


Abstract
The general transcription factor TFIIB is a key component in the eukaryotic RNA polymerase II (RNAPII) transcriptional machinery. We have previously shown that a yeast TFIIB mutant (called YR1m4) with four amino acid residues in a species-specific region changed to corresponding human residues affects the expression of genes activated by different activators in vivo. We report here that YR1m4 can interact with several affected activators in vitro. In addition, YR1m4 and other mutants with amino acid alterations within the same region can interact with TATA-binding protein (TBP) and RNAPII normally. However, YR1m4 is defective in supporting activator-independent transcription in assays con-ducted both in vitro and in vivo. We further demonstrate that the interaction between the C-terminal core domain and the N-terminal region is weakened in YR1m4 and other related TFIIB mutants. These results suggest that the intramolecular interaction property of yeast TFIIB plays an important role in transcription regulation in cells.
INTRODUCTION
Accurate transcription initiation by RNA polymerase II (RNAPII) requires a group of proteins called general transcription factors (GTFs). Five such GTFs, TATA-binding protein (TBP), TFIIB, TFIIE, TFIIF and TFIIH, enable RNAPII to recognize gene promoters and participate in the assembly of preinitiation complexes (PICs) (1–4). According to a stepwise model of PIC formation (5), TBP binds to DNA first, followed by the binding of TFIIB, then RNAPII–TFIIF and finally TFIIE and TFIIH. TFIIB interacts with both TBP and RNAPII (6) and is believed to provide a physical link between DNA–TBP and RNAPII–TFIIF complexes. A holoenzyme model proposes that TFIIB may be part of a pre-assembled complex called RNAPII holoenzyme (7), which contains several GTFs in addition to RNAPII. Both genetic and biochemical studies have provided strong evidence that TFIIB also participates in the process of transcription start site selection (8–12). In addition, TFIIB interacts with many activators and, therefore, has been proposed to be a target recruited by activators during transcription activation (13–15).
Analysis of TFIIB primary sequences from different eukaryotes has revealed several structural motifs (9), including a zinc ribbon (16) in the N-terminal region and two imperfect repeats in the C-terminal region. The N-terminal region of TFIIB is highly accessible to protease digestion and is critical for the interaction with RNAPII (6,17–19). The C-terminal region is protease resistant (17,19) and hence referred to as the core domain of TFIIB (cTFIIB). According to a solution structure of cTFIIB (20) and a crystal structure of a TATA–TBP–cTFIIB ternary complex (21), human cTFIIB contains two similarly folded domains, each of which consists of five α-helices. Biochemical studies have suggested an intramolecular interaction between N- and C-domains of human TFIIB (22), an interaction that could be affected by activators. In addition, recent NMR studies have demonstrated that human cTFIIB itself may adopt multiple conformations, which can be altered by interacting with either the N-terminal domain or the activator VP16 (23). However, there is a relative shortage of evidence that demonstrates the biological importance of the conformational properties of TFIIB (see Discussion).
In the yeast Saccharomyces cerevisiae, TFIIB is a monomeric protein of 345 amino acid residues encoded by the essential SUA7 gene (9). Although yeast TFIIB is structurally similar to human TFIIB, it cannot be functionally replaced by human TFIIB in vivo (24,25). Mutational studies of yeast TFIIB revealed a species-specific surface within a solvent-exposed region in the first repeat of cTFIIB (24). Four positions within this region were identified as critical to TFIIB function in vivo (25). A TFIIB mutant, called YR1m4, with amino acid residues changed at these four critical positions to corresponding human TFIIB residues, impaired the expression of genes activated by different activators in vivo (24,25). Thus, the biochemical characterization of this mutant TFIIB will provide important insights into the molecular mechanisms of transcription regulation.
In our efforts to further determine the biochemical properties of YR1m4, we demonstrate that this mutant TFIIB can interact with several activators. We also demonstrate that YR1m4 and other related TFIIB mutants can interact with several individual GTFs, including TBP, TFIIF and RNAPII. We further report that YR1m4 is compromised in supporting activator-independent transcription in both in vivo and in vitro assays. Finally, we demonstrate that an intramolecular interaction between the N- and C-terminal domains of the tested TFIIB mutants is affected, and our experiments reveal a strong correlation between this intramolecular interaction property and activator-independent transcriptional activity in vivo. Our results suggest that the intramolecular interaction of yeast TFIIB plays an important role in transcription regulation.
MATERIALS AND METHODS
Yeast strains
The yeast strains used in the analysis of the expression of endogenous CTS1 gene are listed in Table 1. The SUA7 gene in W303 diploid strain DY5424 (MATa / MATα + / ace2::HIS3 + / swi5::TRP1 ade2-1 can1-100 his3-11,15 leu2-3,112 lys2 trp1-1 ura3-1) was disrupted using a sua7::hisG-URA3-hisG disruptor plasmid, and correct disruption was confirmed by Southern blotting. This diploid was transformed with one of two plasmids, pMA1210 or pYR1m4-LEU2 (24). These plasmids, which have LEU2 markers, express either the wild-type SUA7 protein or the YR1m4 mutant version of SUA7 from the ADH1 promoter. The two diploid strains with the plasmids were sporulated and haploid segregants differing at ACE2, SWI5 and SUA7 were isolated.
Table 1. Yeast strains used in the analysis of expression of the endogenous CTS1 gene.
