

Abstract
Linear DNA undergoes a series of compression and folding events, forming various three‐dimensional (3D) structural units in mammalian cells, including chromosomal territory, compartment, topologically associating domain, and chromatin loop. These structures play crucial roles in regulating gene expression, cell differentiation, and disease progression. Deciphering the principles underlying 3D genome folding and the molecular mechanisms governing cell fate determination remains a challenge. With advancements in high‐throughput sequencing and imaging techniques, the hierarchical organization and functional roles of higher‐order chromatin structures have been gradually illuminated. This review systematically discussed the structural hierarchy of the 3D genome, the effects and mechanisms of cis‐regulatory elements interaction in the 3D genome for regulating spatiotemporally specific gene expression, the roles and mechanisms of dynamic changes in 3D chromatin conformation during embryonic development, and the pathological mechanisms of diseases such as congenital developmental abnormalities and cancer, which are attributed to alterations in 3D genome organization and aberrations in key structural proteins. Finally, prospects were made for the research about 3D genome structure, function, and genetic intervention, and the roles in disease development, prevention, and treatment, which may offer some clues for precise diagnosis and treatment of related diseases.
Keywords: cancer, congenital developmental abnormality, three‐dimensional genome, topologically associating domain
Linear DNA undergoes a series of compression and folding events, resulting in various three‐dimensional (3D) structural units in mammalian cells. These structures play crucial roles in regulating gene expression, cell differentiation, organ development, and disease progression. Here, the 3D genome structure and function were systematically reviewed.
1. INTRODUCTION
The three‐dimensional (3D) genome structure plays a crucial role in determining cell fate and maintaining normal cellular functions. 1 In the late 19th century, the existence of chromatin territory and euchromatin/heterochromatin in the cell nucleus was proposed based on findings from optical microscopes and chromatin dyes, 2 and Hans Winker introduced the term “genome” in 1920. 3 With the development of fluorescent in situ hybridization, the individual chromosomal territories could be directly observed. 4 , 5 Chromosome conformation capture was first reported in 2002, which employs formaldehyde‐induced chromatin cross‐linking and restriction enzyme digestion to connect spatially proximate DNA fragments. 6 The interaction frequency between two specific loci in the genome is detected in a “one versus one” format, using polymerase chain reaction or next‐generation sequencing technology. 6 Circular chromosome conformation capture facilitates the detection of specific DNA fragments' interactions with other genomic regions. Designing primers for the detected fragment achieves a “one versus all” format, allowing for the detection of all genome regions. 7 , 8 In 2009, genome‐wide chromosome conformation capture (Hi‐C) was reported and can detect all chromatin interactions throughout the genome, referred to as “all versus all”. 9 Furthermore, remarkable advancements in optical and electron microscopy technologies have enabled direct observation of chromatin structures. The development of super‐resolution fluorescence imaging techniques, such as stimulated emission depletion and structured illumination microscopy, has been especially notable. 10 By combining these techniques with specific DNA probes, the chromatin conformation of particular genomic regions within individual cells can be observed, providing new insights for exploring gene expression regulation and other intricate biological processes within the cell nucleus. 11 , 12
DNA sequence undergoes a series of complex compressions and folding to ultimately form chromosomes. 13 A chromosome maintains a specific location within the nucleus, denoted as a chromosome territory (CT). 14 These chromosomes are organized in a structured manner within the nucleus and can be classified into euchromatin and heterochromatin based on gene transcriptional activity. 15 , 16 At the subchromosomal level, A/B compartments, with an average size of 3−5 Mb, correspond to euchromatin and heterochromatin, respectively. 17 , 18 Compartments at the megabase level can be further divided into different topologically associating domains (TADs). 19 TAD represents the fundamental units of 3D genome structure and function. 20 , 21 , 22 High‐frequency interactions between enhancers and promoters within TAD regulate cell‐specific expression of development‐related genes. 22 The CCCTC‐binding factor (CTCF) at TAD boundaries isolates aberrant regulatory interference, ensuring efficient transcription. 23 At a smaller scale (<2 Mb), chromatin loops often link enhancers and promoters, playing a crucial role in the regulation of gene transcription. 15 Structural variations or structural protein abnormalities can lead to disease by disrupting TAD structure and affecting cis‐regulatory element function. 24 , 25 , 26 These may be critical pathological mechanisms for many diseases, such as congenital diseases and cancers. 27 , 28
Here, we tried to make a systematic review of the 3D genome structure, function, and relationship with diseases. First, the structural characteristics of mammalian 3D chromatin organization were summarized. Then, the effect and mechanisms of cis‐regulatory elements’ interaction in the3D genome for regulating spatiotemporally specific gene expression, the role and mechanisms of dynamical changes of 3D chromatin conformation during embryonic development were discussed. Subsequently, the pathological mechanisms of some diseases, such as congenital developmental abnormalities and cancer, which attribute to alterations in 3D genome organization and aberrations in key structural proteins, were systematically reviewed. Finally, prospects were made for the research about 3D genome structure, function, and genetic intervention, and the roles in disease development, prevention, and treatment.
2. 3D GENOME STRUCTURE
The hierarchical structure of the 3D genome, from high to low levels, includes CT, A/B compartment, topologically associated domain, and chromatin loop (Figure 1).
FIGURE 1.
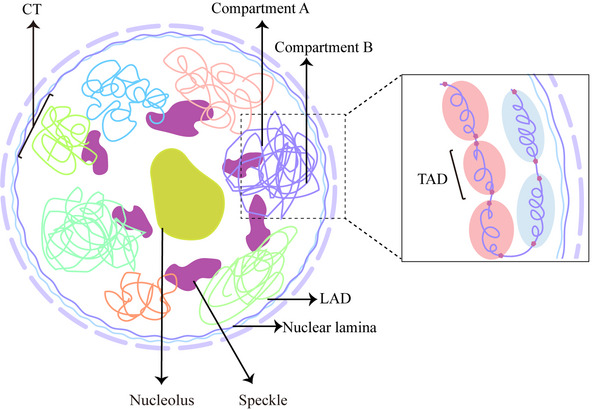
Chromosome 3D structure. In mammalian cells, chromosomes nonrandomly occupy specific regions within the interphase nucleus, known as chromosome territory (CT). Chromosomes can be classified into euchromatin and heterochromatin based on their transcriptional activity and regulatory roles. Euchromatin contains numerous transcriptionally active genes and is loosely arranged, while heterochromatin is tightly organized and primarily comprises transcriptionally silent genes. Heterochromatin at the nuclear periphery interacts with nuclear lamins, giving rise to Lamin‐associated domains (LADs). At the megabase level, the A/B compartment is further subdivided into TAD. Covering approximately 90% of chromatin structure, TAD represents the basic unit of 3D genome function and structure. Within TAD, high‐frequency interactions between enhancers and promoters regulate cell‐specific expression of developmental genes. CTCF, located at TAD boundaries, effectively isolates aberrant regulatory information interference and ensures smooth transcription.
2.1. Chromatin and chromosome territory
In mammalian cells, chromosomes are formed from approximately 2 m of DNA sequences through a series of compressions and folding events. 29 , 30 , 31 Each nucleosome encompasses around 146 base pairs of DNA, giving rise to chromatin fibers that subsequently condense and fold to create chromosomes. 21 , 32 These chromosomes nonrandomly occupy specific regions within the interphase nucleus, known as CT. 13 , 22 Chromosomes with similar characteristics tend to cluster and are influenced by size, gene density, and transcriptional activity. 33 , 34 , 35 The distribution of chromosome territories can vary among different cell types. 36 , 37 , 38 For instance, differences in nuclear shapes between human fibroblasts and lymphocytes result in distinct radial distributions of chromosome territories. 39 Moreover, interactions between CT boundaries may be associated with disease‐related chromosomal translocations. 14
Chromosomes can be classified into euchromatin and heterochromatin based on their transcriptional activity and regulatory roles. 9 Euchromatin, comprising numerous transcriptionally active genes, exhibits a loosely organized structure, whereas heterochromatin, characterized by transcriptionally silent genes, displays a tightly packed arrangement. 40 Heterochromatin at the nuclear periphery interacts with nuclear lamins (lamin A/C, B1, and B2), giving rise to Lamin‐associated domains (LADs). 41 , 42 , 43 , 44 Over 1000 LADs exist in humans and mice, with a median size of approximately 0.5 Mb, comprising 30−40% of the entire genome. 45 , 46 LADs are characterized by heterochromatin histone modifications H3K9me2 and H3K9me3 and predominantly exhibit low or silent gene expression. 47 LADs can be further categorized into constitutive LADs (cLADs) and facultative LADs (fLADs), based on their conservation across cell types. 48 cLADs retain a relatively stable position in the genome and display high conservation among cell types. 46 In contrast, fLADs demonstrate variability across cell types, associate with cell type‐specific gene expression regulation and functions, and experience dynamic changes during the cell cycle, cellular differentiation, and circadian rhythms. 49 , 50
2.2. A/B compartment
At the sub‐chromosomal level, A/B compartments correspond to transcriptionally active and silent chromatin. 51 These compartmental structures have an average size of 3−5 Mb. 17 The A compartment is situated near nuclear speckles regions and modified by active histones, while the B compartment locates at the nuclear periphery and is modified by inactive histones. 15 Sites within the same compartment exhibit more frequent interactions. 15 Highly spatially plastic, compartments undergo extensive A–B switching during embryonic stem cell differentiation. Genes associated with A–B compartment switching demonstrate increased transcriptional activity throughout development. 16
2.3. Topologically associating domain
At the megabase level, A/B compartments are further divided into different TADs. 52 In mammalian genomes, TADs tend to be conserved across species and different cell types, 15 , 53 , 54 covering approximately 90% of chromatin structure and serving as the fundamental unit of 3D genome function and structure. 55 In most cases, TAD formation precedes gene expression. 56 In the human genome, TADs emerge during the eight‐cell stage when the interactions between regulatory elements within TAD are low and gradually increase throughout embryo development. TADs span approximately 0.1–1 Mb, and up to a maximum of 2 Mb, containing one or more genes. 52 Smaller sub‐TAD structures reside within TADs and are associated with tissue‐specific gene expression. 57 , 58 , 59 Although earlier studies have suggested that TADs function as a unit at the cell population level, recent studies indicate that TADs are chromatin structures in individual cells, exhibiting globular conformations and sharp structural domain boundaries. 60 , 61
TADs boundaries are co‐localized with housekeeping genes, cohesin, and CTCF. 53 , 54 CTCF is located at TAD boundaries and effectively compartmentalizes external regulatory information. 53 , 54 As a highly conserved zinc finger protein, CTCF plays crucial roles in transcriptional activation/repression, insulation, and the formation of higher‐order chromatin structures. 62 , 63 Due to the DNA sequences that bind CTCF being asymmetric, CTCF located at TADs boundaries has directionality. 64 , 65 In over 90% of TADs, CTCF sites are situated at boundaries in a convergent orientation. 66 CRISPR‐edited inversions of CTCF binding sequences disrupt TAD structure and cause abnormal gene expression, suggesting that CTCF orientation is important for TAD. 67 Multiple CTCF binding sites at TAD boundaries ensure precise gene expression through synergistic and redundant effects. 68 , 69 , 70 Recent studies showed that six CTCF binding sites exist between Epha4 and Pax3 TADs, and are located adjacent to each other. Deletion of a single CTCF binding site can alter Pax3 expression level without causing observable phenotypes.However, deletion of multiple CTCF binding sites leads to TADs fusion and abnormal finger development in mice. 70 TAD boundaries are highly conserved and remain relatively stable during organ development and tissue differentiation. 16 , 23 , 71 However, human genome analysis suggests that although the vast majority of CTCF loci are conserved, thousands of CTCF loci may be associated with tissue‐specific gene expression. 72 For instance, CTCF function and insulating activity are positively correlated with the level of enhancer–promoter interactions within TAD. 73 CTCF at TAD boundaries is more conserved than CTCF located within TAD, with stronger binding, more open chromatin, and lower DNA methylation levels. 74 Furthermore, TAD boundaries containing a single gene are more conserved than TAD boundaries that contain multiple genes. 75
2.4. Chromatin loop
Chromatin loop is relatively stable, ring‐shaped structure formed by chromatin within 3D space, often containing regulatory elements such as enhancers and promoters. 76 In 2014, Rao and colleagues discovered approximately 10,000 chromatin loops through the analysis of in situ Hi‐C conducted on human lymphoblastoid cells. 15 Most of these loops are conservation across various species and cell types. 15 Genes that form chromatin loops exhibit higher expression levels compared with those did not form chromatin loops, indicating the involvement of chromatin loops in regulating gene expression. 15 Furthermore, an analysis of 24 cell lines derived from three human germ layers revealed that approximately 28% of chromatin loops varied among different cells. 77 These tissue‐specific chromatin loops are strongly associated with the tissue‐specific gene expression. 77
The “loop extrusion” model, mediated by cohesin‐CTCF cooperation, aptly describes the formation of chromatin loops and TADs 78 (Figure 2). In this process, CTCF and cohesin exhibit distinct roles. 78 CTCF acts as a barrier to prevent overextension of cohesin. 79 Cohesin is a ring‐shaped DNA‐entrapping adenosine triphosphatase (ATPase) complex that works as a molecular motor. 80 Human cohesin comprises three subunits: SMC1, SMC2, and Scc. 81 Cohesin moves along the chromatin fiber with a speed of 1.5 kb/s until it interacts with convergently oriented CTCF sites. 82 This process lasts 10−30 min. 83 Scc1 plays an essential role in loop extrusion by binding to several regulators, such as NIPBL and PDS5. 84 The cohesin loading factor NIPBL loads cohesin at specific DNA sites, promoting cohesin translocation across chromosome fibers. 85 , 86 , 87 In addition, NIPBL can activate ATPase to supply energy for the high‐speed movement of cohesin on chromatin fibers. 88 Cohesin release from chromosomes requires PDS5 to recruit WAPL, a cohesin‐releasing factor. 89 The unloading efficiency of WAPL is stronger than that of PDS5, and both participate in the process of cohesin release. 89 , 90 , 91 Cohesin ensures its smooth arrival at the CTCF site by directly interacting with CTCF. 92 The N‐terminal 77‐aa region of CTCF can directly interact with the cohesin Scc1‐SA2, providing a structural and functional basis for the precisely anchoring of cohesin to CTCF binding sites. 93
FIGURE 2.
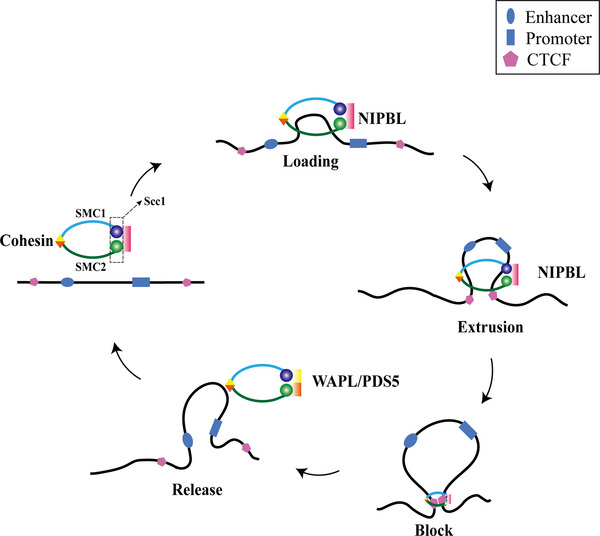
Mechanism of loop extrusion. Cohesin acts as a molecular motor in the “loop extrusion” model: After binding to chromosomes, cohesin moves in two opposite directions along chromatin fibers and extrudes DNA loops until it contacts the target CTCF site in the converging direction. During TAD formation, cohesin undergoes functional changes by interacting with various regulatory factors. The cohesin loading factor, NIPBL, loads cohesin at specific DNA sites and facilitates cohesin's translocation on chromosomal fibers. Cohesin's release from chromosomes requires the PDS5 subunit to recruit the cohesin‐releasing factor, WAPL. The unloading efficiency of WAPL is stronger than that of PDS5, and both participate in the cohesin release process. Finally, cohesin ensures its smooth arrival at the target CTCF site through direct interaction with CTCF.
The functional changes of the aforementioned cohesin subunits or complexes affect the formation of TAD. For example, deletion of the Scc1 subunit leads to the disruption of TAD, 75 , 94 while depletion of WAPL leads to a more than 20‐fold prolongation of the duration of cohesin action, resulting in the formation of a larger chromatin loop, which increases the amount of TAD and/or sub‐TAD. 90 , 95 , 96 , 97
3. MECHANISMS OF 3D GENOME REGULATING GENE EXPRESSION: CIS‐REGULATORY ELEMENTS INTERACTION IN 3D GENOME
In the human genome, protein‐coding sequences constitute merely 2% of the DNA sequence, 98 and gene expression is subject to regulation by various noncoding structures, such as promoters and enhancers within the 3D genome, and does not operate in isolation (Figure 3). 99 , 100
FIGURE 3.
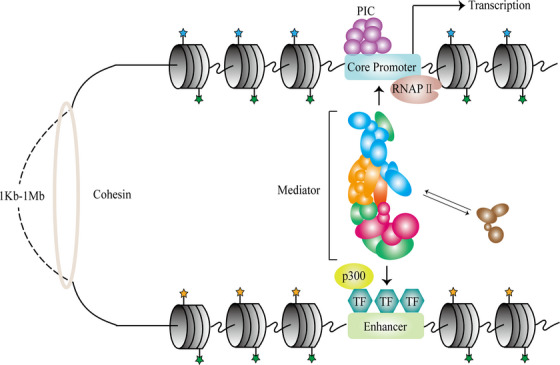
Mechanisms of enhancer–promoter interactions. Within TADs, cis‐regulatory elements play crucial roles in gene expression. Cohesin compresses DNA to form chromatin loops, thereby shortening the distance between enhancers and promoters. The promoter is the transcription start site for mammalian gene expression, determining the location and direction of transcription. The core promoter is a DNA sequence situated approximately 50 bp upstream and downstream of the transcription start site, containing multiple general transcription factor binding sites, and represents the minimal sequence required for initiating gene expression. Enhancers are significant non‐coding elements, typically located in nucleosome‐depleted regions, and contain multiple transcription factor binding sites.
