

Abstract
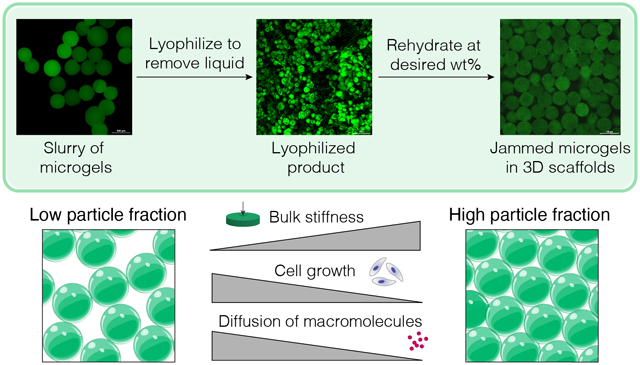
Microporous annealed particle (MAP) hydrogels are porous 3D scaffolds generated by interlinking randomly packed microgels (μgels). Particle fraction, hydrogel stiffness, microparticle shape, and crosslinking chemistry are paramount to the microstructure that microgels make within MAP scaffolds. Of these parameters, control over the particle fraction in MAP scaffolds varies greatly by user and drying technique, leading to inconsistent microarchitectures. These inconsistencies have biological ramifications, as the particle fraction of MAP scaffolds determines the void space within the material which strongly impacts cell growth. Here, we describe a method of freeze-drying microgels that leads to consistent and user-defined particle fractions by weighing the dried microgel powder and reconstituting at known volumes. Though freeze-drying hydrogels typically leads to ice crystal and cryogel formation, we report on mediums that result in freeze-dried microgels that retain their original properties when rehydrated. By rehydrating lyophilized microgels to form MAP scaffolds, we demonstrate that particle fraction controls the bulk scaffold stiffness, but not local microgel stiffness. Further, the particle fraction in MAP scaffolds directly affects cell growth and macromolecular diffusion. Using controlled particle fractions in MAP scaffolds, we can now reproducibly assess mechanical properties, diffusion of macromolecules, and cell responses within user-defined microarchitectures.
Keywords: hydrogel, microparticles, lyophilize, MAP scaffold, porosity
Graphical Abstract
1. INTRODUCTION
Microporous annealed particle (MAP) gels are comprised of spherical microgels that form a bulk, porous scaffold when interlinked. The void space between the interlinked microgels supports accelerated cell growth in vitro [1–4] and infiltration and migration in vivo [5–17]. Prior to MAP scaffold formation, microgel building blocks exist in a suspension. The excess liquid must be removed from the suspension to yield a sufficient particle fraction of microgels to achieve a jammed state [18]. In this jammed state, microgels exhibit shear-thinning properties, allowing them to be injectable, space-filling materials [5, 7, 14] and inks for 3D printing [4, 19]. Upon injection, orthogonal crosslinking reactions can be used to interlink the microgels and form MAP scaffolds [1, 2, 6, 7, 20, 21]. The particle fraction of microgels in the MAP gels is inversely proportional to the void fraction, with loosely packed microgels generating greater void than closely packed microgels [14, 22].
Most users produce water-swollen microgels using reverse emulsions either in batch or within microfluidic devices, as well as via lithography, electrohydrodynamic spraying, and mechanical fragmentation [22, 23]. Purified microgels are then concentrated to induce particle jamming through vacuum-driven filtration [19] or centrifugation [14] coupled with manual removal of buffer via aspiration or with an absorbent material. Although particle fraction is a critical property of MAP scaffolds, there have been few attempts to accurately modulate it to date. Using microgels of varying size or shape has been shown to alter the particle fraction in MAP scaffolds [3, 24, 25] as well as non-annealed granular materials [14, 26]. Additionally, centrifugation at varying speeds has been shown to alter the degree to which microgels are concentrated for generating non-annealed granular materials with different particle fractions [14, 26]. However, our work demonstrates that concentrating spherical microgels via centrifugation results in variable particle fractions. Given that particle fraction itself is a bioactive signal for seeded or infiltrating cells, such as in modulating angiogenic sprouting [14] and non-viral gene transfer [3], our goal was to develop methodology to control the particle fraction of microgels in a reproducible manner without having to alter the microgel size or shape.
Freeze-drying, or lyophilization, is a process which removes aqueous solvent from the system through sublimation to yield a dried product. This method is widely applied to polymers to improve material shelf-life [27]. Lyophilization of crosslinked polymer networks, however, can result in the formation of pores during the freezing process when crystal structures in the solvent form and act as porogens [28]. The interconnected porous network that forms results in cryogels that exhibit different mechanical properties from their nanoporous counterparts [29]. Though in some instances microgel cryogels may be desirable, our goal was to find freeze-drying conditions that do not lead to cryogel formation within microgels.
Here, we describe a method for freeze-drying that allows microgels to retain their original properties after rehydration and leads to consistent particle fractions in MAP scaffolds. With this control over particle fractions in MAP scaffolds, we can now reproducibly assess mechanical properties, diffusion, and cell responses within these user-defined microarchitectures.
2. METHODS
2.1. Microgel production
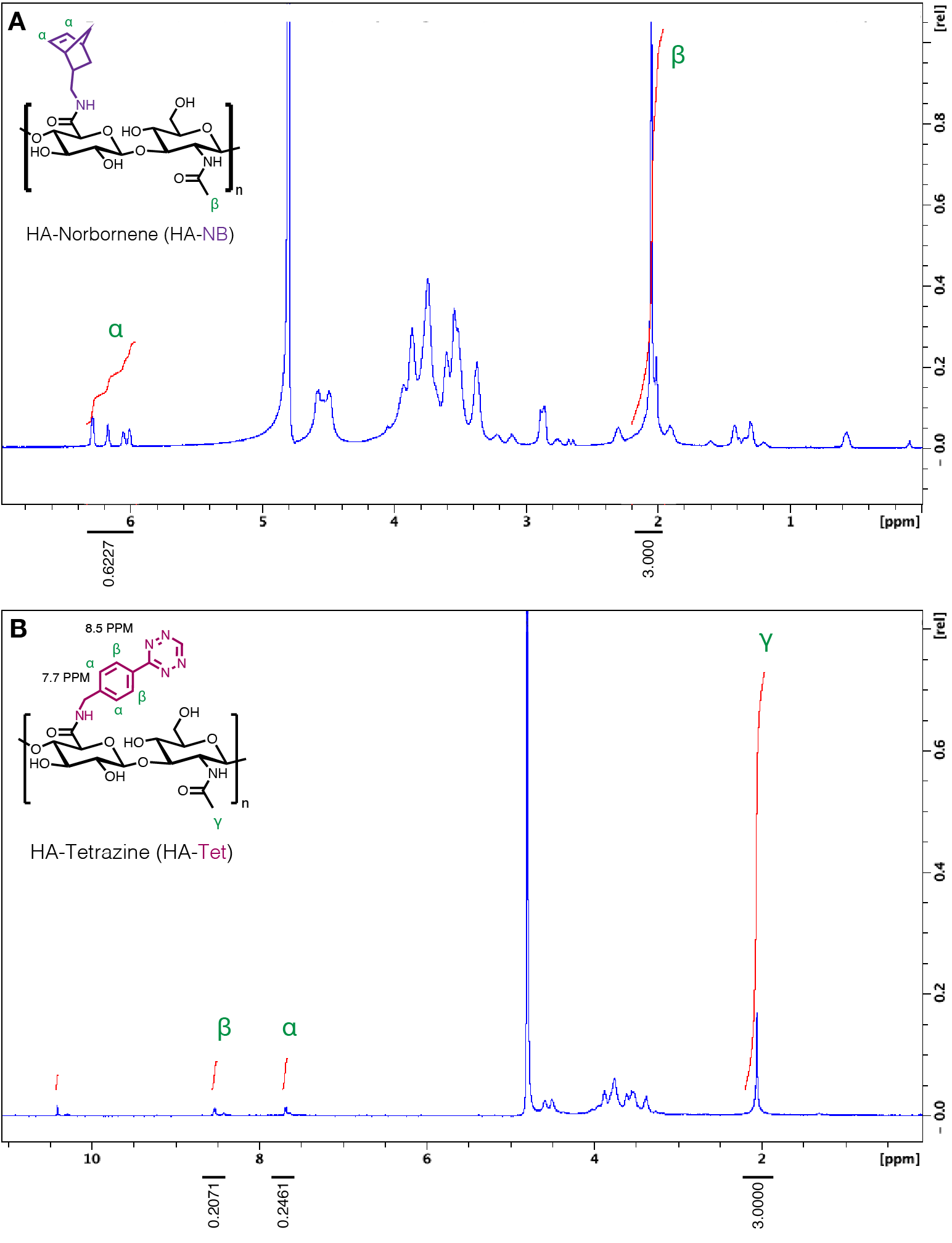
Hyaluronic acid (HA) (Contipro, pharmaceutical grade, 79 kDa) was modified at the carboxylic acid site with 5-norbornene-2-methylamine (NB) (TCI Chemicals) [3]. Approximately 31% of HA repeat units were successfully modified with NB, as determined by proton NMR analysis performed in deuterium oxide. 1H NMR shifts of pendant norbornenes at δ6.33 and δ6.02 (vinyl protons, endo), and δ6.26 and δ6.23 ppm (vinyl protons, exo) were compared to the HA methyl group δ2.05 ppm to determine functionalization. Microfluidic production of HA-NB microgels has been described previously, and these methods were used for both HA-NB and 8arm-PEG-NB (JenKem, 20 kDa) microgel synthesis [27]. In brief, microgels formulated using either HA- NB (3.4 wt% (w/v)) or 8arm-PEG-NB (10 wt%) precursor solution in 0.3 M HEPES buffer with 3.5 mM MMP-cleavable crosslinker (Ac-GCRDGPQGIWGQDRCG-NH2, GenScript), 1.75 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Millipore Sigma), and 9.9 mM lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, TCI Chemicals). The surfactant/oil phase for the microfluidic microgel generation consisted of 5% (v/v) Span-80 (Millipore Sigma) in heavy white mineral oil (Avantor) degassed prior to use. Microgel precursor was sterile filtered using a 0.2 μm filter. Microgel precursor was loaded in a 1 mL syringe alongside a 5 mL syringe of oil/surfactant on a dual-syringe pump (Chemyx) run at 0.3 uL min−1 to generate the particle sizes described in this work. Detailed methods for generating particles of unique sizes with this setup can be found in the work by Wilson, et al. [30]. Microgel production was conducted on a microfluidic device cast with Sylgard 184 polydimethylsiloxane (PDMS) (Dow Corning) prepared and cured according to the manufacturer’s instructions then plasma bonded to a glass slide using a corona plasma gun. Prior to the run, Rain-X was flushed through the device to coat the channels. Microgels fabricated on the device were collected in a 15 mL conical tube with a UV light fixed over the collection tube to facilitate downstream microgel crosslinking at an intensity of 20 mW cm−1.
Microgels were purified by first washing with 2% Pluronic F-127 (Sigma-Aldrich) (w/v) in washing buffer (300 mM HEPES, 50 mM NaCl, 50 mM CaCl2) followed by repeated washes (centrifugation (5,000 RCF) and aspiration of the wash) with washing buffer. Purified microgels were washed with 70% ethanol to sterilize.
2.2. Tet-linker and Tet-fluorophore synthesis
4arm-PEG-Tetrazine (20 kDa) was synthesized as described previously [6]. HA-tetrazine synthesis was adapted from Zhang, et al. using 79 kDa HA with molar equivalents of 1:1:0.25 of HA-repeat units to 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) (Thermo Fisher Scientific) to tetrazine-amine (Chem-Impex) to successfully modify approximately 11% of HA repeat units with tetrazine [31]. All polymer modifications were purified with dialysis (6–8 kDa molecular weight cut-off) and subsequently quantified by proton NMR. 1H NMR shifts of pendant tetrazine groups at δ8.5 (2H) and δ7.7 (2H) (aromatic protons) were compared to the HA methyl group δ2.05 ppm to determine functionalization.
Alexa Fluor 488 C5‐tetrazine (Alexa488‐Tet) was synthesized through two base‐catalyzed thiol‐Michael addition reactions in series, as previously described [6]. Briefly, Alexa Fluor 488 C5 maleimide (Invitrogen) and methyltetrazine-PEG4-maleimide (MW: 514.53 Da) (Kerafast) were added in equimolar amounts to HS-PEG-SH (MW: 3500 Da) (JenKem Technology USA) along with triethylamine (TEA) at 0.5 molar equivalent to Alexa Fluor 488 C5 maleimide. The product was precipitated in diethyl ether after reacting overnight then dried under vacuum. The resulting Alexa488-Tet product was dissolved in dimethylformamide at 1 mg mL−1 and stored at −20 °C. To label purified microgels, Alexa488-Tet was diluted to 0.07 mg mL−1 in 1x PBS. The diluted Alexa488-Tet solution was added to the microgels 1:2 by volume and incubated at 37°C for 30 minutes. The microgels were washed three times with 1x PBS to remove unreacted fluorophore.
