

Abstract
The management of acute hypoxemic respiratory failure (AHRF) in newborns continues to be a clinical challenge with elevated risk for significant morbidities and mortality, especially when accompanied with persistent pulmonary hypertension of the newborn (PPHN). PPHN is a syndrome characterized by marked hypoxemia secondary to extrapulmonary right-to-left shunting across the ductus arteriosus and/or foramen ovale with high pulmonary artery pressure and increased pulmonary vascular resistance (PVR). After optimizing respiratory support, cardiac performance and systemic hemodynamics, targeting persistent elevations in PVR with inhaled nitric oxide (iNO) therapy has improved outcomes of neonates with PPHN physiology. Despite aggressive cardiopulmonary management, a significant proportion of patients have an inadequate response to iNO therapy, prompting consideration for additional pulmonary vasodilator therapy. This article reviews the pathophysiology and management of PPHN in term newborns with AHRF while highlighting both animal and human data to inform a physiologic approach to the use of PH-targeted therapies.
Keywords: Hypoxemia, Persistent pulmonary hypertension of the newbron, Nitric oxide, Sildenafil, Prostacyclin, Milrinone, Bosentan
1. Introduction
The normal fetal circulation is characterized by high pulmonary artery pressure secondary to high pulmonary vascular resistance (PVR) with low pulmonary blood flow (Qp), leading to right-to-left shunting of blood flow across the ductus arteriosus and foramen ovale. This physiology allows for the preferential distribution of combined ventricular output to the placenta, providing efficient gas exchange to enable optimal tissue oxygenation in the developing fetus. Successful transition from intra-uterine to extra-uterine life is dependent upon a rapid and dramatic decrease in PVR to accommodate an 8–10 fold increase in pulmonary blood flow, as tissue oxygenation becomes dependent upon alveolar gas exchange and high flow through the postnatal pulmonary circulation [1]. Infants who fail to achieve or sustain the normal decrease in PVR at birth develop severe hypoxemia, recognized as persistent pulmonary hypertension of the newborn (PPHN).
PPHN is a syndrome associated with diverse neonatal cardiac and pulmonary disorders that are characterized by a common physiology, which includes sustained elevations in PVR causing extrapulmonary right-to-left shunting of blood across the patent ductus arteriosus (PDA), the foramen ovale (PFO), or both, resulting in critical hypoxemia, poorly responsive to inspired oxygen and optimal mechanical ventilation. PPHN affects ~0.2% of live births and is associated with a one-year mortality of 7.6% of all babies [2]. Importantly, the incidence of PPHN is directly associated with the degree of prematurity, with rates as high as 18% in infants born at 22–23 weeks gestational age [3]. This review discusses selected aspects of perinatal pulmonary vascular development and physiology, and the pathophysiology, clinical manifestations and management of PPHN, highlighting the physiologic basis of pulmonary vasodilator therapy in late preterm and term newborns. We will briefly discuss implications for approaches to the growing understanding of PPHN and targeted PH drug therapies in preterm neonates.
2. Fetal pulmonary vascular tone and regulation
To meet the high metabolic demands in the developing fetus, the fetal circulation has adapted to ensure optimal oxygen delivery by directing highly oxygenated blood from the placental circulation to the fetal heart and brain. Blood returning to the heart via placental circulation preferentially streams across the right atrium, through the foramen ovale into the left atrium, and is ejected by the left ventricle to be available for coronary and cerebral circulation. Venous return from the cerebral circulation mixes with coronary venous return and this desaturated blood enters the right ventricle where the majority of right ventricular output is directed across the ductus arteriosus to the placenta via the descending aorta. Animal studies have demonstrated that the fetal lung receives 3–7% of combined ventricular output, increasing as gestational age increases [4]. Human studies indicate a similar progressive increase in Qp throughout gestation, but Doppler ultrasound and phase-contrast magnetic resonance imaging studies estimate Qp to be proportionately greater than Qp reported in sheep, ranging from 11% to 25% [5–8]. Despite marked distal pulmonary vascular growth throughout late gestation, PVR remains high in the late preterm fetus, suggesting an important role for active vasoconstriction in the developing lung before birth.
In utero, high PVR is maintained through various mechanisms including vascular compression by the fluid filled distal lung, hypoxemic vasoconstriction, sustained production of vasoconstrictor mediators (especially the potent agent, endothelin-1 [ET]), limited release of endogenous vasodilators (such as prostacyclin [PGI2] and nitric oxide [NO]), and enhanced myogenic tone, which opposes vasodilation in response to diverse hemodynamic, metabolic and molecular stimuli [9, 10]. Intrauterine mechanisms that limit production of endogenous vasodilators and enhance the production of vasoconstrictors are likely related to diverse maturational changes in endothelial and smooth muscle cell function, and the failure of pulmonary vascular growth and vasodilation at birth likely reflects the impact of perinatal vascular injury [11–16].
2.1. Fetal to newborn transition
With the loss of the low resistance placental circulation and oxygen-breathing at birth, the normal transition from fetal to extrauterine life is characterized by a rise in systemic vascular resistance and fall in PVR, favoring a marked increase in Qp, as tissue oxygenation requires the rapid establishment of the alveolar-capillary gas transfer interface. Alveolar ventilation, increased oxygen tension, and alterations in vasoactive stimuli, especially NO and PGI2, act synergistically to optimize the rapid increase in Qp [17]. Disruptions of the NO–cyclic guanosine monophosphate (cGMP), prostacyclin–cyclic adenosine monophosphate (cAMP), and ET-1 pathways play an important role in the pulmonary vasoconstriction associated with PPHN. At birth, increases in endothelial NO synthase activity increases NO production, stimulating smooth muscle cell soluble guanylate cyclase activity and causing vasodilation via increased cGMP production. The arachidonic acid-prostacyclin pathway also plays an important role in pulmonary vasodilation via activation of adenylate cyclase and subsequent increase in cAMP concentration in the vascular smooth muscle cells. The effects of ET-1 are mediated through two receptors: ETA receptors on smooth muscle cells mediate vasoconstriction, and ETB receptors on endothelial cells, which enable ET-1 clearance from the circulation and further mediate vasodilation, with a relative increase in ETA receptors in experimental models of PPHN [18]. (Fig. 1).
Fig. 1.
