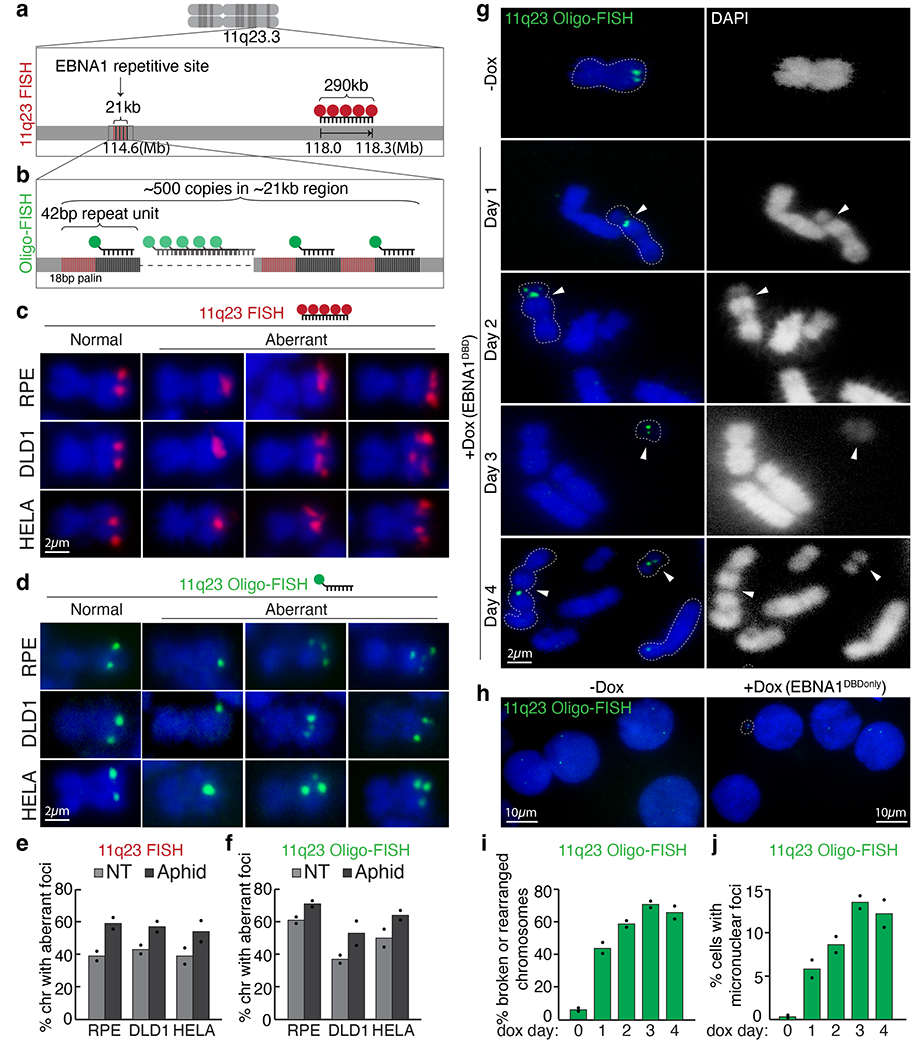
a, Schematic representation of 11q23 FISH using a fluorescently labelled BAC probe (in red) that hybridizes to the indicated genomic locus. b, Schematic representation of oligo-FISH using a fluorescently labelled oligonucleotide (in green) that hybridizes to the sequence adjacent to the 18-bp palindrome as part of the 42-bp repeat unit. c,d, Representative 11q23 FISH (c) and oligo-FISH (d) images showing normal or aberrant foci on mitotic chromosomes in the indicated cell lines with or without 24 h of 0.2 μM aphidicolin. Scale bars, 2 μm. e,f, Quantification of staining in c,d, respectively. For 11q23 FISH with BAC probe, a total of 255 chromosomes were counted for RPE, 236 for DLD1 and 256 for HeLa cells. For 11q23 oligo-FISH, a total of 235 chromosomes were counted for RPE, 202 for DLD1 and 196 for HeLa cells. Data are presented as bars representing mean values from two independent experiments. NT, non treated. g, Representative 11q23 oligo-FISH images of mitotic chromosomes with and without Dox-inducible expression of EBNA1(DBD). Arrowheads indicate oligo-FISH signals that localized at either a gap on centric chromosomes or on small acentric fragments. Dashed lines outline the oligo-FISH-containing chromosomes. DAPI staining is shown as either merged in blue or separately in white. Scale bar, 2 μm. h, Quantification of 11q23 oligo-FISH staining in g. A total of 101 chromosomes were counted on day 0, 98 on day 1, 80 on day 2, 90 on day 3, and 83 on day 4. Data are presented as bars representing mean values from two independent experiments. i, Representative 11q23 oligo-FISH of interphase nuclei with and without Dox-inducible expression of EBNA1(DBD). Scale bars, 10 μm. j, Quantification of staining in i. A total of 800 interphase cells were counted on day 0, 276 on day 1, 253 on day 2, 241 on day 3, and 260 on day 4. Data are presented as bars representing mean values from two independent experiments.
Fragile sites are prone to breakage when challenged with additional stress22,23. Structural and functional studies of EBNA1 binding to the cluster of 18-bp palindromic sequences in the EBV genome have revealed that binding induces a bend in the DNA5,6,29. Additionally, occupancy by EBNA1 and/or potential interacting proteins30 may impede DNA replication and/or other chromatin-associated activities. Recognizing this, we reasoned that clustered EBNA1 binding at an inherently unstable repetitive site may trigger breakage at 11q23. If breakage is left unrepaired in mitosis, the acentric distal fragment can be mis-segregated into micronuclei and the fusogenic centromere-containing proximal fragment subjected to breakage–fusion–bridge cycles that also produce micronuclei31. To test this, doxycycline (Dox)-inducible expression of Flag–EBNA1(DBD) was used to induce EBNA1 foci formation on chromosome 11 in pseudo-diploid DLD1 cells (Extended Data Fig. 6). Remarkably, oligo-FISH revealed that, within 1 day of Dox-induced EBNA1 accumulation, about 40% of mitotic spreads contained 1 or more chromosomes that appeared broken at the EBNA1-binding site, as indicated by a visible gap in the 4′,6-diamidino-2-phenylindole (DAPI) signal (Fig. 3g,h). In subsequent cell cycles, repeat-containing foci appeared on structurally rearranged chromosomal fragments as well as in micronuclei (Fig. 3i,j). Consistent with this, live imaging of cells expressing Clover-tagged EBNA1 (Extended Data Fig. 7a) captured mis-segregation of EBNA1 foci either producing non-diploid (0, 1, 3, 4 or >4) foci in primary nuclei (Extended Data Fig. 7b–d,f) or giving rise to micronuclei (Extended Data Fig. 7e) in daughter cells. As expected, EBNA1 expression did not induce detectable changes at an unrelated repetitive site at chromosome 3q29.
The ataxia telangiectasia mutated (ATM) gene, a tumour suppressor frequently altered in various cancers32, is located 6 Mb proximal to the EBNA1-binding site (Extended Data Fig. 8a). Located 4 Mb distal to the EBNA1-binding site is the mixed lineage leukaemia (MLL) proto-oncogene (Extended Data Fig. 9a), which is rearranged in 70% of infant leukaemias and 10% of adult acute myeloid leukaemias33. Following 1 day of induced EBNA1 expression, about 50% of mitotic spreads contained chromosome 11 fragments in which ATM remained on the centromere-containing fragment proximal to the 11q23 break (Extended Data Fig. 8b,c), and MLL localized on the smaller, acentric fragment consistent with the expected 22-Mb chromosome 11 tip distal to the EBNA1-binding site (Extended Data Fig. 9b,c). As expected, chromosome 11 breakage continued for the next three cell cycles, leading to appearance of dicentric chromosomes formed by ATM-containing proximal fragments. Loss of MLL-containing acentric fragments was also seen in about 15% of mitotic spreads. Indeed, chromosome 11 fragments appeared in micronuclei in about 5% of cells within the first cell cycle of induced EBNA1 expression, increasing to an average of about 10–15% in the subsequent three cell cycles (Extended Data Figs. 8d,f and 9d,f). Micronuclear fragments included ATM (Extended Data Fig. 8d,f), MLL (Extended Data Fig. 9d,f), and 11p15 on the p arm (Extended Data Fig. 10), indicating that the entirety of chromosome 11 is subjected to mis-segregation following EBNA1-induced breakage. Correspondingly, we observed a fivefold increase in cells exhibiting abnormal numbers of intranuclear ATM (Extended Data Fig. 8e,g) or MLL (Extended Data Fig. 9e,g) foci by day 4.
Next, using a set of truncation mutants (Extended Data Fig. 11a–c), we validated that EBNA1-induced breakage at 11q23 is dependent on its DNA-binding domain that is required for binding to the palindromic repeats (Extended Data Fig. 11d–g). Notably, deletion of residues 410 to 460 adjacent to the DNA-binding domain (EBNA1(DBDΔmid); Extended Data Fig. 11a) decreased the frequency of breakage and micronucleation (Extended Data Fig. 11d–g), presumably owing to disrupted interaction with the palindromic repeats and/or diminished recruitment of interacting proteins, such as the ubiquitin-specific protease USP7 (ref. 30), that may contribute to breakage.
Consistent with earlier evidence that fragility of repetitive DNA depends on increased copy number of repeats26,34,35, we next found that the frequency of EBNA1-induced breakage at 11q23 was dose dependent on increasing levels of EBNA1 (Extended Data Fig. 12). As stable EBV latency is maintained by epigenetic repression of viral transcription on the EBV episome36, we examined whether in latent infection, expression of EBNA1 was kept at low levels that prevent rampant breakage of chromosome 11. Indeed, using quantitative immunoblotting (Extended Data Fig. 13a–c), we determined that the baseline level of EBNA1 in latent, EBV-infected Raji or TK6 cells was about half (Extended Data Fig. 13d–f) the level sufficient to trigger breakage of chromosome 11 following Dox-induced (20 ng ml−1 Dox) EBNA1 expression in DLD1 cells (Extended Data Fig. 12). Finally, we used Dox-inducible expression of Flag-EBNA1(DBD) to directly determine the consequence of elevating EBNA1 abundance in latently infected Raji or TK6 cells (Extended Data Fig. 14). Within 1 day of induced expression of EBNA1 (threefold the baseline level for Raji cells and onefold for TK6 cells; Fig. 4a) breakage of chromosome 11 at 11q23 was seen in 34% of Raji cells and 52% of TK6 cells (Fig. 4b–d,f), with a fivefold increase in the appearance of MLL-containing micronuclei observed in TK6 cells (Fig. 4e,g). As expected, cycles of breakage and micronucleation continued for at least 4 days, consistent with evidence from non-EBV-infected DLD1 cells (Fig. 3 and Extended Data Figs. 8 and 9).
Fig. 4 |. Elevated abundance of EBNA1 triggers 11q23 breakage and micronucleation in cells latently infected with EBV.