2.3. Microgel lyophilization
Sterilized microgels were incubated 50% (v/v) in a variety of mediums at a range of concentrations overnight at 4°C. These mediums include: isopropyl alcohol (IPA), ethanol (EtOH), sodium chloride, phosphate-buffered saline (PBS, pH 7.4), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.8), tris(hydroxymethyl)aminomethane (Tris, pH 8.0), 2-(N-morpholino)ethanesulfonic acid (MES, pH 5.5), Dimethylsulfoxide (DMSO), and Acetonitrile (MeCN). The microgels in solution were mixed with a displacement pipette then flash-frozen in liquid nitrogen. Microgels were then lyophilized and kept at room temperature prior to rehydration. For rehydration, microgels were weighed and reconstituted in DiH2O at varying wt% (w/v).
2.4. Characterization of lyophilized microgels (lyo-microgels)
Rehydrated lyo-microgels were imaged on a confocal microscope (Nikon Ti Eclipse with a C2 LED laser light source). Maximum intensity projections were generated from Z-stacks (20x magnification, air objective, 2.5 μm step size) to measure microgel diameter. 3D renderings were created in IMARIS (Bitplane) to measure microgel volume and sphericity. Single 2D slices from Z-stacks were used to assess intra-microgel defects within lyo-microgels. Intensity profiles across the diameter of microgels as well as integrated density measurements of microgel area were measured in ImageJ. IMARIS 3D renderings were also used to assess particle fraction of lyo- microgels at varying wt% in MAP scaffolds.
2.5. Cell viability with lyo-microgels
Lyo-microgels were rehydrated in excess DMEM medium supplemented with 1% penicillin-streptomycin and 10% fetal bovine serum. Non-lyophilized microgels of the same population were incubated in the same medium for >1 hour. Both types of microgels were concentrated via centrifugation. D1 mouse mesenchymal cells (D1 ORL UVA) were mixed with microgels at a concentration of 10,000 cells μL−1. In each well, 5 μL of the cell and microgel mixture was added in a 96-well plate in triplicate, as well as a 2D control with D1 cells only. Viability was assessed after days 1, 3, and 5 using CellTiter-Glo kit (Promega).
2.6. Incubation to assess bacterial growth
Lyo-microgels were incubated from 1–3 days in Luria-Bertani broth (LB, VWR) without antibiotic at 37°C to determine whether the lyophilization process introduced areas for potential contamination. OD600 was measured using a spectrophotometer each day to monitor for signs of growth, compared to freshly diluted microgels in broth.
2.7. Bulk scaffold mechanical characterization
For material characterization, MAP scaffold discs were made by sandwiching 50 μL microgels (84% v/v) and crosslinker (16% v/v) between two glass slides treated with SigmaCote (Sigma-Aldrich) with a 1 mm Teflon spacer around the edges of the slides. Clips were used to secure the slides and was incubated in a petri dish with a wet KimWipe to act as a humidity chamber during inter-particle crosslinking. Once microgels were interlinked, scaffolds were swollen overnight in 1x PBS. Swollen gels were punched with an 8 mm biopsy punch. For Tet-linker comparison studies, a range of crosslinker ratios were used to anneal the non-lyo-microgels. The concentrations of HA-Tet for 2.5, 5, 10, and 20 Tet/HA backbone were 0.016 mM, 0.032 mM, 0.063 mM, and 0.126 mM respectively. The concentrations of 4arm-PEG-Tet for 5, 10, and 20 Tet/HA backbone were 0.24 mM, 0.47 mM, and 0.94 mM respectively. Lyo-microgels were annealed with 0.02 mg mL−1 HA-Tet. Rheological measurements on a parallel plate rheometer (Anton Paar, Physica MCR 301, 8 mm measuring plate) included amplitude, time, and frequency sweeps to measure storage and loss moduli within the linear viscoelastic region (1% strain) as well as study gelation kinetics. Compression testing was performed on a micro-strain analyzer (TA Instruments RSA III, Duke SMIF) at a rate of 0.01 mm s−1. The slope of the resultant stress-strain curves was calculated between 65–80 % strain.
2.8. Local scaffold mechanical characterization with Atomic Force Microscopy (AFM)
Either nanoporous or MAP scaffolds (lyo-microgels annealed with 0.02 mg mL−1 HA-Tet) were adhered to a 15 mm glass coverslip using superglue. A hydrophobic pen created a ring around the scaffold to prevent water from flowing off the coverslip during experimentation. Water was added dropwise to the scaffold to create a dome. A 5 μm spherical probe (0.6 N/m spring constant, Novascan) was used to generate force curves with a 1.5 nN trigger force (Asylum Cypher Atomic Force Microscope, Duke SMIF). Force maps were generated across 30 μm × 30 μm surface of microgels. The resultant Young’s Modulus was approximated using the Hertz model assuming a Poisson ratio of 0.5.
2.9. Cell culture in MAP scaffolds
To create a custom cell culture device for these experiments, a negative mold was 3D printed using a 3D, Form 2 stereolithography printer (Formlabs, Inc., Duke SMIF). The culture wells were composed of a lower cylindrical culture section (4 mm in diameter and 1 mm tall) and a larger cylindrical media reservoir (5 mm in diameter and 3 mm tall) above the lower culturing section able to contain at least 50 μL of media. The wells were cast with PDMS and bonded to glass coverslips then autoclaved for sterilization.
Prior to lyophilization, all microgels were post-modified with 1 mM RGD peptide (Ac-RGDSPGERCG-NH2, GenScript) using thiol-ene click chemistry. Microgels (90% of the working volume) were combined with RGD peptide, LAP photo-initiator, TCEP in HEPES buffer (10% of the working volume) at final concentrations of 1 mM, 1.5 mM, and 1.67 mM respectively. The mixture was irradiated with UV light (20 mW/cm2) for 1 minute. Microgels were washed three times with 0.3 M HEPES then once with 70% EtOH. Microgels for lyophilization were incubated overnight in equal volume 70% EtOH then lyophilized. Lyo-microgels were rehydrated in DMEM medium at a range of wt% (w/v). Non-lyo- microgels were incubated in DMEM medium and concentrated via centrifugation. HA-Tet was dissolved at 0.02 mg mL−1 in DMEM medium for all conditions. Cells were pelleted at the same density for each group, based on 10,000 cell μL−1 MAP with non-lyo-microgels. Microgels (84% v/v) and HA-Tet (16% v/v) were mixed well with the cell pellet and seeded at 5 μL per well. Inter-particle crosslinking occurred at 37°C for 20 minutes prior to the addition of excess DMEM medium.
At time points of 1, 3, and 5 days, the wells were fixed with 4% paraformaldehyde for 30 minutes at room temperature. The wells were washed three times with 1x PBS then blocked with 0.15% triton-x in PBS for 1 hour at room temperature. The wells were stained with DAPI (1:1000) (Sigma-Aldrich) and Alexa Fluor™ 647 Phalloidin (1:40) (Invitrogen) in the same blocking buffer overnight at 4°C. Samples were washed three times with PBS then imaged on a confocal microscope at 20x magnification (Z-stacks at 2.5 μm steps).
2.10. Analysis of cell response
For generating heatmaps of signal intensity, the channel of interest in the Z-slices from confocal Z-stacks were first converted to JPEG image sequences in ImageJ. A MATLAB (The MathWorks, Inc.) script was created to analyze the image sequence (Z-stacks at 2.5 μm step size) by averaging the fluorescent intensity in 150 μm × 150 μm squares to generate heatmaps. The resultant square intensities were averaged across 30 Z-slices for each image to increase the dimension of the analysis of the scaffolds (i.e., each datapoint represents the average of the squares from a Z-slice). In IMARIS software, 3D volume renderings of confocal Z-stacks were used to quantify cell volume (F-actin) and particle fraction. The spot counter tool was used to quantify cell counts using DAPI signal.
2.11. Measuring scaffold diffusion
Glass slides were prepared by treating with SigmaCote (Sigma-Aldrich) and attaching a PDMS cylinder (1 mm tall and 1 mm in diameter) with superglue. 50 μL of microgels and crosslinker was pipetted around and on each PDMS plugs. A 1 mm Nylon spacer was added and topped with a glass slide. Clips were used to secure the slides and was incubated in a petri dish with a wet KimWipe to act as a humidity chamber during inter-particle crosslinking. Once microgels were interlinked, scaffolds were carefully removed and swollen overnight in 1x PBS. Solutions of fluorescent dextran (Alexa-fluor 647), with molecular weights of 10 kDa and 70 kDa, were prepared at concentrations of 0.35 mg mL−1 and 0.05 mg mL−1, respectively. These concentrations were determined experimentally while creating the corresponding standard curves. Standard curves were generated using a range of concentrations from 0.05 to 0.5 mg mL−1 for the 70kDa dextran and 0.001 to 0.05 mg mL−1 for the 10 kDa dextran. 1.5 μL of each dilution was pipetted onto a glass coverslip and imaged. These images were analyzed in ImageJ to determine the average intensity of the image, then a linear fit was used to obtain the equation for the standard curve for each molecular weight. To image diffusion through the scaffolds, they were first carefully dried with a Kimwipe then placed on glass coverslips. 150 μL of PBS was added to ensure they remained hydrated during imaging. 2 μL of the fluorescent dextran solution were pipetted into the center cavity in the scaffold created by the PDMS plugs. A coverslip was then placed on the gel to prevent evaporation, then the gel was imaged at a fixed Z-plane on a confocal microscope (Nikon Ti Eclipse with a C2 LED laser light source) at 0.1 FPS for 30 minutes.
Analysis was performed in ImageJ using the Bio-Formats plugin [32]. Images were converted to grayscale then linear concentration profiles were obtained over the full range of timepoints, extending radially outward from the center. The profiles of relative diffusion were generated by sampling incremental values across time and distance. Values at the time and distance midpoints were recorded for each condition. Under the assumption our gels represent a semi-infinite medium for diffusion, we utilized Equation 1 to estimate the effective diffusion coefficient (Deff) for each gel. Using the normalized concentration values (C/Co), we applied a surface fitting model in MATLAB to fit the parameter Deff to our data which yielded an estimated Deff for each condition. This analysis was performed on the entire dataset (not just the sampled values) across time and distance for each condition.
2.12. Statistical analysis
Pre-processing of data included normalization of fraction of positive pixels and sphericity to the non-lyo condition. Data are presented with error bars representing standard deviation. For mechanical testing, n = 3 for each condition. One-way ANOVA performed for the following yielded P < 0.05 prompting post-hoc analysis: storage moduli, microgel concentration for centrifugation drying, particle fraction, fraction of positive pixels, sphericity, Young’s Moduli, and diffusion coefficient. Tukey’s multiple comparison test was used to compare groups defined by Tet/HA ratio, centrifugation time, gel condition (nanoporous, non-lyo MAP, or lyo-microgel MAP at varying wt%), or lyophilization medium. For in vitro cell viability experiments, n = 3 with each condition ran in triplicate. A paired T-test was performed comparing conditions at each time point, with significance reported at P < 0.05 (*). For 3D cell culture, n = 3 with duplicate gels for each condition. One-way ANOVA with Tukey HSD was performed on the samples at each time point. For all experiments, significance is indicated by *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Statistical analysis was performed with GraphPad Prism software.
3. RESULTS
3.1. MAP scaffold fabrication includes a linear tetrazine linker for improved inter-particle crosslinking efficiency
For our MAP platform, the primary material component was hyaluronic acid (HA) modified with norbornene (NB) functional groups. The NB handle permitted the use of click chemistry schemes for i) forming the microgels and ii) linking them together [3, 6]. We generated HA-NB microgels on a flow-focusing microfluidic device as previously described (Figure 1A) [14]. The average size of HA-NB microgels used throughout this work was approximately 92 μm in diameter with a polydispersity index (PDI) of 1.01, represented by frequency distributions of diameter in Figure S1. To form bulk MAP scaffolds, the unreacted NB handles on HA-NB microgels were involved in a secondary crosslinking reaction with a tetrazine (Tet) linker (Figure 1B).
Figure 1.