| Strain | Description |
|---|---|
| DY5529 | MATα sua7::hisG-URA-hisG + YEp-pADH1-TFIIB(WT) |
| DY5531 | MATa sua7::hisG swi5::TRP1 + YEp-pADH1-TFIIB(WT) |
| DY5533 | MATα sua7::hisG-URA3-hisG ace2::HIS3 + YEp-pADH1-TFIIB (WT) |
| DY5535 | MATa sua7::hisG-URA3-hisG ace2::HIS3 swi5::TRP1 + YEp-pADH1-TFIIB(WT) |
| DY5537 | MATα sua7::hisG-URA3-hisG + YEp-pADH1-TFIIB(YR1m4) |
| DY5539 | MATa sua7::hisG-URA3-hisG swi5::TRP1 + YEp-pADH1-TFIIB(YR1m4) |
| DY5541 | MATα sua7::hisG-URA3-hisG ace2::HIS3 + YEp-pADH1-TFIIB(YR1m4) |
| DY5543 | MATα sua7::hisG-URA3-hisG ace2::HIS3 swi5::TRP1 + YEp-pADH1-TFBII(YR1m4) |
The yeast strain 604 (MATα his3 trp1 ura3-52 leu2 cyh2r LYS2+ Δsua7) has its chromosomal SUA7 gene disrupted with a sua7::hisG-URA3-hisG disruptor plasmid, while cell viability is supported by plasmids expressing either wild-type TFIIB or YR1m4 from the ADH1 promoter. This strain was used for reporter gene assays in Figure 2. Yeast strain FP133 used in preparation of TFIIB depleted whole cell extract is a gift of Dr A. Ponticelli. This strain has a temperature-sensitive allele of TFIIB (12), which permits the preparation of a TFIIB-depleted whole cell extract. The yeast strain used in the artificial recruitment assay is a gift of Dr M. Strubin (26). This strain carries a lacZ reporter gene integrated at the HIS3 locus with a single RFX-binding site upstream of a his3 TATA element. The chromosomal TFIIB gene is deleted and a low level of wild-type TFIIB is provided by pYES2 plasmid when cells are grown in glucose medium.
Figure 2.

The TFIIB mutant YR1m4 is defective in supporting activated gene expression in vivo. (A) A CYC1TATA-lacZ reporter plasmid with (pM1820) (31) or without (pLG670Z) (31) CTS1UAS element was introduced into yeast strain 604 containing a plasmid expressing either wild-type TFIIB or YR1m4. The absolute activities are 1.58, 101.54, 1.0 and 2.68 U β-galactosidase in columns 1–4, respectively. (B) A LexAOP-CYC1TATA-lacZ reporter plasmid was introduced into the same strain as in (A). Plasmids that express either LexA-VP16 or LexA were transformed into the cells and assayed for β-galactosidase activity. The absolute β-galactosidase units are 4.6, 100.5, 2.8 and 6.4 in columns 1–4, respectively. All assays were done using three independent transformants in duplicate. The SDs are <20% in all cases.
Plasmid constructions and recombinant protein purification
DNA fragments were amplified by polymerase chain reactions (PCRs) and cloned into vector pET15b for His6-fusion protein or vector pGEX-KG for glutathione-S-transferase (GST) fusion protein production. DNA fragments used in in vitro transcription and translation were cloned into vector pGEM5 under control of the SP6 promoter. Details of plasmid constructions are available upon request.
Constructs were expressed in Escherichia coli strain BL21 (His6-fusion protein), Xa90 or DH5α (GST-fusion proteins). Cells were grown at 37°C and IPTG was added to a final concentration of 1 mM when the OD600nm of the culture reached 0.4–0.6. Cells were then incubated at 37°C for 3 h and extracts were prepared. For GST-fusion proteins, cell extracts were incubated with glutathione Sepharose-4B beads in buffer B (1× PBS buffer, 1% Triton X-100, 1 mM PMSF, protease inhibitor cocktail tablets added according to the instructions of Boehringer, Indianapolis, IN) at 4°C for 1 h. The beads were washed three times with the same buffer with 1 mM PMSF but without other protease inhibitors. The GST-fusion proteins were eluted with buffer E (20 mM reduced glutathione in 50 mM Tris–HCl, pH 8.0). Alternatively, the beads were incubated with 5 U of thrombin in buffer T (50 mM Tris–HCl, pH 8.0, 0.1% β-mercaptoethanol, 150 mM NaCl, 2.5 mM CaCl2) at room temperature for 2 h to release proteins without GST. Purified proteins were dialyzed against buffer D (20 mM HEPES–KOH, pH 7.5, 20% glycerol, 10 mM EGTA, 10 mM MgSO4, 5 mM DTT). Protein concentrations were determined using a protein assay kit (Bio-Rad, Hercules, CA), and their purity was judged by SDS–PAGE. His6-fusion proteins were purified according to Qiagen (Valencia, CA).
Analysis of expression of endogenous CTS1 gene
Yeast cells were grown on YPD media and RNA was isolated for S1 nuclease protection assays using CTS1 and CMD1 probes as described previously (27).
GST pull-down assay
Glutathione Sepharose beads (40 µl) were washed sequentially with 10 vol of H2O, 3.0 M NaCl, H2O, and binding buffer (40 mM HEPES, pH 7.5, 100 mM KCl, 5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, 0.5 mM PMSF, 0.5% NP-40, 0.25% milk). The beads were suspended in 0.4 ml of binding buffer, the purified GST fusion proteins were added and incubated at 4°C for 2 h. The beads were then washed twice with 10 vol of binding buffer and resuspended in 0.3 ml of binding buffer before the second protein was added. The mixture was incubated at 4°C for 1–3 h. The beads were washed twice with 10 vol of binding buffer, and then once with the binding buffer without milk. Forty-five microliters SDS–PAGE loading buffer was added and the mixture was boiled for 5 min. After centrifugation at 10 000 r.p.m. for 5 min, 15 µl of the supernatant was subjected to SDS–PAGE and assayed by either western blot or autoradiography. The amount of precipitated GST fusion protein, estimated by either western blot or Coomassie brilliant blue staining, was used for normalization as indicated in the figure legends.
In vitro transcription assay
TFIIB-depleted whole-cell extracts were prepared from strain FP133 as described by Bangur et al. (12). The plasmid pJJ470 used in this experiment is a gift from Dr J. Jaehning. It contains a 278 bp G-less cassette under the control of CYC1 TATA box with UASGAL upstream (28).
Artificial recruitment assay
Artificial recruitment assay was conducted according to Gonzalez-Couto et al. (26). Basically, our TFIIB mutant genes were cloned into a Myc-TFIIB vector. The encoded fusion proteins were then assayed for their abilities upon artificially recruited to a promoter (see 26 and text for further details).
