

Abstract
Significant advances in physicochemical properties of polymeric micelles enable optimization of therapeutic drug efficacy, supporting nanomedicine manufacturing and clinical translation. Yet, the effect of micelle morphology on pharmacological efficacy is not adequately addressed. This work addresses this gap by assessing pharmacological efficacy of polymeric micelles with spherical and worm-like morphologies. It is observed that poly(2-oxazoline)-based polymeric micelles can be elongated over time from a spherical structure to worm-like structure, with elongation influenced by several conditions, including the amount and type of drug loaded into the micelles. The role of different morphologies on pharmacological performance of drug loaded micelles against triple-negative breast cancer and pancreatic cancer tumor models is further evaluated. Spherical micelles accumulate rapidly in the tumor tissue while retaining large amounts of drug; worm-like micelles accumulate more slowly and only upon releasing significant amounts of drug. These findings suggest that the dynamic character of the drug–micelle structure and the micelle morphology play a critical role in pharmacological performance, and that spherical micelles are better suited for systemic delivery of anticancer drugs to tumors when drugs are loosely associated with the polymeric micelles.
Keywords: critical micelle concentration, micelle morphology, pharmacokinetics, polymeric micelles, tumor accumulation
1. Introduction
Polymeric micelles formed by amphiphilic block copolymers exhibit large potential as platforms for solubilization and delivery of poorly soluble therapeutic compounds. Several polymeric-micelle drugs are approved for clinical use and others are in the pipeline for clinical evaluation and approval worldwide. Unique traits of polymeric micelles include ease of self-assembly, universality with respect to drug compatibility, and ability to address major challenges in pharmaceutics, such as poor drug solubility. However, micelles’ dynamic character also limits in vivo testing and therefore understanding of their performance. Despite considerable progress in this field, understanding of how the physicochemical properties (stability, size, shape, extent of drug loading, and drug release)[1–3] relate to the pharmacological performance remains elusive.
Kataoka and colleagues demonstrated that tumor accumulation of polymeric micelles depends on particle size and tumor desmoplasticity;[2] smaller polymeric micelles show better accumulation and penetration into more desmoplastic tumor and possess improved antitumor activity. A study by Discher and colleagues emphasized the importance of micelle morphology; filament, worm-like micelles display greater longevity in plasma circulation, whereas micelle disassembly into smaller spherical particles results in faster clearance.[1] Yet, neither of these studies related micellar pharmacological performance to their drug loading and release characteristics—a critical aspect of drug efficacy. In the former study, drug was covalently bound to the micelles and slowly released as a result of ligand exchange, a process proceeding in biological milieu over several days. In the latter, micelles were free of the drug. Subsequent attempts to prepare micelles that maintain filament morphology while retaining pharmacologically relevant drug loadings have been unsuccessful.[4]
A useful and potentially universal embodiment of this technology is the polymeric micelle system that may: solubilize drugs without the need for chemical drug modification, have a drug-loading capacity of at least 10% by mass, and release drug within at least 12 h (i.e., slow release). The current study advances our understanding of the pharmacological performance of such micelles by using poly(2-oxazoline) (POx) block copolymers. These block copolymers form polymeric micelles that incorporate many structurally diverse drugs at high loadings. The differential, noncovalent interactions of the block copolymers with the drugs lead to the formation of micelles with two distinct morphological and stabilization characteristics: those that stabilize small spherical micelles, and those that allow the formation of worm-like micelles, even at high drug loadings. We also examined the complex interplay between micelle circulation, drug release, and the ability of different morphologies to reach their site of action while carrying the loaded drug. To do so, we carried out a comprehensive analysis that includes physiochemical structural measurements, molecular interactions and modeling, drug release, in vivo pharmacokinetics (PK), and antitumor activity in animal models. Our findings indicate that formulating drugs into small polymeric micelles is beneficial to their pharmacological performance; their geometries are conducive to increased colloidal stability, formulation reproducibility, and drug delivery to tumors.
2. Results
2.1. Multiple Morphologies in Blank Polymeric Micelles
To understand how micellar morphologies influence drug loading and pharmacological performance, we prepared micellar structures with varying morphologies. This was accomplished by self-assembly of amphiphilic block copolymers above a critical micelle concentration (CMC). Micellar structures contain a hydrophobic core that is shielded from the ambient aqueous solution by a corona of hydrophilic blocks. Micellar structures can exhibit different shapes, including spheres, rods, worm-like structures, and bilayers which can subsequently develop into vesicles.[5,6] In particular, polymeric micelles can undergo 1D growth to form long worm-like aggregates. This behavior is captured in the four synthesized A-B-A triblock copolymers (T1–T4; Figures S1 and S2, Supporting Information), in which hydrophilic poly(2-methyl-2-oxazoline) A-blocks vary in length and hydrophobic poly(2-butyl-2-oxazoline) B-blocks are kept constant at 20 repeating units (r.u.). Micelles were prepared from these copolymers using a thin film hydration method; resulting structures were observed using transmission electron microscopy (TEM). We also examined them at two time intervals: 1 and 72 h after formation. After 1 h, all micelles exhibited small spherical structures (Figure 1a). After 72 h, the population gradually shifted to worm-like morphologies (Figure 1b and Figure S3a, Supporting Information). Further, the elongation process from spherical to worm-like micelles strongly correlated to the change in particle size and size distribution, as shown by dynamic light scattering (DLS; Figure 1c). Spherical particles examined after 1 h exhibited particle sizes in the 10–30 nm range with a narrow size distribution. As micelles elongated, particle size and polydispersity index (PDI) value increased simultaneously, indicating the co-existence of spherical micelles and worm-like micelles. Micelles continued to elongate until reaching worm-like structures 200 nm or larger and exhibiting a narrow size distribution. Thus, DLS provides a convenient tool for predicting the morphology of the micelles since we observe only two types of micelles clearly defined in TEM.
Figure 1.
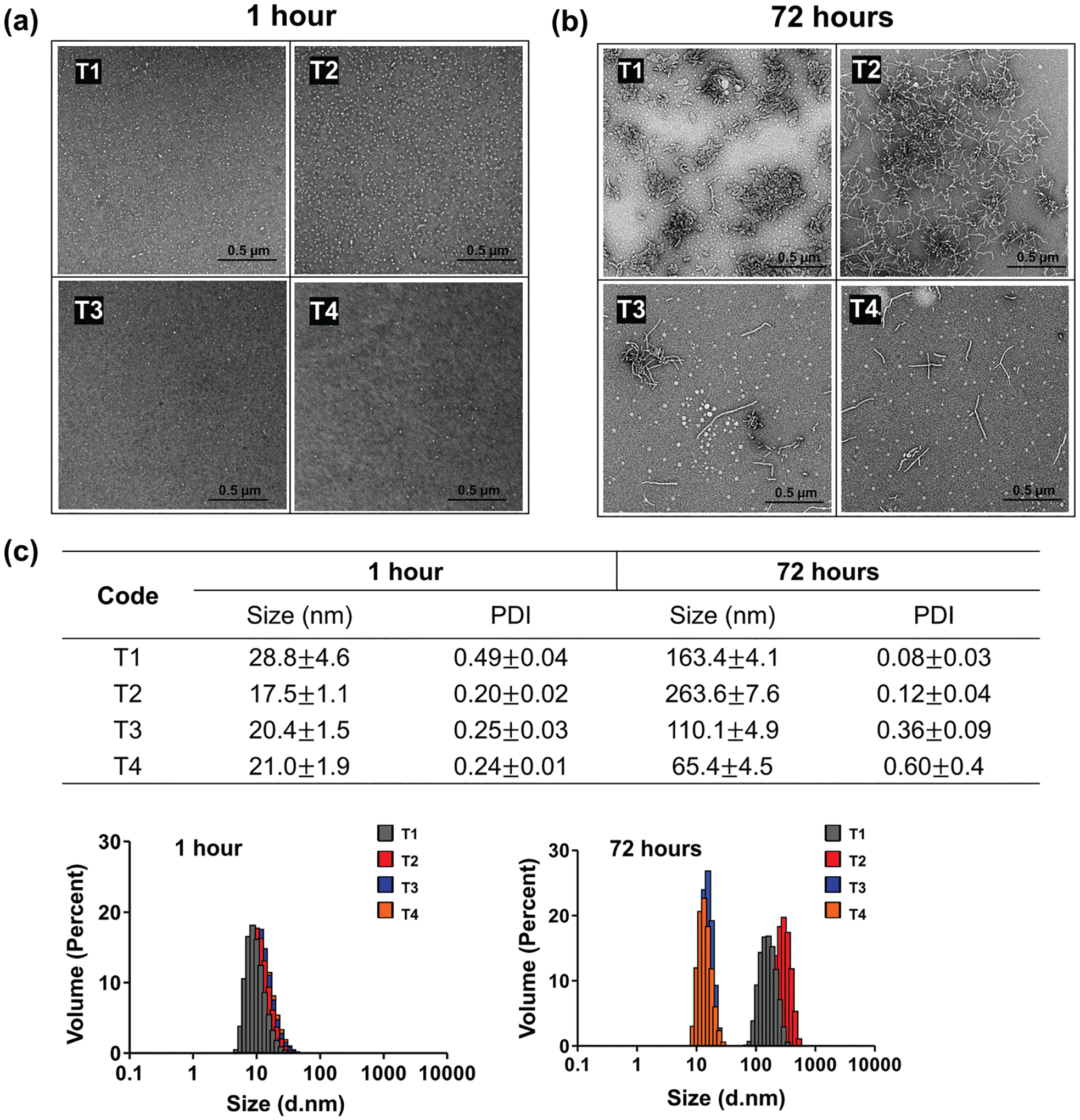
a,b) TEM images and c) DLS size (DZ), polydispersity (PDI), and volume weighted size distribution of T1–T4 based micelles at 1 and 72 h. Polymer concentrations are 10 mg mL−1 in distilled water. Errors shown represent three standard deviations of uncertainty.
Elongation and growth were predominantly dependent on the A block length; the T1 and T2 polymers completely converted to nonspherical or worm-like micelles after 72 h, whereas T3 and T4 transitioned to a mixture of morphologies. With the T1 having the shortest A block (10 r.u.) micelles grew three-dimensionally and anisotropically. With the T2 having longer A block (20 r.u.) micelles elongated in a 1D manner resulting in worm-like structures. In the case of the T3 and T4, having even longer A blocks (30 and 40 r.u., respectively), some of the micelles elongated in a 1D manner but many structures remained spherical even after 72 h. These results are consistent with the well-known principles for polymeric micelle formation.[5,7] The repulsion of the hydrophilic chains strongly influences the growth and the resulting morphology of the polymeric micelles. The T1 and T2 likely cannot ensure sufficient repulsion in the shell to counteract elongation. An increase in the hydrophilic block length led to an increase of the repulsion due to the excluded volume effect within the shell. This favored spherical micelles, where the surface area per one hydrophilic chain is greater and led to partial inhibition of the spherical-to-worm-like structural transitions in T3 and T4. Despite being thermodynamically favored, the worm-like micelle formation was kinetically limited, taking up to a couple of days before completion in T2.
We also investigated micelle elongation with respect to the polymer concentration, temperature, and salt concentration. Generally, elongation was either favored by increasing the polymer concentration and temperature or inhibited by increasing the ionic strength (Figure S3b, Supporting Information). As the polymer concentration increased, the elongation rate increased probably due to more frequent micelle collisions. As the temperature increased, the elongation accelerated probably because of increased hydrophobic interactions. Finally, addition of salt can favor the formation of spherical micelles due to dehydration of polymer core and increased repulsion of the chains in the shell. Altogether, these parameters could provide control over micelle stability and morphology, yielding a more predictable behavior during the formation process.
2.2. Drug Dependent Effects on Micelle Morphology
Previously, we showed that POx micelles can exhibit different morphologies when loaded with different drugs. We observed two types of behavior: the first, exemplified by paclitaxel, inhibited the formation of worm-like micelle architecture and stabilized the compact micelle spheres to retain a narrow size distribution;[4] the second, exemplified by etoposide, was unable to prevent the formation of elongated worm-like micelles even at high drug loadings.[8] For simplicity, we refer henceforth to the first behavior as “worm-inhibiting,” and to the second behavior as “worm-permissive.” As a proof of concept, we randomly selected a set of 20 poorly soluble drugs and tested their loading efficiency (LE) and loading capacity (LC) in our POx micelles (Table S1, Supporting Information). Drugs with high LC were first encapsulated by POx micelles at various loading ratios (polymer/drug w/w ratios of 10/1, 10/2, 10/4, and 10/8). Then, the sphere-to-worm-like transition processes were monitored over time as done before drug-loading. For comparison we also varied the temperature at 4 and 25 °C, which strongly affected the transition, and used the T2 block copolymer with relatively shorter hydrophilic blocks to increase the propensity of the transitions (Figure 2).
