

Summary
The heme-regulated kinase HRI is activated under heme/iron deficient conditions; however, the underlying molecular mechanism is incompletely understood. Here, we show that iron deficiency-induced HRI activation requires the mitochondrial protein DELE1. Notably, mitochondrial import of DELE1 and its subsequent protein stability are regulated by iron availability. Under steady state conditions, DELE1 is degraded by the mitochondrial matrix-resident protease LONP1 soon after mitochondrial import. Upon iron chelation, DELE1 import is arrested, thereby stabilizing DELE1 on the mitochondrial surface to activate the HRI-mediated integrated stress response (ISR). Ablation of this DELE1-HRI-ISR pathway in an erythroid cell model enhances cell death under iron-limited conditions, suggesting a cell protective role for this pathway in iron-demanding cell lineages. Our findings highlight mitochondrial import regulation of DELE1 as the core component of a previously unrecognized mitochondrial iron responsive pathway that elicits stress signaling following perturbation of iron homeostasis.
Keywords: HRI, DELE1, LONP1, integrated stress response, iron, mitochondria, mitochondrial import, mitochondrial proteostasis, erythroid cells
eTOC blurb
Sekine and Houston et al. identified DELE1 as an iron-responsive molecule at mitochondria that has cell protective roles via HRI-ISR pathway activation in iron-deficient conditions. The mitochondrial import and subsequent degradation of DELE1 were regulated by intracellular iron availability, demonstrating the general importance of import coupled-stress sensing mechanisms at mitochondria.
Graphical Abstract
Introduction
The cytosolic kinase HRI is highly expressed in erythroid cells and governs the stress response called integrated stress response (ISR) during terminal erythropoiesis1–3. Under stress conditions, HRI phosphorylates the α subunit of the translation initiation factor eIF2 (eIF2α), thereby attenuating global mRNA translation, while activating the transcription factor ATF4, a master regulator of the transcriptional response for the ISR. Early studies using rabbit reticulocyte lysates revealed that the addition of hemin (heme) to the lysates reverses HRI-mediated translational inhibition 4–7. Subsequent in vitro studies using purified HRI demonstrated that the direct binding of heme on HRI modulated the conformation of HRI thereby suppressing its kinase activity 8–11. These findings led to a current model of HRI regulation in which HRI is activated through dissociation of the inhibitory heme from HRI when intracellular levels of heme decline. HRI knockout (KO) mice do not exhibit significant erythroid abnormalities under standard dietary condition 12. However, when challenged with an iron deficient diet, these mice develop a characteristic anemia with globin aggregation in red blood cells 12. One potential explanation for these iron-dependent in vivo effects is that iron deficiency concomitantly decreases heme levels, and this fall in heme is the mechanism of HRI activation. However, such conjectures have remained unproven.
It has been shown that various types of mitochondrial stress, such as membrane potential loss, inhibition of OXPHOS, and perturbation of mitochondrial proteostasis, activate the ISR 13–15. Recent cell genetic screens have demonstrated that HRI is required for the mitochondrial stress-triggered ISR activation, and identified the mitochondria resident proteins DELE1 and OMA1 as upstream regulators of HRI 16,17. Upon treatment with various mitochondrial stressors the mitochondrial protease OMA1 cleaves DELE1, and the cleaved form of DELE1 is released into the cytosol where it interacts with HRI, activating the ISR. Recent studies have indicated the importance of this pathway in in vivo models of mitochondrial cardiomyopathy 18–21. However, the functional link between this newly identified mitochondria-dependent pathway and the classic heme-dependent HRI regulation model remains unclear.
Accumulating evidence indicates that mitochondrial import of certain proteins is precisely regulated in response to mitochondrial stress. Importantly, this regulation is often used to signal mitochondrial stress to other subcellular compartments such as the cytosol and the nucleus, which constitutes the “mitochondrial import-coupled stress response”. A well characterized example of this is the Parkinson’s Disease (PD)-linked mitochondrial kinase PINK1, whose import arrest and stabilization on the outer mitochondrial membrane (OMM) upon mitochondrial depolarization triggers Parkin-mediated mitophagy 22–24. Furthermore, it has been shown in C. elegans that suppression of the mitochondrial import of the transcription factor ATFS-1 by proteotoxic stress leads to the translocation of ATFS-1 to the nucleus, which is central for the transcriptional response known as the mitochondrial unfolded protein response (mtUPR) 25.
Here, we show another example of the mitochondrial import-coupled stress response. We found that DELE1 is an iron responsive protein whose mitochondrial import and subsequent protein stability are regulated by intracellular iron availability. The full-length form of DELE1, stabilized by iron deficiency, interacted with and activated HRI on the mitochondrial surface, suggesting that mitochondria are utilized as a site for triggering the ISR when iron homeostasis is perturbed. Our findings point to a hitherto unappreciated mitochondrial-based iron-monitoring system and also underscore the importance of mitochondrial protein import regulation as a dynamic, stress-sensing mechanism.
Results
DELE1 is a short-lived protein that is degraded by LONP1 after mitochondrial import
Previous studies have suggested that the protein stability of DELE1 is post-translationally regulated in cultured cells 26,27. To monitor endogenous DELE1 protein stability, we performed cycloheximide (CHX)-chase experiments using HEK293T cells expressing an endogenously HA-tagged DELE1, which overcame the difficulty of detecting endogenous DELE1 using the currently available antibodies 16,17. Consistent with the recent study 27, DELE1 underwent rapid degradation within minutes of CHX treatment (Figure 1A). In contrast, expression levels of other mitochondrial proteins such as TOM40, TIM50 and TIM23 did not change during this chase period (Figure 1A). This suggests that DELE1 is actively degraded under basal conditions. The proteasome inhibitor MG132 and the autophagy inhibitor Bafilomycin A1 did not enhance DELE1 expression (Figure 1B), while these inhibitors accumulated their known substrates (the cleaved form of PINK1 for the proteasome and p62 for autophagy) 22,28, indicating that DELE1 is likely degraded independent of the proteasome and macroautophagy. We thus speculated that mitochondrial proteases may be involved in its basal degradation and conducted a small scale screen using siRNAs for reported mitochondrial proteases (Figures 1C and 1D) 29. This targeted screen found that knockdown of LONP1, the matrix-localized ATP-dependent serine protease 30, had the most pronounced effects on DELE1 (Figure 1D). LONP1 siRNAs increased DELE1 at steady state, and almost completely prevented DELE1 degradation during the CHX-chase (Figure 1E). We also observed partial restoration of DELE1 expression in cells with knockdown of AFG3L2 and SPG7, that comprise the matrix-facing m-AAA protease (Figure 1D) 29. In contrast, none of the siRNAs for the inner mitochondrial membrane (IMM) or intermembrane space (IMS) proteases affected DELE1 stability (Figure 1C). These observations suggest that DELE1 is degraded in the mitochondrial matrix predominantly in a LONP1-dependent manner.
Figure 1. DELE1 is a short-lived protein that is degraded by LONP1 after mitochondrial import.
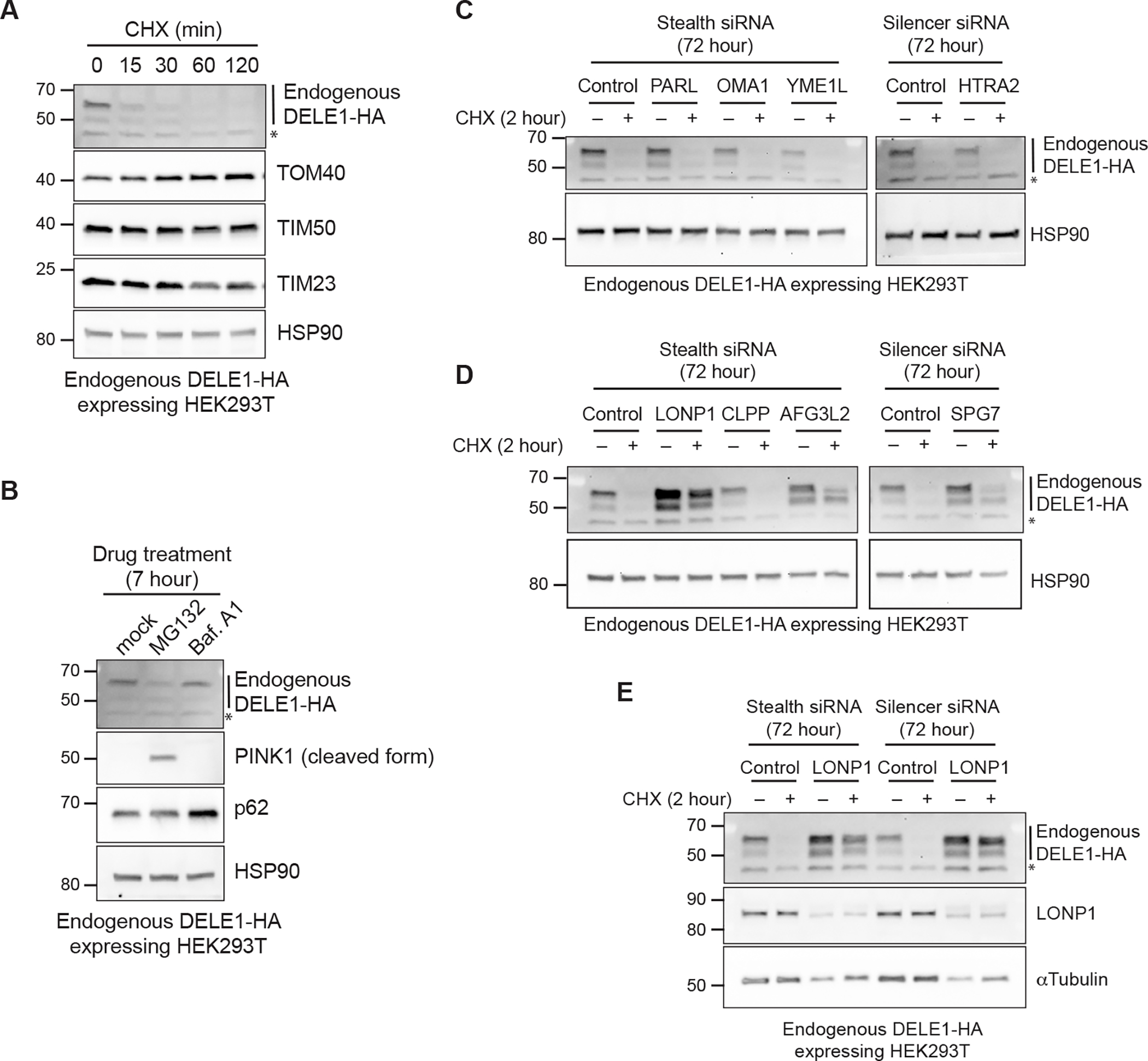
(A) Endogenous DELE1-HA expressing HEK293T cells were treated with cycloheximide (CHX) for the indicated time periods. Cell lysates were analyzed by immunoblotting (IB) with the indicated antibodies. HSP90 is shown as a loading control.
(B) IB for lysates of endogenous DELE1-HA HEK293T cells treated with MG132 or Bafilomycin A1 (Baf. A1) for 7 hours.
(C-E) IB for lysates of endogenous DELE1-HA HEK293T cells transfected with the indicated siRNAs for 72 hours and treated with CHX for the last 2 hours before harvest. *; non-specific bands.
Iron deficiency stabilizes DELE1
LONP1 is known to function as a quality control protease that degrades misfolded and oxidatively damaged proteins in the mitochondrial matrix 30. It has also been shown that LONP1 is actively involved in various mitochondrial processes through the regulated proteolysis of specific substrates. Considering that DELE1 is an activator of the heme-responsive kinase HRI, we tested whether heme and iron-related stimuli affected LONP1-mediated degradation of DELE1. We found that two iron chelators, deferoxamine (DFO) and deferiprone (DFP) increased DELE1 protein levels (Figure 2A). These compounds have been shown to decrease both cytosolic and mitochondrial iron levels 31,32, and we confirmed their effects by monitoring the increase in the cytosolic iron-responsive protein IRP2 33 (Figure 2A). In contrast, neither heme (hemin) nor a heme biosynthesis inhibitor (succinyl acetone, SA) affected DELE1 expression, while the heme-responsive proteins ALAS1 and HMOX1 were increased by SA and hemin, respectively (Figure 2A). Whereas the mitochondrial uncoupler CCCP induced the cleavage of DELE1 as previously reported 16,17, DFO and DFP treatment appeared to promote stabilization of DELE1, including its full-length form (Figure 2A). Levels of a tetracycline (tet)-inducible DELE1-HA in HeLa cells was also enhanced in a DFO dose-dependent manner, similar to the response of IRP2 (Figure 2B). CHX-chase experiments revealed that pre-treatment with DFO or DFP significantly stabilized the full-length form of endogenous DELE1 following a CHX-chase (Figure 2C). This was also observed for exogenously expressed DELE1-HA in DELE1 KO HEK293T cells (Figures 2D and S1A). In contrast, pre-treatment with CCCP stabilized DELE1, but as the cleaved form (Figure 2C). It is noteworthy that for unknown reasons, the iron deficiency-induced accumulation of endogenously HA-tagged DELE1 was more prominently observed when cells were seeded at a lower cell density (Figure S1B). Nevertheless, CHX-chase experiments clearly demonstrate that under iron-deficient conditions, DELE1 is stabilized at any cell density (Figure S1B). Collectively, this implies that full-length DELE1 stability is regulated by iron availability.
Figure 2. Iron deficiency stabilizes DELE1.
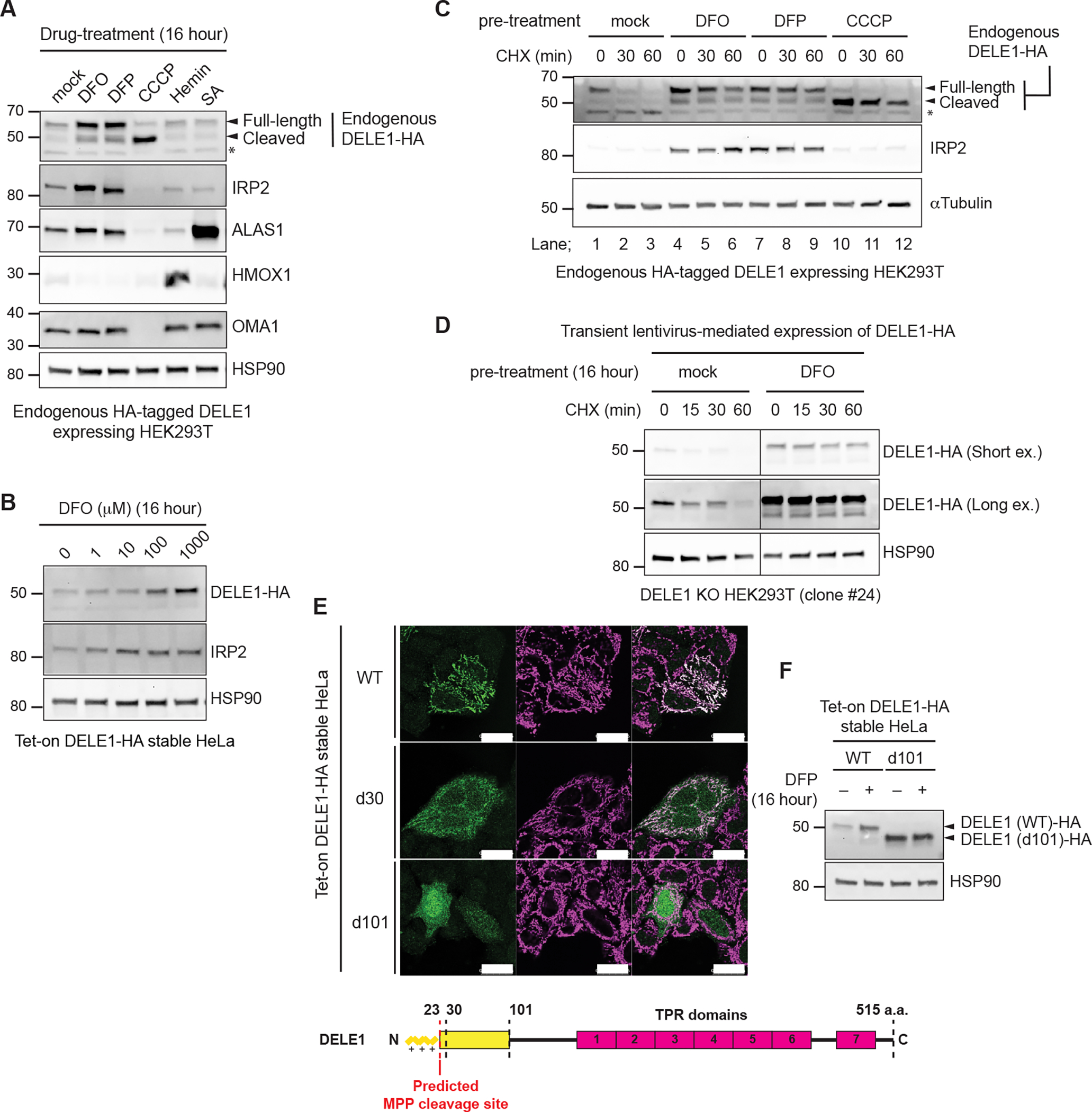
(A) Endogenous DELE1-HA HEK293T cells were treated with Deferoxamine (DFO), Deferiprone (DFP), CCCP, Hemin or succinyl acetone (SA) for 16 hours. The lysates were analyzed by immunoblotting (IB) with the indicated antibodies.