3.1. Promoter
The promoter serves as the starting point for mammalian gene expression and determines the location and direction of transcription. 101 The core promoter is a DNA sequence located approximately 50 base pairs upstream and downstream of the transcription start site, containing multiple universal transcription factor start sites, and is the minimal sequence that initiates gene expression. 102 , 103 The promoter has some transcriptional activity, but it is relatively weak. 104 , 105 In specific cases, promoters can act as enhancers to regulate gene expression by interacting with other promoters. 106 , 107 The transcription preinitiation complex(PIC) assembles at the promoter and binds to the promoter sequence, a prerequisite for gene transcription. 108 , 109 The structure and function of the PIC vary between stages, and the PIC consists of universal transcription factors (TFIIA, TFIIB, TFIID, TFIIE, TFIIF, TFIIH), RNA polymerase II (RNAP II), and other factors in most cases. 110 These general transcription factors play distinct roles in regulating gene expression, with TFIID recognizing and binding to promoter DNA sequences and TFIIB directing other transcription factors and RNAPII to bind to promoter sequences. 111 RNAPII undergoes three stages of transcription initiation, pause, and pause release at the promoter, 112 with RNAPII pausing after about 60 bases of transcription and restarting transcription. 113 The exact mechanism of this process is unclear, and current studies suggest that RNAPII transcription is influenced by various factors. For example, p300/CBP promotes RNAPII transcription initiation and pause release, whereas TFIID is associated with RNAPII pause. 114 , 115
3.2. Enhancer
Enhancers are important noncoding elements that regulate the tissue‐specific expression of genes. 116 Active enhancers typically reside in nucleosome‐deficient regions and contain multiple transcription factor binding sites. 117 , 118 The regulatory function of enhancers is generally direction‐independent, with enhancers separated from their cognate promoters by approximately 104−106 base pairs. 119 Cohesin facilitates enhancer–promoter interactions by shortening the distance between enhancers and promoters through chromatin loop formation. 120 A single enhancer can act directly with its cognate promoter to regulate target gene expression, 121 while multiple enhancers can act on the same target gene and regulate its transcription through synergistic or cumulative effects. 122 , 123 , 124 In addition, superenhancers span longer DNA sequences, often containing multiple common enhancers within them, and can drive higher expression levels of target genes with increased activity. 125 Shadow enhancers are a class of enhancers that partially or completely overlap in expression patterns. 126 During mammalian development, especially under conditions of physiological or genetic stress, these enhancers with similar expression patterns are able to resist genetic variation and keep relatively stable gene expression levels. 127 These seemingly “redundant” enhancers provide a guarantee of accurate gene expression. 128
3.3. Mechanisms of enhancer–promoter interactions
Chromatin loops can shorten the distance between enhancers and promoters, but the interaction between them also requires the involvement of transcription factors and multiple cofactors. 129 Enhancer sequences contain multiple transcription factor binding sites and are able to specifically bind to a variety of transcription factors. Pioneer transcription factors are able to efficiently degrade nucleosomes by binding to DNA sequences through an alpha helix structure, 130 resulting in a chromatin‐open state. 131 , 132
Transcription factors recruit p300/CBP and mediator complexes. 133 P300/CBP, a transcriptional activator with acetyltransferase activity, directly activates enhancers through histone acetylation. 134 The mediator complex serves as a functional hub between the enhancer and the promoter and consists of four parts: head, middle, tail, and kinase module (CDK8 module). 135 , 136 This complex is capable of transmitting genetic information from the enhancer to the promoter. 137 First, the tail of the mediator interacts with transcription factors at the enhancer, whereas its head and middle of the mediator are involved in RNAP II recruitment and assembly of the transcriptional preinitiation complex. 138 The kinase module (CDK8) phosphorylates the carboxy terminus of RNAP II to facilitate transcription initiation. 139 In conjunction with transcription factors and cofactors, enhancers deliver genetic information to the promoter for precise expression of target genes.
4. MECHANISMS OF DYNAMICAL CHANGE OF 3D GENOME AMONG DIFFERENT FUNCTIONAL STATES
The 3D genome undergoes dynamic changes during cell differentiation and organ development to regulate interactions between cis‐regulatory elements, exhibiting diverse functional states. In this section, the mechanisms of these dynamic changes in the 3D genome are summarized.
4.1. Chromatin compartment switches
During mammalian cell differentiation, chromatin compartments exhibit dynamic changes at various stages and significantly influence the spatiotemporal‐specific expression of development‐related genes. 140 Throughout the differentiation of human embryonic stem cells (hESCs), substantial transitions transpire between compartments A and B. 141 , 142 , 143 The number of genomes participating in A–B compartment switches varies among cell types, and the number of TADs involved declines as differentiation progresses. 144 , 145 For instance, when ESCs differentiate into skeletal muscle progenitors, approximately 20% of the genome participates in the A–B compartment switches. In contrast, only 6.5% of genomes are involved in compartmentalization when skeletal muscle progenitor cells further differentiate. 144 , 146 Among the 10 marker genes associated with preadipocyte differentiation, nine are observed to convert from compartment B to A during differentiation. 147 Similarly, Prdn1 and Atf4, related to plasma cell fate determination, are transferred to euchromatin, whereas Ebf1, an inhibitor of their differentiation, transitions from euchromatin to heterochromatin. 148
4.2. Dynamical change of TAD
During mammalian development, TAD facilitates precise gene expression by providing a suitable environment while minimizing interference from external regulatory information. 149 TAD‐D (∼250 kb) and TAD‐E (∼500 kb) are adjacent TADs within the mouse X chromosome inactivation center, containing mutually repressed genes Tsix and Xist, respectively. 54 Xist is associated with X chromosome inactivation, while Tsix represses the expression of Xist. 150 In male mice, embryonic stem cell differentiation (mESCs), Tsix is transcriptionally active, but Xist is barely expressing. 151 In female mESCs, although Tsix is also transcriptionally active, Xist transcript levels gradually increase as mESCs further differentiate, eventually leading to X chromosome inactivation. 152 In male mESCs, a 40 kb inversion across the TAD‐D and TAD‐E boundaries places the promoters of Tsix and Xist inside each other's TAD, respectively. This alteration in the regulatory environment enables enhancers to interact with nontarget promoters, causing promoters that should regulate Tsix gene expression to be ectopically activated within TAD‐E, ultimately leading to aberrant Xist expression in male mESCs. 56
The HoxD gene clusters locate between two adjacent TADs: the telomeric side (T‐DOM) and the centromeric side (C‐DOM). During mammalian limb development, HoxD genes are sequentially regulated by spatiotemporal‐specific enhancers within these two TADs. 153 , 154 , 155 The T‐DOM TAD contains multiple enhancers that regulate proximal limb development by controlling the expression of Hoxd9, Hoxd10, and Hoxd11 genes. 156 In contrast, during subsequent distal limb development, enhancers within T‐DOM are silenced, whereas enhancers within C‐DOM are activated. 157 Enhancer II1, one of the enhancers in C‐DOM responsible for distal limb development, plays a crucial role in tetrapod distal limb development by binding to the transcription factor Hox13. When the enhancer II1 sequence is inserted into T‐DOM, its activity is almost entirely suppressed, even though the transcription factor Hox13 remains explicitly bound to the sequence within enhancer II1. 158 , 159 The regulatory activity of enhancer II1 in the distal limb is restored only after most of the sequence at the mitotic end of T‐DOM (Mtx2‐II1‐T‐DOM) is deleted. 160 Thus, promoter and enhancer activities relate to the chromatin environment during gene expression. 157 TADs, as higher‐order chromatin structures, provide an appropriate regulatory environment for enhancer–promoter interactions, ensuring that development‐related genes are expressed in a cell‐ or tissue‐specific manner.
During cell differentiation, sub‐TADs gradually form within TADs, promoting cis‐regulatory elements to precisely regulate target gene expression. 161 , 162 Runx1, a transcription factor, is associated with hematopoietic development and is situated within a 1.1 Mb TAD, which forms prior to Runx1 gene expression. 163 , 164 Analysis of mESCs reveals the emergence of two sub‐TADs within the Runx1 TAD, promoting tissue‐specific enhancer–promoter interactions. 165 Similarly, observation of α‐globin protein motifs within erythrocytes at different stages of differentiation in mice shows the appearance of sub‐TADs during cell differentiation. 166 , 167
Furthermore, a progressive increase in chromatin accessibility, accompanied by a corresponding rise in the frequency of enhancer–promoter interactions within TADs, is observed during the differentiation of various cell types. 166 For instance, due to changes in enhancer activity, frequent chromatin loop reconnections are observed during the differentiation from preadipocytes to adipocytes. 168
5. ROLE OF DYNAMICAL CHANGE OF 3D GENOME IN GAMETOGENESIS AND EARLY EMBRYOGENESIS
Meiosis plays a crucial role in germ cell development, facilitating the maturation of sperm and oocytes. 169 , 170 Throughout this process, germ cells undergo morphological transformations, and their chromosomes experience intricate 3D structural alterations, ultimately leading to highly differentiated germ cells. 171 In this section, the dynamic changes of the 3D genome in gametogenesis and early embryogenesis are systematically reviewed.
5.1. Role of dynamical change of 3D genome in spermatogenesis
Meiosis I and II enable the development of spermatogonia into spermatozoa, 172 involving homologous chromosome separation and sister chromatid separation to form round spermatids. 169 , 173 Subsequently, these round spermatids undergo a series of complex, dynamic changes to generate mature spermatozoa. 174 During meiotic prophase, although A/B compartments are still present, TAD disappears due to alterations in cohesin activity in mouse spermatocytes. 175 , 176 , 177 In postmeiotic stages, the level of genomic compartmentalization in round spermatocytes gradually increases. Mature spermatocytes exhibit chromatin higher‐order structures such as compartments and TADs, demonstrating frequent remote interactions between TADs. 178 , 179 Comparing Hi‐C data from mature sperm cells, fibroblasts, and ESCs reveals considerable similarity in their 3D genomes. 180 As protamine replaces over 85% of histone loci in mature spermatozoa, chromatin becomes highly compressed, leading to a more than 10‐fold reduction in sperm cell nuclear volume compared with fibroblasts. 180 In contrast, retained histones are highly enriched at the promoters of crucial developmental genes. For instance, nucleosomes modified by active histones were observed at the transcription start sites of genes such as EVX1/2 and ID1. 174 , 181 Moreover, studies on mouse sperm reveal that mature sperm contain over 10,000 enhancers and over 500 superenhancers. 180 , 182 The vast majority of these are identical to those in mESCs, suggesting that regulatory elements are ready for tissue differentiation and cell fate decisions as early as in the sperm. 177
5.2. Role of dynamical change of 3D genome in oogenesis
In contrast to sperm development, oocyte formation begins during the embryonic stage. 183 , 184 Primordial germ cells, originating from the proximal epiblast, differentiate into oogenic cells within the embryonic gonads. These oogenic cells undergo a series of differentiations, eventually forming primary oocytes that enter meiosis I prophase. Upon reaching the diplotene stage of meiosis I, the primary oocytes constitute primordial follicles, which are subsequently stored in the ovary. During puberty, the primordial follicles gradually transition hormonally into germinal vesicle oocytes. 185 These germinal vesicle oocytes continue to undergo hormone‐stimulated meiosis and remain suspended in the metaphase of meiosis II until fertilization. 185
During oogenesis, the 3D genome structure undergoes a series of dynamic changes, characterized by the gradual disappearance of compartment, TAD, and loop. 186 , 187 Meiosis I oocytes and germinal vesicle oocytes exhibit similar numbers of TADs, whereas metaphase II (MII) oocytes display an almost complete absence of TADs and A/B compartments, resulting in a uniform chromatin folding pattern. 188
5.3. Role of dynamical change of 3D genome in zygote
After fertilization, two transcriptionally quiescent gametes fuse to form a zygote, characterized by a more relaxed chromatin structure. 189 , 190 , 191 Transcription remains inactive in zygotes and early embryos. 189 , 192 Concurrent with zygotic genome activation (ZGA), gene expression is activated, and an extensive reorganization of the 3D genome structure occurs. 193 , 194 , 195 Mouse embryo studies reveal a progressive increase in TAD intensity and the extent of chromatin remote interactions at the two‐cell stage. 196 , 197 In contrast, human embryos exhibit compartments and TADs as early as the eight‐cell stage, with continuous enhancement throughout development. 181 , 198 Although the onset of ZGA often coincides with TAD formation in many species, recent research indicates that TAD formation associates with DNA replication and that inhibiting ZGA does not impede TAD formation. 195
6. ROLE OF DYNAMICAL CHANGE OF 3D GENOME IN ORGAN DEVELOPMENT
In addition to playing a crucial role in gametogenesis, dynamic changes in the 3D genome are also important for regulating organ development.
6.1. Role of dynamical change of 3D genome in brain development
The mammalian brain, a highly complex organ, is primarily composed of neurons and neuroglial cells. 199 Glial cells include astrocytes, oligodendrocytes, and microglia. 200 Neurons can be classified as either excitatory or inhibitory. 201 , 202 Apart from microglia, all other cells are derived from neural progenitor cells (NPCs), which strictly regulate the spatiotemporal distribution of gene expression to modulate physiological functions such as memory, cognition, and emotion. 203 , 204
During brain development, ESCs first differentiate into NPCs, which subsequently differentiate into neurons and further into various cell subtypes. 199 Throughout this process, chromatin experiences extensive reorganization and compaction. 205 Initially, the chromatin of ESCs is globally accessible, but as differentiation progresses, it becomes increasingly condensed, reducing accessibility. 206 Concurrently, at the compartment level, the A compartment size decreases by approximately 5% compared with ESCs as neural cell differentiation continues. 16 , 71 Despite this change, TAD boundaries do not exhibit significant alterations; the TAD landscape remains stable in NPCs, neurons, and glial cells. 207 As differentiation ensues, TAD sizes undergo a slight increase, interaction levels between TADs in the A compartment decrease, and those in the B compartment increase. 71
Additionally, human‐specific brain structures, such as the formation of the subplate layer, are associated with the development of human‐specific TADs and chromatin loops. 208 , 209 These human‐specific TADs are generally smaller than conserved TADs between species due to chromatin structure alterations, and their boundary CTCF binding strengths are marginally weaker. 208 Human‐specific enhancers play a crucial role in brain development by effectively regulating target gene tissue‐specific expression. 210 , 211 , 212 For instance, the human‐specific enhancer EOMES controls FGFR2 expression, which is associated with NPCs proliferation. 206 EPHA7 is regulated by an upstream human‐specific enhancer, and its inactivation impacts the development of human cortical pyramidal cells, 208 ARHGAP11B collaborates with a human‐specific enhancer located 500 kb away, contributing to the expansion of the human neocortex. 213
6.2. Role of dynamical change of 3D genome in cardiac development
The heart, originating from the mesoderm, has chromatin regulatory mechanisms in its development process that have long captivated researchers. Primarily composed of cardiomyocytes, the heart derives these cells from cardiac progenitor cells, which in turn arise from distinct subgroups of cardiac mesodermal cells. 214
Cardiomyocyte‐specific compartments form early during heart development and differentiation. 215 Studies reveal that in adult mouse cardiomyocytes, approximately 66.7% of cardiac‐specific genes reside in compartment A. 216 During cardiomyocyte differentiation, around 20% of the genome undergoes compartment conversion, with genes transitioning from compartment B to A demonstrating greater cardiac specificity. 217 , 218 , 219 Moreover, as hESCs or human induced pluripotent stem cells (hiPSCs) differentiate into cardiomyocytes, a decrease in the number of TADs, loss of boundaries, and reduction in intensity are observed. 218 Roughly 70% of TADs remain stable throughout development, suggesting their importance in heart development through the promotion of enhancer–promoter interactions and regulation of developmental gene expression. For instance, Handsdown (Hdn) and Hand2, which are located within the same TAD and closely related to heart development, play a significant role in cardiomyocyte differentiation and heart development. 220 , 221 Hdn encodes a lncRNA and modulates Hand2 transcription by inhibiting its upstream enhancer activity. Hdn deficiency results in an abnormal increase in Hand2 expression, leading to the thickening of the right ventricular wall. 222 Furthermore, structural protein abnormalities associated with TAD formation may cause heart development abnormalities of varying degrees. 214 , 223 , 224 For example, STAG2, one of the cohesin subunits, has its deficiency affecting the proliferation and migration of secondary heart field progenitor cells, leading to delayed heart development and morphological defects. 225 A heterozygous deletion of the adhesion loading factor NIPBL may induce atrial septal defects. 226 , 227
6.3. Role of dynamical change of 3D genome in blood system development
The development and differentiation of hematopoietic stem cells (HSCs) provide a valuable model for 3D genomics research. During embryonic development, HSCs initially appear within the principal arteries and subsequently migrate to the liver. 228 Before birth, HSCs relocate to the bone marrow, where they remain for an extended period. In adult mouse bone marrow, more than 70% of HSCs exist in a quiescent differentiation state. 229
Studies have demonstrated that as ESCs differentiate into HSCs, the spatial arrangement of TADs experiences dynamic shifts coinciding with the transition of A–B compartments. 230 , 231 Within TADs, chromatin accessibility progressively increases, and sub‐TADs form gradually. 165 , 166 Furthermore, research on mouse embryos and adult mouse HSCs has revealed that compartments and TADs are predominantly conserved, with a mere 5% of compartments and 12% of TADs undergoing alterations. 232 In adult mouse HSCs, both compartmentalization and TAD boundary strength augment, whereas chromatin accessibility declines. Approximately 52% of enhancer–promoter interaction levels vary, correlating with differential gene expression. 232
Erythropoiesis refers to the biological process wherein hematopoietic stem cells and progenitor cells differentiate into mature red blood cells. 233 Erythroid progenitor cells proliferate and undergo a sequence of morphological transformations, ultimately yielding enucleated reticulocytes. 234 Throughout erythroid cell differentiation, widespread chromatin condensation occurs, and both chromatin accessibility and transcriptional activity diminish. 235 , 236 Heterochromatin significantly compacts, and H3K9me3 is repositioned, leading to numerous long‐range interactions. 237 Approximately 58% of TADs are disrupted, and TAD boundaries weaken considerably during the final stages of erythroid development. TADs with active chromatin modifications are partially conserved, and GATA1 is thought to participate in the preservation of TADs. 237
6.4. Role of dynamical change of 3D genome in immune system development
B cells and T cells both derive from hematopoietic stem cells in the bone marrow. 238 Within this environment, common lymphoid cells either differentiate into B cells and innate lymphoid‐like cells or migrate to the thymus, where T cell differentiation is initiated. The development of B and T cells is intimately associated with the recombination of antigen gene loci.