Figure 2.
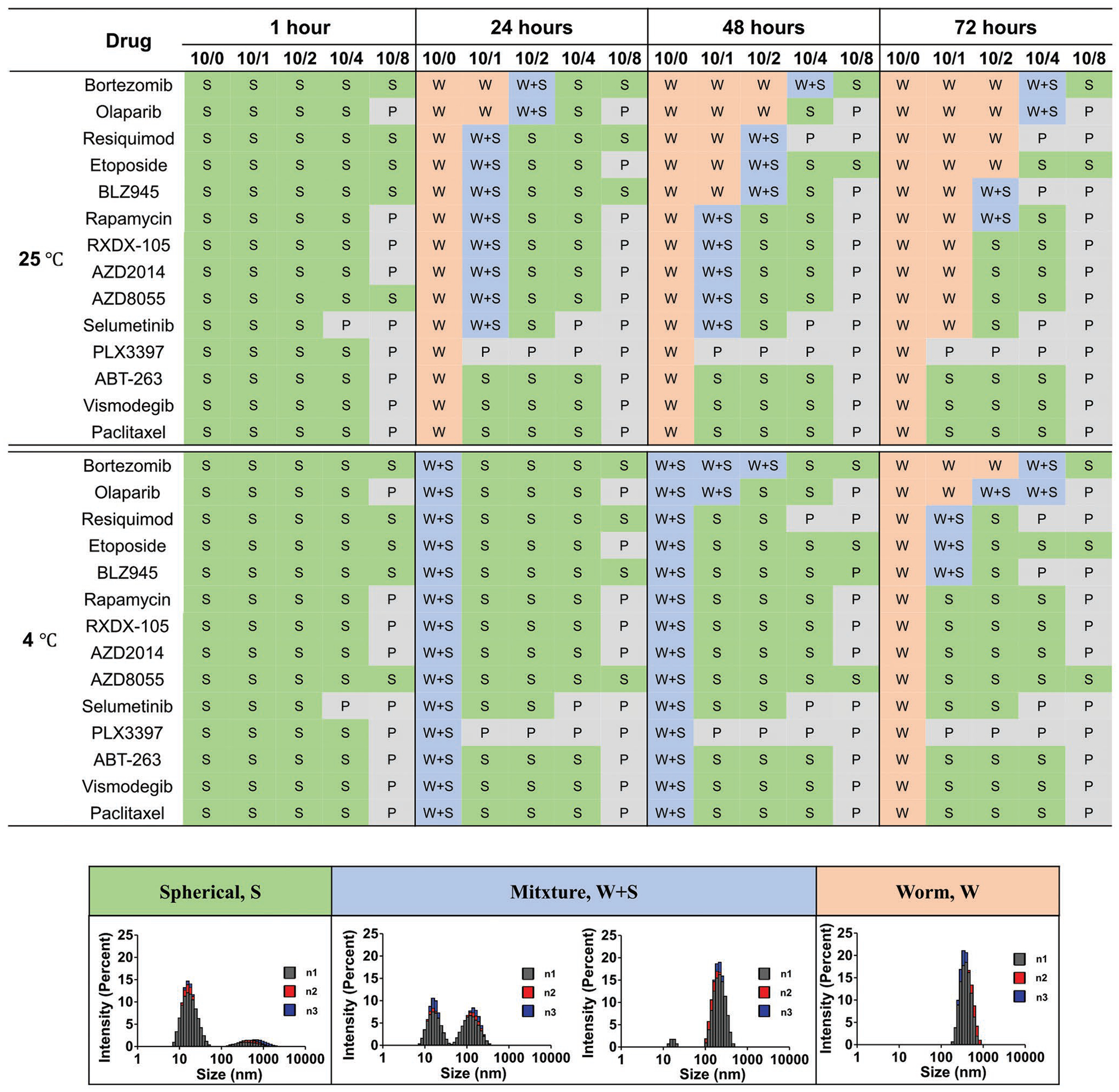
Monitoring the sphere-to-worm-like transition process of T2-based polymeric micelles at various drug loading ratios (polymer/drug w/w ratios of 10/1, 10/2, 10/4, and 10/8) over time. The matrix denotes the resulting morphologies: S, spherical micelles; W+S, mixture of worms and spheres; W, worm-like micelles; and P, precipitation. Increased drug loading resulted in slowing or inhibition of the sphere-to-worm transition. The inset presents the intensity weighted distribution.
We next examined the role of size and synthesis conditions on micellar structure. DLS was used as an indicator of resulting morphologies, which were then validated by cryo-TEM (Figures S4 and S5, Supporting Information). Generally, sphere-to-worm transitions were slowed or inhibited as drug loading increased. This was evidenced by the reduced width of peaks in the distribution of hydrodynamic diameters. Drugs exhibited differential ability to inhibit worm formation. For example, at room temperature, blank spherical micelles become elongated into worm structures after a 24-h incubation. [The sphere-to-worm transition rarely happened at low temperature (4 °C).] Micelles loaded with bortezomib, olaparib, etoposide, or resiquimod showed a similar or slightly slower transition to worm-like structure, which was the most pronounced at 72 h. Other drugs, such as BLZ945, rapamycin, RXDX105, AZD2014, AZD8055, and selumetinib, slightly inhibited the sphere-to-worm transition (especially at higher loading) but did not abolish the transition. In contrast, vismodegib, paclitaxel, and ABT-263 inhibited elongation at a mass fraction of as little as 10%. Therefore, we unveiled two distinct groups of drugs: those that are worm-permissive under certain conditions (even at high drug loadings) and those that are worm-inhibiting and stabilize the compact spherical micelle morphology. Notably for most of these drugs, the formation of worms and in many cases precipitates at high loadings were suppressed by using a block copolymer with longer hydrophilic blocks, such as T3 (Figure S6, Supporting Information). Interestingly, for the worm-inhibiting drugs ABT-263, paclitaxel, and vismodegib, maximal solubilization in T3 was twofold more than that of the same drugs in T2 (8 mg mL−1 vs 4 mg mL−1). Solubility of the worm-permissive drugs did not differ for these polymers.
2.3. Analysis of Micelle Structure by Small-Angle Neutron Scattering (SANS)
To better understand the morphology and effects of different drugs in POx micelles, SANS data were collected on a series of drug concentrations while keeping the block copolymer concentration constant (10 mg mL−1 T2 in D2O). Figure 3 shows the scattering data and fits at different drug loadings. A polydisperse core–shell spherical model for the single micelles in solution provides a consistently reasonable fit for the medium to high scattering angle data. Both vismodegib and paclitaxel stabilize core–shell spherical particles at lower drug concentrations, while worm-like larger clusters still contribute to scattering in the presence of olaparib and etoposide up to 2 mg mL−1 of drug.
Figure 3.
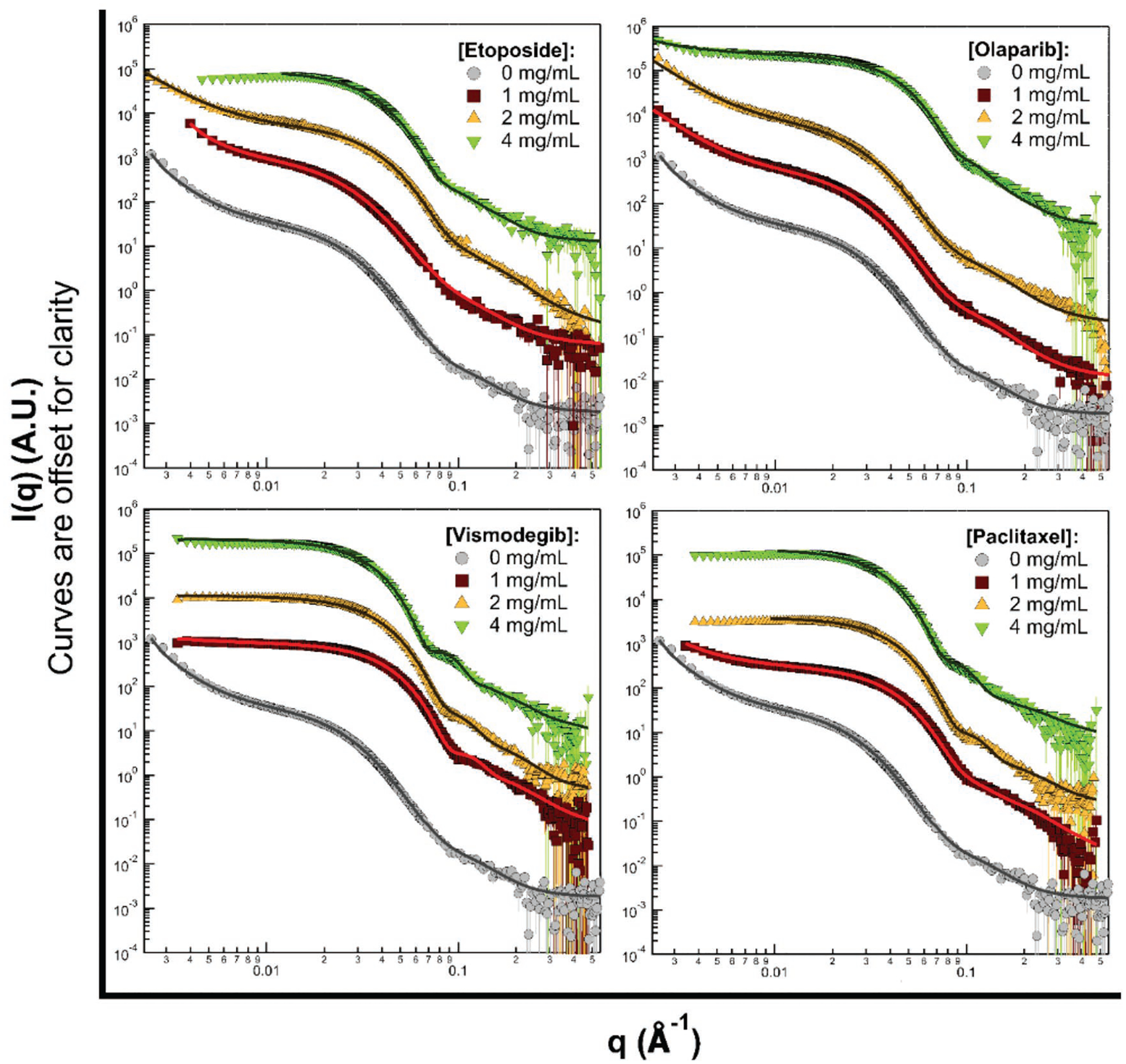
SANS data (symbols) measured for solutions of 10 mg mL−1 of T2 with various drug loading ratios, as labeled. The continuous lines show fits to the data, where the model function was smeared with the instrument resolution information prior to fitting. Error bars correspond to one standard deviation of uncertainty.
A comparative plot of the average core radius is shown in Figure 4, shell thickness and neutron scattering length densities (SLD) of micelles, as a function of the drug concentration (see Table S2, Supporting Information, for a list of the fit parameters). Although the data fitting can only provide a semi-quantitative measurement, given the polydispersity of the samples and the presence of larger particles with dimensions outside the scope of the SANS data measured, the results are consistent with our findings for the worm-permissive and worm-inhibiting drugs. The calculated average diameter of blank T2 sphere micelles in solution is 14.2 ± 1.1 nm (where 1.1 nm corresponds to one standard deviation [SD] of uncertainty). With increasing drug concentration, all T2 solutions show a growth in the core radius, with no significant variation in the core SLD. When compared to the worm-permissive drugs, paclitaxel and vismodegib show more pronounced core size increases and smaller shell thickness. The observed shell SLD with drug loading is consistent with increased hydration of the shell. For the worm-permissive drugs, the shell SLD appears to increase proportionally to drug concentration. At high loads, the increase in SLD reflects the smearing of the core–shell particle structure as the composition changes with drug incorporation, where the high SLD of the shell is consistent with a heavily hydrated outer layer and low polymer density. Overall, the variations observed in core size, shell thickness, and SLDs reflect rearrangements of the micelle composition and density in the presence of increasing amounts of drug.