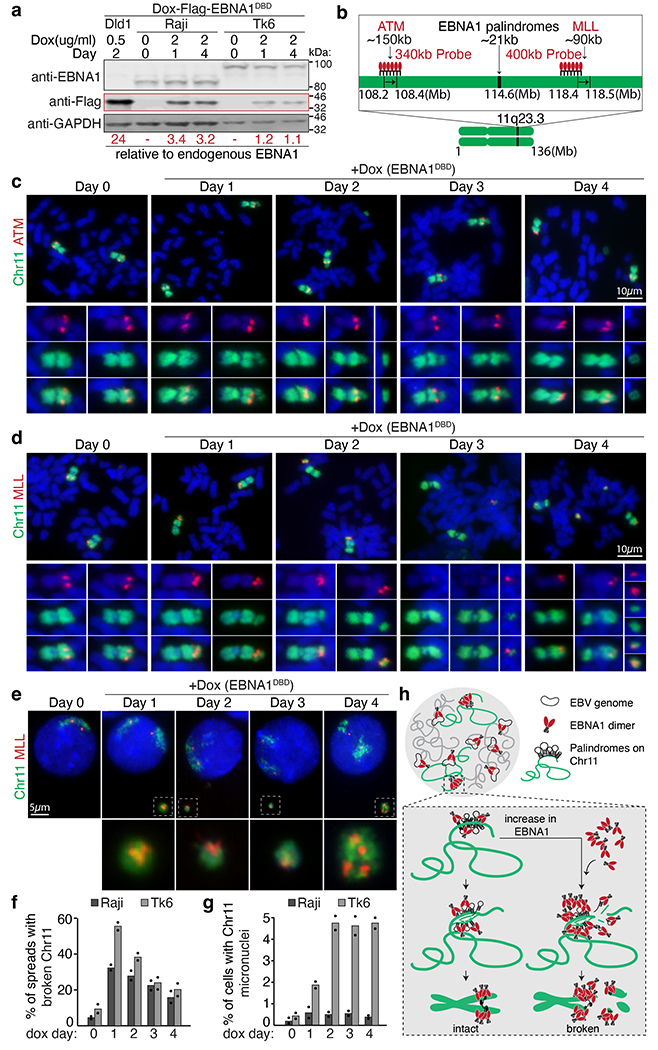
a, Representative immunoblots of Raji cells and TK6 cells expressing Flag–EBNA1(DBD) at day 0, 1 and 4. Numbers under the blots indicate levels of Dox-induced Flag-EBNA1(DBD) relative to endogenous EBNA1, calculated using Dox-induced Flag–EBNA1(DBD) in DLD1 cells previously determined to be 24-fold (Extended Data Fig. 13). A total of 1.5 Å~ 105 Raji or TK6 cells and 1.0 Å~ 105 DLD1 cells were loaded in each lane. Immunoblot was carried out three times, with standard deviations below 10%. For gel source data, see Supplementary Fig. 1. b, Schematic of dualcoloured FISH using a BAC probe against whole chromosome 11 (green) and a BAC probe (in red) against either ATM or MLL. c,d, Representative FISH images showing breakage of chromosome 11 distal to ATM (c) or proximal to MLL (d). Scale bars, 10 μm. e, Representative FISH images showing micronucleation of MLL-containing chromosome 11 fragments. Scale bar, 5 μm. f, Quantification of the frequency of 11q23 breakage. For Raji cells, 105 mitotic spreads were counted on day 0, 122 on day 1, 107 on day 2, 98 on day 3, and 98 on day 4. For TK6 cells, 109 mitotic spreads were counted on day 0, 90 on day 1, 162 on day 2, 81 on day 3, and 67 on day 4. Data are presented as bars representing mean values from two independent experiments. g, Quantification of the frequency of micronucleation. For Raji cells, 700 cells were counted on day 0, 801 on day 1, 905 on day 2, 510 on day 3, and 810 on day 4. For TK6 cells, 1,840 cells were counted on day 0, 646 on day 1, 676 on day 2, 738 on day 3, and 632 on day 4. Data are presented as bars representing mean values from two independent experiments. h, Schematic of the proposed role of EBNA1.
Following an initial break at 11q23, chromosome shattering, termed chromothripsis37, is expected to take place in the resultant micronuclei38–40, producing additional fragments along the entirety of chromosome 11 that are either lost or stabilized by re-ligation into centromere-containing fragments. Indeed, EBNA1-induced chromosome 11 breakage and micronucleation of proximal and distal fragments are the expected cytological hallmarks that would precede chromothriptic shattering, as previously established following a Cas9-induced double-stranded chromosome break41. Such structural variants are thought to frequently drive genomic instability during tumorigenesis, of which some can be detected as clustered rearrangements in end-stage tumours37,42. Therefore, EBNA1-binding-induced breakage at 11q23 is expected to rapidly generate structural variations of both the p and q arms of chromosome 11.
Nasopharyngeal carcinoma (NPC) exhibits the most consistent association with EBV among all cancer types43. Indeed, it has been proposed that EBV infection can precede malignant transformation in NPC development44. We examined a set of 78 previously generated whole-genome sequencing data of NPCs, all of which were annotated as EBV+ on the basis of immunohistochemical staining45,46. Remarkably, 63 out of 78 NPCs (81%) exhibited structural variations on chromosome 11. Notably, structural variants per megabase were particularly enriched on chromosome 11 compared to the rest of the genome (P value = 0.000015; Extended Data Fig. 15a), with chromosomes 3 and 11 having the highest numbers of translocations (Extended Data Fig. 15b). Furthermore, 32 out of 63 NPCs with rearrangements in chromosome 11 exhibited clustered structural rearrangements on chromosome 11 within 100 kb from one another (Extended Data Fig. 15c,d).
Finally, we examined whole-genome sequencing data for 2,439 cancers across 38 tumour types from the Pan-Cancer Analysis of Whole Genomes project47. Through application of computational pathogen-detection pipelines to the available genomics data48, EBV was detected in tumour or adjacent normal samples of 165 patients. Tumours with detectable EBV exhibited an enrichment of chromosome 11 structural variations when compared with EBV− tumours (odds ratio = 1.40; P value = 0.015; Fisher’s exact test; Extended Data Fig. 15e). Moreover, 100% (18 out of 18) of head and neck cancers designated as EBV positive harboured rearrangements on chromosome 11 (Extended Data Fig. 15f). Although the small number of EBV+ samples available within each cancer type limits statistical power, tumours with detectable EBV harboured a higher median proportion of structural variations along chromosome 11 (Extended Data Fig. 15g,h).
Since the initial discovery of 51 fragile sites in human chromosomes almost 40 years ago25, we have now identified an example of fragility induced by binding of EBNA1, the sequence-specific DNA-binding protein encoded by a virus to which almost the entire human population has been exposed. Our discovery of EBV-like 18-bp imperfect palindromic repeats, whose binding by EBNA1 triggers breakage at 11q23, identifies a previously unappreciated link between EBV and structural variations along chromosome 11. We propose that in cells infected with latent EBV, chromosome 11 is likely to be broken by elevated levels of EBNA1 expressed after reactivation from latency49,50 (Fig. 4h). Consistent with this, a history of reactivation of latent EBV has previously been proposed as a major risk factor for the development of tumours, especially nasopharyngeal cancers51. Combined with the ability of EBNA1 to inhibit p53 (ref. 11), breakage of EBNA1-bound palindromic repeats at 11q23 would be predicted to generate inheritable structural variations along chromosome 11, which analyses of whole-genome sequencing reveal to be enriched in cancers with detectable EBV. Beyond that, it is possible that development of tumours designated as EBV negative at present may have included a ‘hit-and-run’52 event of EBNA1-induced breakage at 11q23 without retention of the viral genome in the established tumour.
Plasmids
Retroviral plasmid MSCV-N-Flag-EBNA1 (Addgene #37954) was used to amplify and sub-clone all constructs containing EBNA1. Retroviral plasmid pWZL-EGFP (Addgene #12269) was used to express EBNA1 with an N-terminal Myc tag. Dox-inducible lentiviral plasmid pCW-Dox-Cas9 (Addgene #50661) was used to mutate and express 3xFlag-tagged nuclease-dead Cas9 (dCas9). Lentiviral plasmid pLH-sgRNAplus (Addgene #75388) was used to clone and express guides to label repetitive sites at 11q23 and 3q29 with dCas9. px330–U6-Chimeric-BB-CBh-hSpCas9 and pCDNA5-H1-sgRNA were used to transiently express Cas9 and sgRNA to cut repetitive site at 11q23. Dox-inducible lentiviral plasmid pCW-Dox-Cas9 (Addgene #50661) was used as backbone to express dox-inducible Flag-EBNA1DBD.
Cell lines
All cell lines were cultured in Glutamax-DMEM (Life Technologies) supplemented with 10% fetal bovine serum, at 5% CO2 and 3% O2. All cell lines have been routinely tested to be free of mycoplasma contamination. Origins of non EBV-infected cell lines are as follows: DLD1 and U2OS cells were obtained from ATCC; RPE cells were obtained from David Pellman (Dana Farber Cancer Center); HeLa S3 cells are previously described53. Origins of EBV-infected cell lines are as follows: Raji cells were obtained from Sigma; Daudi cells and Tk6 cells were obtained from ATCC. All plasmid transfections for transient expression were performed using TransIT-LT1 (Mirus). For retroviral production, HEK293T cells were co-transfected with retroviral plasmid and packaging plasmid pCL-10A1 (Novus Biologicals) using TransIT-VirusGEN (Mirus). For lentiviral production, HEK293T cells were co-transfected with lentiviral plasmid and packing plasmids PD2G and PsPAX2 using TransIT-VirusGEN (Mirus). In brief, to stably integrate drug-selectable viral expression plasmids into target cells, cells were grown in supernatant containing either retrovirus or lentivirus for one day and subsequently selected with either puromycin, hygromycin, or blasticidin on the following day.
CRISPR labeling system
Either DLD1 cells or U2OS cells were transduced with lentivirus to stably integrate a puromycin-resistant construct encoding 3xFlag-tagged dCas9 and a hygromycin-resistant construct encoding sgRNA, followed by selection in media containing puromycin and hygromycin. Surviving cells were then transduced with retrovirus to stably integrate a blasticidin-resistant construct encoding Myc-tagged EBNA1DBDonly, followed by selection in media containing blasticidin. Triple-selected cells were then used to perform immunofluorescence using anti-Flag and anti-Myc antibodies.
CRISPR cutting system
Either HeLa cells or U2OS cells were co-transfected with px330–U6-Chimeric-BB-CBh-hSpCas9 and pCDNA5-H1-sgRNA, followed by transient selection in media containing puromycin for two days. Surviving cells were then grown in regular media for one week and transduced with retrovirus to stably integrate a construct encoding Flag-EBNA1. Cells were then used to perform immunofluorescence using anti-Flag.
Immunofluorescence
Immunofluorescence (IF) was performed as described previously54. Briefly, cells grown on coverslips were fixed for 5 min in 2% paraformaldehyde, incubated in blocking solution (1 mg/ml BSA, 3% goat serum, 0.1% Triton X-100, 1mM EDTA in PBS) for 30 minutes, followed by incubation with primary antibodies in blocking solution for 1 hour. Primary antibodies used in this study were as follows: anti-Myc (9B11, Cell signaling, mouse monoclonal), anti-Flag (F1804, Sigma-Aldrich, mouse monoclonal), Secondary antibodies used were Alexa 488, Alexa 555, and Alexa 647 (Molecular probes, Life Technologies). Cells were then washed in PBS three times and counterstained with 4.6-diamidino-2-phenylindole (DAPI). Coverslips were mounted on glass slides using ProLong Gold antifade (Sigma). Images were acquired on a DeltaVision elite system (Applied Precision, GE) at 100x magnification (5x 0.5um z-sections). Quick projections were generated using the softWoRx program.
Live cell imaging
Three days following transduction with Clover-Flag-EBNA1, cells were imaged using a CQ1 spinning disk confocal system (Yokogawa Electric Corporation) with 40x magnification at 37°C and 5% CO2. Image acquisition and data analysis were performed using CQ1 software and ImageJ, respectively. Images of 20 fields/well at 8 × 3-μm z-sections were acquired at 10 minutes intervals for 48 hours. EBNA1 foci through mitosis were scored as either symmetric inheritance or asymmetric inheritance into daughter cell nuclei.
Fluorescence In Situ Hybridization (FISH)
All FISH images were acquired on a DeltaVision elite system (Applied Precision, GE) at 100x magnification (5x 0.5um z-sections). Quick projections were generated using the softWoRx program. Cells were plated in 10cm plates the night before harvesting. The next day, ~80% confluent cells were incubated for 30 minutes with 0.2ug/ml colcemid (KaryoMax, Thermo Fisher). Cells were trypsinized, harvested, resuspended in 0.075M KCl at 37 degree for 15 minutes, fixed in methanol/acetic acid (3:1), and stored in fixative until use.
