

Abstract
RNA-dependent protein kinase (PKR) is an interferon-induced, RNA-activated enzyme that phosphorylates and inhibits the function of the translation initiation factor eIF-2. PKR is activated in vitro by binding RNA molecules with extensive duplex structure. To further define the nature of the RNA regulation of PKR, we have prepared and characterized site-specifically modified proteins consisting of the PKR 20 kDa RNA-binding domain (RBD). Here we show that the two cysteines found naturally in this domain can be altered by site-directed mutagenesis without loss of RNA binding affinity or the RNA-regulated kinase activity. Introduction of cysteine residues at other sites in the PKR RBD allows for site-specific modification with thiol-selective reagents. PKR RBD mutants reacted selectively with a maleimide to introduce a photoactivatable crosslinking aryl azide at three different positions in the protein. RNA crosslinking efficiency was found to be dependent on the amino acid modified, suggesting differences in access to the RNA from these positions in the protein. One of the amino acid modifications that led to crosslinking of the RNA is located at a residue known to be an autophosphorylation site, suggesting that autophosphorylation at this site could influence the RNA binding properties of PKR. The PKR RBD conjugates described here and other similar reagents prepared via these methods are applicable to future studies of PKR–RNA complexes using techniques such as photocrosslinking, fluorescence resonance energy transfer and affinity cleaving.
INTRODUCTION
RNA-dependent protein kinase (PKR) was originally identified as a protein that causes translation inhibition in reticulocyte lysates treated with RNA (1). It was later recognized that PKR is a key component of the interferon signaling system, a collection of pathways that lead to growth inhibition in a number of different cell lines in response to viral infection (2). PKR is activated by binding to double-stranded RNA (dsRNA) molecules of greater than ~30 bp. Once activated, PKR undergoes autophosphorylation reactions at multiple serine and threonine residues (3,4). Activated PKR also phosphorylates and blocks the function of the translation initiation factor eIF-2, inhibiting continued initiation of protein synthesis (5). Recent discoveries of PKR-activating structures present in human mRNA (6), viral genomic RNA (7) and RNAs selected from a sequence-randomized library that do not have 30 bp duplex regions (8) indicate that RNA structural motifs more complex than simple, perfectly matched duplexes can activate the enzyme. Furthermore, single-stranded viral RNAs have been identified that fold into structures capable of binding and inhibiting PKR (9). Therefore, there are multiple RNA structures that bind PKR and the complexes formed can be functionally distinct.
PKR is 68 kDa with an ~20 kDa N-terminal RNA-binding domain (RBD) and a C-terminal protein kinase domain (Fig. 1). The RBD is composed of two copies of the dsRNA-binding motif (dsRBM I and dsRBM II), a sequence motif found in many dsRNA-binding proteins (10). These proteins bind dsRNA in a largely sequence-independent fashion and are involved in a myriad of biological processes such as RNA editing (11), RNA trafficking (12), RNA processing (13), transcriptional regulation (14) and the interferon antiviral response (15). The solution structure of the 20 kDa RBD of PKR has been determined by NMR spectroscopy (16). Each dsRBM consists of an α-β-β-β-α structure where the two α-helices are packed onto one side of a three-stranded β-sheet. The two dsRBMs are separated by a 22 residue linker of poorly defined structure. Although there have been no reports of high resolution structural data on a PKR–RNA complex, Ryter and Schultz have solved the structure of a single dsRBM from the Xenopus laevis protein Xlrbpa bound to a short RNA duplex (17). The protein bound 16 bp of dsRNA along one face of the helix, crossing the major groove and contacting the two adjacent minor groove sites. Double-stranded RNA specificity arises from numerous contacts between the protein and the 2′-OH groups in the minor groove (18). How dsRBM proteins like PKR bind more complex RNAs and the mechanism of RNA-mediated kinase regulation of PKR are poorly understood at this time.
Figure 1.

Domain structure of PKR. The dsRBMs and the kinase subdomains of PKR are shown as shaded and black boxes, respectively. Amino acid numbering is given above the diagram.
In our ongoing work related to understanding the RNA regulation of PKR, we have prepared site-specifically modified proteins consisting of the PKR 20 kDa RBD. These modified proteins have functional groups introduced at different positions to help define features of PKR–RNA complexes. Here we show that PKR RBD mutants react selectively with a thiol-specific reagent to introduce a photoactivatable crosslinker. RNA crosslinking efficiency was found to be dependent on the position of modification, implying differences in proximity to the RNA for these positions in the protein. Interestingly, one of the amino acid modifications that led to crosslinking of the RNA is located at a serine known to be an autophosphorylation site (4), suggesting that autophosphorylation at this site could influence the RNA binding properties of PKR.