Figure 4.
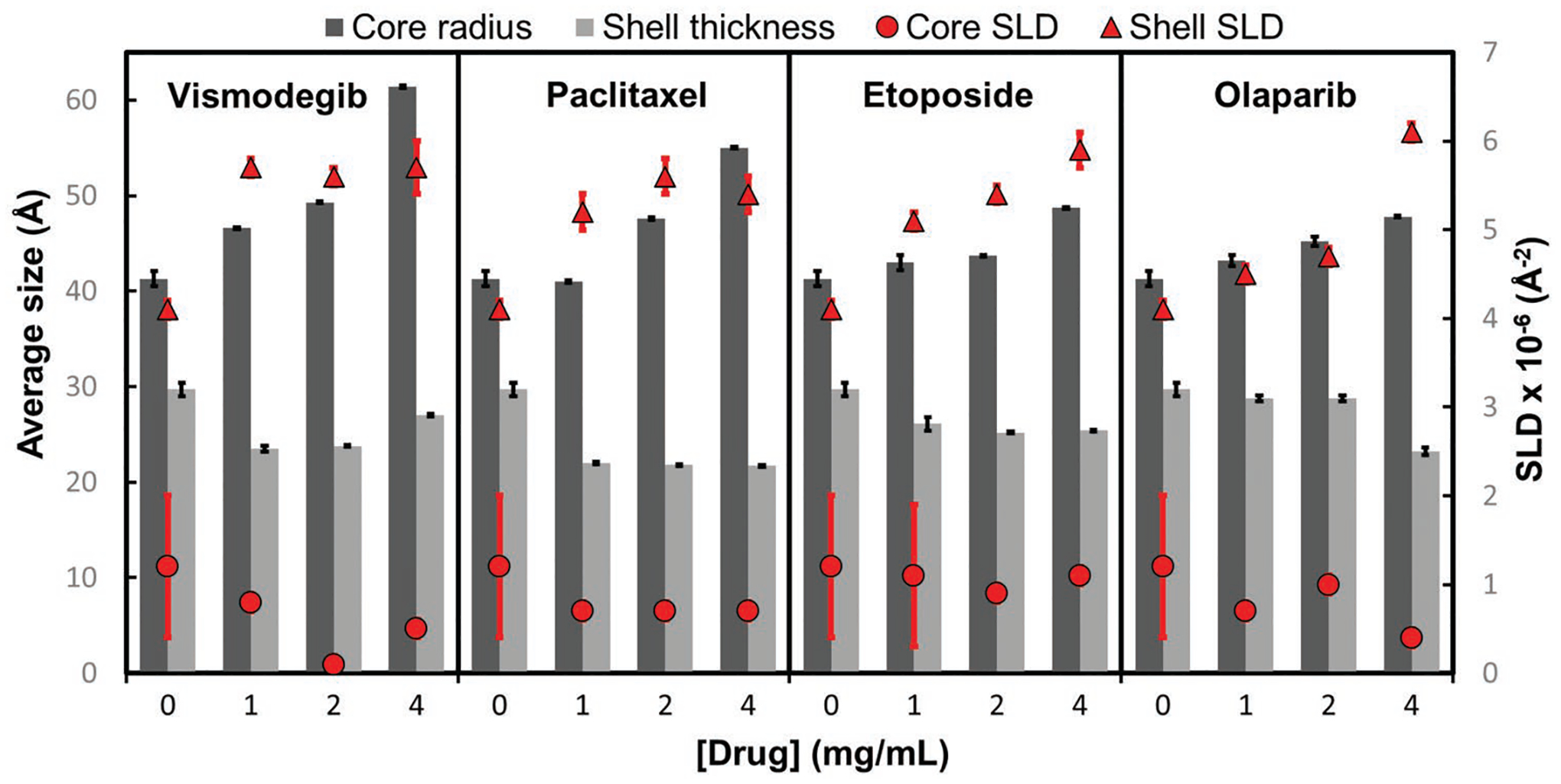
Fitted parameters for SANS data of T2 micelles in the presence of various drug concentrations. The dark grey bars represent the average core radius of the micelles in the sample, the light gray bars show the average shell thickness, red circles show the micelle core SLD, and red squares show the shell SLD. For comparison, the calculated neutron SLD for pure drug solutions in 100% D2O are: 2.929 × 10−6 Å−2 for Etoposide (d = 1.6 g cm−3), 2.797 × 10−6 Å−2 for olaparib (d = 1.4 g cm−3), 2.694 × 10−6 Å−2 for paclitaxel (d = 1.4 g cm−3), and 2.845 × 10−6 Å−2 for vismodegib (d = 1.4 g cm−3). Error bars correspond to one standard deviation of uncertainty.
2.4. Assessing Drug Localization in the Micelles by Molecular Dynamics (MD) Simulation
To understand drug locations within the micellar structures, we performed MD simulations of drug-loaded T2 micellar structures. Simulations of etoposide and paclitaxel loaded into POx micelles were performed at various polymer and drug molar ratios (50:50, 50:100, and 50:200) corresponding to the mass ratios (10/1, 10/2, and 10/4) used throughout this work (Figure 5). We observed self-aggregation of polymers in every simulation. At every drug loading, most etoposide molecules were located at the center of the micelles, indicating that etoposide binds to the hydrophobic micelle core. In contrast, paclitaxel was located initially in the peripheral areas of micelles at lower loading, potentially coming in contact with both core- and shell-forming blocks. As drug loading increased, drug placement in the core also increased. This simulation suggests large differences in the drug–polymer interactions displayed by worm-permitting and worm-inhibiting drugs.
Figure 5.
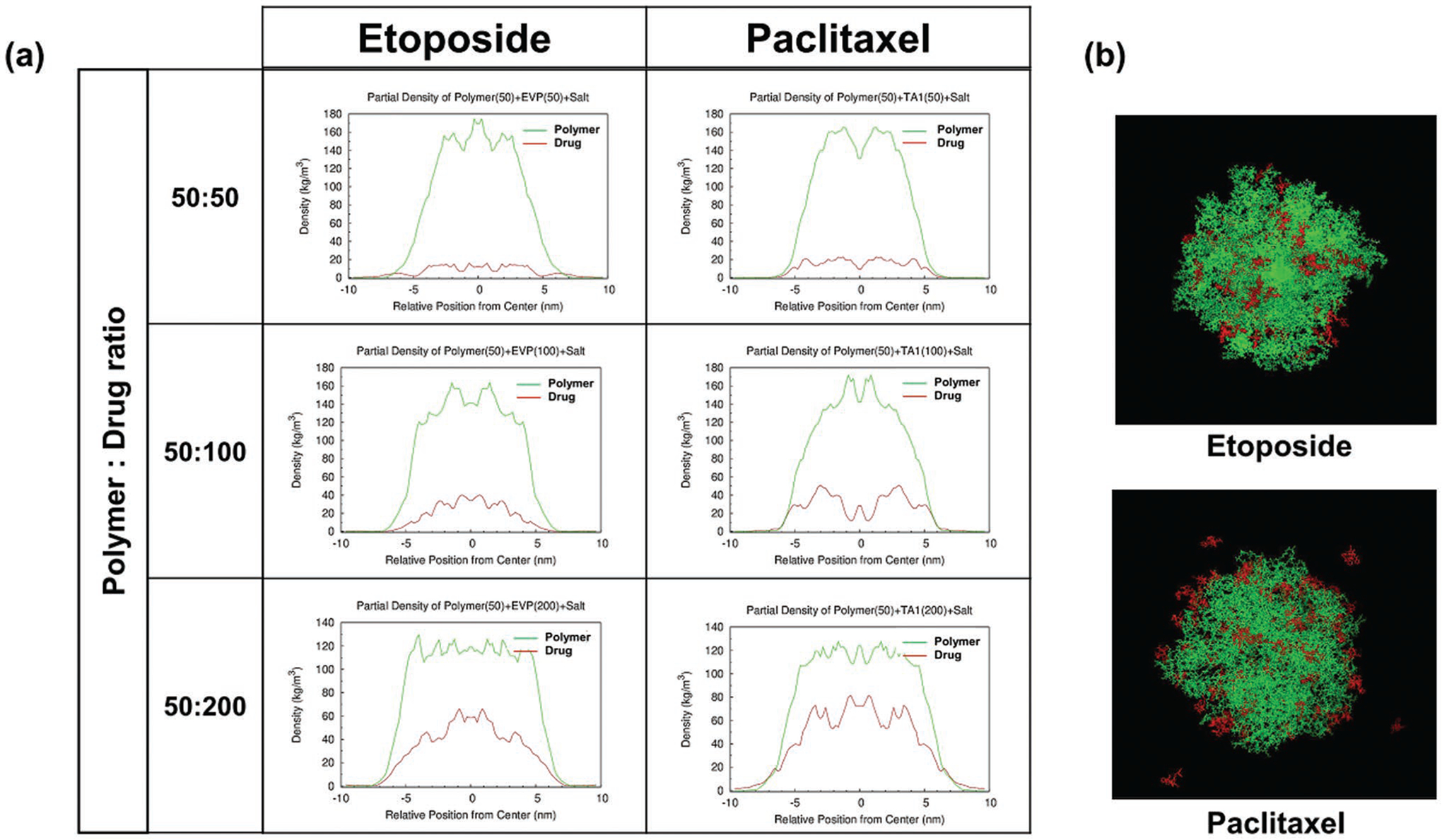
MD simulation of etoposide and paclitaxel in T2 micelles: a) the drug (red) and polymer (green) density profiles from the center of the micelles, and b) simulated snapshots of the drug-loaded micelles. The 50 polymer chains and 50, 100, and 200 drug molecules were simulated for a total of 100 ns.
2.5. Effect of Drugs on the Stability of the Micelles by DLS Measurements
For the first time, we demonstrate here the differential effects of drugs on the stability of morphologically distinct micelles formed from the same block copolymer. We determined the effective CMC* by measuring DLS count rates upon dilution at 1 and 72 h (Figure S7, Supporting Information). By this method CMC* of the blank worm-like micelles of T2 was approximately six times smaller than that of spherical micelles for the same polymer (T2) (Figure 6). However, there is an inherent limitation of the method in comparing CMC* between different morphologies because the DLS intensity signal is more sensitive to particles of larger size. Therefore, we focused on comparing the CMC* of drug loaded micelles within the same morphology. For worm-permissive olaparib and etoposide, the drug loading resulted in a small decrease in CMC* for spherical micelles (Figure 6). In contrast, worm-inhibiting drugs (paclitaxel and vismodegib) decreased the CMC* of spherical micelles to a greater extent. At high drug loadings, the apparent CMC* of spherical micelles both at 1 and 72 h was as low as that of the worm-like micelles in the absence of drug. Thus, worm-inhibiting drugs appear to stabilize the spherical micelle structure upon dilution.
Figure 6.