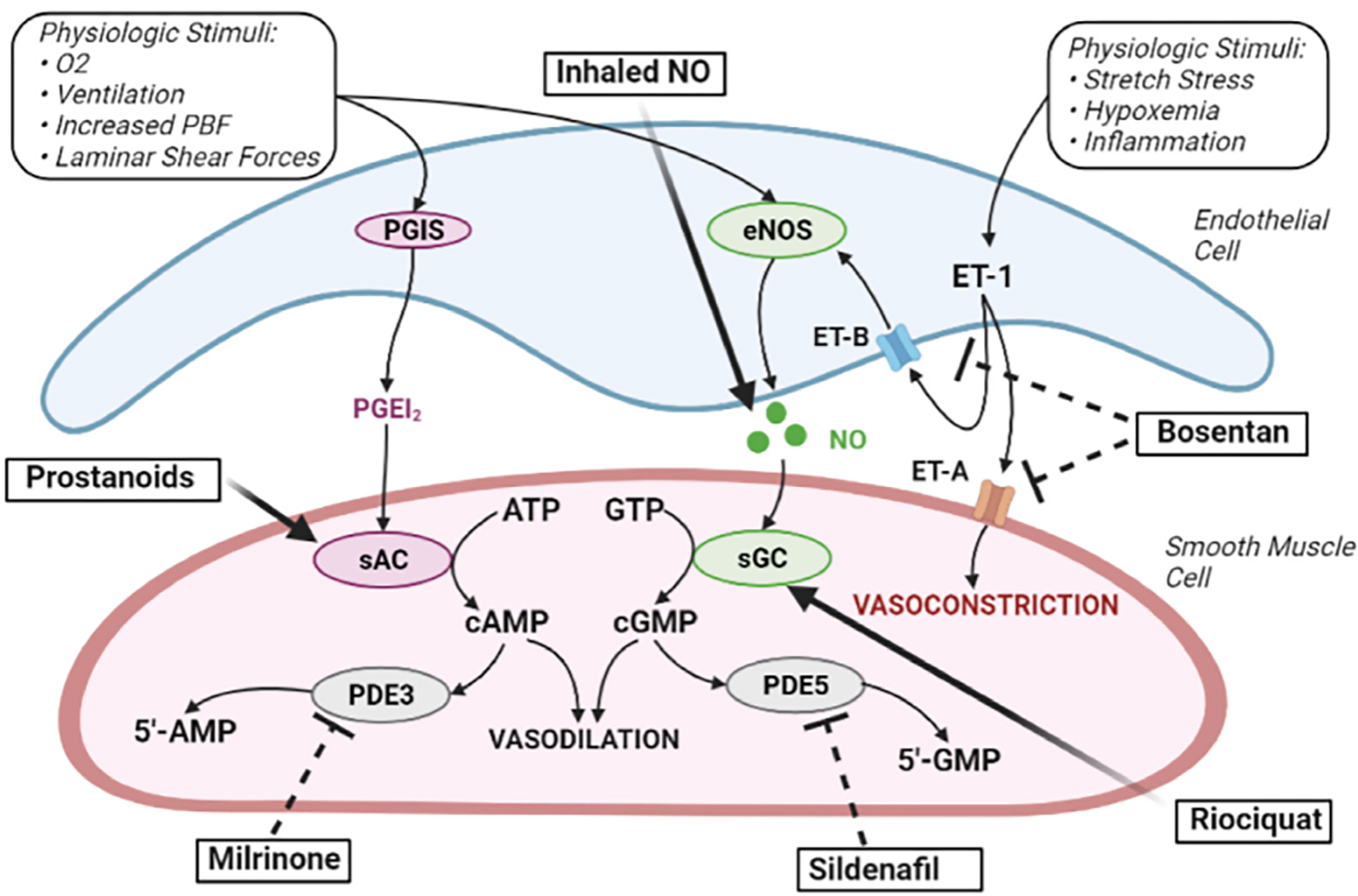
Pulmonary Hypertension Drug Therapies Target Endothelial Cell Derived Pulmonary Vasodilator Pathways. Nitric oxide (NO) and prostacyclin (PGI2) signaling pathways regulate pulmonary vascular tone in the developing lung. ATP, Adenosine triphosphate; cAMP, cyclic AMP; cGMP, cyclic GMP; COX-1, cyclooxygenase 1; GTP, guanosine triphosphate; NOS, nitric oxide synthase; PDE3, phosphodiesterase 3; PDE5, phosphodiesterase 5; PBF, pulmonary blood flow; PGIS, prostacyclin synthase. (Created with BioRender.com)
3. Clinical manifestations and the diagnosis of PPHN
PPHN is characterized by failure of the fetal circulation to transition at birth resulting in sustained elevations in PVR. Persistent elevations in PVR lead to increased extrapulmonary right-to-left shunting of deoxygenated blood across the PFO and PDA, reduced Qp and refractory hypoxemia. It is imperative that the clinician recognize that not all newborns with acute hypoxemic respiratory failure (AHRF) have PPHN physiology. The diagnosis of PPHN in the setting of AHRF in term or preterm neonates is confirmed most commonly by echocardiographic (ECHO) evidence of extrapulmonary right-to-left shunting and increased pre- and post-ductal gradients in PaO2 or saturation.
While diverse mechanisms potentially contribute to PPHN and can evolve over time, PPHN has traditionally been classified into 1 of 3 categories: (1) maladaptation: pulmonary vessels have normal structure and number but have abnormal vasoreactivity, which is the most common type, including respiratory distress syndrome (RDS), sepsis, and meconium aspiration syndrome (MAS); (2) excessive muscularization: increased smooth muscle cell thickness and distal extension of muscle to vessels that are normally not muscularized, resulting from antenatal injuries including chronic intrauterine hypoxemia and idiopathic PPHN, and (3) vascular underdevelopment: lung hypoplasia associated with decreased pulmonary artery number, including congenital diaphragmatic hernia (CDH), oligohydramnios associated pulmonary hypoplasia, and multiple developmental lung disorders (Table 1).
Table 1.
Systems based approach to persistent pulmonary hypertension of the newborn.
| Systems Based Approach to PPHN Etiology | |
|---|---|
|
| |
| Pulmonary | Cardiovascular |
|
| |
| • Respiratory Distress Syndrome • Congenital Pneumonia • Sepsis • Meconium Aspiration • Antenatal Asphyxia • Pulmonary Hypoplasia (Oligohydramnios) • Idiopathic • Congenital Diaphragmatic Hernia • Developmental Lung Disorders • Trisomy 21 • TBX-4 Mutations • Alveolar Capillary Dysplasia (FOXF1) • CRH Receptor-1 • Congenital Surfactant Deficiencies (ABCA3,SP-B/C) |
• Left Ventricular Dysfunction • Total Anomalous Pulmonary Venous Return • Structural Cardiac Disease • Aortic Coarctation • Interrupted Aortic Arch • Transposition of the Great Arteries • Ebstein’s Anomaly • Mitral Stenosis • Pulmonary Atresia • Glycogen Storage Disease II (Pompe’s Disease) • Hepatic Arteriovenous Malformations (AVM) • Cerebral AVMs (Vein of Galen) • Pulmonary Vein Stenosis • Premature DA Closure |
| Other | |
| • Neuromuscular Disease • Inborn Errors of Metabolism • Maternal Medication Use • Selective Serotonin Reuptake Inhibitors (SSRI) • Aspirin/NSAID • Polycythemia • Thrombocythemia |
|
TBX-4, T-Box Transcription Factor-4 gene; FOXF1, Forkhead Box F1 gene; CRH, Cortisol-releasing Hormone; ABCA3, ATP-binding Cassette A3; SP, Surfactant Protein; NSAID, Nonsteroidal Anti-inflammatory Drug.
Although helpful, this paradigm is incomplete, as these categories are imprecise and there can be multiple overlapping features between groups and clinical causes of PPHN are often multifactorial. For example, PPHN often complicates the course of newborns with CDH, in which mechanisms of elevated PVR can include decreased lung vascular growth, hypertensive arterial remodeling, and abnormal vasoreactivity [19]. In addition, left ventricular systolic or diastolic dysfunction may contribute to high pulmonary artery pressure secondary to left atrial and pulmonary venous hypertension in CDH, as well as other diseases [20].
Infants with PPHN often present with labile hypoxemia and clinical evidence of right-to-left extrapulmonary shunting, demonstrated as a gradient in pre-ductal and post-ductal oxygen saturations greater than 10% [21]. Labile hypoxemia is the result of abrupt increases in PVR leading to increased right-to-left shunting. Acutely reducing PVR with pulmonary vasodilator drug therapy is fundamental in maintaining systemic arterial oxygen saturations by reducing extrapulmonary shunting. Past studies have clearly shown the need to first optimize mechanical ventilation and cardiac performance before applying PH-targeted drug therapies such as inhaled NO (iNO) [22]. Initial evaluation including a careful physical examination and chest radiography can be helpful in differentiating PPHN from other etiologies of hypoxemic respiratory failure. Physical findings are often subtle including a loud second heart sound or a systolic murmur of tricuspid regurgitation, though this murmur is not uncommon in the first days of a neonate’s life. In general, the degree of hypoxemia is disproportionate to the severity of radiographic evidence of lung disease in primary PPHN. Chest radiographs remain useful and can help differential parenchymal lung disease such as RDS or MAS from other non-pulmonary etiologies of PPHN. Typical radiographic findings in idiopathic PPHN include pulmonary vascular oligemia, normal or slight hyperinflation and a lack parenchymal infiltrates.