Gel shift assay of DB complex
The TATA-containing DNA probe was a 42-bp fragment that contains the adenoviral major late promoter TATA box (29). The reaction buffer contains 20 mM HEPES (pH 7.5), 50 mM KCl, 10 mM KOAc, 7.5 mM MgCl2, 2 mM DTT, 200 µg/ml BSA. The reaction mixture (20 µl) was incubated at room temperature for 30 min and loaded onto a 5% native polyacrylamide gel (29:1) immediately. The gel was run at room temperature in 1× TBE buffer at 150 V. The gel was then dried and subjected to autoradiography.
RESULTS
The TFIIB mutant YR1m4 is defective in supporting activated gene expression in vivo
During the course of the analysis of our yeast TFIIB mutant YR1m4 in vivo, we observed that cells containing YR1m4 exhibited an aggregated phenotype (data not shown), suggesting a defect in cell separation. This phenotype resembles that of cells lacking the CTS1 gene, which encodes an endochitinase that hydrolyzes chitin in Saccharomyces (30). To determine whether the expression of the endogenous CTS1 gene is affected in YR1m4 cells, we carried out an S1 nuclease protection assay using mRNA isolated from different isogenic yeast strains (Fig. 1). Our results demonstrate that the endogenous CTS1 gene was expressed at a lower level in YR1m4 cells when compared to that in wild-type cells (Fig. 1A, lane 1 versus 2). Northern analysis indicates that ACE2 mRNA level in YR1m4 cells was unaffected (data not shown). Consistent with previous studies (30), our S1 assay results also demonstrate that the CTS1 expression is activated by ACE2 and independent of the homologous activator SWI5. Figure 1B is a summary of our S1 assay results with CTS1 expression levels normalized to an internal control.
Figure 1.
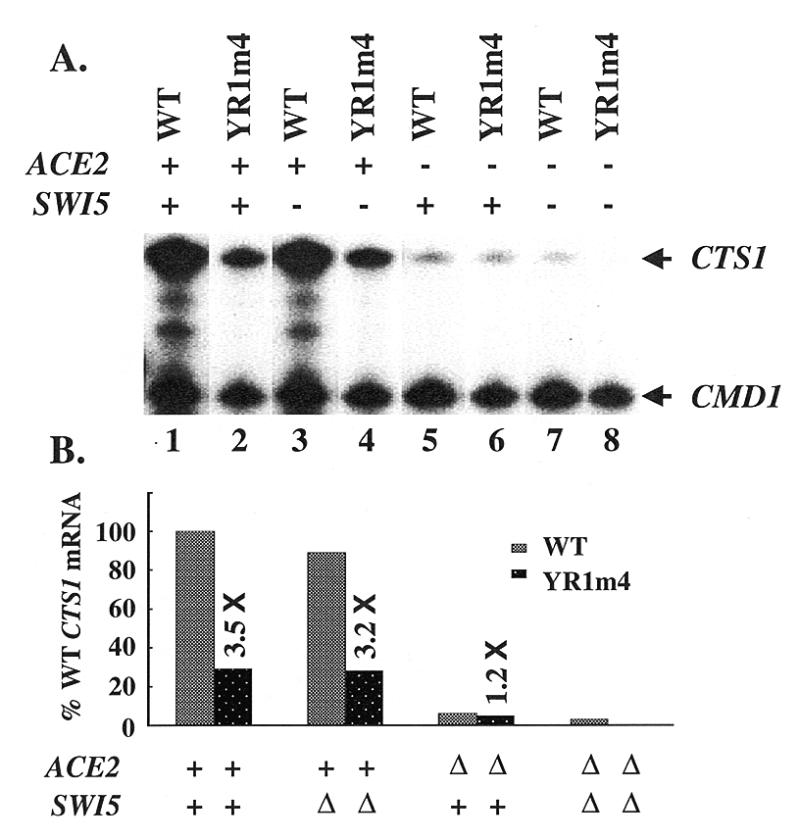
The expression of endogenous CTS1 gene is decreased in YR1m4 cells. (A) S1 nuclease protection analysis of mRNA isolated from isogenic yeast strains that contain either wild-type TFIIB or YR1m4 and differ at ACE2 and SWI5. CMD1 was used as an internal control (31). (B) Relative CTS1 mRNA levels. Activities shown were quantitated by ImageQuant v1.2 and normalized with the CMD1 level. The values are 100, 29, 89, 28, 6, 5, 3, 0.3 in columns 1–8, respectively.
We also analyzed the activity of a CYC1-lacZ reporter gene under the control of the CTS1 upstream activation sequence (UAS) (31). Our β-galactosidase assay results shown in Figure 2A (columns 1 and 2) demonstrate that ACE2 increased the transcription activity by 64.3-fold in wild-type cells but only 2.7-fold in YR1m4 cells (Fig. 2A, columns 3 and 4). In a similar reporter gene assay, transcription activation by an activator containing the transcription activation domain (TAD) of VP16 was also shown to be decreased. LexA-VP16 fusion protein activated transcription by 21.8-fold in wild-type cells (Fig. 2B, columns 1 and 2) but only 2.3-fold in YR1m4 cells (Fig. 2B, columns 3 and 4). In addition, our previous studies have demonstrated that several other artificial reporter genes under the control of different UASs were also affected to various extents in YR1m4 cells (25). Together, these results show that YR1m4 has a defect in supporting activated transcription in vivo.
Interaction between YR1m4 and activators
TFIIB has been proposed to be a direct target for many transcriptional activators, primarily by virtue of protein–protein interaction assays conducted in vitro (13,14). To determine whether the interaction between TFIIB and the activators ACE2 and VP16 is affected by YR1m4 mutations, we carried out GST pull-down assays. In these assays, bacterially expressed and immobilized GST fusion proteins, containing either wild-type TFIIB or YR1m4, were used to precipitate activator proteins generated in an in vitro transcription/translation system. Our results shown in Figure 3 demonstrate that ACE2 and GAL4-VP16 can interact with both wild-type TFIIB and YR1m4 (Fig. 3A and B), suggesting that the residues mutated in YR1m4 do not directly interfere with the interaction between TFIIB and these activators. Another activator, native GAL4 with its own TADs, whose activation was only slightly affected in a reporter gene assay in vivo (<1.5-fold decrease) (25), can also interact with both wild-type TFIIB and YR1m4 in a similar assay (Fig. 3C). Among the tested activators that showed a decreased activity in YR1m4 cells, only TAD1 of ADR1 (32), which activates the expression of the ADH2 gene, exhibited a defect in interacting with YR1m4 (C.Denis, personal communication). Together, these experiments suggest that the inability of YR1m4 to interact with activators cannot be the only molecular defect accounting for its inability to support transcription efficiently.
