

Summary
Single-cell RNA-sequencing (scRNA-seq) is becoming a ubiquitous method in profiling the cellular transcriptomes of both malignant and non-malignant cells from the human brain. Here, we present a protocol to isolate viable tumor cells from human ex vivo glioblastoma cultures for single-cell transcriptomic analysis. We describe steps including surgical tissue collection, sectioning, culturing, primary tumor cells inoculation, growth tracking, fluorescence-based cell sorting, and population-enriched scRNA-seq. This comprehensive methodology empowers in-depth understanding of brain tumor biology at the single-cell level.
For complete details on the use and execution of this protocol, please refer to Ravi et al.1
Subject areas: Cell Biology, Cell Isolation, Single Cell, Flow Cytometry/Mass Cytometry, Cancer, Sequencing, RNAseq, Microscopy, Model Organisms, Molecular Biology, Tissue Engineering
Graphical abstract
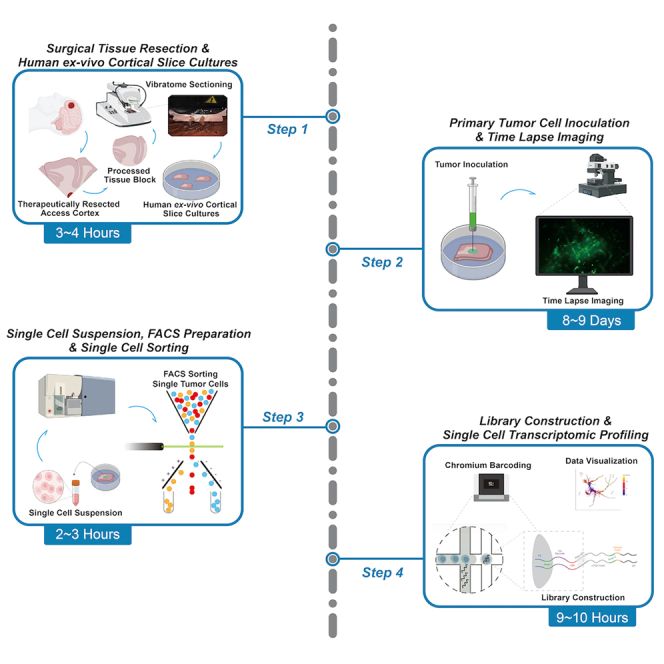
Highlights
-
•
Describes methods to re-purpose therapeutically discarded human cortical tissue
-
•
Protocol is adaptable to brain tissue from different ages of human or rodent subjects
-
•
Outlines steps for isolating single-tumor cells from human ex vivo cortical cultures
-
•
Details quality control measures and library preparation for Illumina sequencing
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Single-cell RNA-sequencing (scRNA-seq) is becoming a ubiquitous method in profiling the cellular transcriptomes of both malignant and non-malignant cells from the human brain. Here, we present a protocol to isolate viable tumor cells from human ex vivo glioblastoma cultures for single-cell transcriptomic analysis. We describe steps including surgical tissue collection, sectioning, culturing, GBM inoculation, growth tracking, fluorescence-based cell sorting, and population-enriched scRNA-seq. This comprehensive methodology empowers in-depth understanding of brain tumor biology at the single-cell level.
Before you begin
This protocol describes the specific steps required to carry out single-cell RNA sequencing from human organotypic cortical glioblastoma models. Transcriptomic profiling of all cell types on a single-cell level is a powerful tool to understand the intra-tumoral heterogeneity of these tumors. The readers can target any specific cell populations in the microenvironment by customizing FACS sorting markers.
Institutional permissions
The local ethics committee of the University of Freiburg approved data evaluation, imaging procedures and experimental design (protocol 1000020/09 and 472/15_160880). Human brain tissue specimens were obtained with informed consent under a Declaration of Helsinki, as requested by the local ethics committee (No. 187/04). All methods were carried out in accordance with the approved guidelines. Written informed consent was obtained from all patients. The studies were approved by an institutional review board.