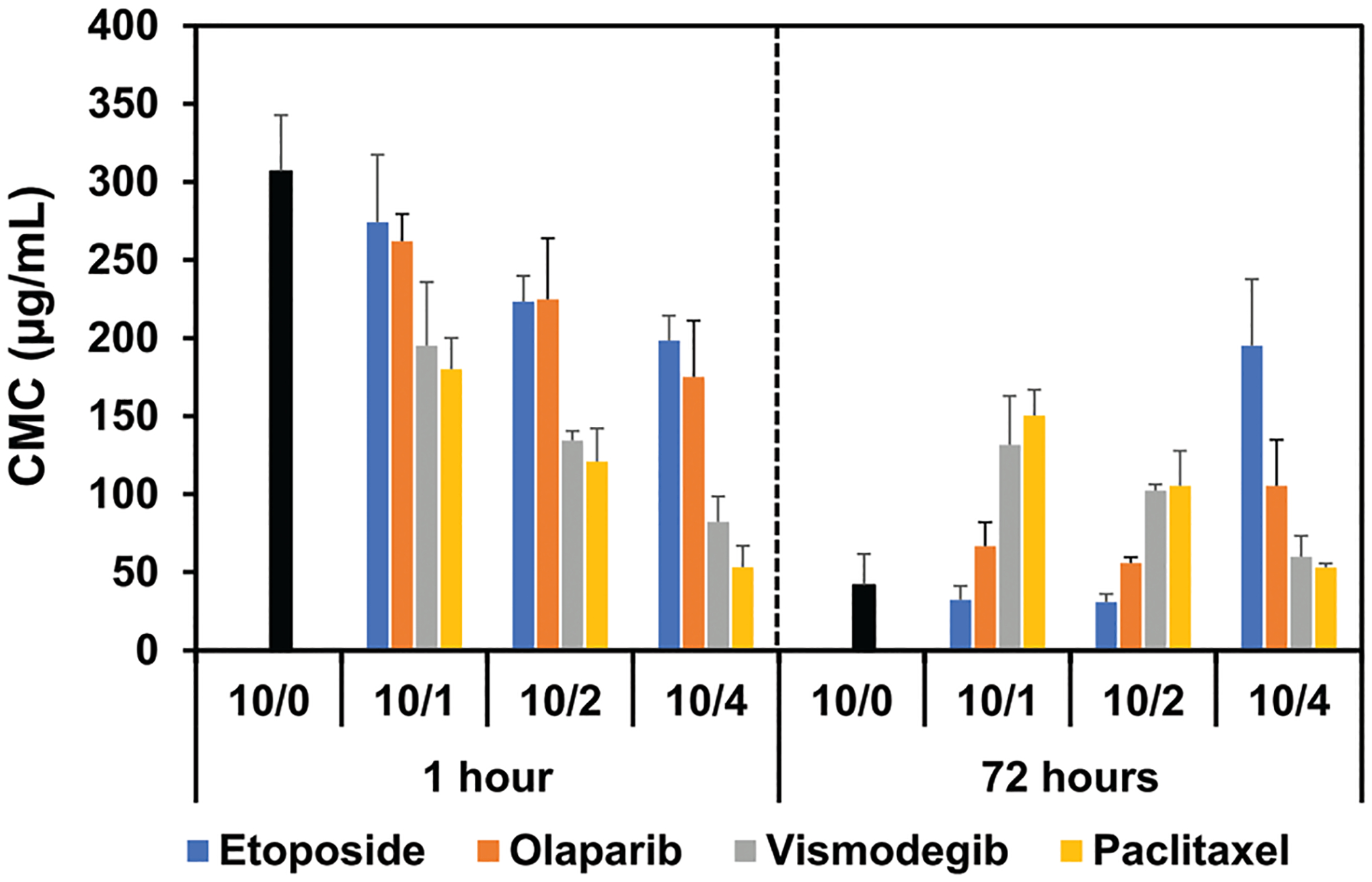
Effective CMC* of polymeric micelles at various drug loading ratios (polymer/drug ratios of 10/0, 10/1, 10/2, and 10/4) determined by DLS at 1 and 72 h after formation at 25 °C. Error bars correspond to three standard deviation of uncertainty.
2.6. Probing Drug Effects on the Micelle Microenvironment by Reichardt’s Dye (RD)
To investigate the changes of the micellar microenvironment upon the loading of different drugs we used 2,6-diphenyl-4-(2,4,6-triphenyl-N-pyridino)phenolate, better known as RD, which exhibits strong solvatochromatic effects in its absorbance spectrum.[9] Generally, the RD incorporation in the blank, drug-free micelles resulted in a decrease in polarity due to the relative hydrophobic environment maintained by the 2-butyl-2-oxazoline core. This manifested in the absorbance spectrum as an increase in the dye λmax compared to its absorbance in solution with polar solvents (Figure S8a,b, Supporting Information). As the worm-inhibiting drugs paclitaxel and vismodegib were solubilized in the micelles, the dye environment became more polar, which led to profound decreases of the λmax even at low drug loadings. In contrast, the worm-permissive drugs bortezomib, marimastat, resiquimod, and olaparib had little or no effect on the polarity, as evidenced by the lack of or slight changes in the λmax. Interestingly, etoposide (worm-permissive) had an intermediate effect, causing relatively smaller λmax changes at low loading and changes comparable to those exhibited by paclitaxel and vismodegib at high loadings (Figure 7). Drugs also had strikingly different effects on the absorbance intensity of the RD, which characterizes the intermolecular interactions between the dye and surrounding molecules. The absorbance intensity of the dye decreased in the absence of drug upon solubilization in the micelles, likely due to hydrogen bonding and ion-dipole interactions of the dye with the polymer repeating units (Figure S8c, Supporting Information). Worm-permissive drugs either did not change the absorbance intensity (such as olaparib, resiquimod, and etoposide) or further decrease it (such as bortezomib and marimastat) (Figure 7).
Figure 7.
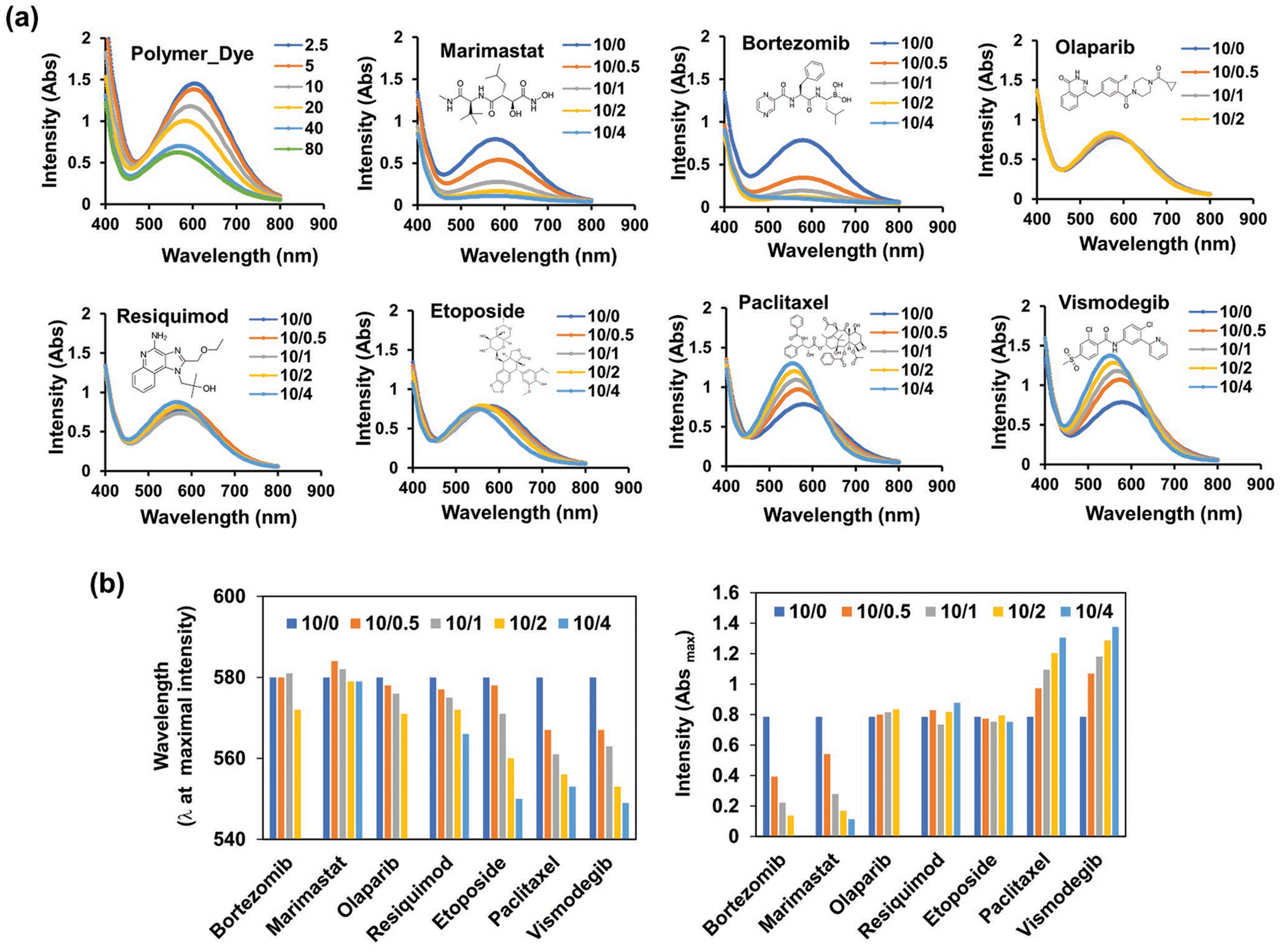
Characterization of micelle microenvironment by Reichardt’s dye (RD). a) UV–vis absorption spectra of RD in blank micelles (polymer/RD ratios of 2.5/1, 5/1, 10/1, 20/1, 40/1, and 80/1) and drug-loaded micelles (polymer/dye/drug ratios of 10/0.5/0, 10/0.5/1, 10/0.5/2, and 10/0.5/4). b) Wavelength and intensity of RD obtained from UV–vis absorption spectra.
This behavior potentially results from differential molecular interactions of RD with the polymer and drugs. The betaine-containing RD can participate in ion–dipole and hydrogen bonding interactions with adjacent non-ionic molecules containing peptide-bond motifs[10] and can likely engage in similar interactions with the poly(2-oxazoline) repeating units. Consequently, the absorbance intensity of RD is suppressed upon its incorporation in the micelles. When marimastat and bortezomib containing repeated amide bonds are added, the absorbance intensity further decreases—likely a result of direct interactions with RD. Other worm-permissive drugs such as etoposide, resiquimod, and olaparib lacking the amide bonds have no effect on the absorbance intensity of RD. Worm-inhibiting drugs paclitaxel and vismodegib contain numerous electronegative and hydrogen bond-accepting groups that may strengthen their interaction with the poly(2-oxazoline). Notably, NMR spectra previously suggested that paclitaxel can interact with the amide bond motif of both poly(2-methyl-2-oxasoline) and poly(2-butyl-2-oxasoline).[11] Therefore, such drugs can bind to the same binding sites that RD binds to and may result in the displacement of the dye if their binding affinity exceeds that of RD. This displacement of RD with the drugs would explain the observed increase in the absorbance intensity (Figure S9, Supporting Information).
2.7. Preparing Drug-Loaded Spherical and Worm-Like Micelles for Injections
Using this knowledge, we were able to alter the morphology of polymeric micelles while keeping drug loading constant, enabling us to thoroughly evaluate the effect of morphology on drug pharmacological performance. We selected olaparib, a poly(ADP-ribose) polymerase (PARP) inhibitor approved for treatment of advanced ovarian and metastatic breast cancer,[12] as a model drug and formulated it into T2 polymeric micelles. Micelles were incubated at room temperature and monitored in a similar fashion to all other micelles to record any changes in particle size and morphology (Figure 8a,b). Over time, olaparib micelles elongated until forming worm-like structures by 72 h; there were no changes in drug concentration or loading during elongation (Figure 8c). Then, we examined whether changes in morphology affected the drug-release profiles in the micelles—a critical parameter for drug delivery. Notably, different morphologies of olaparib-loaded micelles exhibited identical in vitro drug release profiles (Figure 8d). However, size and shape differences resulted in varying micellar mobility in a diffusion-constrained environment (e.g., agarose gel). A model experiment illustrated the improved penetration of such spherical micelles in high viscosity systems (Figure S10, Supporting Information). Finally, olaparib loaded micelles were exposed to bovine serum albumin (BSA), and micelle elongation process was monitored over time. Although BSA significantly delayed the elongation of spherical micelles it had no effect on the size of worm-like micelles (Table S3, Supporting Information). Therefore, we cannot expect that serum proteins will promote the micelle morphology transition in the blood. Moreover, the olaparib loaded spherical micelles would likely retain their morphology for at least 6 to 24 h. This period can be even longer as micelles will be diluted after injection which slows down the spherical micelle elongation (Figure S3b, Supporting Information). Based on this, we proceeded with evaluating olaparib formulations of micelles with different morphologies in an animal model.
Figure 8.