(B) IB for lysates of tet-on DELE1-HA stable HeLa cells treated with the indicated concentration of DFO for 16 hours. Doxycycline was added 8 hours prior to DFO treatment to induce the expression of DELE1-HA.
(C) IB for lysates of endogenous DELE1-HA HEK293T cells pre-treated with either DFO, DFP for 16 hours, or CCCP for 1.5 hours, and subsequently subjected to the CHX chase at the indicated time periods.
(D) HEK293T DELE1 knockout (KO) cells (clone #24) were transiently infected with lentivirus expressing DELE1-HA. After 24 hours, cells were treated with DFO for 16 hours and were subsequently subjected to the CHX chase at the indicated time periods. Cell lysates were analyzed by IB.
(E) Immunocytochemistry (ICC) for Tet-on DELE1 (wild-type, WT)-HA, DELE1 (delta 30, d30)-HA, DELE1 (delta 101, d101)-HA stable HeLa cells. Scale bars; 25 μm. A schematic representation of the domain structure of DELE1 is shown at the bottom.
(F) IB for lysates of tet-on DELE1 WT-HA or DELE1 d101-HA stable HeLa cells were treated with DFP for 16 hours. Doxycycline was added 8 hours prior to DFO treatment to induce the expression of DELE1 WT-HA or DELE1 d101-HA.
See also Figure S1.
DELE1 has a predicted mitochondrial targeting sequence (MTS) at its N-terminus and the MTS cleavage site is predicted to be located after amino acid 23 by the web tool MitoFates (Figure 2E) 34, which has been experimentally verified 27. We confirmed that tet-induced DELE1 wild-type (WT) is localized to mitochondria (Figure 2E). Consistent with previous reports 16,17, a DELE1 mutant that lacked the predicted MTS (DELE1 d30) partially prevented mitochondrial localization of DELE1, but deletion of the N-terminal 101 amino acids of DELE1 (DELE1 d101) almost completely blocked mitochondrial localization (Figure 2E). These results suggest that mitochondrial localization of DELE1 is regulated by a longer N-terminal region encompassing the predicted MTS. Unlike DELE1 WT, the DELE1 d101 mutant lost the ability to accumulate in response to iron chelation (Figure 2F), suggesting that DELE1 responds to iron deficiency at mitochondria.
DELE1 is stabilized on mitochondria by an iron deficiency-dependent mitochondrial import arrest
Treatment of iron chelators significantly decreased mitochondrial ISC-containing proteins including SDHB and LIAS (Figure S2A), suggesting that iron deprivation affected the mitochondrial ISC biosynthesis pathway. It has been reported that misfolded SDHB due to the failure of ISC insertion undergoes degradation by LONP1 35. Indeed, LONP1 knockdown restored the expression of SDHB as well as that of LIAS in iron-chelated cells (Figure S2A), suggesting that LONP1 mediates the degradation of these ISC proteins. Thus, it appears that LONP1 is still active for degrading misfolded ISC proteins under iron deficient conditions, although DELE1 appears to escape from LONP1-mediated degradation.
CCCP treatment induces cleavage of DELE1 by OMA1 and that cleaved DELE1 is released into the cytosol 16,17. We confirmed this observation in our tet-on DELE1-HA HeLa cells (Figure 3A). In contrast, we found that DELE1 was detected predominantly on mitochondria after treatment with DFO (Figure 3A), indicating that DELE1 does not substantially translocate to the cytosol during iron deficiency. This is consistent with the observation that unlike CCCP, iron chelators do not induce cleavage of DELE1 to the same extent as CCCP but rather stabilizes the full-length form of DELE1 (Figures 2A–2C). Cell fractionation analysis of HEK293T cells expressing endogenously HA-tagged DELE1 also revealed that in iron-deficient conditions, the stabilized, full-length DELE1 was retained in the mitochondrial fraction (Figure S2B). Moreover, under iron-deficient conditions, when cells were subsequently exposed to CCCP, the full-length form of DELE1 was able to be converted into the OMA1-cleaved form, indicating that at least during iron deficiency the OMA1 cleavage site (around reside 142 of DELE1) 17 is located in the IMS (Figure S2C). To further determine the precise localization of DELE1 within mitochondria before and after iron chelation, we performed a protease protection assays after a CHX chase by using crude mitochondria isolated from endogenously HA-tagged DELE1 expressing HEK293T cells (Figure 3B). When de novo protein synthesis was blocked with CHX, the balance of DELE1 shifted in favor of its MPP-cleaved form and it was largely protected from proteinase K in isotonic (SEM) buffer, as expected. In contrast, under iron deficiency, a substantial amount of the stabilized DELE1 showed a higher sensitivity to proteinase K in isotonic buffer (Figure 3B), suggesting that the C-terminal region of these DELE1 is exposed outside of the OMM after iron deficiency. We thus hypothesized that the mitochondrial import of DELE1 may be arrested in response to iron deficiency. To monitor the entrance of the N-terminus of DELE1 into the matrix, the TEV sequence (that is recognized and cleaved by TEV protease) was inserted after the predicted MTS of DELE1 [DELE1(TEV30)] (Figure 3C). We expressed DELE1(TEV30) with or without a matrix-targeted TEV protease (Su9-TEV protease) 36 in DELE1 KO HEK293T cells, and subsequently treated the cells with DFO. In the absence of Su9-TEV protease, the N-terminal MTS of DELE1 should be cleaved by the endogenous MTS-cleaving peptidase (MPP) in the matrix 27. In the presence of Su9-TEV protease, the N-terminal 36 amino acids of DELE1 should be cleaved by Su9-TEV protease (Figure 3C). Indeed, DELE1(TEV30) appeared as a doublet under steady state conditions when resolved on an SDS-PAGE 7.5% acrylamide gel; the upper band was the full-length form and the lower band was either the MPP- or TEV-cleaved form (Figures 3D and S2D). Following iron chelation with DFO, cleavage of DELE(TEV30)-HA by matrix MPPs or TEV was partially blocked (Figure 3D). These observations suggest that under iron deficient conditions the N-terminal region of DELE1 reaches the matrix less readily than in the steady state due to mitochondrial import arrest (Figure 3C). By separating endogenous DELE1 protein on SDS-PAGE at high resolution, we also confirmed that iron chelators stabilized the full-length form, but not the MPP-cleaved form of endogenous DELE1, whereas LONP1 knockdown resulted in stabilization of both forms of DELE1 (Figures 3E and 3F). These results suggest that this iron deficiency-induced mitochondrial import arrest of DELE1 may enable DELE1 to escape from MPP-mediated MTS cleavage and subsequent LONP1-mediated degradation, thereby promoting the stabilization of full-length DELE1 (Figure 3C). Although our observations suggested that the stabilized, full-length DELE1 passed through the IMS and OMM (Figures S2C and 3B), stable interactions between DELE1 and TOM/TIM import channels were not observed, in contrast to PINK1 import arrest (Figure S2E) 37–40.
Figure 3. DELE1 is stabilized on mitochondria by an iron deficiency-dependent mitochondrial import arrest.
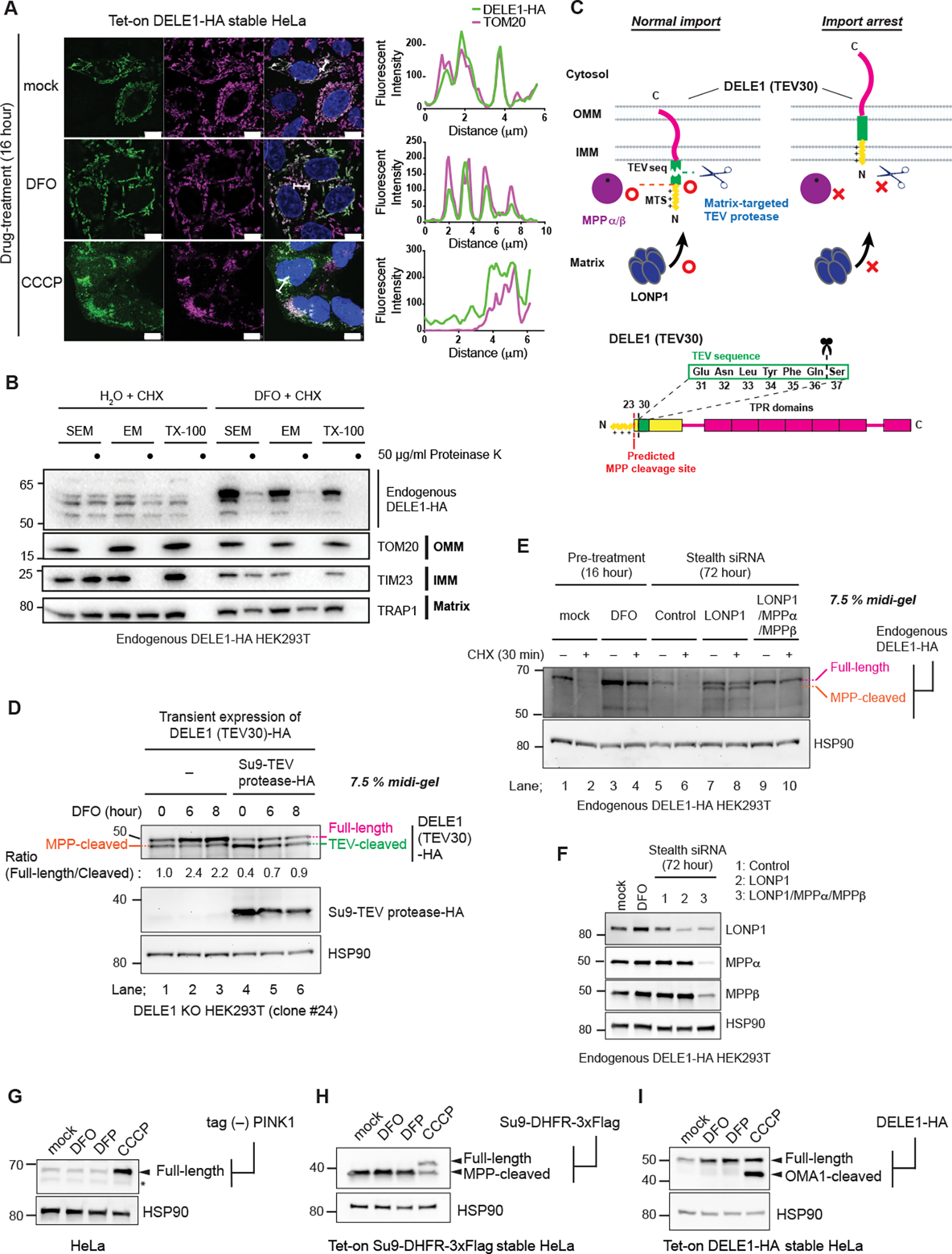
(A) Tet-on DELE-HA stable HeLa cells were treated with DFO or CCCP for 16 hours. The subcellular localization of DELE1-HA was determined by immunocytochemistry (ICC). Scale bars; 10 μm. Line profiles for the indicated fluorescent intensities determined along the white lines are shown to the right. (B) Crude mitochondrial fractions were isolated from Endogenous DELE1-HA HEK293T cells that were treated with CHX for 30 min with or without the pre-treatment with DFO for 16 hours, and were subjected to in vitro proteinase K protection assay at the indicated proteinase K concentration in the indicated buffer conditions. Sensitivities of endogenous DELE1-HA and other mitochondrial marker proteins to proteinase K were determined by immunoblotting (IB).
(C) Schematic representation for the TEV cleavage assay. Suppression of the proteases-mediated cleavage of DELE1 by import arrest (upper panel). Insertion of the TEV cleavage sequence after the predicted MPP cleavage site of DELE1 [DELE1(TEV30)] (lower panel).
(D) DELE1 KO cells were transfected with the TEV sequence-inserted DELE1 (DELE1(TEV30)-HA) with or without the matrix-targeted TEV protease (Su9-TEV protease-HA). After 24 hours, cells were treated with DFO for the indicated time periods. Cell lysates were separated in an SDS-PAGE 7.5% polyacrylamide Midi gel and analyzed by IB.
(E) IB for lysates of endogenous DELE1-HA HEK293T cells treated with CHX for 30 min with or without pre-treatment of DFO for 16 hours (lane 1–4), or transfected with the indicated siRNAs for 72 hours, and subsequently treated with CHX for 30 min (lane 5–10).
(F) Knockdown efficiency of indicated proteins in (E) determined by IB.
(G to I) HeLa cells transiently transfected with tag (−) PINK1 (G), Tet-on Su9-DHFR-3xFlag stable HeLa cells (H), or Tet-on DELE1-HA stable HeLa cells (I), were treated with DFO, DFP or CCCP for 16 hours. Doxycycline was added 8 hours prior to DFO treatment to induce the expression of Su9-DHFR-3xFlag (H) or DELE1-HA (I). Cell lysates were analyzed by IB. *; non-specific bands.
See also Figure S2.
Mitochondrial membrane potential across the IMM provides a driving force for the mitochondrial import of MTS-containing proteins 41. Consistently, mitochondrial depolarization by CCCP therefore induced the accumulation of the full-length (MPP-uncleaved) form of PINK1 23,24 and Su9-DHFR, a model protein for MTS-mediated import (Figures 3G and 3H). Although DELE1 is cleaved by OMA1 under CCCP-treated conditions, the total amount of DELE1 protein increased (Figures 2A and 3I). This indicates that like other mitochondrial proteins with a canonical MTS, the mitochondrial import of DELE1 depends on mitochondrial membrane potential. In contrast, while DELE1 was stabilized by iron deficiency, both PINK1 and Su9-DHFR were insensitive to this perturbation (Figures 3G and 3H). Thus, iron chelation-dependent import arrest is not a general means of regulation for MTS-containing proteins and may be specific for DELE1.
DELE1 activates an HRI-mediated ISR following iron deficiency
We next examined the possibility that stabilized DELE1 in response to iron deficiency activates the HRI-ISR pathway. Treatment with the iron chelators DFO or DFP increased phosphorylation of eIF2α and expression of ATF4 in a time- and concentration-dependent manner (Figures 4A and 4B). Flow cytometric quantitative analyses using an ATF4 reporter indicated DFO-dependent translational upregulation of ATF4 (Figure 4C) 17. Furthermore, the increase in ATF4 expression was attenuated by ISRIB, a chemical inhibitor of the ISR (Figure S3A) 42,43. The time course of ATF4 accumulation by iron chelation was slower than that induced by CCCP (Figure 4A). However, ATF4 accumulation followed the induction of IRP2 and was well correlated with the stabilization of DELE1, as well as with the degradation of mitochondrial and cytosolic ISC proteins (Figure S3B). Concomitant iron supplementation using high concentrations of ferric ammonium citrate (FAC) appeared to counteract the effects of cytosolic and mitochondrial iron chelation by DFO, as indicated by suppression of IRP2 accumulation and LIAS degradation, as well as induction of mitochondrial Ferritin (FTMT) (Figure 4D). FAC treatment also abrogated DFO-induced stabilization of endogenous DELE1 and ATF4 activation (Figure 4D). In contrast, the heme biosynthesis inhibitor SA did not activate the ISR at least within the time points investigated (Figure 4E). Addition of hemin to the culture media substantially increased the expression of HMOX1 44 under DFO-treated conditions, but it did not overcome DFO-induced iron deficiency (monitored by IRP2) (Figure 4F). Under this condition, hemin supplementation less efficiently suppressed the iron chelation-induced stabilization of endogenous DELE1 and accumulation of ATF4 compared to FAC treatment (Figure 4F). Collectively, these results suggest that iron is the primary factor in DFO-induced DELE1 stabilization and ISR activation.
Figure 4. DELE1 activates an HRI-mediated ISR following iron deficiency.
