

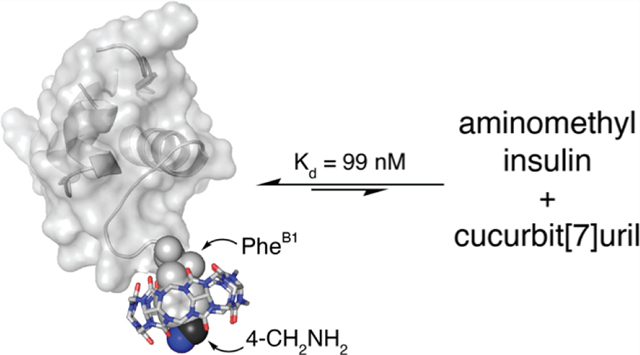
Abstract
Advancements in the molecular recognition of insulin by nonantibody-based means would facilitate the development of methodology for the continuous detection of insulin for the management of diabetes mellitus. Herein, we report a novel insulin derivative that binds to the synthetic receptor cucurbit[7]uril (Q7) at a single site and with high nanomolar affinity. The insulin derivative was prepared by a four-step protein semisynthetic method to present a 4-aminomethyl group on the side chain of the PheB1 position. The resulting aminomethyl insulin binds to Q7 with an equilibrium dissociation constant value of 99 nM in neutral phosphate buffer, as determined by isothermal titration calorimetry. This 6.8-fold enhancement in affinity versus native insulin was gained by an atom-economical modification (−CH2NH2). To the best of our knowledge, this is the highest reported binding affinity for an insulin derivative by a synthetic receptor. This strategy for engineering protein affinity tags induces minimal change to the protein structure while increasing affinity and selectivity for a synthetic receptor.
Graphical Abstract
INTRODUCTION
This study describes the synthesis and characterization of a semisynthetic analogue of human insulin that binds to the synthetic receptor cucurbit[n]uril (Q7) with high nanomolar affinity. The convenient and continuous detection of human insulin in real time remains an elusive goal for the management of diabetes mellitus. Current methods for insulin detection use monoclonal antibodies, which are insufficiently stable for applications involving continuous detection.1 By contrast, synthetic receptors are inexpensive and chemically robust, making them an attractive alternative to antibody and aptamer-based assays. However, synthetic receptors usually lack the binding affinity and selectivity required to detect proteins at physiologically relevant concentrations and in complex mixtures such as serum.2 The current study takes a significant step toward that goal using cucurbit[7]uril (Q7).
Q7 (Figure 1) is a water-soluble, synthetic organic macrocycle that binds to a wide range of small molecules with Kd values ranging from millimolar to attomolar (10−18 M) in aqueous solution.3–5 The hydrophobic cavity and C=O lined portals of Q7 stabilize intermolecular complexes with guests through electrostatic and hydrophobic interactions. Q7 is known to bind selectively to N-terminal aromatic residues via hydrophobic inclusion of the nonpolar side chain within the Q7 cavity and chelation of the N-terminal ammonium group by negatively charged C=O oxygens on the Q7 portal.6 Importantly, Q7 binds human insulin rapidly, reversibly, and site-selectively at the Phe residue located at the N-terminal position of the B-chain (i.e., PheB1), with an equilibrium dissociation constant (Kd) value of 670 nM in neutral phosphate buffer.7 Moreover, Q7 can bind selectively to proteins containing an N-terminal Phe, including human insulin and human growth hormone, within simple and complex mixtures, including human serum.2 These properties combined with the known biocompatibility of Q7 make this receptor attractive for applications involving the continuous detection of proteins.8
Figure 1.