ECHO is the tool of choice to diagnose and monitor infants with PPHN. Timely ECHO is critical to address the infant’s underlying physiology, including confirming extrapulmonary shunting across the PDA or PFO, and excluding the diagnosis of structural heart disease. ECHO evidence of PPHN should be demonstrated by evidence of bidirectional or predominantly right-to-left shunting across the PFO and/or PDA.
Structural heart disease presenting as PPHN may include total anomalous pulmonary venous return, and lesions with ductal-dependent systemic circulation, such as severe aortic coarctation and hypoplastic left heart syndrome. Without ECHO, empiric use of iNO or other PH-targeted therapies to decrease PVR may lead to systemic hypoperfusion, worse hypoxemia and increased pulmonary edema, especially in the setting of LV dysfunction which can delay initiation of targeted interventions.
Additional information can be obtained by careful assessments of other echocardiographic metrics. These include tricuspid regurgitant peak velocity (TRJV), right ventricular systolic time intervals, and septal flattening, but have not been demonstrated to reliably predict pulmonary artery pressures [23–25]. Decreased right ventricular function in the setting of PPHN is associated with more severe disease and can be secondary to persistent afterload, hypoxemia, inflammation or other mechanisms [26–28]. It is essential to recognize the presence of left ventricular dysfunction, suggested by the presence of right-to-left shunting across the PDA with left-to-right shunting across the PFO, to prioritize optimization of cardiac performance prior to the use of pulmonary vasodilators. In the setting of LV dysfunction, PH-targeted drugs may lead to worsening pulmonary edema, and these infants may be best managed with LV support and reducing systemic afterload. PH drug therapy should follow steps to first enhance lung recruitment, avoid overdistension or atelectasis, and to support systemic hemodynamics and left ventricular function.
4. Management of PPHN
4.1. Approach to respiratory support
Initial care of newborns with evidence of PPHN requires optimal lung recruitment with mechanical ventilation to establish adequate gas exchange and optimize functional residual capacity (FRC). Specific ventilator strategies are partly dependent upon the blend of underlying airways and lung parenchymal disease, but failure to achieve an optimal FRC can worsen PVR because of low lung volumes or marked hyperinflation. Achieving optimal lung volumes decreases ventilation-perfusion mismatch while attempting to avoid ventilator-induced lung injury (VILI) from phasic stretch, especially in the setting of low FRC. Some infants may benefit from high frequency oscillatory or jet ventilation to increase lung recruitment and maintain FRC while avoiding stretch-induced injury to the lung, which can in itself decrease right-to-left extrapulmonary shunting and improve oxygenation without requiring additional PH-targeted drug therapy [22,29,30].
4.2. Oxygen therapy
As O2 delivery is imperative for maintenance of tissue metabolism and has pulmonary vasodilator effects, it remains the first line therapy for PPHN. O2 causes selective pulmonary vasodilation through direct activation of calcium-activated K+ channels and enhanced NO production after birth [31,32]. However, even relatively brief periods of hyperoxia may contribute to oxidative stress, impair NO-cGMP signaling, augment inflammation and exacerbate pulmonary vascular injury [33]. In addition to improving pulmonary hemodynamics, cardiac output and ventilation-perfusion matching, the ability of iNO and other PH-targeted drugs to lower the need for higher levels of supplemental oxygen may provide an indirect “lung protective” effect.
5. PH-targeted drug therapies
5.1. Inhaled nitric oxide
Optimal management of PPHN involves selective pulmonary vasodilation without worsening systemic hemodynamics. Inhaled nitric oxide (iNO) readily diffuses from alveoli into pulmonary smooth muscle cells causing pulmonary vasodilation with minimal systemic consequences because of its short half-life (2–6s) and rapid inactivation by avid NO-hemoglobin binding in the circulation [22,34]. Inhaled NO is a rapid and selective vasodilator secondary to the ability to deliver this small gas molecule via inhalation, both invasively and non-invasively, to air spaces approximating the pulmonary vasculature causing decreased intrapulmonary right-to-left shunting and improved V/Q matching [35]. Inhaled NO which has repeatedly been shown to be safe and effective in reducing the need for ECMO, including large placebo-controlled multicenter trials, remains the only United States Food and Drug Administration (FDA) approved specific pulmonary vasodilator therapy for late preterm and term infants with AHRF and PPHN [36]. Initial RCTs demonstrated a decreased need for ECMO support in late preterm and term infants with severe PPHN and an oxygenation index (OI) of 25–40 [37,38]. A subsequent large trial did not report reductions in ECMO use or death with earlier use of iNO in infants with moderate respiratory failure (median OI of 20) [39].
Past work suggests that an appropriate starting dose in term neonates is 20 parts per million (ppm) (Table 2). Higher doses rarely add much clinical benefit in patients with poor responses but have been used in some patients [40]. The most common reason for failure to respond to iNO is inadequate lung recruitment and measures to improve lung volume, including changes in ventilator strategy, initiation of high frequency ventilation or surfactant administration, should be addressed prior to dose alterations. Once an infant demonstrates stable hemodynamics accompanied by maintained oxygenation with decreased fraction of inspired oxygen (FiO2)requirements it is reasonable to progressively wean the dose at 2–4 h intervals to a goal of 5 ppm. At 5 ppm the dose should be gradually decreased by 1 ppm at similar time intervals. Abrupt changes in iNO dose can result in “rebound vasoconstriction,” causing acute hypoxemia, which can be managed by returning the iNO dose to the prior level, and repeating trials at lower doses on subsequent days. Weaning can generally be accomplished in 4–5 days. Prolonged need for iNO therapy without resolution of disease should lead to a more extensive evaluation to determine whether other developmental lung diseases or worsening cardiac function are contributing to persistent elevations in PVR (Table 1).
Table 2.
Pulmonary hypertension targeted drug therapies.
| Drug | Dose | Mechanism of Action | Adverse Reactions | Indications |
|---|---|---|---|---|
|
| ||||
| Inhaled Nitric Oxide | Inhalation 5–20 ppm | - Activates sGC in vascular smooth muscle cells - Selective Pulmonary Vasodilator |
- Rebound PH with rapid discontinuation - Methemoglobinemia with higher doses |
Hypoxemic respiratory failure with PPHN physiology |
| Sildenafil | Intravenous - Loading Dose: 0.4 mg/kg over 3 h - Maintenance: 0.067 mg/kg/h Enteral - Initial Dose 0.5 mg/kg/dose followed by increasing to 1–2 mg/kg every 6–8 h |
- Selective PDE5 inhibition increases vascular smooth muscle cGMP levels - Pulmonary Vasodilator |
- Hypotension (particularly with loading dose) - Hypoxemia |
PPHN refractory to inhaled nitric oxide therapy and other conventional therapies |
| Milrinone | Intravenous - Initial Infusion Dose: 0.2–0.33 μg/kg per minute - Dose Titration: Increase by 0.33 μg/kg per minute to a maximum of 1 μg/kg per minute |
- PDE3 inhibition increases vascular smooth muscle cAMP levels - Improves cardiac performance through pulmonary vasodilation, systemic afterload reduction, and enhanced lusitropy |
- Hypotension - Thrombocytopenia - Arrhythmias |
Left ventricular dysfunction associated with PPHN |
| Bosentan | Enteral 1 –2 mg/kg every 12 h |
- Dual ETA and ETB receptor inhibitor | - Hepatotoxicity: Monitor LFTs monthly - Edema - Anemia - Teratogenic |
PPHN refractory to iNO and other therapies Chronic PH (CDH, BPD) |
| Prostanoids | Treprostinil (Remodulin) - Initial Infusion Dose: 1.25 ng/kg per minute - Dose titration: Slowly increase to 10 ng/kg per minute *Can be delivered subcutaneously in the same dosing range Epoprostenol (Flolan) - Inhaled: 20–100 ng/kg per minute. Start at 2 ng/kg per minute and increase to 20 ng/kg per minute within 3 h - Continuous Intravenous Infusion: 1–2 ng/kg per minute, incremental increases as tolerated |
- Activates sAC in vascular smooth muscle cells - Pulmonary Vasodilator |
Ventilation–perfusion mismatch may complicate use in the setting of lung disease. Nonselective vasodilator, can cause systemic hypotension Risk of rebound PH with sudden withdrawal Flushing, diarrhea | PPHN refractory to iNO and other therapies |
sCG, soluble guanylate cyclase; PH, pulmonary hypertension; PPHN, persistent pulmonary hypertension of the newborn; PDE5, phosphodiesterase 5; cGMP, cyclic GMP; PDE3, phosphodiesterase 3; cAMP, Cyclic AMP; ET, endothelin; LFT, liver function test; iNO, inhaled nitric oxide; BPD, bronchopulmonary dysplasia; CDH, congenital diaphragmatic hernia; sAC, soluble adenylate cyclase.
Findings from past RCTs highlight that iNO at studied doses has minimal toxicity in late preterm and term infants. In the presence of elevated oxygen concentrations, NO is converted to a cytotoxic small molecule, nitrogen dioxide (NO2). Similarly, methemoglobinemia results from avid binding and metabolism of NO with hemoglobin, but methemoglobin levels are typically low when used at recommended doses.
5.2. Sildenafil
Phosphodiesterase type 5 (PDE5) is a cGMP-specific phosphodiesterase isoenzyme that catalyzes the catabolism of cGMP to its inactive metabolite, 5′-GMP, in vascular smooth muscle, which can augment pulmonary vasoconstriction and limit responsiveness to exogenous iNO therapy [41,42]. Various animal studies have demonstrated that hyperoxia leads to increased PDE5 activity, further highlighting the importance of judicious oxygen therapy use [42–44].
Sildenafil (Revatio), available as intravenous (IV) and enteral forms, is the primary PDE5 inhibitor used for the treatment of PH. Sildenafil is a useful adjuvant therapy in acute PPHN for infants with an inadequate response to iNO or with the lack of iNO availability, especially in resource limited countries. As with iNO therapy, sildenafil should be used cautiously in infants with LV dysfunction, because of a high risk for pulmonary edema, and may be most beneficial in combination with agents targeting cardiac performance or reducing LV afterload. Two trials demonstrated the early use of sildenafil improved oxygenation and outcomes in late preterm and term infants who were not treated with concurrent iNO therapy [45,46]. In a recent multicenter trial, IV sildenafil did not demonstrate a decreased need or duration of iNO therapy, though the treatment effect may have been diluted by inclusion of infants with a lower OI at enrollment [47]. Apart from acute PPHN, enteral sildenafil is a potentially useful agent in cases of chronic pulmonary hypertension such as congenital diaphragmatic hernia (CDH) or BPD-associated PPHN [48,49].
Sildenafil is generally well tolerated when administered at recommended doses with the most common side effect being systemic hypotension. IV dosing consists of an initial loading dose of 0.4 mg/kg over 3 h, followed by a continuous infusion of 0.067 mg/kg/hr [46]. Oral sildenafil therapy most often consists of an initial dose of 0.5 mg/kg, followed by increases to 1–2 mg/kg every 6–8 h [36]. (Table 2).
Sildenafil undergoes near complete first pass hepatic metabolism via hepatic cytochrome P450 (CYP) enzymes, specifically, CYP3A, in adults, with limited renal excretion [50]. While various pharmacologic studies demonstrate appropriate therapeutic levels are reached using studied dosing regimens, significant variability exists in neonatal pharmacokinetics [51–54]. Compared to adults, infants demonstrate higher clearance rates and larger volume of distribution, resulting in variable sildenafil concentrations. Sildenafil dosing should be adjusted clinically as serum levels fluctuate over time because of clearance rates increasing with chronological age and secondary to drug-drug interactions via changes in CYP activity. Particular attention should be paid with co-administration of fluconazole, an inhibitor of CYP3A4 inhibitor, which decreased sildenafil clearance by 47–59% in various models [53–55].
5.3. Milrinone
Inhibition of phosphodiesterase 3 (PDE3), a cAMP specific phosphodiesterase isoenzyme that localizes to cardiac myocytes and vascular smooth muscle cells, offers a unique opportunity in the management of PPHN to improve Qp though optimization of cardiac function by reducing PVR and decreasing LV afterload. Milrinone is a PDE3 inhibitor that enhances intracellular cAMP signaling through inhibition of PDE3, which may lead to improved myocyte contractility, improved lusitropy, and vascular smooth muscle relaxation [56,57]. Unlike iNO, milrinone is not selective for the pulmonary circulation and may impair myocardial perfusion through the reduction of systemic vascular resistance and arterial blood pressure, but these systemic effects may be helpful in PPHN with LV dysfunction. Some neonates with PPHN have ECHO evidence of paradoxical left-to-right atrial level shunting, reflecting the potential contribution of pulmonary venous hypertension from increased left atrial pressure associated with LV hypoplasia or diastolic dysfunction. In this setting, milrinone may serve a unique purpose by improving LV performance secondary to systemic afterload reduction, while augmenting pulmonary vasodilation [58,59]. The addition of milrinone therapy in late preterm and term neonates with PPHN who are poorly responsive to iNO can reduce extrapulmonary shunting, improve oxygenation and augment the response to iNO [58,60].
Initial milrinone infusions range from 0.2 to 0.33 mcg/kg/min with the ability to titrate to 1 mcg/kg/min as tolerated hemodynamically [59,61,62]. (Table 2) Given the long half-life of milrinone, numerous studies have evaluated the use of a loading dose to rapidly obtain therapeutic concentrations perioperatively for congenital cardiac surgery in neonates; however, limited pharmacologic data exists to guide the use of a loading dose in PPHN [63,64]. Milrinone clearance in neonates is decreased compared to older children and significant variability exists between individual infants and across gestational ages [61,65]. We caution the use of a loading dose due to the uncertain and variable clearance rates of milrinone, placing infants at risk for acute and prolonged systemic hypotension, particularly when renal clearance is further impaired such as during therapeutic hypothermia for hypoxic-ischemic encephalopathy or significant renal injury [66].
5.4. Endothelin receptor antagonists
Endothelin (ET-1) is a robust vasoconstrictor and elevated concentrations have been found in neonates with PPHN [67]. In a fetal sheep model of PPHN, chronic in utero ET receptor blockade decreased pulmonary artery pressure, right ventricular hypertrophy, distal muscularization of small pulmonary arteries, and improved the fall in PVR at birth [68–70]. Bosentan is an oral dual ET receptor antagonist approved for children and adults with chronic pulmonary hypertension and has been used as a treatment for infants with severe PH with poor responsiveness to iNO and sildenafil [71]. Its use as an acute pulmonary vasodilator for infants with PPHN continues to evolve. In a pilot study, bosentan improved oxygenation in term neonates compared to placebo in a setting where iNO was unavailable [72]. However, a RCT failed to show that the early addition of bosentan to iNO therapy conferred clinical benefit [73]. Bosentan should only be used after screening for liver disease with serum hepatic transaminases and liver ultrasound, and serum enzyme levels should be monitored during therapy (Table 2).
No pharmacologic studies have examined bosentan in the neonate. Bosentan undergoes primary hepatic metabolism and similar to adults, the half-life in young children is 4–5 h, though this is likely to be variable in neonates. Simultaneous use of sildenafil and bosentan results in decreased sildenafil concentrations and elevated bosentan concentrations [74–76].
5.5. Prostanoid therapy
Prostacyclin (PGI2), is a potent vasodilator that increases intracellular cAMP through activation of adenylate cyclase. IV PGI2 agents are well established in the management of older children and adults with pulmonary arterial hypertension, but until recently, there has been less experience reported in PPHN. Concerns include the risk of systemic hypotension, the potential for worsening ventilation-perfusion mismatch in neonates with parenchymal lung disease, and the need for central venous access. However, increasing case series have shown that the addition of systemic prostanoid therapy may be of benefit in PH refractory to other PH-targeted therapies (Table 2). A recent observational study reported a decreased OI and need for ECMO in iNO-refractory PPHN with the addition of IV epoprostenol [77].
Past studies have suggested that intratracheal or aerosolized PGI2 can improve oxygenation and appears well tolerated in neonates with refractory PPHN [78–80]. However; in the critically ill neonate, the use of inhaled PGI2 has been limited because of problems with inconsistent delivery, airways irritation, and bronchoconstriction. Newer PGE2 analogs and preparations are emerging as possible therapeutic options, especially in severe PPHN or PPHN associated with developmental lung diseases, including IV epoprostenol (Flolan), intermittently inhaled Iloprost (Ventavis), and Treprostinil (Remodulin) administered subcutaneously or IV (Table 2).
5.6. Soluble guanylate cyclase activators
The lack of response to iNO in some infants may result from impaired NO-sGC signaling [15,81]. Exposure to high oxygen tension results in the oxidation of sGC and decreased ability to produce sGC-mediated vasodilation leading to the investigation of direct sGC activators. In fetal lambs with experimental PPHN, cinaciguat, a sGC activator, increased Qp, decreased PVR, and resulted in sustained cGMP production from isolated pulmonary endothelial cells under oxidative stress [82]. Riociguat, an oral sGC activator, approved for use in adults with chronic PH, may evolve as a novel therapy for refractory PPHN in the setting of prolonged hyperoxia exposure [82–84]. Sildenafil must be discontinued with initiation of riociguat because of the risk of severe systemic hypotension.
6. Approach to iNO resistant disease
The use of combined pulmonary vasodilator therapy is shaped by the underlying pathophysiologic process, anticipated clinical course, and evolving interactions of the cardio-pulmonary axis of individual patients. It is imperative to reiterate that while critical to the management of PPHN, successful use of vasodilator therapy is dependent upon optimizing supportive measures including systemic hemodynamics and pulmonary recruitment. Both changes in oxygenation and hemodynamics should be serially assessed to guide the addition, titration, and weaning of pulmonary vasodilators.
Following confirmation of pulmonary hypertensive physiology without significant LV dysfunction, iNO is the preferred initial therapy. An adequate response to iNO is demonstrated as a decrease in OI of >10% or increase in PaO2 of >15%. As noted above, an inadequate response to iNO is often due to decreased alveolar delivery secondary to suboptimal pulmonary recruitment and changes in ventilation including high frequency ventilation should be considered prior to initiation of additional pulmonary vasodilators. Higher concentrations of iNO are unlikely to be beneficial and increase the risk of adverse effects [40]. Reassessment of systemic hemodynamics and serial ECHOs are useful in guiding therapy when oxygenation remains minimally response to iNO.
Limited data exists to support dual vasodilator therapy in PPHN, but careful consideration of augmenting iNO therapy with IV sildenafil, inhaled PGI2, or IV PGI2 are reasonable options for infants whose OI remains >20 in the acute setting without systemic hypotension and who have normal left ventricular function. IV milrinone at a dose of 0.2 mcg/kg/min may be helpful to improve Qp by decreasing pulmonary venous pressures in the setting of LV dysfunction. Fluid resuscitation or inotropic support should be anticipated due to the nonspecific properties of systemically administered vasodilators.
Minimal clinical data exists to support the use of bosentan or soluble guanylate cyclase activators in acute PPHN. Due to the limited data and variable response of individual patients, the inability to wean iNO with the addition of a second therapy or lack of improvement by ECHO, may be an indication for the addition of a third vasodilator agent. Refractory PH should be approached from a multidisciplinary approach involving neonatologists, pulmonologists, and cardiologists with experience in pediatric PH.
For infants who initially stabilized with iNO alone, but develop evidence of rebound PH, or are at risk for chronic PH (cPH), the addition of PO sildenafil may be helpful to facilitate weaning from iNO and invasive mechanical ventilation. There is minimal evidence to guide weaning of pulmonary vasodilators in the setting of combined therapy. With improvement in oxygenation and weaning of inotropic support, weaning of individual therapies should be considered. With near normalization or normalization of cardiac function and PH on serial ECHOs, therapies can be gradually weaned with close clinical monitoring.
7. PPHN in preterm infants with AHRF
The incidence of PPHN has an inverse relationship to the degree of prematurity [3]. Although AHRF and the need for respiratory support after birth is common in premature neonates, the diagnosis of PPHN still requires ECHO confirmation of right-to-left extrapulmonary shunting. Despite RCTs suggesting the safety of iNO use in preterm infants, the use of iNO in preterm infants is not recommended for those with AHRF in the absence of documented PPHN physiology.
Inhaled NO has an evolving and controversial role in the treatment of early PPHN for infants <34 weeks’ GA with variable iNO use ranging from 2% to 9% in extremely preterm infants 22–30 weeks’ GA [85]. A recent observational study demonstrated that iNO therapy is equally effective in improving oxygenation in preterm and term neonates [86]. While iNO has not been shown to decrease BPD in RCTs, none of these trials included ECHO documentation of PPHN physiology [87]. However, multiple case series that studied the effects of iNO in preterm infants with PPHN have shown improved oxygenation, especially in infants with suspected lung hypoplasia from prolonged premature rupture of membranes or oligohydramnios, leading to recommendations for selective use in preterm infants with proven PPHN [88,89].
When used at lower doses (5–10 ppm) in premature neonates, multiple RCTs have not demonstrated an increased risk for severe intraventricular hemorrhage (IVH) or bleeding, with some data showing a reduction in severe IVH [90,91]. As with term infants, caution is needed with rapid decreases in iNO therapy, rebound pulmonary vasoconstriction may worsen pulmonary and systemic hemodynamics.
8. Summary
The use of preclinical animal models of pulmonary hypertension has significantly contributed to our understanding of transitional neonatal physiology and the development of pulmonary hypertension therapies. Despite advances in our ability to care for infants with PPHN, these infants remain at significant risk for morbidity and mortality. While iNO is an effective therapy for many neonates, there are some with an inadequate response, emphasizing the complexity of pulmonary artery and smooth muscle signaling and diversity of disease phenotypes. Thus, ongoing research is needed to better understand adjunctive therapies both in preterm and term neonates.
Practice points
PPHN is a common clinical problem affecting preterm and term neonates
Successful optimization of oxygenation, cardiac performance, and systemic perfusion rely on strategies that include adequate lung recruitment, careful use of oxygenation, selected use of surfactant, and cardiotonic therapies, rather than PH-targeted drug therapy alone
iNO use is a safe, selective and often effective vasodilator therapy for PPHN management but should not be used without confirming the diagnosis of PPHN in the setting of AHRF
Sildenafil is a useful adjunctive therapy in neonates poorly responsive to iNO or in resource-limited settings
Milrinone should be considered in the setting of incomplete responsiveness to iNO and with evidence of severe right or left ventricular dysfunction complicating PPHN, and only in the setting of stable systemic hemodynamics
Partial responsiveness to or the prolonged need for aggressive therapies with evidence of sustained PH should lead to early consideration of genetic studies to diagnose developmental lung diseases
iNO should be considered in premature neonates with clinical evidence of PPHN and historical features suggestive of pulmonary hypoplasia.
Research directions
The use of preclinical studies including whole animal and in vitro models will continue to be beneficial in understanding cellular mechanisms of PPHN and help in identifying additional therapeutic modalities.
Ongoing research is needed to better understand the efficacy and safety of diverse pulmonary vasodilator drugs as well as novel targets for treatments to enhance lung vascular remodeling and growth in both preterm and term neonates.
References
- [1].Dawes GS, Mott JC, Widdicombe JG, Wyatt DG. Changes in the lungs of the newborn lamb. J Physiol 1953;121:141–62. 10.1113/jphysiol.1953.sp004936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Steurer MA, et al. Persistent pulmonary hypertension of the newborn in late preterm and term infants in California. Pediatrics 2017;139:e20161165. 10.1542/peds.2016-1165. [DOI] [PubMed] [Google Scholar]
- [3].Nakanishi H, Suenaga H, Uchiyama A, Kusuda S, Neonatal Research Network J. Persistent pulmonary hypertension of the newborn in extremely preterm infants: a Japanese cohort study. Arch Dis Child Fetal Neonatal Ed 2018;103:F554–61. 10.1136/archdischild-2017-313778. [DOI] [PubMed] [Google Scholar]
- [4].Rudolph AM, Heymann MA. Circulatory changes during growth in the fetal lamb. Circ Res 1970;26:289–99. 10.1161/01.res.26.3.289. [DOI] [PubMed] [Google Scholar]
- [5].Rasanen J, Wood DC, Weiner S, Ludomirski A, Huhta JC. Role of the pulmonary circulation in the distribution of human fetal cardiac output during the second half of pregnancy. Circulation 1996;94:1068–73. 10.1161/01.cir.94.5.1068. [DOI] [PubMed] [Google Scholar]
- [6].Mielke G, Benda N. Cardiac output and central distribution of blood flow in the human fetus. Circulation 2001;103:1662–8. 10.1161/01.cir.103.12.1662. [DOI] [PubMed] [Google Scholar]
- [7].Sutton MS, Groves A, Macneill A, Sharland G, Allan L. Assessment of changes in blood flow through the lungs and foramen ovale in the normal human fetus with gestational age: a prospective Doppler echocardiographic study. Heart 1994;71:232–7. 10.1136/hrt.71.3.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Prsa M, et al. Reference ranges of blood flow in the major vessels of the normal human fetal circulation at term by phase-contrast magnetic resonance imaging. Circulation: Cardiovasc Imag 2014;7:663–70. 10.1161/circimaging.113.001859. [DOI] [PubMed] [Google Scholar]
- [9].Gao Y, Raj JU. Regulation of the pulmonary circulation in the fetus and newborn. Physiol Rev 2010;90:1291–335. 10.1152/physrev.00032.2009. [DOI] [PubMed] [Google Scholar]
- [10].Lakshminrusimha S, Steinhorn RH. Pulmonary vascular biology during neonatal transition. Clin Perinatol 1999;26:601–19. [PubMed] [Google Scholar]
- [11].Abman SH, Accurso FJ. Acute and chronic fetal pulmonary hypertension alter pulmonary vasoreactivity. Chest 1988;93:117S–9S. 10.1378/chest.93.3_supplement.117s-a. [DOI] [PubMed] [Google Scholar]
- [12].Abman SH, Accurso FJ. Acute effects of partial compression of ductus arteriosus on fetal pulmonary circulation. Am J Physiol 1989;257:H626–34. 10.1152/ajpheart.1989.257.2.H626. [DOI] [PubMed] [Google Scholar]
- [13].Accurso FJ, Alpert B, Wilkening RB, Petersen RG, Meschia G. Time-dependent response of fetal pulmonary blood flow to an increase in fetal oxygen tension. Respir Physiol 1986;63:43–52. 10.1016/0034-5687(86)90029-0. [DOI] [PubMed] [Google Scholar]
- [14].Storme L, Rairigh RL, Parker TA, Kinsella JP, Abman SH. In vivo evidence for a myogenic response in the fetal pulmonary circulation. Pediatr Res 1999;45: 425–31. 10.1203/00006450-199903000-00022. [DOI] [PubMed] [Google Scholar]
- [15].Villamor E, et al. Chronic intrauterine pulmonary hypertension impairs endothelial nitric oxide synthase in the ovine fetus. Am J Physiol 1997;272:L1013–20. 10.1152/ajplung.1997.272.5.L1013. [DOI] [PubMed] [Google Scholar]
- [16].Abman SH, Shanley PF, Accurso FJ. Failure of postnatal adaptation of the pulmonary circulation after chronic intrauterine pulmonary hypertension in fetal lambs. J Clin Invest 1989;83:1849–58. 10.1172/jci114091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Abman SH, Chatfield BA, Hall SL, McMurtry IF. Role of endothelium-derived relaxing factor during transition of pulmonary circulation at birth. Am J Physiol 1990;259:H1921–7. 10.1152/ajpheart.1990.259.6.H1921. [DOI] [PubMed] [Google Scholar]
- [18].Dunbar Ivy D, Le Cras TD, Horan MP, Abman SH. Increased lung preproET-1 and decreased ETB-receptor gene expression in fetal pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 1998;274:L535–41. 10.1152/ajplung.1998.274.4.l535. [DOI] [PubMed] [Google Scholar]
- [19].Mous DS, Buscop-Van Kempen MJ, Wijnen RMH, Tibboel D, Rottier RJ. Changes in vasoactive pathways in congenital diaphragmatic hernia associated pulmonary hypertension explain unresponsiveness to pharmacotherapy. Respir Res 2017;18. 10.1186/s12931-017-0670-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kinsella JP, et al. The left ventricle in congenital diaphragmatic hernia: implications for the management of pulmonary hypertension. J Pediatr 2018;197:17–22. 10.1016/j.jpeds.2018.02.040. [DOI] [PubMed] [Google Scholar]
- [21].Lakshminrusimha S, Konduri GG, Steinhorn RH. Considerations in the management of hypoxemic respiratory failure and persistent pulmonary hypertension in term and late preterm neonates. J Perinatol : official journal of the California Perinatal Association 2016;36(Suppl 2):S12–9. 10.1038/jp.2016.44. [DOI] [PubMed] [Google Scholar]
- [22].Kinsella JP, Abman SH. Clinical approach to inhaled nitric oxide therapy in the newborn with hypoxemia. J Pediatr 2000;136:717–26. [PubMed] [Google Scholar]
- [23].Mourani PM, Sontag MK, Younoszai A, Ivy DD, Abman SH. Clinical utility of echocardiography for the diagnosis and management of pulmonary vascular disease in young children with chronic lung disease. Pediatrics 2008;121:317–25. 10.1542/peds.2007-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Skinner JR, et al. Right heart pressure determination by Doppler in infants with tricuspid regurgitation. Arch Dis Child 1993;69:216–20. 10.1136/adc.69.2.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Groh GK, et al. Doppler echocardiography inaccurately estimates right ventricular pressure in children with elevated right heart pressure. J Am Soc Echocardiogr 2014;27:163–71. 10.1016/j.echo.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kinsella JP, McCurnin DC, Clark RH, Lally KP, Null DM Jr. Cardiac performance in ECMO candidates: echocardiographic predictors for ECMO. J Pediatr Surg 1992;27:44–7. 10.1016/0022-3468(92)90102-d. [DOI] [PubMed] [Google Scholar]
- [27].Malowitz JR, et al. Right ventricular echocardiographic indices predict poor outcomes in infants with persistent pulmonary hypertension of the newborn. Eur Heart J Cardiovasc Imaging 2015;16:1224–31. 10.1093/ehjci/jev071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Breinig S, et al. Echocardiographic parameters predictive of poor outcome in persistent pulmonary hypertension of the newborn (PPHN): preliminary results. Pediatr Cardiol 2021;42:1848–53. 10.1007/s00246-021-02677-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Clark RH, Yoder BA, Sell MS. Prospective, randomized comparison of high-frequency oscillation and conventional ventilation in candidates for extracorporeal membrane oxygenation. J Pediatr 1994;124:447–54. 10.1016/s0022-3476(94)70374-4. [DOI] [PubMed] [Google Scholar]
- [30].Kuluz MA, et al. Preliminary observations of the use of high-frequency jet ventilation as rescue therapy in infants with congenital diaphragmatic hernia. J Pediatr Surg 2010;45:698–702. 10.1016/j.jpedsurg.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cornfield DN, Reeve HL, Tolarova S, Weir EK, Archer S. Oxygen causes fetal pulmonary vasodilation through activation of a calcium-dependent potassium channel. Proc Natl Acad Sci U S A 1996;93:8089–94. 10.1073/pnas.93.15.8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cornfield DN. Developmental regulation of oxygen sensing and ion channels in the pulmonary vasculature. Adv Exp Med Biol 2010;661:201–20. 10.1007/978-1-60761-500-2_13. [DOI] [PubMed] [Google Scholar]
- [33].Farrow KN, et al. Hyperoxia increases phosphodiesterase 5 expression and activity in ovine fetal pulmonary artery smooth muscle cells. Circ Res 2008;102:226–33. 10.1161/circresaha.107.161463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Frostell C, Fratacci MD, Wain JC, Jones R, Zapol WM. Inhaled nitric oxide. A selective pulmonary vasodilator reversing hypoxic pulmonary vasoconstriction. Circulation 1991;83:2038–47. 10.1161/01.cir.83.6.2038. [DOI] [PubMed] [Google Scholar]
- [35].Kinsella JP, Parker TA, Ivy DD, Abman SH. Noninvasive delivery of inhaled nitric oxide therapy for late pulmonary hypertension in newborn infants with congenital diaphragmatic hernia. J Pediatr 2003;142:397–401. 10.1067/mpd.2003.140. [DOI] [PubMed] [Google Scholar]
- [36].Abman SH, et al. Pediatric pulmonary hypertension: guidelines from the American heart association and American thoracic society. Circulation 2015;132:2037–99. 10.1161/CIR.0000000000000329. [DOI] [PubMed] [Google Scholar]
- [37].Clark RH, et al. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. Clinical Inhaled Nitric Oxide Research Group. N Engl J Med 2000;342:469–74. 10.1056/NEJM200002173420704. [DOI] [PubMed] [Google Scholar]
- [38].Inhaled nitric oxide in full-term and nearly full-term infants with hypoxic respiratory failure. N Engl J Med 1997;336:597–604. 10.1056/nejm199702273360901. [DOI] [PubMed] [Google Scholar]
- [39].Konduri GG, et al. A randomized trial of early versus standard inhaled nitric oxide therapy in term and near-term newborn infants with hypoxic respiratory failure. Pediatrics 2004;113:559–64. 10.1542/peds.113.3.559. [DOI] [PubMed] [Google Scholar]
- [40].Tworetzky W, et al. Inhaled nitric oxide in neonates with persistent pulmonary hypertension. Lancet 2001;357:118–20. 10.1016/S0140-6736(00)03548-0. [DOI] [PubMed] [Google Scholar]
- [41].Hanson KA, et al. Chronic pulmonary hypertension increases fetal lung cGMP phosphodiesterase activity. Am J Physiol 1998;275:L931–41. [DOI] [PubMed] [Google Scholar]
- [42].Farrow KN, et al. SOD and inhaled nitric oxide normalize phosphodiesterase 5 expression and activity in neonatal lambs with persistent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2010;299:L109–16. 10.1152/ajplung.00309.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Farrow KN, et al. Hyperoxia increases phosphodiesterase 5 expression and activity in ovine fetal pulmonary artery smooth muscle cells. Circ Res 2008;102:226–33. 10.1161/CIRCRESAHA.107.161463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Farrow KN, et al. Brief hyperoxia increases mitochondrial oxidation and increases phosphodiesterase 5 activity in fetal pulmonary artery smooth muscle cells. Antioxidants Redox Signal 2012;17:460–70. 10.1089/ars.2011.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Baquero H, Soliz A, Neira F, Venegas ME, Sola A. Oral sildenafil in infants with persistent pulmonary hypertension of the newborn: a pilot randomized blinded study. Pediatrics 2006;117:1077–83. 10.1542/peds.2005-0523. [DOI] [PubMed] [Google Scholar]
- [46].Steinhorn RH, et al. Intravenous sildenafil in the treatment of neonates with persistent pulmonary hypertension. J Pediatr 2009;155:841–7. 10.1016/j.jpeds.2009.06.012.e841. [DOI] [PubMed] [Google Scholar]
- [47].Pierce CM, et al. Efficacy and safety of IV sildenafil in the treatment of newborn infants with, or at risk of, persistent pulmonary hypertension of the newborn (PPHN): a multicenter, randomized, placebo-controlled trial. J Pediatr 2021;237:154–61. 10.1016/j.jpeds.2021.05.051.e153. [DOI] [PubMed] [Google Scholar]
- [48].Mourani PM, Sontag MK, Ivy DD, Abman SH. Effects of long-term sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J Pediatr 2009;154:379–84. 10.1016/j.jpeds.2008.09.021.e372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Keller RL, et al. Abnormal vascular tone in infants and children with lung hypoplasia: findings from cardiac catheterization and the response to chronic therapy. Pediatr Crit Care Med 2006;7:589–94. 10.1097/01.PCC.0000244401.53189.CB. [DOI] [PubMed] [Google Scholar]
- [50].Hyland R, Roe EG, Jones BC, Smith DA. Identification of the cytochrome P450 enzymes involved in the N-demethylation of sildenafil. Br J Clin Pharmacol 2001;51:239–48. 10.1046/j.1365-2125.2001.00318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ahsman MJ, et al. Sildenafil exposure in neonates with pulmonary hypertension after administration via a nasogastric tube. Arch Dis Child Fetal Neonatal Ed 2010;95:F109–14. 10.1136/adc.2009.168336. [DOI] [PubMed] [Google Scholar]
- [52].Rhee S-J, et al. Population pharmacokinetic analysis of sildenafil in term and preterm infants with pulmonary arterial hypertension. Sci Rep 2022;12. 10.1038/s41598-022-11038-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Cochius-den Otter SCM, et al. Pharmacokinetic modeling of intravenous sildenafil in newborns with congenital diaphragmatic hernia. Eur J Clin Pharmacol 2020;76:219–27. 10.1007/s00228-019-02767-1. [DOI] [PubMed] [Google Scholar]
- [54].Gonzalez D, et al. Population pharmacokinetics of sildenafil in extremely premature infants. Br J Clin Pharmacol 2019;85:2824–37. 10.1111/bcp.14111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mukherjee A, Dombi T, Wittke B, Lalonde R. Population pharmacokinetics of sildenafil in term neonates: evidence of rapid maturation of metabolic clearance in the early postnatal period. Clin Pharmacol Ther 2009;85:56–63. 10.1038/clpt.2008.177. [DOI] [PubMed] [Google Scholar]
- [56].Chen B, et al. Regulation of phosphodiesterase 3 in the pulmonary arteries during the perinatal period in sheep. Pediatr Res 2009;66:682–7. 10.1203/pdr.0b013e3181bce574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Silver PJ, et al. Phosphodiesterase isozyme inhibition, activation of the cAMP system, and positive inotropy mediated by milrinone in isolated Guinea pig cardiac muscle. J Cardiovasc Pharmacol 1989;13:530–40. [PubMed] [Google Scholar]
- [58].James AT, Corcoran JD, McNamara PJ, Franklin O, El-Khuffash AF. The effect of milrinone on right and left ventricular function when used as a rescue therapy for term infants with pulmonary hypertension. Cardiol Young 2016;26:90–9. 10.1017/S1047951114002698. [DOI] [PubMed] [Google Scholar]
- [59].Chang AC, Atz AM, Wernovsky G, Burke RP, Wessel DL. Milrinone: systemic and pulmonary hemodynamic effects in neonates after cardiac surgery. Crit Care Med 1995;23:1907–14. 10.1097/00003246-199511000-00018. [DOI] [PubMed] [Google Scholar]
- [60].McNamara PJ, Laique F, Muang-In S, Whyte HE. Milrinone improves oxygenation in neonates with severe persistent pulmonary hypertension of the newborn. J Crit Care 2006;21:217–22. 10.1016/j.jcrc.2006.01.001. [DOI] [PubMed] [Google Scholar]
- [61].McNamara PJ, Shivananda SP, Sahni M, Freeman D, Taddio A. Pharmacology of milrinone in neonates with persistent pulmonary hypertension of the newborn and suboptimal response to inhaled nitric oxide. Pediatr Crit Care Med 2013;14:74–84. 10.1097/PCC.0b013e31824ea2cd. [DOI] [PubMed] [Google Scholar]
- [62].Paradisis M, et al. Population pharmacokinetics and dosing regimen design of milrinone in preterm infants. Arch Dis Child Fetal Neonatal Ed 2007;92:F204–9. 10.1136/adc.2005.092817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zuppa AF, et al. Population pharmacokinetics of milrinone in neonates with hypoplastic left heart syndrome undergoing stage I reconstruction. Anesth Analg 2006;102:1062–9. 10.1213/01.ane.0000198626.67391.34. [DOI] [PubMed] [Google Scholar]
- [64].Bailey JM, et al. A population pharmacokinetic analysis of milrinone in pediatric patients after cardiac surgery. J Pharmacokinet Pharmacodyn 2004;31:43–59. 10.1023/b:jopa.0000029488.45177.48. [DOI] [PubMed] [Google Scholar]
- [65].Giaccone A, et al. Milrinone pharmacokinetics and pharmacodynamics in neonates with persistent pulmonary hypertension of the newborn. Am J Perinatol 2017;34:749–58. 10.1055/s-0036-1597996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bischoff AR, Habib S, Mcnamara PJ, Giesinger RE. Hemodynamic response to milrinone for refractory hypoxemia during therapeutic hypothermia for neonatal hypoxic ischemic encephalopathy. J Perinatol 2021;41:2345–54. 10.1038/s41372-021-01049-y. [DOI] [PubMed] [Google Scholar]
- [67].Rosenberg AA, et al. Elevated immunoreactive endothelin-1 levels in newborn infants with persistent pulmonary hypertension. J Pediatr 1993;123:109–14. 10.1016/s0022-3476(05)81552-5. [DOI] [PubMed] [Google Scholar]
- [68].Ivy DD, Le Cras TD, Horan MP, Abman SH. Increased lung preproET-1 and decreased ETB-receptor gene expression in fetal pulmonary hypertension. Am J Physiol 1998;274:L535–41. [DOI] [PubMed] [Google Scholar]
- [69].Ivy DD, et al. Prolonged endothelin A receptor blockade attenuates chronic pulmonary hypertension in the ovine fetus. J Clin Invest 1997;99:1179–86. 10.1172/JCI119274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ivy DD, et al. Chronic intrauterine pulmonary hypertension alters endothelin receptor activity in the ovine fetal lung. Pediatr Res 1996;39:435–42. 10.1203/00006450-199603000-00010. [DOI] [PubMed] [Google Scholar]
- [71].Abman SH, et al. Pediatric pulmonary hypertension: guidelines from the American heart association and American thoracic society. Circulation 2015;132:2037–99. 10.1161/CIR.0000000000000329. [DOI] [PubMed] [Google Scholar]
- [72].Mohamed WA, Ismail M. A randomized, double-blind, placebo-controlled, prospective study of bosentan for the treatment of persistent pulmonary hypertension of the newborn. J Perinatol 2012;32:608–13. 10.1038/jp.2011.157. [DOI] [PubMed] [Google Scholar]
- [73].Steinhorn RH, et al. Bosentan as adjunctive therapy for persistent pulmonary hypertension of the newborn: results of the randomized multicenter placebo-controlled exploratory trial. J Pediatr 2016;177:90–6. 10.1016/j.jpeds.2016.06.078.e93. [DOI] [PubMed] [Google Scholar]
- [74].Barst RJ, et al. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther 2003;73:372–82. 10.1016/s0009-9236(03)00005-5. [DOI] [PubMed] [Google Scholar]
- [75].Beghetti M, et al. Pharmacokinetic and clinical profile of a novel formulation of bosentan in children with pulmonary arterial hypertension: the FUTURE-1 study. Br J Clin Pharmacol 2009;68:948–55. 10.1111/j.1365-2125.2009.03532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Burgess G, Hoogkamer H, Collings L, Dingemanse J. Mutual pharmacokinetic interactions between steady-state bosentan and sildenafil. Eur J Clin Pharmacol 2008;64:43–50. 10.1007/s00228-007-0408-z. [DOI] [PubMed] [Google Scholar]
- [77].Ahmad KA, et al. Intravenous epoprostenol improves oxygenation index in patients with persistent pulmonary hypertension of the newborn refractory to nitric oxide. J Perinatol 2018;38:1212–9. 10.1038/s41372-018-0179-7. [DOI] [PubMed] [Google Scholar]
- [78].Bindl L, Fahnenstich H, Peukert U. Aerosolised prostacyclin for pulmonary hypertension in neonates. Arch Dis Child Fetal Neonatal Ed 1994;71:F214–6. 10.1136/fn.71.3.f214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Soditt V, Aring C, Groneck P. Improvement of oxygenation induced by aerosolized prostacyclin in a preterm infant with persistent pulmonary hypertension of the newborn. Intensive Care Med 1997;23:1275–8. 10.1007/s001340050498. [DOI] [PubMed] [Google Scholar]
- [80].Kelly LK, Porta NFM, Goodman DM, Carroll CL, Steinhorn RH. Inhaled prostacyclin for term infants with persistent pulmonary hypertension refractory to inhaled nitric oxide. J Pediatr 2002;141:830–2. 10.1067/mpd.2002.129849. [DOI] [PubMed] [Google Scholar]
- [81].Steinhorn RH, Russell JA, Morin 3rd FC. Disruption of cGMP production in pulmonary arteries isolated from fetal lambs with pulmonary hypertension. Am J Physiol 1995;268:H1483–9. 10.1152/ajpheart.1995.268.4.H1483. [DOI] [PubMed] [Google Scholar]
- [82].Chester M, et al. Cinaciguat, a soluble guanylate cyclase activator, augments cGMP after oxidative stress and causes pulmonary vasodilation in neonatal pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2011;301:L755–64. 10.1152/ajplung.00138.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Chester M, et al. Cinaciguat, a soluble guanylate cyclase activator, causes potent and sustained pulmonary vasodilation in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 2009;297:L318–25. 10.1152/ajplung.00062.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Spreemann T, Bertram H, Happel CM, Kozlik-Feldmann R, Hansmann G. First-in-child use of the oral soluble guanylate cyclase stimulator riociguat in pulmonary arterial hypertension. Pulm Circ 2018;8:1–6. 10.1177/2045893217743123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Handley SC, et al. Inhaled nitric oxide use in preterm infants in California neonatal intensive care units. J Perinatol 2016;36:635–9. 10.1038/jp.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Nelin L, et al. Use of inhaled nitric oxide in preterm vs term/near-term neonates with pulmonary hypertension: results of the PaTTerN registry study. J Perinatol 2022;42:14–8. 10.1038/s41372-021-01252-x. [DOI] [PubMed] [Google Scholar]
- [87].Askie LM, et al. Inhaled nitric oxide in preterm infants: an individual-patient data meta-analysis of randomized trials. Pediatrics 2011;128:729–39. 10.1542/peds.2010-2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Abman SH, et al. Pediatric pulmonary hypertension. Circulation 2015;132:2037–99. 10.1161/cir.0000000000000329. [DOI] [PubMed] [Google Scholar]
- [89].Kinsella JP, et al. Recommendations for the use of inhaled nitric oxide therapy in premature newborns with severe pulmonary hypertension. J Pediatr 2016;170:312–4. 10.1016/j.jpeds.2015.11.050. [DOI] [PubMed] [Google Scholar]
- [90].Kinsella JP, et al. Inhaled nitric oxide in premature neonates with severe hypoxaemic respiratory failure: a randomised controlled trial. Lancet 1999;354:1061–5. 10.1016/s0140-6736(99)03558-8. [DOI] [PubMed] [Google Scholar]
- [91].Kinsella JP, et al. Early inhaled nitric oxide therapy in premature newborns with respiratory failure. N Engl J Med 2006;355:354–64. 10.1056/nejmoa060442. [DOI] [PubMed] [Google Scholar]