We hereby remind the readers who will carry out the same experiment to follow the advice of their local ethics committee and get an approval from their institutional review board before you begin.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| rLV.EF1ZsGreen1-9 | Takara Bio | 0037VCT |
| Biological samples | ||
| Human neocortical access tissue/GBM tissue samples | Department of Neurosurgery, Medical Centre, University of Freiburg | N/A |
| Experimental models: Cell lines | ||
| BTSC_#233 | Uniklinik Freiburg | Primary Cell Line |
| Chemicals, peptides, and recombinant proteins | ||
| SPRIselect reagent | Beckman Coulter | B23318 |
| Bovine serum albumin (BSA) | Sigma-Aldrich | A3059 |
| 4′,6-Diamidino-2-phenylindole (DAPI) | Merck KGaA | 32670#5MG |
| Hibernate™ -A Medium | Gibco | 12087586 |
| N-methyl-D-Glucamin (NMDG) | Sigma-Aldrich | M2004 |
| Antibiotic-Antimycotic (100×) | Gibco | 15240062 |
| Neurobasal™ Medium | Gibco | 11570556 |
| L-Glutamine | Gibco | 25030149 |
| B-27 Supplement, serum-free (50×) | Gibco | 11530536 |
| MgSO4 | Sigma-Aldrich | M3409 |
| HEPES | Sigma-Aldrich | H0887 |
| D-Glucose | Sigma-Aldrich | RNBG7039 |
| Potassium chloride (KCl) | Merck KGaA | 1.04936.0500 |
| Potassium dihydrogen phosphate (KH2PO4) | Merck KGaA | 1.04873.0250 |
| Sodium chloride (NaCl) | VWR | 27810.295 |
| Disodium hydrogen phosphate dihydrate (Na2HPO4) | VWR | 28029.292 |
| 4% Paraformaldehyde solution | Thermo Fisher Scientific | J61899.AK |
| O.C.T. Tissuetek | Sakura | SA62550-01/-12 |
| Incidin™ Plus | Ecolab | 3011520 |
| Fetal bovine serum (FBS) | PAN Biotech | 1502 |
| Ethylenediaminetetraacetic acid (EDTA) | Carl Roth | 8043.1 |
| Critical commercial assays | ||
| KAPA SYBR FAST qPCR Master Mix | Roche | KK4600 |
| Chromium Next GEM Single Cell 3ʹ GEM, Library & Gel Bead Kit v3.1 | 10× Genomics | PN-1000121 |
| Chromium Next GEM Chip G Single Cell Kit | 10× Genomics | PN-1000120 |
| Single Index Kit T Set A | 10× Genomics | PN-1000213 |
| Fragment Analyzer 5200 HS NGS Kit | Agilent | DNF-474 |
| Qubit 1× dsDNA HS Kit | Thermo Fisher Scientific | Q33231 |
| NextSeq 500/550 High Output Kit v2.5 (150 cycles) | Illumina | 20024907 |
| NextSeq 500/550 High Output kit v2.5 (75 cycles) | Illumina | 20024906 |
| Neural Tissue Dissociation Kit (T) | Miltenyi Biotec | 130-093-231 |
| Software and algorithms | ||
| R/R Studio software | Posit | https://posit.co/downloads/ |
| Cell Ranger analysis pipeline | 10× Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/installation |
| Cranial Map Neuro Navigation Cart 2 | Stryker Corporation | N/A |
| Other | ||
| Leica VT1200 semi-automatic vibratome | Leica | 14912000001 |
| Millipore insert | Merck KGaA | PIHP03050 |
| 6-well plate | Greiner BIO-ONE | 657160 |
| Cryostar™ NX70 cryostat | Epredia | 15380755 |
| NIO laser imaging system | Invenio Imaging | 01-0224 |
| Hamilton micro syringe | Hamilton | 80330 |
| EVOS M7000 microscope | Thermo Fisher Scientific | AMF7000 |
| EVOS onstage incubator | Thermo Fisher Scientific | AMC1000 |
| C-Tube | Miltenyi Biotech | 130-093-237 |
| Chromium controller | 10× Genomics | N/A |
| Qubit™ 4 Fluorometer | Thermo Fisher Scientific | Q33238 |
| 5200 Fragment Analyzer | Agilent | M5310AA |
| BD FACSAria™ Fusion | BD Biosciences | N/A |
| BD FACSAria™ III | BD Biosciences | N/A |
| MoFlo Astrios EQ | Beckman Coulter | N/A |
Materials and equipment
Preparation Medium (PM) for Slicing – 160 mL
| Reagent | Final concentration | Amount |
|---|---|---|
| Hibernate Medium | N/A | 147.72 mL |
| D-Glucose (1 M) | 13 mM | 2.08 mL |
| NMDG (1 M) | 30 mM | 6 mL |
| L-Glutamine (200 mM) | 0.5 mM | 1 mL |
| Antibiotic-Antimycotic (100×) | 2% | 3.2 mL |
| Total | N/A | 160 mL |
CRITICAL: Following sterile filtration, it is recommended to divide the 160 mL preparation medium into 50 mL aliquots to prevent contamination. These aliquots should then be stored at a temperature of −20°C to maintain the integrity of the medium. It is crucial to avoid freeze-thaw cycles as they can compromise the quality of the medium. Moreover, it is essential to use the aliquoted medium within 20 days of preparation to ensure its effectiveness. Each sectioning procedure with a vibratome requires approximately 100 mL of the preparation medium. Therefore, it's important to plan the required amount of medium carefully to avoid any shortages during the experimental procedure.
Note: To prepare 1M N-Methly D-Glucamin take 1.95 g of the NMDG powder and mix it in 40 mL of Sterile H20. Please filter the solution before using.