FISH using fluorescently-labeled BAC probes (Metasystems):
Fixed samples dropped onto glass slides were coated with fluorescently labeled BAC probes (Metasystems), denatured at 80°C for 10min, and incubated at 37°C overnight. Following overnight hybridization, slides were then washed in 2xSSC + 0.1% Tween, 0.2xSSC, and water, followed by mounting with anti-fade ProLong Gold with DAPI (Invitrogen).
Metasystems BAC probes used in the manuscript are as follows:
Fragility of 11q23: XL MLL plus Break Apart Probe (D-5060-100-OG)
Chromosome 11 paint: XCP 11 Green (D-0311-050-FI)
Breakage distal to ATM: XL ATM/11cen (D-5102-100-OG)
Breakage proximal to MLL: XL KMT2A BA (D5090-100-OG)
Oligo-FISH using fluorescently labeled DNA oligo (Integrated DNA Technologies):
fixed samples dropped onto glass slides were coated with hybridization solution (50% formamide, 10% dextran sulfate in 2x SSC) containing 20nM Cy3 end-labeled oligo denatured at 80°C for 3min and incubated at room temperature for 2 hours. Slides were then washed in 2xSSC + 0.1% Tween, 0.2xSSC, and water, followed by mounting with anti-fade ProLong Gold with DAPI (Invitrogen).
Sequences of IDT oligos used are as follows:
11q23 repetitive site: 5’-ATAAGTATTGCCTCG-3’
3q29 repetitive site: 5’-GATATAGTGAAGCTCC-3’
Genomic landscape of EBV-like repetitive sequences
An approximate pattern matching approach was applied to identify the motifs that resemble the 18 bp EBV consensus palindromic sequence (3’-GGGTAGCATATGCTACCC-5’) in the human genome (GRCh38). The analysis was conducted by first splitting the genome into 0.1Mb bins excluding the gap regions. For each 0.1Mb region, all 18 bp sequences were identified that had at most 6 mismatches compared to the EBV binding consensus sequence. Further filtering was applied to account for the palindromic nature of the EBV consensus sequence. Sequences were considered palindromic if the first 7 bases (1-7 bp) were the reverse complement of the last 7 bases (18-12 bp) with at most 2 base mismatches. To visualize the distribution of the matching 18 bp sequences, we generated genome-wide and zoom-in plots for the repetitive site in chromosome 11 using the karyoploteR package in the R programming language. The consensus motif of the 18 bp matching and the 24 bp spacer sequences interspersed between the consecutive 18 bp repetitive sequences were generated using ggseqlogo in R.
A similar approach was adopted to identify and extract the 18 bp repetitive and the 12 bp and 24 bp spacer sequences in the EBV (NC_007605) and chimpanzee (PanTro6) genomes, respectively. To determine if the repetitive site is conserved evolutionarily, the UCSC genome browser was used to visualize the pairwise alignment of the repetitive site in the human genome (chr11:114,604,212-114,625,620) to that of other primates.
Repeat landscape across different populations using long read sequencing:
To explore the repeat landscape of the 11q23 region across different populations, we applied an approximate pattern matching approach to identify the motifs that resemble the 18 bp EBV consensus palindromic sequence (3’-GGGTAGCATATGCTACCC-5’) across GRCh38 aligned PacBio long-read sequenced genomes of 2 individuals (HG003 and HG004) of Ashkenazim descent and 2 individuals (HG006, HG007) of Chinese descent. The analysis was conducted by first extracting all the reads that mapped to the 11q23 region (chr11:110,600,000-121,300,000). For every read, all 18 bp sequences were identified that had at most 5 mismatches compared to the EBV binding consensus sequence. Any duplicate matches found across different reads at the same 18 base pair (bp) genomic location were removed, Further filtering was applied to account for the palindromic nature of the EBV consensus sequence. Sequences were palindromic if the first 7 bases (1-7 bp) were the reverse complement of the last 7 bases (18-12 bp) with at most 2 base mismatches. To visualize the distribution of the EBV-like repeat sequences across the 4 different individuals, we generated zoom-in plots for the repetitive site in 11q23 using the karyoploteR package in the R programming language.
The PacBio sequence datasets used in this study are publicly available and can be found from the following ftp links:
HG003:
ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/HG003_NA24149_father/PacBio_MtSinai_NIST/PacBio_minimap2_bam/HG003_PacBio_GRCh38.bam
HG004:
ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/HG004_NA24143_mother/PacBio_MtSinai_NIST/PacBio_minimap2_bam/HG004_PacBio_GRCh38.bam
HG006:
ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/ChineseTrio/HG006_NA24694-huCA017E_father/PacBio_MtSinai/PacBio_minimap2_bam/HG006_PacBio_GRCh38.bam
HG007:
ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/ChineseTrio/HG007_NA24695-hu38168_mother/PacBio_MtSinai/PacBio_minimap2_bam/HG007_PacBio_GRCh38.bam
Quantification of experimentally induced levels of EBNA1 expression
Three cell lines latently infected with EBV were used to assess baseline expression of endogenous EBNA1: Daudi cells and Raji cells derived from EBV-infected Burkitt’s lymphoma, and Tk6 cells established from B-lymphoblasts immortalized by EBV. Since the dox-induced allele of Flag-EBNA1DBD lacks the N-terminal residues 40-65 recognized by the anti-EBNA1 antibody, we first determined that a Flag-tagged EBNA1ΔGAGR recognized by the anti-EBNA1 antibody is expressed at 8-fold the level of latent EBNA1 in Raji and Tk6 cells, and 14-fold in Daudi cells. Using this Flag-EBNA1ΔGAGR allele as normalization, we used anti-Flag antibody to determine the levels of dox-induced Flag-EBNA1DBD relative to latent EBNA1 levels. From this, we found that the minimum level of dox-induced Flag-EBNA1DBD (20ng/ml doxycycline) sufficient to induce breakage in DLD1 cells is a mere doubling (2-fold) of the baseline level of EBNA1 in Raji cells and Tk6 cells.
Examination of 11q genomic rearrangements in whole-genome sequenced Nasopharyngeal carcinoma (NPC)
To determine the landscape of genomic rearrangements in 78 whole-genome sequenced NPC samples, we analyzed the Manta SV calls that were provided as part of the original publications45,46. For every sample, the total number of large-scale genomic rearrangements events including deletions, insertions, duplications, inversions, and translocations on chromosome 11 were determined and normalized by the size of the chromosome 11. The distribution of the normalized chromosome 11 genomic rearrangements was compared with that of other chromosomes using a Two-sided Mann-Whitney U rank test. To determine if translocations were enriched in chromosome 11, we counted the total number of translocations between each pair of chromosomes across all samples and normalized it by the total number of samples.
Associations between EBV and 11q genomic rearrangements
The available EBV annotations48 were used for all cancer samples. As previously suggested, a more stringent criteria of 1,000 reads per million extracted reads (PMER) was applied for stomach cancers55. Together with the consensus somatic mutation calls from the official PCAWG release47, we identified a total of 2,439 PCAWG samples that had annotation for EBV and contained at least two structural variants. From these samples, 165 out of the 2,439 PCAWG samples were classified as EBV+ as they had EBV reads found in either their adjacent-normal or tumor genomes48. To determine if the chromosome 11 aberrations were statistically enriched in EBV+ samples, a Fisher’s exact test was first conducted across all PCAWG samples. Further, cancer-specific analyses were performed within tumor types that had at least 15 EBV+ samples. Only plots for cancer types with statistically significant results (P-value ≤ 0.05) are shown.
Extended Data
Extended Data Figure 1. EBNA1 localization is enriched at a single genomic locus in the endogenous human genome.
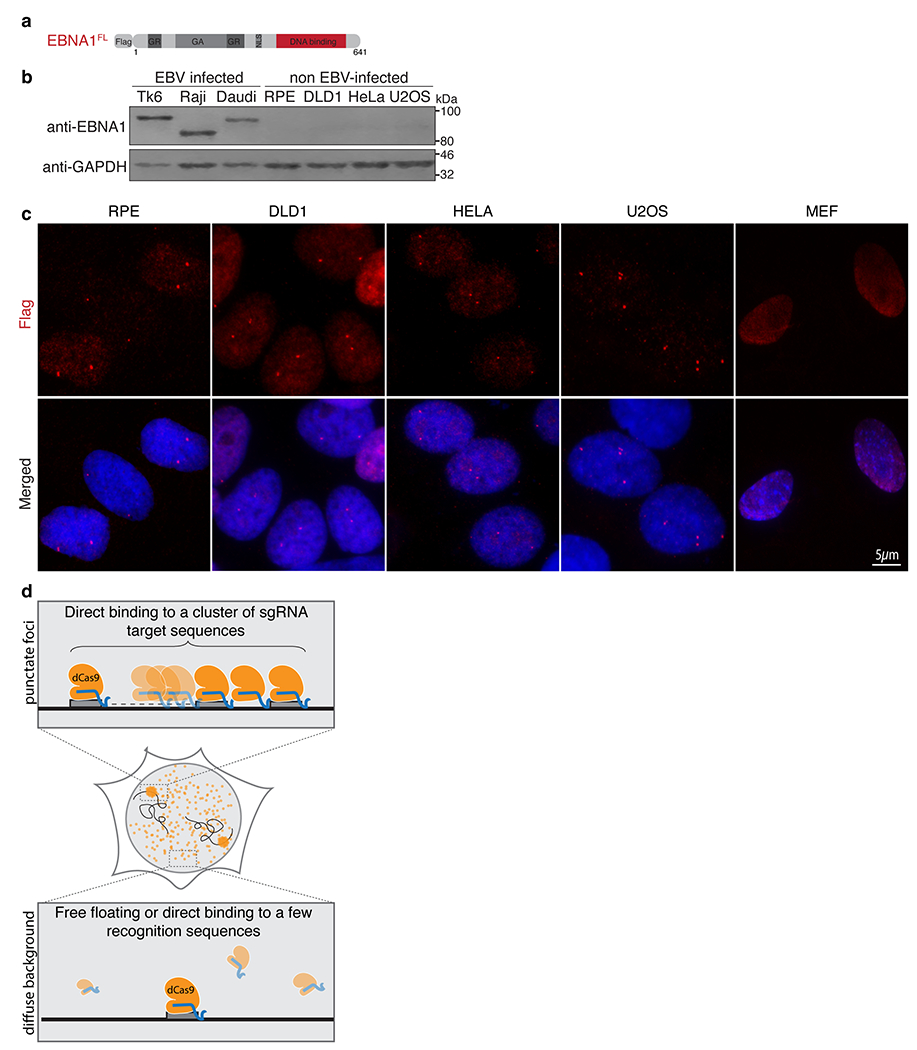
(a) Schematic representation of the Flag-tagged allele of full length EBNA1. (b) anti-EBNA1 (Santa Cruz sc-81581) immunoblot of the indicated cell lines. Immunoblots were repeated 3 times with similar results. (c) Representative anti-Flag immunofluorescence images of Flag-EBNA1FL foci in the indicated cell lines as quantified in Figure 1d from three independent experiments. TK6 cells are established from B-lymphocytes immortalized with EBV. Raji cells and Daudi cells are derived from EBV-infected Burkitt’s Lymphoma. RPEs are hTERT-immortalized primary retinal pigment epithelial cells. DLD1s are derived from colon cancer. HeLas are derived from cervical cancer. U2OS cells are derived from osteosarcoma. MEFs are mouse embryonic fibroblasts. (d) Schematic representation of sequence-specific enrichment of dCas9 (yellow) in complex with a single sgRNA (blue) targeting a sequence (grey) clustered at a repetitive site.
Extended Data Figure 2. The human genome contains a cluster of EBV-like 18bp imperfect palindromic sequences at 11q23.

Number of copies (y-axis) of 18bp imperfect palindromic sequences with up to 6 variant and 2 asymmetric nucleotides as plotted per 0.1 mega-base region across the human reference genome GRCh38 (a) and number of copies of 18bp imperfect palindromic sequences with up to 5 variant and 2 asymmetric nucleotides as plotted per 10 kilo-base region at 11q23 region for long-read sequenced genomes of two individuals of Ashkenazi descent (b) and two individuals of Chinese descent (c).