Figure 3.
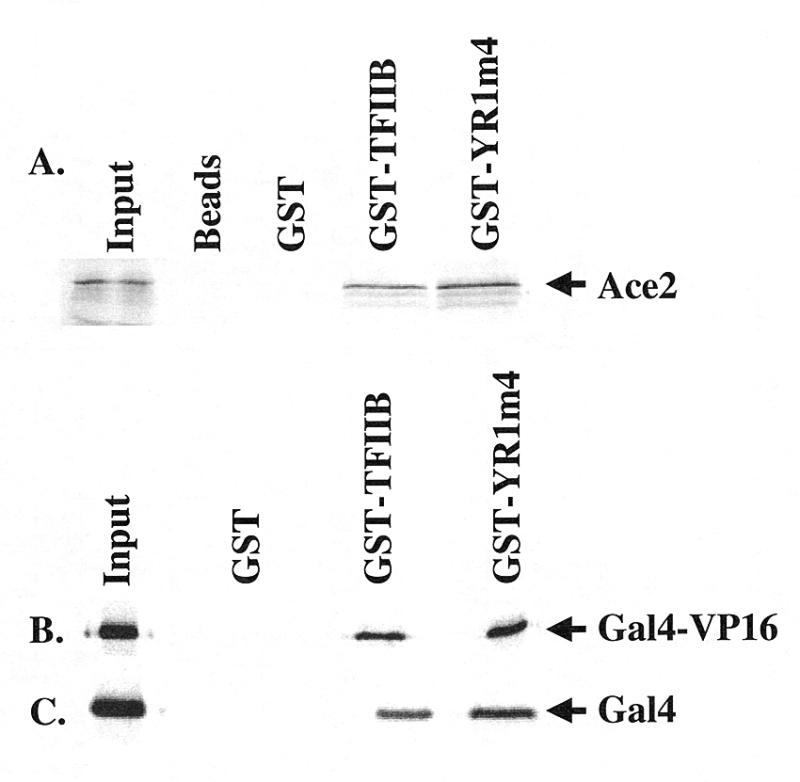
YR1m4 interacts with activators ACE2, GAL4-VP16 and GAL4. Aliquots of 5 µg of purified GST, GST-TFIIB or GST-YR1m4 were incubated with 40 µl of glutathione Sepharose beads as described in Materials and Methods. The activators ACE2, GAL4-VP16 and full-length GAL4 were in vitro transcribed and translated as 35S-labeled proteins and 5 µl of lysate were used in each reaction. The precipitated proteins were applied to SDS–PAGE; 0.1 µl of lysate was used as input shown in the figure.
YR1m4 is defective in supporting activator-independent transcription in vitro and in vivo
To search for molecular defects associated with YR1m4, we carried out transcription assays in vitro. A TFIIB-depleted yeast whole cell extract was prepared from the strain FP133 (12) and supplemented with recombinant TFIIB, either wild-type or YR1m4. The extract itself does not support transcription due to the lack of sufficient amount of TFIIB (Fig. 4, lane 2). As previously reported (12), wild-type TFIIB can restore transcription efficiently (lane 3), which can be further increased by addition of GAL4-VP16 (lane 4); our assay was carried out using high template DNA concentrations, which, as previously reported (28), reduce the effect of activation. When YR1m4 was used in the same assay, it exhibited a severe defect in supporting both non-activated (lane 5) and activated transcription (lane 6).
Figure 4.
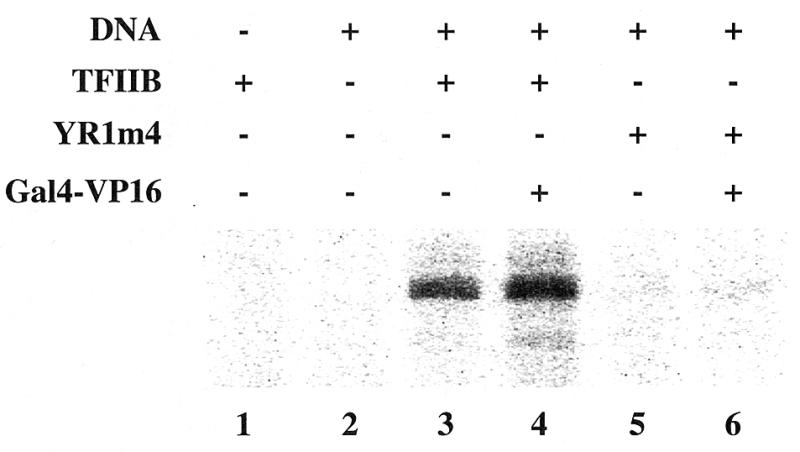
YR1m4 is defective in supporting transcription in vitro. In vitro transcription assay was performed with a TFIIB-depleted whole cell extract (12). This extract is unable to support transcription without the addition of recombinant TFIIB protein (lane 2). An aliquot of 2 µl of extract (~80 µg of protein) was used in the reaction. G-less cassette plasmid pJJ470 (100 ng) (28) were used as template. Purified recombinant TFIIB protein (50 ng), either wild type or YR1m4, was supplemented as indicated. Activated transcription was assayed upon the addition of 10 ng of purified GAL4-VP16 fusion protein.