Growth Medium (GM) for Slice Culture – 160 mL
| Reagent | Final concentration | Amount |
|---|---|---|
| Neurobasal Medium | N/A | 147.80 mL |
| D-Glucose (1 M) | 13 mM | 2.08 mL |
| Magnesium Sulfate (1 M) | 2 mM | 320 μL |
| L-Glutamine (200 mM) | 0.5 mM | 1 mL |
| HEPES (1 M) | 15 mM | 2.4 mL |
| B-27 Serum free (50×) | 2% | 3.2 mL |
| Antibiotic-Antimycotic (100×) | 2% | 3.2 mL |
| Total | N/A | 160 mL |
PBS (10×) – 1 L
| Reagent | Final concentration | Amount |
|---|---|---|
| Milli-Q Water | N/A | 1 L |
| KCl | 26.8 mM | 2.0 g |
| KH2PO4 | 14.7 mM | 2.0 g |
| NaCl | 1.396 M | 80.0 g |
| Na2HPO4 | 64.6 mM | 11.5 g |
| Total | N/A | 1 L |
Note: The PBS solution is prepared in the absence of Mg2+ or Ca2+ ions. A long-term storage at room temperature (25°C) is allowed.
FACS Buffer – 100 mL
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS (1×) | N/A | 97 mL |
| FBS (100%) | 2% | 2 mL |
| EDTA (0.5 M) | 5 mM | 1 mL |
| Total | N/A | 100 mL |
Note: We recommend to prepare FACS buffer shortly before use instead of keeping it in store for a long term. During the experiments, the FACS buffer should be kept on ice.
Step-by-step method details
Tissue collection, transport, sectioning, and preservation
Timing: 3–4 h
Here we describe best practices to collect, preserve and carry out the slicing of neocortical access tissue obtained from the resection of brain tumors and from epilepsy surgery. The neocortical access tissue used is not required for neuropathological diagnostics and would otherwise be discarded during the usual course of surgery. Depending on the point of entry and the extent of resection, the neurosurgeon removes a piece of neocortical access tissue, using the shortest non-eloquent trajectory as usual. The dissection is ideally performed with a scalpel and a sharp dissector, with only minimal cauterization and avoiding mechanical damage.1,2,3,4,5 The anatomical location of the resected tissue is recorded in the neuronavigation software (Cranial Map Neuro Navigation Cart 2; Stryker). In the operating theater, the resected tissue is immediately immersed in pre-cooled (0°C) and oxygenated preparation medium (95% O2, 5% CO2) and transported to the laboratory on ice as quickly as possible (<5 min).
-
1.Set up of the tissue processing platform, tissue sectioning, and tissue culture.
-
a.Keep the cortical tissue always immersed in preparation medium saturated with carbogen (95% O2, 5% CO2) at 0 ± 4°C before tissue processing.
-
b.Process the resected tissue.
-
i.Prepare a cooled metal platform (0° ± 4°C) that is generously flooded with preparation medium.
-
ii.Extract the resected tissue from the transport chamber and place it on the cooled platform (Figure 1B).
-
iii.Dissect away all visibly cauterized and damaged parts.
-
iv.Further dissect the processed tissue block into smaller blocks (size from 0.5–1 cm3) for sectioning (Figure 1B).Note: We recommend that each processed and dissected smaller block of tissue contains portions of the neocortex as well as underlying white matter.
-
i.
-
c.Section the prepared tissue blocks into 300 μm slices with a dimension of 6 mm by 6 mm using a tissue vibratome (Leica V1200 vibratome) at a speed of 0.16 mm/s, in preparation medium.Note: The temperature of the sectioning chamber is monitored and is maintained between 0–4°C with the help of crushed ice. During sectioning, the vibratome containing the preparation medium must be bubbled using carbogen.
-
d.Process the resected tissue.Note: The temperature of the sectioning chamber is monitored and is maintained between 0–4°C with the help of crushed ice. During sectioning, the vibratome containing the preparation medium must be bubbled using carbogen.
-
e.Transfer 300 μm tissue sections onto Millipore inserts in 6-well plates, with 1.2 mL of growth medium in each well.Note: The first two tissue sections from each block are generally discarded due to lack of consistency of section thickness (>300 μm).Note: Each block of tissue allows for the generation of 20–25 tissue sections for technical replicates, allowing for the usage of tissue sections across multiple experimental paradigms.Note: We recommend a maximum of 3 sections/well. Increase in the number of sections/well requires an increase in the frequency of medium exchange due to increased cytokine/chemokine release during culture. The medium can be collected for further experimental workplan with ELISA to detect metabolites from within the culture medium such as cytokines and neurotransmitters, as has been previously described by us.2CRITICAL: For proper pre-recovery of tissue sections before culture, it is essential to transfer them using a glass pasteur pipette into ice-cold incubation medium in 60 mm × 15 mm petri dishes that are saturated with carbogen. The sections should be allowed to incubate for 10–15 min before being placed onto the inserts for culture. This step is critical to ensure the viability of the tissue sections during the subsequent culture process.