Extended Data Figure 3. 11q23 repetitive site containing 18bp imperfect palindromes is evolutionarily conserved amongst the Great Apes.
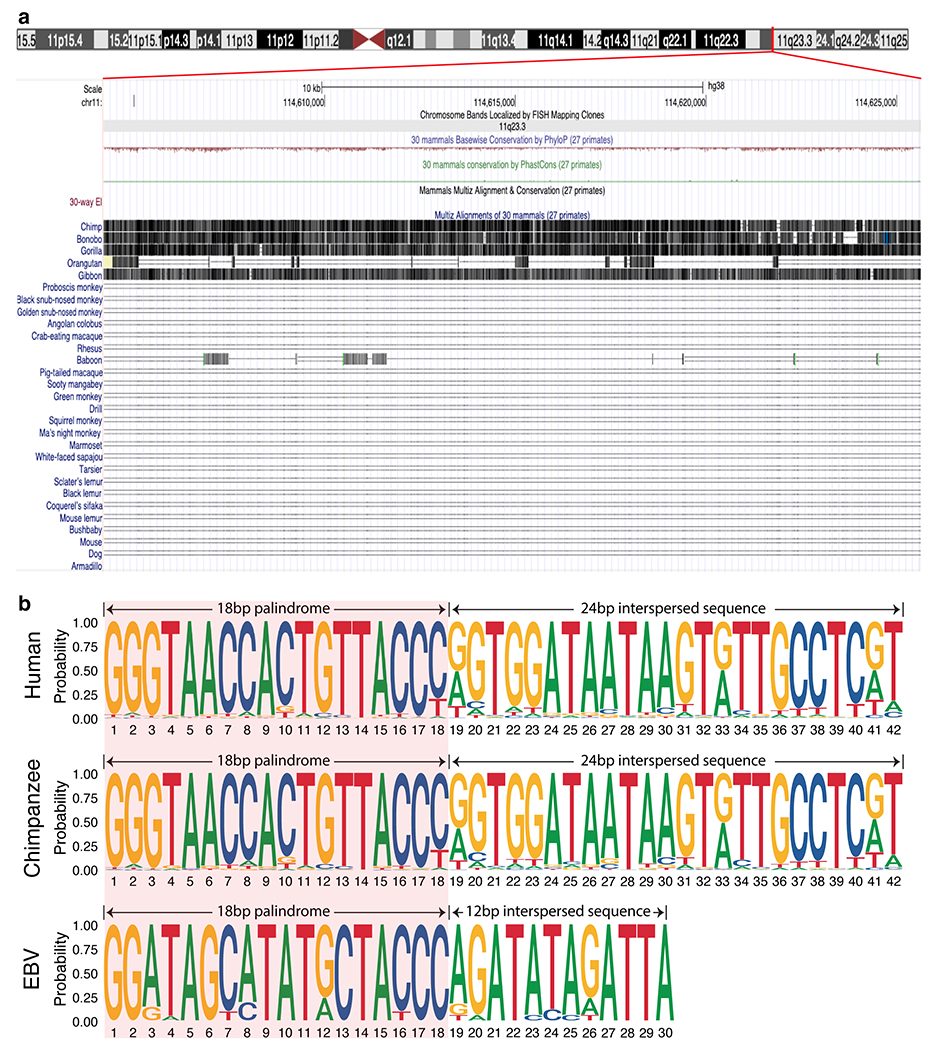
(a) Pairwise sequence alignment of the 11q23 repetitive site (chr11:114,604,212-114,625,620) in the human genome against 30 mammalian genomes. Vertical lines with darker grayscale color represent higher alignment quality, whereas a horizontal single or double line represents a lack of sequence homology between the human and the aligned genome. (b) Comparison of the consensus motif logos of the repeat sequence from the human, chimpanzee, or EBV genome showing that while the 18bp palindromes are conserved amongst all three species, the interspersed sequences are distinct between the primates and EBV.
Extended Data Figure 4. CRISPR labeling and CRISPR cutting approaches demonstrating localization of EBNA1 at the cluster of 18bp imperfect palindromic sequences at 11q23.
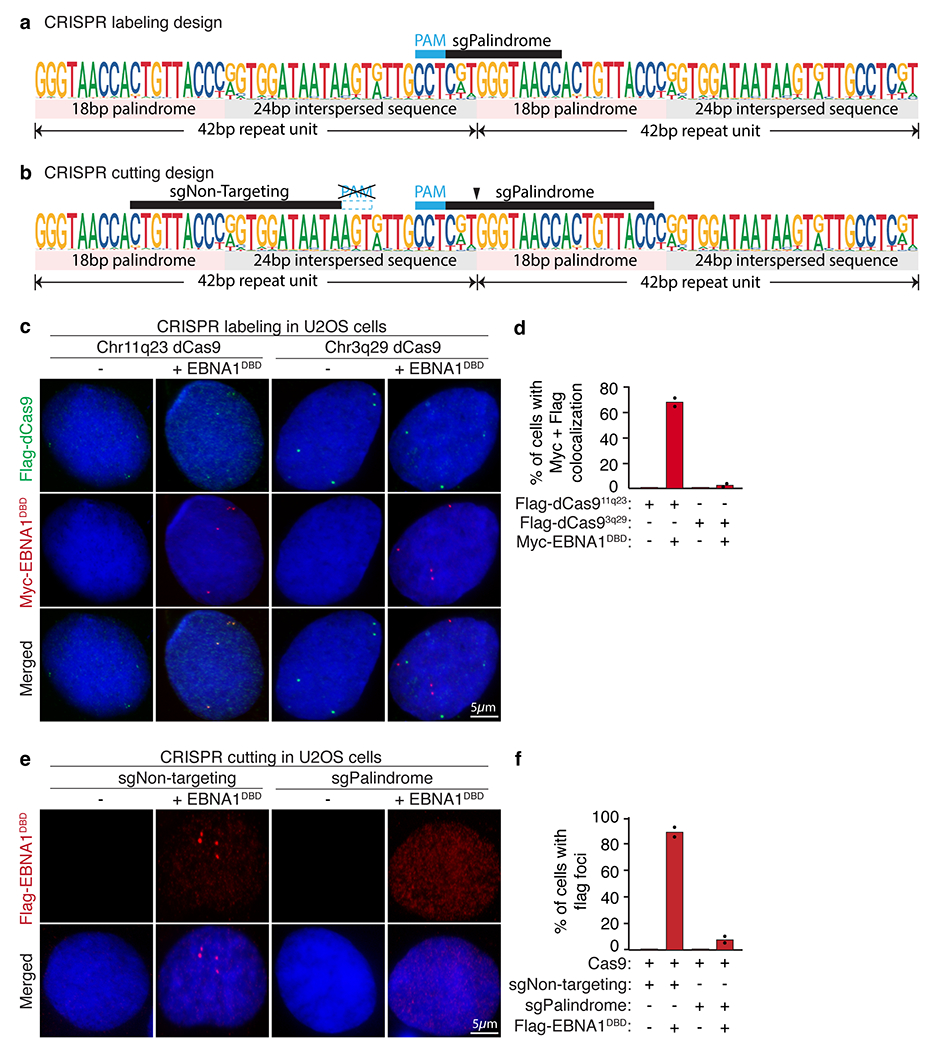
(a) Schematic of the consensus sequence of the 42bp repeat unit showing the sgRNA sequence (sgPalindrome) used in the CRISPR labeling approach. (b) Schematic of the consensus sequence of the 42bp repeat unit showing the sgRNA sequences (sgNon-Targeting and sgPalindrome) used in the CRISPR cutting approach. (c) Representative anti-Flag (in green) and anti-Myc (in red) IF of U2OS cells stably expressing Flag-dCas9 directed by the indicated sgRNAs with or without colocalization with Myc-EBNA1DBD as quantified in (d). (e) Representative anti-Flag IF of Flag-EBNA1DBD in U2OS cells treated with Cas9 and the indicated sgRNAs as quantified in (f).
Extended Data Figure 5. Repetitive DNA at 3q29 forms aberrant structures on mitotic chromosomes.
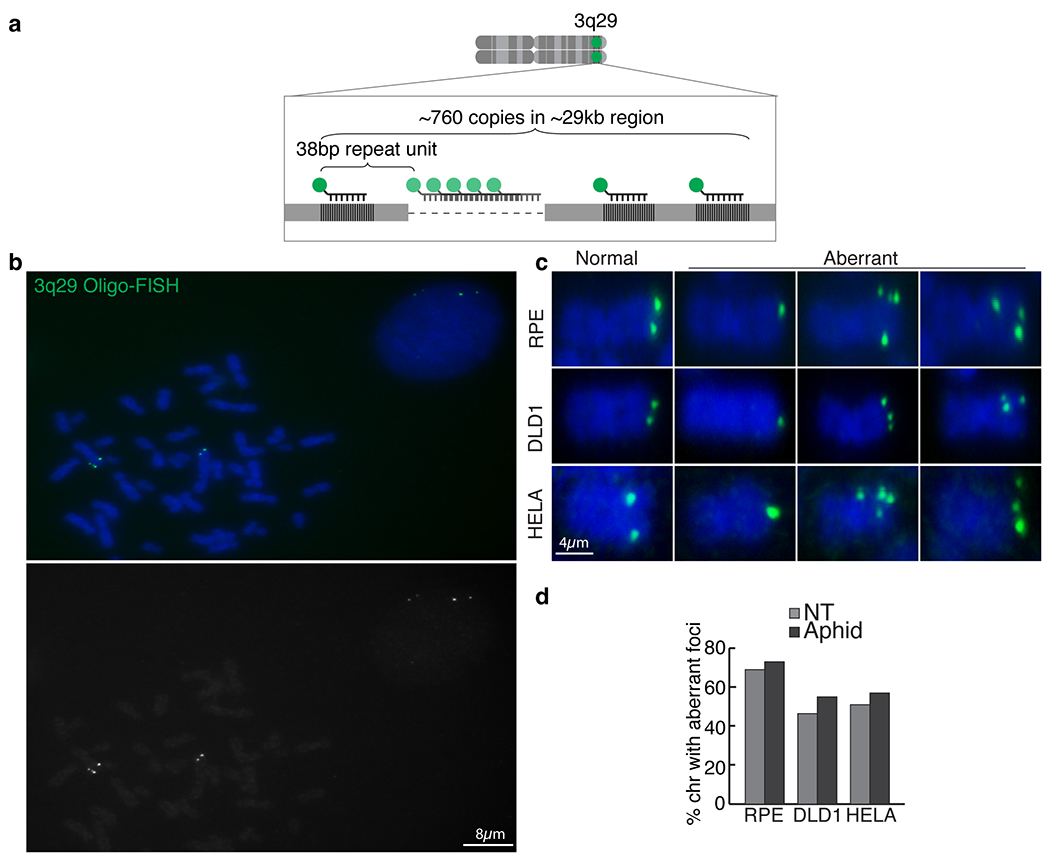
(a) Schematic representation of the oligo-FISH approach using a fluorescently labeled oligo (in green) to target a portion of the repeat sequence clustered at 3q29. (b) Representative oligo-FISH (c) Zoomed-in representative oligo-FISH showing normal appearance (two dots) or fragile appearance (single dot, multiple dots) on mitotic chromosomes in the indicated cell lines as quantified in (d) with or without 0.2uM aphidicolin treatment for 24 hours.
Extended Data Figure 6. Dox-inducible expression of EBNA1DBD in DLD1 cells.