We also used an artificial recruitment assay (26) to further characterize the behavior of YR1m4 in supporting activator-independent transcription in vivo. In this experiment, a fusion protein, Max-RFX, was expressed in yeast cells. This protein contains the leucine zipper of Max fused to a DNA-binding protein RFX, which can recognize a RFX binding site (X) placed upstream of a lacZ reporter gene. The leucine zipper of c-Myc was attached to TFIIB at the C-terminus. When expressed in yeast cells, TFIIB–Myc fusion protein is recruited by Max-RFX through the hetero-dimerization interaction between Myc and Max, leading to the expression of the nearby reporter gene lacZ (Fig. 5A). Since no conventional DNA-binding activator is involved in this artificial recruitment assay, it measures the activator-independent function of TFIIB, reflecting its intrinsic properties (M.Strubin, personal communication). In this experiment, a non-functional TFIIB mutant, C48S, was used as a negative control, which showed only background activity in this assay as previously reported (26). Our results shown in Figure 5B demonstrate that YR1m4 has only 19% activity compared with that of wild-type TFIIB. It is interesting to note that, in the absence of both activators SWI5 and ACE2, the expression of the endogenous CTS1 gene in YR1m4 cells is decreased compared with that in wild-type cells (Fig. 1A, lanes 7 and 8). Together, our experiments suggest that YR1m4 is defective in activator-independent transcription both in vitro and in vivo. Interestingly, the defect of YR1m4 in supporting activator-independent transcription in vivo is more severe on endogenous (Fig. 1A, compare lanes 7 and 8) or integrated (Fig. 5) reporter genes than on reporter genes carried on replication plasmids (Fig. 2A and B, compare columns 1 and 3) (24), possibly reflecting differences in promoter accessibility.
Figure 5.

In vivo assay of the activator-independent transcriptional activity. (A) A schematic diagram depicting the artificial recruitment assay. See text and Gonzalez-Couto et al. (26) for further details. X, RFX binding site; Tx, transcription; IIB, TFIIB. (B) Relative activator-independent transcriptional activities of TFIIB derivatives. The level of β-galactosidase activity upon the recruitment of wild-type TFIIB was assigned the number of 100. C48S, a single point mutation that abolishes the interaction between TFIIB and RNAPII (26), was used as a negative control. VP16-RFX, which contains a conventional activation domain VP16 fused to RFX, was used as a positive control.
We further tested in the artificial recruitment assay several other TFIIB mutants that have amino acid alterations in the same region mutated in YR1m4 but exhibit varying ability to support cell growth (24,25). Our experiments revealed a good correlation between their activity in this artificial recruitment assay and their ability to support cell growth. These findings further support the idea that the physiological phenotype associated with our TFIIB mutants results from their defects in supporting transcription.
Interaction between TFIIB and other GTFs
Since TFIIB is able to interact with several other GTFs, including TBP (5) and RNAPII (6,33), it is possible that YR1m4 and other related yeast TFIIB mutants fail to support activator-independent transcription because they are defective in interacting with these factors. To test this idea directly, the interaction between TFIIB and both TBP and RNAPII was analyzed in GST pull-down experiments. As shown in Figure 6A, all the tested TFIIB mutants can interact with in vitro translated 35S-labeled TBP. These mutants can also interact with RNAPII normally (Fig. 6B and C).
Figure 6.
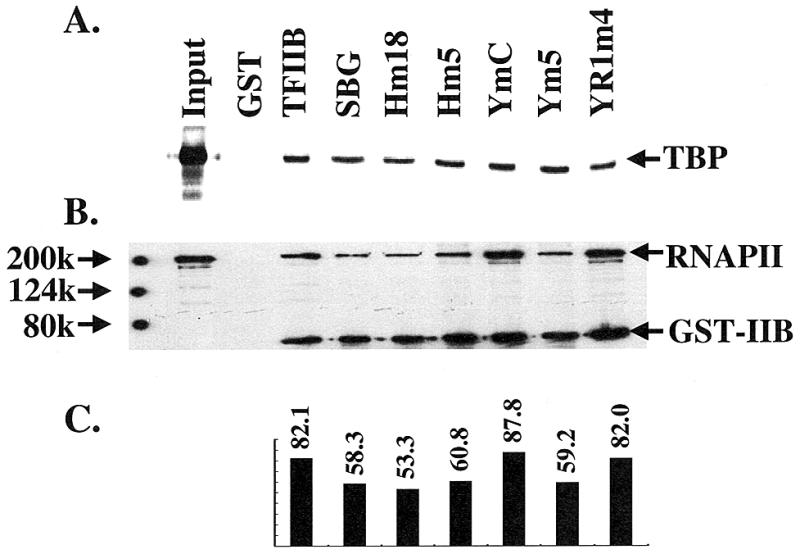
YR1m4 can interact with TBP and RNAPII in vitro. (A) GST-TFIIB derivatives can pull down TBP. 35S-labeled yeast TBP was synthesized in an in vitro transcription and translation system. See Materials and Methods and legend to Figure 3 for further details. (B) TFIIB–RNAPII interaction. Partially purified yeast RNAPII, kindly provided by Dr M. Sayre, was used in the pull-down experiment. The immobilized proteins were assayed by western blot using antibodies against the largest subunit of RNAPII (8WG16 from BABCO) and the GST (B-14, Santa Cruz Biotechnology). Input represents one-twentieth of the RNAPII used in the experiments. (C) The relative amount of immobilized RNAPII normalized by the amount of precipitated GST fusion proteins. Although there are light differences in the relative amounts of precipitated RNAPII, such differences do not correlate with the in vivo activity of the tested TFIIB derivatives.
We also performed a gel shift experiment to detect the formation of a DNA–TBP–TFIIB (DB) complex. This analysis allows the detection of TFIIB–TBP interaction on DNA. The results shown in Figure 7 demonstrate that the ability of YR1m4 to form the DB complex is not impaired. In fact, such complex formation is enhanced slightly (2–3-fold) for YR1m4; we currently do not fully understand the molecular basis for this observation, but it might be related to either the conformation properties of YR1m4 (see below) or subtle effects on the interaction with TBP–DNA complex although the mutated amino acids do not face TBP or DNA (21).
Figure 7.
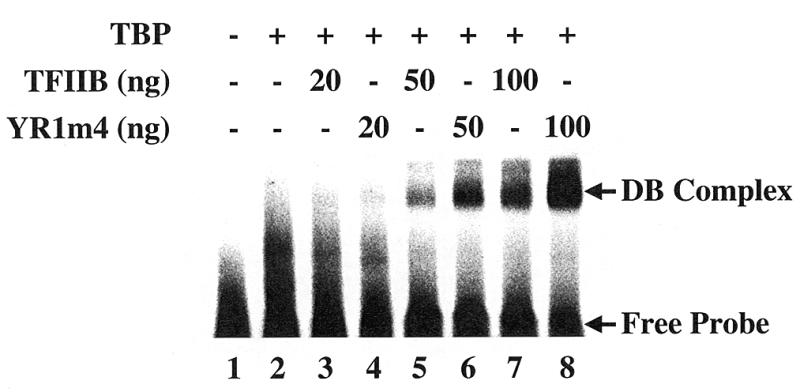
DB complex formation with wild-type TFIIB and YR1m4. The reactions contained 50 fmol of 32P-labeled 42-bp DNA fragments containing the ADMLP-2 TATA box. 20 ng of purified yeast TBP was added except in lane 1.