MATERIALS AND METHODS
General
Distilled, deionized water was used for all aqueous reactions and dilutions. Biochemical reagents were obtained from Sigma/Aldrich unless otherwise noted. Restriction enzymes and nucleic acid modifying enzymes were purchased from Stratagene, Boehringer-Mannheim or New England Biolabs. Oligonucleotides were prepared on a Perkin Elmer/ABI Model 392 DNA/RNA synthesizer with β-cyanoethyl phosphoramidites. 5′-Dimethoxytrityl-protected 2′-deoxyadenosine, 2′-deoxyguanosine, 2′-deoxycytidine and thymidine phosphoramidites were purchased from Perkin Elmer/ABI. [γ-32P]ATP (3000 Ci/mmol) was obtained from DuPont NEN. Storage phosphor autoradiography was carried out using imaging plates obtained from Kodak. A Molecular Dynamics STORM 840 was used to obtain all data from phosphor imaging plates. N-(4-Azido-2,3,5,6-tetrafluorobenzyl)-3-maleimidylpropionamide (TFPAM-3) was purchased from Molecular Probes. All NMR and crystal structures were visualized on a Silicon Graphics O2 work station running Insight II (Biosym).
Construction of the PKR RBD single cysteine mutants
Mutants of the PKR RBD (amino acids 1–184) were obtained as glutathione S-transferase (GST) fusion proteins by expression in Escherichia coli using derivatives of the bacterial expression plasmid pGEX-2T (Pharmacia). An expression plasmid for the cysteine-free PKR RBD was constructed using the Altered Sites II in vitro mutagenesis system (Promega). Cysteines 121 and 135 in dsRBM II were each mutated to valine with the following primers: C121V, 5′-ATGCACCCCCGATGCTACCTGTTCATAATTTAC-3′; C135V, 5′-TCTTTCTGTCCCA-TTTTGACTTTATAATGAAATCCTT-3′. Underlined nucleotides represent Cys→Val mutations and all mutations were confirmed by DNA sequencing. PCR mutagenesis was used to introduce single cysteine mutations in the resulting construct. The following primers were used: E13C (5′ mutagenic oligo primer), 5′-GCGTGAggatccGAAGAAATGGCTGGTGATC-TTTCAGCAGGTTTCTTCATGGAGTGTCTTAATACA-3′; S33C (mutagenic oligo), 5′-ATCATGTGGAGGTCCACAA-TTAGGCAGTTCTTG-3′; E29C (mutagenic oligo), 5′-TCCTGAATTAGGCAGACATTGATATTTAAGTAC-3′; PKR RBD template 5′ primer, 5′-GCGTGAggatccGAAGAAATG-GCTGGTGATCTT-3′; PKR RBD template 3′ primer, 5′-TCA-CGCGGTACCTTAAGTAGCAAAAGAACCAGAGGA-3′. Lower case letters represent incorporated restriction sites (E13C BamHI and PKR RBD template 5′ primer BamHI) and underlined nucleotides represent introduced cysteine mutations. All single cysteine mutations were verified by DNA sequencing.
Expression and purification of PKR RBD proteins
BL-21 E.coli (Pharmacia) transformed with the necessary expression plasmid were grown to an OD600 of between 0.4 and 0.6 at 37°C in LB medium supplemented with ampicillin (100 µg/ml). Isopropylthiogalactopyranoside (IPTG) was added to a final concentration of 0.3 mM and the cells were allowed to grow for an additional 4 h at 37°C. The cells were collected by centrifugation and resuspended in phosphate-buffered saline (PBS), 1 mM PMSF and 100 µg/ml lysozyme. These cell suspensions were frozen at –80°C for 12 h followed by thawing at room temperature for 1 h. DNase I was added to a final concentration of 25 µg/ml and the cell lysates were centrifuged at 16 000 g for 0.5 h at 4°C. The clarified lysates were incubated with glutathione–Sepharose (Pharmacia) for 3 h at 4°C. Unbound proteins were removed by successive washes with 20 mM Tris–HCl, pH 8.3, 1 mM DTT, 1 mM PMSF. The affinity matrix with bound GST–RBD was equilibrated by washing with thrombin cleavage buffer (120 mM Tris–HCl, pH 8.6, 150 mM NaCl, 7 mM CaCl2). The GST domain of the fusion protein was removed by cleavage with thrombin. Thrombin (0.12 U/µl) (Pharmacia) was added and the cleavage reaction was allowed to proceed for 24 h at room temperature. The supernatant containing PKR RBD was removed and the remaining matrix was washed with storage buffer (25 mM Tris–HCl, pH 7.0, 10 mM NaCl). Each protein fraction was then loaded into a 10 000 Da molecular weight cut-off Slide-A-Lyzer cassette (Pierce) and dialyzed overnight into storage buffer. The purity of each recombinant PKR RBD protein was estimated to be >95% based on analysis of overloaded Coomassie blue stained protein gels. Protein concentration was determined by the Bio-Rad protein assay with bovine serum albumin (BSA) as the protein standard (Bio-Rad). All protein samples were aliquoted into 20–30 µl fractions and stored at –20°C.