(Top left) Chemical formula of Q7; (bottom left) schematic of the binding of N-terminal Amf to Q7, showing inclusion of the aromatic side chain and interaction of the α-ammonium and side-chain ammonium groups with opposite portals of Q7 (shown in red); (right) model of the Q7·AM-insulin complex based on the crystal structure of the Q7·insulin complex (PDB ID3Q6E).7
Although Q7 binds insulin at submicromolar concentrations, applications under physiological conditions would require a higher binding affinity. Prior work demonstrated that a noncanonical amino acid residue, 4-aminomethylphenylalanine (Amf, Figure 1), can increase the affinity for Q7.9 Substitution of Phe with Amf in the tripeptide sequence Phe-Gly-Gly resulted in a 330-fold increase in binding affinity to Q7 despite having only two additional heavy atoms.9 Increasing the Q7· insulin binding affinity would be necessary for the detection of insulin in the physiological range (30 pM to 2.0 nM),10,11 and we were interested in learning whether the gain in affinity would translate to proteins containing Amf. Importantly, the aminomethylation of insulin is a minimal modification with low propensity to significantly modify its structure or activity. Therefore, we pursued the synthesis and characterization of an insulin analogue that substitutes Phe at position B1 with Amf, with the goal of creating a minimally modified insulin analogue, aminomethyl insulin (AM-insulin, Figure 1) with strengthened affinity for Q7.
RESULTS AND DISCUSSION
Experimental Considerations.
Although insulin is a relatively small protein, it is difficult to modify because it comprises two polypeptide chains and three disulfide bonds. Moreover, the correct folding of the A and B chains of insulin is dependent on the presence of the C peptide, which is excised posttranslationally. These structural features present substantial challenges to the modification of insulin. With this in mind, we considered three different approaches to the synthesis of AM-insulin.
Total synthesis offers the most control over the synthetic process, and the convergent total synthesis of insulin has been reported.12 The availability of Amf suitably protected for solid-phase peptide synthesis makes this route attractive due to the ease with which Phe could be substituted with Amf at the appropriate stage of the synthesis. The total synthesis of insulin, however, is a many-step procedure that necessarily results in low overall yield and a high cost/scale ratio. Therefore, we sought an alternative approach.
Native chemical ligation is a process whereby a peptide fragment containing a C-terminal thioester is coupled to a peptide fragment containing an N-terminal Cys residue via a peptide bond.13 Insulin contains a Cys residue at the B7 position, and thus, we pursued the expression of an insulin mutant lacking the first six residues of the B chain of insulin in Pichia pastoris, a yeast commonly used for the expression of pharmaceutical-grade insulin.14 In principle, the resulting des(B1−B6)-insulin could then be coupled to a hexapeptide variant of insulin B1−B6 containing an N-terminal Amf residue and C-terminal thioester group. We experienced difficulty in the expression of the soluble des(B1−B6)-insulin variant, however, and therefore sought an alternative approach.
The semisynthesis of AM-insulin from native insulin is a particularly attractive strategy due to the availability of the inexpensive, commercially available starting material, pharmaceutical-grade human insulin, and established synthetic protocols for the substitution of the B1 position as reported by Offord and co-workers in the 1970s and 1980s.15 They reported that the Phe residue at B1 can be selectively substituted with other residues in a four-step synthesis with a relatively high yield and scale. Therefore, we pursued this route and were able to synthesize pure AM-insulin on a half-gram scale with a single chromatographic purification.
Semisynthesis and Characterization of AM-Insulin.
We designed a four-step semisynthesis (Figure 2) of the target AM-insulin (1) starting from recombinant human insulin inspired by the approach reported by Offord and co-workers.15 They described the selective protection of the α- and ε-amino groups at GlyA1 and LysB29, respectively, with a tert-butyloxycarbamate (Boc) group via addition of excess tert-butyloxycarbonyl azide (or di-tert-butyloxydicarbonate) to a dimethyl sulfoxide solution of native insulin in the presence of base. The selective protection of these groups leaves the N-terminal α-amino group at PheB1 unprotected. PheB1 can then be removed selectively by Edman degradation. An appropriately protected Amf can then be coupled to the free α-amine at the resulting N-terminal ValB2 residue. The modified protein can then be deprotected using a strong acid. Purification is employed after the final step of the synthesis.
Figure 2.