In addition to TBP and RNAPII, we also tested the interaction between TFIIB and several other GTFs, including TFIIF, and two subunits of TFIIH, Tfb2 and Tfb3 (34). The Tfb2 subunit of TFIIH cannot interact with either wild-type or mutant TFIIB. TFIIF and Tfb3 are able to interact with TFIIB, but no difference was observed between wild-type TFIIB and YR1m4 (data not shown). The fact that the interaction between TFIIB and the tested GTFs is not affected by these mutations suggests that this region of yeast TFIIB is not directly involved in interacting with these factors individually. However, we cannot exclude the possibility that YR1m4 fails to directly interact with other GTFs not included in our tests.
Decreased intramolecular interaction in yeast TFIIB mutants
An intramolecular interaction between the N-terminal domain and core domain has been observed in human TFIIB (22). Such an interaction has also been observed in yeast TFIIB as reported by Wu and Hampsey (15) and Bangur et al. (35). To determine whether the mutations in YR1m4 alter the intramolecular interaction, TFIIB core domain (amino acids 128–345) was expressed in E.coli as a GST fusion protein and used to precipitate in vitro synthesized 35S-labeled N-terminal domain (amino acids 1–135). Our results (Fig. 8A) show that, unlike wild-type TFIIB (lane 3), YR1m4 exhibits a weakened intramolecular interaction (lane 4). A titration experiment further confirmed that the intramolecular interaction is weakened in YR1m4 compared with wild-type TFIIB (Fig. 8B and C).
Figure 8.
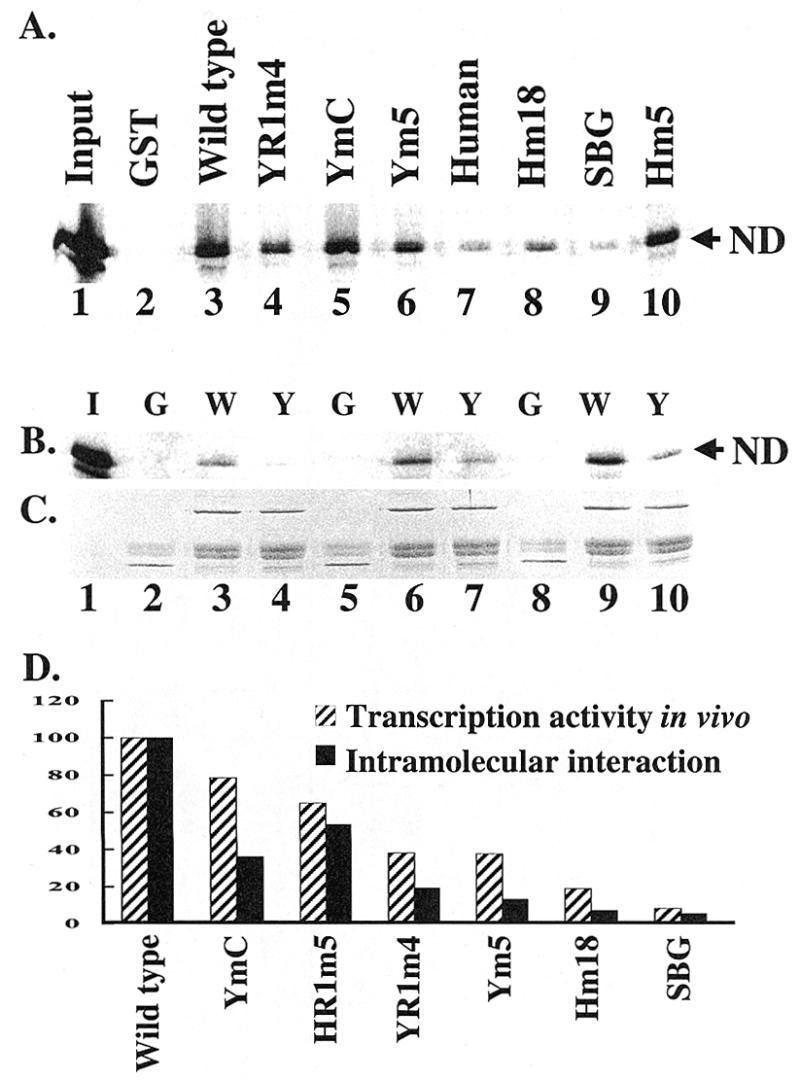
The intramolecular interaction is altered in YR1m4 and other related TFIIB mutants. (A) The C-terminal domain of yeast TFIIB (amino acid residues 128–345) was expressed as a GST fusion protein to pull down in vitro synthesized 35S-labeled N-terminal domain (amino acid residues 1–135). The experimental procedures were the same as described in the legend of Figure 3. The human TFIIB core domain (amino acid residues 124–315) was also included in this experiment, which failed to interact with yeast TFIIB N-domain. ND, N-terminal domain. Input represents one-twentieth of the lysate used in the pull down experiments. (B) Purified GST (5 µg), C-terminal domain of yeast TFIIB or YR1m4 was incubated with different amounts of 35S-labeled N-terminal domain (2, 4 and 8 µl of lysate in lanes 2–4, 5–7 and 8–10, respectively). G, GST; W, wild-type TFIIB C-terminal domain GST fusion; Y, YR1m4 C-terminal domain GST fusion; I, input, which represents one-tenth of the lysate used in lanes 8–10. (C) The amount of precipitated GST fusion proteins was estimated by SDS–PAGE and Coomassie blue staining. (D) Correlation between the intramolecular interaction property (A) and in vivo transcription activity (Fig. 5B). The amount of N-terminal domain pulled down by the C-terminal domain of yeast TFIIB was assigned the number 100.