-
f.Refresh the growth medium at periodic intervals to ensure maximal viability: The first medium refresh must be carried out 6 h post plating, followed by medium refreshes every 48 h, until the end of the culture period.Note: For infiltration within the cortical tissue sections is assessed using conventional histopathological staining methods (e.g., Hematoxylin and Eosin (H&E)) or alternatively using label-free simulated Raman histology (SRH).Note: For H&E staining and immunohistochemistry, tissue sections are embedded in O.C.T. (Tissue-TEK, SA62550-01/-02) and re-sectioned using a cryotome (NX70, ThermoFischer Scientific) for a final thickness of 10 μm.Note: For SRH imaging, no re-sectioning is required. An entire 300 μm tissue section is briefly (15 min) fixed in 4% paraformaldehyde (PFA) and subsequently washed in PBS. The section is then mounted on a specialized microscopy slide (NIO Slide, Invenio Imaging #PRT 01-0224). Imaging is performed using the NIO Laser Imaging System (Invenio Imaging) as described6,7 and tumor infiltration is assessed over the neocortical depth (Figure 1C).CRITICAL: Sterile processing guidelines must be upheld during the entire protocol to ensure tissue viability, avoiding any/all sources of contamination. To ensure proper tissue processing, it is necessary to clean the vibratome with 100% alcohol and wash all spatulas and scalpels with Incidin™. The vibratome can be placed outside the flowhood during this process. And lastly, to prevent any potential contamination, it is mandatory to wear both lab coats and face masks. Additionally, preparation and growth medium should be sterile filtered.
-
a.
Figure 1.

Neurosurgical cortical tissue sampling and pathological evaluation
(A) Illustration of tissue dissection for tissue culture experiments.
(B) The example of a neocortical access tissue sample from a neurosurgical operation (left) which is processed and cut into blocks (0.5–1 cm3, right), Scale bar: 1 cm.
(C) Stimulated Raman histology of a representative slice of non-infiltrated neocortical access tissue (top row) vs. a slice of infiltrated (arrow) access tissue (bottom row) from the same sample. Note, that slices from a visibly tumor infiltrated block would not be used for tissue culture experiments. WM: white matter, Boxes and Scale bar: 500 μm.
Primary Glioblastoma cell inoculation, tumor growth, and monitoring
This section describes the isolation of tumor cells from our human organotypic cortical glioblastoma (GBM) model for single-cell transcriptional profiling.
Note: It is necessary to ensure the viability of the cultured sections using electrophysiology or immunostaining protocols, during the culture period.2
-
2.Generate fluorescently labeled GBM cells.Note: Here we use patient derived GBM cells. Cells were transduced with lentiviral particles (rLV.EF1.ZsGreen1-9 (0037VCT), Takarabio; Clonetech). The concentration of viral particles required was calculated according to the manufacturer’s instructions (M.O.I. = 10, 109 TU/mL).
-
a.Seed 5 × 105 cells per well and incubated overnight in an incubator at 37°C and 5% CO2.
-
b.Prepare the transduction mix by adding the required volume of thawed viral particles and Hexadimethrine bromide (Polybrene, 800 μg/mL).
-
c.Refresh culture medium after 24 h.
-
d.Assess the quality of transduction after 48 h using microscopy.
-
a.
-
3.Inoculate 1 μL of GBM cells at the interface of gray and white matter using a 10 μL Hamilton syringe (80330; Hamilton Company, Bonaduz, Switzerland). Inoculate fluorescently tagged GBM cells into cultured sections.Note: We suggest to carry out this step 48 h post tissue plating to allow the tissue to recover from the trauma of sectioning.
-
a.Trypsinize GBM cells and dilute the suspension to ass concentration of 20,000 cells/μL.
-
b.Inoculate 1 μL of GBM cells at the interface of gray and white matter using a 10 μL Hamilton syringe (80330; Hamilton Company, Bonaduz, Switzerland).CRITICAL: Inoculation using the Hamilton micro syringe must be performed delicately to avoid damage to the tissue section.CRITICAL: To prevent cross contamination, the Hamilton micro syringe must be washed using methanol, ethanol, and then PBS ∼20 times, between inoculations.Note: To obtain reliable and informative results when injecting primary tumor cells into a tissue section of 6mm-by-6mm dimension, it is essential to optimize the injection volume. Based on our experience, we recommend injecting no more than 1 μL (20,000 cells/μL) of primary tumor cells to prevent compromising the viability of the cortical tissue sections. Injecting excessive cells can cause cell expansion and migration, leading to inaccuracies in the experimental results.
-
a.
-
4.
Rinse the sections with growth medium immediately after GBM inoculation.
Note: If any clog is noticed in the Hamilton syringe. It is advisable to flush the syringe with 100% ethanol.
-
5.