Throughout B cell development, the dimensions, quantity, and positions of the majority of TADs remain relatively constant. 239 However, significant gene loci associated with B cell fate determination exhibit dynamic changes. The Ebf1 gene locus encodes an essential B cell transport protein and serves a crucial regulatory function in B cell differentiation. 240 At the pluripotent stage, the Ebf1 locus resides in the transcriptionally repressed B compartment. 241 , 242 As cells transition from pre‐pro‐B cells to pro‐B cells, Ebf1 relocates to the transcriptionally active A compartment, playing a role in B cell fate determination. 239 During the differentiation of B cells into plasma cells, Ebf1 is repositioned to the heterochromatin region. 148
T cell development relies on Bcl11b expression. In the pluripotent stage, Bcl11b is situated within the B compartment, with its activation closely tied to the non‐coding RNA ThymoD. 243 , 244 ThymoD facilitates local DNA demethylation through transcription, promoting CTCF‐DNA binding and cohesin protein recruitment. This process contributes to Bcl11b‐TAD formation and strengthens the interactions between enhancers and promoters. 245
7. 3D GENOME STRUCTURAL VARIATIONS AND DISEASE
The 3D genome structure is pivotal in gene expression regulation. Pathological conditions can lead to extensive reorganization of the higher‐order chromatin structure, which affects the expression of functional genes. The switching of aberrant A/B compartments at the subchromosomal level is closely linked to the onset and progression of the disease. Chromatin structural variations (SVs) at the megabase scale modify the TAD structure and interactions between regulatory elements, resulting in abnormal gene expression levels. Additionally, the formation of disease‐specific chromatin loops alters enhancer–promoter interactions, exacerbating gene expression dysfunction and contributing to disease development. This section systematically reviews the dynamic reorganization of higher‐order chromatin structures at different levels during the onset and progression of diseases. This may provide valuable insights into the complex relationship between chromatin conformation and diseases (Table 1).
TABLE 1.
3D genome structural variations and disease.
| Chromatin Structure | Gene Locus | Diseases | Pathogenic mechanism | Reference |
|---|---|---|---|---|
| Compartment |
Sugcct Fgfr2 |
Nonalcoholic fatty liver disease | Approximately 33% of the genome undergoes A/B compartment conversion. Sugct and FGFR2 are activated in association with Non‐alcoholic fatty liver disease. | 251 |
| Compartment |
WNT5A CDK44 |
Prostate cancer | The metastatic potential and invasiveness of prostate cancer are closely linked to dynamic genome alterations. Forty‐eight genes have shifted from B to A compartments and activated. | 252 |
| Compartment |
HNF4A NR5A2 |
HBV infection | HBV DNA integration into the human genome leads to the remodeling of the 3D genome structure. During this process, dynamic changes occur in the A/B compartments, including HNF4A and NR5A2. | 256 |
| Topologically associating domain | Sox9 | Pierre‐Robin sequence | Deletions of the long‐range enhancer clusters of Ec1.45 and/or Ec1.25 in the Sox9‐TAD result in decreased expression levels of Sox9 and cause. | 267 |
| Topologically associating domain | Ihh | Abnormal Ossification of the Skull | Deletions of enhancers i2–i9 decreased the expression levels of Ihh which lead to abnormal ossification of the skull. Whereas duplication of i2–i9 increased Ihh expression. | 281 |
| Topologically associating domain | HoxD | 2q31 syndrome | Sequence duplications or deletions within HoxD‐TAD resulted in a reduction to similar levels of enhancer–promoter interactions. | 288 |
| Topologically associating domain | Sox9 | Sex reversal/disorder of sex development | Deletion of the Sox9 upstream enhancer eSR‐A(Enh13) results in sex reversal, whereas a sequence duplication involving this enhancer results in a disorder of sex development. | 268 , 277 |
| Topologically associating domain | Pmp22 | Hereditary neuropathy/Charcot Marie tooth. | Deletion of Pmp22‐SE, an upstream enhancer of PMP22, leads to hereditary neuropathy. Sequence duplications involving this enhancer are associated with Charcot‐Marie‐Tooth disease. | 271 |
| Topologically associating domain | Pitx1 | Hindlimb deformity | The enhancer Pen tissue specifically regulates the Pitx1 expression in hindlimb development and aberrant Pitx1 expression by Pen deletion results in hindlimb deformity. | 319 |
| Topologically associating domain |
Epha4 Pax3 |
Brachydac‐tyly | A heterozygous deletion of 1.75‐1.9 Mb on 2q35 spanning the TAD boundaries of EphA4 and Pax3 resulted in TAD fusion. In this fused TAD, an enhancer originally regulating EphA4 interacts with the Pax3 promoter. | 258 |
| Topologically associating domain | MYC‐BDME | T‐cell acute lymphoblastic leukemia | Abnormal MYC transcript levels in T‐cell acute lymphoblastic leukemia patients result from MYC‐TAD fusion with adjacent TADs, causing aberrant MYC promoter interactions with enhancers BDME. | 293 |
| Topologically associating domain | CFTR‐WNT2 | Intestinal neoplasia | A 121.1 kb heterozygous deletion on 7q31.2 located in the border between CFTR and WNT2. Consequently, the enhancer (located in introns 1, 10, and 11), which initially regulates the tissue‐specific expression of CFTR, interacts with the WNT2 promoter. | 297 |
| Topologically associating domain |
CDKN2A MIR31HG |
Pancreatic ductal carcinoma | The proto‐oncogene MIR31HG‐TAD is adjacent to CDKN2A‐TAD, and sequence deletions involving TAD boundaries can result in the fusion of the two TADs. The proto‐oncogene MIR31HG promoter is aberrantly regulated by the enhancer, resulting in an increase in its expression level. | 299 |
| Topologically associating domain |
Sox9 Kcnj2 |
Cooks Syndrome | Duplications spanning the Sox9‐TAD and Kcnj‐TAD give rise to “neo‐TAD”., and result in an enhancer otherwise regulating Sox9 expression incorrectly interacting with the Kcnj2 promoter in the “neo‐TAD”. | 304 |
| Topologically associating domain |
YPEL2 GDPD1 |
Retinitis Pigmentosa | Sequence duplications spanning YPEL2, GDPD1 result in the creation of neo‐TAD, and enhancers originally located in YPEL2‐TAD ectopically regulate GDPD1 expression. | 307 |
| Topologically associating domain | GPR101RBMX | X‐linked acrogigantism | A 600 kb sequence duplication on chromosome Xq26.3 led to a neo‐TAD. Within the neo‐TAD, the GPR101 promoter interacted with the enhancer eRBMX from within the TAD on the centromeric side and up‐regulated the GPR101 transcript levels. | 300 |
| Topologically associating domain |
SHH MNX1 |
Complete pulmonary hypoplasia | Sequence duplications spanning the SHH‐TAD and telomeric TAD generated 2 consecutive neo‐TAD. In the neo‐TAD, MACS1, an enhancer that originally regulates the tissue‐specific expression of SHH interacts with the MNX1 promoter. | 301 |
| Topologically associating domain |
EGFR LINC01446 |
Glioblastoma | In glioblastoma multiforme, a sequence duplication between ZINCO1446 and EGFR on 7p11.2 creates a neo‐TAD, resulting in EGFR being aberrantly activated. | 311 |
| Topologically associating domain | TFAP2A | Branchiooculofacial syndrome | An 89 Mb inversion with a breakpoint within TAD disconnects the TFAP2A gene from enhancers such as Enh100 and Enh105, which resulted in haploinsufficient expression of the TFAP2A gene in human neural crest cells. | 315 |
| Topologically associating domain | Pitx1 | Liebenberg syndrome | Pitx1 is regulated by enhancers RA4 and RA5, and enhancer Pen regulates tissue‐specific expression of Pitx1. A fragment inversion of 113 Kb containing Pen and RA4 places pen in the position of RA4, causing Pitx1 to be transcriptionally activated erroneously by Pen in forelimb development. | 320 |
| Topologically associating domain |
Wnt6 Epha4 |
F syndrome | The heterozygous inversion of 1.1 Mb sequence in 2q36 region crosses the Wnt6‐TAD and Epha4‐TAD boundary caused the Epha4 enhancer to incorrectly regulate wnt6 expression. | 258 |
| Topologically associating domain | PRDM6SNCAIP | Medulloblastoma | The PRDM6 gene, situated at 5q23, is located approximately 600 kb downstream from SNCAIP. Owing to chromosomal inversion, the super‐enhancer initially responsible for regulating SNCAIP interacts with PRDM6. | 324 |
| Topologically associating domain | MEF2C | 5q14.3 microdeletion syndrome | A complex translocation of 1Mb between 5q14.3 and 3q24 results in a disrupted TAD structure that disconnecting MEF2C from its cognate enhancers. | 329 |
| Topologically associating domain |
Kcnj2 Kcnj16 |
Cooks syndrome | The translocation on chr17 disrupts the interaction of the KCNJ2 promoter with its cognate enhancer IMR90. | 312 |
| Topologically associating domain |
GATA2 EVI1 |
Acute myeloid leukemia | Translocations between 3q21 and 3q26.2 have been observed in patients with acute myeloid leukemia, repositioning an enhancer located upstream of the GATA2 gene within the telomeric side TAD. | 335 |
| Topologically associating domain |
MYB QKI |
Angiocentric glioma | Translocations between 2q35 and 13q14 resulting in interaction of the FOXO1 enhancer B116Z with the Pax3 promoter have been observed in rhabdomyosarcoma cell lines. | 337 |
| Topologically associating domain | CCND1IGH | Malignant B cell lymphomas | CCND1 expression levels are increased more than 500‐fold in malignant B cell lymphomas. The t(11; 14) (q13; q32) causes a superenhancer that should intrinsically regulate IGH expression to interact with the CCND1, ultimately leading to a significant increase in CCND1 expression levels. | 333 |
| Chromatin loop |
DDX60L CXXL13 |
Systemic lupus erythematosus | In Systemic lupus erythematosus patients, 391 disease‐specific chromatin loops are present, encompassing crucial inflammation‐ and immunity‐related genes, including DDX60L and CXCL13. | 373 |
| Chromatin loop |
NPPA NPPB |
Dilated cardiomyopa‐thy | During Dilated cardiomyopathy development, enhancer–promoter loop dynamic remodeling occurs extensively across the genome in response to rapid transcription. | 376 |
| Chromatin loop | LIPC | Pancreatic cancer | In metastatic pancreatic cancer cells, the number of chromatin loops increases LIPC expression is modulated by Enhancer 3 and Enhancer 4, while tissue‐specific chromatin loops form progressively during pancreatic cancer distant metastasis. | 378 |
| Chromatin loop | MYCN | Acute myeloid leukemia | Disease‐specific chromatin loop formation has been observed in AML, involving oncogenes. The specific interaction between the MYCN promoter and enhancers situated 650 kb downstream is related to AML onset. | 379 |
7.1. Compartment and disease
The abnormal conversion of A/B compartments under pathological conditions may result in dysregulated gene expression, playing a crucial role in the onset and progression of diseases. 246 Nonalcoholic fatty liver disease (NAFLD) is a chronic liver disease primarily observed in obese individuals, characterized by hepatic fat accumulation. 247 NAFLD increases the risk of liver cirrhosis and hepatoma. 248 Studies on NAFLD‐induced mice have revealed that around 33% of the genome undergoes A/B compartment conversion. Sugct and Fgfr2 are examples of genes transferred to the A compartment, linked to lipid metabolism disorders and tumor development potential, respectively. 249 , 250 , 251 This finding enhances our understanding of NAFLD pathogenesis, disease progression, and the pathological mechanisms that make patients susceptible to liver cancer. 251 The metastatic potential and invasiveness of prostate cancer are also intimately connected to dynamic genome alterations. Recent in vitro investigations on prostate cancer metastasis have revealed that during cancer progression, there is a widespread genome compartment conversion, which results in significant changes in the nuclear chromatin activation environment, leading to increased mixing and interaction in the A compartment. 252 Forty‐eight genes have been transferred from transcriptionally repressed B compartments to A compartments and activated, including genes associated with prostate cancer progression and poor prognoses, such as WNT5A, CDK44, and TMPRS22. These research findings reveal potential key factors in the pathogenesis and progression of prostate cancer. 252
Additionally, viruses can reshape the host's 3D genome structure, which affects gene expression and is closely linked to disease progression. The Hepatitis B virus (HBV) is a leading cause of hepatocellular carcinoma. 253 Integration of HBV DNA into the human genome results in the remodeling of the 3D genome structure. 254 , 255 During this process, there are dynamic changes in the A/B compartments, mainly in the genomic regions on chromosomes 9, 13, and 21. Most genes are moved from transcriptionally repressed B compartments to A compartments, where they are subsequently activated. 256 These regions have many transposable elements that promote viral DNA replication. A smaller subset of regions moves from the A compartment to the B compartment, including enhancers and genes associated with the regulation of inflammatory responses. 256 Research on human cells infected with SARS‐CoV‐2 shows that about 30% of the genome undergoes compartment switching, which is accompanied by a decrease in chromatin interaction levels within the A compartment. This may be linked to the high incidence of acute sequelae resulting from SARS‐CoV‐2 infection. 257
7.2. TAD and disease
Approximately 5% of the human genome exhibits structural variation. 258 SVs include both balanced and unbalanced rearrangements, which lead to altered expression levels of functional genes by impacting the TAD structure. This results in aberrant enhancer–promoter interactions, contributing to the development of numerous human diseases (Table 1). 259 Copy number variation comprises chromatin deletions and duplications, whereas inversions and translocations represent balanced rearrangements. 25 This process is distinct from chromatin remodeling during cell cycle progression, which occurs in the nucleus without concomitant changes. 173 , 260 Copy number variants within the same TAD can alter gene expression by influencing the amount of regulatory elements or the frequency of interactions. 261 Structural variations between TADs can affect gene expression by disrupting enhancer–promoter interactions or creating ectopic interactions (Figure 4).
FIGURE 4.
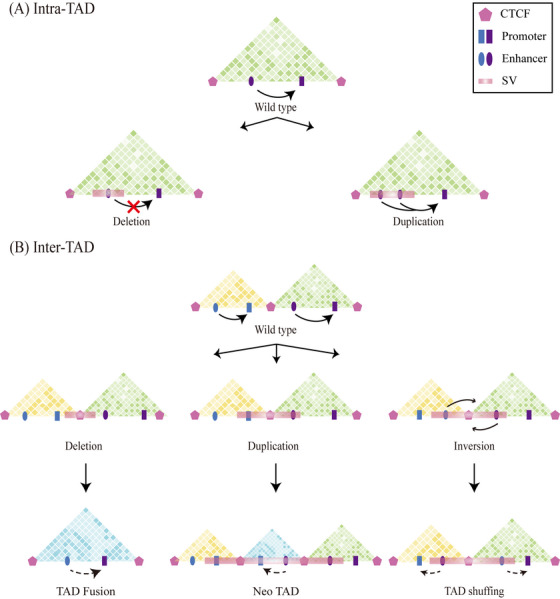
Structural variation located between TAD. (A) Sequence deletions or duplications within TADs primarily affect regulatory functions by altering the number of enhancers and causing abnormal expression of target genes. (B) Sequence deletions at the boundaries result in the merging of adjacent TADs into one, forming a new TAD, referred to as “TAD fusion.” Duplication of DNA sequences containing boundary elements can create a “neo‐TAD.” The “neo‐TAD” is situated between the original TADs, wherein the interacting enhancers and promoters originate from different initial TADs, and their interactions do not interfere with the enhancer–promoter interactions within the original TADs. Chromosomal inversions occurring between adjacent TADs alter the position and/or orientation of DNA segments, placing genes and/or regulatory elements in different chromosomal contexts, and causing pathological effects known as “TAD shuffling”.