To determine whether the intramolecular interaction defect associated with YR1m4 is biologically relevant, we further analyzed the intramolecular interaction properties of several other related TFIIB mutants with varying phenotypic severity (24,25). Our experiments showed that all the related TFIIB mutants that were tested exhibited a weakened intramolecular interaction compared with wild-type TFIIB. More importantly, the intramolecular interaction properties of these mutants correlate well with their in vivo transcription activity detected in the artificial recruitment assay (Fig. 8D) as well as their ability to support cell growth (24,25). These results further illustrate the biological importance of intramolecular interaction of TFIIB.
We performed a partial proteolysis assay to determine whether protease sensitivity is altered in YR1m4. Either the recombinant TFIIB protein with HA attached to the C-terminus or the in vitro synthesized TFIIB protein was subjected to Staphylococcus aureus V8 protease digestion. Under our experimental conditions, no significant difference was observed between wild-type TFIIB and YR1m4 (data not shown). As reported previously (35), the major V8 protease cleavage site is located in a hinge region between the N-terminal zinc ribbon domain and the core domain. It is possible that, in the absence of all the other factors that interact with TFIIB in the PIC, the altered intramolecular interaction of YR1m4 affects its conformation without generating a new protease sensitivity profile.
DISCUSSION
The experiments described in this report were designed to further analyze the molecular properties of a mutant yeast TFIIB, YR1m4, which is defective in supporting activated transcription in vivo (24,25 and this report). Our biochemical studies show that YR1m4 is not defective in interacting directly with several affected activators. Furthermore, despite its ability to interact with several GTFs tested individually, this mutant TFIIB is severely compromised in supporting activator-independent transcription in vitro. In addition, we show that in an artificial recruitment assay, YR1m4 is defective in supporting activator-independent transcription in vivo. The intramolecular interaction between the N-terminal domain and core domain of this mutant is reduced in comparison with wild-type TFIIB. Our experiments reveal a strong correlation between the intramolecular interaction property and the activator-independent transcriptional activity in vivo (Fig. 8D) as well as the ability to support cell growth (24,25). Our results suggest that this intramolecular interaction of TFIIB plays an important role in proper transcription control in cells.
Most of the yeast TFIIB mutants reported so far by our and other laboratories cause slow cell growth phenotypes (9,12,24,36). Our previous studies (25), as well as the current analysis, suggest that the slow growth phenotype of YR1m4 cells is due to a decreased expression of specific genes. While some TFIIB mutants were shown to have activation-specific defects (15), others were reported to be defective in supporting basal transcription in vitro (12,36). It has been shown that several mutant TFIIB molecules that are defective in interacting with certain activators fail to support activated transcription in vitro (14). Thus, it has been proposed that the direct recruitment of TFIIB by these activators can lead to an increased level of transcription (13,14). For YR1m4, we observed that most activators, except TAD1 of ADR1 (C.Denis, personal communication), could directly interact with the mutant TFIIB protein. These results suggest that the transcription defect associated with YR1m4 may result from a generally defective TFIIB and, in fewer cases, an impaired direct interaction between activators and YR1m4. Our finding that YR1m4 exhibits a weakened intramolecular interaction and is compromised in supporting activator-independent transcription further illustrates a general defect of this mutant protein.
Biochemical and structural studies of human TFIIB suggest that conformational changes of TFIIB may contribute to transcription regulation in vitro (22,23). It was reported that the activation domain of VP16 could disrupt the intramolecular interaction between the N-terminal domain and the core domain of human TFIIB (22). Recent NMR studies of human cTFIIB suggest that VP16 and the N-terminal domain of TFIIB may compete with each other by interacting with the same or an overlapping surface on cTFIIB (23). Although the structure of full-length TFIIB protein is currently unavailable, NMR studies suggest that human TFIIB may adopt multiple conformations, resulting from both intramolecular interaction and the interaction with other factors, including GTFs and activators, as well as RNAPII (19). These studies suggest that TFIIB may undergo conformational changes in transcription.
Recent studies have described two yeast TFIIB mutants that affect the conformation properties of the protein (15,35). While both of those mutants contain amino acid alterations at the N-terminus (S53P and R64E), YR1m4 contains mutations in the core domain. In addition, YR1m4 exhibits biochemical and physiological properties that are different from those of the two reported mutants. In particular, while the S53P mutant protein described by Wu and Hampsey (15) shows a major conformational defect only in the presence of the activator protein PHO4, YR1m4 has an intramolecular interaction defect affecting both activated and activator-independent transcription in vivo and in vitro. While intramolecular interaction in the R64E mutant protein described by Bangur et al. is enhanced (35), this interaction is weakened in YR1m4. Interestingly, despite their opposite effects on intramolecular interaction, both R64E and YR1m4 are defective in supporting transcription, although transcription defect for R64E was detected only in vitro but not in vivo (12). Taken together, these studies suggest that efficient transcription and activation require a well-balanced intramolecular interaction of TFIIB, an interaction that may facilitate dynamic conformational changes of the protein.
We propose that, during PIC formation, yeast TFIIB is in a conformation that is stabilized by its intramolecular interaction. Upon PIC assembly, interactions with other proteins including activators may compete with and eventually break the intramolecular interaction of TFIIB, thus facilitating a subsequent step(s) of transcription initiation. According to this model, the weakened intramolecular interaction of YR1m4 is disfavored for PIC formation, even though it can interact with other GTFs individually. Our model is supported by a recent observation that one of the currently analyzed TFIIB mutants with a mutation in the species-specific region, C149R (YmC), has a defect in PIC formation (37). Regardless of the exact events taking place during transcription initiation and activation, our study underscores the importance of intramolecular interaction of TFIIB in controlling transcription in cells.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs M. Sayre, R. Young, R. Kornberg, J. Jaehning, M. Strubin and A. Ponticelli for providing us with valuable materials, and Drs M. Barton, I. Cartwright, C. Denis, M. Strubin and members of our laboratories for discussions and comments on the manuscript. This publication was made possible by NIH grants GM48624 to D.J.S. and GM56215 to J.M., as well as NIH grant P30 ES06096 to University of Cincinnati.