Four-step semisynthesis of AM-insulin from native insulin. Abbreviations: Msc-ONp, methyl sulfonylethyl p-nitrophenyl carbonate; Et3N, triethylamine; r.t., room temperature; PITC, phenyl isothiocyanate; TFA, trifluoroacetic acid; NEM, N-ethyl morpholine.
Boc-protecting groups are commonly used in peptide synthesis, but their removal requires a strong acid, such as trifluoroacetic acid. Geiger et al. reported an alternative procedure involving the methylsulfonylethylcarbamate (Msc)-protecting group,16 which uses milder conditions (dilute NaOH) and shorter exposure time (∼5 min) than required for Boc deprotection.17 Therefore, we chose the Msc-protecting group for the semisynthesis of AM-insulin.
In the first step, GlyA1 and LysB29 were selectively Msc-protected using methylsulfonylethyl p-nitrophenyl carbonate (Msc-ONp, Scheme S1)18 to produce diMsc-insulin (2) according to a literature procedure (see Supporting Information for experimental details).18 Before optimization, the reaction produced a mixture of monoMsc-insulin, diMsc-insulin, and triMsc-insulin. The production of monoMsc-insulin is problematic because in subsequent steps the additional unprotected free amine would be susceptible to reaction. TriMsc-insulin is not desired, but it is inert to the conditions of Edman degradation and peptide coupling and can therefore be tolerated more easily than monoMsc-insulin. The reaction was therefore optimized to minimize the production of monoMsc-insulin using 6.6 equiv of Msc-ONp in order to yield a mixture containing a 4.6:1.0 ratio of diMsc-insulin to the triMsc-insulin side product and negligible monoMsc-insulin, as determined by reversed phase high-performance liquid chromatography (RP-HPLC) traces at 220 nm. Therefore, the crude mixture contained 82% diMsc-insulin (Figures S3−S5). In the second step, Edman degradation under standard conditions was carried out on the crude mixture of diMsc-insulin and triMsc-insulin to produce a mixture of the target des(PheB1)-diMsc-insulin (3) and residual triMsc-insulin impurity, with 90% conversion of 2 ⇒ 3.
We needed an appropriately protected form of aminomethyl-phenylalanine to couple onto the newly created α-amine at the N-terminal ValB2. In order to align with protection of the α- and ε-amino groups at GlyA1 and LysB2 and to simplify deprotection, we decided to use the Msc group. We produced Msc-Phe(4-CH2NH-MSc)-OSu (6, Figure 2, Scheme S1) in two steps from Msc-ONp in 68% overall yield (Figures S1 and S2). The crude Msc-Phe(4-CH2NH-MSc)-OSu mixture was added to the crude product mixture from the synthesis of des(PheB1)-diMsc-insulin (3) under basic conditions to produce tetraMsc-AM-insulin (4), with 79% conversion of 3 ⇒ 4 (Figures S8 and S9). Deprotection of tetraMsc-AM-insulin (4) was accomplished under basic conditions to produce the target product AM-insulin (5), with 69% conversion of 4 ⇒ 5 (Figures S10 and S11).
In each step, the extent of conversion was estimated by the analysis of the crude reaction mixture using RP-HPLC traces at 220 nm, with product identity confirmed by electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS). The overall reaction yield for the four steps was 40%. The target was purified by reversed-phase preparative HPLC to yield pure AM-insulin (5, Figures S10–S14). The yield and purity of each step of the semisynthesis were sufficiently high that only one chromatographic purification step was necessary, allowing us to conveniently scale up to produce 0.6 g AM-insulin (5). This is a significant advantage over the previously reported semisynthetic methods involving insulin, which involved manual column chromatography, dialysis, or immunoadsorption and which are more time-consuming and technically demanding when purification is performed after each step. Moreover, early semisynthetic methods used thinlayer chromatography to monitor reactions,15 so it is perhaps not surprising that RP-HPLC and ESI-TOF-MS allow us to more carefully analyze reaction mixtures.