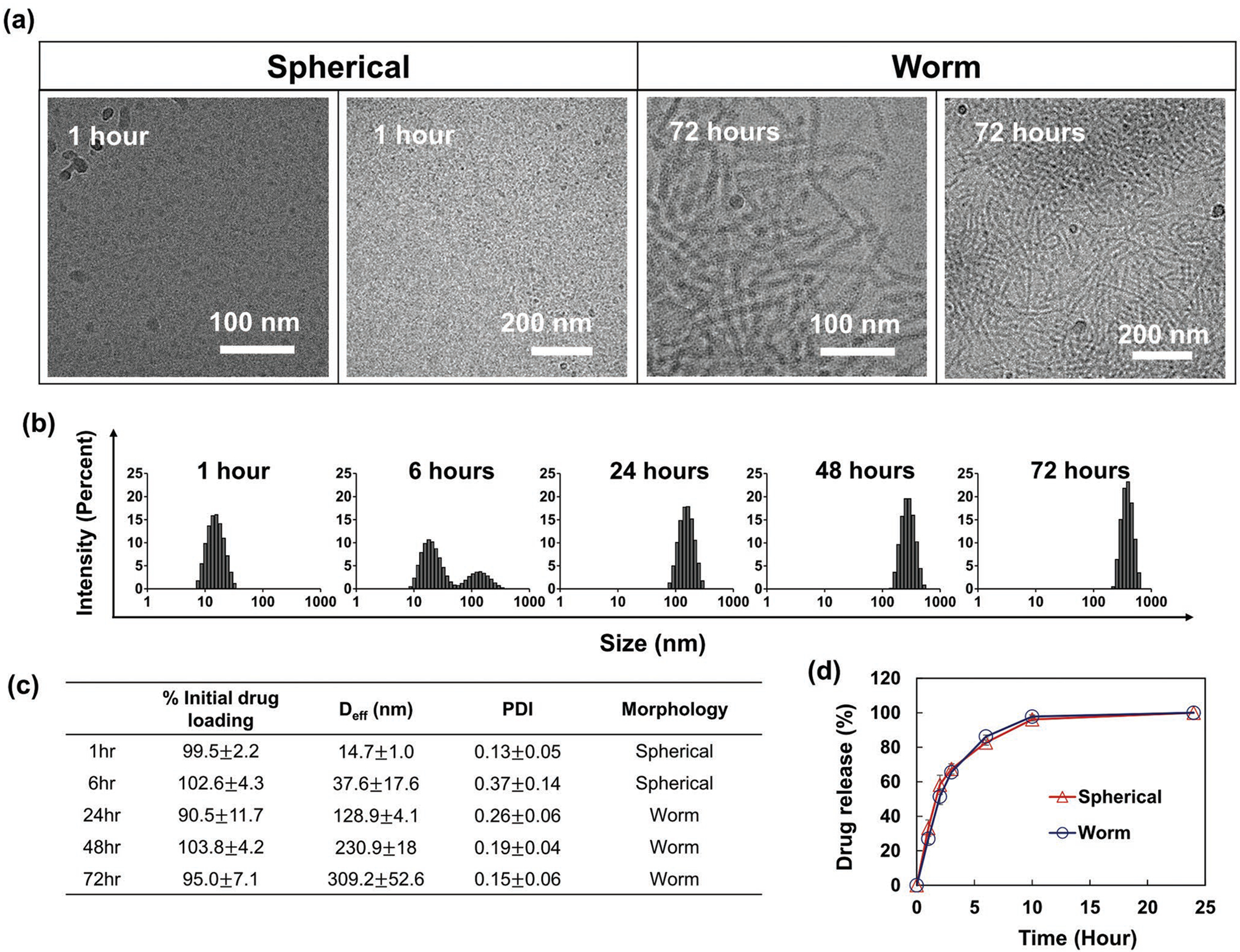
Preparation and characterization of olaparib-loaded micelles (polymer/drug ratio of 10/1) with different morphologies. a) Cryo-TEM images of olaparib-loaded micelles at 1 and 72 h after hydration. Change of b) intensity weighted size distribution and c) particle size, PDI, and drug concentration of olaparib-loaded micelles over time. d) Olaparib release profile from sphere- and worm-like micelles. Olaparib-loaded micelles were prepared using the thin film hydration method. While keeping drug loading content, the olaparib micelles elongated until forming worm-like structure by 72 h. Olaparib micelles did not exhibit significant changes in the drug release profile. Errors shown correspond to three standard deviations of uncertainty.
2.8. Dependence of Antitumor Effect of a Micellar Drug on the Micelle Morphology
We evaluated the antitumor activity of olaparib against a tumor model (BRCA1-mutant human breast carcinoma xenograft MDA-MB-436; Figure 9a). Olaparib formulations in the spherical and worm-like micelles were administered intravenously (IV) at 10 and 20 mg kg−1, which correspond to 1/4 and 1/2 of the drug maximum tolerated dose (MTD; 40 mg kg−1) with a frequency of twice a week. Olaparib loaded in worm-like micelles showed significant antitumor efficacy by delaying tumor growth at the higher drug dose. These micelles also displayed a small effect at the lower dose, but differences were not statistically significant between drug treatment and saline (0.9% NaCl) control groups. When the same drug was administered in spherical micelles, however, we observed significant antitumor activity at both drug doses (10 and 20 mg kg−1). Interestingly, the drug loaded in the spherical micelles was significantly more active than the drug loaded in the worm-like micelles, even at the same doses. Notably, neither the blank spherical micelles nor the free olaparib (20 mg kg−1) displayed inhibition of the tumor growth.
Figure 9.
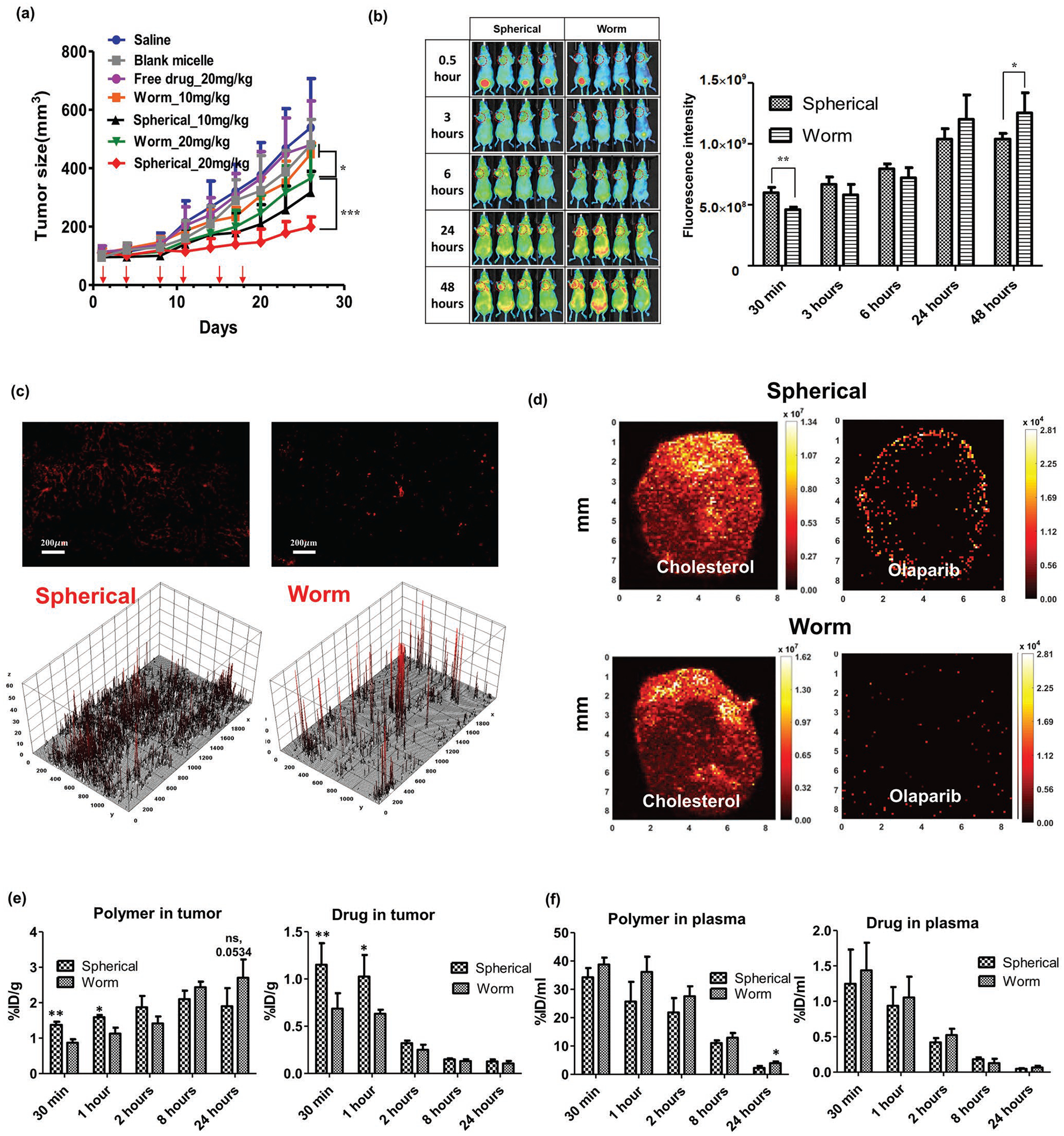
Antitumor activity, biodistribution, and PK of olaparib-loaded micelles (polymer/drug ratio of 10/1) in a tumor model (BRCA1-mutant human breast carcinoma xenograft MDA-MB-436). a) Tumor growth inhibition after six injections of olaparib-loaded micelles at 1/4 and 1/2 of the drug MTD (40 mg kg−1), delivered twice a week (shown by arrows) (n = 4–5). b) Non-invasive fluorescent imaging of tumor model after a single injection of the AF-647 labeled micelles. c) Visualization and 3D surface plots (analyzed in Image J) of the distribution AF-647 labeled micelles in tumor sections 1 h after injection (see Figure S11, Supporting Information, for blood vessel staining). d) IR-MALDESI MSI analysis of olaparib in tumor sections from mice following a single dose of olaparib-loaded micelles (10 mg kg−1) at 1 h. Quantification of the polymer and drug concentrations in e) tumor and f) plasma (n = 4–5). Statistical difference: *p < 0.05, **p < 0.01, ***p < 0.001.
To test the generality of our observation for another drug and tumor model, we used selumetinib, a MEK inhibitor, approved for treatment of neurofibromatosis type 1,[13,14] which is in clinical trials for pancreatic cancers.[14] We loaded this drug in the spherical and worm-like micelles of T2 using similar formulation strategy as described above (Figure S12, Supporting Information) and evaluated the resulting formulations in the human pancreatic carcinoma xenograft BXPC-3 model. The results reinforced the effect of the micelle morphology on the drug performance. In this case selumetinib with the worm-like micelles was not active while the same dose in spherical micelles produced significant inhibition of the tumor growth (Figure S13a, Supporting Information).
2.9. Effect of the Micelle Morphology on the PK and Tumor Distribution of the Drug
AF-647 labeled micelles with different morphology were IV-injected into nude mice bearing MDA-MB-436 tumors; the distribution of the micelles was assessed by high-resolution fluorescent imaging of the tumor-bearing mice (Figure 9b). Micelles gradually accumulated at the tumor site over time for all formulations, although there were notable differences in accumulation kinetics (Figure 9b). In comparison to worm-like micelles, spherical micelles accumulated in the tumor more rapidly, at an early time (e.g., 30 min, 1 h). However, after longer formation periods, the tumor accumulation of worm-like micelles gradually increased and exceeded the accumulation of spherical micelles after 24 h. The AF-647-labeled micelles were visualized in tumor sections 1 h after injection (Figure 9c). Both formulations accumulated in the tumor tissue but with different localization patterns. Overall, spherical micelles showed an even distribution and weak and scattered signal throughout the tumor tissue. Worm-like micelles, in contrast, were more localized and displayed a strong signal in a small area within tumor tissue (Figure S11, Supporting Information, for blood vessel staining). Similar difference was observed for selumetinib loaded spherical and worm-like micelles in the second tumor model in BXPC-3 bearing mice (Figure S12b, Supporting Information).