Additionally, recent studies have shown that structural variation is a significant mechanism contributing to cancer. 262 , 263 Genomic rearrangements cause transcriptional dysregulation of proto‐oncogenes and oncogenes by changing interactions between cis‐regulatory elements in somatic cells, ultimately leading to abnormal proliferation and differentiation of somatic cells. 264 , 265 , 266
7.2.1. Deletions and duplications within TADs lead to diseases
Copy number variations within TADs significantly modulate gene expression by changing the quantity of regulatory elements or the frequency of interactions between them. Such changes can cause the dysregulation of gene expression, contributing to various human diseases and developmental disorders (Figure 4A). 267 , 268 , 269 , 270 , 271 , 272
Sox9 is located within a 2 Mb TAD, 273 , 274 containing multiple tissue‐specific enhancers that participate in mammalian sex determination, craniomaxillofacial development, and chondrogenesis. 268 , 275 , 276 , 277 Located 1.45 and 1.25 Mb upstream of the Sox9 gene, remote enhancer clusters Ec1.45 and Ec1.25 regulate its tissue‐specific expression in the mandibular process and the first branchial arch region. 267 Deletion of Ec1.45 and/or Ec1.25 in the Sox9‐TAD leads to reduced Sox9 expression levels, causing Pierre–Robin sequence, 267 a group of craniomaxillofacial developmental malformations such as mandibular dysplasia, cleft palate, and tongue recession. 278 , 279 Specifically, deletion of Ec1.45 in the human genome can cause a decrease of over 50% in the expression level of Sox9, 267 whereas simultaneous deletion of both appears to have a more substantial impact on Sox9 expression. 280
The Ihh gene participates in skeletal development, and its expression is regulated by at least nine enhancers (i1–i9) located within Ihh‐TAD. 281 Deletion of Ihh in mouse models causes joint fusion and skeletal shortening, 282 whereas Ihh duplication is associated with finger deformities and premature closure of cranial sutures. 283 , 284 Deleting enhancers i2–i9 within Ihh‐TAD in mice results in a 98% reduction of Ihh mRNA expression levels, leading to abnormal cranial ossification, reduced bone cortex, and shortened extremities. 281 In contrast, duplication of i2–i9 enhances Ihh‐TAD endointeraction, increasing the expression of Ihh to five‐fold in the head and limbs, which results in premature closure of the cranial suture and syndactyly. 281
It is worth noting that not all intra‐TAD fragment duplications result in increased enhancer–promoter interactions and target gene expression levels within the TAD. Alterations in enhancer–promoter distances can influence interaction frequency and, as a result, impact gene expression. 285 2q31 syndrome, characterized by facial malformations, mental retardation, and limb deformities, 286 can result from sequence duplication or deletion within HoxD‐TAD. 287 , 288 This occurs because sequence duplication increases the distance between the enhancer and gene, which weakens the interaction between the HoxD promoter and its enhancer, resulting in a 50% downregulation of the HoxD gene cluster transcript levels. In this case, sequence duplication within HoxD‐TAD leads to similar changes in gene expression levels as deletion. 288
7.2.2. Deletions between TADs lead to diseases
Copy number variations across TAD boundaries can disrupt chromosomal rearrangements of boundary elements. Deletions at the boundaries can merge adjacent TADs, resulting in a “TAD fusion”, 258 and can lead to abnormal gene expression levels as enhancers from different TADs interact with promoters (Figure 4B). 289 Structural variation has been identified as a crucial mechanism underlying cancer development. 262 , 263 Genomic rearrangements can cause transcriptional dysregulation of proto‐oncogenes and oncogenes by altering interactions between cis‐regulatory elements in somatic cells, leading to abnormal proliferation and differentiation. 264 , 265 , 266
Congenital diseases
An example of chromosomal rearrangement that impacts gene expression is the Wnt6/Ihh/Epha4/Pax3 locus located on chromosome 2q35‐36. A heterozygous deletion of 1.75–1.9 Mb in the 2q35 region results in short‐fingered malformation in humans and mice. This deletion disrupts the TAD boundary between Epha4 and Pax3, leading to TAD fusion and producing an 800 kb fused TAD. 258 Within this fused TAD, the enhancer that initially regulated Epha4 interacts with the Pax3 promoter, causing an increased expression level of Pax3 and a decreased expression level of Epha4, ultimately leading to the development of short‐fingered malformations.
Not all deletions across TAD boundaries lead to TAD fusion. In the mouse genome, adjacent motifs Sox9‐Kcnj show that deleting only the CTCF locus at the boundary does not result in TAD fusion. TAD fusion occurs only after deleting all four CTCF loci within the TADs. Only deleting all four CTCF loci within the TADs leads to TAD fusion, but it does not significantly affect gene expression. 290 The limited impact on gene expression resulting from small deletions may be due to the redundancy of CTCF sites in the TADs. This redundancy mechanism helps maintain the structural and functional stability of TADs and ensures precise gene expression.
Cancers
The oncogene MYC is a critical downstream target gene of Notch1 signaling. 238 , 291 Its expression levels are frequently elevated in patients with T‐cell acute lymphoblastic leukemia (T‐ALL). 292 Recent studies have shown that deletions of TAD boundaries in T‐ALL patients result in the fusion of MYC‐TAD with adjacent TADs, leading to abnormal interactions between the enhancer BDME/BENC and the MYC promoter within the fused TAD. This results in increased expression of MYC and the development of T‐ALL. 293
CFTR and WNT2 genes are located in adjacent TADs on chromosome 7. Research on patients with intestinal neoplasia pedigrees has shown that a 121.1 kb heterozygous deletion on 7q31.2 disrupts the border between CFTR and WNT2 and deletes the CFTR promoter sequence. 294 Consequently, the enhancer located in introns 1, 10, and 11 that originally regulates the tissue‐specific expression of CFTR interacts with the WNT2 promoter, resulting in increased levels of WNT2 expression and decreased levels of CFTR expression in this pedigree. 294 , 295 , 296 This dysregulation is associated with the development of intestinal adenocarcinoma and small intestinal neuroendocrine tumors. 295 , 297
The majority of patients with pancreatic ductal carcinoma present a homozygous deletion of CDKN2A. 298 Recent studies have shown that MIR31HG‐TAD is adjacent to CDKN2A‐TAD in various pancreatic ductal carcinoma cell lines. Deletions in the boundary regions of the two TADs result in their fusion. In the fused TADs, the MIR31HG promoter is abnormally regulated by the enhancer, leading to increased expression levels. 299
7.2.3. Duplications between TADs lead to diseases
Duplication of DNA sequences containing boundary elements can result in the formation of “neo‐TADs”, 300 , 301 , 302 which are located between the original TADs. The enhancers and promoters within these “neo‐TADs” originate from different initial TADs, but their interactions do not interfere with the enhancer–promoter interactions in the original TADs (Figure 4B).
Congenital diseases
Cooks syndrome is linked to chromosomal duplications and is characterized by nail hypoplasia and short‐fingered malformations. 303 The duplication of the Sox9‐TAD and Kcnj‐TAD creates a “neo‐TAD”, as evidenced by RNA‐seq results from mouse limbs at various developmental stages that show no changes in the expression levels of Sox9 and Kcnj16, but an increase in Kcnj2 expression. 304 This rise in Kcnj2 expression is a result of misinteraction between the enhancer that originally regulated Sox9 expression and the Kcnj2 promoter within the “neo‐TAD”.
Retinitis pigmentosa is a common inherited retinal disease characterized by progressive peripheral vision loss and night blindness, which can lead to blindness in severe cases. 305 The disease is associated with a chromosome 17q22 duplication spanning the YPEL2 and GDPD1 genes. YPEL2 is highly expressed in the brain and retina, 306 whereas GDPD1 encodes glycerophosphodiesterase and is predominantly expressed in the brain and testis. 307 This duplication leads to the formation of new interaction domains, where enhancers originally located in YPEL2‐TAD regulate GDPD1 expression ectopically. 307 Elevated GDPD1 expression levels can cause dysregulation of lipid metabolism, a pathogenic factor contributing to photoreceptor cell inactivation and the development of retinitis pigmentosa. 308 , 309 , 310
Cancer
EGFR and LINC01446 are situated in adjacent TADs on chromosome 7p11.2. In cell lines derived from glioblastoma patients, a sequence duplication was observed between EGFR and LINC01446, generating a “neo‐TAD” between the two adjacent TADs. Within the “neo‐TAD,” the enhancer initially intended to regulate LINC01446 expression aberrantly interacts with the promoter responsible for regulating EGFR expression, leading to an increase in EGFR expression levels. This is one of the pathological mechanisms that contribute to the development of glioblastoma. 311
7.2.4. Inversion between TADs leads to diseases
Balanced chromosomal rearrangements, including inversions and translocations, occur between spatially adjacent or separated TADs, changing the position and/or orientation of DNA segments. 27 Balanced rearrangements cause pathological effects by placing genes and/or regulatory elements in different chromosomal environments, a phenomenon known as “TAD shuffling”. 312 TADs that experience balancing rearrangements result in abnormal gene expression due to disrupted enhancer–promoter interactions, enhancer hijacking effects, or positional effects (Figure 4B). 27
Congenital diseases
The mechanisms described above can explain the etiology of chromosomal inversions causing congenital diseases such as branchiooculofacial syndrome and Liebenberg syndrome. Branchiooculofacial syndrome is a rare developmental defect caused by heterozygous deletions or mutations in the TFAP2A gene. 313 , 314 In patients with branchiooculofacial syndrome, an 89Mb inversion was found with a breakpoint located in the TAD, which disconnects the TFAP2A gene from enhancers such as Enh100 and Enh105, leading to haploinsufficient expression of TFAP2A in human neural crest cells. 315
Liebenberg syndrome is characterized by a heterozygous leg‐arm transformation, where the upper limb exhibits morphological features typically seen in the lower limb. 316 , 317 Pitx1 is expressed in the hindlimbs, pituitary gland, and first‐gill arch, and gene rearrangements involving Pitx1 are associated with the disorder. 318 , 319 During limb development, Pitx1 is regulated by enhancers RA4 and RA5, in both the anterior and posterior limbs. 320 However, enhancer Pen specifically regulates Pitx1 expression in the hindlimbs, and there is no interaction with Pitx1 during forelimb development. 318 , 320 In Liebenberg syndrome, an inversion of a 113‐kb fragment containing enhancers Pen and RA4 results in the placement of Pen in the position of RA4. This misplacement causes Pitx1 to be activated by Pen during forelimb development, leading to the development of phenotypes such as radial curvature, patellar heterotaxy, and shortened ulnar hawk in adult mice. 320
Cancers
Medulloblastoma is a highly malignant tumor of the central nervous system that commonly occurs in children. 321 , 322 , 323 Recent studies suggest that chromosomal inversions may significantly contribute to the development of this disease. 324 The PRDM6 gene, encoding a histone transferase, is located approximately 600 kb downstream from SNCAIP at 5q23. Chromosomal inversion leads to the interaction between the super‐enhancer responsible for regulating SNCAIP and PRDM6, resulting in a significant increase of approximately 20‐fold in PRDM6 expression levels. 262 , 324
7.2.5. Translocations between TADs lead to diseases
Congenital diseases
Chromosomal translocations can also cause developmental defects through pathological mechanisms akin to inversions. MEF2C is considered one of the crucial pathogenic genes in the 5q14.3 microdeletion syndrome, 325 , 326 which is associated with developmental brain malformations, epilepsy, and intellectual deficits. 327 , 328 A 1 Mb complex translocation between 5q14.3 and 3q24 disrupts the TAD structure, disconnecting MEF2C from the enhancer that regulates its expression. 329 Chromosomal translocations reduce MEF2C expression to 50% compared with controls, 329 a level comparable to that observed in humans with MEF2C heterozygous deletions. 330
Cancers
A chromosomal translocation involving 11q13 and 14q32 has been identified in multiple myeloma and mantle cell lymphoma, and is associated with the CCND1 and IGH gene loci. 331 The proto‐oncogene CCND1, situated on chromosome 11, encodes a cell cycle protein, while the IGH gene is located on chromosome 14. 332 This translocation allows the superenhancer, which initially regulates IGH, to aberrantly activate CCND1 expression, leading to an up to 500‐fold increase in its expression levels. 333
Acute myeloid leukemia (AML) is linked to aberrant expression of the stem cell factor EVI1. Chromosomal translocations involving 3q21 and 3q26.2 have been observed in AML patients. 334 These rearrangements permit the upstream enhancer of the GATA2 gene to relocate and aberrantly interact with the EVI1 promoter, leading to increased EVI1 expression and GATA2 haploinsufficiency. As a result, hematopoietic stem cell growth and differentiation are impeded, contributing to the development of AML. 335
Angiocentric glioma, a low‐grade malignant glioma, is associated with the MYB‐QKI rearrangement. 336 The proto‐oncogene MYB and the oncogene QKI reside at 6q23.3 and 6q25.3, respectively. Chromosomal translocations lead to the enhancers Q3SE1 and Q3SE2, which initially regulate QKI transcription, aberrantly activating the proto‐oncogene MYB expression. Consequently, the fusion protein MYB‐QKI, a proto‐oncoprotein, contributes to the development of angiocentric glioma. 337
Chromatin remodeling can contribute to cancer development by altering the local 3D genomic structure. Human papillomavirus (HPV) genes can integrate into the human genome, facilitating cervical cancer development through TAD structure remodeling. 338 , 339 PEG3 and CCDC16, genes co‐located within the same TAD on chromosome 19, are impacted by this alteration. 340 , 341 HPV reshapes the TAD structure, dividing it into two unequal TADs. As a result, enhancers initially regulating PEG3 aberrantly activate the proto‐oncogene CCDC16 expression, contributing to cervical cancer development. 338
7.2.6. Abnormalities in structural proteins lead to diseases
The “loop extrusion” model, mediated by CTCF, cohesin, and their regulators, effectively elucidates TAD formation and remote enhancer–promoter interactions within TAD. 342 Previous research has demonstrated distinct functions of cohesin and CTCF in TAD formation. 78 , 79 Abnormalities in cohesin or CTCF function can result in aberrant gene expression due to alterations in higher chromatin structure. Mutations in genes encoding cohesin subunits or regulatory factors can disrupt TAD formation and chromatin interactions, affecting normal gene expression. 343 These mutations, which impact cohesin activity and function, are termed “cohesinopathies”. 344 Cornelia de Lange syndrome (CdLS) is characterized by intellectual disability, microcephaly, growth retardation, and upper limb deformities. 345 CdLS pathogenesis is linked to cohesin dysfunction. 344 , 345 NIPBL mutations are found in 65% of CdLS patients. 346 , 347 Normally, the cohesin loader NIPBL introduces cohesin into the promoter of highly expressed genes, 84 , 348 , 349 facilitating its movement along chromatin fibers. 87 , 350 In primary fibroblasts derived from CdLS patients, cohesin is still loaded at specific sites, but its chromatin fiber binding stability is reduced. 351 The NIPBL mutation decreases cohesin mobility and ultimately affects the DNA loop extrusion process and TAD formation. 351
CTCF is involved in forming chromosomal higher‐order structures and plays a crucial role in cell differentiation and apoptosis. 352 , 353 , 354 Located at TAD boundaries, CTCF avoids ectopic contact with enhancers while promoting high‐frequency enhancer–promoter interactions within the TAD. However, CTCF is highly sensitive to methylation levels, 355 , 356 and abnormally elevated methylation levels at CTCF‐DNA binding sites cause disruption of the TAD boundaries, 357 leading to a decrease in enhancer–promoter interactions within the TAD and an increase in inter‐TAD interactions. 78 This mechanism was first identified in the study of isocitrate dehydrogenase (IDH) mutant gliomas. IDH mutant glioma patients have increased levels of DNA methylation, 357 , 358 resulting in ectopic interaction between the enhancer located 50 kb upstream of FIP1L1 and the proto‐oncogene PDGFRA promoter, leading to a three‐fold increase in the expression level of proto‐oncogene PDGFRA and promoting glioma cell proliferation. 359 Similarly, in regions of pathogenic short tandem repeats, local DNA methylation levels elevate and affect local CTCF binding sites. 360 , 361 Short tandem repeats comprise repeats of three or more base pairs in a DNA sequence. They make up approximately 1% of the human genome and are generally non–pathogenic. 362 Recent Hi‐C data suggested that in various congenital disorders, such as fragile X syndrome, Friedreich's ataxia, and Huntington's disease, pathogenic short tandem repeat sequences are located at the TAD boundary. 363 , 364 , 365 , 366 These pathogenic short tandem repeat regions have abnormally elevated local methylation levels and alter the TAD boundaries, contributing to many congenital disorders. 367 , 368 For example, FMR1 has been identified as the pathogenic gene in fragile X syndrome. 369 Disruption of the FMR1‐TAD boundary due to increased local methylation in the genome of Fragile X syndrome patients results in disrupted FMR1 interaction with telomere orientation cognate enhancers and decreased FMR1 expression. 370
7.3. Chromatin loop and disease
During disease development, chromatin loops experience reprogramming in a cell‐specific manner, regulating gene expression. Systemic lupus erythematosus (SLE) is an autoimmune disease frequently involving multiple organs. 371 In CD4+ T cells derived from SLE patients, 391 disease‐specific chromatin loops are present, encompassing crucial inflammation‐related and immunity‐related genes, including DDX60L and CXCL13. 372 , 373 Notably, the DDX60L locus contains two disease‐specific chromatin loops, whose formation is associated with histone modifications in the promoter region mediated by transcription factor SPI1. 372 , 373
Dilated cardiomyopathy (DCM) is a leading cause of heart failure. 374 , 375 Recent studies have revealed that during DCM development, enhancer–promoter loop dynamic remodeling occurs extensively across the genome in response to rapid transcription under cardiac stress conditions. 376 Chromatin loops reprogramming, directly driven by transcription factor HAND1, results in elevated expression levels of DCM‐associated pathogenic genes. 376 For instance, the NPPA‐AS1 promoter possesses enhancer functions, interacting with NPPA and NPPB promoters during DCM development and leading to the co‐transcription of NPPA and NPPB. 376
Distant metastasis in pancreatic cancer significantly contributes to its poor prognosis. 377 Recent investigations suggest that pancreatic cancer distant metastasis correlates with epigenetic alterations. 378 In metastatic pancreatic cancer cells, the number of chromatin loops increases, along with the emergence of cell‐specific chromatin loops. LIPC, a gene promoting pancreatic cancer metastasis, is implicated in tumor cell migration and invasion. 378 LIPC expression is modulated by Enhancer 3 and Enhancer 4, while tissue‐specific chromatin loops form progressively during pancreatic cancer distant metastasis, enhancing LIPC expression. 378 Additionally, disease‐specific chromatin loop formation has been observed in AML, involving oncogenes such as MYCN, WT1, and RUNX1. 379 For example, a specific interaction between the MYCN promoter and enhancers situated 650 kb downstream is related to AML onset. 379
8. CONCLUSION AND PERSPECTIVE
The 3D genome structure and its functions have long been a focal point. In recent years, significant advances have been made in this area due to the rapid development of chromatin conformation capture techniques and super‐resolution fluorescence imaging technologies. In this review, the structural hierarchy of the 3D genome, the effect and mechanisms of cis‐regulatory element interactions in the 3D genome for regulating spatiotemporally specific gene expression, the role and mechanisms of dynamic changes in 3D chromatin conformation during embryonic development, and the pathological mechanisms of diseases such as congenital developmental abnormalities and cancer, which are attributed to alterations in 3D genome organization and aberrations in key structural proteins, were systematically discussed. In summary, the 3D genome structure plays crucial roles in cell differentiation and disease development by regulating spatiotemporal gene expression, which may offer some clues for precise diagnosis and treatment of related diseases.
Nevertheless, further research is needed to understand the fundamental principles of 3D genome organization and the relationship between 3D genome structure and spatiotemporal gene expression. The recent development of single‐cell chromatin conformation capture techniques and genome architecture mapping technologies has enabled a more in‐depth exploration of the structural and functional features of the 3D genome. This also includes the mechanisms by which chromatin higher‐order structures regulate cell‐type‐specific gene expression, thereby shedding light on the impact of the genome's spatial organization on cell differentiation and fate determination.
Furthermore, SVs and abnormalities in structural proteins can influence the function of cis‐regulatory elements, leading to atypical gene expression and, consequently, various diseases. Advancements in chromatin conformation capture techniques and transcriptomics will facilitate a deeper understanding of the pathological mechanisms underlying developmental defects and cancer. This will provide new theoretical insights and research directions for prenatal screening and precision diagnosis and introduce novel therapeutic targets for treating a more comprehensive range of congenital diseases and malignant tumors.
AUTHOR CONTRIBUTIONS
H.L. wrote the manuscript and reviewed the literature. H.T. wrote the manuscript and drew the pictures. M.Y. reviewed the literature and drew the pictures. G.L. wrote the manuscript and reviewed the literature. Q.B. selected and revised the article. G.D. selected and revised the article. D.W. selected and proofread the article. J.D. selected, proofread, and revised the article. All authors read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interest.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (82071097) and the Shanghai Pujiang Program (2020PJD026). Key Laboratory of Oral Biomedical Research of Zhejiang Province Foundation (2021M007), and the Research Project of Shanghai Municipal Health Commission (20234Z0006).
Liu H, Tsai H, Yang M, et al. Three‐dimensional genome structure and function. MedComm. 2023;4:e326. 10.1002/mco2.326
Contributor Information
Gang Ding, Email: dinggang@wfmc.edu.cn.
Dandan Wu, Email: 114067@sh9hospital.org.cn.
Jiewen Dai, Email: daijiewen@163.com.
DATA AVAILABILITY STATEMENT
Data are available upon request from the authors.
REFERENCES
- 1. Stadhouders R, Filion GJ, Graf T. Transcription factors and 3D genome conformation in cell‐fate decisions. Nature. 2019;569(7756):345‐354. [DOI] [PubMed] [Google Scholar]
- 2. Cremer T, Cremer M. Chromosome Territories. Cold Spring Harbor Perspectives in Biology. 2010;2(3):a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mohanta TK, Mishra AK, Al‐Harrasi A. The 3D Genome: from Structure to Function. Int J Mol Sci. 2021;22(21):11585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cremer T, Cremer C. Rise, fall and resurrection of chromosome territories: a historical perspective. Part I. The rise of chromosome territories. European Journal of Histochemistry: EJH. 2006;50(3):161‐176. [PubMed] [Google Scholar]
- 5. Cremer T, Cremer C. Rise, fall and resurrection of chromosome territories: a historical perspective. Part II. Fall and resurrection of chromosome territories during the 1950s to 1980s. Part III. Chromosome territories and the functional nuclear architecture: experiments and models from the 1990s to the present. European Journal of Histochemistry: EJH. 2006;50(4):223‐272. [PubMed] [Google Scholar]
- 6. Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295(5558):1306‐1311. [DOI] [PubMed] [Google Scholar]
- 7. Simonis M, Klous P, Splinter E, et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture‐on‐chip (4C). Nat Genet. 2006;38(11):1348‐1354. [DOI] [PubMed] [Google Scholar]
- 8. Zhao Z, Tavoosidana G, Sjölinder M, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra‐ and interchromosomal interactions. Nat Genet. 2006;38(11):1341‐1347. [DOI] [PubMed] [Google Scholar]
- 9. Lieberman‐Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long‐range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li M, Gan J, Sun Y, et al. Architectural proteins for the formation and maintenance of the 3D genome. Sci China Life Sci. 2020;63(6):795‐810. [DOI] [PubMed] [Google Scholar]
- 11. Hafner A, Boettiger A. The spatial organization of transcriptional control. Nat Rev Genet. 2023;24(1):53‐68. [DOI] [PubMed] [Google Scholar]
- 12. Jerkovic I, Cavalli G. Understanding 3D genome organization by multidisciplinary methods. Nat Rev Mol Cell Biol. 2021;22(8):511‐528. [DOI] [PubMed] [Google Scholar]
- 13. Oudelaar AM, Higgs DR. The relationship between genome structure and function. Nat Rev Genet. 2021;22(3):154‐168. [DOI] [PubMed] [Google Scholar]
- 14. Fritz AJ, Barutcu AR, Martin‐Buley L, et al. Chromosomes at Work: organization of Chromosome Territories in the Interphase Nucleus. J Cell Biochem. 2016;117(1):9‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rao SS, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665‐1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dixon JR, Jung I, Selvaraj S, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518(7539):331‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dekker J, Mirny L. The 3D Genome as Moderator of Chromosomal Communication. Cell. 2016;164(6):1110‐1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mirny LA, Imakaev M, Abdennur N. Two major mechanisms of chromosome organization. Curr Opin Cell Biol. 2019;58:142‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krietenstein N, Abraham S, Venev SV, et al. Ultrastructural Details of Mammalian Chromosome Architecture. Mol Cell. 2020;78(3):554‐565. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zheng H, Xie W. The role of 3D genome organization in development and cell differentiation. Nat Rev Mol Cell Biol. 2019;20(9):535‐550. [DOI] [PubMed] [Google Scholar]
- 21. Misteli T. The Self‐Organizing Genome: principles of Genome Architecture and Function. Cell. 2020;183(1):28‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rowley MJ, Corces VG. Organizational principles of 3D genome architecture. Nat Rev Genet. 2018;19(12):789‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Phillips‐Cremins JE, Sauria ME, Sanyal A, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153(6):1281‐1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krijger PH, de Laat W. Regulation of disease‐associated gene expression in the 3D genome. Nat Rev Mol Cell Biol. 2016;17(12):771‐782. [DOI] [PubMed] [Google Scholar]
- 25. Spielmann M, Mundlos S. Structural variations, the regulatory landscape of the genome and their alteration in human disease. Bioessays. 2013;35(6):533‐543. [DOI] [PubMed] [Google Scholar]
- 26. Stevens TJ, Lando D, Basu S, et al. 3D structures of individual mammalian genomes studied by single‐cell Hi‐C. Nature. 2017;544(7648):59‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Finn EH, Misteli T. Molecular basis and biological function of variability in spatial genome organization. Science. 2019;365(6457):eaaw9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ibrahim DM, Mundlos S. Three‐dimensional chromatin in disease: what holds us together and what drives us apart? Curr Opin Cell Biol. 2020;64:1‐9. [DOI] [PubMed] [Google Scholar]
- 29. Furlong EEM, Levine M. Developmental enhancers and chromosome topology. Science. 2018;361(6409):1341‐1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kempfer R, Pombo A. Methods for mapping 3D chromosome architecture. Nat Rev Genet. 2020;21(4):207‐226. [DOI] [PubMed] [Google Scholar]
- 31. McCord RP, Kaplan N, Giorgetti L. Chromosome conformation capture and beyond: toward an integrative view of chromosome structure and function. Mol Cell. 2020;77(4):688‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421(6921):448‐453. [DOI] [PubMed] [Google Scholar]
- 33. Shah S, Takei Y, Zhou W, et al. Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell. 2018;174(2):363‐376. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boyle S, Rodesch MJ, Halvensleben HA, Jeddeloh JA, Bickmore WA. Fluorescence in situ hybridization with high‐complexity repeat‐free oligonucleotide probes generated by massively parallel synthesis. Chromosome Research: An International Journal on the Molecular, Supramolecular and Evolutionary Aspects of Chromosome Biology. 2011;19(7):901‐909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Osborne CS, Chakalova L, Brown KE, et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet. 2004;36(10):1065‐1071. [DOI] [PubMed] [Google Scholar]
- 36. Wang Y, Qu Z, Fang Y, et al. Chromosome territory reorganization through artificial chromosome fusion is compatible with cell fate determination and mouse development. Cell discovery. 2023;9(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA. Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol. 1999;145(6):1119‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cremer M, von Hase J, Volm T, et al. Non‐random radial higher‐order chromatin arrangements in nuclei of diploid human cells. Chromosome Research: An International Journal on the Molecular, Supramolecular and Evolutionary Aspects of Chromosome Biology. 2001;9(7):541‐567. [DOI] [PubMed] [Google Scholar]
- 39. Bolzer A, Kreth G, Solovei I, et al. Three‐dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 2005;3(5):e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sexton T, Yaffe E, Kenigsberg E, et al. Three‐dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148(3):458‐472. [DOI] [PubMed] [Google Scholar]
- 41. Manzo SG, Dauban L, van Steensel B. Lamina‐associated domains: tethers and looseners. Curr Opin Cell Biol. 2022;74:80‐87. [DOI] [PubMed] [Google Scholar]
- 42. van Steensel B, Belmont AS. Lamina‐associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell. 2017;169(5):780‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Briand N, Collas P. Lamina‐associated domains: peripheral matters and internal affairs. Genome Biol. 2020;21(1):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bizhanova A, Kaufman PD. Close to the edge: heterochromatin at the nucleolar and nuclear peripheries. Biochimica et Biophysica Acta Gene Regulatory Mechanisms. 2021;1864(1):194666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. van Bemmel JG, Pagie L, Braunschweig U, et al. The insulator protein SU(HW) fine‐tunes nuclear lamina interactions of the Drosophila genome. PLoS One. 2010;5(11):e15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Meuleman W, Peric‐Hupkes D, Kind J, et al. Constitutive nuclear lamina‐genome interactions are highly conserved and associated with A/T‐rich sequence. Genome Res. 2013;23(2):270‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet. 2009;41(2):246‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Madsen‐Østerbye J, Abdelhalim M, Baudement MO, Collas P. Local euchromatin enrichment in lamina‐associated domains anticipates their repositioning in the adipogenic lineage. Genome Biol. 2022;23(1):91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Peric‐Hupkes D, Meuleman W, Pagie L, et al. Molecular maps of the reorganization of genome‐nuclear lamina interactions during differentiation. Mol Cell. 2010;38(4):603‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Robson MI, de Las Heras JI, Czapiewski R, et al. Tissue‐specific gene repositioning by muscle nuclear membrane proteins enhances repression of critical developmental genes during myogenesis. Mol Cell. 2016;62(6):834‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aboelnour E, Bonev B. Decoding the organization, dynamics, and function of the 4D genome. Dev Cell. 2021;56(11):1562‐1573. [DOI] [PubMed] [Google Scholar]
- 52. Ji X, Dadon DB, Powell BE, et al. 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell. 2016;18(2):262‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nora EP, Lajoie BR, Schulz EG, et al. Spatial partitioning of the regulatory landscape of the X‐inactivation centre. Nature. 2012;485(7398):381‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Symmons O, Pan L, Remeseiro S, et al. The Shh topological domain facilitates the action of remote enhancers by reducing the effects of genomic distances. Dev Cell. 2016;39(5):529‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. van Bemmel JG, Galupa R, Gard C, et al. The bipartite TAD organization of the X‐inactivation center ensures opposing developmental regulation of Tsix and Xist. Nat Genet. 2019;51(6):1024‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Montavon T, Soshnikova N, Mascrez B, et al. A regulatory archipelago controls Hox genes transcription in digits. Cell. 2011;147(5):1132‐1145. [DOI] [PubMed] [Google Scholar]
- 58. Williamson I, Lettice LA, Hill RE, Bickmore WA. Shh and ZRS enhancer colocalisation is specific to the zone of polarising activity. Development. 2016;143(16):2994‐3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Andrey G, Schöpflin R, Jerković I, et al. Characterization of hundreds of regulatory landscapes in developing limbs reveals two regimes of chromatin folding. Genome Res. 2017;27(2):223‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Beagan JA, Phillips‐Cremins JE. On the existence and functionality of topologically associating domains. Nat Genet. 2020;52(1):8‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bintu B, Mateo LJ, Su JH, et al. Super‐resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science. 2018;362(6413):eaau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ohlsson R, Renkawitz R, Lobanenkov V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001;17(9):520‐527. [DOI] [PubMed] [Google Scholar]
- 63. Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137(7):1194‐1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Arnould C, Rocher V, Finoux AL, et al. Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature. 2021;590(7847):660‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Murayama Y. DNA entry, exit and second DNA capture by cohesin: insights from biochemical experiments. Nucleus (Austin, Tex). 2018;9(1):492‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vian L, Pękowska A, Rao SSP, et al. The energetics and physiological impact of cohesin extrusion. Cell. 2018;173(5):1165‐1178. e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91(1):35‐45. [DOI] [PubMed] [Google Scholar]
- 68. Paliou C, Guckelberger P, Schöpflin R, et al. Preformed chromatin topology assists transcriptional robustness of Shh during limb development. Proc Natl Acad Sci U S A. 2019;116(25):12390‐12399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Williamson I, Kane L, Devenney PS, et al. Developmentally regulated Shh expression is robust to TAD perturbations. Development. 2019;146(19):dev179523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Anania C, Acemel RD, Jedamzick J, et al. In vivo dissection of a clustered‐CTCF domain boundary reveals developmental principles of regulatory insulation. Nat Genet. 2022;54(7):1026‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bonev B, Mendelson Cohen N, Szabo Q, et al. Multiscale 3D genome rewiring during mouse neural development. Cell. 2017;171(3):557‐572. e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen H, Tian Y, Shu W, Bo X, Wang S. Comprehensive identification and annotation of cell type‐specific and ubiquitous CTCF‐binding sites in the human genome. PLoS One. 2012;7(7):e41374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ren G, Jin W, Cui K, et al. CTCF‐mediated enhancer‐promoter interaction is a critical regulator of cell‐to‐cell variation of gene expression. Mol Cell. 2017;67(6):1049‐1058. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Khoury A, Achinger‐Kawecka J, Bert SA, et al. Constitutively bound CTCF sites maintain 3D chromatin architecture and long‐range epigenetically regulated domains. Nat Commun. 2020;11(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wu HJ, Landshammer A, Stamenova EK, et al. Topological isolation of developmental regulators in mammalian genomes. Nat Commun. 2021;12(1):4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ding T, Zhang H. Novel biological insights revealed from the investigation of multiscale genome architecture. Computational and Structural Biotechnology Journal. 2023;21:312‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Grubert F, Srivas R, Spacek DV, et al. Landscape of cohesin‐mediated chromatin loops in the human genome. Nature. 2020;583(7818):737‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zuin J, Dixon JR, van der Reijden MI, et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci U S A. 2014;111(3):996‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schwarzer W, Abdennur N, Goloborodko A, et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature. 2017;551(7678):51‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell. 1997;91(1):47‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Higashi TL, Eickhoff P, Sousa JS, et al. A Structure‐Based Mechanism for DNA Entry into the Cohesin Ring. Mol Cell. 2020;79(6):917‐933. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ganji M, Shaltiel IA, Bisht S, et al. Real‐time imaging of DNA loop extrusion by condensin. Science. 2018;360(6384):102‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gabriele M, Brandão HB, Grosse‐Holz S, et al. Dynamics of CTCF‐ and cohesin‐mediated chromatin looping revealed by live‐cell imaging. Science. 2022;376(6592):496‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Petela NJ, Gligoris TG, Metson J, et al. scc2 is a potent activator of Cohesin's ATPase that promotes loading by binding Scc1 without Pds5. Mol Cell. 2018;70(6):1134‐1148. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ciosk R, Shirayama M, Shevchenko A, et al. Cohesin's binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol Cell. 2000;5(2):243‐254. [DOI] [PubMed] [Google Scholar]
- 86. Kanke M, Tahara E, Nishiyama T. Cohesin acetylation and Wapl‐Pds5 oppositely regulate translocation of cohesin along DNA. EMBO J. 2016;35(24):2686‐2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Collier JE, Lee BG, Roig MB, et al. Transport of DNA within cohesin involves clamping on top of engaged heads by Scc2 and entrapment within the ring by Scc3. Elife. 2020;9:e59560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Murayama Y, Uhlmann F. Biochemical reconstitution of topological DNA binding by the cohesin ring. Nature. 2014;505(7483):367‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ouyang Z, Zheng G, Tomchick DR, Luo X, Yu H. Structural basis and IP6 requirement for Pds5‐dependent cohesin dynamics. Mol Cell. 2016;62(2):248‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tedeschi A, Wutz G, Huet S, et al. Wapl is an essential regulator of chromatin structure and chromosome segregation. Nature. 2013;501(7468):564‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rowland BD, Roig MB, Nishino T, et al. Building sister chromatid cohesion: smc3 acetylation counteracts an antiestablishment activity. Mol Cell. 2009;33(6):763‐774. [DOI] [PubMed] [Google Scholar]
- 92. Xiao T, Wallace J, Felsenfeld G. Specific sites in the C terminus of CTCF interact with the SA2 subunit of the cohesin complex and are required for cohesin‐dependent insulation activity. Mol Cell Biol. 2011;31(11):2174‐2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Pugacheva EM, Kubo N, Loukinov D, et al. CTCF mediates chromatin looping via N‐terminal domain‐dependent cohesin retention. Proc Natl Acad Sci U S A. 2020;117(4):2020‐2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wutz G, Várnai C, Nagasaka K, et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017;36(24):3573‐3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kueng S, Hegemann B, Peters BH, et al. Wapl controls the dynamic association of cohesin with chromatin. Cell. 2006;127(5):955‐967. [DOI] [PubMed] [Google Scholar]
- 96. Gassler J, Brandão HB, Imakaev M, et al. A mechanism of cohesin‐dependent loop extrusion organizes zygotic genome architecture. EMBO J. 2017;36(24):3600‐3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Haarhuis JHI, van der Weide RH, Blomen VA, et al. The cohesin release factor WAPL restricts chromatin loop extension. Cell. 2017;169(4):693‐707. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Schoenfelder S, Fraser P. Long‐range enhancer‐promoter contacts in gene expression control. Nat Rev Genet. 2019;20(8):437‐455. [DOI] [PubMed] [Google Scholar]
- 99. Heintzman ND, Hon GC, Hawkins RD, et al. Histone modifications at human enhancers reflect global cell‐type‐specific gene expression. Nature. 2009;459(7243):108‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Long HK, Prescott SL, Wysocka J. Ever‐changing landscapes: transcriptional enhancers in development and evolution. Cell. 2016;167(5):1170‐1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Haberle V, Stark A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat Rev Mol Cell Biol. 2018;19(10):621‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Andersson R, Sandelin A. Determinants of enhancer and promoter activities of regulatory elements. Nat Rev Genet. 2020;21(2):71‐87. [DOI] [PubMed] [Google Scholar]
- 103. Smith E, Shilatifard A. Enhancer biology and enhanceropathies. Nat Struct Mol Biol. 2014;21(3):210‐219. [DOI] [PubMed] [Google Scholar]
- 104. Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13(9):613‐626. [DOI] [PubMed] [Google Scholar]
- 105. Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome‐wide predictions. Nat Rev Genet. 2014;15(4):272‐286. [DOI] [PubMed] [Google Scholar]
- 106. Diao Y, Fang R, Li B, et al. A tiling‐deletion‐based genetic screen for cis‐regulatory element identification in mammalian cells. Nat Methods. 2017;14(6):629‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Dao LTM, Galindo‐Albarrán AO, Castro‐Mondragon JA, et al. Genome‐wide characterization of mammalian promoters with distal enhancer functions. Nat Genet. 2017;49(7):1073‐1081. [DOI] [PubMed] [Google Scholar]
- 108. Lenhard B, Sandelin A, Carninci P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat Rev Genet. 2012;13(4):233‐245. [DOI] [PubMed] [Google Scholar]
- 109. Juven‐Gershon T, Hsu JY, Kadonaga JT. Perspectives on the RNA polymerase II core promoter. Biochem Soc Trans. 2006;34(Pt 6):1047‐1050. [DOI] [PubMed] [Google Scholar]
- 110. Schier AC, Taatjes DJ. Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev. 2020;34(7‐8):465‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Sainsbury S, Bernecky C, Cramer P. Structural basis of transcription initiation by RNA polymerase II. Nat Rev Mol Cell Biol. 2015;16(3):129‐143. [DOI] [PubMed] [Google Scholar]
- 112. Adelman K, Lis JT. Promoter‐proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13(10):720‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kwak H, Fuda NJ, Core LJ, Lis JT. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science. 2013;339(6122):950‐953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Narita T, Ito S, Higashijima Y, et al. Enhancers are activated by p300/CBP activity‐dependent PIC assembly, RNAPII recruitment, and pause release. Mol Cell. 2021;81(10):2166‐2182. e6. [DOI] [PubMed] [Google Scholar]
- 115. Fant CB, Levandowski CB, Gupta K, et al. TFIID enables RNA polymerase II promoter‐proximal pausing. Mol Cell. 2020;78(4):785‐793. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Rickels R, Shilatifard A. Enhancer logic and mechanics in development and disease. Trends Cell Biol. 2018;28(8):608‐630. [DOI] [PubMed] [Google Scholar]
- 117. Panigrahi A, O'Malley BW. Mechanisms of enhancer action: the known and the unknown. Genome Biol. 2021;22(1):108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Neumayr C, Haberle V, Serebreni L, et al. Differential cofactor dependencies define distinct types of human enhancers. Nature. 2022;606(7913):406‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hua P, Badat M, Hanssen LLP, et al. Defining genome architecture at base‐pair resolution. Nature. 2021;595(7865):125‐129. [DOI] [PubMed] [Google Scholar]
- 120. Robson MI, Ringel AR, Mundlos S. Regulatory landscaping: how enhancer‐promoter communication is sculpted in 3D. Mol Cell. 2019;74(6):1110‐1122. [DOI] [PubMed] [Google Scholar]
- 121. Attanasio C, Nord AS, Zhu Y, et al. Fine tuning of craniofacial morphology by distant‐acting enhancers. Science. 2013;342(6157):1241006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Torbey P, Thierion E, Collombet S, et al. Cooperation, cis‐interactions, versatility and evolutionary plasticity of multiple cis‐acting elements underlie krox20 hindbrain regulation. PLoS Genet. 2018;14(8):e1007581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Oudelaar AM, Davies JOJ, Hanssen LLP, et al. Single‐allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nat Genet. 2018;50(12):1744‐1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Choi J, Lysakovskaia K, Stik G, et al. Evidence for additive and synergistic action of mammalian enhancers during cell fate determination. Elife. 2021;10:e65381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Moorthy SD, Davidson S, Shchuka VM, et al. Enhancers and super‐enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res. 2017;27(2):246‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Perry MW, Boettiger AN, Bothma JP, Levine M. Shadow enhancers foster robustness of Drosophila gastrulation. Curr Biol. 2010;20(17):1562‐1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wunderlich Z, Bragdon MD, Vincent BJ, White JA, Estrada J, DePace AH. Krüppel expression levels are maintained through compensatory evolution of shadow enhancers. Cell Rep. 2015;12(11):1740‐1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kvon EZ, Waymack R, Gad M, Wunderlich Z. Enhancer redundancy in development and disease. Nat Rev Genet. 2021;22(5):324‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zabidi MA, Stark A. Regulatory enhancer‐core‐promoter communication via transcription factors and cofactors. Trends Genet. 2016;32(12):801‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Fernandez Garcia M, Moore CD, Schulz KN, et al. Structural features of transcription factors associating with nucleosome binding. Mol Cell. 2019;75(5):921‐932. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Zaret KS. Pioneering the chromatin landscape. Nat Genet. 2018;50(2):167‐169. [DOI] [PubMed] [Google Scholar]
- 132. Armstrong JA, Emerson BM. NF‐E2 disrupts chromatin structure at human beta‐globin locus control region hypersensitive site 2 in vitro. Mol Cell Biol. 1996;16(10):5634‐5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kulaeva OI, Nizovtseva EV, Polikanov YS, Ulianov SV, Studitsky VM. Distant activation of transcription: mechanisms of enhancer action. Mol Cell Biol. 2012;32(24):4892‐4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Weinert BT, Narita T, Satpathy S, et al. Time‐resolved analysis reveals rapid dynamics and broad scope of the CBP/p300 acetylome. Cell. 2018;174(1):231‐244. e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Robinson PJ, Trnka MJ, Bushnell DA, et al. Structure of a complete mediator‐RNA polymerase II pre‐initiation complex. Cell. 2016;166(6):1411‐1422. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Chen X, Yin X, Li J, et al. Structures of the human mediator and mediator‐bound preinitiation complex. Science. 2021;372(6546):eabg0635. [DOI] [PubMed] [Google Scholar]
- 137. Schilbach S, Hantsche M, Tegunov D, et al. Structures of transcription pre‐initiation complex with TFIIH and mediator. Nature. 2017;551(7679):204‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Soutourina J. Transcription regulation by the mediator complex. Nat Rev Mol Cell Biol. 2018;19(4):262‐274. [DOI] [PubMed] [Google Scholar]
- 139. Allen BL, Taatjes DJ. The mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16(3):155‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Schmitt AD, Hu M, Jung I, et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 2016;17(8):2042‐2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Orsztynowicz M, Lechniak D, Pawlak P, et al. Changes in chromosome territory position within the nucleus reflect alternations in gene expression related to embryonic lineage specification. PLoS One. 2017;12(8):e0182398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Stadhouders R, Vidal E, Serra F, et al. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet. 2018;50(2):238‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Chu X, Wang J. Microscopic Chromosomal structural and dynamical origin of cell differentiation and reprogramming. Advanced Science (Weinheim, Baden‐Wurttemberg, Germany). 2020;7(20):2001572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zhang N, Mendieta‐Esteban J, Magli A, et al. Muscle progenitor specification and myogenic differentiation are associated with changes in chromatin topology. Nat Commun. 2020;11(1):6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kai Y, Li BE, Zhu M, et al. Mapping the evolving landscape of super‐enhancers during cell differentiation. Genome Biol. 2021;22(1):269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Neems DS, Garza‐Gongora AG, Smith ED, Kosak ST. Topologically associated domains enriched for lineage‐specific genes reveal expression‐dependent nuclear topologies during myogenesis. Proc Natl Acad Sci U S A. 2016;113(12):E1691‐E1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. He M, Li Y, Tang Q, et al. Genome‐wide chromatin structure changes during adipogenesis and myogenesis. International journal of biological sciences. 2018;14(11):1571‐1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Bortnick A, He Z, Aubrey M, et al. Plasma cell fate is orchestrated by elaborate changes in genome compartmentalization and inter‐chromosomal hubs. Cell Rep. 2020;31(1):107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Boltsis I, Grosveld F, Giraud G, Kolovos P. Chromatin conformation in development and disease. Front Cell Dev Biol. 2021;9:723859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Galupa R, Heard E. X‐chromosome inactivation: new insights into cis and trans regulation. Curr Opin Genet Dev. 2015;31:57‐66. [DOI] [PubMed] [Google Scholar]
- 151. Sousa EJ, Stuart HT, Bates LE, et al. Exit from naive pluripotency induces a transient x chromosome inactivation‐like state in males. Cell Stem Cell. 2018;22(6):919‐928. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Nesterova TB, Senner CE, Schneider J, et al. Pluripotency factor binding and Tsix expression act synergistically to repress Xist in undifferentiated embryonic stem cells. Epigenetics Chromatin. 2011;4(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Andrey G, Montavon T, Mascrez B, et al. A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science. 2013;340(6137):1234167. [DOI] [PubMed] [Google Scholar]
- 154. Woltering JM, Noordermeer D, Leleu M, Duboule D. Conservation and divergence of regulatory strategies at Hox Loci and the origin of tetrapod digits. PLoS Biol. 2014;12(1):e1001773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Amândio AR, Beccari L, Lopez‐Delisle L, et al. Sequential in cis mutagenesis in vivo reveals various functions for CTCF sites at the mouse HoxD cluster. Genes Dev. 2021;35(21‐22):1490‐1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Bolt CC, Lopez‐Delisle L, Mascrez B, Duboule D. Mesomelic dysplasias associated with the HOXD locus are caused by regulatory reallocations. Nat Commun. 2021;12(1):5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Rodríguez‐Carballo E, Lopez‐Delisle L, Willemin A, et al. Chromatin topology and the timing of enhancer function at the HoxD locus. Proc Natl Acad Sci U S A. 2020;117(49):31231‐31241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Beccari L, Yakushiji‐Kaminatsui N, Woltering JM, et al. A role for HOX13 proteins in the regulatory switch between TADs at the HoxD locus. Genes Dev. 2016;30(10):1172‐1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Desanlis I, Kherdjemil Y, Mayran A, et al. HOX13‐dependent chromatin accessibility underlies the transition towards the digit development program. Nat Commun. 2020;11(1):2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Bolt CC, Lopez‐Delisle L, Hintermann A, et al. Context‐dependent enhancer function revealed by targeted inter‐TAD relocation. Nat Commun. 2022;13(1):3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Sivakumar A, de Las Heras JI, Schirmer EC. Spatial genome organization: from development to disease. Front Cell Dev Biol. 2019;7:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Shukla V, Cetnarowska A, Hyldahl M, Mandrup S. Interplay between regulatory elements and chromatin topology in cellular lineage determination. Trends Genet. 2022;38(10):1048‐1061. [DOI] [PubMed] [Google Scholar]
- 163. Gao L, Tober J, Gao P, Chen C, Tan K, Speck NA. RUNX1 and the endothelial origin of blood. Exp Hematol. 2018;68:2‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yzaguirre AD, de Bruijn MF, Speck NA. The role of runx1 in embryonic blood cell formation. Adv Exp Med Biol. 2017;962:47‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Owens DDG, Anselmi G, Oudelaar AM, et al. Dynamic Runx1 chromatin boundaries affect gene expression in hematopoietic development. Nat Commun. 2022;13(1):773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Oudelaar AM, Beagrie RA, Gosden M, et al. Dynamics of the 4D genome during in vivo lineage specification and differentiation. Nat Commun. 2020;11(1):2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Oudelaar AM, Beagrie RA, Kassouf MT, Higgs DR. The mouse alpha‐globin cluster: a paradigm for studying genome regulation and organization. Curr Opin Genet Dev. 2021;67:18‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Siersbæk R, Madsen JGS, Javierre BM, et al. Dynamic rewiring of promoter‐anchored chromatin loops during adipocyte differentiation. Mol Cell. 2017;66(3):420‐435. e5. [DOI] [PubMed] [Google Scholar]
- 169. Hunter N. Meiotic recombination: the essence of heredity. Cold Spring Harb Perspect Biol. 2015;7(12):a016618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Gray S, Cohen PE. Control of meiotic crossovers: from double‐strand break formation to designation. Annu Rev Genet. 2016;50:175‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Clermont Y. Kinetics of spermatogenesis in mammals: seminiferous epithelium cycle and spermatogonial renewal. Physiol Rev. 1972;52(1):198‐236. [DOI] [PubMed] [Google Scholar]
- 172. Lin Z, Tong MH. m(6)A mRNA modification regulates mammalian spermatogenesis. Biochimica et Biophysica Acta Gene Regulatory Mechanisms. 2019;1862(3):403‐411. [DOI] [PubMed] [Google Scholar]
- 173. Vara C, Paytuví‐Gallart A, Cuartero Y, et al. The impact of chromosomal fusions on 3D genome folding and recombination in the germ line. Nat Commun. 2021;12(1):2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, Cairns BR. Distinctive chromatin in human sperm packages genes for embryo development. Nature. 2009;460(7254):473‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Alavattam KG, Maezawa S, Sakashita A, et al. Attenuated chromatin compartmentalization in meiosis and its maturation in sperm development. Nat Struct Mol Biol. 2019;26(3):175‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Patel L, Kang R, Rosenberg SC, et al. Dynamic reorganization of the genome shapes the recombination landscape in meiotic prophase. Nat Struct Mol Biol. 2019;26(3):164‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Luo Z, Wang X, Jiang H, et al. Reorganized 3D genome structures support transcriptional regulation in mouse spermatogenesis. iScience. 2020;23(4):101034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Wang Y, Wang H, Zhang Y, et al. Reprogramming of meiotic chromatin architecture during spermatogenesis. Mol Cell. 2019;73(3):547‐561. e6. [DOI] [PubMed] [Google Scholar]
- 179. Vara C, Paytuví‐Gallart A, Cuartero Y, et al. Three‐dimensional genomic structure and cohesin occupancy correlate with transcriptional activity during spermatogenesis. Cell Rep. 2019;28(2):352‐367. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Battulin N, Fishman VS, Mazur AM, et al. Comparison of the three‐dimensional organization of sperm and fibroblast genomes using the Hi‐C approach. Genome Biol. 2015;16(1):77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Yamaguchi K, Hada M, Fukuda Y, et al. Re‐evaluating the localization of sperm‐retained histones revealed the modification‐dependent accumulation in specific genome regions. Cell Rep. 2018;23(13):3920‐3932. [DOI] [PubMed] [Google Scholar]
- 182. Jung YH, Sauria MEG, Lyu X, et al. Chromatin states in mouse sperm correlate with embryonic and adult regulatory landscapes. Cell Rep. 2017;18(6):1366‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Oktem O, Urman B. Understanding follicle growth in vivo. Hum Reprod. 2010;25(12):2944‐2954. [DOI] [PubMed] [Google Scholar]
- 184. Zhu Y, Yu J, Gu J, et al. Relaxed 3D genome conformation facilitates the pluripotent to totipotent‐like state transition in embryonic stem cells. Nucleic Acids Res. 2021;49(21):12167‐12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Du Z, Zheng H, Kawamura YK, et al. Polycomb group proteins regulate chromatin architecture in mouse oocytes and early embryos. Mol Cell. 2020;77(4):825‐839. e7. [DOI] [PubMed] [Google Scholar]
- 186. Flyamer IM, Gassler J, Imakaev M, et al. Single‐nucleus Hi‐C reveals unique chromatin reorganization at oocyte‐to‐zygote transition. Nature. 2017;544(7648):110‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Silva MCC, Powell S, Ladstätter S, et al. Wapl releases Scc1‐cohesin and regulates chromosome structure and segregation in mouse oocytes. J Cell Biol. 2020;219(4):e201906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Tian GG, Hou C, Li J, Wu J. Three‐dimensional genome structure shapes the recombination landscape of chromatin features during female germline stem cell development. Clin Transl Med. 2022;12(6):e927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Ing‐Simmons E, Rigau M, Vaquerizas JM. Emerging mechanisms and dynamics of three‐dimensional genome organisation at zygotic genome activation. Curr Opin Cell Biol. 2022;74:37‐46. [DOI] [PubMed] [Google Scholar]
- 190. Collombet S, Ranisavljevic N, Nagano T, et al. Parental‐to‐embryo switch of chromosome organization in early embryogenesis. Nature. 2020;580(7801):142‐146. [DOI] [PubMed] [Google Scholar]
- 191. Von Stetina JR, Orr‐Weaver TL. Developmental control of oocyte maturation and egg activation in metazoan models. Cold Spring Harb Perspect Biol. 2011;3(10):a005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Xu R, Li C, Liu X, Gao S. Insights into epigenetic patterns in mammalian early embryos. Protein & cell. 2021;12(1):7‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Sankar A, Lerdrup M, Manaf A, et al. KDM4A regulates the maternal‐to‐zygotic transition by protecting broad H3K4me3 domains from H3K9me3 invasion in oocytes. Nat Cell Biol. 2020;22(4):380‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Hug CB, Vaquerizas JM. The birth of the 3D genome during early embryonic development. Trends Genet. 2018;34(12):903‐914. [DOI] [PubMed] [Google Scholar]
- 195. Hug CB, Grimaldi AG, Kruse K, Vaquerizas JM. Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell. 2017;169(2):216‐228. e19. [DOI] [PubMed] [Google Scholar]
- 196. Du Z, Zheng H, Huang B, et al. Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature. 2017;547(7662):232‐235. [DOI] [PubMed] [Google Scholar]
- 197. Jung YH, Kremsky I, Gold HB, et al. Maintenance of CTCF‐ and transcription factor‐mediated interactions from the Gametes to the early mouse embryo. Mol Cell. 2019;75(1):154‐171. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Chen X, Ke Y, Wu K, et al. Key role for CTCF in establishing chromatin structure in human embryos. Nature. 2019;576(7786):306‐310. [DOI] [PubMed] [Google Scholar]
- 199. Dileep V, Tsai LH. Three‐dimensional chromatin organization in brain function and dysfunction. Curr Opin Neurobiol. 2021;69:214‐221. [DOI] [PubMed] [Google Scholar]
- 200. Allen NJ, Barres BA. Neuroscience: glia ‐ more than just brain glue. Nature. 2009;457(7230):675‐677. [DOI] [PubMed] [Google Scholar]
- 201. Darmanis S, Sloan SA, Zhang Y, et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci U S A. 2015;112(23):7285‐7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Zeng H, Sanes JR. Neuronal cell‐type classification: challenges, opportunities and the path forward. Nat Rev Neurosci. 2017;18(9):530‐546. [DOI] [PubMed] [Google Scholar]
- 203. LaBeau J. Neuroscience for clinicians: evidence, models, and practice by C. Alexander Simpkins and m. Annellen Simpkins. Am J Clin Hypn. 2014;57(1):84‐86. [DOI] [PubMed] [Google Scholar]
- 204. Liu W, Zhong W, Chen J, Huang B, Hu M, Li Y. Understanding regulatory mechanisms of brain function and disease through 3D genome organization. Genes (Basel). 2022;13(4):586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Le Gros MA, Clowney EJ, Magklara A, et al. Soft X‐ray tomography reveals gradual chromatin compaction and reorganization during neurogenesis in vivo. Cell Rep. 2016;17(8):2125‐2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. de la Torre‐Ubieta L, Stein JL, Won H, et al. The dynamic landscape of open chromatin during human cortical neurogenesis. Cell. 2018;172(1‐2):289‐304. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Fraser J, Ferrai C, Chiariello AM, et al. Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol Syst Biol. 2015;11(12):852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Luo X, Liu Y, Dang D, et al. 3D Genome of macaque fetal brain reveals evolutionary innovations during primate corticogenesis. Cell. 2021;184(3):723‐740. e21. [DOI] [PubMed] [Google Scholar]
- 209. Kishi Y, Gotoh Y. Regulation of chromatin structure during neural development. Frontiers in Neuroscience. 2018;12:874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Lu L, Liu X, Huang WK, et al. Robust Hi‐C maps of enhancer‐promoter interactions reveal the function of non‐coding genome in neural development and diseases. Mol Cell. 2020;79(3):521‐534. e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Emera D, Yin J, Reilly SK, Gockley J, Noonan JP. Origin and evolution of developmental enhancers in the mammalian neocortex. Proc Natl Acad Sci U S A. 2016;113(19):E2617‐E2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Song M, Pebworth MP, Yang X, et al. Cell‐type‐specific 3D epigenomes in the developing human cortex. Nature. 2020;587(7835):644‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Won H, de la Torre‐Ubieta L, Stein JL, et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 2016;538(7626):523‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Bertero A, Rosa‐Garrido M. Three‐dimensional chromatin organization in cardiac development and disease. J Mol Cell Cardiol. 2021;151:89‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Nothjunge S, Nührenberg TG, Grüning BA, et al. DNA methylation signatures follow preformed chromatin compartments in cardiac myocytes. Nat Commun. 2017;8(1):1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Chapski DJ, Rosa‐Garrido M, Hua N, Alber F, Vondriska TM. Spatial principles of chromatin architecture associated with organ‐specific gene regulation. Frontiers in Cardiovascular Medicine. 2018;5:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Bertero A, Fields PA, Ramani V, et al. Dynamics of genome reorganization during human cardiogenesis reveal an RBM20‐dependent splicing factory. Nat Commun. 2019;10(1):1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Zhang Y, Li T, Preissl S, et al. Transcriptionally active HERV‐H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat Genet. 2019;51(9):1380‐1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Montefiori LE, Sobreira DR, Sakabe NJ, et al. A promoter interaction map for cardiovascular disease genetics. Elife. 2018;7:e35788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Werber M, Wittler L, Timmermann B, Grote P, Herrmann BG. The tissue‐specific transcriptomic landscape of the mid‐gestational mouse embryo. Development. 2014;141(11):2325‐2330. [DOI] [PubMed] [Google Scholar]
- 221. Grote P, Wittler L, Hendrix D, et al. The tissue‐specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell. 2013;24(2):206‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Ritter N, Ali T, Kopitchinski N, et al. The lncRNA locus handsdown regulates cardiac gene programs and is essential for early mouse development. Dev Cell. 2019;50(5):644‐657. e8. [DOI] [PubMed] [Google Scholar]
- 223. Rajderkar S, Mann JM, Panaretos C, et al. Trim33 is required for appropriate development of pre‐cardiogenic mesoderm. Dev Biol. 2019;450(2):101‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. George RM, Firulli AB. Epigenetics and heart development. Front Cell Dev Biol. 2021;9:637996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. De Koninck M, Lapi E, Badía‐Careaga C, et al. Essential roles of cohesin STAG2 in mouse embryonic development and adult tissue homeostasis. Cell Rep. 2020;32(6):108014. [DOI] [PubMed] [Google Scholar]
- 226. Kawauchi S, Calof AL, Santos R, et al. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/‐) mouse, a model of Cornelia de Lange syndrome. PLoS Genet. 2009;5(9):e1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Muto A, Calof AL, Lander AD, Schilling TF. Multifactorial origins of heart and gut defects in nipbl‐deficient zebrafish, a model of Cornelia de Lange syndrome. PLoS Biol. 2011;9(10):e1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Ema H, Nakauchi H. Expansion of hematopoietic stem cells in the developing liver of a mouse embryo. Blood. 2000;95(7):2284‐2288. [PubMed] [Google Scholar]
- 229. Passegué E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202(11):1599‐1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Cuartero S, Stik G, Stadhouders R. Three‐dimensional genome organization in immune cell fate and function. Nat Rev Immunol. 2023;23(4):206‐221. [DOI] [PubMed] [Google Scholar]
- 231. Qiu Y, Huang S. CTCF‐mediated genome organization and leukemogenesis. Leukemia. 2020;34(9):2295‐2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Chen C, Yu W, Tober J, et al. Spatial genome re‐organization between fetal and adult hematopoietic stem cells. Cell Rep. 2019;29(12):4200‐4211. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood. 2011;118(24):6258‐6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Ji P, Murata‐Hori M, Lodish HF. Formation of mammalian erythrocytes: chromatin condensation and enucleation. Trends Cell Biol. 2011;21(7):409‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Shearstone JR, Pop R, Bock C, Boyle P, Meissner A, Socolovsky M. Global DNA demethylation during mouse erythropoiesis in vivo. Science. 2011;334(6057):799‐802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Bartholdy B, Lajugie J, Yan Z, et al. Mechanisms of establishment and functional significance of DNA demethylation during erythroid differentiation. Blood Advances. 2018;2(15):1833‐1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Li D, Wu F, Zhou S, Huang XJ, Lee HY. Heterochromatin rewiring and domain disruption‐mediated chromatin compaction during erythropoiesis. Nat Struct Mol Biol. 2023;30(4):463‐474. [DOI] [PubMed] [Google Scholar]
- 238. Scourzic L, Salataj E, Apostolou E. Deciphering the complexity of 3D chromatin organization driving lymphopoiesis and lymphoid malignancies. Front Immunol. 2021;12:669881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Lin YC, Benner C, Mansson R, et al. Global changes in the nuclear positioning of genes and intra‐ and interdomain genomic interactions that orchestrate B cell fate. Nat Immunol. 2012;13(12):1196‐1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Boya R, Yadavalli AD, Nikhat S, Kurukuti S, Palakodeti D, Pongubala JMR. Developmentally regulated higher‐order chromatin interactions orchestrate B cell fate commitment. Nucleic Acids Res. 2017;45(19):11070‐11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Hagman J, Ramírez J, Lukin K. B lymphocyte lineage specification, commitment and epigenetic control of transcription by early B cell factor 1. Curr Top Microbiol Immunol. 2012;356:17‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Boller S, Grosschedl R. The regulatory network of B‐cell differentiation: a focused view of early B‐cell factor 1 function. Immunol Rev. 2014;261(1):102‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Yui MA, Rothenberg EV. Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol. 2014;14(8):529‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Kueh HY, Yui MA, Ng KK, et al. Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat Immunol. 2016;17(8):956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Isoda T, Moore AJ, He Z, et al. Non‐coding transcription instructs chromatin folding and compartmentalization to dictate enhancer‐promoter communication and T cell fate. Cell. 2017;171(1):103‐119. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Barutcu AR, Lajoie BR, McCord RP, et al. Chromatin interaction analysis reveals changes in small chromosome and telomere clustering between epithelial and breast cancer cells. Genome Biol. 2015;16:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Younossi ZM. Non‐alcoholic fatty liver disease ‐ A global public health perspective. J Hepatol. 2019;70(3):531‐544. [DOI] [PubMed] [Google Scholar]
- 248. Yan T, Yan N, Wang P, et al. Herbal drug discovery for the treatment of nonalcoholic fatty liver disease. Acta Pharmaceutica Sinica B. 2020;10(1):3‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Umano GR, Caprio S, Di Sessa A, et al. The rs626283 variant in the MBOAT7 gene is associated with insulin resistance and fatty liver in Caucasian obese Youth. Am J Gastroenterol. 2018;113(3):376‐383. [DOI] [PubMed] [Google Scholar]
- 250. Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: clinical impact. J Hepatol. 2018;68(2):268‐279. [DOI] [PubMed] [Google Scholar]
- 251. Xu L, Yin L, Qi Y, Tan X, Gao M, Peng J. 3D disorganization and rearrangement of genome provide insights into pathogenesis of NAFLD by integrated Hi‐C, Nanopore, and RNA sequencing. Acta Pharmaceutica Sinica B. 2021;11(10):3150‐3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. San Martin R, Das P, Dos Reis Marques R, et al. Chromosome compartmentalization alterations in prostate cancer cell lines model disease progression. J Cell Biol. 2022;221(2):e202104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Moreau P, Cournac A, Palumbo GA, et al. Tridimensional infiltration of DNA viruses into the host genome shows preferential contact with active chromatin. Nat Commun. 2018;9(1):4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Yang B, Li B, Jia L, et al. 3D landscape of Hepatitis B virus interactions with human chromatins. Cell Discovery. 2020;6(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Tang D, Zhao H, Wu Y, et al. Transcriptionally inactive hepatitis B virus episome DNA preferentially resides in the vicinity of chromosome 19 in 3D host genome upon infection. Cell Rep. 2021;35(13):109288. [DOI] [PubMed] [Google Scholar]
- 256. Guo M, Yao Z, Jiang C, Songyang Z, Gan L, Xiong Y. Three‐dimensional and single‐cell sequencing of liver cancer reveals comprehensive host‐virus interactions in HBV infection. Front Immunol. 2023;14:1161522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Wang R, Lee JH, Kim J, et al. SARS‐CoV‐2 restructures host chromatin architecture. Nature Microbiology. 2023;8(4):679‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Lupiáñez DG, Kraft K, Heinrich V, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene‐enhancer interactions. Cell. 2015;161(5):1012‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Lupiáñez DG, Spielmann M, Mundlos S. Breaking TADs: how alterations of chromatin domains result in disease. Trends Genet. 2016;32(4):225‐237. [DOI] [PubMed] [Google Scholar]
- 260. Spielmann M, Lupiáñez DG, Mundlos S. Structural variation in the 3D genome. Nat Rev Genet. 2018;19(7):453‐467. [DOI] [PubMed] [Google Scholar]
- 261. Spielmann M, Mundlos S. Looking beyond the genes: the role of non‐coding variants in human disease. Hum Mol Genet. 2016;25(R2):R157‐R165. [DOI] [PubMed] [Google Scholar]
- 262. Dubois F, Sidiropoulos N, Weischenfeldt J, Beroukhim R. Structural variations in cancer and the 3D genome. Nat Rev Cancer. 2022;22(9):533‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. McCord RP, Balajee A. 3D genome organization influences the chromosome translocation pattern. Adv Exp Med Biol. 2018;1044:113‐133. [DOI] [PubMed] [Google Scholar]
- 264. Deng S, Feng Y, Pauklin S. 3D chromatin architecture and transcription regulation in cancer. J Hematol Oncol. 2022;15(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Peng A, Peng W, Wang R, Zhao H, Yu X, Sun Y. Regulation of 3D organization and its role in cancer biology. Front Cell Dev Biol. 2022;10:879465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Xu Z, Lee DS, Chandran S, et al. Structural variants drive context‐dependent oncogene activation in cancer. Nature. 2022;612(7940):564‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Long HK, Osterwalder M, Welsh IC, et al. Loss of extreme long‐range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell. 2020;27(5):765‐783. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Croft B, Ohnesorg T, Hewitt J, et al. Human sex reversal is caused by duplication or deletion of core enhancers upstream of SOX9. Nat Commun. 2018;9(1):5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Gonen N, Futtner CR, Wood S, et al. Sex reversal following deletion of a single distal enhancer of Sox9. Science. 2018;360(6396):1469‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Pantera H, Hu B, Moiseev D, et al. Pmp22 super‐enhancer deletion causes tomacula formation and conduction block in peripheral nerves. Hum Mol Genet. 2020;29(10):1689‐1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Pantera H, Moran JJ, Hung HA, Pak E, Dutra A, Svaren J. Regulation of the neuropathy‐associated Pmp22 gene by a distal super‐enhancer. Hum Mol Genet. 2018;27(16):2830‐2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Rouco R, Bompadre O, Rauseo A, et al. Cell‐specific alterations in Pitx1 regulatory landscape activation caused by the loss of a single enhancer. Nat Commun. 2021;12(1):7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Bagheri‐Fam S, Barrionuevo F, Dohrmann U, et al. Long‐range upstream and downstream enhancers control distinct subsets of the complex spatiotemporal Sox9 expression pattern. Dev Biol. 2006;291(2):382‐397. [DOI] [PubMed] [Google Scholar]
- 274. Gordon CT, Tan TY, Benko S, Fitzpatrick D, Lyonnet S, Farlie PG. Long‐range regulation at the SOX9 locus in development and disease. J Med Genet. 2009;46(10):649‐656. [DOI] [PubMed] [Google Scholar]
- 275. Lee YH, Saint‐Jeannet JP. Sox9 function in craniofacial development and disease. Genesis. 2011;49(4):200‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Lefebvre V, Dvir‐Ginzberg M. SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect Tissue Res. 2017;58(1):2‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Kim GJ, Sock E, Buchberger A, et al. Copy number variation of two separate regulatory regions upstream of SOX9 causes isolated 46,XY or 46,XX disorder of sex development. J Med Genet. 2015;52(4):240‐247. [DOI] [PubMed] [Google Scholar]
- 278. Robin P. A fall of the base of the tongue considered as a new cause of nasopharyngeal respiratory impairment: Pierre Robin sequence, a translation. 1923. Plast Reconstr Surg. 1994;93(6):1301‐1303. [PubMed] [Google Scholar]
- 279. Tan TY, Kilpatrick N, Farlie PG. Developmental and genetic perspectives on Pierre Robin sequence. Am J Med Genet C Semin Med Genet. 2013;163c(4):295‐305. [DOI] [PubMed] [Google Scholar]
- 280. Benko S, Fantes JA, Amiel J, et al. Highly conserved non‐coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet. 2009;41(3):359‐364. [DOI] [PubMed] [Google Scholar]