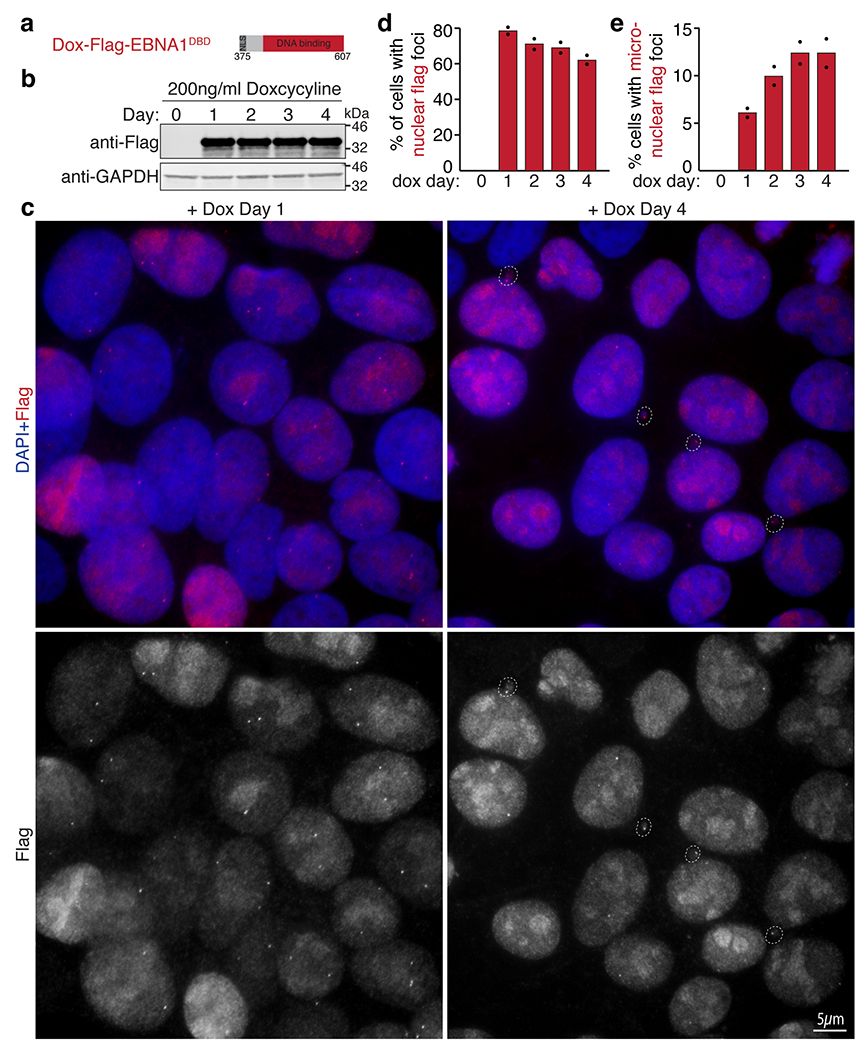
(a) Schematic of the dox-inducible Flag-EBNA1DBD allele encoding the indicated amino acid residues of EBNA1 stably integrated in DLD1 cells. (b) Anti-Flag and anti-GADPDH western blots showing expression of Flag-EBNA1DBD induced with 200ng/ml doxycycline at the indicated days post induction. (c) Representative anti-Flag immunofluorescence images showing nuclear foci on day 1 and appearance of micronuclear foci on Day 4 as quantified in (d-e). A total of 830 cells were quantified. Data are presented as bars representing mean values from two independent experiments.
Extended Data Figure 7. Live imaging of the inheritance of EBNA1 foci through cell division.
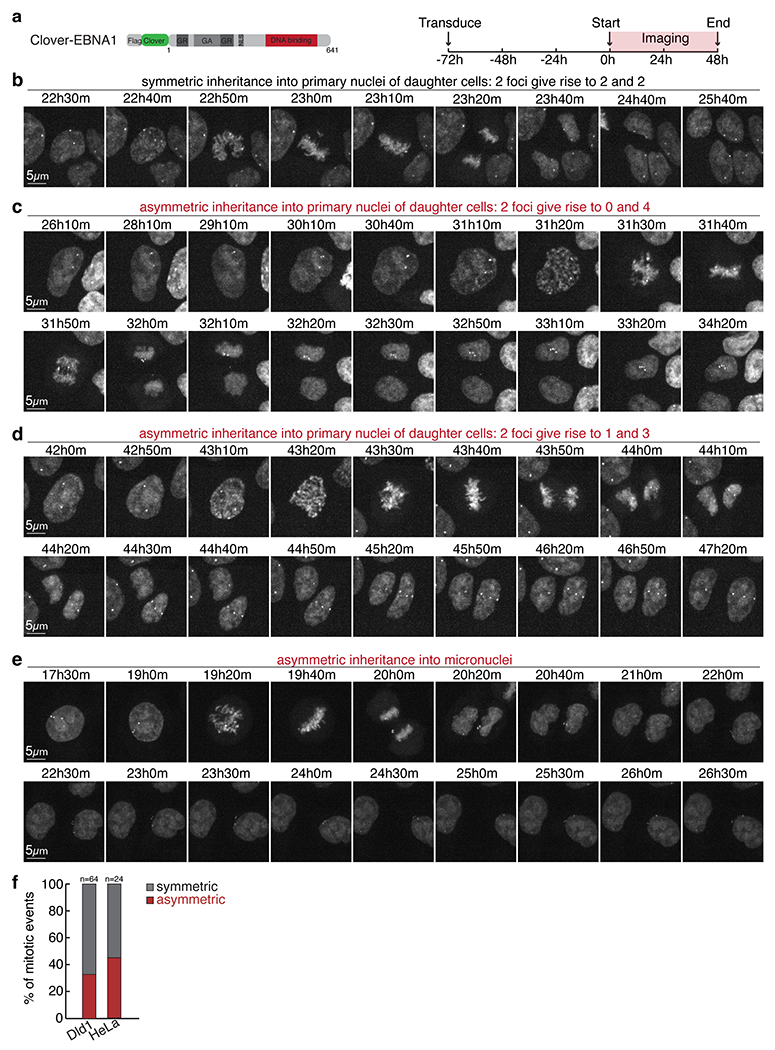
(a) Schematic of the clover-tagged allele of EBNA1 used for live-imaging at 10min intervals for 48 hours starting at day 3 following transduced expression in Dld1s or HeLas. Still images capturing either symmetric inheritance (b) or asymmetric inheritance of EBNA1 foci into primary nuclei (c-d) or micronuclei (e) of daughter cells as quantified in (f). 64 mitotic events were scored for DLD1 cells, 24 mitotic events for HeLa cells.
Extended Data Figure 8. EBNA1-induced breakage of chromosome 11 produces an ATM-containing proximal fragment that undergoes micronucleation.
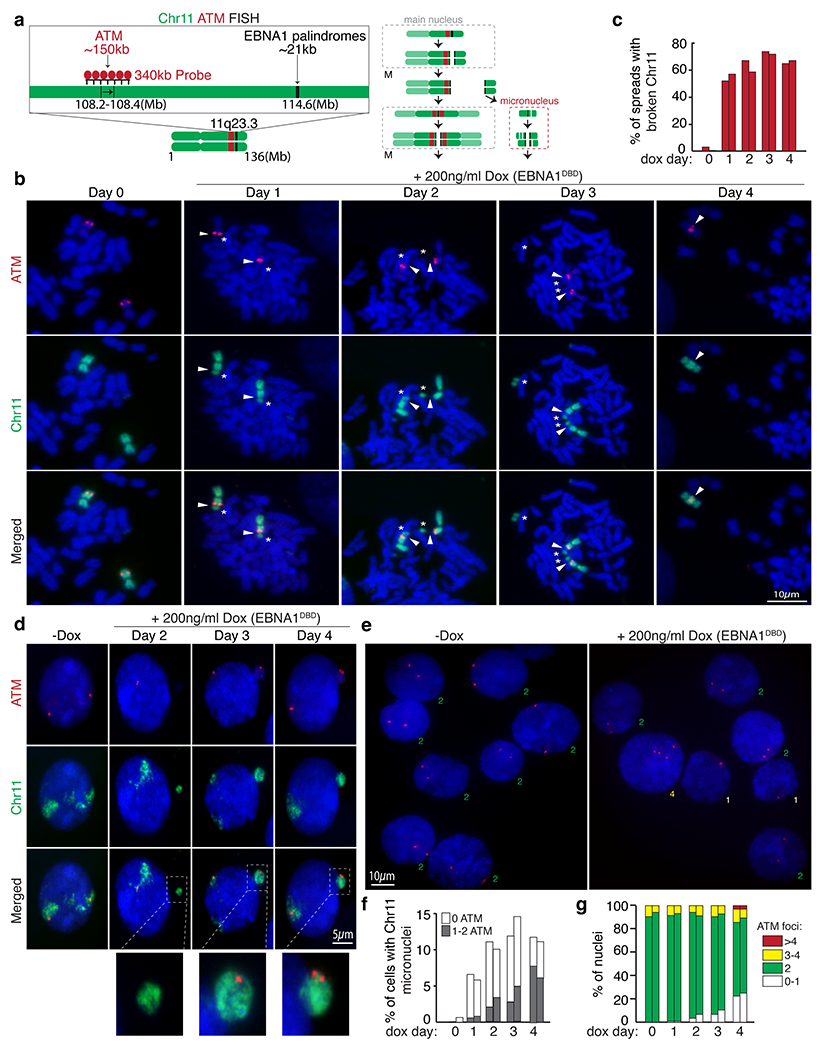
(a) Schematic of dual-colored FISH using whole chromosome 11 probe (green) and probe against ATM (red). (b) Representative FISH images of DLD1 cells showing intact chromosome 11’s (Day 0) and broken chromosome 11 fragments containing ATM proximal to the site of breakage at day 1 to 4 post dox induction. (c) Data represent the percentage of spreads with broken chromosome 11 from two independent experiments. 62 mitotic spreads were quantified on Day 0, 53 on Day 1, 48 on Day2, 58 on Day 3, and 61 on Day 4. (d) Representative FISH images showing micronucleation of chromosome 11 fragments with or without ATM foci. (f) Stacked bars represent the percentage of cells with chromosome 11 micronuclei with the indicated number of ATM foci from two independent experiments. 310 cells were quantified on Day 0, 220 cells on Day 1, 190 on Day 2, 214 on Day 3, and 198 on Day 4. (e) Representative FISH images showing ATM foci in primary nuclei. (g) Stacked bars represent the percentage of primary nuclei with the indicated number of ATM foci from two independent experiments. 800 cells were quantified on Day 0, 900 cells on Day 1, 750 on Day 2, 950 on Day 3, and 780 on Day 4.
Extended Data Figure 9. EBNA1-induced breakage of chromosome 11 produces an MLL-containing distal fragment that undergoes micronucleation.