Monitor the tumor growth every 24 h, using a humidified and temperature-controlled incubating microscopic platform (EVOS M7000 with onstage incubator, Thermo Fisher Scientific, USA).
Note: Culture conditions are maintained as before GBM inoculations.
Figure 2.

Representative images of tumor inoculation
The inoculation of tumor cells is checked under EVOS M7000 microscope.
(A and B) (A) Failed Tumor Inoculation and (B) Successful Tumor Inoculation in human cortical slice cultures. Scale bar: 0.02 inch.
Figure 3.

Inoculated GBM growth pattern live monitored using EVOS microscope
(A–E) Representative live imaging of GBM inoculated cortical tissue at 0,1,3,5,7 days post inoculation (DPI). Scale bar: 0.02 inch.
(F) Immunostaining showing tumor cells interacting with the microenvironment: Tumor cells are tagged with ZsGreen, GFAP was used to mark Astrocytes, DAPI was used to mark nucleus. Scale bar: 50 μm.
Tissue preparation for FACS sorting of viable single cells
This section describes the methods including tissue dissociation, single-cell suspension and FACS sorting from cortical sections for single-cell sequencing.
Note: A minimum 3 of 300 μm thick tissue sections are required for obtaining sufficient viable cells to proceed with 10× Chromium Single Cell Library Preparation protocol.
-
6.Tissue dissociation (using Neural Tissue Dissociation Kit) and single-cell suspension.
-
a.Prepare the enzyme mix 1 (200 μL Enzyme T in 1750 μL Buffer X for every 500 mg of tissue).
-
b.Prepare the enzyme mix 2 (10 μL Enzyme A in 20 μL Buffer Y for every 500 mg of tissue).
-
c.Transfer tissue into C-tube, add enzyme mix 1 and incubate at 37°C for 5 min.
-
d.Dissociate the sample within the C-tube for 2 min at 37°C under slow and continuous rotation.
-
e.Add enzyme mix 2, incubate at 37°C for 5 min.
-
f.Repeat the dissociation step as described in step 6 d.
-
g.Centrifuge sample for 1 min at 350 g, 4°C.
-
h.Remove supernatant and resuspend pellet in 9 mL growth medium.
-
i.Prepare 50 mL falcon tube with a 100 μm strainer at the opening. Apply 1 mL growth medium to moisten the strainer.
-
j.Filter the resuspended pellet solution from step 6 h bit by bit through the strainer tube setup from step 6 i.
-
k.Collect and transfer the sample into a 15 mL falcon tube.
-
a.
-
7.FACS preparation and DAPI staining.
-
a.Centrifuge the suspension in a 15 mL falcon tube at 300 g and 4°C for 10 min.
-
b.Aspirate the supernatant leaving approximately 100 μL in the falcon and resuspend the pellet gently.
-
c.Add the resuspended sample to a FACS tube filled with FACS buffer, and centrifuge at 300g and 4°C for 5 min.
-
d.Repeat steps 7 b and 7 c to wash the samples, aspirate the supernatant and keep approximately 100 μL. Simultaneously, prepare FACS incubation buffer with DAPI (DFIB) by diluting DAPI into FACS buffer for a final concentration of 1 μg/mL.
-
e.Add 450 μL DFIB mix from step 7 d into the washed sample and resuspend gently. This step needs to be carried out on ice, avoiding direct light.
-
f.Collect 70,000 cells that are DAPI- and ZsGreen+ by collecting two sets of 35,000 cells each into pre-coated Eppendorf tubes.
-
a.
Note: To ensure optimal collection efficiency and cell viability, pre-coat the Eppendorf tubes with 4% BSA overnight at 4°C, and fill each tube with 100 μL of FACS collection buffer before use.
-
8.Single-cell barcoding preparation.
-
a.Measure and confirm one set of collected samples with a 1000 μL pipette, and further filter with 70 μm FlowMi Cell Strainers.
-
b.Centrifuge the filtered sample at 300 g and 4°C for 10 min. Aspirate the supernatant, leaving a final volume of 45 μL.CRITICAL: The viability of the sorted cells is determined by microscopic observation. The second collection tube of 35,000 cells per condition is used to check the viability of cells. Resuspend the pellet and use 10 μL of collected cell suspension for a 1:10 dilution of in total 100 μL solution. Mix 10 μL of diluted GBM cells with 10 μL 0.4% Trypan Blue. After a 5 min incubation in room temperature, observe under the microscope count live/dead cells and calculate the viability.
-
c.Resuspend the pellet from step 8 b in the 45 μL supernatant. Use 43.2 μL to proceed 10× Genomics Single Cell Library Preparation protocol (CG000204 Rev D) and Illumina Sequencing protocol (# 15048776 v09).
-
a.
Single-cell library construction and sequencing
In this section, we briefly describe the single-cell library preparation and sequencing procedure. Detailed step-by-step protocol could be found from 10× Genomics (CG000204 Rev D) and Illumina Inc. (# 15048776 v09).