We also analyzed the spatial distribution of olaparib across sections of the tumor using mass spectrometry imaging (MSI) based on infrared matrix-assisted laser desorption electrospray ionization (IR-MALDESI). With this approach, we were able to visualize olaparib signal abundance and to compare its distribution to that of an endogenous cholesterol (Figure 9d). One hour after administration the spherical micelles delivered considerably more olaparib compared to the worm-like micelles, and olaparib in the spherical micelles appeared to be more uniformly distributed through the tumor. As expected, the signal abundance of cholesterol presented in all cell membranes was similar across all samples. This result was reinforced with selumetinib in spherical and worm-like micelles that revealed very similar difference in drug distribution in BXPC-3 tumor model (Figure S13c, Supporting Information).
Next, we carried out a PK study using tissue sampling with radiolabeled 3H-olaparib and AF-647-labeled micelles. We found differences in delivery to tumors of both the polymer and the drug when loaded in spherical or in worm-like micelles. At early time points (1 and 2 h), we observed an increased accumulation of polymer administered as spherical micelles in the tumor compared to worm-like micelles (Figure 9e). This trend reversed over time, and accumulation of polymer in worm-like micelles increased steadily; however, these differences were not statistically significant. Similar trends were seen for the drug, with significantly higher tumor accumulation of olaparib in spherical micelles at 2 h, compared to worm-like micelles. A maximum concentration (Cmax) of the drug was observed in the tumor at 0.5 h (i.e., much earlier than the Cmax of the polymer); this value was higher for spherical micelles compared to worm-like micelles (2.92 μg g−1 vs 1.75 μg g−1; p-value < 0.05) (Table 1). At later time points, this difference in drug accumulation for the two micellar morphologies became insignificant. Ultimately, the tumor AUCall remained higher for the drug in spherical micelles due to the increased delivery of drug at an earlier stage (Table 1). The drug PK in the plasma were also similar for both spherical and worm-like micelles, whereas polymer PK in the plasma displayed a slight increase in Cmax and statistically significant difference in AUC for worm-like micelles compared to spherical micelles (Figure 9f). Therefore, both the polymer and the drug loaded in the spherical micelles distributed faster to the tumors, whereas distribution in the plasma during a period of 24 h was comparable between the two morphologies, or even higher for worm-like micelles (Table S4, Supporting Information).
Table 1.
PK parameters of polymer and drug in tumor and plasma after a single injection of olaparib (10 mg kg−1) in spherical (S) and worm-like (W) micelles (polymer/drug ratio of 10/1).
| Location | Parametera) | Polymer | Drug | ||||
|---|---|---|---|---|---|---|---|
| Spherical | Worm | Statisticsb) | Spherical | Worm | Statisticsb) | ||
| Tumor | Tmax [h] | 8.0 | 24.0 | – | 0.5 | 0.5 | – |
| Cmax [μg mL−1] | 64.1 | 81.5 | ns | 2.9 | 1.7 | ** | |
| AUCall [h × μg mL−1] | 1466.4 | 1638.9 | ns | 13.8 | 11.4 | * | |
| Plasma | Tmax [h] | 0.5 | 0.5 | – | 0.5 | 0.5 | – |
| Cmax [μg mL−1] | 1105.5 | 1238.9 | ns | 3.2 | 3.7 | ns | |
| AUCall [h × μg mL−1] | 7999.6 | 9796.0 | * | 14.7 | 14.6 | ns | |
Tmax: time of the Cmax; Cmax: maximal observed concentration; AUCall: area under the curve from the time of dosing to the time of the last observation;
Significance S versus W groups: *p < 0.05, **p < 0.01.
See Table S4 (Supporting Information) for detailed analysis of the polymer and drug exposure and clearance.
3. Discussion
From the perspective of nanomedicine manufacturing and clinical translation, it is critical to understand how nanoparticle morphology affects both polymer and drug biodistribution and therapeutic efficacy. Of particular relevance to this work, the distribution and clearance of polymeric micelles is reported to vary according to micellar morphology.[1,15,16] However, most studies are limited to the study of in vivo fate of the nanoparticles themselves and do not include data on the fate of the encapsulated drug. Therefore, work elucidating the latter process and demystifying the correlation between micelle morphology, drug efficacy, and biodistribution is critical to enabling novel pharmacological discoveries.
Here, we investigated the elongation of poly(2-oxazoline)-based polymeric micelles from spherical to worm structure. Formation of the core–shell spherical micelles assembled into elongated worm-like large clusters was consistent across the cryo-TEM, DLS, and SANS observations. We found that the process of elongation is kinetically controlled and governed by a variety of conditions including the molecular mass of the polymer, polymer concentration, temperature, and ionic strength. The elongation process is also related to the drug loading of the micelles. As a general rule, when the amount of the added drug increased, the sphere-to-worm transition became slower or was completely inhibited. In view of this elongation and inhibiting process, we classified drugs as “worm-inhibiting” or “worm-permissive” groups.
The differences in the behavior of worm-inhibiting and worm-permissive drugs could be attributed to their specific interactions with the block copolymer, as characterized by the RD study. Considering that all the drugs used for the screening study are poorly soluble, we hypothesized that, in addition to hydrophobic interactions, other specific interactions between drug and polymer (e.g., hydrogen bonding or dipole-dipole interactions) may play a role in micelle stabilization. To assess the effects of these interactions on the micelle stability, we determined the changes of effective CMC* at various drug loadings. The CMC* gradually decreased as the drug loading amount increased. However, worm-inhibiting drugs decreased the CMC* of the spherical micelles upon loading to a greater extent. This suggests that worm-inhibiting drugs are more potent in stabilizing the spherical micelles.
The striking differential effects of the worm-inhibiting and worm-permissive drugs need further explanation. The MD simulations suggest a difference in the localization of these drugs within the micelles. The tendency of a worm-inhibiting drug, paclitaxel, to occupy the outer area within the micelle is indicative of decreased interfacial energy of the micelle in the presence of this drug. This alone may be an important factor favoring the stabilization of the spherical micelles by the worm-inhibiting drugs since the micelle self-assembly is driven by the minimization of the interfacial free energy. In addition, micelle shapes are strongly dependent on the steric repulsion of the hydrophilic chains in the shell and the stretching of the hydrophobic chains in the core.[17] These parameters can change differentially because of interactions of different drugs with either core- or shell-forming blocks.
The stabilization of the spherical micelle morphology implies that the worm-inhibiting drugs “prefer” spherical micelles as their environment. Based on this consideration, the use of the block copolymers that form spherical micelles offers the opportunity to increase loading capacity with respect to worm-inhibiting drugs. This seems to be the case of ABT-263, paclitaxel, and vismodegib, all of which display a twofold increase in solubility in the T3 copolymer (forming the spherical micelles) relative to worm-forming T2 copolymer. No such difference was observed for worm-permissive drugs. The difference can be understood if one assumes that a worm-inhibiting drug “expends” part of the free energy of interaction with worm-forming T2 copolymer to stabilize the spherical morphology. No such expenditure occurs upon drug solubilization with T3 copolymer forming stable spherical micelles. In other words, the chemical potential of such drug is lower when it interacts with the copolymer forming spherical micelles providing more favorable solubilization environment. The worm-permissive drugs are more “morphologically neutral,” i.e., there is no or less differential gain for their incorporation in either copolymer.
Overall, the knowledge and methodology acquired in this study enabled us to evaluate the role that morphology plays on the drug pharmacological performance of spherical and worm-like micelles. Morphology of polymeric micelles can be effectively controlled by tuning the type of polymer, molecular mass, and drug-loading ratio. Our findings suggest that morphology plays a critical role in the tumor accumulation behavior of micellar drug. Provided that spherical micelles are able to accumulate more rapidly, the tumor is exposed to micelles while they retain large amounts of drug. These results are consistent with other studies that show that worm-like micelles have longer circulation in the plasma,[1,16] which can lead to increased exposure and eventual accumulation in tumors. In contrast, worm-like micelles accumulate in tumors more slowly and only after they have released significant amounts of drug. As a result, less drug is delivered into the tumor by worm-like micelles. Although the overall systemic drug exposure is lower with spherical micelles, they result in more efficient tumor exposure and a twofold increase in Cmax partly due to the rapid tumor accumulation. Depending on the drug mechanism, both the exposure and/or Cmax can drive pharmacological performance.[18] Therefore, this result correlates with the increase in the antitumor effect of the drug administered by spherical micelles compared to worm-like micelles. The difference in antitumor activity is the result of the morphological transformations alone provided that all other parameters (e.g., drug regimen, doses, loading in the micelles, drug release rates, and the concentration and composition of the polymer) remained unchanged. The faster penetration of spherical micelles into tumors is likely due to their smaller size compared to worm-like micelles. These results confirm the decisive role that particle size plays in facilitating micelle entry and drug delivery to tumors.[2,15]
Other studies suggested the importance of nanoparticle shape in drug delivery; some of the benefits of leveraging particle nanoparticle morphologies include increased plasma circulation of rod-like nanoparticles,[1,16,19] improved cell-binding-specificity of antibody-decorated to rod-like nanoparticles,[20] and increased MTD of drugs in rod-like micelles.[21] Our findings suggest that the dynamic character of the drug-micelle structure along with the micelle morphology (and inherent size) play a pivotal role in pharmacological performance. In particular, the drug release rates are important and the changes in drug retention within the range of several hours could drastically influence pharmacological performance. This discovery sets our study apart from other nanoparticle studies, wherein drugs were either not included or were covalently conjugated.
Here, we demonstrated that spherical micelles facilitate systemic delivery of anticancer drugs to tumors when the drugs are loosely associated with the polymeric micelles. Our observations indicate that the polymeric micelle shape can be adjusted to a great extent by altering the block copolymer length, and that selection of a block length that favors the formation of spherical micelles can play a pivotal role in the formulation of pharmaceutical drugs. This is critical for improving drug loading, PK, and tumor distribution, and for ensuring long-term stability and reproducibility of formulations (e.g., particle size, polydispersity, and shape)—for which elongation over time is adverse.
4. Experimental Section
Materials:
All chemicals for polymer synthesis were purchased from Sigma-Aldrich (St. Louis, MO). Triblock copolymers of P[MeOx-b-BuOx-b-MeOx] with different degrees of polymerization of the blocks were synthesized by living cationic ring-opening polymerization, as described previously.[22,23] For the synthesis of polymers, methyl trifluoromethanesulfonate (MeOTf), 2-methyl-2-oxazoline (MeOx), and 2-n-butyl-2-oxazoline (BuOx) were dried by refluxing over calcium hydride (CaH2) under inert nitrogen gas and subsequently distilled. Blocks were sequentially synthesized using MeOTf as the initiator and terminated by piperidine. 1H NMR spectrum was acquired using an INOVA 400 system and analyzed using the MestReNova (11.0) software. Spectra were calibrated using MeOD solvent signals (4.78 ppm). Number-average molecular masses (Mn) were determined using samples taken upon completion of each block and final copolymer, based on the ratio of 1H NMR signals of the initiator (CH3, 2.86–2.88, 2.97–3.03 ppm) and the repeating units of block backbone (C2H4, 3.40–3.70). Molecular mass distribution of the polymer was measured by gel permeation chromatography (GPC) on a GPC-max VE2001 system; GPC data were used to determine the polymer PDI.
Bortezomib, olaparib, etoposide, rapamycin, ABT-263, vismodegib, and paclitaxel were purchased from LC Laboratories (Woburn, MA). BLZ945, RXDX-105, and PLX3397 were purchased from MedKoo Biosciences (Morrisville, NC). Resiquimod, AZD2014, and AZD8055 were purchased from Selleckchem (Houston, TX). All other chemicals were obtained from Fisher Scientific INC. (Fairlawn, NJ). All reagents used were of analytical grade.
MDA-MB-436 cells were obtained from UNC Lineberger Tissue Culture Facility and were cultured in a DMEM medium (Gibco 11965–092) supplemented with 10% FBS and 1% penicillin-streptomycin. The Cell Counting Kit (CCK-8) was purchased from Dojindo Molecular Technologies (Rockville, MD). Nude mice were purchased from UNC DLAM animal facility.
Preparation and Characterization of POx-Based Polymeric Micelles:
POx micelles were prepared by the thin-film hydration method described previously.[24] Briefly, stock solutions containing the polymer and the drugs in ethanol were mixed together at the pre-determined ratios and ethanol was then completely evaporated. The resulting thin film was hydrated with normal saline (0.9% NaCl). The size distribution of micelle formulations resulting from various conditions was monitored over time using the DLS technique on a Zetasizer Nano ZS (Malvern Instruments Ltd., UK). The DLS particle sizes are reported as intensity average diameter (Dz).