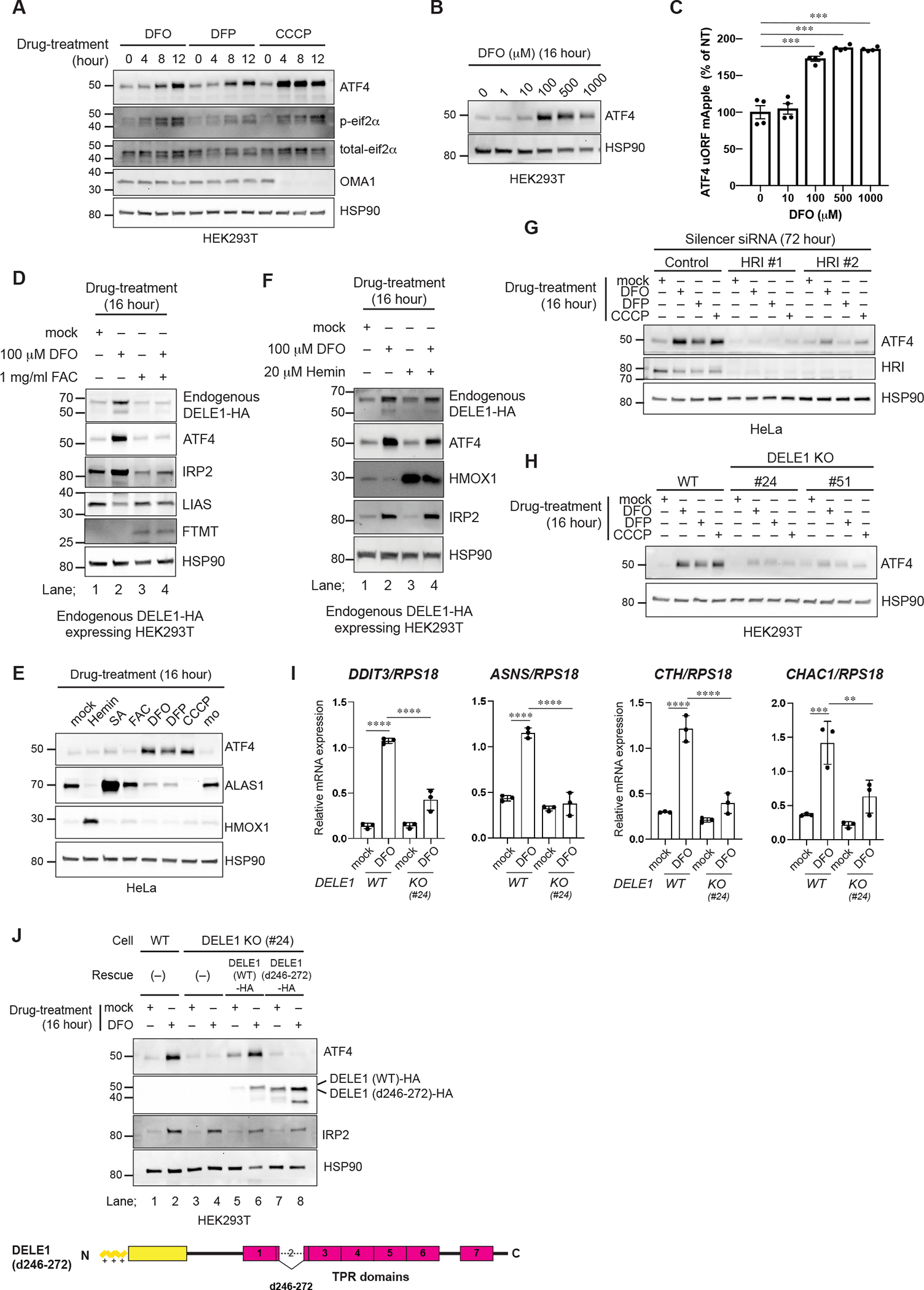
(A, B) HEK293T cells were treated with DFO, DFP or CCCP for the indicated time periods (A) or were treated with the indicated concentrations of DFO for 16 hours (B). Cell lysates were analyzed by immunoblotting (IB) with the indicated antibodies.
(C) ATF4 reporter (ATF4 uORF mApple)-expressing HeLa cells were treated with the indicated concentrations of DFO for 16 hours and were subjected to flow cytometry analysis. Data are shown as mean ± S.D. (N=4). ***P = 0.0001 (One-way ANOVA followed by Dunnett’s multiple comparison’s test).
(D) IB for lysates of endogenous DELE1-HA HEK293T cells treated with DFO with or without ferric ammonium citrate (FAC) for 16 hours.
(E) IB for lysates of HeLa cells treated with the indicated compounds for 16 hours.
(F) IB for lysates of endogenous DELE1-HA HEK293T cells treated with DFO for 16 hours in the presence or absence of hemin.
(G) IB for lysates of HeLa cells transfected with indicated siRNAs for 72 hours, and treated with DFO, DFP, or CCCP for the last 16 hours.
(H) IB for lysates of HEK293T WT or DELE1 KO cell lines (clone #24 and #51) treated with DFO, 1 mM DFP, or CCCP for 16 hours. The lysates were analyzed by IB.
(I) Quantitative PCR for ISR target gene expression in HEK293T WT or DELE1 KO (clone #24) cells treated with DFO for 16 hours. Shown are mean ± S.D. (N=3). **** P<0.0001, *** P<0.001, and ** P<0.01 (One-way ANOVA followed by Turkey’s multiple comparison).
(J) DELE1(WT)-HA or DELE1(d246–272)-HA was expressed in DELE1 KO HEK293T cells (clone #24) through lentivirus infection for 40 hours. Cells were treated with DFO for the last 16 hours before harvest. Cell lysates were analyzed by IB.
See also Figure S3.
Moreover, knockdown experiments revealed that both HRI and DELE1 were required for DFO and DFP-induced ISR activation in HeLa cells (Figures 4G, S3C and S3D). Genetic ablation of DELE1 by CRISPR-Cas9 genome editing in HEK293 cells (Figure S1A) suppressed iron chelation-induced ATF4 activation (Figures 4H and S3E), as well as the transcriptional activation of classical ISR target genes including DDIT3, ASNS, CTH and CHAC1 (Figure 4I). Exogenous reconstitution of DELE1 WT rescued the ISR activation in DELE1 KO cells, while this was not observed when a DELE1 mutant lacking the TPR2 domain was expressed (Figure 4J). The latter observation is consistent with previous studies showing that the TRP2 domain of DELE1 is required for activation of HRI under mitochondrial stress 16,17. Thus, the DELE1-HRI pathway is required for the activation of ISR in iron-deficient cells.
DELE1 on mitochondrial surface activates HRI
Iron chelators appear to activate the DELE1-HRI pathway in a mechanism distinct from how HRI is activated during mitochondrial stress caused by CCCP. Indeed, unlike CCCP, neither DFO nor DFP induced mitochondrial depolarization (Figure 5A). OMA1 is a stress-responsive protease whose proteolytic activity is enhanced by mitochondrial stress-inducing agents including CCCP 45,46. Stress-dependent OMA1 activation occurs through self-cleavage that eventually leads to the degradation of OMA1 36,47. Consistent with the absence of mitochondrial depolarization (Figure 5A), iron chelators appeared not to induce OMA1 activation, which was monitored by the processing of OMA1 itself, as well as its known substrates OPA1 and PGAM5 45,46,48,49 (Figure 5B). Moreover, knockdown of OMA1 did not suppress iron chelation-induced ISR activation in HEK293T or HeLa cells (Figures 5C and 5D), indicating that OMA1 is not required for iron-deficiency-induced HRI activation.
Figure 5. DELE1 on mitochondrial surface activates HRI.
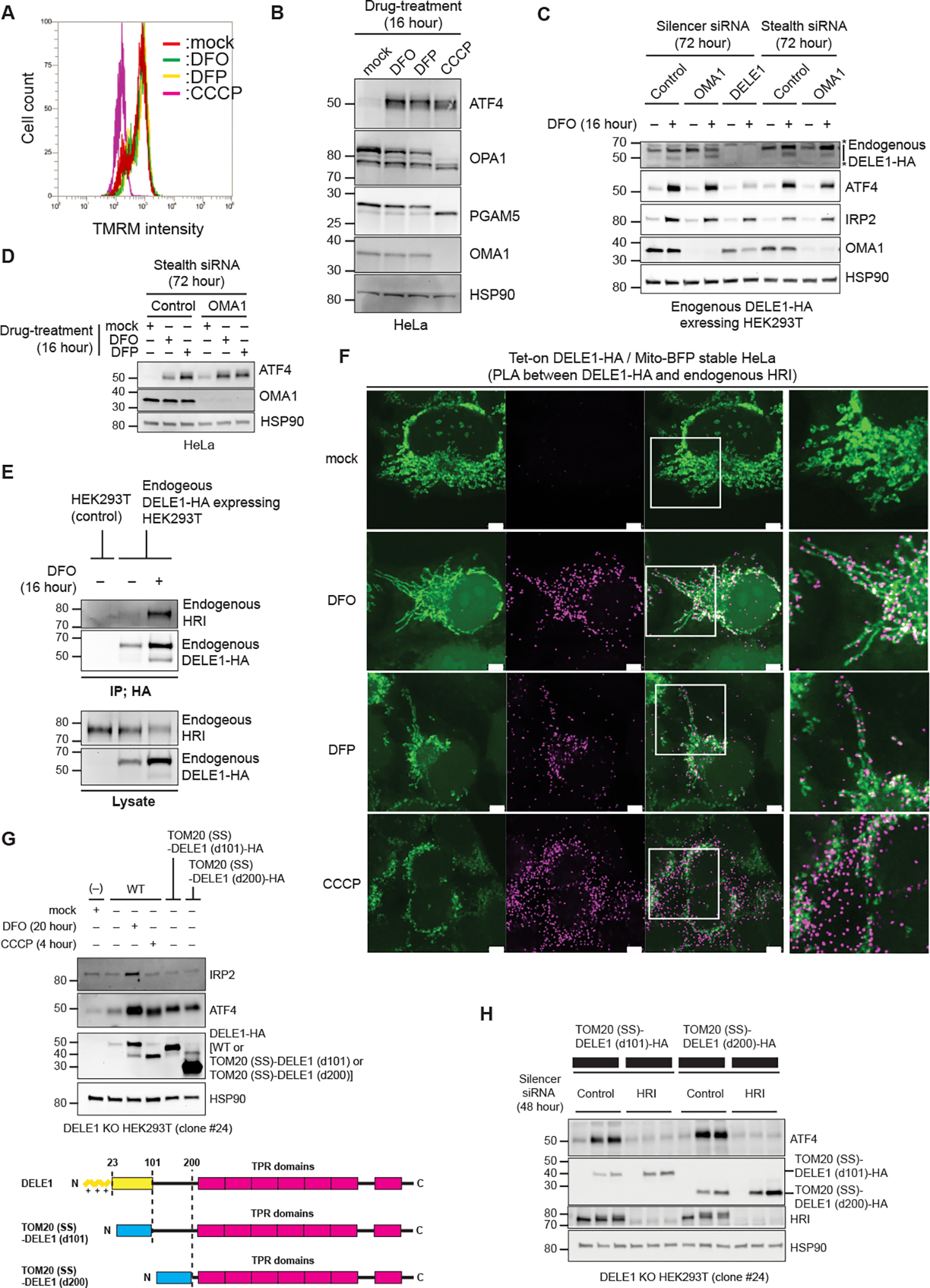
(A) Flow cytometry analysis to determine mitochondrial membrane potential in HeLa cells treated with either DFO, DFP or CCCP for 16 hours, and with TMRM for the last 15 min.
(B) HeLa cells were treated with either DFO, DFP or CCCP for 16 hours. Cell lysates were analyzed by immunoblotting (IB) with the indicated antibodies.
(C, D) IB for lysates of endogenous DELE1-HA HEK293T cells (C) or HeLa cells (D) transfected with indicated siRNAs for 72 hours, and treated with DFO or DFP for the last 16 hours before harvest. *; non-specific bands.
(E) Endogenous DELE1-HA was immunoprecipitated with anti-HA antibody from the indicated HEK293T cells with or without 16 hours of DFO treatment. The samples were subjected to IB. IP; immunoprecipitation.
(F) Proximity Ligation Assay (PLA) between overexpressed DELE1-HA and endogenous HRI. Tet-on DELE1-HA stable HeLa cells that stably express Mito-BFP were treated with DFO or DFP for 20 hours, or CCCP for 4 hours, under Doxycycline-added conditions, and were subjected to PLA. White arrow heads indicate representative PLA signals that were observed exactly on or close proximity to mitochondria. Scale bars; 5 μm. Magnified merge images of the squared area are shown to the right. Low magnification images are shown in Figure S4A.
(G) DELE1-HA or the OMM-tethered DELE1 [TOM20(SS)-DELE1(d101)-HA] or TOM20(SS)-DELE1(d200)-HA] was expressed in DELE1 KO HEK293T cells (clone #24) through lentivirus infection for 44 hours. Cells were treated with DFO for the last 20 hours or CCCP for the last 4 hours before harvest. Cell lysates were analyzed by IB (upper panel). Schematic representation of TOM20(SS)-DELE1(d101)-HA and TOM20(SS)-DELE1 (d200)-HA (lower panel). The OMM-targeting signal of TOM20 is shown as a blue box.
(H) IB for lysates of DELE1 KO HEK293T cells transfected with indicated siRNAs for 48 hours, and infected with the OMM-tethered DELE1-HA-expressing lentiviruses for the last 24 hours before harvest.
See also Figure S4.
Our immunoprecipitation assay revealed that the increase in DELE1 upon iron chelation coincided with more HRI binding to DELE1 (Figure 5E). We examined the subcellular localization of the iron deficiency-induced interaction between HRI and DELE1 using a proximity ligation assay (PLA) (Figures 5F and S4A). We observed robust increases in PLA signals after DFO, DFP, or CCCP treatment (Figure S4A). In contrast, no PLA signals were observed without expression of DELE1 (Figure S4A). These results indicate that PLA signals were derived from the stimuli-dependent interaction between HRI and DELE1. Strikingly, in iron-deficient conditions, the PLA signal was predominantly observed on, or in close proximity to, mitochondrial strings (Figure 5F). In contrast, the PLA signal was diffusely scattered over the cytosol under CCCP-treated conditions (Figure 5F). These results suggest that DELE1 interacts with HRI predominantly on the mitochondrial surface under iron deficient conditions, while this interaction occurs mainly in the cytosol under CCCP-treated conditions.
We thus considered that the mitochondrial import arrest of DELE1 (Figures 3B–3E) resulted in retention of the C-terminal TPR domains on the mitochondrial surface, allowing for the folding of the TPR domains able to subsequently interact with and activate cytosolic HRI. To test if the cytosolic exposure of the DELE1 TPR domains on the mitochondrial surface is sufficient for HRI activation, we tethered DELE1 to the OMM by fusing DELE1(d101 or d200) with a signal sequence (SS) of the OMM-localized protein TOM20 (Figures 5G and S4B), and examined whether this fusion protein can activate the HRI-ISR pathway. Both OMM-tethered DELE1s [TOMM20(SS)-DELE1(d101) and TOMM20(SS)-DELE1(d200)] were more stable than DELE1 WT under steady state conditions (Figure S4C), which is consistent with our observation that DELE1 is degraded in the matrix (Figures 1D and 1E). Also, unlike DELE1 WT, we did not observe iron deficiency-induced stabilization of these DELE1 mutants (Figure S4C). In agreement with the ability of cytosolic full-length DELE1 to bind to HRI and trigger the ISR 27, expression of the OMM-tethered DELE1 alone induced ATF4 expression without iron chelation (Figure 5G). Furthermore, knockdown of HRI abrogated this ATF4 induction (Figure 5H). These results indicate that the OMM-tethered DELE1 can act as a constitutively active form for ISR induction, supporting our hypothesis that in iron deficiency, the exposure of the DELE1 TPR domains on the OMM is sufficient to trigger the HRI-ISR pathway.
Determination of critical factors that are required for the iron deficiency-induced ISR activation
We next sought to identify critical domains in DELE1 that are required for the iron deficiency-induced DELE1 import arrest and subsequent HRI-ISR activation. Recent data reveal that an α-helix located between 80–106 amino acids (a.a.) (Figure 6A) hampers the efficient translocation of DELE1 into the matrix, and thus promotes its OMA1-mediated cleavage in the IMS under certain settings of mitochondrial stress 27. A DELE1 mutant that lacks these amino acids, DELE1(d80-106), stabilized after iron deficiency and rescued DFO-induced ISR activation in DELE1 KO cells (Figure S5A), suggesting that this region is dispensable for the iron-deficient DELE1 response. We also created several other DELE1 mutants including a mutant whose MTS was substituted with PINK1’s MTS, PINK1(MTS)-DELE1(d101), and deletion mutants lacking the intermediate region between the DELE1 MTS and TPR domains, DELE1(d102-200) and DELE1(d200-220), or lacking the entire TPR domains, DELE1(dTPR1-7) (Figure S5B). Unexpectedly, all these DELE1 mutants were able to accumulate in response to iron deficiency (Figures S5C and S5D), suggesting that iron deficiency-induced DELE1 stabilization may not be solely regulated through a single domain or motif in DELE1. Nevertheless, we identified DELE1(d102-200) as a unique mutant that did not rescue the iron deficiency-induced ISR activation (Figure 6B). DELE1 (d102-200) localized in mitochondria before and after iron chelation (Figure S5E). Unlike other DELE1 mutants lacking TPR domains, DELE1(d102-200) did not lose its ability to bind HRI when expressed in the cytosol following deletion of the MTS (Figure 6C). However, DELE1(d102-200) lost the ability to interact with HRI in a DFO-dependent manner (Figure 6D). These observations suggest that the TPR domains in this mutant may not be fully exposed into the cytosol under iron deficient conditions. The 102–200 region of DELE1 does not have any known domains or motifs except for the reported OMA1 cleavage site 17, and the latter half of this region is predicted to be disordered (Figure 6A). We thus speculated that the length of this intermediate region between the MTS and TPR domains might be important for DELE1 passing through the IMS and the OMM and fully exposing the TPR domains to the cytosol when DELE1’s import is arrested (Figure 6G). Consistent with this notion, 50 a.a. deletion either in the first half or the latter half of the 102–200 a.a. [DELE1(d102-150) or DELE1(d150-200)] was able to rescue iron deficiency-induced ISR activation in DELE1 KO cells (Figure 6E). Furthermore, replacement of the 102–200 region of DELE1 with different length of amino acid linkers (20, 50, and 100 a.a.) derived from an intrinsically disordered region of Nucleolin (Figure S5F) 50 demonstrated that a length of more than 50 a.a. within this region was sufficient for this response (Figure 6F). These results support our notion that in the setting of iron deficiency, DELE1 import arrest enables the protein to cytosolically expose its TPR domains, thereby facilitating interaction with HRI on the mitochondrial surface (Figure 6G).
Figure 6. Determination of critical factors that are required for the iron deficiency-induced ISR activation.
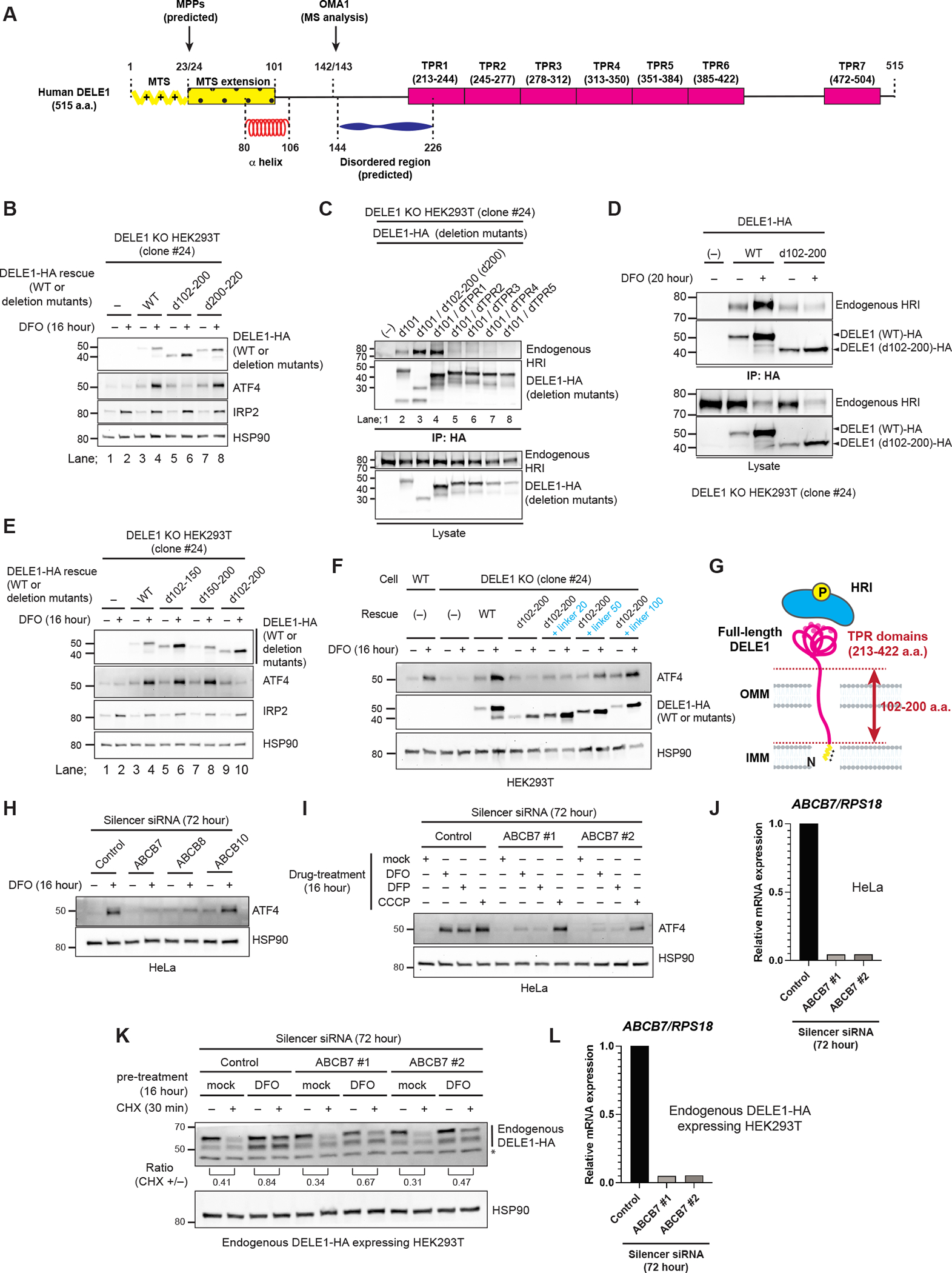
(A) A schematic representation of DELE1 domain structures. See text for details.
(B) DELE1 (WT) or DELE1 deletion mutants that lacked indicated regions were expressed in DELE1 KO HEK293T cells through lentivirus infection for 40 hours. Cells were treated with DFO for the last 16 hours before harvest. Cell lysates were analyzed by immunoblotting (IB).
(C) The indicated DELE1 deletion mutants were transiently transfected in DELE1 KO HEK293T cells. After 24 hours, cells were lysed and each DELE1 was immunoprecipitated with anti-HA antibody. The samples were subjected to IB. IP; immunoprecipitation.
(D) DELE1 WT or d102-200 were transiently infected in DELE1 KO HEK293T cells (clone #24). Cells were subsequently treated with DFO for the last 20 hours before harvest. Each DELE1 was immunoprecipitated with anti-HA antibody. Samples were subjected to IB.
(E, F) DELE1 WT or DELE1 deletion mutants that lacked indicated regions (E) or linker-inserted DELE1(d102-200) mutants (F) were expressed in DELE1 KO HEK293T cells through lentivirus infection for 40 hours. Cells were treated with DFO for the last 16 hours before harvest. Cell lysates were analyzed by IB.
(G) A model for the role of 102–200 a.a. region of DELE1 in HRI activation during iron deficiency. See text for details.
(H) IB for lysates of HeLa cells transfected with indicated siRNAs for 72 hours, and treated with DFO for the last 16 hours.
(I) IB for lysates of HeLa cells transfected with indicated siRNAs for 72 hours, and treated with DFO, DFP, or CCCP for the last 16 hours.
(J) Knockdown efficiency of ABCB7 in (I) determined by quantitative PCR (qPCR).
(K) Endogenous DELE1-HA HEK293T cells were transfected with indicated siRNAs for 72 hours. Cells were treated with DFO for 16 hours, followed by the CHX chase for the last 30 min before harvest. Cell lysates were analyzed by IB. *; non-specific bands.
(L) Knockdown efficiency of ABCB7 in (K) determined by qPCR.
Next, we tried to identify external factors that are involved in the iron deficiency-induced ISR activation. Recent studies have revealed that LONP1 physically and functionally associates with mitochondrial import machineries 51,52. Thus, we examined the possibility that LONP1 is involved in the iron deficiency-induced ISR activation through regulating the DELE1 import status. However, we did not see any defects in iron deficiency-induced ISR activation following LONP1 knockdown (Figure S6A). As previously reported 16, the LONP1 knockdown slightly activated the ISR at steady state (Figure S6A), which may imply the presence of mitochondrial proteotoxic stress in LONP1-deficient cells, as LONP1 is a key quality control protease 30.
It has been suggested that the mitochondrial stress-activated ISR in mammalian cells is a functional counterpart of mtUPR in C. elegans 14,53,54 that is activated by mitochondrial proteotoxic stress-induced mitochondrial import arrest of the transcription factor ATFS-1 25,55. HAF-1, the IMM-resident ATP-binding cassette (ABC) transporter, was identified as a critical factor for the mitochondrial import arrest of ATFS-1 55,56. In mammals, there are three IMM-resident ABC transporters (ABCB7, ABCB8 and ABCB10) 57,58. To date, the exact substrates of these transporters have not been fully determined, but, intriguingly, all of these transporters are reported to play critical roles in the intracellular and mitochondrial metabolism of iron-containing cofactors including ISC and heme 59–65. We thus examined whether these IMM ABC transporters are also involved in the iron depletion-induced ISR activation. Knockdown of ABCB7 or ABCB8, but not ABCB10, attenuated iron deficiency-induced ATF4 expression (Figure 6H). Because ABCB8 knockdown severely impaired cell growth, we focused our subsequent efforts on ABCB7. Notably, ABCB7 was selectively required for the ISR activation by iron chelators DFO and DFP but not for that induced by CCCP (Figure 6I and 6J). Furthermore, CHX-chase experiments revealed that stabilization of DELE1 in response to iron chelation was less efficient in ABCB7 knockdown cells (Figures 6K and 6L). Moreover, similar results were obtained by the ablation of ISCU 66, a matrix-resident essential scaffold component of the ISC assembly (Figures S6B and S6C). As previously reported 65,67, we observed retardation of mitochondrial and/or cytosolic ISC biosynthesis after 72 hours of siRNA treatments for ABCB7 or ISCU as evident by decreases in mitochondrial and/or cytosolic ISC-containing proteins (Figure S6D). These results suggest that with iron deficiency, a potential cross-talk may exist between ISC biosynthesis and the DELE1-HRI-ISR pathway, presumably through modulating the mitochondrial DELE1 import status.
DELE1-HRI-ISR pathway protects erythroid cells against iron deficiency-induced cell death
Lastly, to address the physiological significance of this iron-deficiency-activated DELE1-HRI pathway, we focused on erythroid cells. These cells incorporate large amounts of iron for hemoglobin synthesis, and therefore require tight regulation of intracellular iron homeostasis 68. We employed murine erythroleukemia (MEL) cells as a well-established in vitro model of erythroid differentiation, and generated DELE1 or HRI gene-deleted MEL cells using CRISPR (Figure S7A). Culturing MEL cells with medium containing 2% DMSO rapidly induces erythroid-like differentiation, as evident by elevated expression of erythroid marker genes within 24 hours 69. It has been previously demonstrated that iron limitation by DFO significantly suppresses erythroid differentiation of MEL cells 69. We found that DFO treatment during the initial 24 hours of DMSO-stimulated differentiation increased ATF4 expression, suggesting that iron-deficiency activates the ISR in differentiating MEL cells (Figure 7A). This DFO-induced ATF4 expression was markedly suppressed in DELE1 KO as well as HRI KO MEL cells when compared with WT cells (Figures 7A and 7B). Exogenous expression of DELE1 recovered ATF4 induction in iron-deficient DELE1 KO MEL cells (Figure 7C). Furthermore, RNA sequencing analyses using these MEL cell lines revealed that upregulation of ISR target genes (such as ATF4, DDIT3, TRIB3, MTHFD2, and SLC3A2) by iron chelation was significantly attenuated in both DELE1 KO and HRI KO cells (Figures 7D and 7E and Table S1). This indicates that the DELE1-HRI pathway is required for iron-deficiency-induced ISR activation in differentiating MEL cells. Moreover, we found that DELE1 KO and HRI KO cells exhibited higher cell death following 48 hours of DFO treatment (Figure 7F). Collectively, these findings suggest that the DELE1-HRI mediated ISR plays a crucial role in protecting erythroid cells from cell death following a perturbation of iron homeostasis.
Figure 7. DELE1-HRI-ISR pathway protects the erythropoietic cell lineage against ferroptosis.
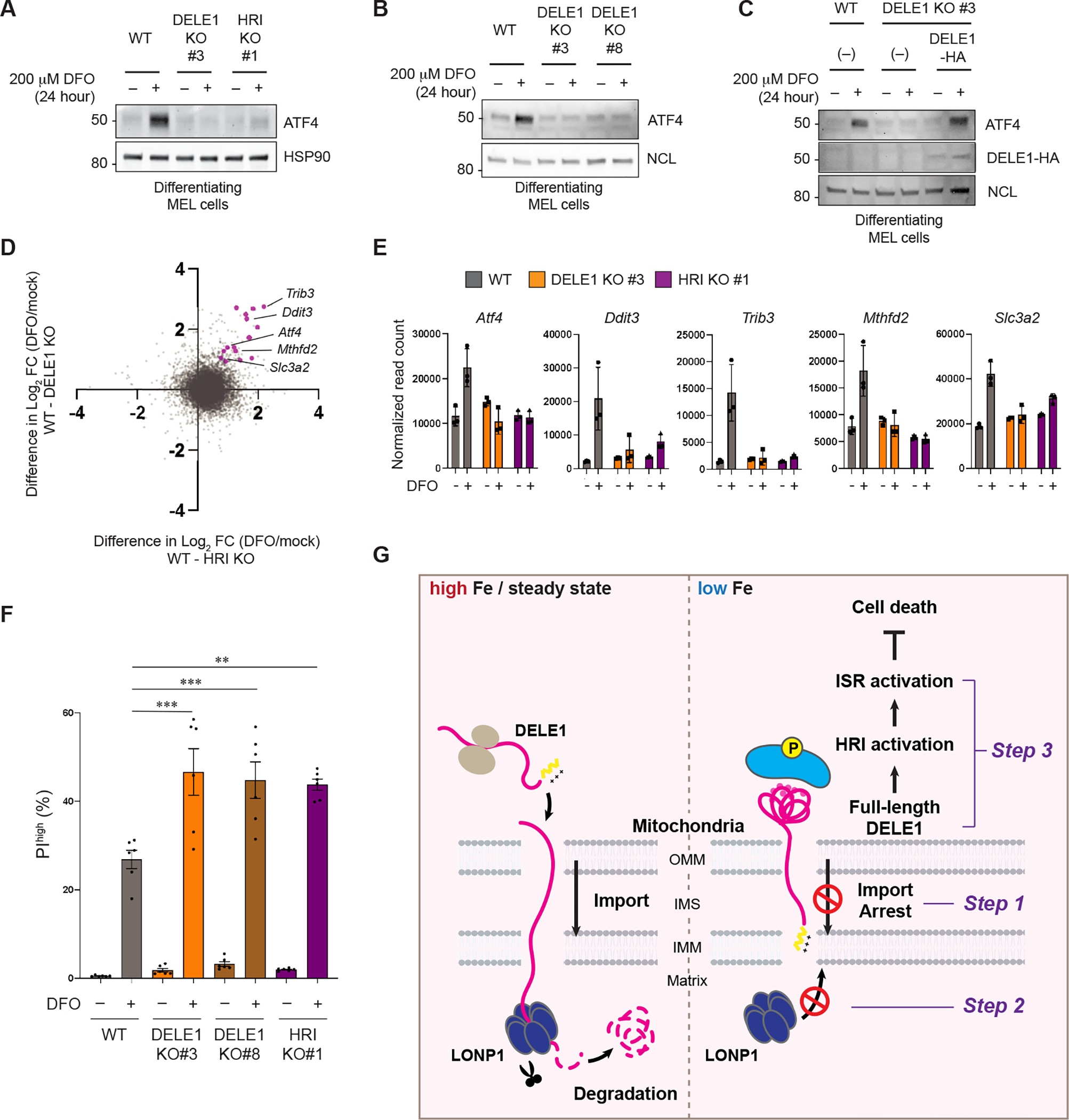
(A, B) MEL wildtype (WT), two independent DELE1 KO (clone #3 and clone #8), and HRI KO (clone #1) cells were cultured with 2% DMSO-containing medium in the presence or absence of DFO for 24 hours. Cell lysates were analyzed by immunoblotting (IB) with the indicated antibodies. HSP90 in (A) and NCL in (B) are shown as loading controls.
(C) IB for lysates of WT, DELE1 KO, or DELE1(WT)-HA-transfected DELE1 KO MEL cells. Cells were treated with DFO for the last 24 hours before harvest.
(D) MEL WT, DELE1 KO, and HRI KO cells were cultured with 2% DMSO-containing medium in the presence or absence of DFO for 24 hours. Cell lysates were subjected to RNA sequencing analysis. Differences between WT and HRI KO (X axis) or WT and DELE1 KO (Y axis) in log2 fold changes (FC) of each gene expression before and after iron chelation (DFO/mock) are plotted. Representative ISR target genes are shown in magenta.
(E) Relative mRNA expression of representative ISR target genes selected from the RNA sequencing data.
(F) MEL WT, DELE1 KO (clone #3 and #8), and HRI KO (clone #1) cells were cultured with 2% DMSO-containing medium in the presence or absence of DFO for 48 hours. Dead cells were determined by flow cytometry for Propidium iodide (PI) positive cells. Data are shown as mean ± S.D. (N=5–6). ***P<0.001 and **P<0.01 (One-way ANOVA followed by Turkey’s multiple comparison).
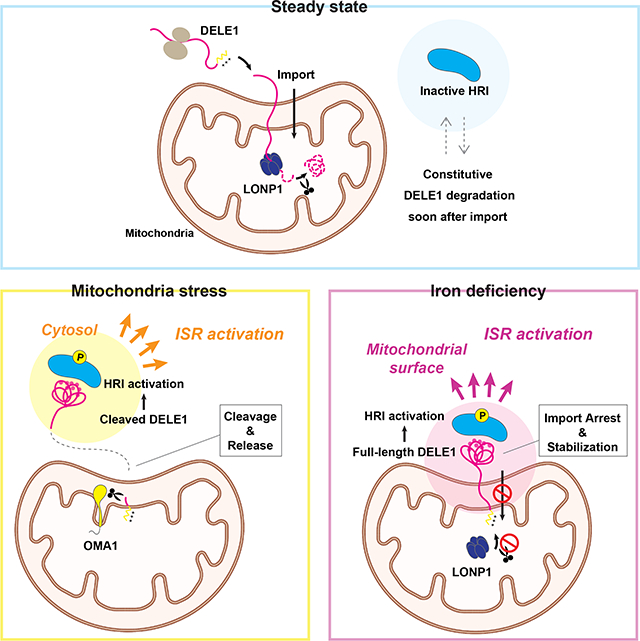
(G) A proposed model of the iron-deficiency-induced mitochondrial import regulation of DELE1 and the subsequent activation of the HRI-ISR pathway. See text for details.
See also Figure S7.
Discussion
Based on these findings, we propose a following model of DELE1-mediated HRI activation under low iron conditions (Figure 7G). Under steady state conditions where enough iron is available, DELE1 is constitutively degraded by the matrix-resident protease LONP1 soon after mitochondrial import, thereby maintaining low steady-state levels of DELE1 expression (Figure 7G, left). When intracellular iron availability is limited, the mitochondrial import of DELE1 is arrested (Figure 7G, right, step 1). This iron deficiency-induced mitochondrial import arrest of DELE1 enables DELE1 to escape from LONP1-mediated degradation (Figure 7G, right, step 2). As a consequence, full-length DELE1 is stabilized at mitochondria with its C-terminal domain exposed to the cytosol, and thereby allowing the TPR domains to fold and interact with HRI (Figure 7G, right, step 3). The stabilized, full-length form of DELE1 passes through the IMS and the OMM through an unknown mechanism. Importantly, the iron deficiency-induced activation of DELE1-HRI-ISR pathway does not require OMA1-mediated DELE1 cleavage (Figures 5C and 5D). Recent work uncovered that mitochondrial import stress (e.g., knockdown of mitochondrial import machineries or overexpression of mitochondrial precursor proteins) activates DELE1-HRI-ISR pathway 27. Of note, ISR activation in this context also does not require OMA1, and it can be mediated by full-length DELE1 that accumulates in the cytosol. Therefore, DELE1 appears to activate the HRI-ISR pathway without OMA1-mediated cleavage under certain conditions when its C-terminal TPR domain has physical access to cytosolic HRI.
Recent studies have revealed the importance of mitochondrial import regulation in sensing various mitochondrial perturbations and triggering appropriate stress responses, as represented by the PINK1/Parkin-mediated mitophagy triggered by PINK1 import arrest 22 and the mtUPR in C. elegans triggered by ATFS-1 import arrest 25. Notably, expression of both proteins is kept at a low level in the absence of stress by constitutive degradation. In both cases, the stress-dependent import arrest enables them to escape from this degradation to elicit a stress response. The constitutive degradation of these proteins appears to be an energy-consuming process but allows a rapid response to stress sensed within mitochondria. Similar to PINK1 and ATFS-1, DELE1 import arrest underscores this general mechanism of mitochondrial stress sensing, in this case to sensing intracellular iron deficiency to activate the ISR. Particularly, our findings demonstrated the mechanistic similarity between the ATFS-1-dependent mtUPR and the DELE1-dependent ISR: namely, the requirement of LONP1 for steady state degradation of the stress sensor and ABC transporters for the stress-dependent activation.
Similar to ATFS-1, a previous report suggests that the DELE1-HRI-ISR pathway is activated by mitochondrial proteotoxic stress. LONP1 deletion or mitochondrial chaperone inhibitor-mediated ISR activation was shown to be dependent on DELE1 16. It is noteworthy that iron chelation may also cause mitochondrial proteotoxic stress. Under iron-limited conditions, the biosynthesis of ISC is impaired, generating unfolded or misfolded ISC proteins in the mitochondrial matrix, which can become substrates for LONP1 (Figure S2A) 35. Therefore, the accumulation of misfolded ISC proteins by iron deficiency may increase the substrate burden on LONP1, thereby decreasing the ability of LONP1 to degrade DELE1. This could be an alternative explanation for how iron deficiency-induces DELE1 stabilization. Although we do not exclude this possibility, our results support the notion that iron chelation-induced DELE1 stabilization does not solely result from the loss of LONP1 function. First, LONP1 knockdown and iron chelation stabilize different species of DELE1 (Figure 3E). LONP1 knockdown stabilized both the full-length and the MPP-cleaved form of DELE1 while iron chelation predominantly stabilized the full-length form. Therefore, iron deficiency prevents DELE1 to reach the mitochondrial matrix regardless of the LONP1 activity status. Second, although LONP1 knockdown cells exhibited slightly higher ATF4 levels at steady state, we still observed iron chelator-dependent induction of ATF4 in these cells (Figure S6A). These observations indicate the existence of an additional layer of regulation, namely the DELE1 import regulation, leading to more robust activation of ISR under iron deficient conditions, compared to LONP1 inactivation alone.
There are few examples of proteins whose mitochondrial import is regulated by iron. In our study, iron deficiency-dependent import arrest was observed only for DELE1, but not for two other MTS-containing proteins we tested (Figures 3G and 3H). A recent report identified that FTMT, a mitochondrial ferritin, may also be subject to import arrest in iron deficiency, and that this regulation may be related to its role in iron depletion-induced mitophagy 32. It has been demonstrated that ATFS-1 is activated by P. aeruginosa infection, in addition to the mitochondrial proteotoxic stress 70. Intriguingly, the exposure of worms to P. aeruginosa strains lacking siderophore, a bacterial toxic compound that has an iron-chelating activity, resulted in less mtUPR activation than that induced by wild-type P. aeruginosa 70. Thus, it is tempting to speculate that ATFS-1 import might also be sensitive to bacterial toxin-mediated iron chelation, and possibly to other iron-deficient conditions.
We observed that the ablation of ABCB7 and ISCU, components involved in ISC metabolism, attenuated iron deficiency-induced ISR activation and DELE1 stabilization (Figures 6I–6L and S6B–S6C). Given lack of ABCB7 and ISCU impaired the maturation of mitochondrial ISC-containing proteins (Figure S6D), it is tempting to speculate that their common target, mitochondrial ISC, regulates the import of DELE1. However, as intracellular iron-cofactor metabolism pathways are highly interconnected, several alternative mechanisms are possible. Loss-of-function mutations in ABCB7 cause X-linked sideroblastic anemia with ataxia, highlighting its importance in erythrocyte maturation 64. Consistently, ABCB7 was also reported to be involved in heme biosynthesis through its interaction with ABCB10 and FECH, two critical components in heme metabolism 65. However, the contribution of heme in DELE1 import regulation may not be significant, as the effect of hemin on iron deficiency-induced DELE1 stabilization was modest (Figure 4F) and knockdown of ABCB10 did not suppress ISR activation by DFO (Figure 6H). Alternatively, it is possible that perturbed intracellular and/or mitochondrial iron metabolism in ABCB7- or ISCU-deficient cells may affect the DELE1 stability. It has been reported that the ablation of ABCB7 (and ABCB8) results in mitochondrial iron overload 59,65,71. The ablation of ISCU also affects the intracellular iron metabolism 67. Nevertheless, either scenario supports our conclusion that DELE1 responds to changes in intracellular iron availability.
A pro-survival role for the HRI-ISR pathway in erythroid cells has been well established 12. Analysis of HRI KO mice fed an iron-deficient diet has indicated that the HRI-mediated ISR suppresses translation of globin in order to prevent toxic aggregation of heme-deficient globin under heme-limited conditions caused by iron deficiency, thereby protecting erythroid progenitors. By using DMSO-stimulated MEL cells as an erythroid cell model, we also indicated a cell protective role for the DELE1-HRI-ISR pathway in the setting of iron deficiency (Figure 7F). It has been reported that the DELE1-HRI-ISR pathway also act as a cell death promoter in some contexts. For example, the deletion of DELE1 or HRI prevented cell death under mitochondrial stress induced by CCCP 16. We observed DFO-induced ISR activation was relatively weak and slow compared to the CCCP-induced ISR (Figure 4A). We also showed different subcellular localizations of the DELE1-HRI interaction between iron chelation (mitochondrial surface) and CCCP (the cytosol) (Figure 5F). Thus, temporal and spatial regulation of this pathway might be important for determining cellular outcomes under various stress conditions.
The iron-dependent mitochondrial import regulation of DELE1 and its critical role in the activation of HRI we described in this study represent a previously unrecognized mitochondrial-based iron sensing mechanism connecting mitochondria to the cytosolic ISR. Mitochondria are the major iron containing subcellular compartment, as they harbor the biosynthetic pathways for two major iron cofactors, heme and ISC. Therefore, it is not surprising that mitochondria would have their own iron monitoring system, in addition to the well-characterized iron homeostasis mechanism governed by cytosolic IRP1/2 33.
Limitations of the study
Due to technical difficulties of compartment specific iron depletion, it has not been determined whether mitochondrial or cytoplasmic iron deficiency alone is sufficient to cause DELE1 import arrest. Our attempts using DELE1 deletion and chimera mutants indicated the complexity of the mechanism of DELE1 import arrest. To further delineate this mechanism, identification of DELE1 interacting molecules as well as targets of iron or other iron-containing small molecules during this process will be required. Although we demonstrated that iron chelators predominantly stabilized the DELE1 full-length form, we also observed accumulation of a small fraction of a DELE1 short-form, which had a similar molecular size to the OMA1-cleaved form (Figure 2A), but was generated in an apparent OMA1-independent manner (Figure 5C). As this DELE1 short-form was released into the cytosol (Figure S2B), we cannot rule out the partial contribution of this form to iron deficiency-induced ISR activation.
STAR Methods
Resource availability
Lead Contact
Further information and requests for reagents will be fulfilled by Lead Contact Shiori Sekine (sekine@pitt.edu).
Materials availability
All constructs or cell lines generated in this study are available from the lead contact upon request and completion of a Material Transfer Agreement.
Data and code availability
All RNA seq data have been deposited at SRA under the accession number PRJNA970328. Raw images were deposited to Mendeley Data (DOI: 10.17632/7p8w52928v.1). All data are publicly available as of the date of publication. Accession numbers and DOI are listed in the key resources table.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HA | Cell Signaling | 2367S; RRID:AB_10691311 |
| TOM40 | Proteintech | 18409-1-AP; RRID:AB_2303725 |
| TIM50 | Santa Cruz | sc-393678; RRID:AB_2714191 |
| TIM23 | BD Biosciences | 611222; RRID:AB_398754 |
| HSP90 | Santa Cruz | sc-7947; RRID:AB_2121235 |
| HSP90 | Cell Signaling | 4874S; RRID:AB_2121214 |
| PINK1 | Cell Signaling | 6946S; RRID:AB_11179069 |
| p62 | Abcam | ab56416; RRID:AB_945626 |
| LONP1 | Proteintech | 66043-1-lg; RRID:AB_2881494 |
| LONP1 | Novus Biologicals | NBP1-81734; RRID:AB_11002818 |
| αTubulin | Sigma | T6199; RRID:AB_477583 |
| IRP2 | Cell Signaling | 37135S; RRID:AB_2799110 |
| ALAS1 | Abcam | ab154860 |
| HMOX1 | GeneTex | GTX101147; RRID:AB_1950502 |
| OMA1 | Santa Cruz | sc-515788; RRID:AB_2905488 |
| TOM20 | Santa Cruz | sc-17764; RRID:AB_628381 |
| MPPα | Sigma | HPA021648; RRID:AB_1855491 |
| MPPβ | Proteintech | 16064-1-AP; RRID:AB_2167122 |
| M2 | Sigma | F1804; RRID: AB_262044 |
| ATF4 | Cell Signaling | 11815S; RRID:AB_2616025 |
| phospho-eif2α | Cell Signaling | 3398; RRID:AB_2096481 |
| total-eif2α | Cell Signaling | 5324; RRID:AB_10692650 |
| LIAS | Proteintech | 11577-1-AP; RRID:AB_2135972 |
| SDHB | Proteintech | 10620-1-AP; RRID:AB_2285522 |
| PPAT | Proteintech | 15401-1-AP; RRID:AB_2166532 |
| FTMT | Abcam | ab66111 |
| OPA1 | BD Biosciences | 612607; RRID:AB_399889 |
| PGAM5 | Thermo Fisher Scientific | PA5-57894; RRID:AB_2645430 |
| HRI | Proteintech | 20499-1-AP; RRID:AB_10697665 |
| ISCU | Proteintech | 14812-1-AP; RRID:AB_2280362 |
| NCL | Cell Signaling | 14574; RRIS:AB_2798519 |
| TRAP1 | Cell Signaling | 92345: RRID:AB_2800183 |
| Bacterial and virus strains | ||
| N/A | ||
| Biological samples | ||
| N/A | ||
| Chemicals, peptides, and recombinant proteins | ||
| Cycloheximide (CHX) | Cayman Chemical | 14126 |
| MG132 | Sigma | C2211 |
| Bafilomycin A1 (Baf. A1) | Cayman Chemical | 11038 |
| Deferoxamine (mesylate) (DFO) | Cayman Chemical | 14595 |
| Deferiprone (DFP) | Cayman Chemical | 20387 |
| CCCP | Sigma | C2759 |
| Hemin chloride | Cayman Chemical | 16487 |
| Succinyl acetone (SA) | MedChemExpress | HY-W010184 |
| Proteinase K | Sigma | P6556 |
| Ferric ammonium citrate (FAC) | Sigma | RES20400-A7 |
| Erastin | Cayman Chemical | 17754 |
| trans-ISRIB | Cayman Chemical | 16258 |
| Doxycycline | Sigma | D9891 |
| DSP (dithiobis(succinimidyl propionate)) | Thermo Fisher Scientific | 22585 |
| X-tremeGENE 9 DNA transfection reagent | Roche | 6365787001 |
| Lipofectamine RNAi max transfection reagent | Thermo Fisher Scientific | 13778075 |
| Tetramethylrhodamine, Methyl Ester, Perchlorate (TMRM) | Thermo Fisher Scientific | T668 |
| Critical commercial assays | ||
| Duolink In Situ PLA Probe Starter Kit | Sigma | DUO92101 |
| Deposited data | ||
| Raw images | This study | Mendeley Data (DOI: 10.17632/7p8w52928v.1) |
| RNA seq data | This study | SRA (accession number: PRJNA970328) |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-3216 |
| HeLa | ATCC | CCL-2.2 |
| Endogenous DELE1-HA HEK293T | Fessler et al., 2020 | N/A |
| Tet-on DELE1-HA stable HeLa | Houston et al., 2021 | N/A |
| Tet-on Su9-DHFR-3xFlag stable HeLa | Sekine et al., 2019 | N/A |
| DELE1 KO HEK293T clone #24 | This study | N/A |
| DELE1 KO HEK293T clone #51 | This study | N/A |
| ATF4_uORF_mApple-stable HeLa | This study | N/A |
| Tet-on DELE1-HA / Mito-BFP stable HeLa | This study | N/A |
| MEL | Millipore Sigma | F4N |
| DELE1 KO MEL clone #3 | This study | N/A |
| DELE1 KO MEL clone #8 | This study | N/A |
| HRI KO MEL clone #1 | This study | N/A |
| Experimental models: Organisms/strains | ||
| N/A | ||
| Oligonucleotides | ||
| Stealth RNAi Negative Control Med GC Duplex #2 | Thermo Fisher Scientific | 12935112 |
| Silencer Select Negative Control #1 siRNA | Thermo Fisher Scientific | 4390843 |
| Stealth RNAi PARL | Thermo Fisher Scientific | HSS183080 |
| Stealth RNAi OMA1 | Thermo Fisher Scientific | HSS132976 |
| Stealth RNAi YME1L | Thermo Fisher Scientific | HSS116546 |
| Stealth RNAi HtrA2 | Thermo Fisher Scientific | HSS146986 |
| Stealth RNAi LONP | Thermo Fisher Scientific | HSS113887 |
| Silencer Select LONP #1 | Thermo Fisher Scientific | s17902 |
| Silencer Select LONP #2 | Thermo Fisher Scientific | s17903 |
| Stealth RNAi CLPP | Thermo Fisher Scientific | HSS112033 |
| Stealth RNAi AFG3L2 | Thermo Fisher Scientific | HSS116886 |
| Stealth RNAi SPG7 | Thermo Fisher Scientific | HSS110137 |
| Stealth RNAi MPPa | Thermo Fisher Scientific | HSS118357 |
| Stealth RNAi MPPb | Thermo Fisher Scientific | HSS114169 |
| Silencer Select HRI #1 | Thermo Fisher Scientific | s25822 |
| Silencer Select HRI #2 | Thermo Fisher Scientific | s25823 |
| Silencer Select DELE1 #1 | Thermo Fisher Scientific | s18966 |
| Silencer Select DELE1 #2 | Thermo Fisher Scientific | s18967 |
| Silencer Select ABCB7 #1 | Thermo Fisher Scientific | s855 |
| Silencer Select ABCB7 #2 | Thermo Fisher Scientific | s857 |
| Silencer Select ABCB8 | Thermo Fisher Scientific | s22104 |
| Silencer Select ABCB10 | Thermo Fisher Scientific | s23857 |
| Silencer Select ISCU #1 | Thermo Fisher Scientific | s23909 |
| Silencer Select ISCU #2 | Thermo Fisher Scientific | s23908 |
| Recombinant DNA | ||
| pLVX-puro-DELE1-HA | Houston et al., 2021 | N/A |
| pLVX-puro-DELE1 (TEV30)-HA | This study | N/A |
| pcDNA3.1 (+) Su9-TEV protease-HA | Sekine et al., 2019 | N/A |
| pRetroX-Tight-Pur-DELE1-HA | Houston et al., 2021 | N/A |
| pRetroX-Tight-Pur-DELE1(d30) -HA | This study | N/A |
| pRetroX-Tight-Pur-DELE1(d101)-HA | This study | N/A |
| pRetroX-Tight-Pur-pSu9-DHFR-tev-3xFlag | Sekine et al., 2019 | N/A |
| pCMV (d1) PINK1 (Tag-) | Okatsu et al., 2012 | N/A |
| pLVX-puro-PINK1-HA | This study | N/A |
| lentiCRISPR v2-hygro-human DELE1 (Guide#1) | This study | N/A |
| lentiCRISPR v2-hygro-human DELE1 (Guide#2) | This study | N/A |
| pXG237 | Guo et al., 2020 | Addgene #141281 |
| pLVX-puro-DELE1(d246-272)-HA | This study | N/A |
| pLVX-puro-TOMM20(ss)-DELE1(d101)-HA | This study | N/A |
| pLVX-puro-TOMM20(ss)-DELE1(d200)-HA | This study | N/A |
| pLVX-puro-DELE1(d102-200)-HA | This study | N/A |
| pLVX-puro-DELE1(d200-220)-HA | This study | N/A |
| pLVX-puro-DELE1(d101)-HA | This study | N/A |
| pLVX-puro-DELE1(d101/d102-200)-HA | This study | N/A |
| pLVX-puro-DELE1(d101/dTPR1)-HA | This study | N/A |
| pLVX-puro-DELE1(d101/dTPR2)-HA | This study | N/A |
| pLVX-puro-DELE1(d101/dTPR3)-HA | This study | N/A |
| pLVX-puro-DELE1(d101/dTPR4)-HA | This study | N/A |
| pLVX-puro-DELE1(d101/dTPR5)-HA | This study | N/A |
| pLVX-puro-DELE1(d102-150)-HA | This study | N/A |
| pLVX-puro-DELE1(d150-200)-HA | This study | N/A |
| pLVX-puro-DELE1(d102-200 + linker 20)-HA | This study | N/A |
| pLVX-puro-DELE1(d102-200 + linker 50)-HA | This study | N/A |
| pLVX-puro-DELE1(d102-200 + linker 100)-HA | This study | N/A |
| lentiCRISPR v2-puro-mouse DELE1(Guide#1) | This study | N/A |
| lentiCRISPR v2-puro-mouse DELE1 (Guide#2) | This study | N/A |
| lentiCRISPR v2-puro-mouse HRI (Guide#1) | This study | N/A |
| lentiCRISPR v2-puro-mouse HRI (Guide#2) | This study | N/A |
| Mito-BFP | Friedman et al., 2011 | Addgene #49151 |
| pLVX-puro-DELE1(d80-106)-HA | This study | N/A |
| pLVX-puro-PINK1(MTS)-DELE1(d101)-HA | This study | N/A |
| pLVX-puro-DELE1(dTPR1-7)-HA | This study | N/A |
| pLVX-puro | Clontech | 632164 |
| pRetroX-Tight-Pur | Clontech | 632104 |
| pRetroX-Tet-On Advanced | Clontech | 632104 |
| lentiCRISPR v2-puro | Sanjana et al., 2014 | Addgene #52961 |
| lentiCRISPR v2-hygro | Stringer et al., 2019 | Addgene #98291 |
| Software and algorithms | ||
| BioRender | N/A | https://app.biorender.com/user/signin |
| Other | ||
| N/A | ||
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
HEK293T and HeLa cells were cultured in DMEM (Gibco) supplemented with 10% FBS (VWR Life Science), 10 mM HEPES (Gibco), 1 mM Sodium pyruvate (Gibco), non-essential amino acids (Gibco) and GlutaMAX (Gibco). MEL cells (F4N, Sigma) were cultured in RPMI Medium 1640 with L-Glutamine and 25 mM HEPES (Gibco) supplemented with 10% FBS and 1 x Penicillin Streptomycin Solution (Corning). MEL cell differentiation was induced by using this RPMI medium with 2% Dimethyl sulfoxide (DMSO) (Santa Cruz).