AM-insulin was characterized by circular dichroism (CD) spectroscopy (Figure 3). The similarity of the CD spectra of AM-insulin and native insulin implies homology in their global structures and suggests that addition of the aminomethyl group imposes a minimal change to the protein structure.
Figure 3.

Overlay of UV circular dichroism spectra of insulin (open ovals) and AM-insulin (filled ovals) at 25 °C in 10 mM sodium phosphate, 1 mM lysine, 4 mM EDTA, pH 7.0.
Characterization of AM-Insulin Binding.
The binding of Q7 to AM-insulin was confirmed by ESI-TOF-MS (Figure 4). Analysis of an aqueous solution containing an equimolar mixture of Q7 and AM-insulin revealed one major peak corresponding to the sum of the two masses, indicating a 1:1 complex in the gas phase. The thermodynamics of Q7 binding to AM-insulin were characterized by isothermal titration calorimetry (ITC) (Figure 5 and Table 1) in 10 mM sodium phosphate, pH 7.0. These results show that Q7 binds to AM-insulin with a 1:1 Q7:protein complex stoichiometry in a neutral aqueous solution, which corroborates the MS data, and with an observed Kd value of 99 (±6) nM, which is 6.8-fold higher affinity than that reported previously for native insulin (Kd = 670 ± 160 nM).7 This gain in affinity is driven by a 4.4 kcal/mol increase in the exothermicity of binding, which is offset by a 3.3 kcal/mol increase in the unfavorable entropic contribution to the free energy of binding (−TΔS).
Figure 4.

Deconvoluted ESI-TOF mass spectra of aqueous samples of (a) AM-insulin, calc m/z 5837.76, and (b) an equimolar mixture of AM-insulin and Q7, calc m/z 7000.10.
Figure 5.

Representative ITC data of the titration of AM-insulin with Q7 at 300 K in 10 mM sodium phosphate, pH 7.0.
Table 1.
Thermodynamic Data for Q7 Binding
| guest | Kd (nM)d | ΔHd (kcal mol−1) |
−TΔSe (kcal mol−1) |
|---|---|---|---|
| AM-insulin | 99 (±6) | −15.2 (±0.1) | 5.6 (±0.9) |
| insulina | 670 (±160) | −10.8 (±0.5) | 2.3 (±0.4) |
| change due to AMb | 6.8-fold | −4.4 | 3.3 |
| Amf-Gly-Glyc | 0.95 (±0.15) | −14.2 (±0.2) | 1.8 (±0.2) |
| Phe-Gly-Glyc | 310 (±80) | −13.4 (±0.4) | 4.4 (±0.3) |
| change due to AMb | 330-fold | −0.8 | −2.6 |
| Gly-Amf-Glyc | 510 (±20) | −8.2 (±0.1) | −0.5 (±0.1) |
| Gly-Phe-Glyc | 4300 (±100) | −9.8 (±0.1) | 2.4 (±0.1) |
| change due to AMb | 8.4-fold | 1.6 | −2.9 |
| AM-phenylalaninec | 460 (±10) | −4.2 (±0.1) | −4.5 (±0.1) |
| phenylalaninec | 8700 (±1100) | −7.6 (±0.2) | 0.7 (±0.1) |
| change due to AMb | 41-fold | 3.4 | −5.2 |
Data from ref 7.
Change due to aminomethylation was calculated as the quotient of Kd values or the difference in ΔH or −TΔS values.
Data from ref 9.
Mean values measured from at least three ITC experiments at 300 K in 10 mM sodium phosphate, pH 7.0.
Entropic contributions to the free energy of binding were calculated from the Kd and ΔH values, with error values propagated from those of Kd and ΔH.