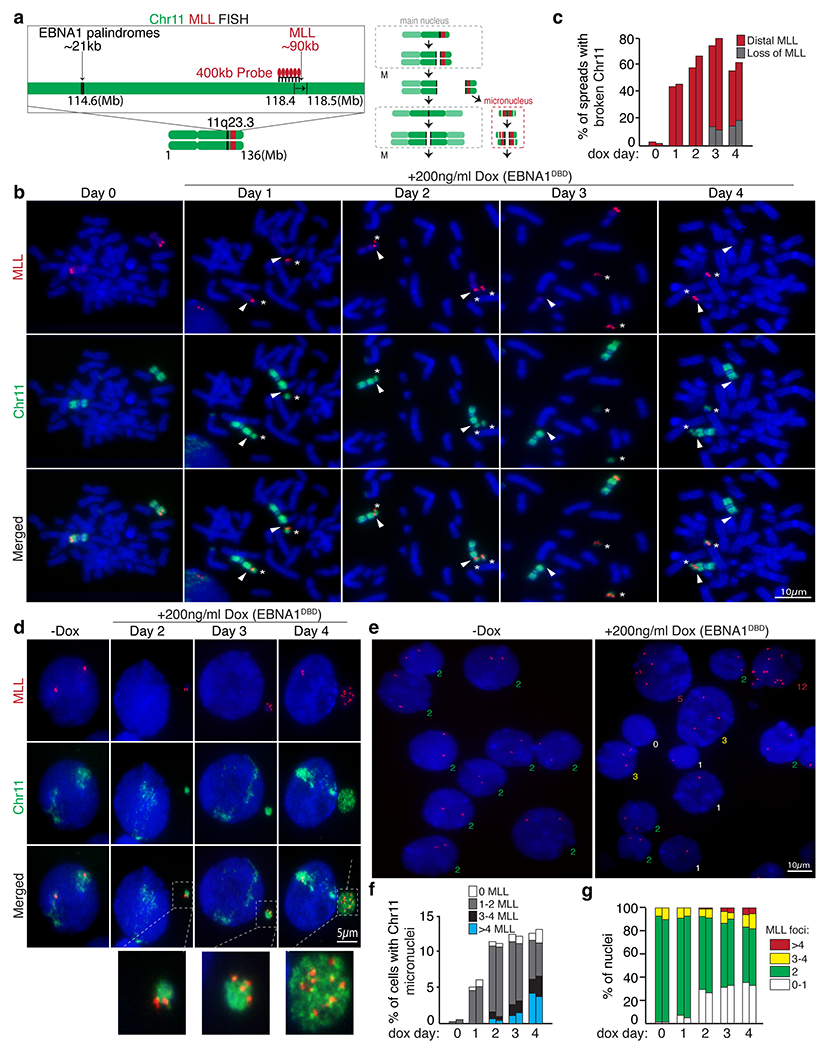
(a) Schematic of dual-colored FISH using whole chromosome 11 probe (green) and probe against MLL (red). (b) Representative FISH images of DLD1 cells showing intact chromosome 11’s (Day 0) and broken chromosome 11 fragments containing MLL distal to the site of breakage at day 1 to 4 post dox induction. (c) Data represent the percentage of spreads with broken chromosome 11 from two independent experiments. 71 mitotic spreads were quantified on Day 0, 49 on Day 1, 50 on Day2, 53 on Day 3, and 71 on Day 4. (d) Representative FISH images showing micronucleation of chromosome 11 fragments with or without MLL foci. (f) Stacked bars represent the percentage of cells with chromosome 11 micronuclei with the indicated number of MLL foci from two independent experiments. 400 cells were quantified on Day 0, 210 cells on Day 1, 235 on Day 2, 190 on Day 3, and 180 on Day 4. (e) Representative FISH images showing MLL foci in primary nuclei. (g) Stacked bars represent the percentage of primary nuclei with the indicated number of MLL foci. 950 cells were quantified on Day 0, 880 cells on Day 1, 980 on Day 2, 1100 on Day 3, and 950 on Day 4.
Extended Data Figure 10. EBNA1-induced micronucleation of chromosome 11 includes the p-arm.
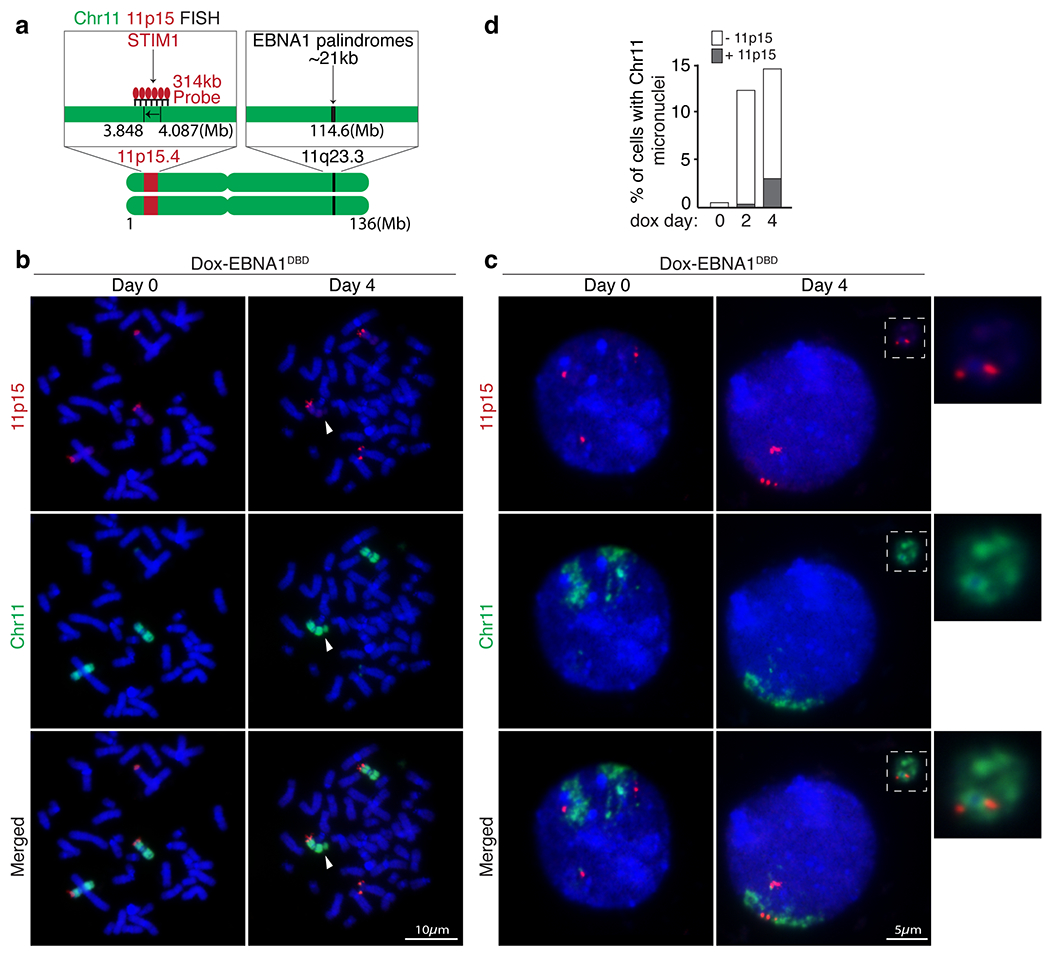
(a) Schematic of dual-colored FISH using whole chromosome 11 probe (green) and probe against 11p15 (red) on the p-arm. (b) Representative FISH images of DLD1 cells showing 11p15 on the p-arm of either intact chromosome 11’s (Day 0) or broken chromosome 11 fragments (Day 4) upon induced expression of EBNA1. (c) Representative FISH images showing that EBNA1-induced micronucleation of chromosome 11 (Day 4) includes 11p15 on the p-arm. (d) Stacked bars represent the percentage of cells with chromosome 11 micronuclei with or without 11p15. 400 cells were quantified on Day 0, 310 cells on Day 2 and 380 cells on Day 4.
Extended Data Figure 11. EBNA1-induced breakage at 11q23 is dependent on the DNA binding domain.
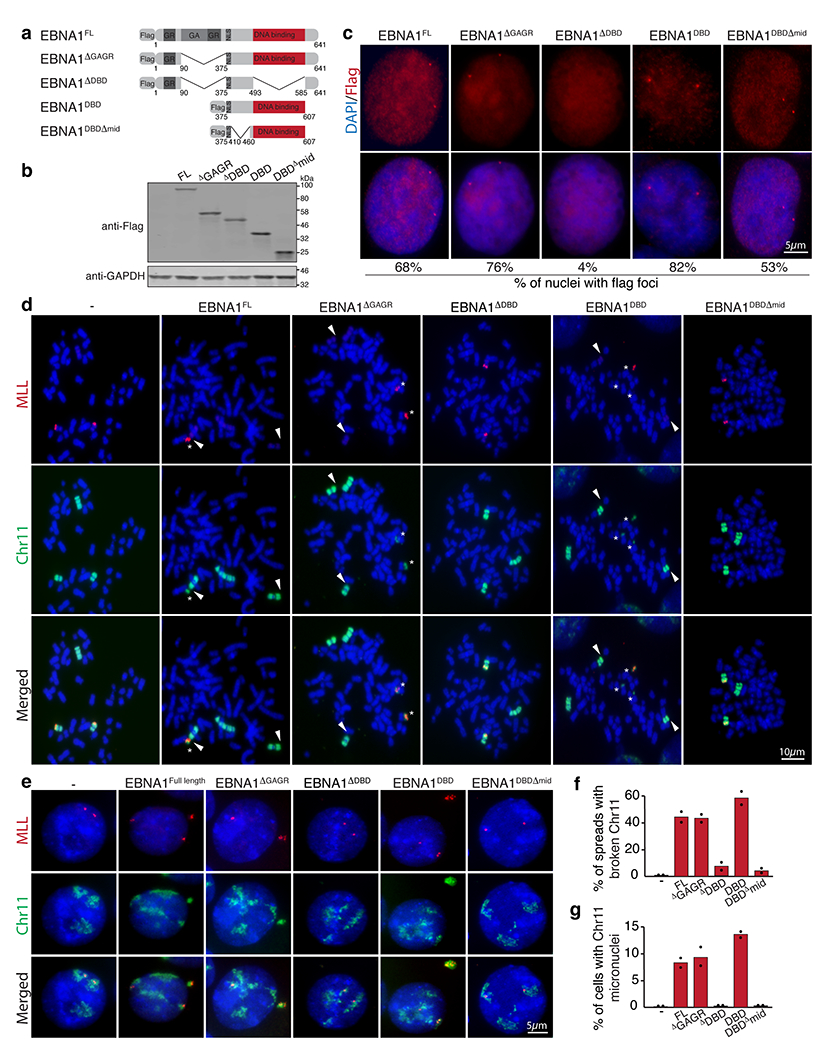
(a) Schematic of the Flag-tagged alleles of EBNA1 expressed in HeLa cell that harbor two copies of chromosome 11 and a derivative chromosome 11 lacking 11q23. (b) anti-Flag immunoblot of cells expressing the indicated alleles. (c) Anti-Flag immunofluorescence showing localization of the indicated EBNA1 alleles. Numbers indicate the percentage of nuclei with flag foci. (d) Representative dual colored FISH showing chromosome 11’s in HeLa cells, including two copies of chromosome 11 (in green) harboring MLL (in red) and one derivative chromosome 11 lacking MLL. White arrows indicate the site of breakage proximal to MLL as quantified in (f). 48 mitotic spreads were quantified for non-transduced HeLas, 65 for HeLas expressing EBNA1FL, 98 for HeLas expressing EBNA1ΔGAGR, 40 for HeLas expressing EBNA1ΔDBD, 69 for HeLas expressing EBNA1DBD, and 82 for HeLas expressing EBNA1DBDΔmid. Data are presented as bars representing mean values from two independent experiments. (e) Representative FISH images of interphase nuclei showing micronucleation of chromosome 11 fragments containing MLL as quantified in (g). 210 cells were quantified for non-transduced HeLas, 150 for HeLas expressing EBNA1FL, 195 for HeLas expressing EBNA1ΔGAGR, 170 for HeLas expressing EBNA1ΔDBD, 230 for HeLas expressing EBNA1DBD, and 210 for HeLas expressing EBNA1DBDΔmid. Data are presented as bars representing mean values from two independent experiments.
Extended Data Figure 12. EBNA1 abundance has a dose-dependent effect on the frequency of breakage at 11q23 in DLD1 cells.