-
9.GEM generation and barcoding.
-
a.Prepare Master Mix on ice.
-
b.Assemble Chromium Next GEM Chip G.
-
c.Load Chromium Next GEM Chip G row labeled 1 with 70 μL Master Mix + cell suspension.
-
d.Load Chromium Next GEM Chip G row labeled 2 with 50 μL Gel Beads.
-
e.Load Chromium Next GEM Chip G row labeled 3 with 45 μL Partitioning Oil.
-
f.Run the Chromium Controller (about 18 min).
-
g.Immediately transfer GEMs and proceed GEM-RT Incubation.Store at 4°C for up to 72 h or −20°C for up to 1 week, or proceed to the next step.
-
h.Add 125 μL Recovery Agent without pipetting mix or vortexing.
-
i.Carefully discard Recovery Agent and Partitioning Oil from the bottom of the tubes.
-
a.
-
10.Post GEM-RT Clean-up and cDNA Amplification.
-
a.Prepare Dynabeads Cleanup Mix and proceed to cleanup.
-
b.Wash samples 2 times in 80% ethanol using a magnetic separator.
-
c.Prepare Elution Solution I based on the protocol.
-
d.Immediately add Elution Solution I, incubate and transfer supernatant to new strip tubes using a magnetic separator.
-
e.Prepare cDNA Amplification Mix, mix with samples.
-
f.Incubate in a thermal cycler following 10× Genomics protocol for cDNA Amplification.Store at 4°C for up to 72 h or −20°C for up to 1 week, or proceed to the next step.
-
g.Add SPRIselect and incubate for a cDNA cleanup.
-
h.Remove supernatant after magnetic separation and wash 2 times with 80% ethanol.
-
i.Resuspend beads in Buffer EB, incubate and magnetic separate again.
-
j.Collect and transfer 40 μL samples to a new tube strip.Store at 4°C for up to 72 h or −20°C for up to 4 weeks, or proceed to the next step.
-
k.Run 1 μL of sample (Dilution Factor 1:10) on Fragment Analyzer with High Sensitivity NGS (1–6000 bp) kits for cDNA quality control.
-
a.
-
11.Gene expression library construction.
-
a.Prepare a thermal cycler and Fragmentation Mix.
-
b.Transfer 10 μL cDNA samples and proceed the following part of protocol.
-
c.Add Buffer EB and Fragmentation Mix to sample strip, pipette mix.
-
d.Transfer samples into the pre-cooled thermal cycler and proceed incubation.
-
e.Apply SPRIselect (0.6×), incubate, magnetic separate and collect supernatant.
-
f.Apply SPRIselect (0.8×), incubate, magnetic separate and collect beads.
-
g.Wash 2 times with 80% ethanol, resuspend in Buffer EB and collect 50 μL supernatant using a magnetic separator.
-
h.Prepare Adaptor Ligation Mix and well mix with samples.
-
i.Set up a thermal cycler and proceed incubation following 10× Genomics protocol.
-
j.Apply SPRIselect (0.8×), incubate, magnetic separate and collect beads.
-
k.Wash 2 times with 80% ethanol, resuspend in Buffer EB and collect 30 μL supernatant using a magnetic separator.
-
l.Prepare Sample Index PCR Mix and add the mix to sample tubes.
-
m.Add individual sample index (i7 Multiplex Index, Plate SI-GA) to tubes and pipette mix.
-
n.Incubate in a thermal cycler with proper cycle numbers based on cDNA quality control results from step 10 k.Store at 4°C for up to 72 h, or proceed to the next step.
-
o.Apply SPRIselect (0.6×), incubate, magnetic separate and collect supernatant.
-
p.Apply SPRIselect (0.8×), incubate, magnetic separate and collect beads.
-
q.Wash 2 times with 80% ethanol, resuspend in Buffer EB and collect 35 μL supernatant using a magnetic separator.Store at 4°C for up to 72 h or −20°C for long-term storage.
-
r.Run 1 μL of sample (Dilution Factor 1:10) on Fragment Analyzer with High Sensitivity NGS (1–6000 bp) kits for final library quality control.
-
a.
-
12.Library normalization, denaturation, dilution, and sequencing.
-
a.Normalize each single library to a “starting concentration” and combine single libraries to a library mixture.
-
b.Denature combined library with a corresponding amount (see the table from Illumina protocol) of 0.2 N NaOH and 200 mM Tris-HCl pH 7.
-
c.Dilute the denatured library to the final concentration with HT1.CRITICAL: The entire library construction is recommended to proceed on ice as possible to minimize the degradation of samples.Note: A Phix control for the final sequencing library could be planned and included if needed.
-
d.Load the samples in cartridges and run Illumina NextSeq 550 with the following cycle numbers (Read 1 – i7 – i5 – Read 2: 28–8 – 0–56 cycles).
-
e.Following sequencing, the Cell Ranger analysis pipeline (Version 7.1.0, Manufacturer) will align reads and count barcodes and UMIs to map transcripts in singe cells.
-
f.The primary output of Cell Ranger is a count matrix, consisting of columns of every cell barcode and rows for all measured features, such as genes for transcriptome analysis.
-
g.The final files were exported to R for further analysis and visualization using standard pipelines.
-
a.
Figure 4.

Representative library quality control results using Fragment Analyzer
(A–C) Representative cDNA library (A), final library (B) quality control performed using Fragment Analyzer. As well as low quality cDNA library (C) quality control result using Fragment Analyzer.
Expected outcomes
This protocol will provide a comprehensive single-cell RNA sequencing data set from the isolated glioblastoma cells. Given the expected yield of approximately 35,000 to 70,000 single cells from three 300 micron tissue sections, we anticipate generating a robust and detailed map of the transcriptomic landscape of these cells.
In addition, the negative flow-through collection should yield a rich diversity of non-glioblastoma cell types including neurons, oligodendrocytes, oligodendrocyte progenitor cells, astrocytes, microglia, etc., This will enable the study of the brain’s microenvironment and the interactions between glioblastoma cells and other cell types, offering new insights into disease pathogenesis and possible therapeutic targets.
When this protocol is adapted for the enrichment of specific cell types, it should provide reliable and reproducible outcomes regarding the selected target cell population. The resultant RNA sequencing data will enhance our understanding of the individual cell types and their unique roles within the human ex-vivo cortical cultures. This will empower researchers to investigate the roles and interactions of specific cell types in more detail, and potentially unveil previously unrecognized cell states or functions. Details of scRNA-seq analysis can be found in our recent publication Ravi, et al., 2022 Cancer Cell.1
Limitations
Single-cell sequencing has emerged as a powerful tool to understand the complexity and heterogeneity of biological systems. However, several challenges and limitations are associated with this approach, particularly when studying human ex-vivo cortical cultures. Obtaining viable single cells from such cultures can be challenging due to factors such as tissue quality, time constraints, and technical variability. Moreover, single-cell sequencing protocols may result in the loss of certain cell types or a decrease in the number of cells due to several filtering steps during the procedure. The complexity and heterogeneity of human ex-vivo cortical cultures pose additional challenges, including the identification and isolation of specific cell populations for single-cell sequencing. Furthermore, the variability in the culture conditions, the genetic background of the donors, can affect the accuracy and reproducibility of the single-cell sequencing data.
Therefore, careful considerations and quality controls are required when designing single-cell sequencing experiments to obtain meaningful and reproducible results from human ex-vivo cortical cultures.
Troubleshooting
Problem 1
Unequal slice thickness during sectioning or blocks detaching from vibratome plate (Related to step 1 b).
Potential solution
-
•
Make sure tissue blocks are correctly glued onto the preparation table.
-
•
Remove blood clots from tissue blocks before gluing them onto the vibratome plate.
-
•
Remove the arachnoidal membrane and blood vessels from the pial surface of the block.
-
•
Rather detach and re-mount a block than try to section a sub optimally attached tissue block.
-
•
Consider changing cutting direction from perpendicular to orthogonal to the pial surface.
Problem 2
Reduced viability of ex-vivo human cortical cultures (Related to step 1 e).
Potential solution
-
•
Ensuring proper hydration of human ex-vivo cortical sections is a critical consideration for successful culture. To this end, it is recommended to add 30 μL of growth medium on top of the sections during medium changes to prevent tissue desiccation.
-
•
We recommend checking for bacterial or fungal contamination in the incubator. Use sterile solutions and equipment, work in a laminar flow hood, and perform rigorous cleaning and disinfection of the work area and equipment.
-
•
Make sure that the sections are not thicker than 300 μm as it will significantly affect the viability of the cultures.
Problem 3
Drifting of tissue samples during live cell imaging (Related to step 5).
Potential solution
-
•
Optimizing live tissue imaging of tissue sections in 3D culture experiments requires careful attention to the liquid environment. Specifically, we recommend adding no more than 30 μL of medium on top of the section and limiting the medium below the insert to 1.2 mL. Excess liquid can cause section drift, compromising image registration with the previous time point and undermining the quality of live tissue imaging.
Problem 4
Low output of ZsGreen tagged GBM cells from FACS Sorting (Related to step 7 f).
Potential solution
-
•
Inappropriate working temperature, and extended working times are significant contributing factors to low single-cell viability. Low single-cell viability, in turn, leads to a low output of ZsGreen tagged cells. To address this issue, it is recommended that users reduce the number of samples used per run. The optimal number of samples for each run is four. By reducing the number of samples, users can maintain the appropriate working temperature, and shorten the working time, which will result in higher single-cell viability.