These results were surprising when compared to the previously reported 330-fold increase in Q7 binding affinity for the tripeptide Amf-Gly-Gly (Kd = 0.95 ± 0.15 nM) versus Phe-Gly-Gly (310 ± 80 nM) (shown in Table 1 for reference).9 In that study, we reasoned that the exceptional gain in affinity was due to the simultaneous and synergistic interaction of Q7 with the N-terminal ammonium group, the side-chain ammonium group, and the neighboring amide bond. By contrast, the Q7 binding affinity gained by adding an aminomethyl group to insulin is more similar to that observed in other prior work, including an 8-fold gain in Q7 binding affinity for the tripeptide Gly-Amf-Gly compared to that for Gly-Phe-Gly and a 19-fold gain in Q7 binding affinity for 4-aminomethyl phenylalanine compared to that for phenylalanine (shown in Table 1 for reference). In these two other contexts, however, the affinity gain was driven by entropy to offset the loss in enthalpy.
In trying to understand the factors responsible for the significant difference in Q7 binding affinity for AM-modified insulin versus AM-modified Phe-Gly-Gly, we first considered the differences in the accessibility of the Q7 binding site. Insulin is a folded protein, but previously reported X-ray crystallographic data showed that the N-terminus of the insulin B-chain unfolds in order to accommodate Q7 binding.7 It is possible that an AMF residue at position B1 behaves significantly differently from the analogous insulin PheB1 residue, perhaps reducing the propensity of the AMF side chain to bind deeply within the Q7 cavity and precluding simultaneous interaction of the α-ammonium and side-chain ammonium groups with opposite portals of Q7. If this were a major factor, then we would expect Q7 to bind more stably to a peptide analogue of the N-terminus of the AM-insulin B-chain than to AM-insulin. To address this hypothesis, the tetrapeptide Amf-Val-Asn-Gln and its canonical parent, Phe-Val-Asn-Gln (i.e., insulin B1–B4), were synthesized, and their binding characteristics with Q7 were measured by 1H NMR, ITC, and ESI-TOF-MS (Table 2 and Figures S15–S18, S23, S24, S27, and S28). For both peptides, the stoichiometry of binding was confirmed by ESI-TOF-MS and ITC to be 1:1 Q7:peptide. Interestingly, the binding affinities of Q7 for Amf-Val-Asn-Gln (Kd = 80 nM) and AM-insulin (Kd = 99 nM) are very similar, and the binding affinities of Q7 for Phe-Val-Asn-Gln (Kd = 250 nM) and native insulin (Kd = 670 nM) are also similar. These results suggest that the tetrapeptides are good models for insulin binding by Q7. The NMR data show large upfield chemical shift perturbation of all aromatic signals for Amf-Val-Asn-Gln, akin to that observed previously for Amf-Gly-Gly and aminomethyl phenylalanine,9 which suggests that the side chain is able to bury deeply within the Q7 cavity. Taken together, these data suggest that binding site accessibility is likely not the main factor responsible for the significant difference in Q7 binding affinity for AM-insulin versus Amf-Gly-Gly.
Table 2.
Thermodynamic Data for Q7 Binding to Tetrapeptides
| guest | Kd (nM)d | ΔHd (kcal mol−1) |
−TΔSe (kcal mol−1) |
|---|---|---|---|
| Amf-Gly-Asn-Gln | 50 (±20) | −16.9 (±0.7) | 6.8 (±0.4) |
| Phe-Gly-Asn-Gln | 140 (±30) | −15.6 (±0.1) | 6.1 (±0.1) |
| change due to AMa | 2.8-fold | −1.3 | 0.7 |
| Amf-Val-Asn-Gln | 80 (±16) | −15.7 (±0.1) | 6.0 (±0.5) |
| Phe-Val-Asn-Gln | 250 (±97) | −14.5 (±0.3) | 5.3 (±0.5) |
| change due to AMa | 3.1-fold | −1.2 | 0.7 |
| AM-insulinb | 99 (±6) | −15.2 (±0.1) | 5.6 (±0.9) |
| insulinc | 670 (±160) | −10.8 (±0.5) | 2.3 (±0.4) |
| change due to AMa | 6.8-fold | −4.4 | 3.3 |
Change due to aminomethylation was calculated as the quotient of Kd values or the difference in ΔH or −TΔS values.
Shown again for reference.
Data from ref 7.