REFERENCES
- 1.Hampsey M. (1998) Microbiol. Mol. Biol. Rev., 62, 465–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tjian R. and Maniatis,T. (1994) Cell, 77, 5–8. [DOI] [PubMed] [Google Scholar]
- 3.Ptashne M. and Gann,A. (1997) Nature, 386, 569–577. [DOI] [PubMed] [Google Scholar]
- 4.Orphanides G., Lagrange,T. and Reinberg,D. (1996) Genes Dev., 10, 2657–2683. [DOI] [PubMed] [Google Scholar]
- 5.Buratowski S., Hahn,S., Guarente,L. and Sharp,P.A. (1989) Cell, 56, 549–561. [DOI] [PubMed] [Google Scholar]
- 6.Ha I., Roberts,S., Maldonado,E., Sun,X., Kim,L.U., Green,M. and Reinberg,D. (1993) Genes Dev., 7, 1021–1032. [DOI] [PubMed] [Google Scholar]
- 7.Conaway R.C., Reines,D., Garrett,K.P., Powell,W. and Conaway,J.W. (1996) Methods Enzymol., 273, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berroteran R.W., Ware,D.E. and Hampsey,M. (1994) Mol. Cell. Biol., 14, 226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinto I., Ware,D.E. and Hampsey,M. (1992) Cell, 68, 977–988. [DOI] [PubMed] [Google Scholar]
- 10.Li Y., Flanagan,P.M., Tschochner,H. and Kornberg,R.D. (1994) Science, 263, 805–807. [DOI] [PubMed] [Google Scholar]
- 11.Pinto I., Wu,W.H., Na,J.G. and Hampsey,M. (1994) J. Biol. Chem., 269, 30569–30573. [PubMed] [Google Scholar]
- 12.Bangur C.S., Pardee,T.S. and Ponticelli,A.S. (1997) Mol. Cell. Biol., 17, 6784–6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Y.S., Ha,I., Maldonado,E., Reinberg,D. and Green,M.R. (1991) Nature, 353, 569–571. [DOI] [PubMed] [Google Scholar]
- 14.Roberts S.G., Ha,I., Maldonado,E., Reinberg,D. and Green,M.R. (1993) Nature, 363, 741–744. [DOI] [PubMed] [Google Scholar]
- 15.Wu W.H. and Hampsey,M. (1999) Proc. Natl Acad. Sci. USA, 96, 2764–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu W., Zeng,Q., Colangelo,C.M., Lewis,M., Summers,M.F. and Scott,R.A. (1996) Nat. Struct. Biol., 3, 122–124. [DOI] [PubMed] [Google Scholar]
- 17.Barberis A., Muller,C.W., Harrison,S.C. and Ptashne,M. (1993) Proc. Natl Acad. Sci. USA, 90, 5628–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buratowski S. and Zhou,H. (1993) Proc. Natl Acad. Sci. USA, 90, 5633–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malik S., Lee,D.K. and Roeder,R.G. (1993) Mol. Cell. Biol., 13, 6253–6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bagby S., Kim,S., Maldonado,E., Tong,K.I., Reinberg,D. and Ikura,M. (1995) Cell, 82, 857–867. [DOI] [PubMed] [Google Scholar]
- 21.Nikolov D.B., Chen,H., Halay,E.D., Usheva,A.A., Hisatake,K., Lee,D.K., Roeder,R.G. and Burley,S.K. (1995) Nature, 377, 119–128. [DOI] [PubMed] [Google Scholar]
- 22.Roberts S.G. and Green,M.R. (1994) Nature, 371, 717–720. [DOI] [PubMed] [Google Scholar]
- 23.Hayashi F., Ishima,R., Liu,D., Tong,K.I., Kim,S., Reinberg,D., Bagby,S. and Ikura,M. (1998) Biochemistry, 37, 7941–7951. [DOI] [PubMed] [Google Scholar]
- 24.Shaw S.P., Wingfield,J., Dorsey,M.J. and Ma,J. (1996) Mol. Cell. Biol., 16, 3651–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaw S.P., Carson,D.J., Dorsey,M.J. and Ma,J. (1997) Proc. Natl Acad. Sci. USA, 94, 2427–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez-Couto E., Klages,N. and Strubin,M. (1997) Proc. Natl Acad. Sci. USA, 94, 8036–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McBride H.J., Yu,Y. and Stillman,D.J. (1999) J. Biol. Chem., 274, 21029–21036. [DOI] [PubMed] [Google Scholar]
- 28.Woontner M., Wade,P.A., Bonner,J. and Jaehning,J.A. (1991) Mol. Cell. Biol., 11, 4555–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ziff E.B. and Evans,R.M. (1978) Cell, 15, 1463–1475. [DOI] [PubMed] [Google Scholar]
- 30.Dohrmann P.R., Butler,G., Tamai,K., Dorland,S., Greene,J.R., Thiele,D.J. and Stillman,D.J. (1992) Genes Dev., 6, 93–104. [DOI] [PubMed] [Google Scholar]
- 31.Dohrmann P.R., Voth,W.P. and Stillman,D.J. (1996) Mol. Cell. Biol., 16, 1746–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiang Y.C., Komarnitsky,P., Chase,D. and Denis,C.L. (1996) J. Biol. Chem., 271, 32359–32365. [DOI] [PubMed] [Google Scholar]
- 33.Fang S.M. and Burton,Z.F. (1996) J. Biol. Chem., 271, 11703–11709. [DOI] [PubMed] [Google Scholar]
- 34.Feaver W.J., Henry,N.L., Wang,Z., Wu,X., Svejstrup,J.Q., Bushnell,D.A., Friedberg,E.C. and Kornberg,R.D. (1997) J. Biol. Chem., 272, 19319–19327. [DOI] [PubMed] [Google Scholar]
- 35.Bangur C.S., Faitar,S.L., Folster,J.P. and Ponticelli,A.S. (1999) J. Biol. Chem., 274, 23203–23209. [DOI] [PubMed] [Google Scholar]
- 36.Pardee T.S., Bangur,C.S. and Ponticelli,A.S. (1998) J. Biol. Chem., 273, 17859–17864. [DOI] [PubMed] [Google Scholar]
- 37.Ranish J.A., Yudkovsky,N. and Hahn,S. (1999) Genes Dev., 13, 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]