The amount of drug loaded in polymeric micelles was analyzed by reversed-phase high-pressure liquid chromatography (HPLC; Agilent Technologies 1200 series) performed using a Nucleosil C18, 5 μm column (L × I.D. 250 mm × 4.6 mm). Each sample was diluted 50 times in a prescribed mobile phase (methanol/water volume percentage of 70%/30%, with 0.01 trifluoroacetic acid for bortezomib; and acetonitrile/water volume percentage of 50%/50%, with 0.01 trifluoroacetic acid for other drugs). Following this step, 10 μL of diluted sample were injected into the HPLC while the flow rate was kept at 1.0 mL min−1 and the column temperature was kept at 40 °C.
For TEM image acquisition, we used a LEO EM910 TEM operating at 80 kV (Carl Zeiss SMT Inc., Peabody, MA). Diluted samples were deposited onto a copper grid containing a carbon film and stained with 1% uranyl acetate before imaging. For Cryo-TEM image acquisition, cryo-grids were prepared by rapid immersion into an ethane/propane mixture (40%/60%) using a Vitrobot Mark IV (Thermo Fisher Scientific) set to 22 °C and 95% humidity. C-Flat TEM grids (ProtoChips) were rendered hydrophilic by indirect oxygen plasma cleaning (25% oxygen, 75% argon) using a Tergeo-EM system (PIE Scientific LLC). Cryo-grids were imaged using a 200 kV Talos Artica G3 (Thermo Fisher Scientific) equipped with a Gatan K3 direct-electron detector (Gatan).
Screening of Micelle Elongation:
The effect of various conditions including polymer concentration (1.25–80 mg mL−1), temperature (4, 25, and 37 °C), and salt concentration (up to ≈4% NaCl) on micelle elongations were investigated by monitoring the change of particle size and size distribution over time using DLS (Zetasizer Nano ZS; Malvern Instruments Ltd., UK). To examine drug-loading effects on micelle elongation, drugs were formulated into polymeric micelles at predetermined drug loading ratios (polymer/drug w/w ratios of 10/0, 10/1, 10/2, 10/4, and 10/8); the change of particle size and size distribution were monitored over time. The morphology of each formulation was classified into three categories based on particle size and size distribution: 1) spherical micelles (S), 2) mixtures of spherical and worm micelles (W+S), and 3) worm micelles (W). If micelles were destabilized and precipitated, samples were classified as precipitate (P).
Small Angle Neutron Scattering:
To perform SANS measurements, blank and drug-loaded micelle solutions were prepared in D2O containing 10 mg mL−1 of T2. All samples were degassed before SANS measurements; these were examined using the NG7 and NGB30 systems[25] at the National Institute of Standards and Technology Center for Neutron Research (NCNR). Data collection was performed at 25 °C. A neutron wavelength λ of 6 Å and three sample-to-detector distances were used. A high-resolution lens configuration (λ = 8.09 Å) provided the lower scattering angle data. SANS data were collected in the range of momentum transfer 0.002 < q (Å−1) < 0.55, where q is defined as follows
| (1) |
where the scattering angle is given by 2θ. Data were reduced and analyzed using the SANS macro routines and analysis packages, respectively, developed for IGOR at the NCNR.[26] Raw counts were normalized to a common monitor count and corrected for empty cell counts, ambient background counts, and nonuniform detector response. Scattered intensities were corrected to remove incoherent scattering (e.g., from the buffer and from hydrogen) using a flat background subtraction based on the lowest intensity recorded at high q (for each sample).
SANS data were measured for solutions containing a constant concentration of polymer and increasing amounts of drug (polymer/drug w/w ratios of 10/0, 10/1, 10/2, and 10/4; Figure 4). An upturn in scattered intensities for the blank and lower drug concentration micelle solutions was observed at q lower than ≈0.015 Å−1. This is characteristic of larger clusters or aggregates and is consistent with cryo-TEM and light scattering observations. The average size of these larger objects is beyond the angular range measured by SANS. Data were fit with the following functional form
| (2) |
where the first term describes Porod scattering from clusters of micelles, B is the incoherent background, and Imicelle denotes the scattering from individual T2 spherical micelles (blank or drug loaded). With increasing drug loading ratio, single micelles are stabilized, and a flattening of the lower-q upturn is observed. Contributions to scattering from the individual micelles were modeled as follows
| (3) |
where np is number density of micelles with an effective sphere diameter σ. P(q) was modeled using a polydisperse sphere where the ratio between the core radius and the total micellar radius is held constant.[27] P(q) is the particle form factor averaged over the particle size distribution
| (4) |
The single particle scattering amplitude is an average over a Schulz distribution of radii (see Equations (32)–(37) in ref. [27]), where
| (5) |
ρ is the particle SLD and r is the distance between particles. An average structure factor approximation was used
| (6) |
where g(r) is the pair correlation function defined by a hard sphere interparticle potential. The Ornstein-Zernicke equation was solved using a Percus-Yevick closure as described previously.[26,28]
Light Scattering Intensity:
The effective CMC* of poly(2-oxazoline)-based polymeric micelles was determined by DLS (wavelength 663 nm). The light scattered by the samples was detected by an avalanche photodiode at a backscattering angle of 173°. The intensity of scattered light (derived count rates, kilo-counts per second (kcps)) was obtained for blank and drug loaded micelles (1 mg mL−1), diluted in distilled water to varying concentrations. Above a given polymer concentration, the intensity of light scattering proportionally decreased as polymer concentration decreased; above this concentration threshold, there were no significant changes in the intensity of light scattering. This point of deflection (e.g., slope change) in the intensity of light scattering was defined as effective CMC*.[29]
Reichardt’s Dye:
RD and drug-loaded polymeric micelles were prepared using a thin-film hydration method. The polymer, drugs, and Reichardt’s dye were mixed in ethanol at pre-determined ratios (10/0.5/0.5, 10/1/0.5, 10/2/0.5, and 10/4/0.5), after which ethanol was completely evaporated from the mixtures. The resulting thin film was subsequently hydrated using normal saline (0.9% NaCl). After 1 h stabilization at RT, the UV–vis spectra of each sample were recorded. The maximum absorption wavelength and the absorbance intensity values of RD were analyzed.
Molecular Dynamics Simulation:
MD simulations were performed in GROMACS 2020 using an OPLS-AA force field. A total of 50 polymer chains and 50, 100, and 200 drugs were packed randomly in a cubic box and surrounded with water and salt. The structure was annealed to 333 K for the first 50 ns of the simulation to equilibrate the system. The system was then cooled back to 300 K for at least 100 ns. Partial density was computed using the “density” module in GROMACS.
In Vitro Drug Release:
Drug release profiles were obtained using the membrane dialysis method described elsewhere.[23,30] First, the olaparib loaded micelles were diluted in phosphate-buffered saline (PBS) to achieve a drug concentration of 0.1 mg mL−1. Then, the diluted micelle solutions were transferred to floatable Slide-A-Lyzer MINI dialysis devices (100 μL capacity, 3.5 kDa MWCO) and suspended in 30 mL of PBS supplemented with 10% fetal bovine serum (FBS) (to ensure sink conditions). Four devices were used for every time point. At predetermined time points, the samples were collected from the devices and the remaining drug amounts were quantified by HPLC. Drug release profiles were constructed by plotting the percentage released over time.
Estimation of Maximum Tolerated Dose:
A dose-escalation study was used to estimate the MTD before in vivo efficacy studies. Healthy nude mice (8 weeks old, female) were divided into groups of four, with each group subjected to increasing doses of drugs. Olaparib PM (20, 40, and 60 mg kg−1) and normal saline (0.9% NaCl) were injected intravenously using a q4d × 6 regimen. Mice were examined every other day to monitor signs of toxicity, including hunched posture, rough coat, and body weight changes (loss of 15% or greater) in accordance to approved Institutional Animal Care and Use Committee (IACUC) protocols (Protocol No. 19–175).
Tumor Inhibition:
Female athymic nude mice (6–8 weeks) were subcutaneously inoculated in the right flank with MDA-MB-436 cells or BXPC-3 cells (5 × 106 cells in 100 μL of 1:1 mix of PBS and BD Matrigel). When the tumor sizes reached ≈100 mm3, mice were randomized into several groups, and the animals (n = 5) received the following injections (i.v.; q4d × 6) by group: For the MDAMB-436 model: normal saline; 10 mg kg−1 of spherical-shaped olaparib PM; 10 mg kg−1 of worm-shaped olaparib PM; 20 mg kg−1 of spherical-shaped olaparib PM; 20 mg kg−1 of worm-shaped olaparib PM; 20 mg kg−1 of free drug (olaparib dissolved in 5% DMSO with 30% PEG300); or blank micelles. For BXPC-3 model: normal saline; 20 mg kg−1 of spherical-shaped selumetinib PM; 20 mg kg−1 of worm-shaped selumetinib PM. Tumor volume was calculated using the formula (length × width2)/2.
In Vivo Particle Distribution:
Nude mice bearing MDA-MB-436 tumors were IV-injected with a single dose of spherical or worm-shaped olaparib PM micelles (10 mg kg−1) containing an AF-647 conjugated polymer (2.5% of total polymer). After 1 h from sample injections, mice were euthanized. Tumors were then collected and frozen in a cryo-mold filled with an optimal cutting temperature (OCT) compound (Tissue-Tek; Sakura Finetek, Leiden, The Netherlands). Tumor cryosections were obtained at a thickness of 10 μm and tumor vasculatures were stained with anti-CD31 (Abcam, Cambridge, UK) with assistance from Translational Pathology Laboratories. Stained slides were digitally acquired using an Aperio ScanScope XT (Aperio). Polymer and CD31 intensity at tumor sections were visualized by 3D surface plot analysis (ImageJ). The X and Y-axes represent the area of the tumor sections, whereas the Z-axis represents the polymer (Red) and CD31 (Green) intensities.
Pharmacokinetic and Biodistribution Studies:
Female athymic nude mice (6–8 weeks) were subcutaneously inoculated in the right flank with MDA-MB-436 cells (5 × 106 cells in 100 μL of 1:1 mix of PBS and BD Matrigel). PK studies proceeded once tumor sizes reached ≈200 mm3.
For drug PK studies, mice were administered a single dose of spherical or worm-shaped olaparib-loaded micelles (10 mg kg−1) containing 3H-labeled olaparib (200 μCi kg−1) via tail vein. At predetermined time points (0.5, 1, 2, 8, 24, and 48 h), each group of mice (n = 3–4) was euthanized; samples (plasma and tissues) were collected to analyze the olaparib concentration by measuring the radioactivity. A portion of plasma was diluted in mixture of Soluene-350 and isopropyl alcohol (1:1 ratio) and incubated at 60 °C for 2 h. Tissues were weighed and homogenized using a homogenizer Tissuemiser (Fisher Scientific, Pittsburgh, PA) and Soluene-350. A portion of the blood or tissue homogenate was placed in a scintillation vial and mixed with 10 mL of scintillation cocktail (Ultima Gold). Radioactivity was measured using a liquid scintillation counter, Tricarb 4000 (Packard, Meriden, CT).
For polymer PK studies, mice were administered a single dose of spherical or worm-shaped olaparib-loaded micelles (10 mg kg−1) containing an AF-647-conjugated polymer (2.5% of total polymer) via tail vein. Plasma was collected and diluted in saline (0.9% NaCl) and analyzed using fluorescence microscopy. Organs were collected and homogenized in a mixture of 2 mg mL−1 of collagenase and RIPA buffer (1:1 ratio). Polymers in homogenate were extracted using MeOH, and their fluorescence intensity was analyzed using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA) (excitation = 633 nm, emission = 670 nm).
PK data were analyzed using Phoenix Winnonlin modeling software. Noncompartmental analysis (NCA) was performed using sparse sampling; NCA parameters were used as initial estimates for model development. AUC and residual plot analyses were utilized as criteria for determining the best model for the polymers and the drug.