Mean values measured from at least three ITC experiments at 300 K in 10 mM sodium phosphate, pH 7.0.
Entropic contributions to the free energy of binding were calculated from the Kd and ΔH values, with error values propagated from those of Kd and ΔH.
Another factor that could be responsible for the significant difference in Q7 binding affinity for AM-insulin versus AmfGly-Gly is the greater steric hindrance of the isopropyl side chain at the neighboring ValB2 residue in AM-insulin and in Amf-Val-Asn-Gln, compared to Amf-Gly-Gly. We have previously observed an energetic cost due to this steric interaction in the context of Q8-peptide complexes.19 To address this question, we synthesized Amf-Gly-Asn-Gln and Phe-Gly-Asn-Gln as GlyB2 analogues of Amf-Val-Asn-Gln and Phe-Val-Asn-Gln, and we characterized their binding characteristics with Q7 by 1H NMR, ITC, and ESI-TOF-MS (Table 2 and Figures S19−S22, S25, S26, S29, and S30). For both peptides, the stoichiometry of binding was confirmed by ESI-TOF-MS and ITC to be 1:1 Q7:peptide. The binding affinities of Q7 for Amf-Gly-Asn-Gln (Kd = 50 nM) and Amf-Val-AsnGln (Kd = 80 nM) are very similar, and the binding affinities of Q7 for Phe-Gly-Asn-Gln (Kd = 140 nM) and Phe-Val-Asn-Gln (Kd = 250 nM) are also very similar. Again, the side-chain aromatic NMR signals for AMF show a large upfield chemical shift perturbation, indicating binding deep within the cavity of Q7. These results show that Amf-Gly-Asn-Gln binds in the same manner as Amf-Val-Asn-Gln and that steric hindrance at the second position is not the main factor responsible for the significant difference in Q7 binding affinity for AM-insulin versus Amf-Gly-Gly.
The structure–activity studies with tetrapeptides did not yield a conclusive explanation for the limit of binding in the high nanomolar range, but we believe they yielded some useful insights. The remarkable similarity in binding thermodynamics of Q7 for Amf-Val-Asn-Gln and AM-insulin leads us to tentatively rule out the possible effects of folding at the N-terminus of the insulin B-chain, which may include solvent accessibility or stable interaction of AmfB1 with proximal sites on the protein surface. The similarity in the relationships in the thermodynamics of binding of Q7 with Amf-Val-Asn-Gln versus Amf-Gly-Asn-Gln and with Phe-Val-Asn-Gln versus Phe-Gly-Asn-Gln suggests that the thermodynamic effects of the second position are consistent whether the first position is Amf or Phe. The similarity in the relationships in the thermodynamics of binding of Q7 with Amf-Val-Asn-Gln versus Phe-Val-Asn-Gln and with Amf-Gly-Asn-Gln versus Phe-Gly-Asn-Gln suggests that the thermodynamic effects of the first position are consistent whether the second position is Val or Gly. Unfortunately, the binding affinity is still 50-fold weaker for Amf-Gly-Asn-Gln than for Amf-Gly-Gly. The research groups of Brunsveld and Liu have added the entire Phe-Gly-Gly tag to proteins to facilitate numerous applications involving the binding of cucurbit[8]uril (Q8) to proteins,20,21,22,23 and therefore, it may be necessary to add the entire Amf-Gly-Gly tag to the N-terminus of the protein in order to achieve low nanomolar binding affinity for Q7. Although aminomethylation of insulin did not increase the affinity for Q7 by orders of magnitude, the results presented here demonstrate to the best of our knowledge the highest affinity reported for the binding of an insulin derivative with a synthetic receptor.
CONCLUSIONS
This study describes the synthesis of a minimally modified insulin analogue obtained through a four-step semisynthetic approach and characterization of its binding to Q7. This synthetic approach improves upon previous methods via the use of alternative protection strategies and high-resolution analytical techniques, thereby allowing for the optimization of reaction yields and product distributions, and the use of only one purification step. The tetrapeptide analogues of the N-terminus of the B-chain of insulin were demonstrated to be good models of the proteins in terms of Q7 binding. The aminomethylation of insulin increased the affinity of Q7 for insulin by 6.8-fold and resulted in a Kd value of 99 nM. In addition to the gain in affinity, it is important to note that Q7 is highly selective for the AMF residue.9 Therefore, this approach to the engineering of a protein affinity tag imposes a minimal change to the structure of a protein while increasing its affinity and selectivity for a synthetic receptor. The results demonstrated here represent an important step toward the real-time detection of insulin at physiologically relevant concentrations, and more broadly, we anticipate the application of this method to other proteins to improve their isolation and detection.
Supplementary Material
ACKNOWLEDGMENTS
This manuscript is dedicated to the memory of Robin E. Offord, whose groundbreaking research and kind encouragement inspired and advanced this work. The authors gratefully acknowledge financial support from the Welch Foundation (W-0031 and W-1640), the National Institutes of Health (GM126511-01 and GM141708-01), and the National Science Foundation (CHE-1309978, CHE-1726441, and BIO-0718766). We thank Prof. Werner Nau for his kind encouragement.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.2c00501.
Experimental details and mass spectrometry, isothermal titration calorimetry, and nuclear magnetic resonance spectroscopy data for the tetrapeptides (PDF)
Complete contact information is available at:
Contributor Information
Hayden R. Anderson, Department of Chemistry, Trinity University, San Antonio, Texas 78212, United States.
Wei L. Reeves, Department of Chemistry, Trinity University, San Antonio, Texas 78212, United States.
Andrew T. Bockus, Department of Chemistry, Trinity University, San Antonio, Texas 78212, United States
Paolo Suating, Department of Chemistry, Trinity University, San Antonio, Texas 78212, United States.
Amy G. Grice, Department of Chemistry, Trinity University, San Antonio, Texas 78212, United States
Madeleine Gallagher, Department of Chemistry, Trinity University, San Antonio, Texas 78212, United States.
Adam R. Urbach, Department of Chemistry, Trinity University, San Antonio, Texas 78212, United States
REFERENCES
- (1).Luong A-D; Roy I; Malhotra BD; Luong JHT Analytical and Biosensing Platforms for Insulin: A Review. Sens. Actuators Rep 2021, 3, 100028. [Google Scholar]
- (2).Li W; Bockus AT; Vinciguerra L; Isaacs AR; Urbach AR Predictive recognition of native proteins by cucurbit[7]uril in a complex mixture. Chem. Commun 2016, 52, 8537–8540. [DOI] [PubMed] [Google Scholar]
- (3).Barrow SJ; Kasera S; Rowland MJ; del Barrio J; Scherman OA Cucurbituril-Based Molecular Recognition. Chem. Rev 2015, 115, 12320–12406. [DOI] [PubMed] [Google Scholar]
- (4).Lagona L; Mukhopadhyay P; Chakrabarti S; Isaacs L. The Cucurbit[n]uril Family. Angew. Chem., Int. Ed 2005, 44, 4844–4870. [DOI] [PubMed] [Google Scholar]
- (5).Lee JW; Samal S; Selvapalam N; Kim HJ; Kim K. Cucurbituril Homologues and Derivatives: New Opportunities in Supramolecular Chemistry. Acc. Chem. Res 2003, 36, 621–630. [DOI] [PubMed] [Google Scholar]