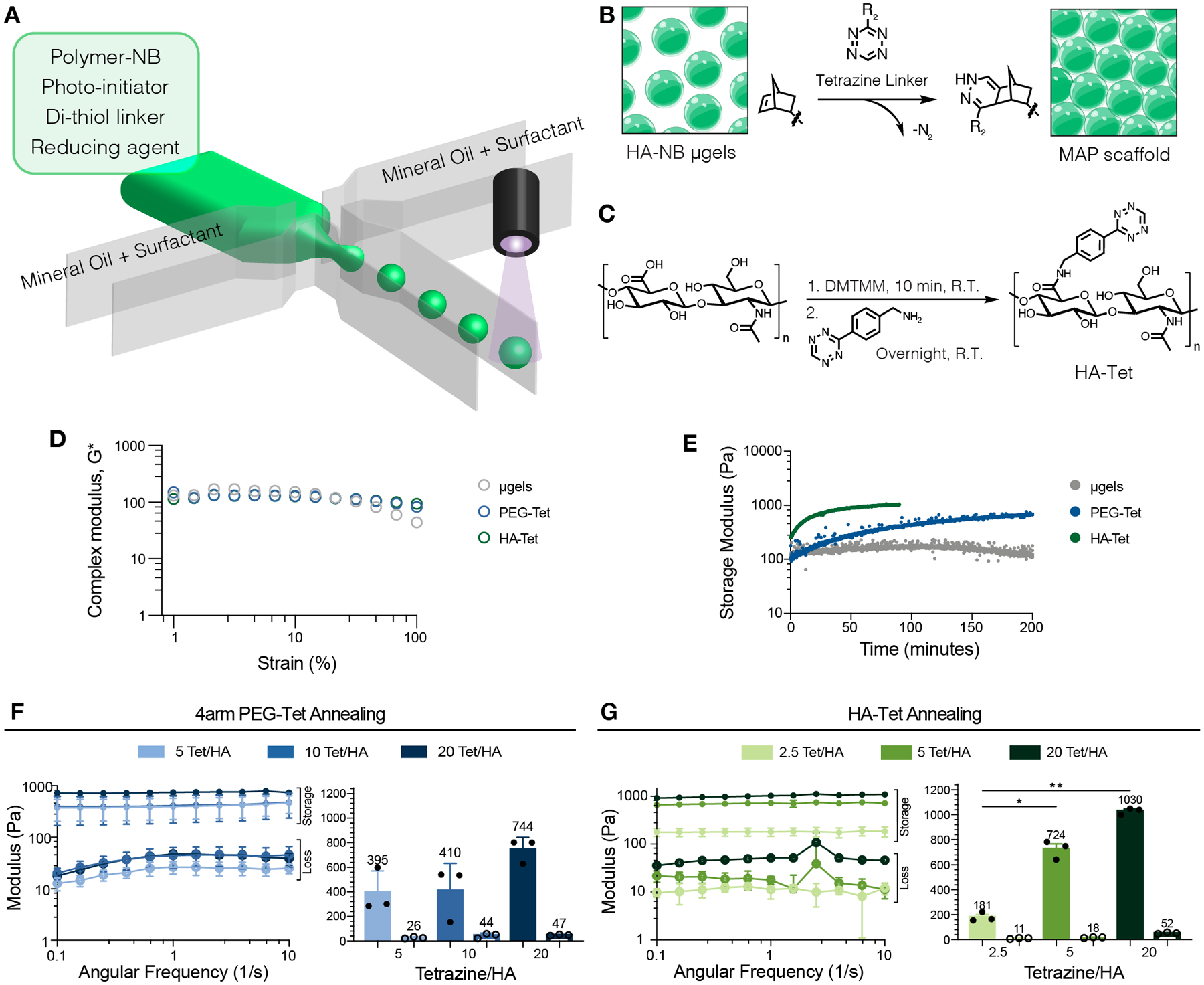
(A) Schematic of microfluidic generation of microgels. (B) Schematic of HA-NB microgels (green) annealed with a tetrazine linker to form MAP scaffolds. (C) Reaction schematic of HA-Tet synthesis. (D) Complex modulus for microgels without Tet- linker (gray), HA-Tet (green), and 4arm-PEG-Tet (blue) linkers across strain. (E) Time-sweep of MAP annealing using non-lyo-microgels without Tet- linker, 4arm-PEG-Tet (blue), and HA-Tet (green) linkers for MAP gelation. (F) Storage moduli (solid) and loss moduli (open) of MAP gels comprised of microgels annealed with 4arm-PEG-Tet determined by frequency sweep. (G) Storage moduli (solid) and loss moduli (open) of MAP gels comprised of non-lyo-microgels annealed with HA-Tet determined by frequency sweep. A one-way ANOVA with Tukey HSD was performed on the samples for rheology testing (n = 3), with significance reported at p < 0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****).
We synthesized a linear HA-tetrazine (HA-Tet) linker by modifying the carboxylic acid group on the HA backbone (Figure 1C) [31]. Proton NMR spectroscopy showed successful modification of 11% of HA repeat units (Figure S2), corresponding to approximately 23 functional groups per HA chain. MAP scaffolds crosslinked with Tet-linkers exhibited a constant complex modulus across a larger range of strain than microgels without inter-particle crosslinking (Figure 1D). The HA-Tet crosslinker exhibited faster gelation (i.e, inter-particle linking of the microgels to form a bulk scaffold) compared to a 4arm PEG-Tet linker (Figure 1E). Bulk MAP hydrogels crosslinked with HA-Tet required approximately 35 min to fully crosslink while the MAP hydrogels crosslinked with 4arm PEG-Tet required more than 75 min, approximately twice as long (Figure 1E). We observed viscoelastic solid behavior (storage modulus > loss modulus) for MAP scaffolds crosslinked with both 4arm PEG-Tet and HA-Tet (Figure 1F–G) across all crosslinking ratios. While bulk MAP scaffold stiffness was shown to increase with increasing Tet for both linkers (Figure 1F–G), we observed differences in the range of stiffness they achieved. While 4arm PEG-Tet required a ratio of 20 Tet/HA to achieve a bulk stiffness of 744 Pa, HA-Tet crosslinking achieved a comparable stiffness (724 Pa) with only a 5 Tet/HA ratio. Moving forward, all MAP scaffolds were annealed using the linear HA-Tet linker.
3.2. Particle fraction is a bioactive cue in MAP scaffolds
Prior to inter-particle crosslinking the microgels with a Tet linker, the process of making MAP scaffolds first involves removal of excess buffer from the microgel slurry (Figure 2A). Centrifugation has been the standard method to separate the microgels from the buffer either over a strainer or with manual removal of the supernatant buffer from the microgel pellet either by aspiration or with an absorbent material. Both the centrifugation speed and time are variable across users of MAP scaffolds. These methods result in a highly variable range of HA-NB microgel concentrations in the final ‘dried’ product, as shown by Figure 2B. In practice, we have also observed disruption of the microgel pellet (not quantified) over time if the supernatant solution is not immediately removed once centrifugation is concluded. After the microgels are concentrated via centrifugation, the variability in microgel concentration is then perpetuated when making MAP scaffolds and is evident in the range of particle fractions generated using this method (Figure 2C).
Figure 2.
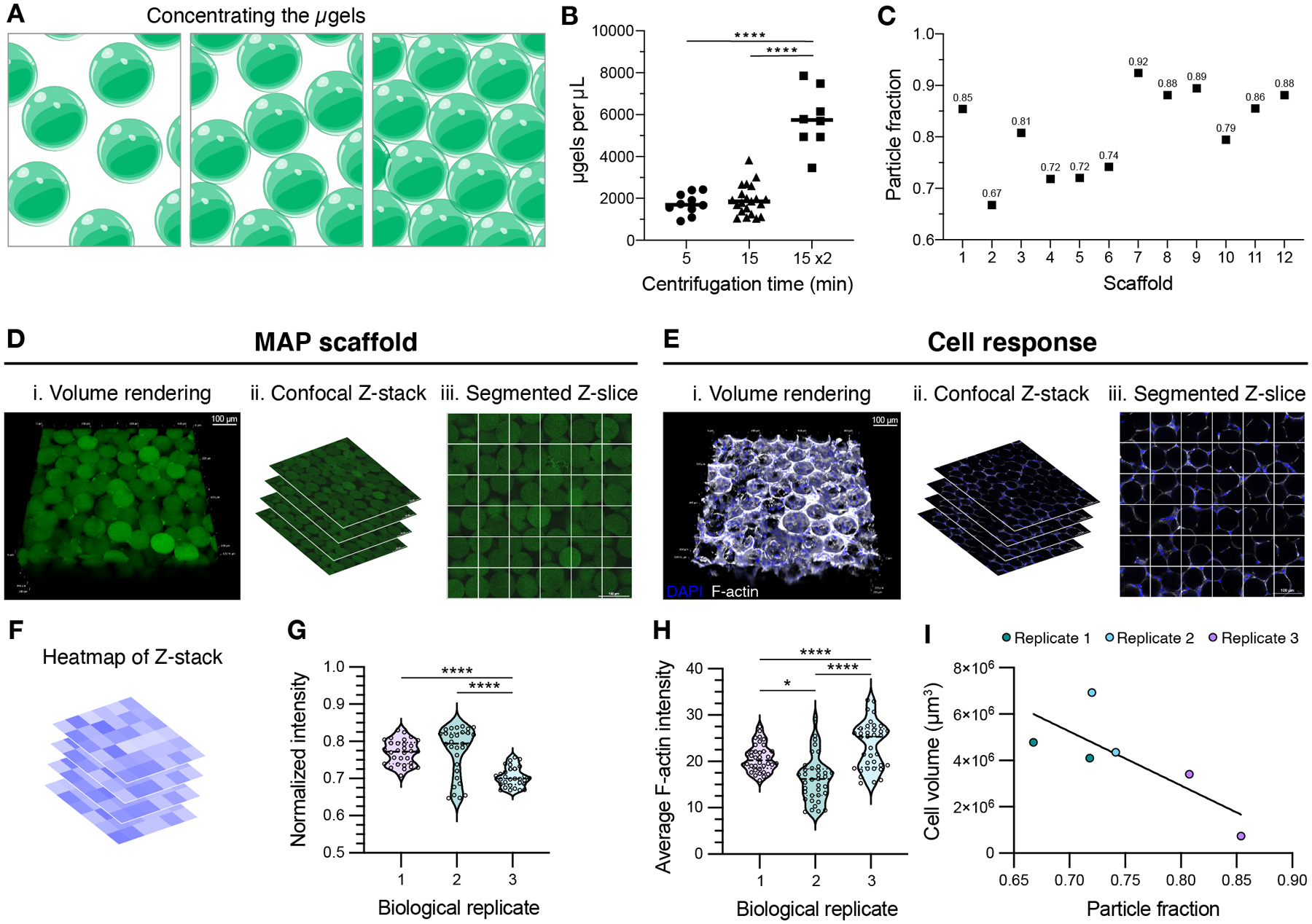
(A) Schematic of microgels being concentrated from the microgel slurry during MAP scaffold preparation. (B) Concentration of microgels in the dried product when using manual buffer removal after centrifugation for 5 minutes, 15 minutes, or 15 minutes twice. A one-way ANOVA with Tukey HSD was performed on the samples dried independently (n = 8), with significance reported at p < 0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****). (C) Using centrifugation for 15 min 2x, the resultant MAP scaffolds boast a range of particle fractions. (D) MAP scaffolds prepared after centrifuging microgels for 15 minutes twice are shown as (i) volume renderings taken from (ii) confocal Z-stacks. (iii) The individual Z-slices from the Z-stacks are segmented to quantify the average intensity of the MAP scaffold channel within each square (scale bar = 100 μm). (E) D1 mouse mesenchymal cells were seeded in MAP scaffolds prepared after centrifuging microgels for 15 minutes twice and are shown as (i) volume renderings taken from (ii) confocal Z-stacks. (iii) The individual Z-slices from the Z-stacks are segmented to quantify the average intensity of the F-actin channel within each square (scale bar = 100 μm). (F) Heatmaps were generated for each Z-slice within the Z-stacks. (G) For MAP scaffolds, the average intensity across the Z-slices is plotted for one sample from each biological replicate. A one-way ANOVA with Tukey HSD was performed on the replicates, with significance reported at p < 0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****). (H) For cell response within these scaffolds, the average F-actin intensity across the Z-slices is plotted for one sample from each biological replicate. A one-way ANOVA with Tukey HSD was performed on the replicates, with significance reported at p < 0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****). (I) Cell volume was determined by IMARIS rendering of the F-actin channel from the confocal Z-stack. Cell volume results were plotted across the corresponding particle fraction from the sample.
While this quantification investigated bulk scaffold particle fraction using volume renderings (Figure 2D), we also observed inconsistencies in different regions of the scaffold where particles would be tightly packed together, or large empty gaps would be present. To assess these inconsistencies, we took the individual Z-slices comprising the confocal Z-stack and segmented each image into 150 μm × 150 μm squares (Figure 2D). The fluorescent intensity of the scaffold (one from each drying replicate) was measured within each square spanning 30 Z-slices from the image (Figure 2F) and normalized to the highest intensity within each slice. Consistent with our observations, the inhomogeneous distribution of particles within the scaffold is captured by the spread of intensity observed across Z-slices (Figure 2G). With this analysis we also see significant differences in the scaffolds from independent drying replicates.
MAP scaffolds generated using these methods are commonly implemented for in vitro studies; however, the variation in particle fraction has yet to be considered as a confounding factor in the cell responses MAP users observe in these scaffolds. To determine if the particle fraction influences cell response, we cultured D1 mouse mesenchymal cells in MAP scaffolds concentrated via centrifugation. Similar to our assessment of the scaffolds, we quantified the cell response both regionally (looking for pockets of high or low cell growth) as well as within the entire scaffold (Figure 2E). Across Z-slices for the drying replicates, there were significant differences in the regional cell response (F-actin intensity), corresponding to pockets of high and low cell growth (Figure 2H). We also observed a correlation between the total cell volume in the scaffolds to the particle fractions within the scaffold (Figure 2I), indicating that the total cell volume is greater in scaffolds with lower particle fractions.
3.3. Lyophilization medium can be changed to either induce or prevent cryogel formation in HA-based microgels
We demonstrated that the particle fraction in MAP scaffolds influences cell growth, but concentrating the microgels via centrifugation failed to provide control over unique particle fractions during MAP scaffold fabrication. We then investigated other methods for concentrating or drying the microgels, such as lyophilization, to provide control over the final particle fraction in MAP scaffolds. It has been shown that lyophilizing polyethylene glycol (PEG) microgels in isopropyl alcohol (IPA) allowed microgels to retain their physical properties after rehydration [12]. We first tested the lyophilization of PEG-based microgels using PEG-NB microgels after they were generated on a microfluidic device as described above. The average diameter of PEG-NB microgels was 118 μm in diameter with PDI of 1.00, represented by a frequency distribution in Figure S1. The microgels (Figure 3A) were incubated in the respective medium overnight, flash-frozen with liquid nitrogen, then freeze-dried (Figure 3B). Lyophilized microgels (lyo-microgels) were rehydrated in excess water and imaged on a confocal microscope to assess cryogel formation within the microgels (Figure 3C). The formation of crystals during the freeze-drying process was not evident in either condition for both maximum intensity projections (Figure 3D) and single Z-slices (Figure 3E) from confocal Z-stacks. Similarly, the intensity profiles across microgel diameter (Figure 3F) show an even distribution of fluorescence (gel), indicating that defects were not observed under these conditions.
Figure 3.
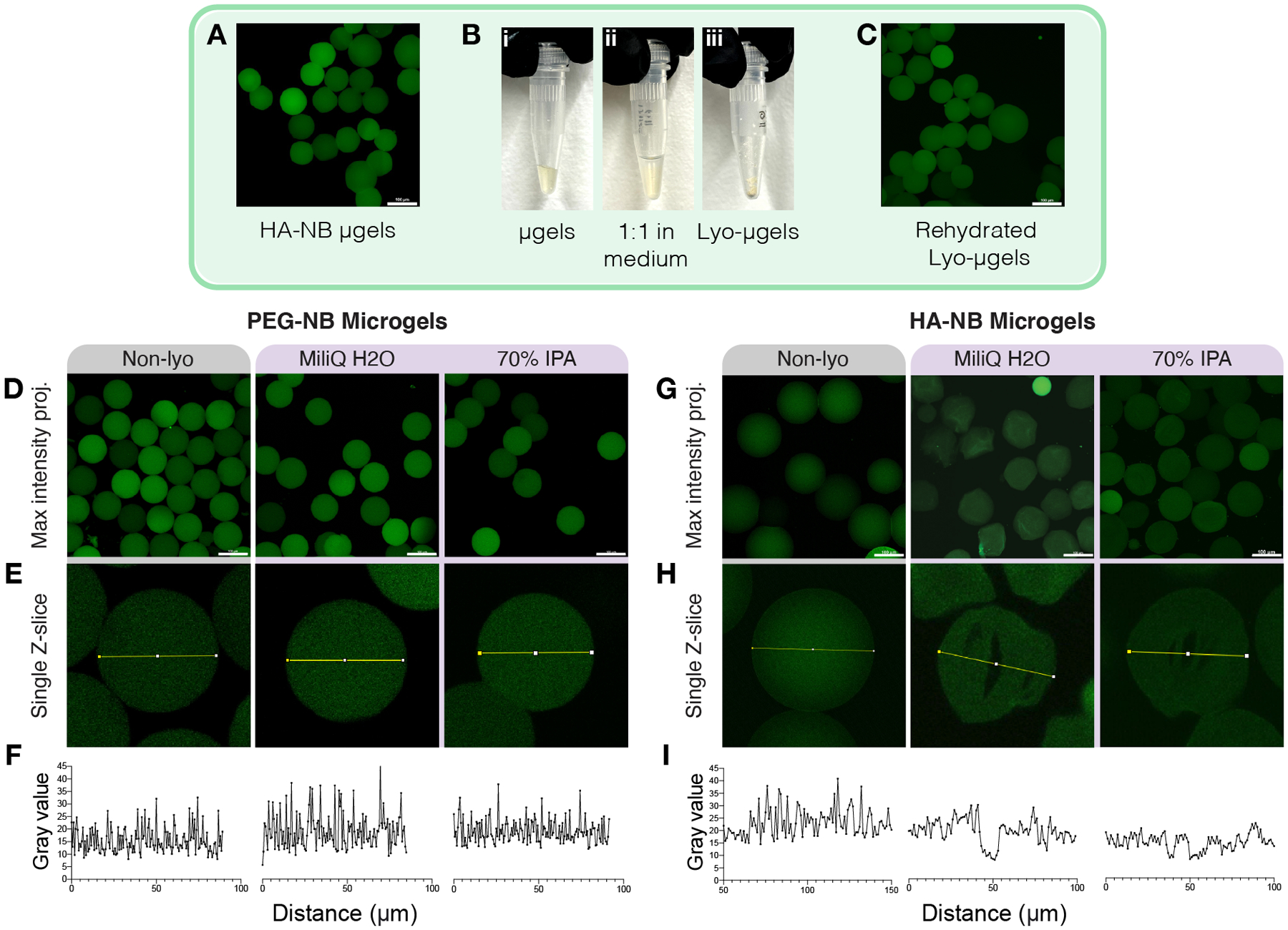
The drying process for microgels (A-B) involves 1:1 incubation in lyophilization medium. (C) Lyo-microgels can then be rehydrated to achieve water-swollen microgels. Microgels fabricated with PEG-NB were lyophilized in water and 70% IPA. Representative images for non-lyophilized (Non-lyo) as well as lyo-microgels for water and 70% IPA conditions shown as (D) maximum intensity projections (scale bar = 100 μm). and (E) single Z-slices from the original Z-stack with intensity profiles measured across the microgels plotted in (F) for each condition. Microgels fabricated with HA-NB were lyophilized in water and 70% IPA. Representative images for non-lyophilized (Non-lyo) as well as lyophilized microgels for water and 70% IPA conditions shown as (G) maximum intensity projections (scale bar = 100 μm). and (H) single Z-slices from the original Z-stack with intensity profiles measured across the microgels plotted in (I) for each condition.
We extended these methods of lyophilization for drying HA-NB microgels first using 70% IPA and water. Maximum intensity projections of the 3D Z-stacks failed to capture the presence of internal defects in the polymer network (Figure 3G) that were otherwise evident in single Z-slices (Figure 3H). The lack of fluorescence inside microgels is indicative of cryogel formation and is demonstrated by the plotted intensity profiles across microgel diameter compared to non-lyophilized (non-lyo) microgels (Figure 3I). We observed internal defects for the microgels lyophilized in both water and 70% IPA. As a result, we expanded our study to include a range of polar protic, ionic, zwitterionic, and polar aprotic mediums with a range of molecule size and at varying concentrations (Table 1) to determine a lyophilization medium for HA-NB microgels that would not induce cryogel formation.
Table 1.
Lyophilization mediums to optimize HA-NB microgel drying were selected to span medium type: polar protic (purple), ionic (pink), zwitterionic (green), and polar aprotic (blue), as well as molecular weight and concentration.
| Medium type | Name | Formula | Molecular weight [Da] |
Concentration [mM or % v/v] |
|---|---|---|---|---|
| Polar protic | Water | H2O | 18.0 | 100% |
| Isopropyl alcohol (IPA) | C3H8O | 60.1 | 50% | |
| 70% | ||||
| Ethanol (EtOH) | C2H5OH | 46.1 | 30% | |
| 50% | ||||
| 70% | ||||
| Ionic | Phosphate-buffered saline (PBS) | NaCl | 58.4 | 137 |
| KCl | 74.6 | 2.7 | ||
| Na2HPO4 | 142.0 | 8 | ||
| K2HPO4 | 174.2 | 2 | ||
| Sodium chloride (NaCl) | NaCl | 58.4 | 50 | |
| 300 | ||||
| 1000 | ||||
| Zwitterionic | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) | C8H18N2O4S | 238.3 | 50 |
| 300 | ||||
| 1000 | ||||
| tris(hydroxymethyl)aminomethane (Tris) | C4H11NO3 | 121.1 | 50 | |
| 300 | ||||
| 1000 | ||||
| 2-(N-morpholino)ethanesulfonic acid (MES) | C6H13NO4S | 195.2 | 50 | |
| 300 | ||||
| 1000 | ||||
| Polar aprotic | Dimethylsulfoxide (DMSO) | C2H6OS | 78.1 | 50% |
| 100% | ||||
| Acetonitrile (MeCN) | C₂H₃N | 41.1 | 30% | |
| 50% |
To quantify cryogel formation for each condition, single Z-slices were analyzed for the fraction of pixels positive for fluorescence within the microgel area (Figure 4A), with positive fluorescence corresponding to hydrogel and the lack of fluorescence indicating defects in the network. Particularly, ionic buffers (e.g., PBS, NaCl) resulted in the largest proportion of defects within microgels (Figure 4B). Maximum intensity projections and single Z-slices for all conditions can be found in Figure S3. The fraction of positive pixels was observed to be the highest, and therefore most comparable to non-lyo microgels, in the 70% EtOH condition. 3D renderings of confocal Z-stacks with IMARIS software were used to analyze the sphericity of microgels after lyophilization. The majority of conditions allowed microgels to maintain comparable sphericity to non-lyo sphericity (Figure 4C). However, six conditions resulted in statistically significant differences in mean sphericity, as highlighted in Figure 4D. Freeze-drying HA-NB microgels with 70% EtOH yielded results for both internal fluorescence and 3D sphericity comparable to non-lyo conditions, prompting our further investigation of microgel properties with and without lyophilization in 70% EtOH for use as a suitable dehydration method without impacting microgel properties.
Figure 4.
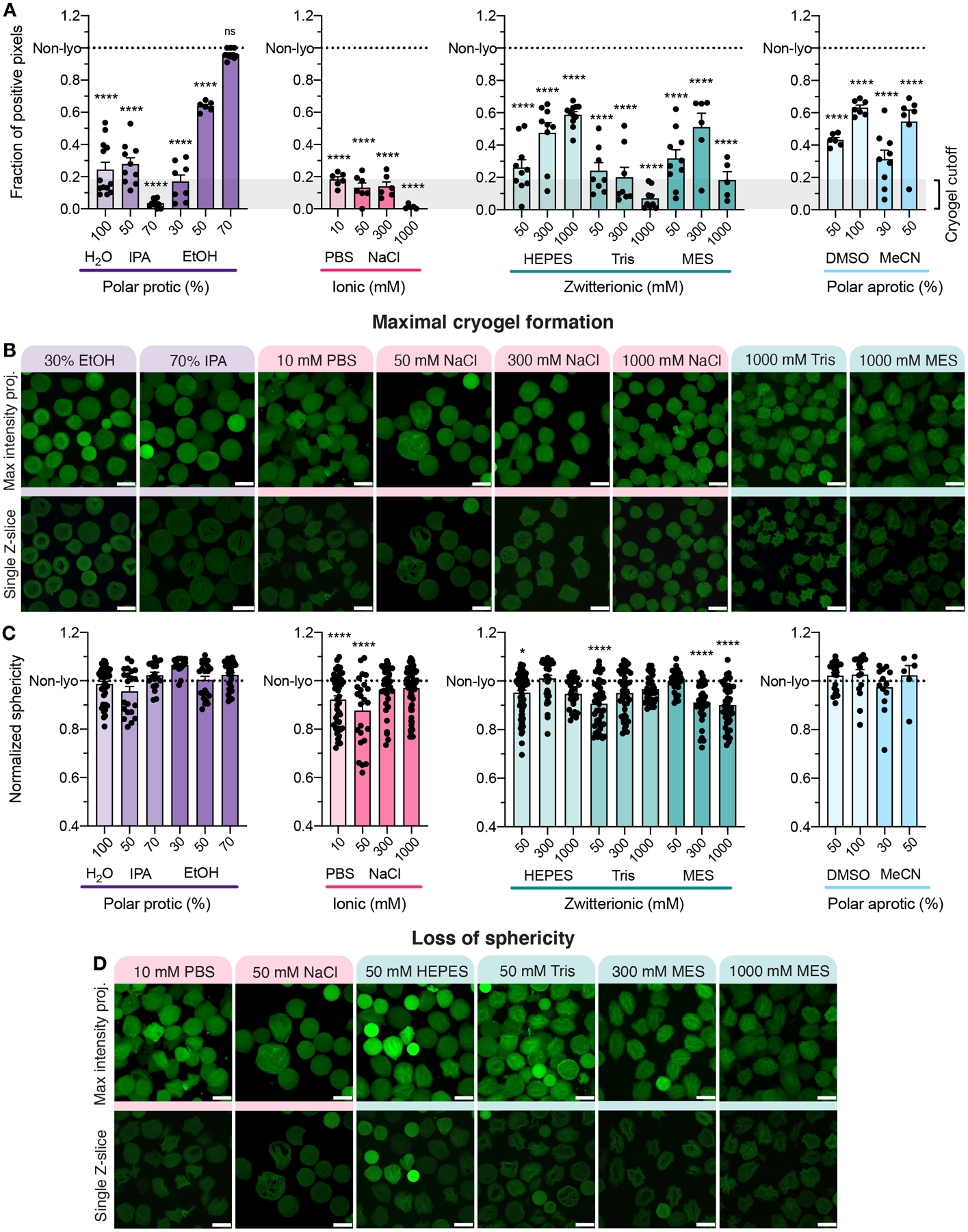
(A) The fraction of pixels positive for fluorescence plotted for each condition separated by polar protic, ionic, zwitterionic, or polar aprotic medium compared to the non-lyo condition represented by the dotted line. A one-way ANOVA with Tukey HSD was performed on the samples comparing condition mean to non-lyo mean (n > 3), with significance reported at p < 0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****). (B) Representative maximum intensity projections and single Z-slices from the original Z-stack for eight lyophilization conditions with less than 20% positive pixels as indicated by the gray box in panel A (scale bar = 100 μm). (C) 3D volume renderings of confocal Z-stacks were made using IMARIS software and analyzed for sphericity. A one-way ANOVA with Tukey HSD was performed on the samples comparing condition mean to non-lyo mean (n > 3), with significance reported at p < 0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****). (D) The medium conditions that significantly varied from non- lyo sphericity are shown as representative maximum intensity projections and single Z-slices from the original Z-stack (scale bar = 100 μm).
3.4. Lyophilization of HA-based microgels in ethanol does not impact particle size or cell viability
To determine if lyophilization with 70% EtOH is compatible with microgels of other material compositions, we tested these methods for PEG-NB microgels. Confocal images of PEG-NB microgels lyophilized in 70% EtOH are shown in Figure 5A–B, which highlight the lack of cryogel formation under these conditions. Similarly, the intensity profile across microgel diameter (Figure 5C) shows an even distribution of fluorescence (gel), indicating that defects were not observed under these conditions. These findings for PEG-NB microgels were consistent with the results for HA-NB microgels (Figure 5D–F).
Figure 5.
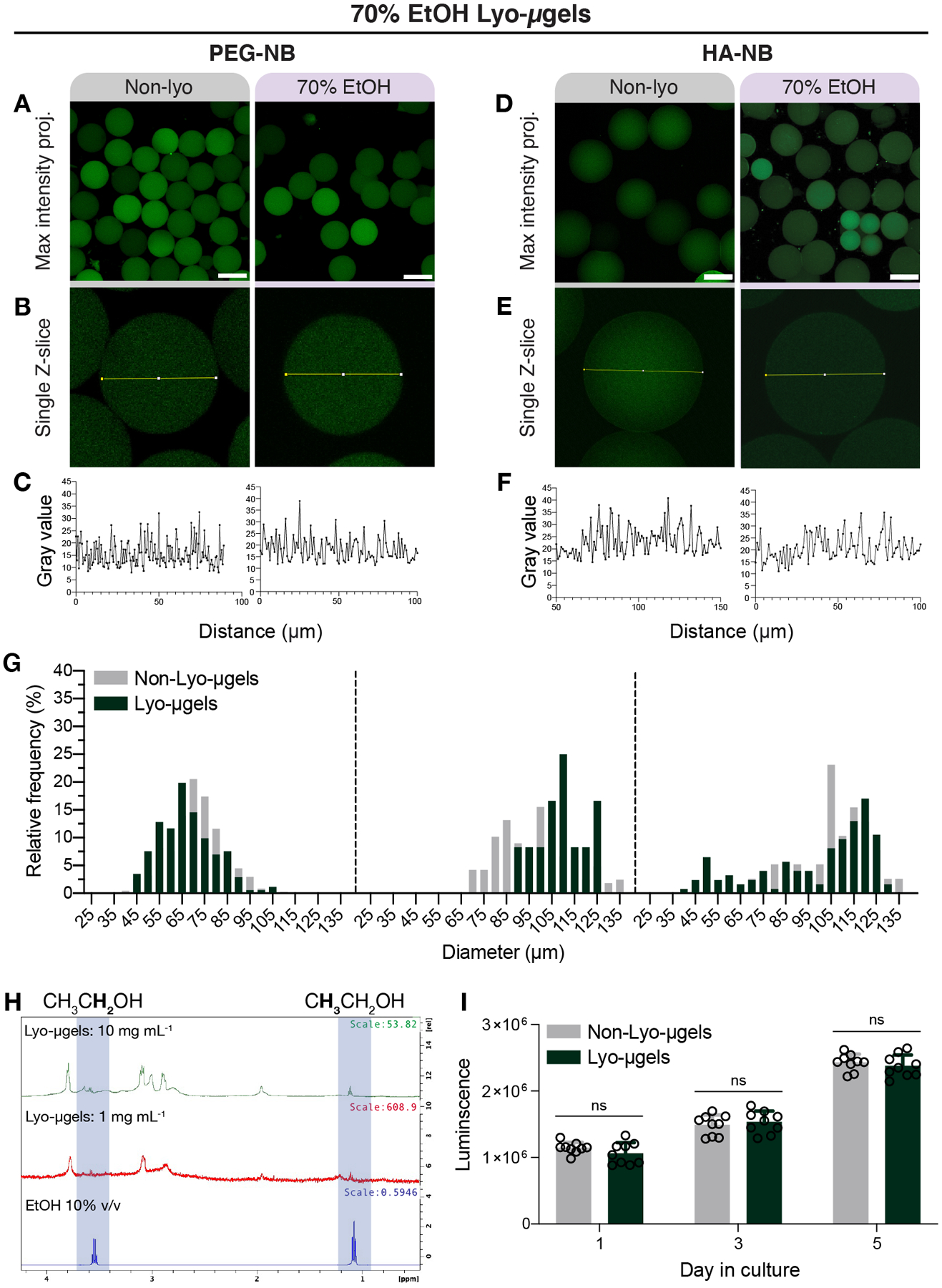
Microgels composed of either PEG or HA can be lyophilized with 70% EtOH and rehydrated. PEG-NB microgels before (Non-lyo) and after lyophilization with 70% EtOH are shown as a maximum intensity projections (A) and (B) single Z-slices within the confocal Z-stack (scale bar = 100 μm). The lack of pore formation within microgels is depicted by the intensity profile across bead diameter (C). HA-NB microgels before (Non-lyo) and after lyophilization with 70% EtOH are shown as a maximum intensity projections (D) and (E) single Z-slices within the confocal Z-stack (scale bar = 100 μm). The lack of pore formation within microgels is depicted by the intensity profile across bead diameter (F). Measurement of HA-NB microgel diameter before and after lyophilization (G) shown as frequency distributions across for the three separate microgel populations. (H) Proton NMR spectra of HA-NB lyo-microgels and ethanol show trace amount of ethanol peaks. (I) Luminescence values correspond to cell viability data for D1 cells cultured with non-lyo (gray) and lyo-microgels (dark green) over 5 days. A paired T-test was performed comparing conditions at each time point (n = 3 with triplicates), with significance reported at p < 0.05 (*).
To further understand how lyophilization of HA-NB microgels in 70% EtOH may affect microgel properties, we included measurements of HA-NB microgel diameter after freeze-drying. The diameter of microgels pre- and post- lyophilization with 70% EtOH resulted in similar frequency distributions across three separate microgel populations (Figure 5E) with no significant difference when comparing mean diameter. After lyophilization, we observed trace amounts of EtOH with proton NMR spectroscopy of lyo-microgels in D2O, as indicated by a triplet of peaks at 1.1 PPM and quartet of peaks at 3.6 PPM corresponding to EtOH methyl and methylene protons (Figure 5F). To determine if cell viability is affected by these trace amounts of residual EtOH, we assessed viability of mouse mesenchymal stem cells seeded with either lyo-microgels or non-lyo-microgels over 5 days of culture (Figure 5G). Across 5 days, there was no significant difference in cell viability when comparing cells in non-lyo and lyo- microgels. An additional benefit of using 70% EtOH for freeze-drying microgels is the maintenance of product sterility by incorporating more than 60% alcohol content.[33, 34] The sterility of lyo- microgels was demonstrated by the lack of bacterial growth after incubating lyo- and non-lyo microgels in LB broth for 3 days (Figure S4). Moving forward, any reference to lyo-microgels were generated using 70% EtOH as the lyophilization medium.
3.5. Controlling rehydration volume of freeze-dried microgels yields reproducible MAP particle fraction
To explore the relationship between lyo-microgel rehydration (wt% MAP) and particle fraction in MAP scaffolds (Figure 6A), lyo-microgels were rehydrated at varying wt% ranging from 3.4–7 wt% lyo-microgels and interlinked with a consistent NB:Tet ratio. 3D renderings of confocal Z-stacks were generated in IMARIS to quantify particle fraction of the rehydrated lyo-microgels. Particle fraction was shown to increase with increasing wt% MAP (Figure 6A–B). Rehydrating the lyo-microgels for MAP scaffold fabrication reduced the standard deviation (indicated by the gray box) of the particle fraction that was observed when using the centrifugation method for concentrating non-lyo-microgels (Figure 6C). We also assessed local pore area which showed that increasing the wt% MAP resulted in an increased frequency of small pores and a decrease in average pore area (Figure 6D).
Figure 6.

(A) Lyo-microgels can be reconstituted at varying final wt% in MAP scaffolds which corresponds to (B-C) increasing particle fraction while non-lyo-microgels concentrated via centrifugation exhibit a larger range of the final particle fraction in MAP (scale bar = 100 μm). The gray box in each condition corresponds to ± standard deviation centered around the mean particle fraction. (D) 2D analysis of pores represented by frequency distributions of pore area for each scaffold type. (E) Storage moduli (solid) and loss moduli (open) of MAP gels comprised of lyo-microgels at varied wt% MAP annealed with HA-Tet determined by frequency sweep. (F) Storage moduli (solid) and loss moduli (open) of MAP gels comprised of lyo-microgels at varied wt% MAP annealed with HA-Tet at an angular frequency of 1 Hz. A one-way ANOVA with Tukey HSD was performed on the samples for rheology testing (n = 3), with significance reported at p < 0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****). Force curves generated with AFM show microgel surface (G) and local stiffness (H). (I) Bulk and local scaffold stiffness of nanoporous gels and MAP gels comprised of lyo-microgels with the same composition as nanoporous gels. A one-way ANOVA with Tukey HSD was performed on the samples (n = 3), with significance reported at p < 0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****).
3.6. Local mechanical properties of lyo-microgels differ from the bulk MAP scaffolds
Independent of crosslinker ratio, the storage modulus of bulk MAP scaffolds increased with increasing wt% MAP, approaching the storage modulus of a 3.4 wt% nanoporous gel (Figure 6E–F). In addition to assessing the bulk scaffold properties, we also probed the stiffness of individual lyo-microgels to understand the effect of freeze-drying with 70% EtOH on particle stiffness. For this purpose, we compared the mechanical properties of MAP gels comprised of lyo-microgels (3.4 wt% MAP) to nanoporous gels (3.4 wt%) prepared from the same precursor as microgels. To assess local stiffness, atomic force microscopy (AFM) was used to probe the nanoporous gel surface as well as the surface of interlinked lyo-microgels within MAP scaffolds. Force maps were generated across a 30 μm × 30 μm area on the microgel surface. Figure 6G depicts a representative surface plot of the microgel surface with the resultant Young’s Modulus (approximated using the Hertz model) shown in Figure 6H. As measured by AFM, the local stiffness of lyo-microgels was not significantly different than the nanoporous gel of the same composition, indicating that freeze-drying with 70% EtOH does not compromise stiffness (Figure 6I). Bulk modulus was then quantified using compression testing of nanoporous and lyo-microgel MAP gels. Both the bulk and local stiffness of nanoporous gels were comparable; however, the bulk stiffness of the MAP gel was significantly less than the local stiffness of the microgels from which it is comprised (Figure 6I).
3.7. Lower particle fraction promotes cell growth in MAP scaffolds
In addition to studying the material properties of these scaffolds, we also assessed the impact of having controlled particle fractions in MAP scaffolds on 3D cell culture. We first studied cell response in MAP scaffolds comprised of either non-lyo or lyo-microgels at 3.4 wt% MAP. Prior to lyophilization, all microgels were post-modified with 1 mM RGD to support integrin binding in vitro. The non-lyophilized control was concentrated using centrifugation to remove the excess media. Lyo-microgels from the same microgel population were freeze-dried and rehydrated in cell media at 3.4 wt%. The non-lyo and lyo conditions were seeded with the same cell density and inter-particle crosslinking ratio of HA-Tet. Confocal Z-stacks of each condition are shown as 3D projections in Figure 7A of scaffolds (green), cell nuclei (blue), and actin filaments (white) across 5 days in culture. While cell counts (Figure 7B) and cell volume (Figure 7C) increased across 5 days for both conditions, greater variability was observed across the non-lyo drying replicates compared to lyo-microgels which boast a controlled particle fraction.
Figure 7.
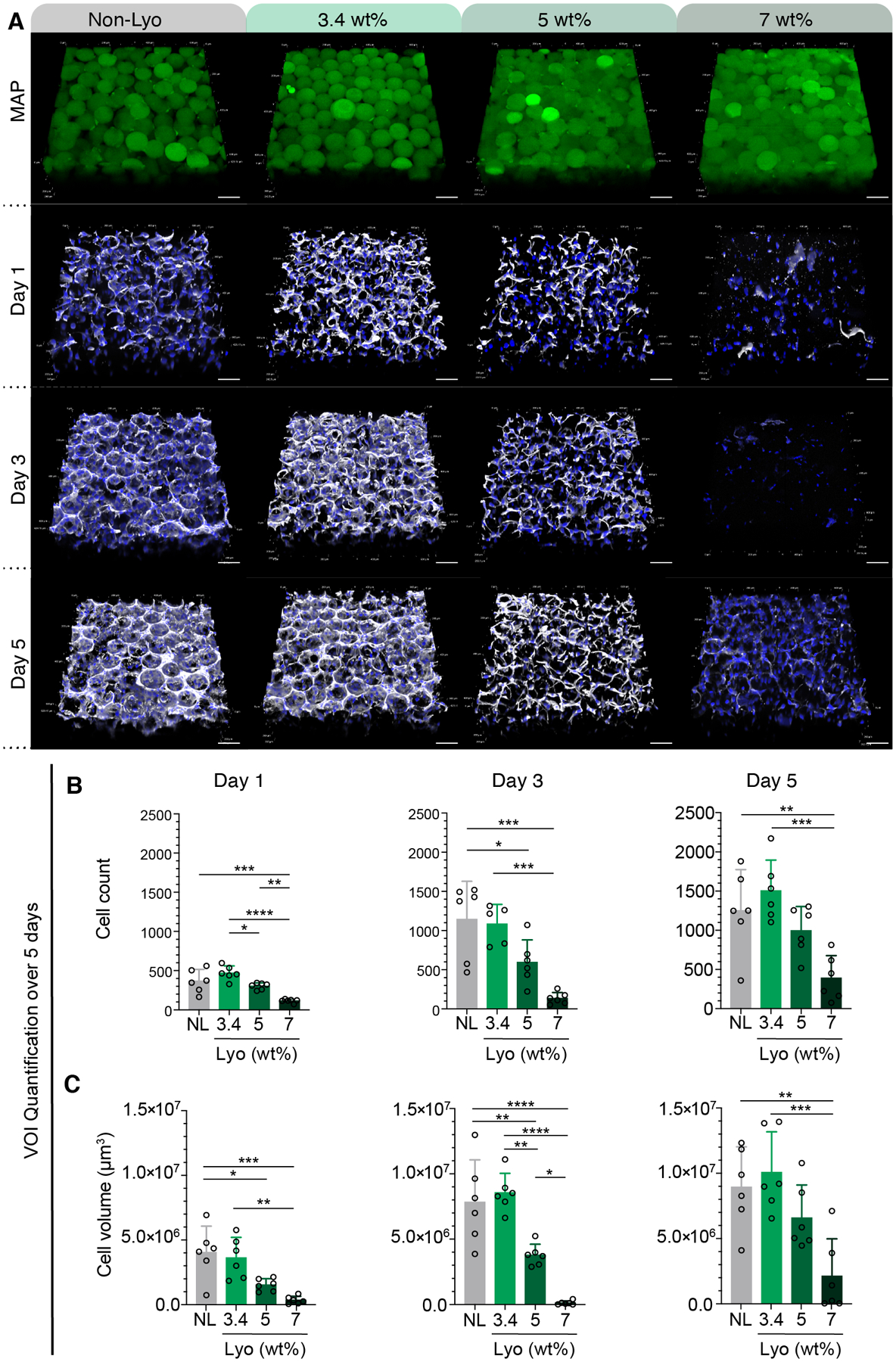
(A) Split channels of confocal Z-stacks for MAP (green) composed of non-lyo-microgels and lyo-microgels reconstituted at 3.4, 5, and 7 wt% cultured with D1 cells (white F-actin, blue DAPI). Scale bar = 100 μm. Quantification of volume of interest (VOI) within each replicate across days 1, 3, 5 for (B) cell count and (C) cell volume as measured by volume renderings in IMARIS software of F-actin staining (white). N=3 with duplicate wells represented by individual datapoints. A one-way ANOVA with Tukey HSD was performed on the samples at each time point with significance reported at p < 0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****).
MAP scaffolds comprised of lyo-microgels rehydrated at 5 and 7 wt% allowed us to achieve consistent particle fractions in MAP scaffolds that could not be achieved with non-lyo microgels. Because of this, we had yet to study cell responses in scaffolds with these high packing fractions. As such, we next created 3D cultures in MAP scaffolds comprised of lyo-microgels at 5 wt% and 7 wt%. As the wt% MAP increased (and therefore the particle fraction), cell counts and cell volume both decreased with increasing particle fraction.
3.8. Controlled particle fraction supports homogenous cell growth in MAP scaffolds
As shown in Figure 2, we observed pockets of high-density and low-density cell growth in the non-lyo MAP scaffolds through confocal microscopy. However, in lyo-microgel MAP scaffolds we observed more homogenous cell distributions. To quantify these observations, the intensity of the F-actin channel (white) in each Z-slice (Figure 8A) was averaged within 150 μm × 150 μm squares (Figure 8C). A representative Z-slice for each condition is shown in Figure 8B with the corresponding heatmap for each image shown in Figure 8D. The average intensity from each square for 30 slices within each Z-stack (Figure 8E) was then averaged to quantify the uniformity of the cell distribution across replicates for each condition (Figure 8F). These plots of the average intensity from each square in the heatmaps shows the large spread of values for the non-lyo condition. In contrast, the uniformity of cell spreading in the lyo-microgel conditions in indicated by the lower range of cell intensity across squares in the heatmap as well as the lower variance amongst all lyo-microgel conditions in Figure 8G. While uniformity is observed in the lyo-microgel conditions at all wt% MAP, the physical restriction of cells as wt% MAP increases is highlighted in higher magnification images showing cells at the scaffold interface (Figure 8H).
Figure 8.
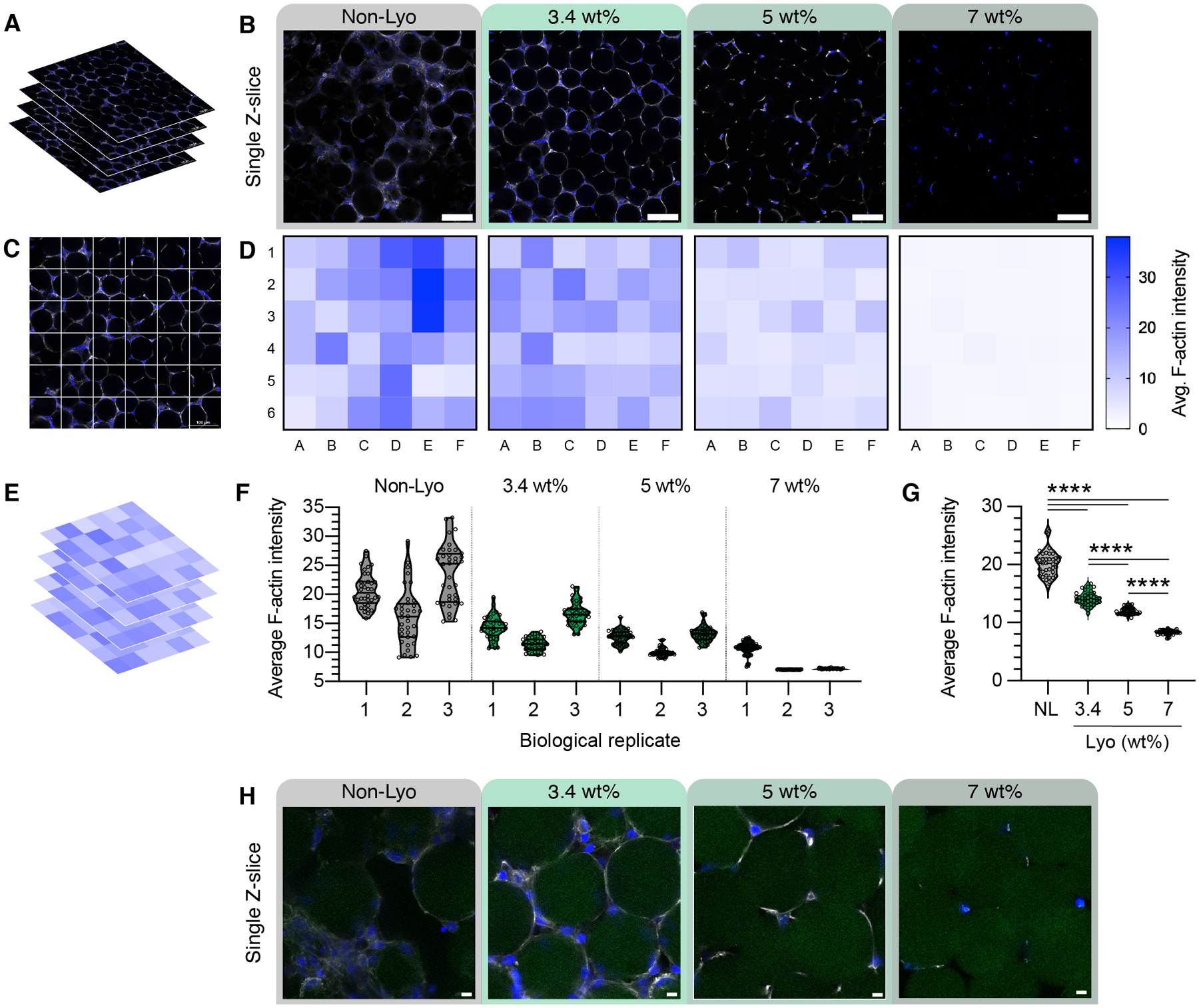
Single Z-slices from confocal Z-stacks (A) are shown in (B) for each condition on Day 5 of culture. The single Z-slices show the cell networks across the field of view (20X magnification, scale bar = 100 μm). (C) The average intensity of the F-actin channel from (A) was averaged within 150 μm × 150 μm squares and can be represented as a heatmap of the image (D). (E) Heatmaps were taken from 30 successive steps in the Z-stacks across replicates, with the averages shown in (F). (G) The average of the three replicates is plotted across conditions. (H) Higher magnification examples show cells at the scaffold interface in a single Z-slice across conditions (scale bar = 10 μm).
3.9. Macromolecule diffusion between microgels is affected by microgel packing density and size
To assess the diffusion of macromolecules in our materials, we cast the hydrogel scaffolds around 1 mm cylindrical spacers then allowed the scaffolds to swell in PBS. Once the spacers were removed, the fluorescently tagged dextran solution was added to the central cavity and imaged over time in a single Z-plane (Figure 9A). An example of this time lapse imaging of diffusion can be found in Supplemental Video 1. Linear concentration profiles were obtained over the full range of timepoints, extending outward radially from the center. The fluorescent intensity was converted to concentration by imaging a standard curve of the fluorescent dextran solutions. Representative diffusion profiles for each condition are shown in Figure 9B–E as relative concentration (concentration at each distance normalized to concentration in the central cavity at time=0) over distance for varying time points.
Figure 9.
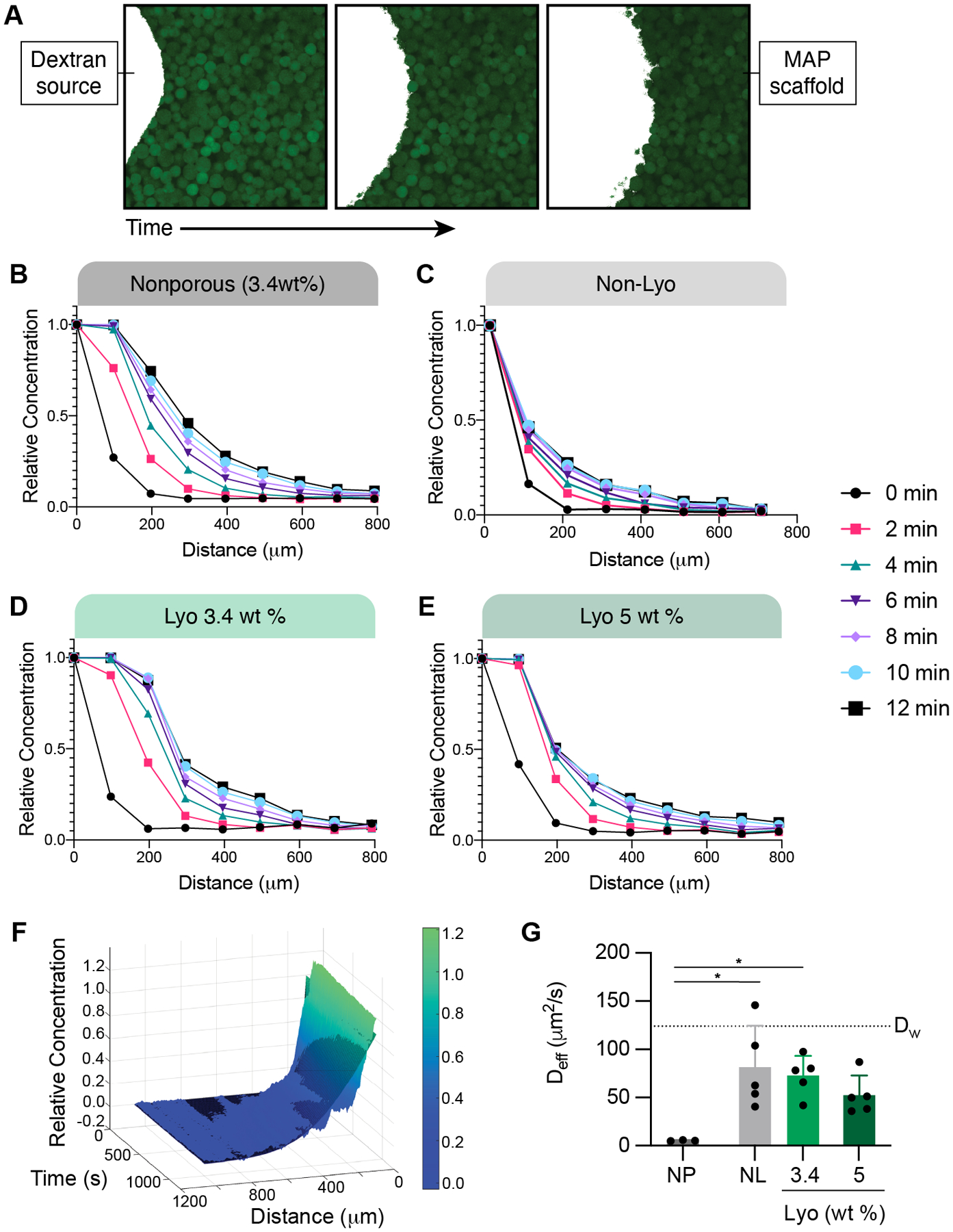
(A) Representative images (single Z-slices) of the fluorescent dextran (white) diffusing through a MAP scaffold (green) over time. Diffusion profiles for (B) 3.4 wt% nanoporous gel, (C) MAP gel with non-lyo-microgels, and MAP gel with (D) 3.4 wt% lyo-microgels and (E) 5 wt% lyo-microgels are presented as the relative concentration of 10 kDa dextran (concentration normalized to initial concentration) over distance for 12 minutes. Representative profiles are for one gel for each condition. (F) Representative example of the non-linear curve fitting (black mesh) for the diffusion profile shown as the relative concentration across time and distance. (G) Estimated effective diffusion coefficient (Deff) of 10 kDa fluorescent Dex for each condition compared to the reported value of free diffusion in water (DW) represented by the dotted line. A one-way ANOVA with Tukey HSD was performed with significance reported at p < 0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****).
Under the assumption our gels represent a semi-infinite medium for diffusion, we utilized Fick’s second law of diffusion to estimate the effective diffusion coefficient (Deff) for each gel (Equation 1). The diffusion coefficient for the 10 kDa fluorescent dextran within each scaffold was quantified by non-linear curve fitting of the normalized concentration values (C/Co) over time and distance (Figure 9F). The estimated Deff for each condition is compared to the values for free diffusion of 10 kDa dextran in water (Dw), as reported experimentally in literature and also approximated using the Stokes-Einstein equation [35].
Equation 1.
Fick’s second law of diffusion to model one-dimensional radial diffusion, with Dex concentration (C), constant Dex source concentration (Co), position (x), diffusivity (D), and time (t).
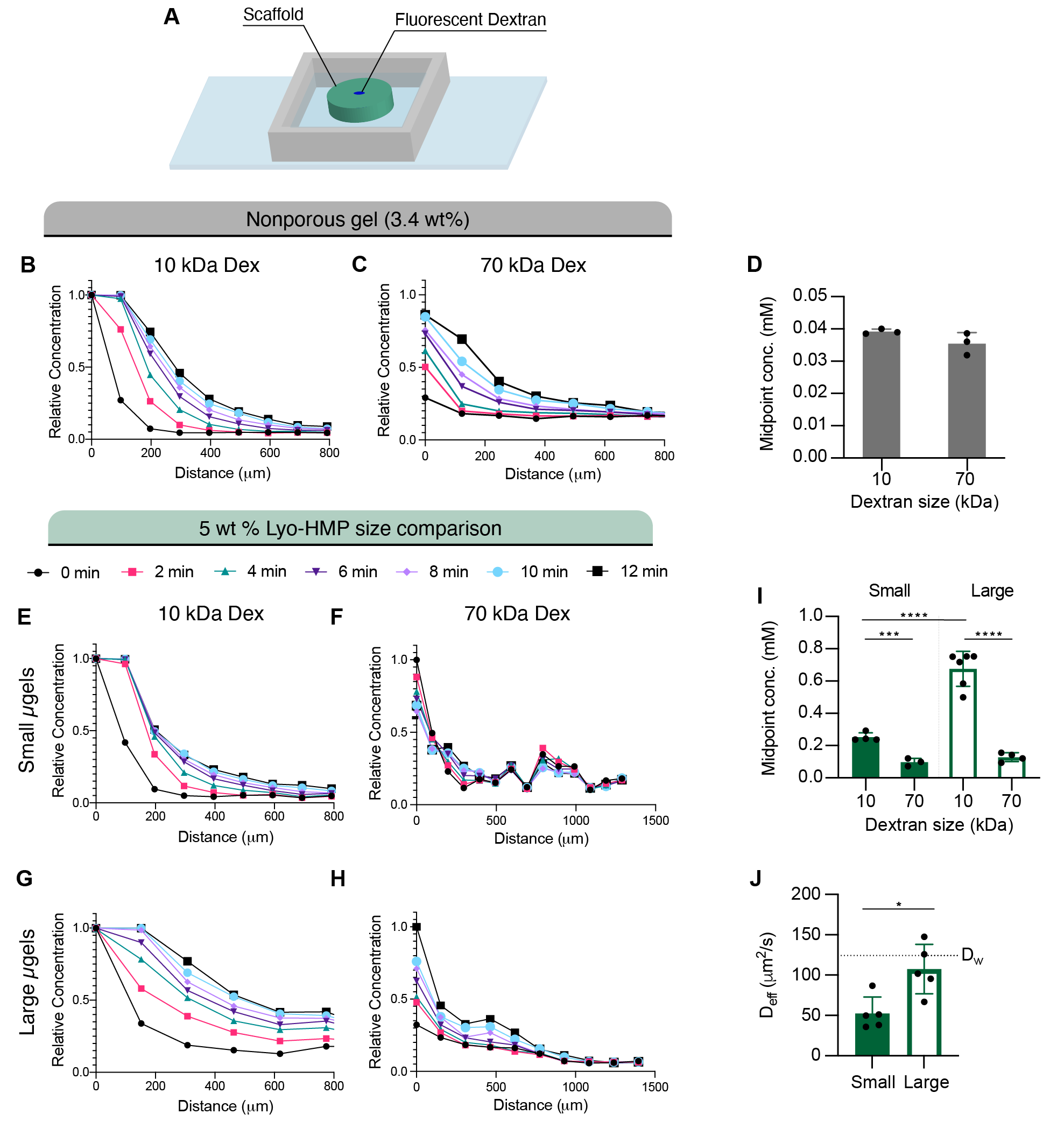
Compared to the MAP scaffolds, we observed significantly less diffusion in nanoporous gels of the same composition as the microgels (3.4 wt%) (Figure 9G). Similarly, diffusion was impeded with increasing particle fraction. Similar trends were also observed when using a larger size Dex (70 kDa) (Figure S5B–F). We then generated MAP scaffolds comprised of two different size populations of lyo-microgels (92 and 296 μm) at equivalent wt% MAP. Diffusion occurred more rapidly in MAP scaffolds comprised of larger microgels compared to smaller microgels (Figure S5G–J) regardless of macromolecule size.
4. DISCUSSION
Tet-NB click chemistry was shown previously to allow control over bulk MAP scaffold stiffness while also being biocompatible upon injection in an ischemic stroke model [6]. The Tet linker used in previous studies was a 4arm-polyethylene glycol-Tetrazine (4arm-PEG-Tet) which inherently limited the active sites of crosslinking to four. The first improvement to our MAP system was to incorporate a linear Tet linker, as an alternative to the star-shaped linker, with more active crosslinking sites to improve the crosslinking kinetics for MAP scaffolds as well as reduce the amount of crosslinker required to achieve comparable mechanical properties. This was achieved with a linear HA-Tet linker that boasts more functional Tet groups than previously reported 4arm-PEG-Tet. HA-Tet supported a greater range of bulk stiffness with the same Tet/HA ratios as the 4arm-PEG-Tet crosslinker. HA-Tet also decreased the time required for MAP scaffold gelation; however, it is worth noting that the tetrazine group on 4arm-PEG-Tet boasts an additional methyl group which improves the stability in aqueous buffer but also has been shown to reduce reactivity which could affect annealing kinetics.
MAP scaffolds generated with microgels concentrated via centrifugation (i.e., non-lyo) exhibited a large range of particle fractions. In vitro studies with non-lyo MAP scaffolds provided evidence that the particle fraction is a bioactive cue that influences cell growth within the scaffolds, with higher particle fractions restricting cell growth. However, concentrating the microgels via centrifugation failed to provide control over the particle fraction. One solution for controlling particle fraction would be to remove all available liquid, such as through freeze-drying, and then reintroduce controlled volumes for rehydrating the dried microgels and achieving a desired particle fraction. Therefore, to reproducibly study cell responses in MAP scaffolds, we investigated lyophilization as a new method for drying microgels and controlling the particle fraction in MAP scaffolds.
A prior report demonstrated that lyophilization of polyethylene glycol (PEG) microgels in IPA allowed microgels to retain their physical properties after rehydration [12]. While we observed this same result for PEG-NB microgels, HA-NB microgels lyophilized in 70% IPA were shown to have internal defects. We demonstrated that implementing different types of lyophilization mediums could either enhance or prevent internal defects from forming within microgels when freeze-dried. For EtOH, HEPES, DMSO, and MeCN, defects were minimized (i.e., increased proportion of fluorescent pixels) as the concentration increased. However, for IPA, NaCl, and Tris buffers, defects were minimized as the concentration decreased. Interestingly, these trends were not consistent among the specific types of mediums except polar aprotic, where both DMSO and MeCN decreased defects with increasing concentration. We also elucidated several mediums for lyophilization that can induce significant internal defects within microgels and could be explored for purposes of cryogel formation. In particular, eight conditions with positive pixels below 20% were highlighted in Figure 4B as maximal cryogel formation conditions for the conditions tested.
Lyo-microgels incubated in 70% EtOH showed minimal internal defects and were the most comparable to non-lyo microgels for both HA-NB and PEG-NB material composition. More than this, freeze-drying microgels using 70% EtOH also allowed HA-NB microgels to retain their original properties (i.e., shape, size, stiffness). By implementing these methods for drying microgels, the dried product could be rehydrated at controllable mg mL−1 concentrations when making MAP scaffolds. Increasing the wt % of lyo-microgels resulted in increasing particle fractions in the MAP scaffolds they comprised. Additionally, higher particle fractions were consistently achieved using higher wt % than could not be achieved with the centrifugation for concentrating microgels.
Users of MAP scaffolds commonly modulate the microgels inter-particle crosslinking chemistry or concentration of the inter-particle crosslinking species to tune the bulk stiffness of MAP scaffolds; however, our methods of generating scaffolds with controlled particle fractions allowed us to investigate how tuning the particle fraction plays a role in bulk scaffold stiffness. In granular hydrogels, mechanical stress is transmitted between touching particles through force chains [36, 37]. Because the particle components of granular materials are responsible the system’s stability under an applied load, we expected particle density to affect the bulk mechanical properties of MAP scaffolds. Specifically, we expected increasing particle density to allow more energy to be stored elastically in the scaffold, corresponding to an elevated storage modulus. To test this hypothesis, we created MAP scaffolds comprised of lyo-microgels at varying wt% MAP and performed rheological testing within the linear viscoelastic region. As we increased the wt % of lyo-microgels, and consequently the particle fraction, the shear storage modulus for MAP scaffolds increased and approached the modulus of a nanoporous gel. These trends were consistent with nanoporous gels in which stiffness increases with increasing the wt% of polymer content [38]. We also investigated the compressive modulus of our material both at the bulk and local scale which showed that the local stiffness of microgels is greater than the bulk scaffold stiffness. These findings were consistent with previous reports of bulk nanoporous and MAP hydrogels exhibiting different mechanical properties [3] due to the mismatched amount of polymer entanglements in the bulk gel versus contact points between microgels in MAP scaffolds [22].
With lyo-microgels, we were able to produce consistent particle fractions in our MAP scaffolds to study the cell response to particle fraction in a controlled manner. We observed the same trends as shown in non-lyo scaffolds with higher particle fractions restricting global cell growth. This was particularly evident in the 7 wt % MAP lyo-microgel scaffolds. By assessing regional cell growth, pockets of high- and low-density cell growth were observed in non-lyo scaffolds; however, lyo-microgel scaffolds supported more homogenous cell growth. These findings demonstrate the implications of particle fraction in MAP scaffolds on cell growth and underscore the inability to replicate this control using the previous methods of concentrating microgels via centrifugation. Using lyo-microgels, MAP users can now eliminate the confounding effect of particle fraction on cell response.
In addition to the role of physical confinement on the cells, we also hypothesized that the diffusion of macromolecules (i.e., nutrients from the media or cell waste) was impeded in the high particle fraction scaffolds. As such, we wanted to assess the impact of particle fraction on macromolecular diffusion within the unique microarchitectures of these scaffolds. Our platform for assessing macromolecular diffusion allowed us to observe the impedance of diffusion in nanoporous gels compared to granular MAP scaffolds. With controlled the particle fractions in the MAP scaffolds we observed slower diffusion as particle fraction increased. Lastly, increasing the size of microgels comprising the MAP scaffolds enabled faster diffusion of macromolecules, likely a result of the larger pores observed between larger particles in granular materials [22].
5. CONCLUSION
In this study we elucidated methods for successfully drying HA-NB microgels without sacrificing material integrity using 70% EtOH as the lyophilization medium. Lyo-microgels generated using 70% EtOH allowed microgels to retain their original size, shape, and stiffness while also minimizing internal defects. Additionally, using 70% EtOH provides an added benefit of sterilization in the process of MAP fabrication. The goal of this work was not only to determine a method for drying microgels that retain material properties, but to also control particle fraction during MAP scaffold formation. Using this method, we eliminated the variability of particle fraction during MAP scaffold formation that arises when using alternative drying methods. Once freeze-dried, lyo-microgels could be weighed and rehydrated with fixed volumes to achieve controlled particle fraction in MAP scaffolds. As we increased the wt% MAP, we generated higher particle fractions in MAP scaffolds than could be achieved using centrifugation to concentrate the microgels. Our findings demonstrate the implications of particle fraction in MAP scaffolds on material properties, cell growth, and diffusion and underscore the inability to replicate this control using the previous methods of concentrating microgels via centrifugation. This work also demonstrates that the molecular architecture of the inter-particle crosslinking polymer affects the kinetics of microgel gelation and the bulk mechanical properties that can be achieved. With controlled particle fractions and efficient inter-particle crosslinking methods, we can now reproducibly assess mechanical properties and diffusion, as well as cell responses within MAP scaffolds.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We would like to thank the National Institutes of Health, the National Institutes of Neurological Disorders and Stroke (1R01NS112940, 1R01NS079691, R01NS094599), and the National Institute of Allergy and Infectious Disease (1R01AI152568). This work was performed in part at the Duke University Shared Materials Instrumentation Facility (SMIF), a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), which is supported by the National Science Foundation (award number ECCS-2025064) as part of the National Nanotechnology Coordinated Infrastructure (NNCI). We would like to thank Katrina L. Wilson for her insights on microgel cryogels as well as her time for training many of the fabrication techniques used in this work.
Footnotes
Supporting Information.
Supplemental figures include the size distributions of microgels used throughout this work, proton NMR spectrum of HA-NB and HA-Tet, representative images for each condition of lyophilized microgels, and images of microgel culture in LB broth to assess bacterial growth. (File type, Word document)
Supplemental video shows the time lapse diffusion of fluorescent dextran in MAP scaffolds. (File type, .mov)
DECLARATION OF INTEREST
ARA and TS have filed a provisional patent on this technology. TS is a founding member and equity owner of Tempo Therapeutics that aims to commercialize MAP technology.
REFERENCES
- [1].Caldwell AS, Campbell GT, Shekiro KMT, Anseth KS, Clickable Microgel Scaffolds as Platforms for 3D Cell Encapsulation, Advanced Healthcare Materials, 6 (2017) 1700254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sideris E, Griffin DR, Ding Y, Li S, Weaver WM, Di Carlo D, Hsiai T, Segura T, Particle Hydrogels Based on Hyaluronic Acid Building Blocks, ACS Biomaterials Science & Engineering 2, 2 (2016) 2034–2041. [DOI] [PubMed] [Google Scholar]
- [3].Truong NF, Kurt E, Tahmizyan N, Lesher-Pérez SC, Chen M, Darling NJ, Xi W, Segura T, Microporous annealed particle hydrogel stiffness, void space size, and adhesion properties impact cell proliferation, cell spreading, and gene transfer, Acta biomaterialia, 94 (2020) 160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Xin SC, David; Garza Jay E.; Gaharwar Akhilesh K.; Alge Daniel L., Clickable PEG hydrogel microspheres as building blocks for 3D bioprinting, Biomaterials Science, 7 (2019) 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Darling N, Sideris Elias, Hamada Naomi, Carmichael S. Thomas, Segura Tatiana Injectable and Spatially Patterned Microporous Annealed Particle (MAP) Hydrogels for Tissue Repair Applications, Advanced Science, 5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Darling NJ, Xi W, Sideris E, Anderson AR, Pong C, Carmichael S Thomas, Segura T, Click by Click Microporous Annealed Particle (MAP) Scaffolds, Advanced Healthcare Materials, 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Griffin DR, Weaver WM, Scumpia PO, Di Carlo D, Segura T, Accelerated wound healing by injectable microporous gel scaffolds assembled from annealed building blocks, Nat Mater, 14 (2015) 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Griffin DR, Archang MM, Kuan C-H, Weaver WM, Weinstein JS, Feng AC, Ruccia A, Sideris E, Ragkousis V, Koh J, Plikus MV, Di Carlo D, Segura T, Scumpia PO, Activating an adaptive immune response from a hydrogel scaffold imparts regenerative wound healing, Nature Materials, 20 (2020) 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Koh J, Griffin DR, Archang MM, Feng AC, Horn T, Margolis M, Zalazar D, Segura T, Scumpia PO, Di Carlo D, Enhanced in vivo delivery of stem cells using microporous annealed particle scaffolds, Small, 15 (2019) 1903147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mealy JE, Chung JJ, Jeong HH, Issadore D, Lee D, Atluri P, Burdick JA, Injectable Granular Hydrogels with Multifunctional Properties for Biomedical Applications, Advanced Materials, 30 (2018) 1705912. [DOI] [PubMed] [Google Scholar]
- [11].Nih LR, Sideris E, Carmichael ST, Segura T, Injection of microporous annealing particle (MAP) hydrogels in the stroke cavity reduces gliosis and inflammation and promotes NPC migration to the lesion, Advanced Materials, 29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Pruett L, Ellis R, McDermott M, Roosa C, Griffin D, Spatially heterogeneous epidermal growth factor release from microporous annealed particle (MAP) hydrogel for improved wound closure, Journal of Materials Chemistry B, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pruett LJ, Jenkins CH, Singh NS, Catallo KJ, Griffin DR, Heparin Microislands in Microporous Annealed Particle Scaffolds for Accelerated Diabetic Wound Healing, Advanced functional materials, 31 (2021) 2104337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Qazi TH, Wu J, Muir VG, Weintraub S, Gullbrand SE, Lee D, Issadore D, Burdick JA, Anisotropic Rod-Shaped Particles Influence Injectable Granular Hydrogel Properties and Cell Invasion, Advanced Materials, (2021) 2109194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sideris E, Kioulaphides S, Wilson KL, Yu A, Chen J, Carmichael ST, Segura T, Particle Hydrogels Decrease Cerebral Atrophy and Attenuate Astrocyte and Microglia/Macrophage Reactivity after Stroke, Advanced Therapeutics, (2019) 2200048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pruett LK, Heather; Martz Teresa; Churnin Ian; Ferrante Sergio; Salopek Lisa; Cottler Patrick; Griffin Donald R.; Daniero James J., Development of a microporous annealed particle hydrogel for long‐term vocal fold augmentation, The Laryngoscope, 130 (2020). [DOI] [PubMed] [Google Scholar]
- [17].Schaeffer C, Pfaff BN, Cornell NJ, Salopek LS, Shan S, Viyar J, Omesiete W, Griffin DR, Cottler PS, DeGeorge BR Jr, Injectable microannealed porous scaffold for articular cartilage regeneration, Annals of Plastic Surgery, 84 (2020) S446–S450. [DOI] [PubMed] [Google Scholar]
- [18].van Hecke M, Jamming of soft particles: geometry, mechanics, scaling and isostaticity, Journal of Physics: Condensed Matter 22 (2009) 033101. [DOI] [PubMed] [Google Scholar]
- [19].Highley CB, Song KH, Daly AC, Burdick JA, Jammed Microgel Inks for 3D Printing Applications, Advanced Science, 6 (2018) 1801076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li F, Truong VX, Fisch P, Levinson C, Glattauer V, Zenobi-Wong M, Thissen H, Forsythe JS, Frith JE, Cartilage tissue formation through assembly of microgels containing mesenchymal stem cells, Acta biomaterialia, 77 (2018) 48–62. [DOI] [PubMed] [Google Scholar]
- [21].Ma T, Gao X, Dong H, He H, Cao X, High-throughput generation of hyaluronic acid microgels via microfluidics-assisted enzymatic crosslinking and/or Diels–Alder click chemistry for cell encapsulation and delivery, Applied Materials Today, 9 (2017) 49–59. [Google Scholar]
- [22].Daly AC, Riley L, Segura T, Burdick JA, Hydrogel microparticles for biomedical applications, Nature Reviews Materials, 5 (2020) 20–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Muir VG, Qazi TH, Shan J, Groll J.r., Burdick JA, Influence of Microgel Fabrication Technique on Granular Hydrogel Properties, ACS Biomaterials Science & Engineering, 7 (2021) 4269–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kurt E, Segura T, Nucleic Acid Delivery from Granular Hydrogels, 11 (2021) 2101867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rommel D, Mork M, Vedaraman S, Bastard C, B. GLP, Kittel Y, Vinokur R, Born N, Haraszti T, De Laporte L, Functionalized Microgel Rods Interlinked into Soft Macroporous Structures for 3D Cell Culture, Advanced Science, 9 (2022) 2103554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Qazi TH, Muir VG, Burdick JA, Methods to Characterize Granular Hydrogel Rheological Properties, Porosity, and Cell Invasion, ACS Biomaterials Science & Engineering, 8 (2022) 1427–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fonte P, Reis S, Sarmento B, Facts and evidences on the lyophilization of polymeric nanoparticles for drug delivery, Journal of Controlled Release, 10 (2016) 75–86. [DOI] [PubMed] [Google Scholar]
- [28].Plieva F, Huiting X, Galaev IY, Bergenståhl B, Mattiasson B, Macroporous elastic polyacrylamide gels prepared at subzero temperatures: control of porous structure, Journal of Materials Chemistry 16 (2006) 4065–4073. [Google Scholar]
- [29].Welzel PB, Friedrichs J, Grimmer M, Vogler S, Freudenberg U, Werner C, Cryogel Micromechanics Unraveled by Atomic Force Microscopy-Based Nanoindentation, Advanced Healthcare Materials, 3 (2014) 1849–1853. [DOI] [PubMed] [Google Scholar]
- [30].Wilson KL, Lesher-Pérez SC, Naffa MM, Kelly SH, Segura T, Stoichiometric Post Modification of Hydrogel Microparticles Dictates Neural Stem Cell Fate in Microporous Annealed Particle Scaffolds, Advanced Materials, (2022) 2201921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang HD, T. K; Xu X; Jia X; Fox JM, Interfacial bioorthogonal crosslinking, ACS Macro Letters, (2014) 727–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Linkert M, Rueden CT, Allan C, Burel JM, Moore W, Patterson A, Loranger B, Moore J, Neves C, MacDonald D, Tarkowska A, Metadata matters: access to image data in the real world, Journal of Cell Biology, 189 (2010) 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Boyce JM, Pittet D, Guideline for Hand Hygiene in Health‐Care Settings: recommendations of the Healthcare Infection Control Practices Advisory Committee and the HICPAC/SHEA/APIC/IDSA Hand Hygiene Task Force, Infection Control and Hospital Epidemiology, 23 (2002) S3–S40. [DOI] [PubMed] [Google Scholar]
- [34].Kampf G, Kramer A, Epidemiologic background of hand hygiene and evaluation of the most important agents for scrubs and rubs, Clinical microbiology reviews, 17 (2004) 863–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Albro MB, Rajan V, Li R, Hung CT, Ateshian GA, Characterization of the Concentration-Dependence of Solute Diffusivity and Partitioning in a Model Dextran-Agarose Transport System, Cellular and Molecular Bioengineering, 2 (2009) 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hurley RC, Hall SA, Andrade JE, Wright J, Quantifying interparticle forces and heterogeneity in 3D granular materials, Physical Review Letters, 117 (2016) 098005. [DOI] [PubMed] [Google Scholar]
- [37].Sun QJ, Feng; Jianguo Liu; Guohua Zhang, Understanding force chains in dense granular materials, International Journal of Modern Physics B 24 (2010) 5743–5759. [Google Scholar]
- [38].Anseth K, Bowman C, Brannon-Peppas L, Mechanical properties of hydrogels and their experimental determination., Biomaterials, 17 (1996) 1647–1657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.