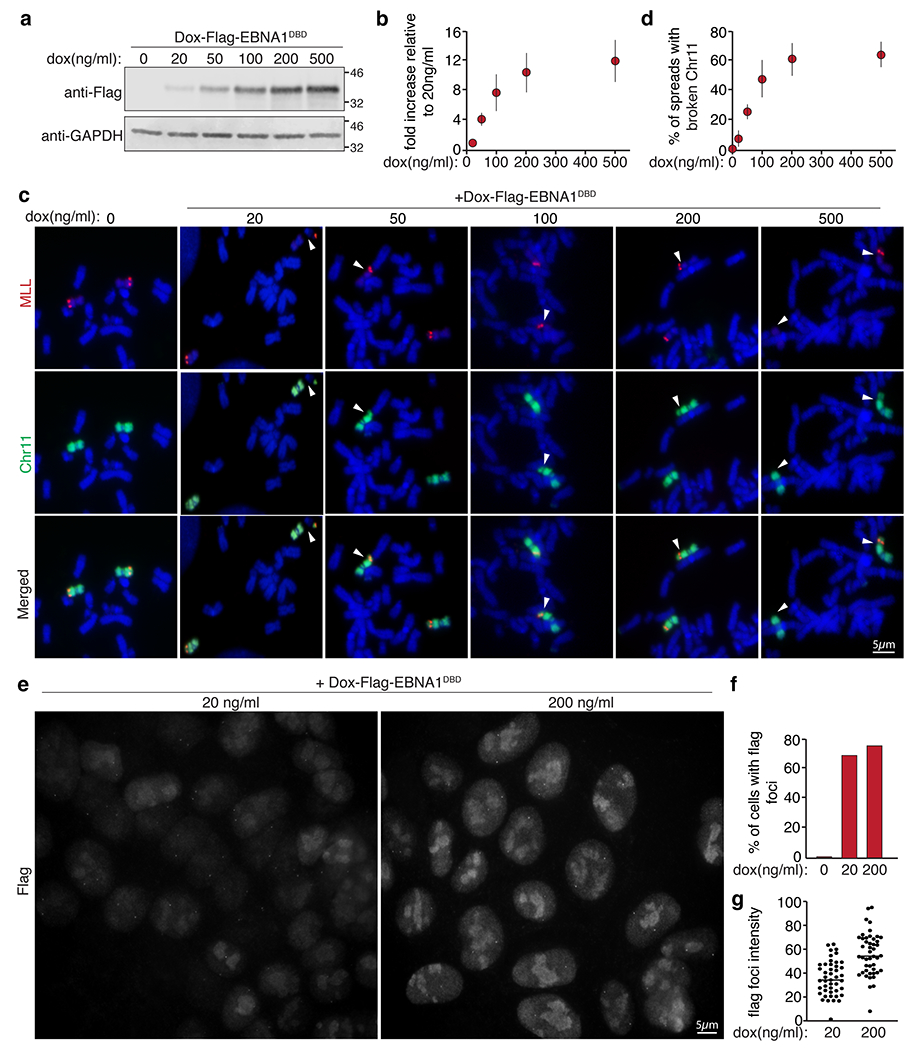
(a) anti-Flag immunoblot of dox-induced Flag-EBNA1DBD expression levels at the indicated doxycycline concentrations as quantified in (b). Data are presented as mean values +/− SEM from three independent experiments. (c) Dual colored FISH of mitotic chromosome 11 (in green) and MLL (in red) showing breakage proximal to MLL at day two following induction with the indicated doxycycline concentrations as quantified in (d). A total of 786 mitotic spreads were counted. Data are presented as mean values +/− SEM from three independent experiments. (e) Representative anti-Flag immunofluorescence images of Flag-EBNA1DBD expression at 20ng/ml or 200ng/ml. (f) Percentage of cells with flag foci. (g) Intensity of flag foci of 90 signals are quantified with horizontal bars representing the mean values.
Extended Data Figure 13. Cell lines latently infected with EBV express EBNA1 levels slightly below the levels required to induce 11q23 breakage.
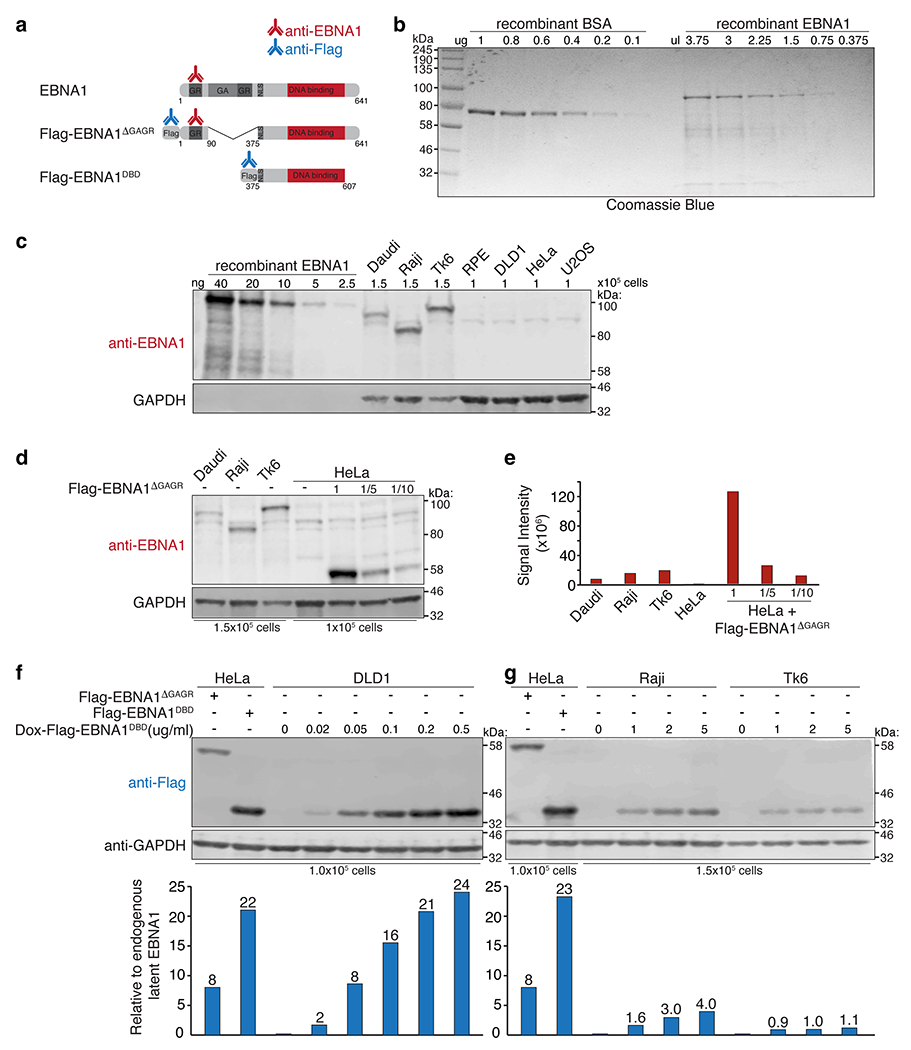
(a) Schematic of the antibodies used to quantify EBNA1. (b) Coomassie blue staining of recombinant BSA next to recombinant EBNA1 (Abcam 138345). (c) anti-EBNA1 (Santa Cruz sc-81581) immunoblot of recombinant EBNA1 next to whole cell lysates of the indicated cell lines. Anti-GAPDH immunoblot shows variation amongst the EBV-infected cell lines when loading the same number of cells. (d) anti-EBNA1 immunoblot of EBNA1 in the indicated EBV-infected cell lines next to HeLa cells expressing Flag-EBNA1ΔGAGR loaded at the indicated dilutions. (e) Bar graph represents signal intensity values normalized to number of cells loaded showing that the 1x Flag-EBNA1ΔGAGR in HeLa cells is expressed at 14-fold relative to baseline EBNA1 in Daudi cells and 8-fold in Raji cells and Tk6 cells. Immunoblots were repeated 3 times where S.D. values were below +/− 10%. (f-g) Anti-Flag immunoblot of HeLa cells, DLD1 cells, Raji cells, and Tk6 cells expressing the indicated Flag-tagged EBNA1 alleles. Bar graph represents fold relative to latent EBNA1 in Raji or Tk6 cells, calculated using the Flag-EBNA1ΔGAGR allele as 8-fold. Immunoblots were each repeated 3 times, S.D. values were below +/− 10%.
Extended Data Figure 14. Elevated levels of EBNA1 trigger breakage at 11q23 in cells latently infected with EBV.
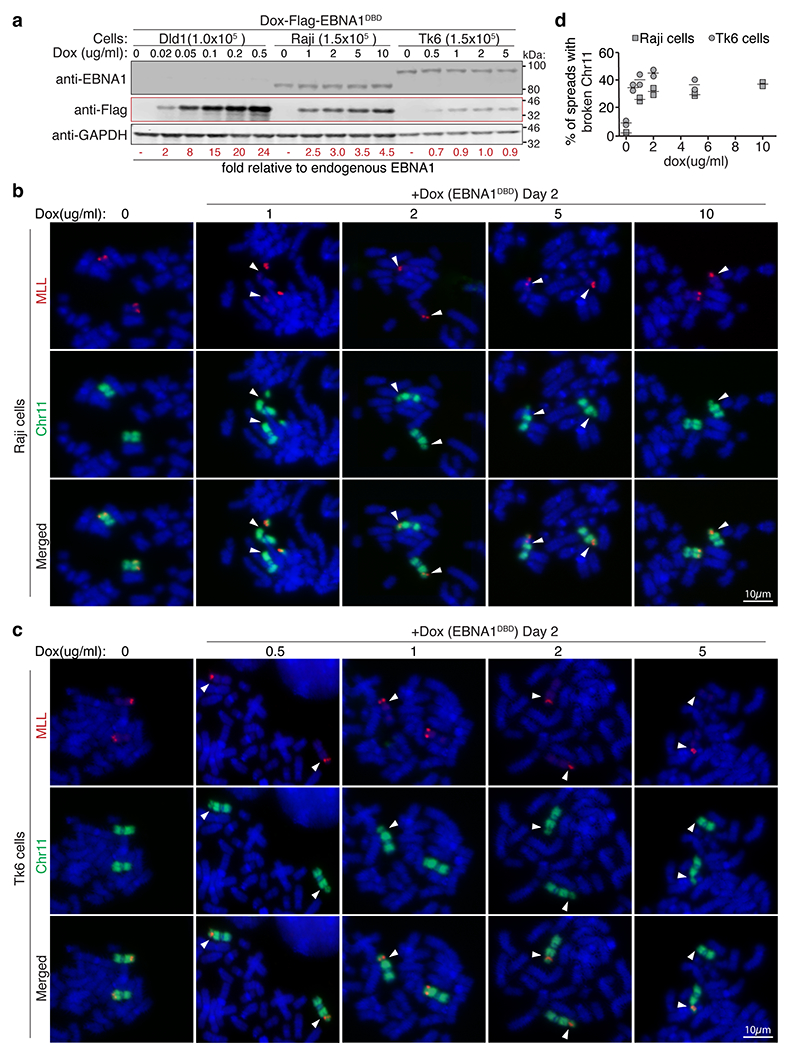
(a) anti-EBNA1 immunoblot verifies the presence of latent EBV genomes in Raji and Tk6 cells where baseline endogenous EBNA1 levels are largely unaffected by induced expression of Flag-EBNA1DBD. Anti-Flag immunoblot shows doxycycline-inducible expression of Flag-EBNA1DBD. Numbers (in red) indicate fold relative to baseline endogenous EBNA1 levels, calculated using Flag-EBNA1DBD levels in DLD1 cells previously determined to be 2, 8, 15, 20, and 24-fold. Immunoblots were repeated 3 times, S.D. Values were below +/− 10%. Representative dual-colored FISH images of chromosome 11 (in green) and MLL (in red) showing 11q23 breakage proximal to MLL (indicated by the white arrows) in either (b) Raji cells or Tk6 cells (c) as quantified in (d) following two days of treatment with the indicated dox concentrations. A total of 228 mitotic spreads were quantified for Raji cells. A total of 284 spreads were quantified for Tk6 cells. Data are presented as horizontal bars representing means from two independent experiments.
Extended Data Figure 15. Prevalence of chromosome 11 structural variations across EBV-infected Nasopharyngeal Carcinoma (NPC) and Pan-Cancer Analysis of Whole Genomes (PCAWG).