Method Details
Transfection and reagents
For RNA interference, 20 nM Stealth siRNAs (Thermo Fisher Scientific) or 5 nM Silencer select siRNAs were transfected using Lipofectamine RNAi max transfection reagent (Thermo Fisher Scientific) during cell seeding. For transient transfection of plasmids, X-tremeGENE 9 DNA Transfection Reagent (Sigma-Aldrich) was used according to the manufacturer’s protocol. All the siRNAs and reagents used in this study are listed in Table S2. Following concentration was used for each drug unless otherwise indicated in the figures: 10 μg/ml cycloheximide; 10 μM MG132; 200 nM Bafilomycin A1 (Baf. A1); 1 mM Deferoxamine (DFO); 1 mM Deferiprone (DFP); 10 μM CCCP (for HEK293T); 20 μM CCCP (for HeLa); 20 μM Hemin; 1 mM succinyl acetone (SA); 1 μg/ml Doxycycline; 20 nM TMRM.
Plasmids
All plasmids construction was performed by PCR amplification (CloneAmp HiFi PCR Premix) using the appropriate primers followed by In-Fusion HD Cloning system (Takara) into the EcoRI site of the pLVX-puro vector (Clontech) or into the BamHI/NotI site of the pRetroX-Tight-puro vector (Clontech). The original vector of the C-terminally HA-tagged Su9-TEV protease was gifted from Dr. Thomas Langer 36, and sub-cloned into the HindIII-XhoI site of pcDNA3.1 (+) 72. The mammalian Tag (−) PINK1 expression vector was gifted from Dr. Noriyuki Matsuda 73. For subcloning of TOM20 (SS)-DELE1 (d101)-HA or TOM20 (SS)-DELE1 (d200)-HA into pLVX-puro vector, the N-terminal 90 bp of human TOMM20 and linker sequence 5’- ggtggatctggaggttctggtgga -3’ were inserted before the DELE1 (d101) or DELE1 (d200)-coding sequence, respectively. For subcloning of PINK1 (MTS)-DELE1 (d101)-HA into pLVX-puro vector, the N-terminal 102 bp of human PINK1 were inserted before the DELE1 (d101)-coding sequence. For replacing the 102–200 region of DELE1 with different length of linkers derived from nucleolin, the nucleotide sequences of human nucleolin coding from V46 to S65 (20 a.a.), to K95 (50 a.a.), or to S145 (100 a.a.) were PCR amplified using the NCL-GFP plasmid 74 as a template, and were inserted into pLVX-puro vector together with the PCR fragments covering DELE1 N-terminal (1–101 a.a.) or C-terminal (201–515 a.a.) regions by In-Fusion assembly. All the plasmids used in this study are listed in key resources table.
Generation of cell lines
The generation of the endogenously HA-tagged DELE1 expressing HEK293T cells was described previously 16. To generate stably transfected cell lines, lentiviruses (for plasmids within pLVX-puro vectors) and retroviruses (for plasmids within pRetroX-Tight-puro vector) were packaged in HEK293T cells. HeLa cells were transduced with viruses with 10 μg/ml polybrene (Sigma) then optimized for protein expression via antibiotics selection. For generating the Tet-on DELE1-HA stable HeLa cell line 75 and the Tet-on Su9-DHFR-3xFlag stable HeLa cell line 72, Retro-X-Tet-on Inducible Expression System (Clontech) was used according to the manufacturer’s instruction. For generating the Tet-on DELE1-HA / Mito-BFP stable HeLa cell line, Tet-on DELE1-HA stable cells were transduced with a previously reported lentiviral Mito-BFP (Addgene plasmid #49151) 76. For generating the ATF4 reporter cell line (ATF4_uORF_mApple-stable HeLa cells), HeLa cells were transduced with a previously reported lentiviral ISR reporter pXG237 containing two ATF4 upstream open reading frames upstream of mApple 17 (Addgene plasmid #141281). A clonal cell line was produced by single-cell sorting into a 96-well plate. For generating the DELE1 KO HEK293T cell lines, the lentiCRISPRv2 system 77–79 was used. Two sgRNAs were designed to target the 5’ and 3’ intron region of Exon2 of the DELE1 gene as shown in Figure S1; 5’ intron region (5’-ggagaccagcagaatcacat-3’) and 3’ intron region (5’-ctcatttcctcccctagtca-3’). After the infection of lentiviruses that express hSpCas9 and DELE1 sgRNAs, infected cells were selected via the treatment with 500 μg/ml puromycin (Sigma) for 24 hours. The selected cells were subjected to single colony isolation in 96-well plates. Genomic DNA of the DELE1 KO clones (#24 and #51) used in this study were extracted with the Genotyping Buffer [100 mM Tris-HCl pH 8.0, 5 mM EDTA, 200 mM NaCl, 0.1% SDS, 0.2 mg/ml proteinase K (Thermo Fisher Scientific)] and PCR amplified using a pair of primers (forward, 5’-gtccaatggcaggagatggt-3’; reverse, 5’-aggctatcaagtaggggcaaag-3’) to confirm the deletion. For generating DELE1 KO and HRI KO MEL cell lines, similar strategies were used with sgRNAs; DELE1 guide #1 (5’-TGAGACCCATCTGAACTGGT-3’), DELE1 guide #2 (5’-CAGGTTCCCACGGTTCAAAG-3’), HRI guide #1 (5’-CTGTCTCACCATCATACTTG-3’), and HRI guide #2 (5’-ATTTAAACACCTGTTTGGAG-3’). All the cell lines used in this study are described in key resources table.
Immunoblotting (IB)
The procedure for IB was previously described 75. Cells were lysed with 1× NuPAGE LDS sample Buffer (Thermo Fisher Scientific) supplemented with 10 mM Dithiothreitol (DTT) (Sigma), and boiled at 98 °C in the bench-top thermal mixer shaking at 1,300 rpm for 15 min. Approximately, 30 – 50 μg of protein per samples were separated on an SDS-PAGE (either 7.5 %, 10 %, or 4–20% gradient) Mini-PROTEAN TGX Precast Gel (Bio-Rad) or Criterion TGX Gel (Bio-Rad) and then transferred to a nitrocellulose membrane (Bio-Rad) or a PVDF membrane (Bio-Rad). The membrane was blocked with Odyssey Blocking Buffer (LI-COR) and incubated with the indicated primary antibodies at 4°C overnight. After washing with PBS-T (PBS + 0.05% Tween-20), the membrane was incubated with HRP-conjugated secondary antibodies (Thermo Fisher Scientific) and washed again with PBS-T. Detection was performed with ECL substrates (Thermo Fisher Scientific) and iBright CL1000 Imaging System (Thermo Fisher Scientific). All the antibodies used in this study are described in key resources table.
Immunocytochemistry (ICC)
ICC was performed as previously described 75. Cells were seeded into Lab-Tek Chambered Coverglass with 4 wells (Thermo Fisher Scientific). Cells were rinsed with PBS and fixed with PBS containing 4% formaldehyde (Thermo Fisher Scientific) for 15 min at room temperature. Cells were permeabilized with PBS containing 0.1% Triton X-100 for 10 min at room temperature. Blocking was performed using PBS containing 2% BSA (2%BSA/PBS) for 30–60 min at room temperature. For immunostaining, cells were incubated with the indicated primary antibodies, diluted in 2%BSA/PBS, overnight at 4C° and with secondary antibodies (Alexa Fluor, Thermo Fisher Scientific), diluted in 2%BSA/PBS, for about 2 hours at room temperature. Cells were imaged using a 63 × /1.4 NA oil immersion objective on Leica SP8 LIGHTNING Confocal Microscope (Leica). All the antibodies used in this study are described in key resources table.
Immunoprecipitation (IP)
For immunoprecipitation, cells were lysed with 1% Triton Buffer [1% Triton-X100, 150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 1 mM EDTA, and protease inhibitors (cOmplete, Sigma)]. After centrifugation, the lysate was incubated with anti-HA Magnetic beads (Pierce) for 20 min at 4C°. After washing the beads with 1% Triton Buffer 4 times, immunoprecipitants were eluted from the beads by boiling for 3 min with 1× NuPAGE LDS sample Buffer (Thermo Fisher Scientific) supplemented with 10 mM Dithiothreitol (DTT) (Sigma) for 3 min. The samples were subjected to the immunoblotting analysis. For DSP cross-linking IP, cells were incubated with 0.1 mM DSP (Thermo Fisher Scientific)-containing ice-cold PBS for 30 min at 4C°. After removing the DSP solution, the cross-linking reaction was stopped by adding 20 mM Tris-HCl (pH 7.4) for 15 min at 4C°. After washing cells with ice-cold PBS for 3 times, cells were lysed with IP lysis buffer [20 mM Tris-HCl pH7.5, 150 mM NaCl, 10 mM EDTA pH8.0, 1% Na-DOC, 1% Triton X-100, and protease inhibitors (cOmplete, Sigma)], and were subjected to immunoprecipitation assay described above.
RNA Isolation and quantitative PCR (qPCR)
Procedures for RNA isolation and subsequent qPCR were described previously 75. All expression levels were normalized to that of RPS18 mRNA. The following qPCR primers were used; RPS18, (forward) 5’-cttccacaggaggcctacac-3’ and (reverse) 5’-cgcaaaatatgctggaacttt-3’; DELE1, (forward) 5’-cccactggaaaggagtgttg-3’ and (reverse) 5’-acccacaggctccctctt-3’; ABCB7, (forward) 5’-ccacacagacccaaaagaag-3’ and (reverse) 5’-caccacccaaaaatcccag-3’; DDIT3, (forward) 5’-TCTTGACCCTGCTTCTCTG-3’ and (reverse) 5’-TCTTCCTCCTCTTCCTCCTG-3’; ASNS, (forward) 5’-GAAGAACACAGATAGCGTGG-3’ and (reverse) 5’-GGCAGAGACAAGTAATAGGAAG-3’; CTH, (forward) 5’-TTCCTGCCACACTTCCAAC-3’ and (reverse) 5’-TGCTGCCACTGCTTTTTC-3’; CHAC1, (forward) 5’-GAGGGATAGTAGGGCATGAG-3’ and (reverse) 5’-CAGACAGACAGAGGGACAAG-3’.
Flow cytometry
To determine mitochondrial membrane potential, 20 nM TMRM (Thermo Fisher Scientific) was directly added to cell culture media and incubated for 15 min. Cells were washed and replaced with normal medium followed by flow cytometry analysis using Attune N×T Acoustic Focusing Cytometer (Thermo Fisher Scientific). To determine cell viability, Propidium iodide (Thermo Fisher Scientific) was added to cell culture media and incubated for 15 min, followed by flow cytometry analysis. For the flow cytometry analysis of the ATF4 reporter cell line (ATF4_uORF_mApple-stable HeLa cells), mApple was measured on an Amnis CellStream flow cytometer (Luminex), using a 561 nm laser for excitation and a 611/31 bandwidth filter for emission. Data was analyzed from the fcs files using a custom Python 3 script and the FlowCytometryTools library.
Crude mitochondrial isolation and protease protection assay
HEK293T were treated with 10 μg/mL cycloheximide (CHX, Sigma-Aldrich, C4859) for 30 min after pre-treatment with 1 mM deferoxamine (DFO, Cayman Chemicals, 14595) or H2O for 16 hours. HEK293T cells were scraped from cell culture plates and resuspended in cold homogenization buffer (220 mM mannitol, 70 mM sucrose, 20 mM HEPES/KOH pH 7.4, 1 mM EDTA). Mitochondria were isolated using a syringe (25G × 5/8””/ ø 0.50 × 16 mm, B. Braun, 4657853) and differential centrifugation: to remove nuclei and intact cells, two centrifugation steps at 700 × g for 10 min and 5 min at 4 °C were performed. The supernatant was then centrifuged at 8000 × g for 15 min at 4 °C. The mitochondrial pellet was resuspended in SEM Buffer and protein concentration was determined by Bradford Assay. For proteinase K digestion, 50 μg mitochondria per reaction were used. After an additional centrifugation step at 8000 × g for 10 min at 4 °C, the mitochondria were resuspended in SEM Buffer (10 mM MOPS-Tris pH 7.2, 250 mM sucrose, 1 mM EDTA), EM buffer (10 mM MOPS-Tris pH 7.2, 1 mM EDTA), or SEM buffer + 0.5% TritonX-100 and incubated on ice for 10 min. For digestion, Proteinase K (50 μg/mL; Sigma-Aldrich, P6556) was added and incubated for 10 min on ice. To stop the reaction, 1 mM final concentration phenylmethansulfonylfluorid (PMSF, Sigma-Aldrich, P7626) was added. After 15 min final incubation, SDS sample buffer was added, and samples were boiled at 95 °C for 10 min.
Proximity Ligation Assay (PLA)
PLA was performed by using Duolink In Situ PLA Probe Starter Kit (DUO92101, Sigma) according to the manufacturer’s instruction and the previously described method 80. For PLA, cells were grown on cover glasses (CAROLINA, 633029). Cells were fixed, permeabilized, and blocked with 0.2% BSA in PBS for 1 hour at room temperature (RT), and then incubated with the following primary antibodies overnight at 4C°; anti-HRI rabbit antibody (Proteintech, 20499–1-AP) and anti-HA mouse antibody (Cell Signaling, 2367). After washing with PBS, samples were incubated with PLA probes (DUO92101, Sigma) for 1 hour at 37C°. After washing with buffer A [0.01 M Tris-HCl (pH 7.4), 0.15 M NaCl, 0.05% Tween 20], samples were incubated with ligation mix (DUO92101, Sigma) for 30 min at 37C°. After washing with buffer A, samples were incubated with polymerase mix (DUO92101, Sigma) for 100 min at 37C°. After sequential washing by buffer B [0.2 M Tris-HCl (pH 7.4), 0.1 M NaCl] and buffer C (10 times dilution of buffer B with water), cover glasses were mounted with ProLong Gold Antifade Mountant (Invitrogen, P36934) onto a glass microscopic slide (VWR, 48311–703). Cells were imaged using a 63 × /1.4 NA oil immersion objective on Leica SP8 LIGHTNING Confocal Microscope (Leica).
RNA-sequencing
Total RNAs were isolated using RNeasy Mini Kit (Qiagen) and reverse-transcribed to cDNA using iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s instruction. RNA sequencing and the data analysis were performed through the commercial service provided by MedGenome, Inc (Foster City, USA). Illumina TruSeq stranded mRNA kit was used to prepare libraries according to the manufacturer’s instructions. The resulting libraries were sequenced on NovaSeq 6000 using paired-end 100 base pair sequencing at the depth of 40M reads per sample.
Quantification and Statistical Analysis
Statistical significances were determined using Prism software (GraphPad Software, Inc.) as indicated in the Figure legends.
Supplementary Material
Supplemental Table 1: RNA seq results of MEL cells, related to Figure 7.
Supplemental Figure S1: Generation of DELE1 knockout (KO) HEK293T cells, related to Figure 2.
Supplemental Figure S2: Determination of DELE1 localization at mitochondria, related to Figure 3.
Supplemental Figure S3: Iron deficiency-induced ISR activation requires DELE1, related to Figure 4.
Supplemental Figure S4: DELE1 interacts with and activates HRI on mitochondrial surface upon iron deficiency, related to Figure 5.
Supplemental Figure S5: Determination of critical domains in DELE1 for iron deficiency-induced ISR activation, related to Figure 6.
Supplemental Figure S6: ISCU deletion attenuates iron deficiency-induced ISR activation, related to Figure 6.
Supplemental Figure S7: Generation of DELE1 or HRI KO MEL cells, related to Figure 7.
Highlights.
Under steady state, DELE1 is degraded by matrix-resident LONP1 soon after import.
DELE1 import is arrested in iron deficiency, which stabilizes it on mitochondria.
The stabilized DELE1 full-length form activates HRI at the mitochondrial surface.
DELE1-HRI-ISR protects erythroid cells from iron deficiency-induced cell death.
Acknowledgements
We thank Dr. Toren Finkel for critical reading of our manuscript, and Dr. Richard J Youle for sharing reagents. We thank Dr. Mo Chen for technical advice on PLA and Dr. Bill Chen for sharing reagents. This work was supported by University of Pittsburgh, Aging Institute Seed (S.S. and Y.S.) and a grant from NIH, NIDDK (R01DK134341, S.S. and Y.S.), as well as the NINDS intramural program (D.P.N.) and grants from the European Research Council (ERC-STG 804182 SOLID, L.T.J.) and German Research Foundation (DFG project JA 2873/2, L.T.J.).
Footnotes
Declaration of interests
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368. 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, and Gorman AM (2016). The integrated stress response. EMBO Rep 17, 1374–1395. 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen JJ, and Zhang S (2019). Heme-regulated eIF2alpha kinase in erythropoiesis and hemoglobinopathies. Blood 134, 1697–1707. 10.1182/blood.2019001915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruns GP, and London IM (1965). The Effect of Hemin on the Synthesis of Globin. Biochem Biophys Res Commun 18, 236–242. 10.1016/0006-291x(65)90746-1. [DOI] [PubMed] [Google Scholar]
- 5.Ranu RS, Levin DH, Delaunay J, Ernst V, and London IM (1976). Regulation of protein synthesis in rabbit reticulocyte lysates: characteristics of inhibition of protein synthesis by a translational inhibitor from heme-deficient lysates and its relationship to the initiation factor which binds Met-tRNAf. Proc Natl Acad Sci U S A 73, 2720–2724. 10.1073/pnas.73.8.2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ranu RS, and London IM (1976). Regulation of protein synthesis in rabbit reticulocyte lysates: purification and initial characterization of the cyclic 3’:5’-AMP independent protein kinase of the heme-regulated translational inhibitor. Proc Natl Acad Sci U S A 73, 4349–4353. 10.1073/pnas.73.12.4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trachsel H, Ranu RS, and London IM (1978). Regulation of protein synthesis in rabbit reticulocyte lysates: purification and characterization of heme-reversible translational inhibitor. Proc Natl Acad Sci U S A 75, 3654–3658. 10.1073/pnas.75.8.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fagard R, and London IM (1981). Relationship between phosphorylation and activity of heme-regulated eukaryotic initiation factor 2 alpha kinase. Proc Natl Acad Sci U S A 78, 866–870. 10.1073/pnas.78.2.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rafie-Kolpin M, Chefalo PJ, Hussain Z, Hahn J, Uma S, Matts RL, and Chen JJ (2000). Two heme-binding domains of heme-regulated eukaryotic initiation factor-2alpha kinase. N terminus and kinase insertion. J Biol Chem 275, 5171–5178. 10.1074/jbc.275.7.5171. [DOI] [PubMed] [Google Scholar]
- 10.Igarashi J, Murase M, Iizuka A, Pichierri F, Martinkova M, and Shimizu T (2008). Elucidation of the heme binding site of heme-regulated eukaryotic initiation factor 2alpha kinase and the role of the regulatory motif in heme sensing by spectroscopic and catalytic studies of mutant proteins. J Biol Chem 283, 18782–18791. 10.1074/jbc.M801400200. [DOI] [PubMed] [Google Scholar]
- 11.Miksanova M, Igarashi J, Minami M, Sagami I, Yamauchi S, Kurokawa H, and Shimizu T (2006). Characterization of heme-regulated eIF2alpha kinase: roles of the N-terminal domain in the oligomeric state, heme binding, catalysis, and inhibition. Biochemistry 45, 9894–9905. 10.1021/bi060556k. [DOI] [PubMed] [Google Scholar]
- 12.Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, Fleming M, Leboulch P, Orkin SH, and Chen JJ (2001). Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J 20, 6909–6918. 10.1093/emboj/20.23.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taniuchi S, Miyake M, Tsugawa K, Oyadomari M, and Oyadomari S (2016). Integrated stress response of vertebrates is regulated by four eIF2alpha kinases. Sci Rep 6, 32886. 10.1038/srep32886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quiros PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, and Auwerx J (2017). Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216, 2027–2045. 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munch C, and Harper JW (2016). Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 534, 710–713. 10.1038/nature18302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fessler E, Eckl EM, Schmitt S, Mancilla IA, Meyer-Bender MF, Hanf M, Philippou-Massier J, Krebs S, Zischka H, and Jae LT (2020). A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 579, 433–437. 10.1038/s41586-020-2076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA, and Kampmann M (2020). Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 579, 427–432. 10.1038/s41586-020-2078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahola S, Rivera Mejias P, Hermans S, Chandragiri S, Giavalisco P, Nolte H, and Langer T (2022). OMA1-mediated integrated stress response protects against ferroptosis in mitochondrial cardiomyopathy. Cell Metab 34, 1875–1891 e1877. 10.1016/j.cmet.2022.08.017. [DOI] [PubMed] [Google Scholar]
- 19.Shammas MK, Huang X, Wu BP, Fessler E, Song IY, Randolph NP, Li Y, Bleck CK, Springer DA, Fratter C, et al. (2022). OMA1 mediates local and global stress responses against protein misfolding in CHCHD10 mitochondrial myopathy. J Clin Invest 132. 10.1172/JCI157504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huynh H, Zhu S, Lee S, Bao Y, Pang J, Nguyen A, Gu Y, Chen C, Ouyang K, Evans SM, and Fang X (2022). DELE1 is protective for mitochondrial cardiomyopathy. J Mol Cell Cardiol 175, 44–48. 10.1016/j.yjmcc.2022.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu S, Nguyen A, Pang J, Zhao J, Chen Z, Liang Z, Gu Y, Huynh H, Bao Y, Lee S, et al. (2022). Mitochondrial Stress Induces an HRI-eIF2alpha Pathway Protective for Cardiomyopathy. Circulation 146, 1028–1031. 10.1161/CIRCULATIONAHA.122.059594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sekine S, and Youle RJ (2018). PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol 16, 2. 10.1186/s12915-017-0470-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, and Youle RJ (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8, e1000298. 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189, 211–221. 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson NS, and Haynes CM (2020). Folding the Mitochondrial UPR into the Integrated Stress Response. Trends Cell Biol 30, 428–439. 10.1016/j.tcb.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harada T, Iwai A, and Miyazaki T (2010). Identification of DELE, a novel DAP3-binding protein which is crucial for death receptor-mediated apoptosis induction. Apoptosis 15, 1247–1255. 10.1007/s10495-010-0519-3. [DOI] [PubMed] [Google Scholar]
- 27.Fessler E, Krumwiede L, and Jae LT (2022). DELE1 tracks perturbed protein import and processing in human mitochondria. Nat Commun 13, 1853. 10.1038/s41467-022-29479-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ueno T, and Komatsu M (2020). Monitoring Autophagy Flux and Activity: Principles and Applications. Bioessays 42, e2000122. 10.1002/bies.202000122. [DOI] [PubMed] [Google Scholar]
- 29.Deshwal S, Fiedler KU, and Langer T (2020). Mitochondrial Proteases: Multifaceted Regulators of Mitochondrial Plasticity. Annu Rev Biochem 89, 501–528. 10.1146/annurev-biochem-062917-012739. [DOI] [PubMed] [Google Scholar]
- 30.Szczepanowska K, and Trifunovic A (2021). Mitochondrial matrix proteases: quality control and beyond. FEBS J. 10.1111/febs.15964. [DOI] [PubMed] [Google Scholar]
- 31.Fujimaki M, Furuya N, Saiki S, Amo T, Imamichi Y, and Hattori N (2019). Iron Supply via NCOA4-Mediated Ferritin Degradation Maintains Mitochondrial Functions. Mol Cell Biol 39. 10.1128/MCB.00010-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hara Y, Yanatori I, Tanaka A, Kishi F, Lemasters JJ, Nishina S, Sasaki K, and Hino K (2020). Iron loss triggers mitophagy through induction of mitochondrial ferritin. EMBO Rep 21, e50202. 10.15252/embr.202050202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hentze MW, Muckenthaler MU, Galy B, and Camaschella C (2010). Two to tango: regulation of Mammalian iron metabolism. Cell 142, 24–38. 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 34.Fukasawa Y, Tsuji J, Fu SC, Tomii K, Horton P, and Imai K (2015). MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol Cell Proteomics 14, 1113–1126. 10.1074/mcp.M114.043083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maio N, Ghezzi D, Verrigni D, Rizza T, Bertini E, Martinelli D, Zeviani M, Singh A, Carrozzo R, and Rouault TA (2016). Disease-Causing SDHAF1 Mutations Impair Transfer of Fe-S Clusters to SDHB. Cell Metab 23, 292–302. 10.1016/j.cmet.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, and Langer T (2014). Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J 33, 578–593. 10.1002/embj.201386474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lazarou M, Jin SM, Kane LA, and Youle RJ (2012). Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22, 320–333. 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, Tanaka K, and Matsuda N (2013). A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem 288, 36372–36384. 10.1074/jbc.M113.509653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akabane S, Watanabe K, Kosako H, Yamashita SI, Nishino K, Kato M, Sekine S, Kanki T, Matsuda N, Endo T, and Oka T (2023). TIM23 facilitates PINK1 activation by safeguarding against OMA1-mediated degradation in damaged mitochondria. Cell Rep, 112454. 10.1016/j.celrep.2023.112454. [DOI] [PubMed] [Google Scholar]
- 40.Sekine S (2020). PINK1 import regulation at a crossroad of mitochondrial fate: the molecular mechanisms of PINK1 import. J Biochem 167, 217–224. 10.1093/jb/mvz069. [DOI] [PubMed] [Google Scholar]
- 41.Wiedemann N, and Pfanner N (2017). Mitochondrial Machineries for Protein Import and Assembly. Annu Rev Biochem 86, 685–714. 10.1146/annurev-biochem-060815-014352. [DOI] [PubMed] [Google Scholar]
- 42.Sekine Y, Zyryanova A, Crespillo-Casado A, Fischer PM, Harding HP, and Ron D (2015). Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 348, 1027–1030. 10.1126/science.aaa6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, et al. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2, e00498. 10.7554/eLife.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Igarashi K, and Sun J (2006). The heme-Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid Redox Signal 8, 107–118. 10.1089/ars.2006.8.107. [DOI] [PubMed] [Google Scholar]
- 45.Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, and Langer T (2009). Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol 187, 1023–1036. 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Head B, Griparic L, Amiri M, Gandre-Babbe S, and van der Bliek AM (2009). Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol 187, 959–966. 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang K, Li H, and Song Z (2014). Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep 15, 576–585. 10.1002/embr.201338240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wai T, Saita S, Nolte H, Muller S, Konig T, Richter-Dennerlein R, Sprenger HG, Madrenas J, Muhlmeister M, Brandt U, et al. (2016). The membrane scaffold SLP2 anchors a proteolytic hub in mitochondria containing PARL and the i-AAA protease YME1L. EMBO Rep 17, 1844–1856. 10.15252/embr.201642698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sekine S, Kanamaru Y, Koike M, Nishihara A, Okada M, Kinoshita H, Kamiyama M, Maruyama J, Uchiyama Y, Ishihara N, et al. (2012). Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. J Biol Chem 287, 34635–34645. 10.1074/jbc.M112.357509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emmott E, and Hiscox JA (2009). Nucleolar targeting: the hub of the matter. EMBO Rep 10, 231–238. 10.1038/embor.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shin CS, Meng S, Garbis SD, Moradian A, Taylor RW, Sweredoski MJ, Lomenick B, and Chan DC (2021). LONP1 and mtHSP70 cooperate to promote mitochondrial protein folding. Nat Commun 12, 265. 10.1038/s41467-020-20597-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsushima Y, Takahashi K, Yue S, Fujiyoshi Y, Yoshioka H, Aihara M, Setoyama D, Uchiumi T, Fukuchi S, and Kang D (2021). Mitochondrial Lon protease is a gatekeeper for proteins newly imported into the matrix. Commun Biol 4, 974. 10.1038/s42003-021-02498-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, and Haynes CM (2016). The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr Biol 26, 2037–2043. 10.1016/j.cub.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eckl EM, Ziegemann O, Krumwiede L, Fessler E, and Jae LT (2021). Sensing, signaling and surviving mitochondrial stress. Cell Mol Life Sci 78, 5925–5951. 10.1007/s00018-021-03887-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, and Haynes CM (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590. 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haynes CM, Yang Y, Blais SP, Neubert TA, and Ron D (2010). The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell 37, 529–540. 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schaedler TA, Faust B, Shintre CA, Carpenter EP, Srinivasan V, van Veen HW, and Balk J (2015). Structures and functions of mitochondrial ABC transporters. Biochem Soc Trans 43, 943–951. 10.1042/BST20150118. [DOI] [PubMed] [Google Scholar]
- 58.Liesa M, Qiu W, and Shirihai OS (2012). Mitochondrial ABC transporters function: the role of ABCB10 (ABC-me) as a novel player in cellular handling of reactive oxygen species. Biochim Biophys Acta 1823, 1945–1957. 10.1016/j.bbamcr.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ichikawa Y, Bayeva M, Ghanefar M, Potini V, Sun L, Mutharasan RK, Wu R, Khechaduri A, Jairaj Naik T, and Ardehali H (2012). Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc Natl Acad Sci U S A 109, 4152–4157. 10.1073/pnas.1119338109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pondarre C, Antiochos BB, Campagna DR, Clarke SL, Greer EL, Deck KM, McDonald A, Han AP, Medlock A, Kutok JL, et al. (2006). The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis. Hum Mol Genet 15, 953–964. 10.1093/hmg/ddl012. [DOI] [PubMed] [Google Scholar]
- 61.Pondarre C, Campagna DR, Antiochos B, Sikorski L, Mulhern H, and Fleming MD (2007). Abcb7, the gene responsible for X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis. Blood 109, 3567–3569. 10.1182/blood-2006-04-015768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hyde BB, Liesa M, Elorza AA, Qiu W, Haigh SE, Richey L, Mikkola HK, Schlaeger TM, and Shirihai OS (2012). The mitochondrial transporter ABC-me (ABCB10), a downstream target of GATA-1, is essential for erythropoiesis in vivo. Cell Death Differ 19, 1117–1126. 10.1038/cdd.2011.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamamoto M, Arimura H, Fukushige T, Minami K, Nishizawa Y, Tanimoto A, Kanekura T, Nakagawa M, Akiyama S, and Furukawa T (2014). Abcb10 role in heme biosynthesis in vivo: Abcb10 knockout in mice causes anemia with protoporphyrin IX and iron accumulation. Mol Cell Biol 34, 1077–1084. 10.1128/MCB.00865-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shimada Y, Okuno S, Kawai A, Shinomiya H, Saito A, Suzuki M, Omori Y, Nishino N, Kanemoto N, Fujiwara T, et al. (1998). Cloning and chromosomal mapping of a novel ABC transporter gene (hABC7), a candidate for X-linked sideroblastic anemia with spinocerebellar ataxia. J Hum Genet 43, 115–122. 10.1007/s100380050051. [DOI] [PubMed] [Google Scholar]
- 65.Maio N, Kim KS, Holmes-Hampton G, Singh A, and Rouault TA (2019). Dimeric ferrochelatase bridges ABCB7 and ABCB10 homodimers in an architecturally defined molecular complex required for heme biosynthesis. Haematologica 104, 1756–1767. 10.3324/haematol.2018.214320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lill R, and Freibert SA (2020). Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu Rev Biochem 89, 471–499. 10.1146/annurev-biochem-013118-111540. [DOI] [PubMed] [Google Scholar]
- 67.Tong WH, and Rouault TA (2006). Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab 3, 199–210. 10.1016/j.cmet.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 68.Ganz T (2013). Systemic iron homeostasis. Physiol Rev 93, 1721–1741. 10.1152/physrev.00008.2013. [DOI] [PubMed] [Google Scholar]
- 69.Crooks DR, Ghosh MC, Haller RG, Tong WH, and Rouault TA (2010). Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood 115, 860–869. 10.1182/blood-2009-09-243105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, and Haynes CM (2014). Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 516, 414–417. 10.1038/nature13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cavadini P, Biasiotto G, Poli M, Levi S, Verardi R, Zanella I, Derosas M, Ingrassia R, Corrado M, and Arosio P (2007). RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood 109, 3552–3559. 10.1182/blood-2006-08-041632. [DOI] [PubMed] [Google Scholar]
- 72.Sekine S, Wang C, Sideris DP, Bunker E, Zhang Z, and Youle RJ (2019). Reciprocal Roles of Tom7 and OMA1 during Mitochondrial Import and Activation of PINK1. Mol Cell 73, 1028–1043 e1025. 10.1016/j.molcel.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 73.Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M, et al. (2012). PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 3, 1016. 10.1038/ncomms2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Houston R, Sekine S, Calderon MJ, Seifuddin F, Wang G, Kawagishi H, Malide DA, Li Y, Gucek M, Pirooznia M, et al. (2020). Acetylation-mediated remodeling of the nucleolus regulates cellular acetyl-CoA responses. PLoS Biol 18, e3000981. 10.1371/journal.pbio.3000981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Houston R, Sekine Y, Larsen MB, Murakami K, Mullett SJ, Wendell SG, Narendra DP, Chen BB, and Sekine S (2021). Discovery of bactericides as an acute mitochondrial membrane damage inducer. Mol Biol Cell 32, ar32. 10.1091/mbc.E21-04-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, and Voeltz GK (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stringer BW, Day BW, D’Souza RCJ, Jamieson PR, Ensbey KS, Bruce ZC, Lim YC, Goasdoue K, Offenhauser C, Akgul S, et al. (2019). A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci Rep 9, 4902. 10.1038/s41598-019-41277-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen M, Horn HT, Wen T, Cryns VL, and Anderson RA (2021). Assessing In Situ Phosphoinositide-Protein Interactions Through Fluorescence Proximity Ligation Assay in Cultured Cells. Methods Mol Biol 2251, 133–142. 10.1007/978-1-0716-1142-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: RNA seq results of MEL cells, related to Figure 7.