IR-MALDESI Imaging of the Spatial Distribution of Drug:
MSI was performed by IR-MALDESI.[31] Harvested tumors were sectioned to a thickness of 10 μm by cryotome and thaw mounted on glass microscope slides. Samples were placed onto the IR-MALDESI sample stage and cooled to −10 °C, allowing a layer of ice to grow on top of the sample that was maintained throughout analysis. An IR laser (λ = 2.94 μm, IR-Opolette 2371; Opotek, Carlsbad, CA) desorbed sample material from 100 μm diameter sampling locations across the surface of the sample. Desorbed neutral molecules were ionized by an electrospray (50/50 mixture of methanol/water (v/v) with 0.2% formic acid) and ions were then sampled into a high resolving power Thermo Fisher Scientific Q Exactive Plus (Bremen, Germany) mass spectrometer. The mass spectrometer was operated in positive ion mode from m/z 200 to 800, with resolving power of 140 000 at m/z 200. Olaparib and selumetinib were identified as protonated molecular ions [M+H+]+ at m/z 435.1821 and m/z 457.0067, respectively. To generate images from mass spectrometry data, raw data were converted to the mzXML format[32] and interrogated using MSiReader.[33]
Statistical Analysis:
The experimental results were expressed as means ± SD. Statistical comparison of data for tumor inhibition was done using a two-way analysis of variance (ANOVA) and followed by Bonferroni post-tests for multiple comparison (n = 4–5). Comparison of particle accumulation in tumor and PK were done by unpaired two-tailed Student’s t-test (n = 3–4). Statistical analyses were performed using the GraphPad Prism software. *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary Material
Acknowledgements
This work was partially supported by the National Cancer Institute (NCI) Alliance for Nanotechnology in Cancer (U54CA198999, Carolina Center of Cancer Nanotechnology Excellence), and NCI’s grant U01CA198910. Animal Studies and IVIS imaging were performed within the UNC Lineberger Animal Studies Core (ASC) Facility at the University of North Carolina at Chapel Hill. The UNC Lineberger Animal Studies Core was supported, in part, by an NCI Center Core Support Grant (CA16086) to the UNC Lineberger Comprehensive Cancer Center. Access to the NGB 30m SANS instrument was provided by the Center for High Resolution Neutron Scattering, a partnership between the National Institute of Standards and Technology and the National Science Foundation under Agreement No. DMR-2010792. Cryogrids were prepared and imaged at the UNC Chapel Hill CryoEM Core. The authors would like to thank the University of North Carolina at Chapel Hill and the Research Computing group for providing computational resources and support that have contributed to these research results. The authors thank C. Santos, M. Ross, and A. Valdivia at MP1U of UNC for helping with the intravenous/intraperitoneal injections. TEM was performed by A. Shankar Kumbhar at the Chapel Hill Analytical and Nanofabrication Laboratory (CHANL), a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), which was supported by the NSF (grant ECCS-1542015) as part of the National Nanotechnology Coordinated Infrastructure (NNCI). The authors thank R. Luxenhofer for very helpful scientific discussion of this paper. The authors acknowledge the support of the National Institute of Standards and Technology, U.S. Department of Commerce, in providing the neutron research facilities used in this work. SCMT is very grateful for the general support and useful discussions with the staff at the NIST Centre for Neutron Research (NCNR), and funding from CNS NIST Cooperative Agreement Nos. 70NANB20H133 and 370NANB17H302.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
A.V.K. is an inventor on patents pertinent to the subject matter of the present contribution and co-founder of DelAqua Pharmaceuticals Inc. having intent of commercial development of POx based drug formulations. MSP discloses potential interest in DelAqua Pharmaceuticals Inc. as a spouse of co-founder.
Contributor Information
Chaemin Lim, Center for Nanotechnology in Drug Delivery and Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Jacob D. Ramsey, Center for Nanotechnology in Drug Delivery and Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Duhyeong Hwang, Center for Nanotechnology in Drug Delivery and Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Susana C. M. Teixeira, Department of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy Street, Newark, DE 19716, USA NIST Center for Neutron Research, National Institute of Standards and Technology, 100 Bureau Drive, Gaithersburg, MD 20899, USA.
Chi-Duen Poon, Research Computer Center University of North Carolina Chapel Hill, Chapel Hill, NC 27599, USA.
Joshua D. Strauss, Department of Biochemistry and Biophysics, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Elias P. Rosen, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Marina Sokolsky-Papkov, Center for Nanotechnology in Drug Delivery and Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Alexander V. Kabanov, Center for Nanotechnology in Drug Delivery and Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA Laboratory of Chemical Design of Bionanomaterials, Faculty of Chemistry, M.V. Lomonosov Moscow State University, Moscow 119992, Russia.
Data Availability Statement
Research data are not shared.
References
- [1].Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE, Nat. Nanotechnol 2007, 2, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K, Nat. Nanotechnol 2011, 6, 815. [DOI] [PubMed] [Google Scholar]
- [3].a) He Z, Wan X, Schulz A, Bludau H, Dobrovolskaia MA, Stern ST, Montgomery SA, Yuan H, Li Z, Alakhova D, Sokolsky M, Darr DB, Perou CM, Jordan R, Luxenhofer R, Kabanov AV, Biomaterials 2016, 101, 296; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hussein YHA, Youssry M, Materials 2018, 11, 688; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rasmussen MK, Pedersen JN, Marie R, Nat. Commun 2020, 11, 2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schulz A, Jaksch S, Schubel R, Wegener E, Di Z, Han Y, Meister A, Kressler J, Kabanov AV, Luxenhofer R, Papadakis CM, Jordan R, ACS Nano 2014, 8, 2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fenyves R, Schmutz M, Horner IJ, Bright FV, Rzayev J, J. Am. Chem. Soc 2014, 136, 7762. [DOI] [PubMed] [Google Scholar]
- [6].a) Hua Z, Pitto-Barry A, Kang Y, Kirby N, Wilks TR, O’Reilly RK, Polym. Chem 2016, 7, 4254; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Patel V, Ray D, Singh K, Abezgauz L, Marangoni G, Aswal VK, Bahadur P, RSC Adv 2015, 5, 87758. [Google Scholar]
- [7].a) Danov KD, Kralchevsky PA, Stoyanov SD, Cook JL, Stott IP, Pelan EG, Adv. Colloid Interface Sci 2018, 256, 1; [DOI] [PubMed] [Google Scholar]; b) Zana R, Kaler EW, Giant Micelles: Properties and Applications, Vol. 140, CRC Press, Boca Raton, FL: 2007. [Google Scholar]
- [8].Wan X, Min Y, Bludau H, Keith A, Sheiko SS, Jordan R, Wang AZ, Sokolsky-Papkov M, Kabanov AV, ACS Nano 2018, 12, 2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Haider MS, Lübtow MM, Endres S, Forster S, Flegler VJ, Böttcher B, Aseyev V, Pöppler A-C, Luxenhofer R, ACS Appl. Mater. Interfaces 2020, 12, 24531; [DOI] [PubMed] [Google Scholar]; b) Reichardt C, Chem. Rev 1994, 94, 2319. [Google Scholar]
- [10].Hachisako H, Ryu N, Hashimoto H, Murakami R, Org. Biomol. Chem 2009, 7, 2338. [DOI] [PubMed] [Google Scholar]
- [11].Grüne M, Luxenhofer R, Iuga D, Brown SP, Pöppler A-C, J. Mater. Chem. B 2020, 8, 6827. [DOI] [PubMed] [Google Scholar]
- [12].a) Robson M, Im S-A, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, Wu W, Goessl C, Runswick S, Conte P, Engl N. J. Med 2017, 377, 523; [DOI] [PubMed] [Google Scholar]; b) Moore K, Colombo N, Scambia G, Kim B-G, Oaknin A, Friedlander M, Lisyanskaya A, Floquet A, Leary A, Sonke GS, Gourley C, Banerjee S, Oza A, González-Martín A, Aghajanian C, Bradley W, Mathews C, Liu J, Lowe ES, Bloomfield R, DiSilvestro P, Engl N. J. Med 2018, 379, 2495. [DOI] [PubMed] [Google Scholar]
- [13].Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, Whitcomb P, Martin S, Aschbacher-Smith LE, Rizvi TA, Wu J, Ershler R, Wolters P, Therrien J, Glod J, Belasco JB, Schorry E, Brofferio A, Starosta AJ, Gillespie A, Doyle AL, Ratner N, Widemann BC, Engl N. J. Med 2016, 375, 2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chung V, McDonough S, Philip PA, Cardin D, Wang-Gillam A, Hui L, Tejani MA, Seery TE, Dy IA, Al Baghdadi T, Hendifar AE, Doyle LA, Lowy AM, Guthrie KA, Blanke CD, Hochster HS, JAMA Oncol 2017, 3, 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yu Q, Roberts MG, Houdaihed L, Liu Y, Ho K, Walker G, Allen C, Reilly RM, Manners I, Winnik MA, Nanoscale 2021, 13, 280. [DOI] [PubMed] [Google Scholar]
- [16].Zhou Z, Ma X, Jin E, Tang J, Sui M, Shen Y, Van Kirk EA, Murdoch WJ, Radosz M, Biomaterials 2013, 34, 5722. [DOI] [PubMed] [Google Scholar]
- [17].Allen C, Maysinger D, Eisenberg A, Colloids Surf., B 1999, 16, 3. [Google Scholar]
- [18].a) Bakshi RP, Nenortas E, Tripathi AK, Sullivan DJ, Shapiro TA, Sci. Transl. Med 2013, 5, 205ra135; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liston DR, Davis M, Clin. Cancer Res 2017, 23, 3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bruckman MA, Randolph LN, VanMeter A, Hern S, Shoffstall AJ, Taurog RE, Steinmetz NF, Virology 2014, 449, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Barua S, Yoo J-W, Kolhar P, Wakankar A, Gokarn YR, Mitragotri S, Proc. Natl. Acad. Sci. USA 2013, 110, 3270; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kolhar P, Anselmo AC, Gupta V, Pant K, Prabhakarpandian B, Ruoslahti E, Mitragotri S, Proc. Natl. Acad. Sci. USA 2013, 110, 10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Christian DA, Cai S, Garbuzenko OB, Harada T, Zajac AL, Minko T, Discher DE, Mol. Pharm 2009, 6, 1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Alves VM, Hwang D, Muratov E, Sokolsky-Papkov M, Varlamova E, Vinod N, Lim C, Andrade CH, Tropsha A, Kabanov A, Sci. Adv 2019, 5, eaav9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hwang D, Dismuke T, Tikunov A, Rosen EP, Kagel JR, Ramsey JD, Lim C, Zamboni W, Kabanov AV, Gershon TR, Sokolsky-Papkov M, Nanomed.: Nanotechnol., Biol. Med 2021, 32, 102345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vinod N, Hwang D, Azam SH, Van Swearingen AED, Wayne E, Fussell SC, Sokolsky-Papkov M, Pecot CV, Kabanov AV, Sci. Adv 2020, 6, eaba5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Glinka CJ, Barker JG, Hammouda B, Krueger S, Moyer JJ, Orts WJ, J. Appl. Crystallogr 1998, 31, 430. [Google Scholar]
- [26].Kline SR, J. Appl. Crystallogr 2006, 39, 895. [Google Scholar]
- [27].Pedersen JS, Schurtenberger P, Macromolecules 1996, 29, 7602. [Google Scholar]
- [28].a) Chen W-R, Butler PD, Magid LJ, Langmuir 2006, 22, 6539; [DOI] [PubMed] [Google Scholar]; b) Percus JK, Yevick GJ, Phys. Rev 1958, 110, 1. [Google Scholar]
- [29].a) Hwang D, Ramsey JD, Makita N, Sachse C, Jordan R, Sokolsky-Papkov M, Kabanov AV, J. Controlled Release 2019, 307, 261; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Topel Ö, Çakır BA, Budama L, Hoda N, J. Mol. Liq 2013, 177, 40. [Google Scholar]
- [30].Lim C, Moon J, Sim T, Won WR, Lee ES, Youn YS, Oh KT, J. Controlled Release 2019, 295, 164. [DOI] [PubMed] [Google Scholar]
- [31].a) Robichaud G, Barry JA, Garrard KP, Muddiman DC, J. Am. Soc. Mass Spectrom 2013, 24, 92; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Robichaud G, Barry JA, Muddiman DC, J. Am. Soc. Mass Spectrom 2014, 25, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kessner D, Chambers M, Burke R, Agus D, Mallick P, Bioinformatics 2008, 24, 2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bokhart MT, Nazari M, Garrard KP, Muddiman DC, J. Am. Soc. Mass Spectrom 2018, 29, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Research data are not shared.