

Abstract
The mechanisms by which FOXP3+ T follicular regulatory (Tfr) cells simultaneously steer antibody formation toward microbe or vaccine recognition and away from self-reactivity remains incompletely understood. To explore underappreciated heterogeneity in human Tfr cell development, function, and localization, we used paired TCRVA/TCRVB sequencing to distinguish tonsillar Tfr cells that are clonally related to natural Tregs (nTfr) from those likely induced from Tfh cells (iTfr). The proteins iTfr and nTfr cells differentially expressed were utilized to pinpoint their in situ locations via multi-plex microscopy and establish their divergent functional roles. In silico analyses and in vitro tonsil organoid tracking models corroborated the existence of separate Treg-to-nTfr and Tfh-to-iTfr developmental trajectories. Our results identify human iTfr cells as a distinct CD38+, GC-resident, Tfh-descended subset that gains suppressive function while retaining the capacity to help B cells, whereas CD38− nTfr cells are elite suppressors primarily localized in follicular mantles. Interventions differentially targeting specific Tfr cell subsets may provide therapeutic opportunities to boost immunity or more precisely treat autoimmune diseases.
One sentence summary:
The origin of human tonsillar T follicular regulatory cells, from Treg or Tfh lineages, predicts TCR repertoire, location, and function.
INTRODUCTION
Germinal centers (GCs) are formed within secondary lymphoid tissues to orchestrate dynamic interactions between T follicular helper (Tfh) cells and GC B cells (1). GCs are remotely controlled by negative feedback from their primary product, systemically circulating affinity-maturated antibodies (2), and locally governed by FOXP3+ T follicular regulatory (Tfr) cells (3). Various Tfr cell-deficient mouse models demonstrate increased production of autoantibodies (4–6) and diminished vaccine responses (7–9). These findings suggest murine Tfr cells are functionally heterogeneous and express a mixture of T cell receptors (TCRs), some recognizing self, others recognizing foreign antigens.
Mouse Tfr cells were originally described as descending solely from thymically-derived FOXP3+ T regulatory cells (Tregs) (7–9) but, more recently, three animal studies have challenged this dogma. One demonstrated that vaccines containing incomplete Freund’s adjuvant could spur mouse FOXP3− naïve CD4+ T cells to differentiate into vaccine-specific Tfr cells (10). A second study utilized longitudinal intravital microscopy to identify accumulation of Tfh-descended FOXP3-expressing T cells in murine GCs two to three weeks after immunization with an Alum-adjuvanted vaccine (11). Finally, TCRA sequence sharing between TCRB-transgenic murine CD4+ subsets indicated Tfr cells are most clonally related to Tregs and, to a slightly lesser degree, Tfh cells (12). Notably, to collect sufficient TCRA mRNA for analysis, material was pooled from multiple draining lymph nodes which likely diluted the clonal relationships of cells from the same GC.
Although their proper function is likely important to immunologic health and their dysfunction a potential contributor to various disease states, few studies have assessed the origins of human Tfr cells (13–17), their intermediate developmental stages or their biologic functions within tissues. Successful human Tfr cell investigations have been impeded by a lack of experimental tools to dynamically study human GC responses. Herein, we describe a series of approaches for studying human tonsillar Tfr cells including use of vaccine-activated tonsillar organoids as model systems to track Treg and Tfh cell contributions to the Tfr pool in vitro. Further, we demonstrate that single cell technologies can clonally distinguish between Tfr cells induced in vivo from Tfh lineage cells (iTfr) and those that are “naturally” derived from Tregs (nTfr). Once identified, we detail key genes and proteins distinguishing these two developmentally distinct populations, including CD38, which is a reliable tonsillar iTfr cell biomarker. Using CD38 as a marker, we catalogue the precise in situ locations of iTfr cells in human tonsil tissues using multiplexed microscopy and sort iTfr from nTfr cells for downstream functional assessments. Together, we have identified underappreciated clonal, transcriptional, functional and positional heterogeneity in the human Tfr pool that reflect distinct precursor-product relationships within both the Treg and Tfh cell lineages.
RESULTS
Differential CD25, CXCR5, and PD1 expression distinguish tonsillar CD4+ T cell subset transcriptomes
To identify viable, human T helper subsets of interest, we isolated CD4+ T cells from excised pediatric tonsils and used CD25 expression to separate CD25hi regulatory cells from CD25− Tfh cells, a strategy employed frequently in published mouse and human studies (Fig. 1A) (18). Among the CD25− population, CXCR5 and PD1 surface expression defined Tfh cells with high purity as assessed separately with intracellular BCL6 and FOXP3 staining. BCL6 expression was universally detected in the CD25−CXCR5+PD1hi Tfh gate (Fig. 1B) with only a very low frequency (<0.1%) of FOXP3-expressing cells identified (fig. S1A). Although a CD25− Tfr subset has been described in mice and humans, less than 10% of the tonsillar CXCR5+FOXP3+ population analyzed lacked CD25 expression and, since none of these highly expressed PD1, they could not be mistaken for Tfh cells (fig. S1B).
Figure 1. In silico models predict Tfh and Treg cells each contribute to the Tfr pool.
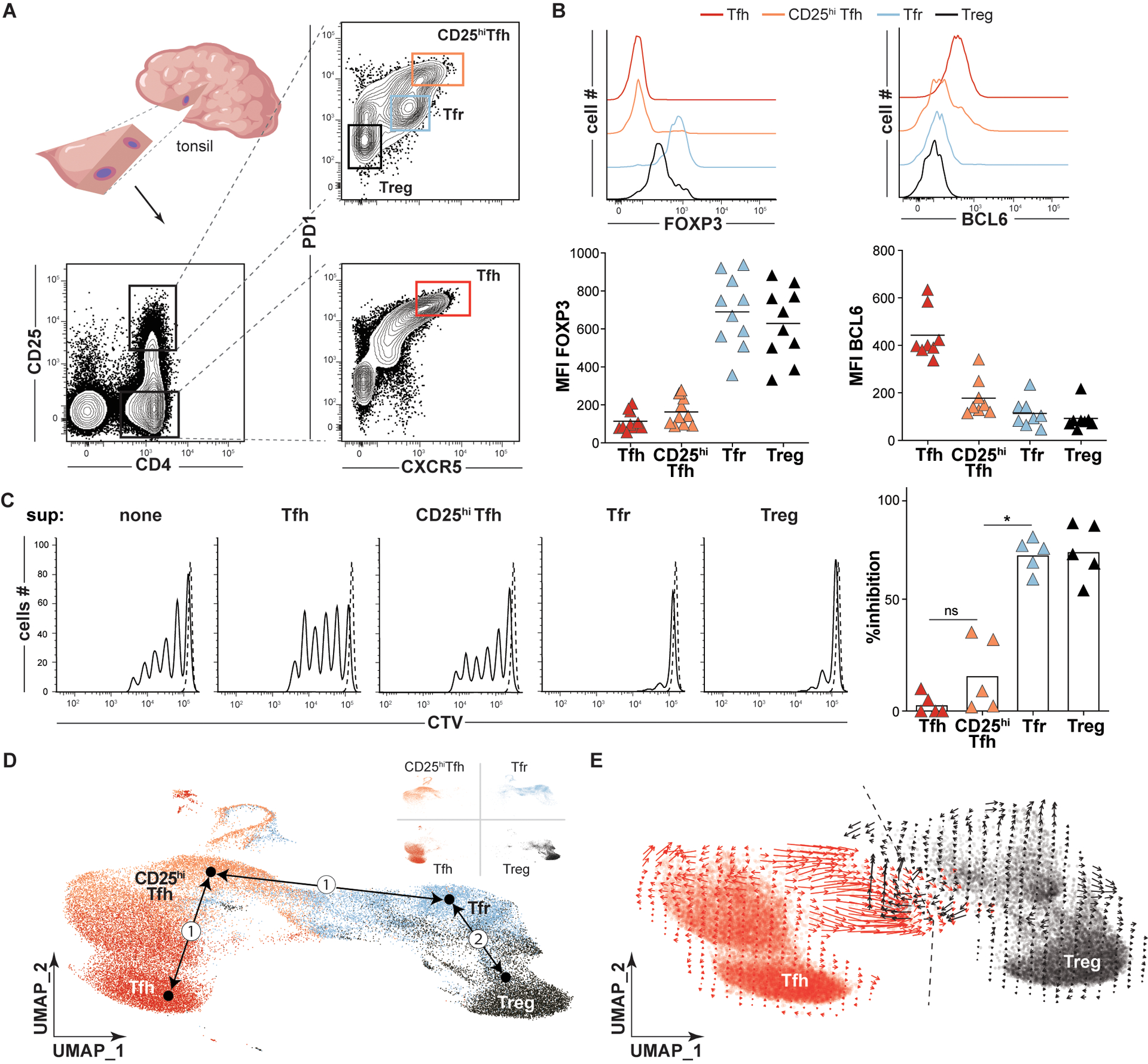
(A) A strategy to sort Tfh (CD4+CD25−CXCR5hiPD1hi, red), CD25hiTfh (CD4+CD25hiCXCR5hiPD1hi, orange), Tfr (CD4+CD25hiCXCR5+PD1int, light blue), and Treg (CD4+CD25hiCXCR5−PD1−, black) cell subsets from a small tonsil wedge is depicted. (B) Histograms and plots display FOXP3 and BCL6 mean fluorescence intensities (MFIs) on indicated T- cell subsets from a representative and all tonsil donors (n=8–10). (C, left) Representative histograms of CTV-labeled T-cell responders stimulated (solid line) or not (dashed line) in coculture with indicated heterologous tonsillar T-cell “suppressor” (sup) subsets are displayed. Bar graph (right) represents mean percent inhibition relative to unstimulated Tresp cells (n=5). (D) Dimensionally reduced single cell RNA-sequencing of TC174 T cell subsets are displayed as a UMAP. Pseudotime cell ordered projections (black arrows) identify two Tfr developmental arcs, (①) to CD25hiTfh and Tfh cells, (②) to Tregs. (E) RNA velocity vectors of only Tfh and Treg cells from TC174 are displayed. An arbitrary dashed centerline denotes convergence. *, P<0.05 by one-way ANOVA.
Within the CD25hi subset were classic CXCR5−PD1lo Tregs, which expressed FOXP3, and two CXCR5-expressing follicular populations. One CXCR5+ subset, which stained PD1 intermediate, corresponded to Tfr cells since it expressed higher FOXP3 than Tregs and could suppress T responder (Tresp) cell proliferation in vitro (Fig. 1C). The other CXCR5+ population was PD1hi and secreted IL-10, but did not express FOXP3 (Fig. 1B). Although a similar CD25hiCXCR5+PD1hi IL-10-secreting tonsillar subset has been attributed suppressive function (19), in our hands, these cells were not effective at inhibiting T responder (Tresp) proliferation in vitro (Fig. 1C) (20). For this reason, we descriptively call this population “CD25hiTfh”.
To explore transcriptional relationships between tonsillar CD4+ T cells, we employed the above strategy to sort live Treg, Tfr, CD25hiTfh and Tfh subsets from two immunocompetent pediatric tonsil donors, TC174 and TC341. Rather than utilizing entire tonsils or tonsil pairs, fewer cells (20,000 per subset) were collected from small tonsil wedge dissections to selectively reduce the number of GCs sampled and maximize clonal overlaps between antigen-expanded lineages (Fig. 1A). Subset-specific single cell RNA-sequencing (sc-RNAseq) libraries were created from sorted cells with an average of 10,372 cells recovered per library (range 8,512–15,598 cells, table S1). Sequenced libraries were dimensionally reduced and visualized in a single two-dimensional Uniform Manifold Approximation and Projection (UMAP) for each tonsil donor (Fig. 1D and fig. S2). In both patient UMAPs, Treg and Tfh cells localized to distant, non-overlapping spaces, while Tfr cells and CD25hiTfh cells filled the intervening space. As expected from our sorting strategy, CXCR5 and PDCD1 (PD1) transcripts were highest in CD25hiTfh and Tfh cells. IL2RA (CD25) transcripts were lowest in Tfh cells but this subset did express IL2RB, the gene encoding the low affinity IL-2 receptor (fig. S3). FOXP3 transcripts were predictably enriched in Treg and Tfr cells. PRDM1 (BLIMP1) and CTLA4 expression were greatest in Tfr, but also high in CD25hiTfh cells. Most transcripts elevated in CD25hiTfh cells encoded activation and/or cell cycle markers (fig. S3). Thus, sorting on variable CD25, CXCR5, and PD1 expression defines four transcriptionally distinct subsets, two that expressed FOXP3 and two that did not. This strategy efficiently captures the large majority of the Tfr cell population while effectively preventing cross contamination between Tfh and Tfr subsets and between Tfh and Treg subsets.
In silico and in vitro models predict Treg and Tfh/CD25hiTfh lineages separately contribute to the Tfr pool
To explore the competing hypotheses that Tfr cells descend from Tregs or from Tfh cells, we set Treg, Tfr, CD25hiTfh and Tfh cell transcriptomes as conceptual differentiation start or end points and used pseudotime analysis (21) to infer developmental trajectories (but not trajectory directions) based on progressive gene expression states. Two Tfr developmental arcs were generated; one connected Tfr cells directly to Tregs and the other connected Tfr to Tfh cells by passing through CD25hiTfh cells (Fig. 1D). To deduce trajectory directions, we compared the relative abundance of nascent (unspliced) and mature (spliced) mRNA of single Tfh and Treg cells (i.e. RNA velocity). Although transcriptionally distinct, Tfh and Treg cell RNA velocities converged along a centerline that ran directly through Tfr transcriptional space (Fig. 1E). Hence, results from transcriptionally-based, in-silico developmental models are consistent with two possible distinct precursor-product relationships for Tfr cells, one shared with Tregs and the other shared with CD25hiTfh and/or Tfh cells.
To test pseudotime inferences and RNA velocity predictions in vitro, tonsillar organoids, which generate vaccine-specific immune responses within characteristic GC light and dark zones (22, 23), were utilized to dynamically track Treg and Tfh lineage cells over time. In separate experiments, 1×104 sorted CellTrace™ Violet (CTV)-stained Tregs or 4×104 sorted carboxyfluorescein succinimidyl ester (CFSE)-stained Tfh cells were incorporated into autologous tonsillar organoids. Organoids were subsequently activated with a human pneumococcal diphtheria toxin-conjugated vaccine adjuvanted with Alum (PCV13, illustrated in Fig. 2A and fig. S4A). After seven to eight days in culture, organoid cells were mechanically dissociated, resuspended and intravitally stained cells re-identified by flow cytometry. Although most CTV+ Tregs did not divide in organoids, the 1–2% that did divide maintained FOXP3 expression and differentially upregulated CXCR5 4–7 fold, a change consistent with Tfr differentiation (fig. S4, B and C). The frequency of divided CFSE+ Tfh cells was greater (52%), with some cells demonstrating ≥ 4 divisions. With each additional division, CD25 mean fluorescence intensities (MFIs) increased, whereas PD1 expression remained stable (Fig. 1B). Also, compared with undivided cells, divided CFSE-stained Tfh cells upregulated key regulatory proteins including BLIMP1, CTLA4, and FOXP3 (Fig. 1C and D). Moreover, CFSE-diluted (i.e., divided), CD25+ cells sorted from day 7 organoids suppressed Tresp proliferation nearly as well as tonsillar Tregs did (Fig. 2E). In contrast, CFSE-bright (i.e., undivided), CD25− cells were poorly suppressive (p<0.001).
Figure 2. Tonsillar organoids model Tfh to Tfr cell development.
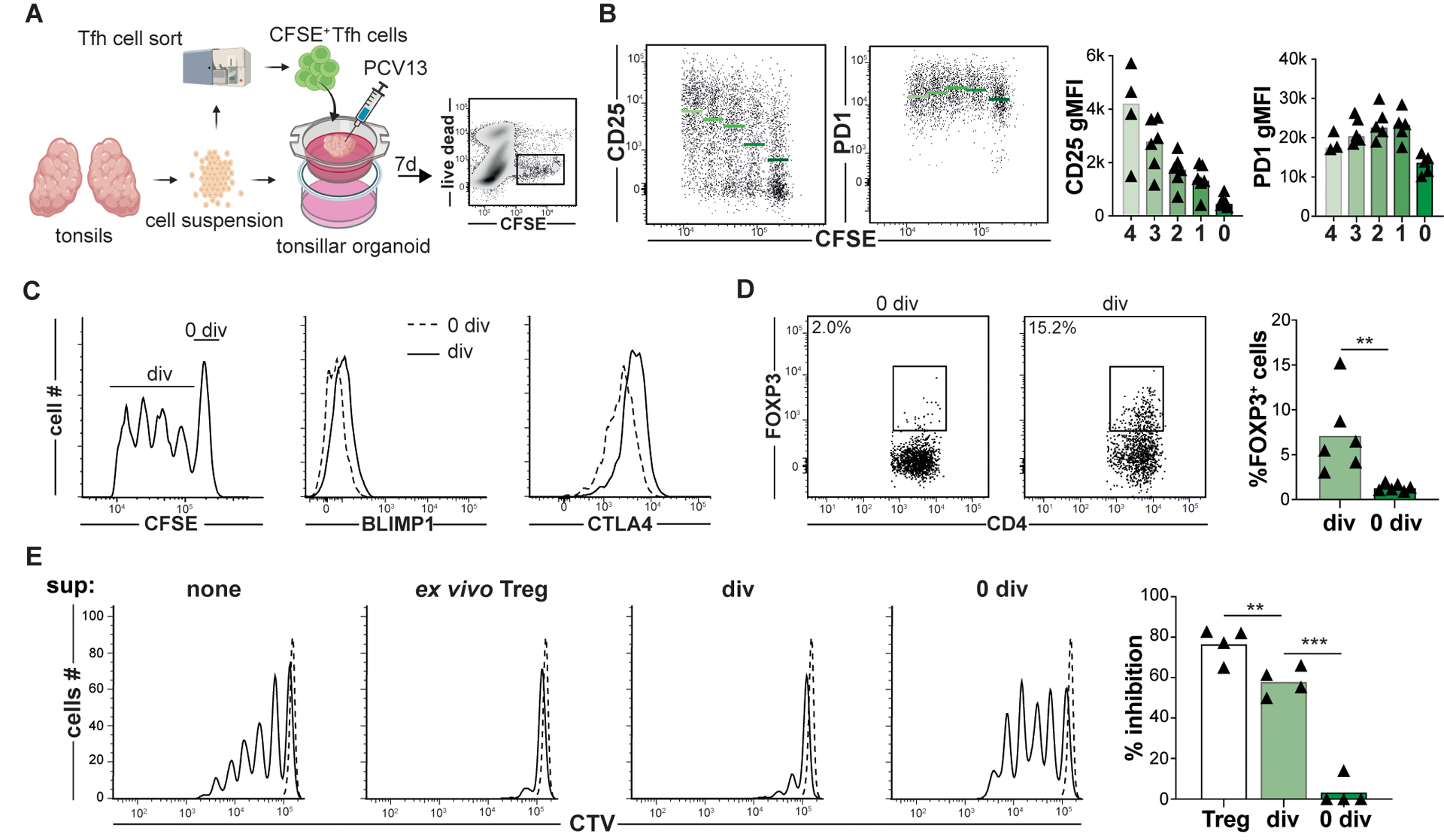
(A) A strategy to track Tfh to Tfr cell development within tonsillar organoids that are activated with pneumococcal conjugated vaccine 13 serotypes (PCV13) is depicted. (B) Day 7 CD25, PD1, (C) BLIMP1, CTLA4 and (D) FOXP3 expression by divided (div) and non-divided (0 div) Tfh cells are displayed. Green bards and horizontal lines indicate mean values (n=6) (E, left) Representative histograms show CTV-labeled T-cell responders (Tresp, CD45RO−CD25−CD4+ T cells) stimulated (solid line) or not (dashed line) in co-culture with either ex vivo Treg cells, or Tfh cells from day 7 organoids that were CD25+CFSEdim or CD25−CFSEbright. Bar graph (right) represents mean percent inhibition relative to unstimulated (n=5). **, P<0.01; ***, P<0.001 by Mann-Whitney U tests (D) and one-way ANOVA (E).
Because IL-2 sensing and CD25 expression seemed pivotal for organoid Tfh cells to transition into Tfr cells, we treated tonsillar organoids with anti-IL2RA (daclizumab) and measured iTfr cell induction on day 7. Compared to untreated organoids, Tfh cells in daclizumab-treated organoids were less divided and expressed less FOXP3 (fig. S5A). Separately, in organoid-free culture, we cultured either activated tonsillar Tfh cells or CD25hiTfh cells with or without recombinant IL-2 (rIL-2). After five days of rIL2 treatment, Tfh cells slowly lost BCL6 expression and upregulated CD25hiTfh-associated proteins including BLIMP1, CD25, CTLA4 and Ki67 but not FOXP3 (fig. S5B and C). In contrast, CD25hiTfh cells treated with rIL2 for five days began expressing FOXP3 (fig. S5C) and were significantly better at suppressing Tresp cell proliferation than ex vivo CD25hiTfh (p<0.01; fig. S5D). Hence, our in vitro models suggest IL-2-responsive tonsillar Tfh can pass through a proliferative CD25hiTfh intermediate stage and seed the Tfr pool.
Tfr clones are related to either Treg or Tfh/CD25hiTfh lineages, but not both
To determine what clonal relationship(s) exist between human tonsillar CD4+ T cell subsets in vivo, paired TCRB and TCRA transcripts were analyzed from each of TC174 and TC341’s respective scRNA-seq libraries. An average of 7,873 cells per library (range 6,484 to 9,596 cells, table S1) were recovered with both a TCRB and TCRA nucleotide sequence. Although no consistent differences in subset V(D)J gene usage patterns (fig. S6 and S7) and predicted CDR3 amino acid length distributions were appreciated (fig. S8), Treg clonal diversity was considerably greater than Tfr, CD25+Tfh and Tfh diversity by several measures (Shannon’s diversity index, inverse Simpson index, Chao1 and abundance-based coverage estimator (ACE); Fig. 3A). These measures likely reflect the higher proportion of non-expanded naïve cells in the Treg pool relative to other studied subsets (fig. S9). For instance, on average 96.2% of Treg clones were comprised of a single cell (range: ±0.3%). This frequency was considerably higher than Tfh (85±1.0%), CD25hiTfh (76.5±1.5%), and Tfr (78±2.0%) subsets (Fig. 3B and fig. S10A). Similarly, the top ten largest Treg clones (which numbered 16.5±4.4 clones due to ranking ties) were comprised of only 10.1±5.5 cells, whereas Tfh, CD25hiTfh, and Tfr top ten clones were bigger, containing 24.1±7.5 cells, 46±14.1 cells, 32.5±7.7 cells, respectively. These data suggest follicular subsets like Tfh, CD25hiTfh, and Tfr are more clonally expanded than Tregs.
Figure 3. The tonsillar Tfr pool overlaps with clonally diverse Tregs and clonally expanded Tfh/CD25hiTfh cells.
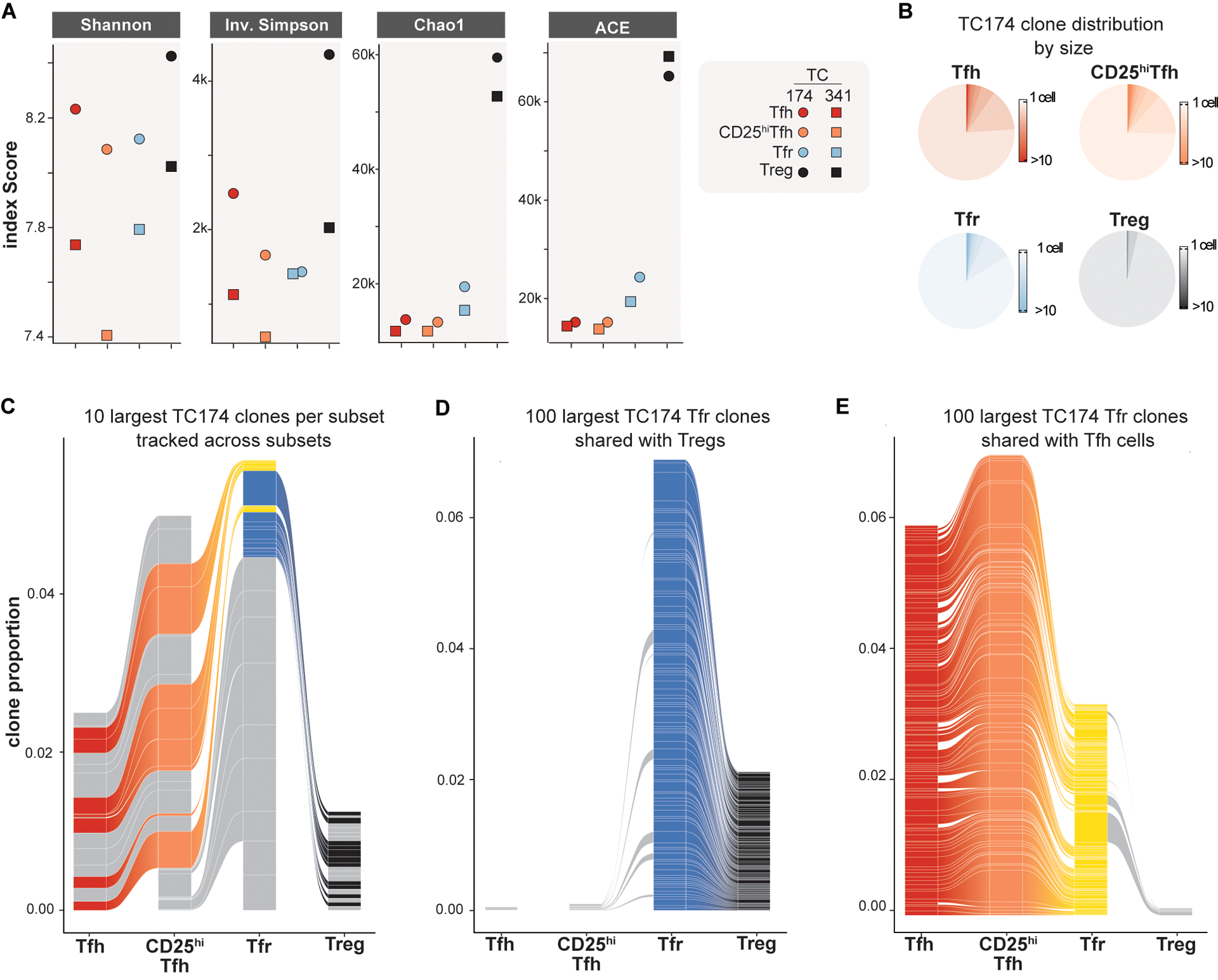
(A) TCRA/TCRB repertoire diversities of Tfh, CD25hiTfh, Tfr and Treg subsets from TC174 and TC341 are measured by indicated indices. (B) Pie charts indicate subset-specific clone size distributions (cells per clone) for TC174. (C) The ten largest clones per TC174 subset are tracked across other subsets. (D) The 100 largest clones shared between TC174 Tfr and Tfh cells and (E) between TC174 Tfr and Tregs are tracked across other subsets. Tfr clones shared with Tfh or Treg subset cells are shaded yellow and royal blue, respectively. Tfh, CD25hiTfh and Treg clones not shared with the Tfr subset and Tfr clones not shared with Tfh or Treg subsets are shaded gray. More than 10 clones (C) or 100 clones (D and E) per subset are displayed to account for ranking ties and clone sharing across subsets.
In mice, the TCRαβ repertoires of thymically-derived Tregs and FOXP3− T helper cells do not overlap (12, 24). Like published murine data, we found clone sharing between Treg and Tfh cells from the same tonsil donor to be minimal (0.3±0.1%), with no overlap among either donor subsets’ top ten largest clones (Fig. 3C, fig. S10B and fig. S11). In contrast, tonsillar Tfh and CD25hiTfh cells from the same donor shared many clones (11.5±0.5%), including most (93.2±1.9%) of the same top ten largest clones reinforcing Tfh and CD25hi cell lineage affiliation. As predicted by work in mice (12), many (29±21%) top ten Treg clones were shared with Tfr cells but none of these were among the largest Tfr cell clones. Instead, some of the largest Tfr clones were shared with either Tfh or CD25hiTfh lineage cells. Moreover, none of the top ten largest clones shared between Tfr and Treg cells overlapped with the top ten largest clones shared between Tfr and Tfh/CD25hiTfh lineage cells (Fig. 3C and fig. S10B), indicating divergent Tfr ancestry.
To determine how comprehensively the Tfr pool could be divided by its clonal relationships with Treg and Tfh/CD25hiTfh lineage cells, we expanded our analysis beyond the top ten to include all Tfr clones (8,207±16 clones). Through this more exhaustive approach, we identified a total of 150±85 clones shared between Tfr and Tregs (Fig. 3D, fig. S10C and fig. S10E) and 318±58 Tfr clones shared between Tfr and Tfh cells (Fig. 3E, fig. S10D and fig. S10E). Importantly, these three subsets only held 5.5±0.5 clones in common, reinforcing that Tfh and Treg cells separately contribute to the Tfr pool. Also notable was the high degree of clone sharing between Tfr, CD25hiTfh and Tfh cells. 50.4±8% of the clones shared by Tfr and Tfh cells were also shared between Tfr and CD25hiTfh cells consistent with a common Tfh to CD25hiTfh to Tfr developmental arc. In contrast, only 7.6±1.7% of Tfr clones shared with Tregs were also shared with CD25hiTfh cells. Hence, corroborating our in silico predictions and in vitro lineage tracing, the majority of shared tonsillar Tfr clones overlapped either Treg lineage or Tfh/CD25hi lineage cells but, importantly, not both populations. These observations provide in vivo evidence that Tregs and Tfh clones separately seed the human tonsillar Tfr pool.
Stringent clonal relationships distinguish Tfr cells with divergent ancestries
Although sort purity in our analysis was uniformly high (fig. S12), we could not exclude the possibility that some observed clonal overlaps between tonsillar CD4+ T cells subsets were the result of unintentional contamination. To reduce experimental noise, we performed a second, more strict analysis which defined a “stringent clone” as ≥ 2 cells in the same donor subset that expressed identical paired TCRA and TCRB nucleotide sequences. This more rigid definition further minimized the clonal overlap between Tregs and Tfh cells, leaving only one stringent clone in the combined TC174 and TC341 dataset (Fig. 4A). Other clonal relationships were strengthened by stringent analysis including Tfr and Treg cells (42 combined stringent clones), Tfr and Tfh cells (73 combined stringent clones), and Tfh cells and CD25hiTfh cells (603 combined stringent clones).
Figure 4. Transcriptomes of stringently defined iTfr and nTfr cells diverge, with cell type origin predicted by CD38/CD38 expression.
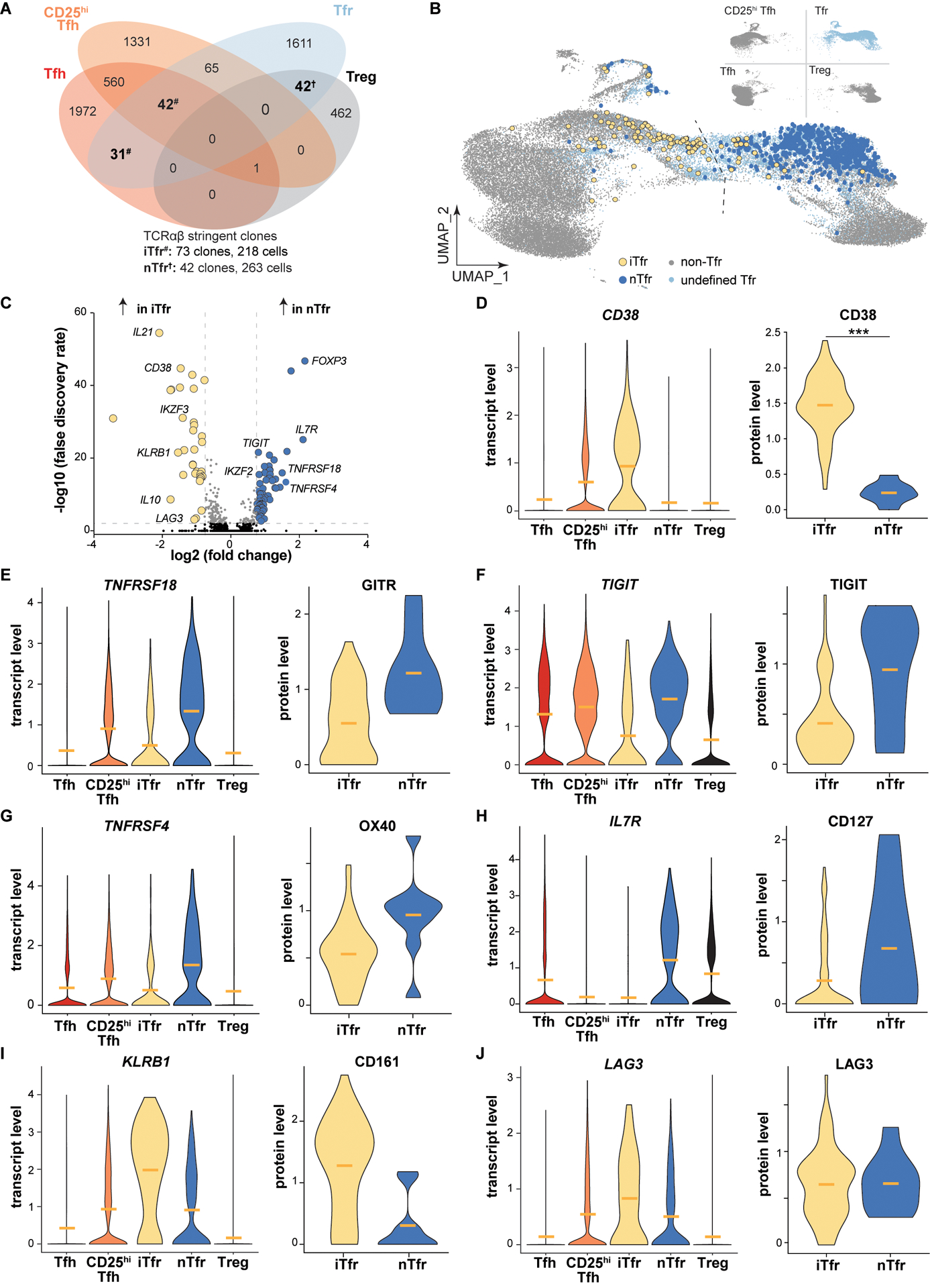
(A) Venn diagram displays overlapping TC174 stringent clones, defined as a clone with ≥2 cells. #, iTfr stringent clones; †, nTfr stringent clones. (B) Positions of stringent TC174 iTfr and nTr cells are displayed overlying a UMAP plot. (C) A volcano plot shows genes differentially expressed between stringently defined iTfr and nTfr cells pooled from TC174 and TC341. (D-J) Violin plots display differential expression of pooled transcripts and corresponding cell surface protein expression on TC341 subsets. Horizontal bars indicate mean values. ***, P<0.001 by Wilcoxon rank sum test with Bonferroni correction for protein expression comparisons.
To group Tfr cells by their clonal relationships with other subsets, Tfr cells that shared stringent clones with Tregs, but not Tfh cells, were designated natural “nTfr” cells, referencing the natural Treg to Tfr differentiation pathway originally described in mice (7–9). Separately we designated Tfr cells that shared stringent clones with Tfh cells, but not Tregs, as induced “iTfr” cells (so named because they were likely induced from Tfh lineage cells). Within tonsil donor-specific transcriptomic UMAP visualizations, most iTfr cells localized closer to Tfh transcriptional space, whereas nTfr cells localized nearer to Tregs (Fig. 4B and fig. S13A). Similarly, few Tfr cells of either variety crossed the centerline previously set by RNA-velocity analysis of TC174 cells (Fig. 4B). Furthermore, the Tfh and CD25hiTfh cells that were stringently related to iTfr clones were broadly distributed across donor-specific UMAPs. Although their TCRs were identical to FOXP3+ iTfr cells, these effector clones often occupied completely separate transcriptional spaces (fig. S13A). In total, these data suggest the stringent clonal relationships between tonsillar Tfr cells and other T helper cell subsets capture lineage-specific gene expression profiles.
Surface CD38 expression effectively distinguishes tonsillar iTfr and nTfr cells
To explore transcriptional differences between iTfr and nTfr cells, we pooled scRNA-seq data from clonally identified iTfr cells (n=218) and nTfr cells (n=263) from both tonsils and found 86 differentially expressed genes (DEGs, Fig. 4C). DEGs upregulated by nTfr cells were connected closely to Treg biology and included FOXP3, ENTPD1 (CD39), TIGIT, TNFRSF4 (OX40), and TNFRSF18 (GITR). Notably IKZF2 (HELIOS), once considered a practical biomarker to distinguish natural, thymically-derived Tregs from induced counterparts, was highly expressed by nTfr cells, but not iTfr cells (25). DEGs upregulated by iTfr cells encoded the follicular cytokine IL21, the regulatory cytokine IL10, AIOLOS (IKZF3), and LAG3 (Fig. 4C). Although FOXP3 transcripts were lower in iTfr cells than nTfr cells, several regulatory transcripts were significantly upregulated in iTfr cells compared to Tfh cells including FOXP3, PRDM1 and IL10 (fig. S13B). iTfr cells also distinctively lacked IL7R transcripts. Of all DEGs encoding cell surface proteins, CD38 was among the most promising candidates to potentially distinguish iTfr from nTfr cells (Fig. 4D). Additionally, DNA-barcoded antibodies targeting 139 TC341 Tfr cell surface molecules identified CD38, over GITR, TIGIT, OX40, CD127, CD161 and LAG3, to be the protein most significantly and consistently upregulated by iTfr cells relative to nTfr cells (p<0.001) (Fig. 4D–J).
To further validate CD38 as a potential iTfr biomarker, we performed a flow cytometric survey of Tfr cells on five to seven additional, unrelated pediatric tonsil donors. Across tonsil donors, CD38 staining was consistently bimodal with approximately 35% of Tfr cells expressing it and 65% not (Fig. 5, A and B), a proportion that matched the ratio of clonally defined nTfr to iTfr cells (Fig. 4A). Consistent with clonally-defined nTfr transcriptomes, CD38−Tfr cells also expressed more CD39, CD25, CTLA4, CD127, GITR, HELIOS, TIGIT, and FOXP3 than CD38+Tfr cells (Fig. 5C–G and fig. S14). Despite lower FOXP3 expression than CD38− Tfr cells, CD38+Tfr cells were clearly and uniformly FOXP3+AIOLOS+LAG3+CD127− cells, a profile matching transcriptomes of clonally-defined iTfr cells (Fig. 5C and fig. S14). ICOS, IL21, and Ki67 MFIs were also all significantly higher in CD38+Tfr cells (p<0.01, p<0.001 and p<0.0001, respectively; Fig. 5C) reflecting a likely developmental relationship with Tfh/CD25hiTfh lineage cells. Hence, differential CD38 expression by tonsillar Tfr cells is associated with broader, lineage-associated immunophenotypes.
Figure 5. Variable CD38 expression predicts distinct and larger Tfr cell immunophenotypes.
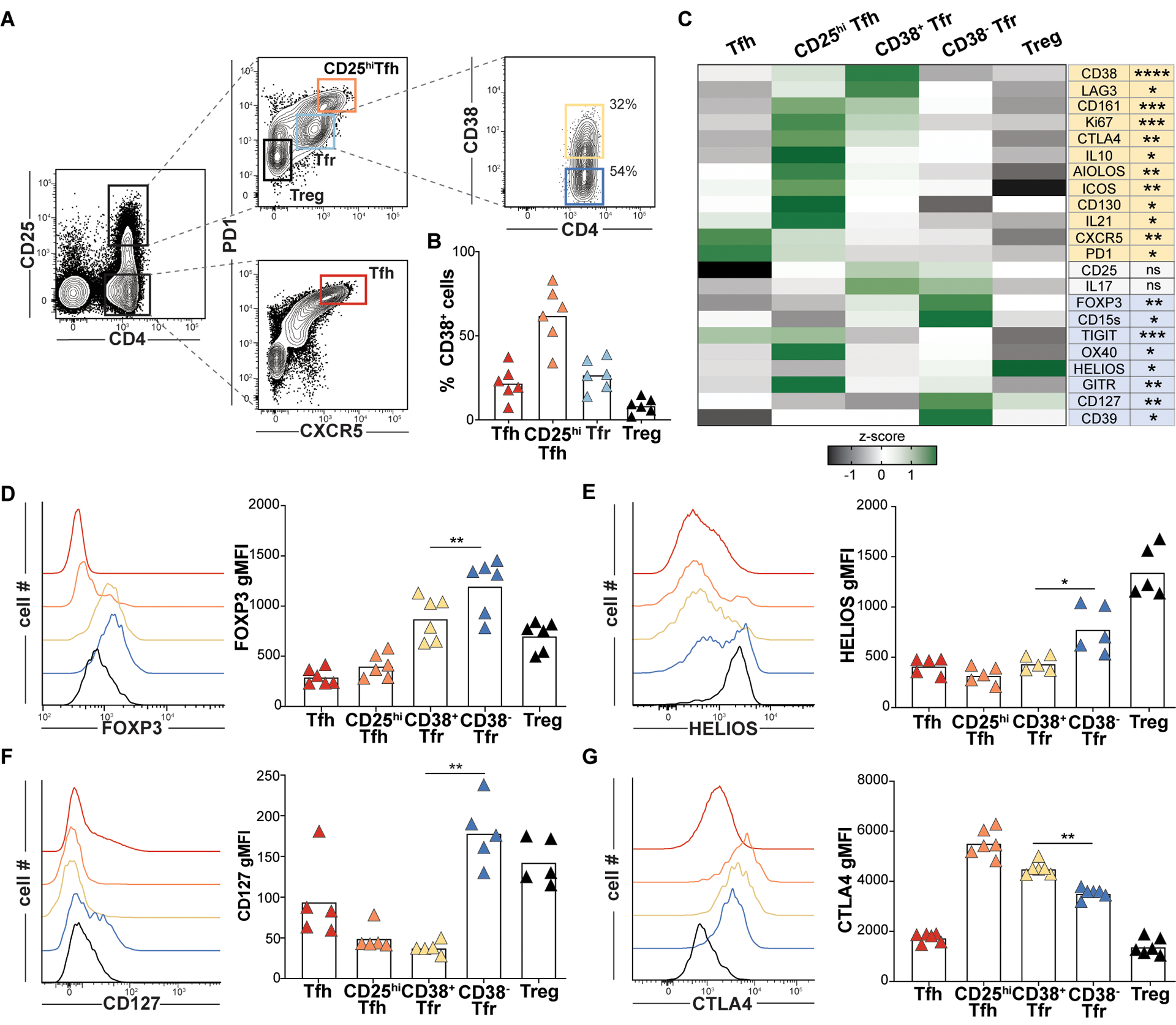
(A) Tfr expression of CD38 from a representative tonsil donor and (B) Indicated T cell CD38 expression frequencies for 6 pediatric tonsil donors. (C) A heat map displays geometric mean fluorescence intensity (gMFI) z-scores associated by T cell subsets from 6 donors. Proteins differentially upregulated in CD38+Tfr (yellow) and CD38−Tfr cells (blue) are shaded. (D-G) Histograms display gMFIs of indicated proteins by T cell subsets from a representative tonsil donor and bar graphs show gMFIs for 5–6 tonsil donors. *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001 by one-way ANOVA.
CD38+Tfr cells are regulatory cells specialized to provide B cell help, while CD38− Tfr cells are elite suppressors
Although FOXP3 transcripts and FOXP3 MFI were significantly higher in nTfr cells and CD38−Tfr cells, respectively, CD38+Tfr cells clearly also expressed FOXP3 (Fig. 5D). In fact, FOXP3 MFIs in CD38+Tfr cells were as high as in bona fide tonsillar Tregs, suggesting a regulatory identity. Additionally, CD38+Tfr cells consistently demonstrated the highest CTLA4 MFI and the greatest frequencies of IL10-secreting cells of any studied regulatory subset (Fig. 5, C and G and Fig. 6A). Indeed, sorted CD38+Tfr cells were as capable of inhibiting Tresp proliferation in vitro as tonsillar Tregs (Fig. 6B). Even stronger suppressive function was demonstrated by CD38−Tfr cells which outperformed all other tested subsets in in vitro assays (p<0.05). Since CD38−Tfr cells displayed lower CTLA4 MFI (Fig. 5G) and less IL10 expression than CD38+Tfr cells (Fig. 6A), their elite suppressive function may derive from one or more other, previously described, regulatory strategies (i.e. ectonucleotidase, IL-2 sink, GITR) (26).
Figure 6. CD38+Tfr cells provide germinal center B cell help, while CD38−Tfr cells are elite suppressors.
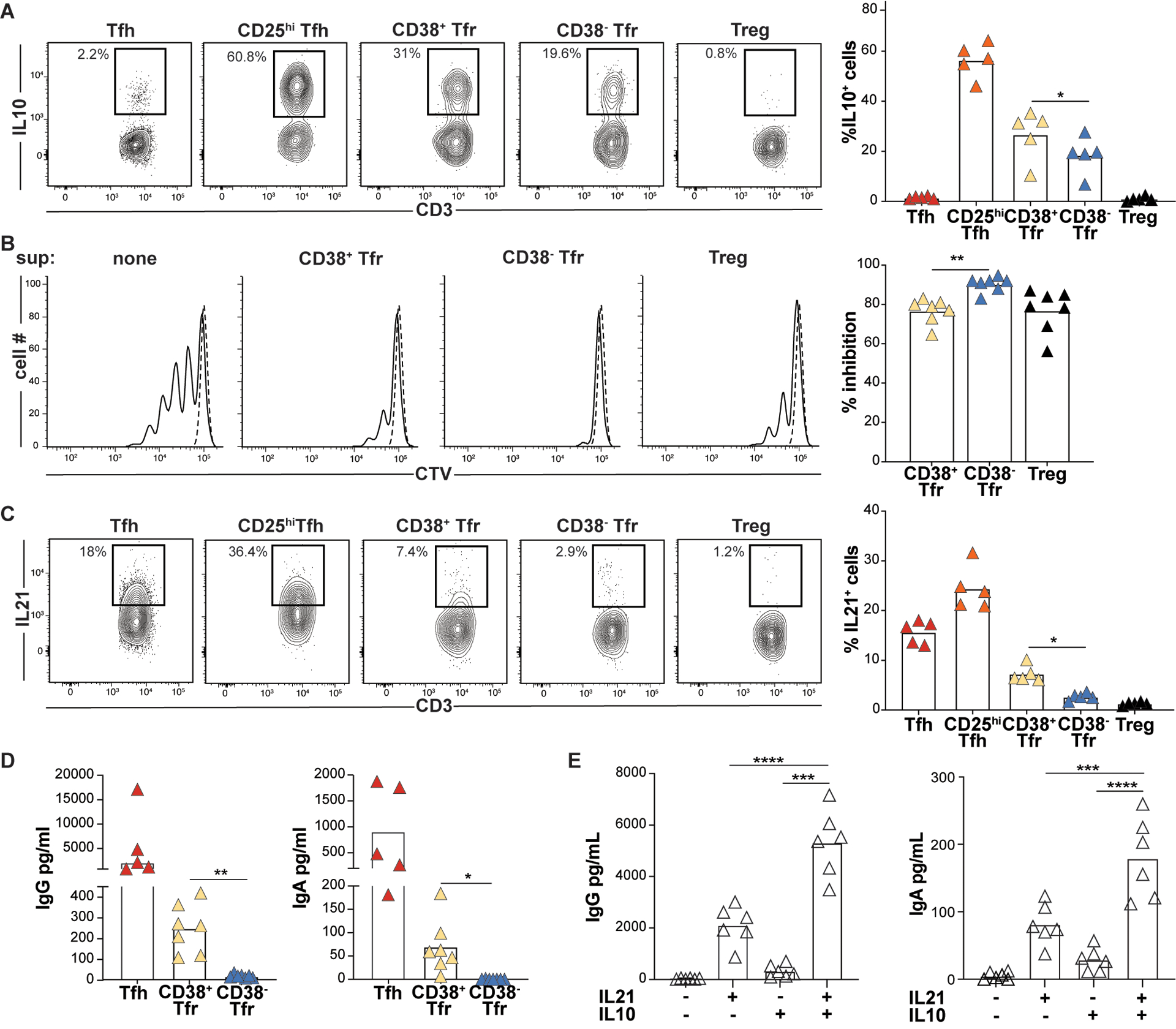
(A) Frequencies of PMA/ionomycin-activated T-subsets expressing IL10 are shown for a representative donor (left) and all available donors (n=5, right). (B) Day 4 Tresp cell proliferation with and without indicated T cell “suppressor” (sup) subsets from a representative donor (left) and all available donors (n=7, right) (C) Frequencies of PMA/ionomycin-activated T-subsets expressing IL21 are shown for a representative donor (left) and all available donors (n=5, right). *, P<0.05; by One-Way ANOVA. Day 7 IgG and IgA supernatant concentrations from GC B cells (D) co-cultured with either Tfh cells, CD38+ or CD38−Tfr cells stimulated with anti-CD3/CD28 beads (n=5) or (E) cultured alone with megaCD40L, recombinant IL21 and/or recombinant IL10 treatment (n=7). *, P<0.05; **, P<0.01; ***, P<0.001 by one-way ANOVA.
Within GCs, Tfh cells induce B cell somatic hypermutation and plasmablast differentiation by secreting IL-21 (27, 28). Since IL21 was the most upregulated iTfr DEG (Fig. 4C), we compared the frequencies of IL21-secreting CD38+ and CD38−Tfr cells across several tonsil donors. Indeed, on average, 2.7 times as many tonsillar CD38+Tfr cells secreted IL21 as CD38−Tfr cells (7.3% vs. 2.7%, p<0.01; Fig. 6C). Moreover, GC B cells co-cultured with autologous CD38+Tfr cells generated significantly higher supernatant IgG (p<0.01) and more IgA (p<0.05) than GC B cells co-cultured with CD38−Tfr cells (Fig. 6D). To determine if IL-10 or IL-21 secretion by CD38+ Tfr cells could account for enhanced IgG and IgA secretion we cultured GC-B cells with and without recombinant IL-10 and/or IL-21. Although IL-10 had little effect by itself, GC B cells treated with a combination of IL-10 and IL-21 secreted significantly more IgG and IgA than those treated with IL-21 alone (p<0.01 for both comparisons; Fig. 6E). Hence, CD38+Tfr cells are regulatory cells that retain and refine their capacity for B cell help through IL-21 and IL-10 secretion, while CD38−Tfr cells are elite suppressors.
CD38+Tfr cells reside inside GCs, CD38−Tfr cells localize to the follicular mantle
Although the follicular dendritic cell lattice attracts Tfh and Tfr cells by maintaining a CXCL13 gradient (29, 30), not all CXCR5-expressing T helper cells are located within GCs. Prior analysis of human lymph nodes indicate most human Tfr cells accumulate within follicles but clearly outside GC borders (31, 32). To identify the precise locations of Tfr cells in human tonsillar tissue, we employed CODEX to sequentially stain and image a DAPI-stained donor tonsil with a panel of 23 oligo-conjugated antibodies. Antibody targets included those relevant to follicular T cell biology and lineage-defining antigens to identify B cell, T cell, follicular dendritic cell, and epithelial populations (Table S2). After image pre-processing (see Materials and Methods) and high-dimensional clustering by marker profiles to identify cell types, a k-nearest neighbors-based, machine-learning platform (33) was employed to assign all tonsillar cells – totaling more than 1.5 million– to one of ten cellular neighborhoods (CNs) (Fig. 7A). Each CN possessed a characteristic cell type composition profile (fig. S15A and B) (33). Tonsil follicles were comprised of three distinct neighborhoods, CN3, CN6, and CN7. CN3 and CN7 primarily contained either GC B cells or Tfh cells, respectively, and corresponded to GC dark and light zones (fig. S15 A-C). CN6 regions surrounded GCs and were comprised largely of naive B cells, a profile consistent with follicular mantles (fig. S15A-C). To determine the spatial distribution of Tfr cells, we searched cells in follicles (i.e. CN3, CN6 and CN7) for FOXP3 and CD4 co-expression (Fig. 7B). Of 1,206 Tfr cells identified in this manner, 1,164 localized to mantles (Fig. 7A and B). The remaining Tfr cells were dispersed among dark zones (n=25) and light zones (n=17; Fig. 7A and B). Tfr locations identified by CODEX were independently confirmed by analyzing five additional tonsil donors with conventional immunofluorescence confocal microscopy. Indeed, most Tfr cells (71%) identified by our secondary analysis were positioned outside of GCs (fig. S16C).
Figure 7. Multiplex imaging identifies CD38+Tfr cells principally within tonsil germinal centers.
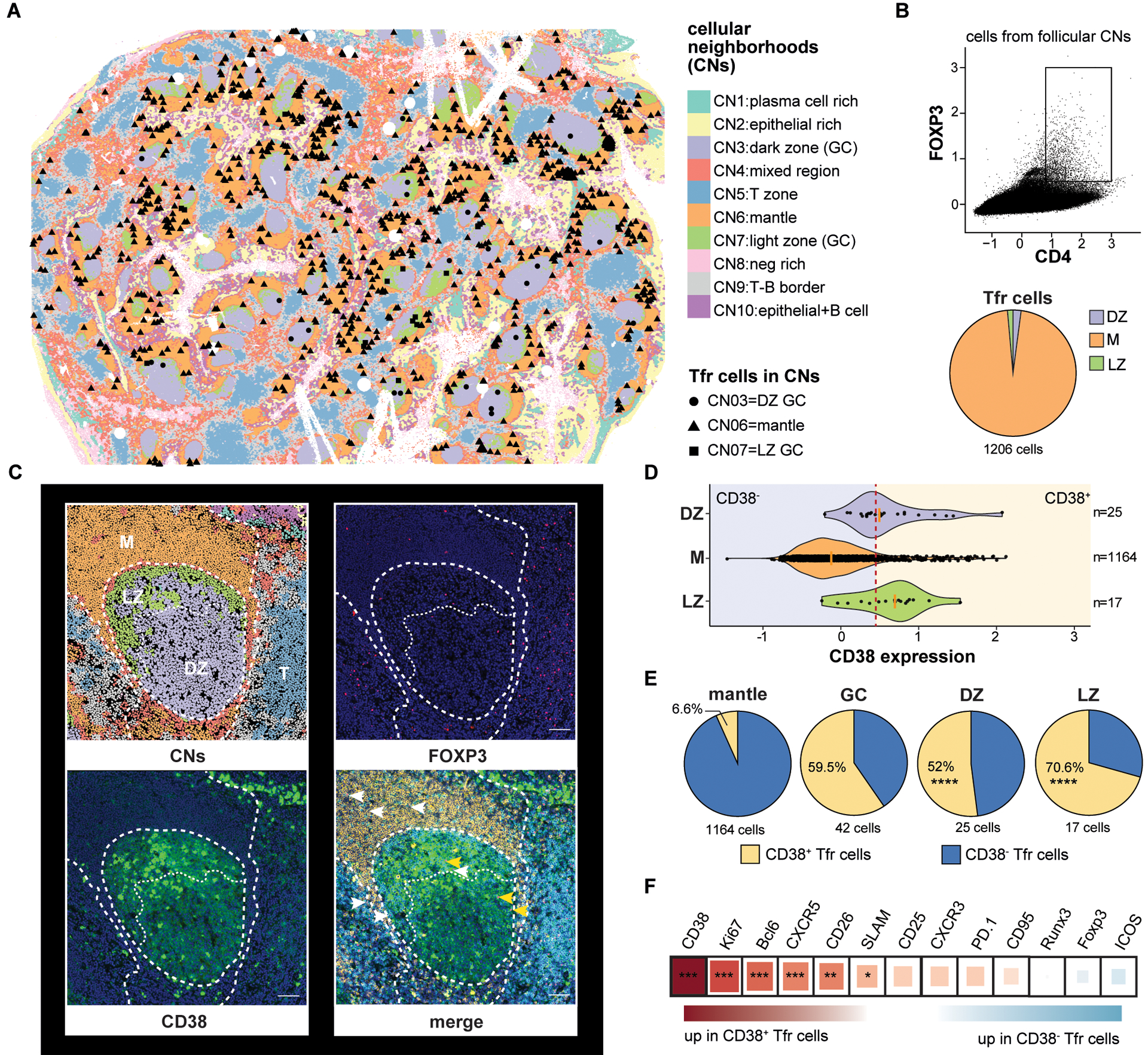
(A) A CODEX-stained human tonsil section is portioned into cell neighborhoods (CNs) and the locations of CD4+FOXP3+ cells in the dark zone (CN3), mantle (CN6), and light zone (CN7) are marked. (B) A CD4+FOXP3+ gate and the proportion of gated cells in each follicular CN is shown. (C) A representative tonsil GC with CNs, FOXP3 (pink) staining, CD38 (green) staining, and CD4 (cyan), IgD (orange), CD38, and FOXP3 overlaid. CD38+ (yellow arrow heads) and CD38− Tfr cells (white arrows heads) positions are indicated (D) Violin plots indicate CD38 expression distribution on CD4+FOXP3+ cells from different CNs. A red dashed line indicates the expression threshold above which Tfr cells were considered CD38+. (E) Proportions of CD38 expressing subsets in indicated follicular CNs. (F) Differential protein expression by CD38+ and CD38−Tfr subsets. Square size and color indicate fold change difference and direction, respectively, by Spearman correlation R coefficient. *, P<0.05 **, P<0.01 ***, P<0.001, ****, P<0.0001 by unpaired Wilcoxon tests or Chi-squared test. Scale bar 200-pixel=64.5μm. DZ, dark zone; LZ, light zone; M, mantle; T, T cell zone.
CD38 is distributed across tonsillar mononuclear cells in a trimodal pattern (fig. S17). At the extremes of expression are naïve B cells, which do not substantively express CD38, whereas plasmablast/plasma cells and highly activated GC B cells stain CD38bright. Intermediate CD38 expressors include a proportion of GC B cells and tonsillar T cells (fig. S17). To assess follicular CD38 distribution in situ, we stained tonsil sections with anti-CD38, anti-FOXP3 and anti-BCL6 antibodies and imaged them with confocal microscopy. CD38bright cells, which likely belonged to the B lymphocyte lineage, were readily identifiable on stained sections, but we also observed CD38-intermediate-expressing Tfr cells, which comprised ~20% of the total Tfr pool (fig. S16, A and B). 80% of CD38+Tfr cells localized within GCs, whereas most (85%) of CD38− Tfr cells were identified outside GCs in a 50μm ring that contained some, but not most, of the follicular mantle (fig. S16, A and C). To determine Tfr subset locations within better-defined mantle, light, and dark zones, we analyzed CD38 expression on a CODEX-stained tonsil section. Unlike conventional confocal microscopy where Tfr cells either clearly expressed CD38 or not, CD38 expression on CODEX-stained Tfr cells appeared more continuous (Fig. 7, C and D). To distinguish between CD38+ and CD38−, cells we set an expression threshold so that the proportion of CD38+ GC-resident Tfr cells identified by CODEX (59.5%; Fig. 7D) closely matched the proportion of counterparts definitively identified by confocal microscopy (58%; fig. S16D). Using this expression threshold, 52% of dark zone and 70.6% of light zone Tfr cells expressed CD38. In contrast, only 6.6% of mantle zone Tfr cells expressed CD38, a significantly different distribution (P<0.0001 by Chi-square tests for both comparisons; Fig. 7E). Hence, tonsillar CD38+Tfr cells are the most common Tfr subset encountered in GCs, whereas CD38−Tfr cells reside mostly in the mantle, closer to Tregs.
To determine if CODEX-identified Tfr cells shared phenotypic features with CD25hiTfh/Tfh or Treg lineage cells, we determined the proteins differentially expressed between CD38+ and CD38−Tfr cells (Fig. 7F). In agreement with flow cytometric analyses, proteins significantly upregulated in CD38+Tfr cells included Ki67, a prominent CD25hiTfh cell marker, and several canonical Tfh cell molecules (BCL6, CXCR5, CD26 and SLAM) (34). In summary, tonsillar CD38+Tfr cells are clonally related to Tfh cells, retain a Tfh-like capacity for GC B-cell help, gain new regulatory functions through differentiation, and outnumber CD38−Tfr cells in GC dark and light zones. In contrast, CD38−Tfr cells are clonally related to Tregs, appear highly specialized to suppress Tresp cell proliferation, and preferentially localize to follicular mantles.
DISCUSSION
Herein, we describe previously unappreciated clonal, functional, and positional heterogeneity within the human tonsillar Tfr cell pool. Using in silico transcriptomic trajectory projections, in vitro tonsillar organoid lineage tracking, and in vivo TCRA/TCRB sequencing, we show human Tfh cells are not a terminally differentiated population. Rather, Tfh cells are capable of proliferating through a CD25hiBLIMP1+ intermediate stage before gaining FOXP3 expression and clear regulatory function as iTfr cells. Despite their considerable transformation, iTfr cells retain critical Tfh cell characteristics, like IL-21 secretion and GC residence that preserve the capacity for, and the opportunity to provide, meaningful follicular B cell help.
The existence of iTfr cells challenges early experiments in the Tfr field that demonstrated adoptively transferred murine Tregs, but not naïve T cells, entered the Tfr pool 7 to 11 days after vaccination (7, 12). Our lineage tracking experiments using tonsillar organoids indicate that one week is likely too brief an interval for human naïve T cells to transition to Tfh cells, then to CD25hiTfh intermediary cells and finally to Tfr cells. Indeed, more recently published longitudinal intra-vital imaging of mouse lymph nodes revealed FOXP3+ cells of Tfh origin accumulated in GCs 14 to 18 days after vaccination and their influx heralded GC contracture (11). Our data suggest iTfr cells may arrive in the light zone at a similar GC life stage to suppress the same TCRαβ-matched Tfh clones they descended from. Since iTfr cells retain the ability to help B cells and permeate the dark zone, they may also maintain follicular output as GCs shrink. In contrast, nTfr cells are elite suppressors primarily positioned outside GCs but within follicular mantles suggesting a gatekeeping role that may include controlling autoreactive T cells at the T-B border.
Using non-biased, multi-omic analysis, we identified CD38 as a practical cell surface marker to divide live, clonally divergent human tonsillar Tfr subsets for downstream analyses, yet it is unclear if this molecule is important for iTfr cell differentiation or function. As CD38-deficient humans have not yet been identified, this line of inquiry may be best addressed by conditional deletion of CD38 from mouse CD4+ T cells, which are known to express CD38 upon activation (35). Although CD38 expression has been primarily described on human myeloid and B cells (36), a subset of GC-resident CD38 expressing CTLA4hi Tfr cells was separately described within human mesenteric lymph nodes (31). Based upon our own microscopy findings, these cells and tonsillar iTfr cells may be analogous in both provenance and function.
While definitive identification of human Tfr cells requires intracellular FOXP3 staining, we did not clonally identify iTfr and nTfr cells nor sort cells for functional analyses using differential FOXP3 expression since fixation/permeabilization would negatively affect mRNA integrity (37) and render cells inviable. Nevertheless, the clonal overlaps we identified between tonsillar Tfr and Treg cells and between Tfr and Tfh cells using cell surface stains were also reported in Foxp3gfp reporter mouse lymph nodes (12). Instead of relying on FOXP3, our sorting strategy relied upon differential CD25, CXCR5, and PD1 expression to capture most Tfr cells without sacrificing purity. One described human Tfr subset definitively excluded by this approach was CD25−Tfr cells (38–40), which comprise <10% of the total tonsillar Tfr pool. Due to their omission, it is unclear if CD25−Tfr cells exist on the iTfr lineage continuum, the nTfr lineage continuum, or represent contributions from additional sources. One clue that suggests a relationship with Tregs is the reportedly high Helios expression by CD25−Tfr cells, a feature of nTfr cells. Future studies using more inclusive strategies will be required to definitively address CD25−Tfr cell developmental origin.
How do Tfh cells initiate their transition to a more regulatory iTfr state? Our data suggests all required factors are present in activated tonsillar organoids yet since organoids contain a diversity of hematopoietic and non-hematopoietic cell types, this hardly narrows the field of candidates. One consideration is IL-2. In our hands, lower doses of IL-2 promote the Tfh to CD25hiTfh and the CD25hiTfh to Tfr transition while CD25 blockade impedes it. Others have shown that high dose IL-2 broadly inhibits murine Tfr cell development while low dose IL-2 permits better nTfr cell differentiation (39). It is possible that IL-2 also exerts dose-dependent effects on human iTfr and/or nTfr cells, a phenomenon that could be potentially exploited for a therapeutic benefit.
Finally, our study sought to describe the origins, functions, and positions of Tfr cells in the secondary lymphoid tissues of healthy humans. Since lymph nodes and spleens are rarely excised from healthy subjects, we utilized tonsils from pediatric donors free of systemic inflammatory or immune deficiency diseases. If iTfr cells are generated in human lymph nodes, as they are in human tonsils and murine lymph nodes (10, 11), iTfr/nTr imbalances may contribute to a spectrum of GC-relevant diseases including disorders caused by autoantibody production (i.e. systemic lupus erythematosus), poor vaccine responses (aging), or both (common variable immune deficiency; CVID). Indeed, increased circulating CD25hiTfh cell frequencies and asymmetrically hyperplastic GCs have been uniformly described in CVID patients with co-morbid autoantibody-mediated cytopenias (20, 41). Application of concepts, models and techniques described herein to diseased patient lymphoid tissues, rather than a continued emphasis on studying their peripheral blood samples, may reveal unexpected pathophysiologies and provide new opportunities to intervene with precision therapies.
MATERIALS AND METHODS
Study design
The main aim of this study was to evaluate the contributions of Treg and Tfh cells to the human tonsillar Tfr pool. The study was conducted by multi-omic sequencing of 82,973 single CD4+ T cells from two immunocompetent tonsil donors. From this larger dataset, TCRAB sequencing overlaps were used to clonally identify iTfr and nTfr cells. CITE-seq was used to determine differentially expressed genes and proteins between iTfr and nTfr cells. Microscopic imaging was performed on 10 tonsil tissue sections from six additional donors to identify the follicular positions of iTfr and nTfr cells. Tonsils from an additional 45 donors were used to generate tonsillar organoid cultures to model in vivo Tfr development and to perform in-vitro analyses that determine differential functional characteristics of Tfr cell subsets.
Tonsillar T CD4+ cell subset preparation and sorting
Fresh tonsils were obtained as discarded surgical waste from de-identified immune- competent children undergoing tonsillectomy to address airway obstruction or recurrent tonsillitis. Tonsil donor mean age was 6 years and 53 % were male. Tissue collection was determined not to be human subjects research by the Children’s Hospital of Philadelphia Institutional Review Board. A single cell suspension of tonsillar mononuclear cells (MNCs) was created by mechanical disruption (tonsils were minced and pressed through a 70-micron cell screen) followed by Ficoll-Paque PLUS density gradient centrifugation (GE Healthcare Life Sciences). CD19-positive cells were removed (StemCell) and CD4+ T cells were enriched with magnetic beads (Biolegend) prior to sorting T-cell subsets on a BD FACSAria™ (BD Bioscience). Dead cells were excluded using LIVE/DEAD stain (Thermo Fisher Scientific). The gating strategy is shown in Fig. 1A.
CD4+ T cell subsets immunophenotyping
Tonsillar enriched CD4+ T cells were immunophenotyped using an LSRFortessa flow cytometer (BD Bioscience) with antibodies listed in table S3. Intracellular staining was performed after fixation and permeabilization with the Foxp3/Transcription Factor Staining Buffer Set (Thermofisher) in accordance with the manufacturer’s instructions. To assess IL-10, IL-17, and IL-21 secretion by tonsillar CD4+, T cell subsets were rested overnight at 37°C then stimulated for 6 h with phorbol 12-myristate 13-acetate (25 ng/mL; Sigma) and ionomycin (1 mg/mL; Sigma) in the presence of Brefeldin A (5 μg/ml; BD Bioscience). Flow cytometric analyses were visualized with FlowJo software (TreeStar).
CITE-Seq and ScRNA-Seq
Viable tonsillar Tfh, CD25hiTfh, Tfr, and Treg cells were sorted from tonsils excised from two immune-competent male patients with obstructive sleep apnea. One patient was four and the other six-years-old. Transcriptome and TCR repertoire for each CD4+ T cell subset was obtained using a ScRNA-Seq approach. A CITE-Seq study was also developed on viable sorted Tfr cells from the 4-year-old donor. For the CITE-Seq sample preparation, Tfr cells were resuspended at 20 × 106 cells/ml in CITE-seq staining buffer (BioLegend) and incubated with Human TruStain FcX Fc Blocking reagent for 10 min at 4°C to block nonspecific antibody binding. Following Fc blocking, cells were incubated with a pool of 139 antibodies conjugated to an antibody-derived tag (ADT), inclusive of seven isotype controls (TotalSeq-C Human Universal Cocktail, anti-human GITR (108–17) and anti-human CD130 (2E1B02); BioLegend) for 30 min at 4°C. Cells were passed through a 40-μm filter and resuspended in 10% FBS RPMI media at 1 × 106 cells/ml. Next-generation sequencing libraries were prepared using the 10× Genomics Chromium Single Cell 5’ Library and Gel Bead kit v1 with Feature Barcoding Technology for Cell Surface Protein, per manufacturer’s “Chromium Single Cell V(D)J Reagent Kits” protocol. Libraries were uniquely indexed using the Chromium i7 Sample Index Kit, pooled, and sequenced on the Illumina NovaSeq 6000 sequencer (v1.5 chemistry) in a paired-end, single indexing run. Sequencing for each gene expression library targeted 20,000 mean reads per cell and each V(D)J library targeted 5,000 read pairs per cell. Downstream sequence processing is described separately in Supplementary Materials.
Tonsillar organoid preparation
Once isolated, MNC were counted and resuspended in organoid media (RPMI with L-glutamine, 10% FBS, 2 mM glutamine, 1X penicillin-streptomycin, 1 mM sodium pyruvate, 1X MEM non-essential amino acids, 10 mM HEPES buffer, and 1 μg/ml of recombinant human B-cell activating factor [BioLegend]) at a concentration of 6 × 107 cells per ml. As previously described by Wagar et al. 6 × 106 MNC were transferred to permeable transwells (0.4-μm pore, 12-mm diameter; Millipore, (24)). Viable sorted Tfh and Treg cells from autologous tonsil donors were stained with carboxyfluorescein diacetate succinimidyl ester (CFSE; Thermo Fisher Scientific) or CellTrace Violet (CTV; Thermo Fisher Scientific), respectively, for tracking. 40 × 103 Tfh cells or 10 × 103 Tregs were added to sepreate organoids. Transwells were then inserted into standard 12-well polystyrene plates containing 1 ml of additional organoid media with or without daclizumab (anti-IL2RA) at 3μg/ml (R&D Biosystem) and placed in an incubator at 37 °C and 5% CO2. Organoid media and drug were replaced every 3 days. On culture day 7, prior Tfh cells were identified according to the CFSE dye and evaluated for FOXP3, CTLA4, CD25, LAG3, and BLIMP1 expression. On culture day 8, prior Tregs were identified according to the CTV dye and evaluated for FOXP3 and CXCR5 expression.
Treg cell suppression assays
CD4+CD45RO−CD25− sorted naive responder T cells (5 × 103) were labeled with CFSE and cocultured with an equal number of either Tfh (ex vivo, day 5 IL-2-primed or CFSE+ sorted tonsillar organoid at day 7), CD25hiTfh (ex vivo or day 5 IL-2-primed), CD38+/CD38−Tfr, or Treg cells. Cultures were activated with anti-CD2/CD3/CD28 coated beads at a bead to cell of ratio of 1:1 (Miltenyi Biotec). Cocultures were stained for viability with the LIVE/DEAD Kit (Thermo Fisher Scientific), and the proliferation of viable responder T cells was determined by CFSE or CTV dilution at culture day 3.5 by flow cytometry.
IL-2 T-cell priming, B-cell/T-cell cocultures and B-cell cultures
2.5 × 104 viable sorted Tfh or CD25hi Tfh cells were activated with CD3/CD28–coated beads (Dynadeads, Sigma) at a ratio of 1 bead per T cell with or without IL-2 at 10ng/ml (R&D system). On day 2 and day 5, differentiation marker expression was measured by flow cytometry.
2.5 × 104 viable CD19+CD21+CD38+IgD− sorted GC B cells were cocultured with an equal number of viable sorted Tfh cells, CD38+ or CD38− Tfr cells. Cocultures were activated with CD3/CD28–coated beads. Separately, 5 × 104 sorted viable GC B cells were cultured with megaCD40L (1μg/ml, Enzo), recombinant IL-21 and/or recombinant IL-10 (both at 25ng/ml, R&D systems). On day 7, culture supernatant IgG, and IgA concentrations were determined by ELISA.
Confocal and multiplex microscopy
Tonsils were frozen in Tissue-Tek O.C.T™ Compound (Sakura Finetek). Multiplex microscopy analyses were done on a 7μm-section tissue using Akoya Phenocycler platform. Tissue was stained following Akoya’s recommendation for staining using a panel described in Table S2. Computational analysis is detailed in Supplementary Materials.
6μm-sections were prepared and mounted on SuperFrost™ Plus glass slides (Fisher Scientific), fixed in cold acetone for 20 min and air dried for standard confocal microscopy analyses. Non-specific antibody binding was avoided using Blocking Reagent™ (Perkin Elmer) for 30 min. Sections were then incubated overnight with anti-human CD38 (EPR4106, abcam), FOXP3-eFluor570 (236A/E7, Invitrogen) BCL6-AF647 (K112–91, BD Bioscience) antibodies at 4 °C overnight. To detected CD38 protein, sections were incubated with anti-rabbit AF488 (Invitrogen) antibodies for 2h at room temperature. After paraformaldehyde 4% fixation (Electron Microscopy Sciences) for 20 min, slides were stained with DAPI (1 μg/ml, Sigma) for 15 min at room temperature and mounted with fluorescent mounting medium (DAKO). Images were acquired using a Leica TCS SP8 Confocal and analyzed with ImageJ software.
Statistical analyses
Prism v9 (GraphPad) was used to perform statistical analyses. Comparisons of two groups were analyzed using non-parametric Mann-Whitney. One-way ANOVA with a follow-up multiple-comparison test (Tukey’s test) was used to compare more than two groups for analyses of immunophenotypic data, functional assays. Chi-square and Spearman correlation R coefficient tests were performed for Tfr cell localization analysis. Non-parametric statistical correlation analysis was performed as previously described (https://github.com/wherrylab/statistics_code) for differential protein expression analysis of CODEX-stained tonsillar tissue. Differential expression analysis was performed in the scRNAseq and CITE-seq data using a Wilcoxon Rank Sum test and a Bonferroni correction to account for multiple comparisons.
Supplementary Material
Acknowledgments:
We thank the subjects and their families. We thank the Children’s Hospital of Philadelphia’s Division of Otolaryngology, Center for Applied Genomics and Flow Cytometry Core for pediatric tonsil samples, next-generation sequencing and technical support, respectively.
Funding:
This work was supported by grants from the National Institutes of Health, National Institute of Allergy and Infectious Diseases AI146026 (NR), National Institute of Allergy and Infectious Diseases AI155577 (EJW), National Institute of Allergy and Infectious Diseases AI115712 (EJW), National Institute of Allergy and Infectious Diseases AI117950 (EJW), National Institute of Allergy and Infectious Diseases AI108545 (EJW)National Institute of Allergy and Infectious Diseases AI082630 (EJW), National Institute of Allergy and Infectious Diseases CA210944 (EJW), National Institute of Allergy and Infectious Diseases AI114852 (RSH), National Heart, Lung, and Blood Institute R38 HL143613 (DAO), National Heart, Lung, and Blood Institute T32 HL 7775–28 (JPB), National Cancer Institute T32 CA009140 (DAO). Additional funding from Parker Institute for Cancer Immunotherapy (EJW), Parker Institute for Cancer Immunotherapy, Parker Bridge Fellow Award (DAO), Gray Foundation (DP), Chan Zuckerberg Initiative Pediatric Networks for the Human Cell Atlas (NR), Gail B. Slap Department of Pediatrics Fellowship Award (WR) and the Sayer family (NR).
Footnotes
Competing Interests:
EJW is a member of the Parker Institute for Cancer Immunotherapy which supported this study. EJW is an advisor for Danger Bio, Marengo, Janssen, Pluto Immunotherapeutics Related Sciences, Rubius Therapeutics, Synthekine, and Surface Oncology. EJW is a founder of and holds stock in Surface Oncology, Danger Bio, and Arsenal Biosciences.
Data and materials availability:
CITE-seq data were deposited and are publicly available through Gene Expression Omnibus under accession number GSE214572. All other data needed to support the conclusions of the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Victora GD, Nussenzweig MC, Germinal centers. Annu. Rev. Immunol 30, 429–457 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Meyer-Hermann M, George LA, Figge MT, Khan M, Goodall M, Young SP, Reynolds A, Falciani F, Waisman A, Notley CA, Ehrenstein MR, Kosco-Vilbois M, Toellner K-M, Germinal center B cells govern their own fate via antibody feedback. J. Exp. Med 210, 457–464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stebegg M, Kumar SD, Silva-Cayetano A, Fonseca VR, Linterman MA, Graca L, Regulation of the Germinal Center Response. Front. Immunol 9, 2469 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu W, Liu X, Lin X, Feng H, Sun L, Li S, Chen H, Tang H, Lu L, Jin W, Dong C, Deficiency in T follicular regulatory cells promotes autoimmunity. J. Exp. Med 215, 815–825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez-Figueroa P, Roco JA, Papa I, Núñez Villacís L, Stanley M, Linterman MA, Dent A, Canete PF, Vinuesa CG, Follicular regulatory T cells produce neuritin to regulate B cells. Cell 184, 1775–1789.e19 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Lu Y, Jiang R, Freyn AW, Wang J, Strohmeier S, Lederer K, Locci M, Zhao H, Angeletti D, O’Connor KC, Kleinstein SH, Nachbagauer R, Craft J, CD4+ follicular regulatory T cells optimize the influenza virus-specific B cell response. J. Exp. Med 218, e20200547 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, Srivastava M, Divekar DP, Beaton L, Hogan JJ, Fagarasan S, Liston A, Smith KGC, Vinuesa CG, Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med 17, 975–982 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, Wang Y-H, Lim H, Reynolds JM, Zhou X, Fan H, Liu Z, Neelapu SS, Dong C, Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med 17, 983–988 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wollenberg I, Agua-Doce A, Hernández A, Almeida C, Oliveira VG, Faro J, Graca L, Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J. Immunol. Baltim Md 1950 187, 4553–4560 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Aloulou M, Carr EJ, Gador M, Bignon A, Liblau RS, Fazilleau N, Linterman MA, Follicular regulatory T cells can be specific for the immunizing antigen and derive from naive T cells. Nat. Commun 7, 10579 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacobsen JT, Hu W, R Castro TB, Solem S, Galante A, Lin Z, Allon SJ, Mesin L, Bilate AM, Schiepers A, Shalek AK, Rudensky AY, Victora GD, Expression of Foxp3 by T follicular helper cells in end-stage germinal centers. Science 373, eabe5146 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maceiras AR, Almeida SCP, Mariotti-Ferrandiz E, Chaara W, Jebbawi F, Six A, Hori S, Klatzmann D, Faro J, Graca L, T follicular helper and T follicular regulatory cells have different TCR specificity. Nat. Commun 8, 15067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim HW, Hillsamer P, Kim CH, Regulatory T cells can migrate to follicles upon T cell activation and suppress GC-Th cells and GC-Th cell-driven B cell responses. J. Clin. Invest 114, 1640–1649 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim HW, Hillsamer P, Banham AH, Kim CH, Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. J. Immunol. Baltim. Md 1950 175, 4180–4183 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Dhaeze T, Peelen E, Hombrouck A, Peeters L, Van Wijmeersch B, Lemkens N, Lemkens P, Somers V, Lucas S, Broux B, Stinissen P, Hellings N, Circulating Follicular Regulatory T Cells Are Defective in Multiple Sclerosis. J. Immunol. Baltim. Md 1950 195, 832–840 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Wen Y, Yang B, Lu J, Zhang J, Yang H, Li J, Imbalance of circulating CD4(+)CXCR5(+)FOXP3(+) Tfr-like cells and CD4(+)CXCR5(+)FOXP3(−) Tfh-like cells in myasthenia gravis. Neurosci. Lett 630, 176–182 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Fonseca VR, Agua-Doce A, Maceiras AR, Pierson W, Ribeiro F, Romão VC, Pires AR, da Silva SL, Fonseca JE, Sousa AE, Linterman MA, Graca L, Human blood Tfr cells are indicators of ongoing humoral activity not fully licensed with suppressive function. Sci. Immunol 2, eaan1487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graca L, Ed., T-follicular helper cells: methods and protocols (Humana Press, New York, NY, 2022). [Google Scholar]
- 19.Cañete PF, Sweet RA, Gonzalez-Figueroa P, Papa I, Ohkura N, Bolton H, Roco JA, Cuenca M, Bassett KJ, Sayin I, Barry E, Lopez A, Canaday DH, Meyer-Hermann M, Doglioni C, de St Groth B. Fazekas, Sakaguchi S, Cook MC, Vinuesa CG, Regulatory roles of IL-10-producing human follicular T cells. J. Exp. Med 216, 1843–1856 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Coz C, Bengsch B, Khanna C, Trofa M, Ohtani T, Nolan BE, Henrickson SE, Lambert MP, Kim TO, Despotovic JM, Feldman S, Fadugba OO, Takach P, Ruffner M, Jyonouchi S, Heimall J, Sullivan KE, Wherry EJ, Romberg N, Common variable immunodeficiency-associated endotoxemia promotes early commitment to the T follicular lineage. J. Allergy Clin. Immunol 144, 1660–1673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL, The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagar LE, Salahudeen A, Constantz CM, Wendel BS, Lyons MM, Mallajosyula V, Jatt LP, Adamska JZ, Blum LK, Gupta N, Jackson KJL, Yang F, Röltgen K, Roskin KM, Blaine KM, Meister KD, Ahmad IN, Cortese M, Dora EG, Tucker SN, Sperling AI, Jain A, Davies DH, Felgner PL, Hammer GB, Kim PS, Robinson WH, Boyd SD, Kuo CJ, Davis MM, Modeling human adaptive immune responses with tonsil organoids. Nat. Med 27, 125–135 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pahl MC, Le Coz C, Su C, Sharma P, Thomas RM, Pippin JA, Cruz Cabrera E, Johnson ME, Leonard ME, Lu S, Chesi A, Sullivan KE, Romberg N, Grant SFA, Wells AD, Implicating effector genes at COVID-19 GWAS loci using promoter-focused Capture-C in disease-relevant immune cell types. Genome Biol. 23, 125 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L, Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity 25, 249–259 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, Shevach EM, Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. Baltim. Md 1950 184, 3433–3441 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt A, Oberle N, Krammer PH, Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol 3, 51 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, Corcoran LM, Godfrey DI, Toellner K-M, Smyth MJ, Nutt SL, Tarlinton DM, IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J. Exp. Med 207, 365–378 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, Verma NK, Smyth MJ, Rigby RJ, Vinuesa CG, IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J. Exp. Med 207, 353–363 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ansel KM, Ngo VN, Hyman PL, Luther SA, Förster R, Sedgwick JD, Browning JL, Lipp M, Cyster JG, A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 406, 309–314 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Gunn MD, Ngo VN, Ansel KM, Ekland EH, Cyster JG, Williams LT, A B-cell-homing chemokine made in lymphoid follicles activates Burkitt’s lymphoma receptor-1. Nature 391, 799–803 (1998). [DOI] [PubMed] [Google Scholar]
- 31.Sayin I, Radtke AJ, Vella LA, Jin W, Wherry EJ, Buggert M, Betts MR, Herati RS, Germain RN, Canaday DH, Spatial distribution and function of T follicular regulatory cells in human lymph nodes. J. Exp. Med 215, 1531–1542 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moysi E, Del Rio Estrada PM, Torres-Ruiz F, Reyes-Terán G, Koup RA, Petrovas C, In Situ Characterization of Human Lymphoid Tissue Immune Cells by Multispectral Confocal Imaging and Quantitative Image Analysis; Implications for HIV Reservoir Characterization. Front. Immunol 12, 683396 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schürch CM, Bhate SS, Barlow GL, Phillips DJ, Noti L, Zlobec I, Chu P, Black S, Demeter J, McIlwain DR, Kinoshita S, Samusik N, Goltsev Y, Nolan GP, Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive Front. Cell 182, 1341–1359.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tardif V, Muir R, Cubas R, Chakhtoura M, Wilkinson P, Metcalf T, Herro R, Haddad EK, Adenosine deaminase-1 delineates human follicular helper T cell function and is altered with HIV. Nat. Commun 10, 823 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandoval-Montes C, Santos-Argumedo L, CD38 is expressed selectively during the activation of a subset of mature T cells with reduced proliferation but improved potential to produce cytokines. J. Leukoc. Biol 77, 513–521 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Camponeschi A, Kläsener K, Sundell T, Lundqvist C, Manna PT, Ayoubzadeh N, Sundqvist M, Thorarinsdottir K, Gatto M, Visentini M, Önnheim K, Aranburu A, Forsman H, Ekwall O, Fogelstrand L, Gjertsson I, Reth M, Mårtensson I-L, Human CD38 regulates B cell antigen receptor dynamic organization in normal and malignant B cells. J. Exp. Med 219, e20220201 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masuda N, Ohnishi T, Kawamoto S, Monden M, Okubo K, Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res. 27, 4436–4443 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ritvo P-GG, Churlaud G, Quiniou V, Florez L, Brimaud F, Fourcade G, Mariotti-Ferrandiz E, Klatzmann D, Tfr cells lack IL-2Rα but express decoy IL-1R2 and IL-1Ra and suppress the IL-1-dependent activation of Tfh cells. Sci. Immunol 2, eaan0368 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Botta D, Fuller MJ, Marquez-Lago TT, Bachus H, Bradley JE, Weinmann AS, Zajac AJ, Randall TD, Lund FE, León B, Ballesteros-Tato A, Dynamic regulation of T follicular regulatory cell responses by interleukin 2 during influenza infection. Nat. Immunol 18, 1249–1260 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wing JB, Kitagawa Y, Locci M, Hume H, Tay C, Morita T, Kidani Y, Matsuda K, Inoue T, Kurosaki T, Crotty S, Coban C, Ohkura N, Sakaguchi S, A distinct subpopulation of CD25- T-follicular regulatory cells localizes in the germinal centers. Proc. Natl. Acad. Sci. U. S. A 114, E6400–E6409 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romberg N, Le Coz C, Glauzy S, Schickel J-N, Trofa M, Nolan BE, Paessler M, Xu ML, Lambert MP, Lakhani SA, Khokha MK, Jyonouchi S, Heimall J, Takach P, Maglione PJ, Catanzaro J, Hsu FI, Sullivan KE, Cunningham-Rundles C, Meffre E, Patients with common variable immunodeficiency with autoimmune cytopenias exhibit hyperplastic yet inefficient germinal center responses. J. Allergy Clin. Immunol 143, 258–265 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R, Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borcherding N, Bormann NL, Kraus G, scRepertoire: An R-based toolkit for single-cell immune receptor analysis. F1000Research 9, 47 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schapiro D, Sokolov A, Yapp C, Chen Y-A, Muhlich JL, Hess J, Creason AL, Nirmal AJ, Baker GJ, Nariya MK, Lin J-R, Maliga Z, Jacobson CA, Hodgman MW, Ruokonen J, Farhi SL, Abbondanza D, McKinley ET, Persson D, Betts C, Sivagnanam S, Regev A, Goecks J, Coffey RJ, Coussens LM, Santagata S, Sorger PK, MCMICRO: a scalable, modular image-processing pipeline for multiplexed tissue imaging. Nat. Methods 19, 311–315 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greenwald NF, Miller G, Moen E, Kong A, Kagel A, Dougherty T, Fullaway CC, McIntosh BJ, Leow KX, Schwartz MS, Pavelchek C, Cui S, Camplisson I, Bar-Tal O, Singh J, Fong M, Chaudhry G, Abraham Z, Moseley J, Warshawsky S, Soon E, Greenbaum S, Risom T, Hollmann T, Bendall SC, Keren L, Graf W, Angelo M, Van Valen D, Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning. Nat. Biotechnol 40, 555–565 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, Black S, Nolan GP, Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 174, 968–981.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.