Supplemental Figure S1: Generation of DELE1 knockout (KO) HEK293T cells, related to Figure 2.
Supplemental Figure S2: Determination of DELE1 localization at mitochondria, related to Figure 3.
Supplemental Figure S3: Iron deficiency-induced ISR activation requires DELE1, related to Figure 4.
Supplemental Figure S4: DELE1 interacts with and activates HRI on mitochondrial surface upon iron deficiency, related to Figure 5.
Supplemental Figure S5: Determination of critical domains in DELE1 for iron deficiency-induced ISR activation, related to Figure 6.
Supplemental Figure S6: ISCU deletion attenuates iron deficiency-induced ISR activation, related to Figure 6.
Supplemental Figure S7: Generation of DELE1 or HRI KO MEL cells, related to Figure 7.
Data Availability Statement
All RNA seq data have been deposited at SRA under the accession number PRJNA970328. Raw images were deposited to Mendeley Data (DOI: 10.17632/7p8w52928v.1). All data are publicly available as of the date of publication. Accession numbers and DOI are listed in the key resources table.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HA | Cell Signaling | 2367S; RRID:AB_10691311 |
| TOM40 | Proteintech | 18409-1-AP; RRID:AB_2303725 |
| TIM50 | Santa Cruz | sc-393678; RRID:AB_2714191 |
| TIM23 | BD Biosciences | 611222; RRID:AB_398754 |
| HSP90 | Santa Cruz | sc-7947; RRID:AB_2121235 |
| HSP90 | Cell Signaling | 4874S; RRID:AB_2121214 |
| PINK1 | Cell Signaling | 6946S; RRID:AB_11179069 |
| p62 | Abcam | ab56416; RRID:AB_945626 |
| LONP1 | Proteintech | 66043-1-lg; RRID:AB_2881494 |
| LONP1 | Novus Biologicals | NBP1-81734; RRID:AB_11002818 |
| αTubulin | Sigma | T6199; RRID:AB_477583 |
| IRP2 | Cell Signaling | 37135S; RRID:AB_2799110 |
| ALAS1 | Abcam | ab154860 |
| HMOX1 | GeneTex | GTX101147; RRID:AB_1950502 |
| OMA1 | Santa Cruz | sc-515788; RRID:AB_2905488 |
| TOM20 | Santa Cruz | sc-17764; RRID:AB_628381 |
| MPPα | Sigma | HPA021648; RRID:AB_1855491 |
| MPPβ | Proteintech | 16064-1-AP; RRID:AB_2167122 |
| M2 | Sigma | F1804; RRID: AB_262044 |
| ATF4 | Cell Signaling | 11815S; RRID:AB_2616025 |
| phospho-eif2α | Cell Signaling | 3398; RRID:AB_2096481 |
| total-eif2α | Cell Signaling | 5324; RRID:AB_10692650 |
| LIAS | Proteintech | 11577-1-AP; RRID:AB_2135972 |
| SDHB | Proteintech | 10620-1-AP; RRID:AB_2285522 |
| PPAT | Proteintech | 15401-1-AP; RRID:AB_2166532 |
| FTMT | Abcam | ab66111 |
| OPA1 | BD Biosciences | 612607; RRID:AB_399889 |
| PGAM5 | Thermo Fisher Scientific | PA5-57894; RRID:AB_2645430 |
| HRI | Proteintech | 20499-1-AP; RRID:AB_10697665 |
| ISCU | Proteintech | 14812-1-AP; RRID:AB_2280362 |
| NCL | Cell Signaling | 14574; RRIS:AB_2798519 |
| TRAP1 | Cell Signaling | 92345: RRID:AB_2800183 |
| Bacterial and virus strains | ||
| N/A | ||
| Biological samples | ||
| N/A | ||
| Chemicals, peptides, and recombinant proteins | ||
| Cycloheximide (CHX) | Cayman Chemical | 14126 |
| MG132 | Sigma | C2211 |
| Bafilomycin A1 (Baf. A1) | Cayman Chemical | 11038 |
| Deferoxamine (mesylate) (DFO) | Cayman Chemical | 14595 |
| Deferiprone (DFP) | Cayman Chemical | 20387 |
| CCCP | Sigma | C2759 |
| Hemin chloride | Cayman Chemical | 16487 |
| Succinyl acetone (SA) | MedChemExpress | HY-W010184 |
| Proteinase K | Sigma | P6556 |
| Ferric ammonium citrate (FAC) | Sigma | RES20400-A7 |
| Erastin | Cayman Chemical | 17754 |
| trans-ISRIB | Cayman Chemical | 16258 |
| Doxycycline | Sigma | D9891 |
| DSP (dithiobis(succinimidyl propionate)) | Thermo Fisher Scientific | 22585 |
| X-tremeGENE 9 DNA transfection reagent | Roche | 6365787001 |
| Lipofectamine RNAi max transfection reagent | Thermo Fisher Scientific | 13778075 |
| Tetramethylrhodamine, Methyl Ester, Perchlorate (TMRM) | Thermo Fisher Scientific | T668 |
| Critical commercial assays | ||
| Duolink In Situ PLA Probe Starter Kit | Sigma | DUO92101 |
| Deposited data | ||
| Raw images | This study | Mendeley Data (DOI: 10.17632/7p8w52928v.1) |
| RNA seq data | This study | SRA (accession number: PRJNA970328) |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-3216 |
| HeLa | ATCC | CCL-2.2 |
| Endogenous DELE1-HA HEK293T | Fessler et al., 2020 | N/A |
| Tet-on DELE1-HA stable HeLa | Houston et al., 2021 | N/A |
| Tet-on Su9-DHFR-3xFlag stable HeLa | Sekine et al., 2019 | N/A |
| DELE1 KO HEK293T clone #24 | This study | N/A |
| DELE1 KO HEK293T clone #51 | This study | N/A |
| ATF4_uORF_mApple-stable HeLa | This study | N/A |
| Tet-on DELE1-HA / Mito-BFP stable HeLa | This study | N/A |
| MEL | Millipore Sigma | F4N |
| DELE1 KO MEL clone #3 | This study | N/A |
| DELE1 KO MEL clone #8 | This study | N/A |
| HRI KO MEL clone #1 | This study | N/A |
| Experimental models: Organisms/strains | ||
| N/A | ||
| Oligonucleotides | ||
| Stealth RNAi Negative Control Med GC Duplex #2 | Thermo Fisher Scientific | 12935112 |
| Silencer Select Negative Control #1 siRNA | Thermo Fisher Scientific | 4390843 |
| Stealth RNAi PARL | Thermo Fisher Scientific | HSS183080 |
| Stealth RNAi OMA1 | Thermo Fisher Scientific | HSS132976 |
| Stealth RNAi YME1L | Thermo Fisher Scientific | HSS116546 |
| Stealth RNAi HtrA2 | Thermo Fisher Scientific | HSS146986 |
| Stealth RNAi LONP | Thermo Fisher Scientific | HSS113887 |
| Silencer Select LONP #1 | Thermo Fisher Scientific | s17902 |
| Silencer Select LONP #2 | Thermo Fisher Scientific | s17903 |
| Stealth RNAi CLPP | Thermo Fisher Scientific | HSS112033 |
| Stealth RNAi AFG3L2 | Thermo Fisher Scientific | HSS116886 |
| Stealth RNAi SPG7 | Thermo Fisher Scientific | HSS110137 |
| Stealth RNAi MPPa | Thermo Fisher Scientific | HSS118357 |
| Stealth RNAi MPPb | Thermo Fisher Scientific | HSS114169 |
| Silencer Select HRI #1 | Thermo Fisher Scientific | s25822 |
| Silencer Select HRI #2 | Thermo Fisher Scientific | s25823 |
| Silencer Select DELE1 #1 | Thermo Fisher Scientific | s18966 |
| Silencer Select DELE1 #2 | Thermo Fisher Scientific | s18967 |
| Silencer Select ABCB7 #1 | Thermo Fisher Scientific | s855 |
| Silencer Select ABCB7 #2 | Thermo Fisher Scientific | s857 |
| Silencer Select ABCB8 | Thermo Fisher Scientific | s22104 |
| Silencer Select ABCB10 | Thermo Fisher Scientific | s23857 |
| Silencer Select ISCU #1 | Thermo Fisher Scientific | s23909 |
| Silencer Select ISCU #2 | Thermo Fisher Scientific | s23908 |
| Recombinant DNA | ||
| pLVX-puro-DELE1-HA | Houston et al., 2021 | N/A |
| pLVX-puro-DELE1 (TEV30)-HA | This study | N/A |
| pcDNA3.1 (+) Su9-TEV protease-HA | Sekine et al., 2019 | N/A |
| pRetroX-Tight-Pur-DELE1-HA | Houston et al., 2021 | N/A |
| pRetroX-Tight-Pur-DELE1(d30) -HA | This study | N/A |
| pRetroX-Tight-Pur-DELE1(d101)-HA | This study | N/A |
| pRetroX-Tight-Pur-pSu9-DHFR-tev-3xFlag | Sekine et al., 2019 | N/A |
| pCMV (d1) PINK1 (Tag-) | Okatsu et al., 2012 | N/A |
| pLVX-puro-PINK1-HA | This study | N/A |
| lentiCRISPR v2-hygro-human DELE1 (Guide#1) | This study | N/A |
| lentiCRISPR v2-hygro-human DELE1 (Guide#2) | This study | N/A |
| pXG237 | Guo et al., 2020 | Addgene #141281 |
| pLVX-puro-DELE1(d246-272)-HA | This study | N/A |
| pLVX-puro-TOMM20(ss)-DELE1(d101)-HA | This study | N/A |
| pLVX-puro-TOMM20(ss)-DELE1(d200)-HA | This study | N/A |
| pLVX-puro-DELE1(d102-200)-HA | This study | N/A |
| pLVX-puro-DELE1(d200-220)-HA | This study | N/A |
| pLVX-puro-DELE1(d101)-HA | This study | N/A |
| pLVX-puro-DELE1(d101/d102-200)-HA | This study | N/A |
| pLVX-puro-DELE1(d101/dTPR1)-HA | This study | N/A |
| pLVX-puro-DELE1(d101/dTPR2)-HA | This study | N/A |
| pLVX-puro-DELE1(d101/dTPR3)-HA | This study | N/A |
| pLVX-puro-DELE1(d101/dTPR4)-HA | This study | N/A |
| pLVX-puro-DELE1(d101/dTPR5)-HA | This study | N/A |
| pLVX-puro-DELE1(d102-150)-HA | This study | N/A |
| pLVX-puro-DELE1(d150-200)-HA | This study | N/A |
| pLVX-puro-DELE1(d102-200 + linker 20)-HA | This study | N/A |
| pLVX-puro-DELE1(d102-200 + linker 50)-HA | This study | N/A |
| pLVX-puro-DELE1(d102-200 + linker 100)-HA | This study | N/A |
| lentiCRISPR v2-puro-mouse DELE1(Guide#1) | This study | N/A |
| lentiCRISPR v2-puro-mouse DELE1 (Guide#2) | This study | N/A |
| lentiCRISPR v2-puro-mouse HRI (Guide#1) | This study | N/A |
| lentiCRISPR v2-puro-mouse HRI (Guide#2) | This study | N/A |
| Mito-BFP | Friedman et al., 2011 | Addgene #49151 |
| pLVX-puro-DELE1(d80-106)-HA | This study | N/A |
| pLVX-puro-PINK1(MTS)-DELE1(d101)-HA | This study | N/A |
| pLVX-puro-DELE1(dTPR1-7)-HA | This study | N/A |
| pLVX-puro | Clontech | 632164 |
| pRetroX-Tight-Pur | Clontech | 632104 |
| pRetroX-Tet-On Advanced | Clontech | 632104 |
| lentiCRISPR v2-puro | Sanjana et al., 2014 | Addgene #52961 |
| lentiCRISPR v2-hygro | Stringer et al., 2019 | Addgene #98291 |
| Software and algorithms | ||
| BioRender | N/A | https://app.biorender.com/user/signin |
| Other | ||
| N/A | ||
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
HEK293T and HeLa cells were cultured in DMEM (Gibco) supplemented with 10% FBS (VWR Life Science), 10 mM HEPES (Gibco), 1 mM Sodium pyruvate (Gibco), non-essential amino acids (Gibco) and GlutaMAX (Gibco). MEL cells (F4N, Sigma) were cultured in RPMI Medium 1640 with L-Glutamine and 25 mM HEPES (Gibco) supplemented with 10% FBS and 1 x Penicillin Streptomycin Solution (Corning). MEL cell differentiation was induced by using this RPMI medium with 2% Dimethyl sulfoxide (DMSO) (Santa Cruz).