-
•
Sections with low tumor growth as determined by live cell imaging should be excluded from tissue sampling. This ensures that the resulting cell suspension will consist of viable and healthy cells, leading to a higher likelihood of successful downstream applications. Additionally, it is crucial to perform regular checks on the Hamilton syringe used for injection to ensure proper functionality. This quality control step is essential for obtaining reliable and reproducible results and will improve the overall success rate of the experiment.
Problem 5
Less viable tumor cells in the single-cell suspension (Related to step 7 b).
Potential solution
-
•
Single-cell suspension has a time dependence for storage. Thus, the downstream processing must be done as soon as possible. In our experimental workflow, we never kept the suspension for more than two weeks at −80°C.
Problem 6
Low yield of genes or transcripts following sequencing (Related to step 12).
Potential solution
-
•
Poor cell quality and mRNA degradation are the most common causes of low transcript recovery in scRNA-seq experiments. To minimize these issues, it is essential to use reagents and consumables that are free of nucleases. In addition, to maintain cell viability, it is crucial to process samples quickly and keep the cells on ice throughout the protocol, minimizing exposure to ambient temperatures.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Vidhya Madapusi Ravi (vidhya.ravi@uniklinik-freiburg.de).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate either specific, unique data sets or code. For detailed information about data sets and codes generated using the protocol described in this study, please refer to Ravi, et al. Cancer Cell 20221 or contact the authors.
Acknowledgments
We express our gratitude to the patients who generously participated in this study, as well as Jonathan Göldner for all the cell culture work and assistance of bio-banking. This work was supported in part by the Freiburg Institute for Advanced Studies (FRIAS) for V.M.R. and Else Kroner-Fresenius Foundation for D.H.H. as well as by MEPHISTO project BMBF (German Ministry of Education and Research, project number 031L0260B, D.H.H. and D.D.).
We thank 10X Genomics and Dietmar Pfeifer for helpful advice. We thank Biorender.com.
Author contributions
V.M.R. designed and optimized the protocol. J.Z. and K.J. carried out slice culture experiments. A.V., J.S., and N.N. carried out Raman experiments. J.B., O.S., N.N., M.P., S.B., C.F., J.S., and D.H.H. formed surgical and neuropathological teams. V.M.R., K.J., and J.Z. wrote the manuscript. V.M.R., K.J., D.H.H, O.S., and J.B. reviewed the manuscript.
Declaration of interests
The authors declare no competing interests.
References
- 1.Ravi V.M., Will P., Kueckelhaus J., Sun N., Joseph K., Salié H., Vollmer L., Kuliesiute U., von Ehr J., Benotmane J.K., et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell. 2022;40:639–655.e13. doi: 10.1016/j.ccell.2022.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Ravi V.M., Joseph K., Wurm J., Behringer S., Garrelfs N., d'Errico P., Naseri Y., Franco P., Meyer-Luehmann M., Sankowski R., et al. Human organotypic brain slice culture: a novel framework for environmental research in neuro-oncology. Life Sci. Alliance. 2019;2:e201900305. doi: 10.26508/lsa.201900305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maier J.P., Ravi V.M., Kueckelhaus J., Behringer S.P., Garrelfs N., Will P., Sun N., von Ehr J., Goeldner J.M., Pfeifer D., et al. Inhibition of metabotropic glutamate receptor III facilitates sensitization to alkylating chemotherapeutics in glioblastoma. Cell Death Dis. 2021;12:723. doi: 10.1038/s41419-021-03937-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ravi V.M., Neidert N., Will P., Joseph K., Maier J.P., Kückelhaus J., Vollmer L., Goeldner J.M., Behringer S.P., Scherer F., et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat. Commun. 2022;13:925. doi: 10.1038/s41467-022-28523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buehler M., Yi X., Ge W., Blattmann P., Rushing E., Reifenberger G., Felsberg J., Yeh C., Corn J.E., Regli L., et al. Quantitative proteomic landscapes of primary and recurrent glioblastoma reveal a protumorigeneic role for FBXO2-dependent glioma-microenvironment interactions. Neuro Oncol. 2023;25:290–302. doi: 10.1093/neuonc/noac169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neidert N., Straehle J., Erny D., Sacalean V., El Rahal A., Steybe D., Schmelzeisen R., Vlachos A., Reinacher P.C., Coenen V.A., et al. Stimulated Raman histology in the neurosurgical workflow of a major European neurosurgical center - part A. Neurosurg. Rev. 2022;45:1731–1739. doi: 10.1007/s10143-021-01712-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Straehle J., Erny D., Neidert N., Heiland D.H., El Rahal A., Sacalean V., Steybe D., Schmelzeisen R., Vlachos A., Mizaikoff B., et al. Neuropathological interpretation of stimulated Raman histology images of brain and spine tumors: part B. Neurosurg. Rev. 2022;45:1721–1729. doi: 10.1007/s10143-021-01711-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate either specific, unique data sets or code. For detailed information about data sets and codes generated using the protocol described in this study, please refer to Ravi, et al. Cancer Cell 20221 or contact the authors.