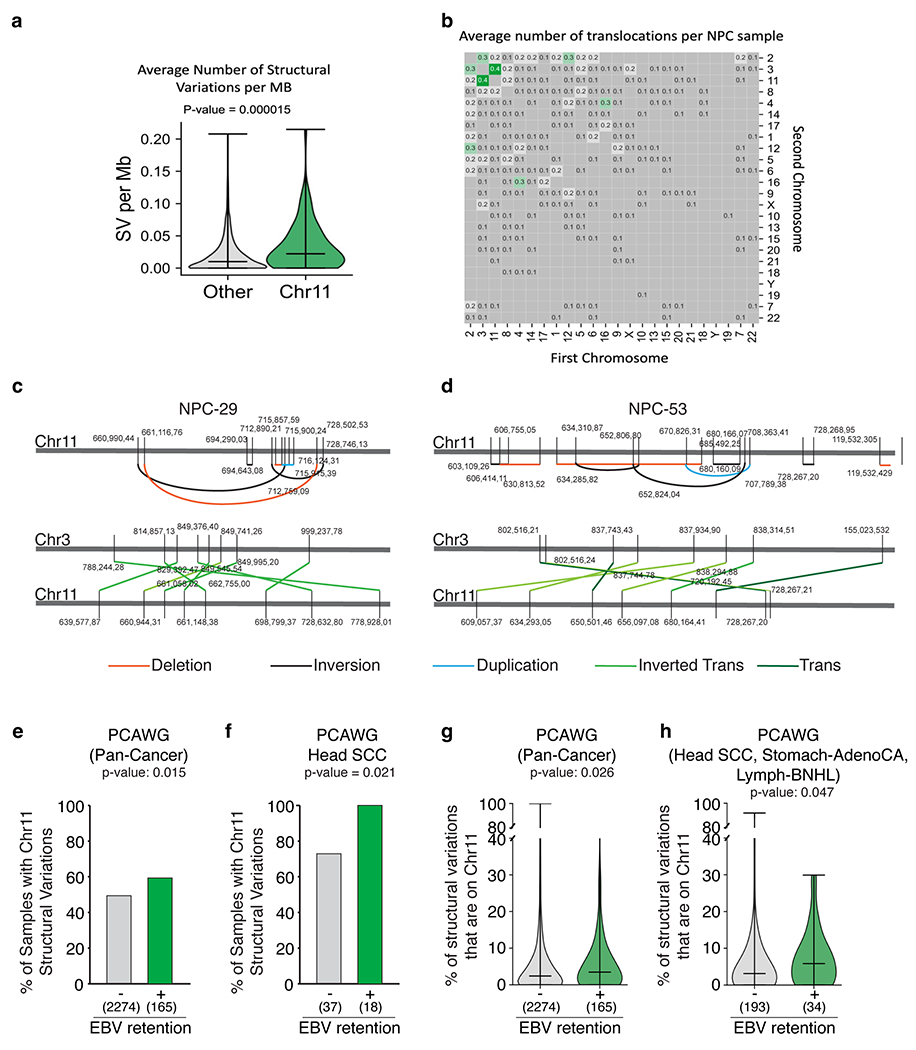
Average number of structural variations (SV) per Mb (a) and translocations (b) of n=78 EBV-infected NPC genomes. Bars represent the minimum, median and the maximum values observed in each violin plot (Two-sided Mann-Whitney U rank test, P-value is indicated, not corrected for multiple hypothesis testing). (c-d) Schematic of clustered structural variations (deletions, inversions, duplications, inverted translocations) on chromosome 11 in NPC genomes. (e-f) Proportion of samples with or without detectable EBV that harbor at least two chromosome 11 structural variations (Two-sided Fisher’s exact test, p-value is indicated, not corrected for multiple hypothesis testing; corrected p-values are 0.08 for pan-cancer and 0.05 for Head-SCC). (g-h) Proportion of structural variations (SV) that are on chromosome 11 in samples with or without detectable EBV. Bars represent the minimum, median and the maximum values observed in each violin plot (Two-sided Mann-Whitney U rank test. P-value is indicated, not corrected for multiple hypothesis testing).
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by grants from the US National Institutes of Health (R35 GM122476 to D.W.C. and R01ES030993-01A1, R01ES032547, and R01CA269919 to L.B.A.). J.S.Z.L. is supported by a postdoctoral fellowship from the Damon Runyon Cancer Research Foundation. L.B.A. is supported by a Packard Fellowship for Science and Engineering. D.W.C. receives salary support from the Ludwig Institute for Cancer Research. Special thanks to Shuvro Prokash Nandi for help with sequencing. The computational analyses reported in this manuscript have utilized the Triton Shared Computing Cluster at the San Diego Supercomputer Center of UC San Diego.
Data availability statement
All datasets used for the structural variation (SV) analyses are publicly available. For the 78 NPC samples, the SV calls are available through supplementary data tables from the following two studies cited in the main text: supplementary data table 6 from https://doi.org/10.1038/ncomms14121 and supplementary data table 5 from https://doi.org/10.1038/s41467-021-24348-6. For the PCAWG samples, the consensus SV calls were downloaded from the ICGC data portal: https://dcc.icgc.org/releases/PCAWG/consensus_sv. The following two files, which are both open access, were downloaded and used for the downstream SV analyses:
References
- 1.Pope JH, Horne MK & Scott W Transformation of foetal human keukocytes in vitro by filtrates of a human leukaemic cell line containing herpes-like virus. Int. J. Cancer 3, 857–866 (1968). [DOI] [PubMed] [Google Scholar]
- 2.Hsu JL & Glaser SL Epstein-barr virus-associated malignancies: epidemiologic patterns and etiologic implications. Crit. Rev. Oncol. Hematol 34, 27–53 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Thorley-Lawson DA & Gross A Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med 350, 1328–1337 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Rawlins DR, Milman G, Hayward SD & Hayward GS Sequence-specific DNA binding of the Epstein-Barr virus nuclear antigen (EBNA-1) to clustered sites in the plasmid maintenance region. Cell 42, 859–868 (1985). [DOI] [PubMed] [Google Scholar]
- 5.Bochkarev A, et al. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein EBNA 1. Cell 83, 39–46 (1995). [DOI] [PubMed] [Google Scholar]
- 6.Bochkarev A, et al. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein, EBNA1, bound to DNA. Cell 84, 791–800 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Sears J, et al. The amino terminus of Epstein-Barr Virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J. Virol 78, 11487–11505 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Leo A, Calderon A & Lieberman PM Control of Viral Latency by Episome Maintenance Proteins. Trends Microbiol. 28, 150–162 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humme S, et al. The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc. Natl. Acad. Sci. U. S. A 100, 10989–10994 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altmann M, et al. Transcriptional activation by EBV nuclear antigen 1 is essential for the expression of EBV’s transforming genes. Proc. Natl. Acad. Sci. U. S. A 103, 14188–14193 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frappier L Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses 4, 1537–1547 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu F, et al. Genome-wide analysis of host-chromosome binding sites for Epstein-Barr Virus Nuclear Antigen 1 (EBNA1). Virol J. 7, 262 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tempera I, et al. Identification of MEF2B, EBF1, and IL6R as Direct Gene Targets of Epstein-Barr Virus (EBV) Nuclear Antigen 1 Critical for EBV-Infected B-Lymphocyte Survival. J. Virol 90, 345–355 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim KD, et al. Epigenetic specifications of host chromosome docking sites for latent Epstein-Barr virus. Nature communications 11, 877 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanda T, Kamiya M, Maruo S, Iwakiri D & Takada K Symmetrical localization of extrachromosomally replicating viral genomes on sister chromatids. J. Cell Sci 120, 1529–1539 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Wang H, et al. CRISPR-Mediated Programmable 3D Genome Positioning and Nuclear Organization. Cell 175, 1405–1417.e1414 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen B, et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ambinder RF, Mullen MA, Chang YN, Hayward GS & Hayward SD Functional domains of Epstein-Barr virus nuclear antigen EBNA-1. J. Virol 65, 1466–1478 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ambinder RF, Shah WA, Rawlins DR, Hayward GS & Hayward SD Definition of the sequence requirements for binding of the EBNA-1 protein to its palindromic target sites in Epstein-Barr virus DNA. J. Virol 64, 2369–2379 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown RE & Freudenreich CH Structure-forming repeats and their impact on genome stability. Curr. Opin. Genet. Dev 67, 41–51 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang G & Vasquez KM Dynamic alternative DNA structures in biology and disease. Nature reviews. Genetics (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durkin SG & Glover TW Chromosome fragile sites. Annu. Rev. Genet 41, 169–192 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Glover TW, Wilson TE & Arlt MF Fragile sites in cancer: more than meets the eye. Nat. Rev. Cancer 17, 489–501 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yunis JJ The chromosomal basis of human neoplasia. Science 221, 227–236 (1983). [DOI] [PubMed] [Google Scholar]
- 25.Yunis JJ & Soreng AL Constitutive fragile sites and cancer. Science 226, 1199–1204 (1984). [DOI] [PubMed] [Google Scholar]
- 26.Yu S, et al. Human chromosomal fragile site FRA16B is an amplified AT-rich minisatellite repeat. Cell 88, 367–374 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Boteva L, et al. Common Fragile Sites Are Characterized by Faulty Condensin Loading after Replication Stress. Cell reports 32, 108177 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sfeir A, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bashaw JM & Yates JL Replication from oriP of Epstein-Barr virus requires exact spacing of two bound dimers of EBNA1 which bend DNA. J. Virol 75, 10603–10611 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malik-Soni N & Frappier L Proteomic profiling of EBNA1-host protein interactions in latent and lytic Epstein-Barr virus infections. J. Virol 86, 6999–7002 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Umbreit NT, et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 368(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JH & Paull TT Cellular functions of the protein kinase ATM and their relevance to human disease. Nat. Rev. Mol. Cell Biol 22, 796–814 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Thirman MJ, et al. Rearrangement of the MLL Gene in Acute Lymphoblastic and Acute Myeloid Leukemias with 11q23 Chromosomal Translocations. N. Engl. J. Med 329, 909–914 (1993). [DOI] [PubMed] [Google Scholar]
- 34.Fu YH, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 67, 1047–1058 (1991). [DOI] [PubMed] [Google Scholar]
- 35.van Wietmarschen N, et al. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 586, 292–298 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lieberman PM Keeping it quiet: chromatin control of gammaherpesvirus latency. Nat Rev Microbiol 11, 863–875 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stephens PJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kato H & Sandberg AA Chromosome pulverization in human cells with micronuclei. J. Natl. Cancer Inst 40, 165–179 (1968). [PubMed] [Google Scholar]
- 39.Zhang CZ, et al. Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ly P, et al. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol 19, 68–75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leibowitz ML, et al. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet 53, 895–905 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cortés-Ciriano I, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet 52, 331–341 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Young LS & Rickinson AB Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4, 757–768 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Pathmanathan R, Prasad U, Sadler R, Flynn K & Raab-Traub N Clonal proliferations of cells infected with Epstein-Barr virus in preinvasive lesions related to nasopharyngeal carcinoma. N. Engl. J. Med 333, 693–698 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Bruce JP, et al. Whole-genome profiling of nasopharyngeal carcinoma reveals viral-host co-operation in inflammatory NF-κB activation and immune escape. Nature communications 12, 4193 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li YY, et al. Exome and genome sequencing of nasopharynx cancer identifies NF-κB pathway activating mutations. Nature communications 8, 14121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zapatka M, et al. The landscape of viral associations in human cancers. Nat. Genet 52, 320–330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sivachandran N, Wang X & Frappier L Functions of the Epstein-Barr virus EBNA1 protein in viral reactivation and lytic infection. J. Virol 86, 6146–6158 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo R, et al. MYC Controls the Epstein-Barr Virus Lytic Switch. Mol. Cell 78, 653–669.e658 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chien YC, et al. Serologic markers of Epstein-Barr virus infection and nasopharyngeal carcinoma in Taiwanese men. N. Engl. J. Med 345, 1877–1882 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Ambinder RF Gammaherpesviruses and “Hit-and-Run” oncogenesis. Am. J. Pathol 156, 1–3 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shoshani O, et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 591, 137–141 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Celli GB & de Lange T DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol 7, 712–718 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets used for the structural variation (SV) analyses are publicly available. For the 78 NPC samples, the SV calls are available through supplementary data tables from the following two studies cited in the main text: supplementary data table 6 from https://doi.org/10.1038/ncomms14121 and supplementary data table 5 from https://doi.org/10.1038/s41467-021-24348-6. For the PCAWG samples, the consensus SV calls were downloaded from the ICGC data portal: https://dcc.icgc.org/releases/PCAWG/consensus_sv. The following two files, which are both open access, were downloaded and used for the downstream SV analyses: