


Abstract
Surface plasmon resonance (BIACORE) was used to determine the kinetic values for formation of the HIV TAR–TAR* (‘kissing hairpin’) RNA complex. The TAR component was also synthesized with the modified nucleoside 2-thiouridine at position 7 in the loop and the kinetics and equilibrium dissociation constants compared with the unmodified TAR hairpin. The BIACORE data show an equilibrium dissociation constant of 1.58 nM for the complex containing the s2U modified TAR hairpin, which is 8-fold lower than for the parent hairpin (12.5 nM). This is a result of a 2-fold faster ka (4.14 × 105 M–1 s–1 versus 2.1 × 105 M–1 s–1) and a 4-fold slower kd (6.55 × 10–4 s–1 versus 2.63 × 10–3 s–1). 1H NMR imino spectra show that the secondary structure interactions involved in complex formation are retained in the s2U-modified complex. Magnesium has been reported to significantly stabilize the TAR–TAR* complex and we found that Mn2+ and Ca2+ are also strongly stabilizing, while Mg2+ exhibited the greatest effect on the complex kinetics. The stabilizing effects of 2-thiouridine indicate that this base modification may be generally useful as an antisense RNA modification for oligonucleotide therapeutics which target RNA loops.
INTRODUCTION
The trans-activation response element (TAR) is a stem–loop structure found at the 5′-end of HIV mRNA that plays a critical transcriptional activation role and is necessary for HIV replication (1–3). The trans-activation by TAR occurs through recognition by the TAT protein, which assembles along with cellular CDK9 and cyclin T1 to form the activation complex (4,5). While the TAR bulge domain has been shown to be sufficient for binding TAT, the apical loop is a key component for formation of the native trans-activation complex composed of TAR, TAT, CDK9 and cyclin T1 (4).
The structure of the free TAR hairpin has been determined by NMR spectroscopy (6,7), as has the structure of TAR complexed with TAT peptides (7,8) and the structure of TAR complexed with arginamide (9). Furthermore, the solution structure of TAR complexed with a complementary hairpin, TAR*, has been determined and represents a model system for understanding RNA loop–loop interactions (10). Other examples of RNA loop–loop interactions are the tRNA anticodon–anticodon complexes studied by Grosjean et al. (11), the HIV dimerization domain (12,13) and the ColE1 complex (14,15). These complexes exhibit equilibrium dissociation constants in the micromolar to nanomolar range; the tRNAs are remarkable in that the complexes are mediated by only 3 bp. For tRNA–tRNA complexes involving A-U base pairs, a dramatic stabilizing effect is seen when uridine at the wobble position is modified to one of the family of 2-thiouridine (s2U) bases commonly found at that sequence location (11,16). The TAR–TAR* complex provides a very high affinity RNA loop–loop interaction that can be used to investigate whether the effects of sulfur modification seen in the tRNA system can be generalized to other RNA loop–loop interactions. The TAR–TAR* system is also a good model system for defining the characteristics of antisense RNA oligonucleotides that target potentially accessible loop regions in RNA (17–19).
The effects of so-called ‘antisense’ modifications on RNA thermodynamic stabilization have been well characterized, with the most predictable modifications being those that stabilize the 3′-endo sugar conformation (20). The sugar modifications are typified by the naturally occurring 2′-O-methyl modification that provides modest RNA duplex stabilization and nuclease resistance (21). Base modifications that stabilize stacking interactions, such as the 5-propyne, are also employed as antisense nucleotides since they have the desirable property of supporting RNase H activity in a deoxynucleotide context, as well as increasing duplex stability (22). The mechanism by which s2U modification provides stabilization likely involves a combination of effects where the s2U nucleoside favors the 3′-endo sugar conformation through interaction with the 2′-hydroxyl and has the additional effect of a more polarizable sulfur that stabilizes stacking with neighboring nucleosides (23,24). Although the number of reported systems is small, replacement of uridine with 2-thiouridine results in dramatic increases in Tm. For the anticodon–anticodon systems described above, a stabilization of 20°C has been reported (16), while a still significant 12°C increase in Tm was seen for a single s2U modification in the stem of an RNA tetraloop hairpin (24). The somewhat smaller, but still significant, stabilization seen in short RNA duplexes demonstrates that the effects are sequence and structurally dependent (25). Unmodified tRNALys anticodon stem–loops fail to bind programmed ribosomes (26), but a single s2U at the wobble position is sufficient to restore most of the affinity (27).
In order to gain some insight into RNA loop–loop interactions and to investigate the general stabilization by s2U in this context, we have used BIACORE to study the kinetics of the TAR–TAR* RNA complex. NMR spectra were also collected on the modified complex to confirm that the structure of modified and unmodified complexes are similar. For this model system, the effects of a single s2U substitution in the TAR hairpin component stabilizes the complex, as seen previously for the anticodon–anticodon complexes, through effects on both the association and dissociation rate constants. Modification by s2U lowers the Kd values to ones where RNA–RNA interactions can begin to compete with native RNA–protein interactions.
MATERIALS AND METHODS
RNA synthesis and purification
The RNA oligonucleotides were synthesized on an Applied Biosystem 394 oligonucleotide synthesizer on a 1 µmol scale using 0.05 M acetonitrile solutions of PAC amidites (PAC A, isopropyl-PAC G, acetyl-C and 5′-biotin phosphoramidite) from Glen Research. The protected phosphoramidite of s2U was synthesized and incorporated into RNA oligonucleotides using the modified oligonucleotide protocol described previously from our laboratory (28). After base deprotection, the dried, protected oligonucleotide was dissolved in 1 ml of neat Et3N·3HF (Aldrich) and stirred at room temperature for 9–12 h (29,30). The reaction was quenched by adding 0.1 ml of water and the RNA was precipitated by adding 10 ml of n-butanol and allowing the solution to stand at –20°C for 6 h. The precipitated RNA was recovered by centrifugation and dried under reduced pressure in a Speed-Vac concentrator. The crude RNA was purified by anion exchange HPLC (31) and dialyzed against 1 M NaCl followed by deionized water and then the RNA lyophilized. The RNA for electrophoresis experiments was 5′-end-labeled using polynucleotide kinase and [γ-32P]ATP (NEN) by standard methods (32) and the nucleotides separated from RNA using a QIAquick (Qiagen) nucleotide removal kit.
Electrophoretic mobility shift assay
RNA oligonucleotides were dissolved in 10 µl of incubation buffer (100 mM NaCl, 10 mM MgCl2, 0.1 mM EDTA, 10 mM Tris–HCl, pH 7.0). The RNA hairpins were annealed individually in the buffer by heating at 80°C for 30 s and cooled immediately on ice. The complementary RNA hairpins were then mixed together and equilibrated at 4°C for 30 min. The sample was then mixed with 2 µl of 40% (w/v) sucrose and loaded immediately onto a 20% polyacrylamide gel at 4°C. The electrophoresis buffer was 100 mM Tris–HEPES, pH 7.8, containing 100 mM NaCl, 10 mM MgCl2 and 0.1 mM EDTA. The effects of Mg2+ on RNA complex stability were determined in this assay by running gels under otherwise identical buffer conditions without MgCl2.
NMR spectroscopy
RNA samples were dissolved in 10 mM phosphate buffer (pH 6.4) containing 10 mM MgCl2, 50 mM NaCl and 0.1mM EDTA to final RNA concentrations of 0.5–1.0 mM. The NMR data were collected at pH 6.4 rather than the pH 7.8 conditions used for gel band shifts in order to minimize exchange broadening of the imino protons. The conditions for NMR and electrophoresis were as used previously for the unmodified TAR–TAR* complex in order to make comparisons relevant (33). The absence of titratable groups in the pH range 6.4–7.8 supports the assumption that the structures observed by NMR, gel band shift and BIACORE experiments are the same under the different buffer conditions. The 1H NMR spectra in H2O were taken on a Varian Unity 500 NMR spectrometer at 10°C using a 1-1 binomial water suppression pulse sequence (34).
BIACORE kinetic analysis
The BIACORE 2000 biosensor, flat carboxymethylated sensor chip C1, NHS/EDC coupling reagents and ethanolamine were from BIACORE AB (Uppsala, Sweden). Kinetic rate constants for TAR16 and TAR16s2U binding to TAR*16 were determined by the technique of surface plasmon resonance (35). Streptavidin (Pierce) was immobilized onto a flat sensor chip (C1) using amine coupling chemistry (36). The immobilization steps were carried out at a flow rate of 20 µl/min in HEPES buffer (20 mM HEPES, 150 mM NaCl, 3.4 mM EDTA and 0.005% P20 surfactant). Two sensor surfaces were simultaneously activated for 7 min with a mixture of NHS (0.05 M) and EDC (0.2 M). Streptavidin was injected at a concentration of ~40 µg/ml in 10 mM sodium acetate, pH 4.4, for 7 min. Ethanolamine (1 M, pH 8.5) was injected for 7 min to block any remaining activated groups. An average of 1000 response units (RU) of streptavidin were immobilized on each flow cell. Biotinylated TAR*16 was injected through flow cell 2 at a concentration of 100 nM until ~30 RU were captured by the streptavidin surface. Flow cell 1 was left with streptavidin only, to be used as a reference surface.
Kinetic binding experiments were performed under conditions that mimic the mobility shift experiments. The same running buffer was used (100 mM NaCl, 10 mM MgCl2, 0.1 mM EDTA and 100 mM Tris–HEPES, pH 7.8) and the instrument was equilibrated at 4°C. Kinetic data were collected by injecting 100 µl of a 200 nM concentration of TAR16 or TAR16s2U at 100 µl/min over flow cells 1 and 2 operating in series. After the injection phase, both surfaces were washed with buffer for 1000 s to monitor dissociation of the bound RNA. To establish the level of experimental noise, each binding study was repeated three times including a blank injection of running buffer. To correct for refractive index changes and instrument noise the response data from the reference surface were subtracted from the responses obtained from the reaction surface. The flow rate was also varied from 100 to 20 µl/min to assess whether the reactions were limited by mass transport.
Kinetic rate constants were determined by fitting the corrected response data to a simple bimolecular interaction model, A + B = AB. The differential rate equations used to describe the reaction model are: d[A]/dt = 0; d[B]/dt = –ka[A][B] + kd[AB]; d[AB]/dt = ka[A][B] – kd[AB]. Kinetic constants ka and kd are the association and dissociation rate constants, respectively, [A] is the concentration of RNA in solution, [B] represents the immobilized TAR*16 and [AB] represents the amount of complex formed during the reactions and is proportional to the response (RU). The rate equations were numerically integrated and the results simultaneously fitted to the association and dissociation phase response data using the non-linear least squares data analysis program CLAMP© (37). To assess the effects of metal ion dependence on binding, biosensor experiments were repeated in buffers containing different amounts of MgCl2, CaCl2 and MnCl2 (0–150 mM).
RESULTS
Electrophoretic mobility shift assays
We initially performed gel mobility shift experiments to get a general assessment of whether modification of TAR16 with s2U would stabilize the complex. Heterodimer formation between TAR16s2U and TAR*16 was shown by the formation of a stable complex with a lower electrophoretic mobility than either hairpin alone. This modified complex was significantly more stable than the unmodified complex previously reported. Both the parent and the modified complexes showed evidence of a second minor species, similar to that reported previously (33). However, neither the NMR experiments nor the BIACORE kinetics indicated the presence of multiple species. The estimated dissociation constant, Kd, of the modified complex is between 1 and 5 nM, as seen in Figure 2, while the Kd of the unmodified TAR–TAR* complex is ~10-fold higher and consistent with the values previously reported (33). Magnesium was reported to increase the Tm of the TAR–TAR* complex by 40°C, but complex formation was still seen in the presence of high monovalent ion concentrations (33). In contrast, under our electrophoresis conditions no shifted complexes were observed without added divalent metals even with NaCl concentrations up to 150 mM. The effects of Mg2+ were confirmed in BIACORE experiments and we extended the divalent metal studies to include both Ca2+ and Mn2+.
Figure 2.
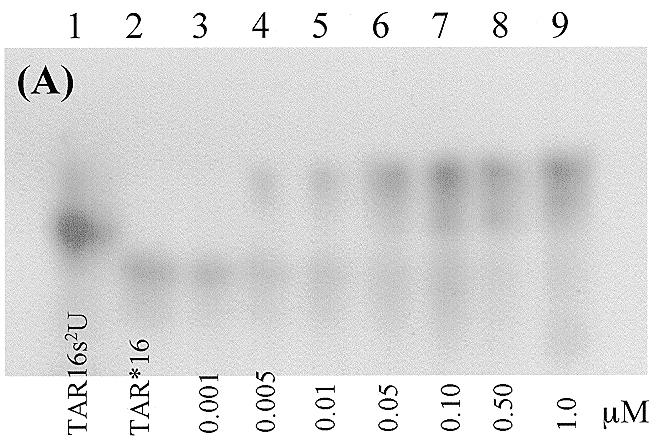
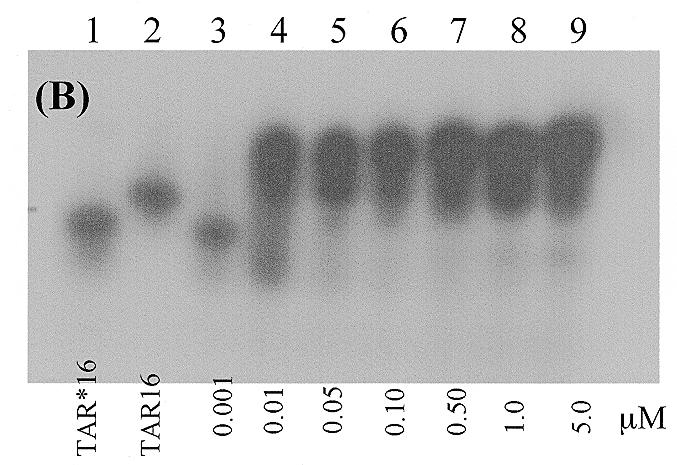
Polyacrylamide gel electrophoresis bandshift assays of RNA–RNA complex formation. The electrophoresis buffer was 100 mM Tris–HEPES, pH 7.8, containing 100 mM NaCl, 10 mM MgCl2 and 0.1 mM EDTA. The concentration series for the two figures are different due to the higher affinity of the TAR16s2U hairpin for its complement. (A) The complex between TAR*16 and TAR16s2U at 4°C. Each lane contains 0.5 nM 32P-labeled TAR*16 except lane 1, which contains 0.5 nM 32P-labeled TAR16s2U only. The concentrations of unlabeled TAR16s2U in lanes 2–9 are 0.0, 0.001, 0.005, 0.01, 0.05, 0.1, 0.5 and 1.0 µM. (B) The complex between TAR*16 and unmodified TAR16 at 4°C. Each lane contains 0.5 nM 32P-labeled TAR*16 except lane 2, which contains 0.5 nM 32P-labeled TAR16 only. The concentrations of unlabeled TAR16 in lanes 3–9 are 0.001, 0.01, 0.05, 0.1, 0.5, 1.0 and 5.0 µM.
NMR imino spectra
1H NMR spectroscopy was used to verify that the modified TARs2U hairpin complex formed via the same secondary base pairing interactions as described for the unmodified TAR–TAR* complex. Figure 3 shows the imino spectral region where resonances from slowly exchanging N–H protons involved in stable hydrogen bonding interactions are detected (33,38). The two component hairpins show the resonances from base pairs in the stem, but the loop imino protons exhibit typical exchange kinetics and no NMR resonances are detected for these residues. The 1H NMR spectrum of the complex is a superposition of these two component spectra plus additional resonances from the loop–loop interaction. The sharp peaks for s2U7, G8, G9 and G10 are clearly resolved in Figure 3C and correspond to those reported for the unmodified complex. The assignments were confirmed by one-dimensional difference NOE experiments (not shown), which allowed us to discriminate between A-U and G-C base pairs and to observe sequential imino to imino connectivites for the resolved resonances. The NOEs we observed from resolved resonances and a chemical shift comparison with the published data for the unmodified complex indicate that the base pairing geometry is the same for the unmodified and the s2U-modified complexes (33).
Figure 3.

1H NMR spectra of the complex formed between TAR16s2U and the complementary TAR*16 RNA hairpin. (A) Imino spectra at 10°C of TAR*16 in 90% H2O buffer, pH 6.4, containing 100 mM NaCl, 10 mM MgCl2, 0.1 mM EDTA. A total of 128 transients were collected on a 0.5 mM sample. (B) Imino spectra at 10°C of TAR16s2U with buffer conditions as in (A). A total of 64 transients were collected on a 0.5 mM sample. (C) Imino spectra at 10°C of the complex formed between the two component hairpins in (A) and (B). A total of 512 transients were collected. The additional numbered peaks correspond to the imino proton resonances that result from base pairs at the kissing hairpin interface.
Kinetic analysis of RNA–RNA interactions
The kinetic binding constants for the RNA kissing loop complexes were determined by immobilizing one of the hairpin loops (TAR*16) onto a streptavidin chip by biotinylating it at the 5′-end. This produced the stable and homogeneous recognition surface that is essential for performing a detailed kinetic analysis. A high flow rate and a low binding capacity surface was used to minimize the affects of mass transport and steric hindrance (37). To collect kinetic binding data, identical concentrations of TAR16 and TAR16s2U (200 nM) were injected over the immobilized TAR*16 and a reference surface simultaneously. Responses from the reference surface were used to correct for refractive index changes and instrument noise, producing high quality sensor data, as shown in Figure 4. Each RNA binding reaction plus a buffer blank was replicated three times. The data from each independent experiment overlap very well, indicating that the TAR*16 RNA surface was stable and could reproducibly bind complementary RNA oligos from solution. Decreasing the flow rate had no effect on the binding rate of either TAR16 or TAR16s2U for TAR*16, demonstrating that the interactions were not limited by mass transport (37).
Figure 4.

Kinetic analysis of RNA kissing loop interactions. The BIACORE running buffer was the same as used for the gel band shift assays: 100 mM NaCl, 10 mM MgCl2, 0.1 mM EDTA and 100 mM Tris–HEPES, pH 7.8. Binding responses were collected for (A) TAR16s2U; (B) TAR16 binding to an immobilized TAR*16 element. The dots represent the experimental data collected for each RNA injected at 200 nM and replicated three times. The injections of a buffer blank are also shown (C). The solid lines represent the best fit to a simple bimolecular reaction model.
A visual comparison of the binding responses obtained for TAR16 and TAR16s2U shows that the s2U-modified oligo forms a more stable complex with the TAR*16 derivatized surface (Fig. 4). It is quite evident that TAR16s2U dissociates much more slowly from the TAR*16 surface, since the TAR16 oligo has dissociated back to baseline in 1000 s, whereas the TAR16s2U oligo has only reached its half-life in that time. A detailed kinetic analysis was performed by globally fitting the association phase and dissociation phase data for each oligo to a simple bimolecular reaction model. The results shown in Figure 4 demonstrate that this model provides an appropriate description of the binding responses for both TAR16 and TAR16s2U. The residual standard deviations from the TAR16 and TAR16s2U data sets were 0.42 and 0.41 RU, respectively, while the replication standard deviation for these data sets was 0.35 RU. This represents a measure of the total experimental noise, taking into account the random noise of the detector, loss of binding activity over time and any other experimental artifacts. The residual standard deviations were only 0.065 RU higher than the replication standard deviation, an indication that the model provides a good description of the data since very little information is left in the residuals (37).
The rate constants returned from analysis of the kinetic data are shown in Table 1 along with the linear approximation standard errors. The association rate for TAR16s2U was approximately twice as fast as that for TAR16, while the dissociation rate was four times slower. Together these rate constants predict an equilibrium dissociation constant for TAR16s2U that was eight times lower than that for TAR16 (1.58 versus 12.5 nM). The electrophoretic mobility shift experiments indicated that the TARs2U–TAR* complex was considerably weaker in the absence of Mg2+. Under our BIACORE conditions, complex formation was detected only in the presence of divalent metals. Increasing the concentration of MgCl2 increased the rate of binding and this increase reached saturation at ~50 mM MgCl2, as shown in Figure 5. Stable complexes were also detected with CaCl2 and MnCl2, but they were somewhat weaker than seen for comparable MgCl2 concentrations. Binding studies in the presence of MgCl2 had both the fastest association rates and the slowest dissociation rates.
Table 1. Kinetic rate and affinity constants determined from BIACORE analysis.
| RNA | ka (M–1 s–1) | kd (s–1) | Kd (nM) |
|---|---|---|---|
| TAR16 | 2.10 × 105 ± 3 × 103 | 2.63 × 10–3 ± 2 × 10–5 | 12.5 ± 0.2 |
| TAR16S2U | 4.14 × 105 ± 4 × 103 | 6.55 × 10–4 ± 6 × 10–6 | 1.58 ± 0.02 |
Figure 5.

MgCl2 dependence of the binding rate and equilibrium constants. TAR16 (solid squares) and TAR16s2U (open squares) were injected over the immobilized TAR*16 element at a concentration of 200 nM containing varying concentrations of MgCl2 (5, 10, 50 and 150 mM) in 100 mM NaCl, 0.1 mM EDTA and 100 mM Tris–HEPES, pH 7.8, at 4°C. The binding responses were analyzed as described in Materials and Methods. (A) Association rates (ka); (B) dissociation rates (kd); (C) equilibrium constants (Kd).
DISCUSSION
The TAR RNA stem–loop in HIV mRNA plays a critical role in HIV replication and has two loop domains where the structure is closely tied to function. The apical loop appears to be relatively unstructured in the free RNA, but can form a tight complex with a complementary RNA hairpin loop (33). The complement to HIV TAR, TAR*, is found within the HIV genomic sequence, but no functional significance has been attributed in vivo to a TAR–TAR* complex. The so-called ‘kissing-hairpin’ motif of the complex is found in several other biologically relevant systems, notably the ColE1 complex and the HIV dimerization domain (12,14).
The TAR system in HIV is an attractive target for therapeutic intervention and the TAR RNA hairpin has been investigated as a target of antisense RNA compounds (17–19). Targeting stable secondary structures such as the TAR hairpin is potentially problematical since the antisense oligonucleotide has to compete with the favorable intramolecular base pairing interactions that form a stable stem. Oligonucleotides that target an accessible loop region such as the TAR loop may be more effective, but a complex mediated by only five base pairs may not be sufficiently stable to compete for proteins that bind the RNA. In the case of TAR, the Kd for the TAR, TAT, cyclin T1 complex is expected to be sub-nanomolar (4) and the Kd for the interaction of TAT peptides with TAR is 16 pM from kinetic measurements (39). Even the conservative estimate of 400 pM obtained from band shifts suggests that oligonucleotides with any of the common modifications would be inefficient competitors for protein–TAR complexes (39).
We found that replacing the single uridine in the loop of TAR with s2U increased the binding affinity for its complementary RNA 8-fold. The NMR spectrum showed that the complex is very stable and that the imino protons from base pairs in the complex have exchange rates similar to those of the stem Watson–Crick base pairs. Despite the increased stability of the complex, we did not see an imino resonance for U6* from the TAR* hairpin, indicating that this potential A-U base pair is still relatively unstable, as seen previously (33). While modification of U6* to s2U would have been logical from a therapeutic perspective, the previous structural data indicated that such a modification would have little effect since that uridine does not form a base pair. Furthermore, we wanted to use the clearly resolved NMR resonance of the U7 imino proton as a measure of stabilization or destabilization in the modified complex. The imino proton at 13.7 p.p.m. gives an AH2 NOE and was assigned to s2U7 by analogy with U7 for the unmodified complex. The chemical shift of the s2U7 imino proton is similar to that of U7 in the unmodified complex and indicates a similar stability and environment. Although the exchange behavior was not surprising given the millimolar RNA concentrations and nanomolar dissociation constants, we were surprised that sulfur modification did not result in a downfield shift of the imino proton. Local shielding and hydrogen bonding differences in tRNAs can result in significantly different imino proton shifts for sulfur-modified uridines (40). The imino protons in s2U-A Watson–Crick pairs are usually found close to 15 p.p.m. rather than the relatively upfield chemical shift of 13.7 seen for s2U7 in the TARs2U–TAR* complex (24,41).
Surface plasmon resonance is a powerful method for determining the kinetics of complex formation in biological systems, but few applications of the method for studying RNA–RNA interactions have been reported. Our BIACORE experiments allowed us to very accurately determine the relative stabilities of two very tight RNA complexes (TAR–TAR* and TARs2U–TAR*) and to investigate how divalent metal ions affect these RNA complexes. The equilibrium dissociation constants from BIACORE are consistent with both the estimates from gel electrophoresis mobility shifts done in our laboratory and with those reported previously (33). The detailed kinetics show that the stabilization of s2U arises from a combination of a faster ka and a slower kd. This two-part effect can be understood by looking at what is known about the effects of s2U modification on nucleoside sugar conformation and a rationalization about the potential effects on hydrogen bonding and base stacking. Compared to uridine, s2U favors the 3′-endo sugar conformation through a steric interaction between the sulfur and the 2′-hydroxyl (23,42). The sulfur also lowers the pKa of the imino proton and would be expected to be a better stacker than uridine. The sugar conformational effect and perhaps also the stacking stabilization within the loop would serve to pre-organize the TARs2U hairpin, resulting in a faster ka. Once the complex is formed, the stacking stabilization, 3′-endo sugar preference and a stronger hydrogen bonding interaction would decrease the kd. The 8-fold difference in Kd values is reasonably well correlated with a 1–2 kcal/mol ΔΔG37 seen for double-stranded RNA stabilization upon a single s2U modification (24,25). The base pairing geometry in the TAR–TAR* system has been shown to be standard Watson–Crick (10) and the stabilization by s2U is consistent with the conclusions of Testa et al. that s2U in a Watson–Crick base pair has a predictable stabilizing effect (25).
The kinetic constants determined in the present BIACORE study are close to the expected values based on the results of earlier studies of modified antisense oligonucleotides. The ka of 4.14 × 105 we observed is 4-fold faster than seen for a very tight binding phosphoramidate antisense oligonucleotide targeted at the TAR stem–loop (18). The phosphoramidate values were determined from band shift assays, so might not be rigorously comparable, but this comparison does indicate that the two methods give comparable kinetic parameters for complexes with similar Kd values (5 nM for the phosphoramidate anti-TAR ODN). Another BIACORE study of PNA hybridization to DNA and RNA also indicated that ka and kd for complexes with Kd values in the low nanomolar range are similar to those that we have measured (43). The lower Kd values that are found upon s2U modification are a combination of effects on both the association and dissociation rates for complex formation, which is consistent with the known effects of sulfur modification on base pairing and base stacking. While we had to modify the putative ‘target’ in the TAR–TAR* system due to the sequence constraints of this system, the increased stability of the complex demonstrates that s2U modification is a viable approach for stabilizing RNA loop–loop interactions and may be generally useful for increasing the binding affinity of antisense oligonucleotides that target biologically important RNA hairpin loops.
Figure 1.

Secondary structures of the RNA hairpins used in this study. (A) TAR16 contains a uridine at position 7 or an s2U-7 for the TAR16s2U oligonucleotide; (B) TAR*16 RNA complementary to the TAR oligo; (C) base pairing geometry of a Watson–Crick s2U-A base pair. The sulfur is in a non-hydrogen bonding position where it can stabilize the base pair through effects on the sugar conformation and through stacking interactions with neighboring base pairs.
Acknowledgments
ACKNOWLEDGEMENTS
This research was supported by the NIH (GM55508). The NMR, MS and oligonucleotide facilities are supported by NIH grants CA42014 and RR06262. We thank Raju K. Kumar for synthesizing the s2U nucleoside phosphoramidite.
REFERENCES
- 1.Fisher A.G., Feinberg,M.B., Josephs,S.F., Harper,M.E., Marselle,L.M., Aldovini,A., Debouk,C., Gallo,R.C. and Wong-Staal,F. (1986) Nature, 320, 367–371. [DOI] [PubMed] [Google Scholar]
- 2.Jones K.A. and Peterlin,B.M. (1994) Annu. Rev. Biochem., 63, 717–743. [DOI] [PubMed] [Google Scholar]
- 3.Steffy K. and Wong-Staal,F. (1991) Microbiol. Rev., 55, 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei P., Garber,M.E., Fang,S.M., Fischer,W.H. and Jones,K.A. (1998) Cell, 92, 451–462. [DOI] [PubMed] [Google Scholar]
- 5.Bieniasz P.D., Grdina,T.A., Bogerd,H.P. and Cullen,B.R. (1998) EMBO J., 17, 7056–7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aboul-ela F., Karn,J. and Varani,G. (1996) Nucleic Acids Res., 24, 3974–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long K.S. and Crothers,D.M. (1999) Biochemistry, 38, 10059–10069. [DOI] [PubMed] [Google Scholar]
- 8.Aboul-ela F., Karn,J. and Varani,G. (1995) J. Mol. Biol., 253, 313–332. [DOI] [PubMed] [Google Scholar]
- 9.Puglisi J.D., Tan,R., Calnan,B.J., Frankel,A.D. and Williamson,J.R. (1992) Science., 257, 76–80. [DOI] [PubMed] [Google Scholar]
- 10.Chang K. and Tinoco,I. (1997) J. Mol. Biol., 269, 52–66. [DOI] [PubMed] [Google Scholar]
- 11.Grosjean H., Houssier,C., Romby,P. and Marquet,R. (1998) In Grosjean,H. and Benne,R. (eds), Modification and Editing of RNA. ASM Press, Washington, DC, pp. 113–133.
- 12.Mujeeb A., Clever,J.L., Billeci,T.M., James,T.L. and Parslow,T.G. (1998) Nature Struct. Biol., 5, 432–436. [DOI] [PubMed] [Google Scholar]
- 13.Paillart J.-C., Skripkin,E., Ehresmann,B., Ehresmann,C. and Marquet,R. (1996) Proc. Natl Acad. Sci. USA, 93, 5572–5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marino J., Gregorian,R., Csankovszki,G. and Crothers,D. (1995) Science, 268, 1448–1454. [DOI] [PubMed] [Google Scholar]
- 15.Gregorian R.S. and Crothers,D.M. (1995) J. Mol. Biol., 248, 968–983. [DOI] [PubMed] [Google Scholar]
- 16.Houssier C., Degee,P., Nicoghosian,K. and Grosjean,H. (1988) J. Biomol. Struct. Dyn., 5, 1259–1266. [DOI] [PubMed] [Google Scholar]
- 17.Boulme F., Perala-Heape,M., Sarih-Cottin,L. and Litvak,S. (1997) Biochim. Biophys. Acta, 1351, 249–255. [DOI] [PubMed] [Google Scholar]
- 18.Boulme F., Freund,F., Moreau,S., Nielsen,P.E., Gryaznov,S., Toulme,J.J. and Litvak,S. (1998) Nucleic Acids Res., 26, 5492–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mestre B., Arzumanov,A., Singh,M., Boulme,F., Litvak,S. and Gait,M.J. (1999) Biochim. Biophys. Acta, 1445, 86–98. [DOI] [PubMed] [Google Scholar]
- 20.Crooke S.T. (1992) Annu. Rev. Pharamacol. Toxicol., 32, 329–376. [DOI] [PubMed] [Google Scholar]
- 21.Cummins L.L., Owens,S.R., Risen,L.M., Lesnik,E.A., Freier,S.M., McGee,D., Guinosso,C.J. and Cook,P.D. (1995) Nucleic Acids Res., 23, 2019–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gutierrez A.J., Matteucci,M.D., Grant,D., Matsumura,S., Wagner,R.W. and Froehler,B.C. (1997) Biochemistry, 36, 743–748. [DOI] [PubMed] [Google Scholar]
- 23.Yokoyama S., Yamaizumi,Z., Nishmura,S. and Miyazawa,T. (1979) Nucleic Acids Res., 6, 2611–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar R.K. and Davis,D.R. (1997) Nucleic Acids Res., 25, 1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Testa S.M., Disney,M.D., Turner,D.H. and Kierzek,R. (1999) Biochemistry, 38, 16655–16662. [DOI] [PubMed] [Google Scholar]
- 26.Von Ahsen U., Green,R., Schroeder,R. and Noller,H. (1997) RNA, 3, 49–56. [PMC free article] [PubMed] [Google Scholar]
- 27.Ashraf S., Sochacka,E., Cain,R., Guenther,R., Makliewicz,A. and Agris,P. (1999) RNA, 5, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar R.K. and Davis,D.R. (1995) J. Org. Chem., 60, 7726–7727. [Google Scholar]
- 29.Wincott F., DiRenzo,A., Shaffer,C., Grimm,S., Tracz,D., Workman,C., Sweedler,D., Gonzalez,C., Scaringe,S. and Usman,N. (1995) Nucleic Acids Res., 23, 2677–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gasparutto D., Livache,T., Bazin,H., Dupla,A., Guy,A., Khordin,A., Molko,D., Roget,A. and Teoule,R. (1992) Nucleic Acids Res., 20, 5159–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sproat B., Colonna,F., Mulla,B., Tsou,D., Andrus,A., Hampel,A. and Vinayak,R. (1995) Nucleosides Nucleotides, 14, 255–273. [Google Scholar]
- 32.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Plainview, NY.
- 33.Chang K.-Y. and Tinoco,I. (1994) Proc. Natl Acad. Sci. USA, 91, 8705–8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hore P.J. (1983) J. Magn. Reson., 55, 283–300. [Google Scholar]
- 35.Myszka D.G. (1997) Curr. Opin. Biotechnol., 8, 50–57. [DOI] [PubMed] [Google Scholar]
- 36.Johnson B. and Lofas,S. (1991) Anal. Biochem., 198, 268–277. [DOI] [PubMed] [Google Scholar]
- 37.Morton T.A. and Myszka,D.G. (1998) Methods Enzymol., 295, 268–294. [DOI] [PubMed] [Google Scholar]
- 38.Varani G. and Tinoco,I. (1991) Q. Rev. Biophys., 24, 479–532. [DOI] [PubMed] [Google Scholar]
- 39.Long K.S. and Crothers,D.M. (1995) Biochemistry, 34, 8885–8895. [DOI] [PubMed] [Google Scholar]
- 40.Griffey R.H., Davis,D.R., Yamaizumi,Z., Nishimura,S., Hawkins,B.L., Poulter,C.D. (1986) J. Biol. Chem., 261, 12074–12078. [PubMed] [Google Scholar]
- 41.Kumar R.K. and Davis,D.R. (1997) Nucleosides Nucleotides, 16, 1469–1472. [Google Scholar]
- 42.Sierzputowska-Gracz H., Sochacka,E., Malkiewicz,A., Kuo,K., Gehrke,C. and Agris,P. (1987) J. Am. Chem. Soc., 109, 7171–7177. [Google Scholar]
- 43.Jensen K.K., Orum,H., Nielsen,P.E. and Norden,B. (1997) Biochemistry, 36, 5072–5077. [DOI] [PubMed] [Google Scholar]