- 281. Will AJ, Cova G, Osterwalder M, et al. Composition and dosage of a multipartite enhancer cluster control developmental expression of Ihh (Indian hedgehog). Nat Genet. 2017;49(10):1539‐1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. St‐Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13(16):2072‐2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Barroso E, Berges‐Soria J, Benito‐Sanz S, et al. Identification of the fourth duplication of upstream IHH regulatory elements, in a family with craniosynostosis Philadelphia type, helps to define the phenotypic characterization of these regulatory elements. Am J Med Genet A. 2015;167a(4):902‐906. [DOI] [PubMed] [Google Scholar]
- 284. Klopocki E, Lohan S, Brancati F, et al. Copy‐number variations involving the IHH locus are associated with syndactyly and craniosynostosis. Am J Hum Genet. 2011;88(1):70‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Zuin J, Roth G, Zhan Y, et al. Nonlinear control of transcription through enhancer‐promoter interactions. Nature. 2022;604(7906):571‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Dimitrov B, Balikova I, de Ravel T, et al. 2q31.1 microdeletion syndrome: redefining the associated clinical phenotype. J Med Genet. 2011;48(2):98‐104. [DOI] [PubMed] [Google Scholar]
- 287. Mitter D, Chiaie BD, Lüdecke HJ, et al. Genotype‐phenotype correlation in eight new patients with a deletion encompassing 2q31.1. Am J Med Genet A. 2010;152a(5):1213‐1224. [DOI] [PubMed] [Google Scholar]
- 288. Montavon T, Thevenet L, Duboule D. Impact of copy number variations (CNVs) on long‐range gene regulation at the HoxD locus. Proc Natl Acad Sci U S A. 2012;109(50):20204‐20211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Huynh L, Hormozdiari F. TAD fusion score: discovery and ranking the contribution of deletions to genome structure. Genome Biol. 2019;20(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Despang A, Schöpflin R, Franke M, et al. Functional dissection of the Sox9‐Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nat Genet. 2019;51(8):1263‐1271. [DOI] [PubMed] [Google Scholar]
- 291. Weng AP, Millholland JM, Yashiro‐Ohtani Y, et al. c‐Myc is an important direct target of Notch1 in T‐cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20(15):2096‐1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Herranz D, Ambesi‐Impiombato A, Palomero T, et al. A NOTCH1‐driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med. 2014;20(10):1130‐1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Kloetgen A, Thandapani P, Ntziachristos P, et al. Three‐dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat Genet. 2020;52(4):388‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Smith EM, Lajoie BR, Jain G, Dekker J. Invariant TAD Boundaries constrain cell‐type‐specific looping interactions between promoters and distal elements around the CFTR locus. Am J Hum Genet. 2016;98(1):185‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Aavikko M, Kaasinen E, Andersson N, et al. WNT2 activation through proximal germline deletion predisposes to small intestinal neuroendocrine tumors and intestinal adenocarcinomas. Hum Mol Genet. 2021;30(24):2429‐2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Yang R, Kerschner JL, Gosalia N, et al. Differential contribution of cis‐regulatory elements to higher order chromatin structure and expression of the CFTR locus. Nucleic Acids Res. 2016;44(7):3082‐3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Lin JR, Wang S, Coy S, et al. Multiplexed 3D atlas of state transitions and immune interaction in colorectal cancer. Cell. 2023;186(2):363‐381. e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Maitra A, Hruban RH. Pancreatic cancer. Annual Review of Pathology. 2008;3:157‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Du Y, Gu Z, Li Z, et al. Dynamic interplay between structural variations and 3D genome organization in pancreatic cancer. Advanced Science (Weinheim, Baden‐Wurttemberg, Germany). 2022;9(18):e2200818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Franke M, Daly AF, Palmeira L, et al. Duplications disrupt chromatin architecture and rewire GPR101‐enhancer communication in X‐linked acrogigantism. Am J Hum Genet. 2022;109(4):553‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Melo US, Piard J, Fischer‐Zirnsak B, et al. Complete lung agenesis caused by complex genomic rearrangements with neo‐TAD formation at the SHH locus. Hum Genet. 2021;140(10):1459‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Liang M, Soomro A, Tasneem S, et al. Enhancer‐gene rewiring in the pathogenesis of Quebec platelet disorder. Blood. 2020;136(23):2679‐2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Kurth I, Klopocki E, Stricker S, et al. Duplications of noncoding elements 5' of SOX9 are associated with brachydactyly‐anonychia. Nat Genet. 2009;41(8):862‐863. [DOI] [PubMed] [Google Scholar]
- 304. Franke M, Ibrahim DM, Andrey G, et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 2016;538(7624):265‐269. [DOI] [PubMed] [Google Scholar]
- 305. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Peng YR, Shekhar K, Yan W, et al. Molecular classification and comparative taxonomics of foveal and peripheral cells in primate retina. Cell. 2019;176(5):1222‐1237. e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. de Bruijn SE, Fiorentino A, Ottaviani D, et al. Structural variants create new topological‐associated domains and ectopic retinal enhancer‐gene contact in dominant retinitis pigmentosa. Am J Hum Genet. 2020;107(5):802‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Friedman JS, Chang B, Krauth DS, et al. Loss of lysophosphatidylcholine acyltransferase 1 leads to photoreceptor degeneration in rd11 mice. Proc Natl Acad Sci U S A. 2010;107(35):15523‐15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Kmoch S, Majewski J, Ramamurthy V, et al. Mutations in PNPLA6 are linked to photoreceptor degeneration and various forms of childhood blindness. Nat Commun. 2015;6:5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Lidgerwood GE, Morris AJ, Conquest A, et al. Role of lysophosphatidic acid in the retinal pigment epithelium and photoreceptors. Biochimica et Biophysica Acta Molecular and Cell Biology of Lipids. 2018;1863(7):750‐761. [DOI] [PubMed] [Google Scholar]
- 311. Yang Q, Jiang N, Zou H, et al. Alterations in 3D chromatin organization contribute to tumorigenesis of EGFR‐amplified glioblastoma. Computational and Structural Biotechnology Journal. 2022;20:1967‐1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Cinque L, Micale L, Manara E, et al. A novel complex genomic rearrangement affecting the KCNJ2 regulatory region causes a variant of Cooks syndrome. Hum Genet. 2022;141(2):217‐227. [DOI] [PubMed] [Google Scholar]
- 313. Milunsky JM, Maher TA, Zhao G, et al. TFAP2A mutations result in branchio‐oculo‐facial syndrome. Am J Hum Genet. 2008;82(5):1171‐1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Milunsky JM, Maher TM, Zhao G, et al. Genotype‐phenotype analysis of the branchio‐oculo‐facial syndrome. Am J Med Genet A. 2011;155a(1):22‐32. [DOI] [PubMed] [Google Scholar]
- 315. Laugsch M, Bartusel M, Rehimi R, et al. Modeling the pathological long‐range regulatory effects of human structural variation with patient‐specific hiPSCs. Cell Stem Cell. 2019;24(5):736‐752. e12. [DOI] [PubMed] [Google Scholar]
- 316. Mennen U, Mundlos S, Spielmann M. The Liebenberg syndrome: in depth analysis of the original family. The Journal of Hand Surgery, European Volume. 2014;39(9):919‐925. [DOI] [PubMed] [Google Scholar]
- 317. Kragesteen BK, Brancati F, Digilio MC, Mundlos S, Spielmann M. H2AFY promoter deletion causes PITX1 endoactivation and Liebenberg syndrome. J Med Genet. 2019;56(4):246‐251. [DOI] [PubMed] [Google Scholar]
- 318. Spielmann M, Brancati F, Krawitz PM, et al. Homeotic arm‐to‐leg transformation associated with genomic rearrangements at the PITX1 locus. Am J Hum Genet. 2012;91(4):629‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Al‐Qattan MM, Al‐Thunayan A, Alabdulkareem I. Liebenberg syndrome is caused by a deletion upstream to the PITX1 gene resulting in transformation of the upper limbs to reflect lower limb characteristics. Gene. 2013;524(1):65‐71. [DOI] [PubMed] [Google Scholar]
- 320. Kragesteen BK, Spielmann M, Paliou C, et al. Dynamic 3D chromatin architecture contributes to enhancer specificity and limb morphogenesis. Nat Genet. 2018;50(10):1463‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Jones DT, Jäger N, Kool M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488(7409):100‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Kool M, Jones DT, Jäger N, et al. Genome sequencing of SHH medulloblastoma predicts genotype‐related response to smoothened inhibition. Cancer Cell. 2014;25(3):393‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Northcott PA, Lee C, Zichner T, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511(7510):428‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Northcott PA, Buchhalter I, Morrissy AS, et al. The whole‐genome landscape of medulloblastoma subtypes. Nature. 2017;547(7663):311‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Zweier M, Gregor A, Zweier C, et al. Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Hum Mutat. 2010;31(6):722‐733. [DOI] [PubMed] [Google Scholar]
- 326. Saitsu H, Igarashi N, Kato M, et al. De novo 5q14.3 translocation 121.5‐kb upstream of MEF2C in a patient with severe intellectual disability and early‐onset epileptic encephalopathy. Am J Med Genet A. 2011;155a(11):2879‐2884. [DOI] [PubMed] [Google Scholar]
- 327. Engels H, Wohlleber E, Zink A, et al. A novel microdeletion syndrome involving 5q14.3‐q15: clinical and molecular cytogenetic characterization of three patients. European Journal of Human Genetics: EJHG. 2009;17(12):1592‐1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Zweier M, Rauch A. The MEF2C‐related and 5q14.3q15 microdeletion syndrome. Molecular Syndromology. 2012;2(3‐5):164‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Redin C, Brand H, Collins RL, et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet. 2017;49(1):36‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Le Meur N, Holder‐Espinasse M, Jaillard S, et al. MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J Med Genet. 2010;47(1):22‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Mikulasova A, Kent D, Trevisan‐Herraz M, et al. Epigenomic translocation of H3K4me3 broad domains over oncogenes following hijacking of super‐enhancers. Genome Res. 2022;32(7):1343‐1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Watson CT, Breden F. The immunoglobulin heavy chain locus: genetic variation, missing data, and implications for human disease. Genes Immun. 2012;13(5):363‐373. [DOI] [PubMed] [Google Scholar]
- 333. Rico D, Kent D, Karataraki N, et al. High‐resolution simulations of chromatin folding at genomic rearrangements in malignant B cells provide mechanistic insights into proto‐oncogene deregulation. Genome Res. 2022;32(7):1355‐1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Suzukawa K, Parganas E, Gajjar A, et al. Identification of a breakpoint cluster region 3' of the ribophorin I gene at 3q21 associated with the transcriptional activation of the EVI1 gene in acute myelogenous leukemias with inv(3)(q21q26). Blood. 1994;84(8):2681‐2688. [PubMed] [Google Scholar]
- 335. Gröschel S, Sanders MA, Hoogenboezem R, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157(2):369‐381. [DOI] [PubMed] [Google Scholar]
- 336. Zhang J, Wu G, Miller CP, et al. Whole‐genome sequencing identifies genetic alterations in pediatric low‐grade gliomas. Nat Genet. 2013;45(6):602‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Bandopadhayay P, Ramkissoon LA, Jain P, et al. MYB‐QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet. 2016;48(3):273‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Cao C, Hong P, Huang X, et al. HPV‐CCDC106 integration alters local chromosome architecture and hijacks an enhancer by three‐dimensional genome structure remodeling in cervical cancer. Journal of Genetics and Genomics = Yi Chuan Xue Bao. 2020;47(8):437‐450. [DOI] [PubMed] [Google Scholar]
- 339. Adeel MM, Jiang H, Arega Y, et al. Structural Variations of the 3D Genome Architecture in Cervical Cancer Development. Front Cell Dev Biol. 2021;9:706375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Adey A, Burton JN, Kitzman JO, et al. The haplotype‐resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500(7461):207‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Shen C, Liu Y, Shi S, et al. Long‐distance interaction of the integrated HPV fragment with MYC gene and 8q24.22 region upregulating the allele‐specific MYC expression in HeLa cells. Int J Cancer. 2017;141(3):540‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016;15(9):2038‐2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Ball AR, Jr., Chen YY, Yokomori K. Mechanisms of cohesin‐mediated gene regulation and lessons learned from cohesinopathies. Biochim Biophys Acta. 2014;1839(3):191‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Norton HK, Phillips‐Cremins JE. Crossed wires: 3D genome misfolding in human disease. J Cell Biol. 2017;216(11):3441‐3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Liu J, Krantz ID. Cohesin and human disease. Annu Rev Genomics Hum Genet. 2008;9:303‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Mannini L, Cucco F, Quarantotti V, Krantz ID, Musio A. Mutation spectrum and genotype‐phenotype correlation in Cornelia de Lange syndrome. Hum Mutat. 2013;34(12):1589‐1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Watrin E, Kaiser FJ, Wendt KS. Gene regulation and chromatin organization: relevance of cohesin mutations to human disease. Curr Opin Genet Dev. 2016;37:59‐66. [DOI] [PubMed] [Google Scholar]
- 348. Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467(7314):430‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Lopez‐Serra L, Kelly G, Patel H, Stewart A, Uhlmann F. The Scc2‐Scc4 complex acts in sister chromatid cohesion and transcriptional regulation by maintaining nucleosome‐free regions. Nat Genet. 2014;46(10):1147‐1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Rhodes J, Mazza D, Nasmyth K, Uphoff S. Scc2/Nipbl hops between chromosomal cohesin rings after loading. Elife. 2017;6:e30000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Garcia P, Fernandez‐Hernandez R, Cuadrado A, et al. Disruption of NIPBL/Scc2 in Cornelia de Lange syndrome provokes cohesin genome‐wide redistribution with an impact in the transcriptome. Nat Commun. 2021;12(1):4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Heath H, Sleutels F, et al. CTCF regulates cell cycle progression of alphabeta T cells in the thymus. EMBO J. 2008;27(21):2839‐2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Soshnikova N, Montavon T, Leleu M, Galjart N, Duboule D. Functional analysis of CTCF during mammalian limb development. Dev Cell. 2010;19(6):819‐830. [DOI] [PubMed] [Google Scholar]
- 354. Splinter E, Heath H, Kooren J, et al. CTCF mediates long‐range chromatin looping and local histone modification in the beta‐globin locus. Genes Dev. 2006;20(17):2349‐2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Bell AC, Felsenfeld G. Methylation of a CTCF‐dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405(6785):482‐485. [DOI] [PubMed] [Google Scholar]
- 356. Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation‐sensitive enhancer‐blocking activity at the H19/Igf2 locus. Nature. 2000;405(6785):486‐489. [DOI] [PubMed] [Google Scholar]
- 357. Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Flavahan WA, Drier Y, Liau BB, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529(7584):110‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Filippova GN, Thienes CP, Penn BH, et al. CTCF‐binding sites flank CTG/CAG repeats and form a methylation‐sensitive insulator at the DM1 locus. Nat Genet. 2001;28(4):335‐343. [DOI] [PubMed] [Google Scholar]
- 361. Mitra I, Huang B, Mousavi N, et al. Patterns of de novo tandem repeat mutations and their role in autism. Nature. 2021;589(7841):246‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Bruneau BG, Nora EP. Chromatin Domains Go on Repeat in Disease. Cell. 2018;175(1):38‐40. [DOI] [PubMed] [Google Scholar]
- 363.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11(4):247‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11(11):786‐799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447(7147):932‐940. [DOI] [PubMed] [Google Scholar]
- 366. Nelson DL, Orr HT, Warren ST. The unstable repeats–three evolving faces of neurological disease. Neuron. 2013;77(5):825‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Edwards JR, Yarychkivska O, Boulard M, Bestor TH. DNA methylation and DNA methyltransferases. Epigenetics Chromatin. 2017;10:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Renda M, Baglivo I, Burgess‐Beusse B, et al. Critical DNA binding interactions of the insulator protein CTCF: a small number of zinc fingers mediate strong binding, and a single finger‐DNA interaction controls binding at imprinted loci. J Biol Chem. 2007;282(46):33336‐33345. [DOI] [PubMed] [Google Scholar]
- 369. Liu XS, Wu H, Krzisch M, et al. Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell. 2018;172(5):979‐992. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Sun JH, Zhou L, Emerson DJ, et al. Disease‐associated short tandem repeats co‐localize with chromatin domain boundaries. Cell. 2018;175(1):224‐238. e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Durcan L, O'Dwyer T, Petri M. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet (London, England). 2019;393(10188):2332‐2343. [DOI] [PubMed] [Google Scholar]
- 372. Grünvogel O, Esser‐Nobis K, Reustle A, et al. DDX60L is an interferon‐stimulated gene product restricting hepatitis C virus replication in cell culture. J Virol. 2015;89(20):10548‐10568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Zhao M, Feng D, Hu L, et al. 3D genome alterations in T cells associated with disease activity of systemic lupus erythematosus. Ann Rheum Dis. 2023;82(2):226‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. 2016;13(6):368‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Merlo M, Cannatà A, Gobbo M, Stolfo D, Elliott PM, Sinagra G. Evolving concepts in dilated cardiomyopathy. Eur J Heart Fail. 2018;20(2):228‐239. [DOI] [PubMed] [Google Scholar]
- 376. Feng Y, Cai L, Hong W, et al. Rewiring of 3D chromatin topology orchestrates transcriptional reprogramming and the development of human dilated cardiomyopathy. Circulation. 2022;145(22):1663‐1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7‐30. [DOI] [PubMed] [Google Scholar]
- 378. Ren B, Yang J, Wang C, et al. High‐resolution Hi‐C maps highlight multiscale 3D epigenome reprogramming during pancreatic cancer metastasis. J Hematol Oncol. 2021;14(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Xu J, Song F, Lyu H, et al. Subtype‐specific 3D genome alteration in acute myeloid leukaemia. Nature. 2022;611(7935):387‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available upon request from the authors.