- (6).Urbach AR; Ramalingam V. Molecular Recognition of Amino Acids, Peptides, and Proteins by Cucurbit[n]uril Receptors. Isr. J. Chem 2011, 51, 664–678. [Google Scholar]
- (7).Chinai JM; Taylor AB; Ryno LM; Hargreaves ND; Morris CA; Hart PJ; Urbach AR Molecular Recognition of Insulin by a Synthetic Receptor. J. Am. Chem. Soc 2011, 133, 8810–8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Das D; Assaf KI; Nau WM Applications of Cucurbiturils in Medicinal Chemistry and Chemical Biology. Front. Chem 2019, 7, 619–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Logsdon LA; Schardon CL; Ramalingam V; Kwee SK; Urbach AR Nanomolar Binding of Peptides Containing Non-canonical Amino Acids by a Synthetic Receptor. J. Am. Chem. Soc 2011, 133, 17087–17092. [DOI] [PubMed] [Google Scholar]
- (10).Melmed S; Polonsky KS; Larsen PR; Kronenberg HM Williams Textbook of Endocrinology, 13th ed.; Elsevier Saunders: Philadelphia, 2016. [Google Scholar]
- (11).Johnson J; Duick D; Chui M; Aldasouqi S. Identifying Prediabetes Using Fasting Insulin Levels. Endocr. Pract 2010, 16, 47–52. [DOI] [PubMed] [Google Scholar]
- (12).Avital-Shmilovici M; Mandal K; Gates ZP; Phillips NB; Weiss MA; Kent SBH Fully Convergent Chemical Synthesis of Ester Insulin: Determination of the High Resolution X-Ray Structure by Racemic Protein Crystallography. J. Am. Chem. Soc 2013, 135, 3173–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dawson PE; Muir TW; Clark-Lewis I; Kent SBH Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- (14).Polez S; Origi D; Zahariev S; Guarnaccia C; Tisminetzky SG; Skoko N; Baralle M. A Simplified and Efficient Process for Insulin Production in Pichia pastoris. PLoS One 2016, 11, No. e0167207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Offord RE Semisynthetic Proteins; John Wiley & Sons Ltd.: New York, 1980. [Google Scholar]
- (16).Geiger R; Obermeier R; Tesser GI Der Methylsulfonyläthyloxycarbonyl-Rest als reversible Aminoschutzgruppe für Insulin. Chem. Ber 1975, 108, 2758–2763. [Google Scholar]
- (17).Ali A; van den Berg RJBHN; Overkleeft HS; Filippov DV; van der Marel G; Codée JDC Methylsulfonylethoxycarbonyl (Msc) and Fluorous Propylsulfonylethoxycarbonyl (Fsc) as Hydroxy-Protecting Groups in Carbohydrate Chemistry. Tetrahedron Lett. 2009, 50, 2185–2188. [Google Scholar]
- (18).Tesser GI; Balvert-Geers IC The Methylsulfonylethoxycarbonyl Group, a New and Versatile Amino Protective Function. Int. J. Pept. Protein Res 1975, 7, 295–305. [DOI] [PubMed] [Google Scholar]
- (19).Hirani Z; Taylor HF; Babcock EF; Bockus AT; Varnado CD Jr.; Bielawski CW; Urbach AR Molecular Recognition of Methionine-Terminated Peptides by Cucurbit[8]uril. J. Am. Chem. Soc 2018, 140, 12263–12269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).van Dun S; Ottmann C; Milroy L-G; Brunsveld L. Supramolecular Chemistry Targeting Proteins. J. Am. Chem. Soc 2017, 40, 13960–13968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang R; Qiao S; Zhao L; Hou C; Li X; Liu Y; Luo Q; Xu J; Li H; Liu J. Dynamic Protein Self-assembly Driven by Host-guest Chemistry and the Folding-unfolding Feature of a Mutually Exclusive Protein. Chem. Commun 2017, 53, 10532–10535. [DOI] [PubMed] [Google Scholar]
- (22).Li X; Bai Y; Huang Z; Si C; Dong Z; Luo Q; Liu J. A Highly Controllable Protein Self-assembly System with Morphological Versatility Induced by Reengineered Host-guest Interactions. Nanoscale 2017, 9, 7991–7997. [DOI] [PubMed] [Google Scholar]
- (23).Li Y; Zhao L; Chen H; Tian R; Li F; Luo Q; Xu J; Hou C; Liu J. Hierarchical Protein Self-assembly into Dynamically Controlled 2D Nanoarrays via Host-guest Chemistry. Chem. Commun 2021, 57, 10620–10623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.