PKR mutagenesis and autophosphorylation assay
A 171 bp region of the PKR RBD expression plasmid encoding the C121V, C135V double mutant was removed by MunI (Stratagene) and NcoI (New England Biolabs) restriction digestion. This fragment of DNA was ligated into MunI–NcoI-digested pET-PKR to generate a FLAG-PKR(C121V, C135V) expression plasmid. FLAG-PKR(C121V, C135V) expression and purification were performed as described (Vuyisich and Beal, submitted for publication). Autophosphorylation assays were performed as follows. Poly(I)·poly(C) RNA at varying concentrations was incubated with FLAG-PKR(C121V, C135V) in 20 mM Tris–HCl (pH 7.6), 10 mM KCl, 10 mM MgCl2 and 10% glycerol for 5 min on ice. [γ-32P]ATP was added to a final concentration of 2 µM and the autophosphorylation reactions were allowed to proceed for 5 min at 30°C. The reactions were placed on ice and subsequently boiled in SDS–PAGE loading buffer. The products were resolved by 10% SDS–PAGE. Labeled proteins were visualized and band intensities quantified using a STORM phosphorimager. Data obtained were plotted as the ratio of PKR autophosphorylation to that measured with no added RNA. Each experiment was carried out in triplicate and plotted values are averages.
PKR RNA ligand preparation
Generation of the RNA transcription template was performed by PCR (8). All required PCR primers and template were synthesized on a Perkin-Elmer DNA synthesizer. T7 RNA polymerase was used to generate the 92 nt RNA. Transcription mixtures were heated to 70°C for 3 min then purified on a 12% denaturing polyacrylamide gel. Bands were visualized via UV shadowing and excised from the gel followed by overnight elution in 0.5 M ammonium acetate, 0.1% SDS, 0.1 mM EDTA. After filtration, the resulting solution was extracted with phenol/chloroform and the RNA was precipitated with ethanol. RNA concentration was determined by measuring the absorbance at 260 nm. For the preparation of 5′-32P-labeled RNA, 100 pmol of transcript was first treated with shrimp alkaline phosphatase (SAP) (Pharmacia) for 2 h at 37°C followed by phenol/chloroform extraction and ethanol precipitation. SAP-treated transcript was purified and isolated from the gel as described above. This dephosphorylated RNA was labeled using polynucleotide kinase (NEB) and [γ-32P]ATP and purified on a 12% denaturing polyacrylamide gel. Labeled RNA was isolated as described above.
Gel mobility shift assays
Dissociation constants for wild-type and E29C PKR RBD proteins binding to the 92 nt RNA ligand were quantified by native gel shift assays (18). In a typical binding experiment, PKR RBD was incubated with 5′-end-labeled RNA in 25 mM HEPES (pH 7.5), 10 mM NaCl, 5% glycerol, 5 mM DTT, 0.1 mM EDTA and 30 µg/ml tRNA for 7 min at room temperature (20 µl final volume). After the incubation was complete, 10 µl of an 80% glycerol solution was added to each binding reaction. PKR RBD/RNA complexes were resolved from free RNA on a 10% (79:1 acrylamide/bisacrylamide) native gel. Data were obtained from the gels using storage phosphor autoradiography and a STORM phosphorimager (Molecular Dynamics). The ratio of RNA bound to unbound RNA was quantified using ImageQuant software (Molecular Dynamics). Dissociation constants for wild type and E29C PKR RBD binding to the 92 nt RNA were calculated using a single-site binding model (equation 1). The fraction of RNA bound (q) as a function of protein concentration was fitted to equation 1, where Θ represents the observed maximum fraction of RNA bound, [RBD] represents protein concentration and Kd is the dissociation constant.
q = Θ {[RBD]/([RBD] + Kd)} 1
Each reported dissociation constant represents the average of three binding experiments ± SD. TFPAM-modified single cysteine mutant PKR RBD binding properties were also analyzed by gel mobility shift. Protein and RNA were incubated at room temperature in the dark for 7 min in 25 mM Tris–HCl (pH 7.0), 10 mM NaCl, 30 µg/ml tRNA. Once the incubation was complete, 10 µl of an 80% glycerol solution was added to the binding reaction. Protein–RNA complexes were resolved on a 10% (79:1 acrylamide/bisacrylamide) native gel.
Conjugation reactions
Conjugation of the PKR RBD cysteine mutants with the thiol-specific photoaffinity reagent TFPAM-3 (Molecular Probes) was carried out by incubating 300 µl of a 100–250 µM PKR RBD solution with 1 mM DTT for 12 h at 4°C. The solution was dialyzed into degassed modification buffer (50 mM HEPES, pH 7.0, 0.5 mM EDTA, 10% glycerol) until the DTT had been removed (3 h). PKR RBD (200 µl solution) was mixed with 3 µl of a 20 mM solution of TFPAM-3 (in DMSO) followed by a 2 h incubation in the dark at room temperature (19). Conjugation reactions were quenched by the removal of excess reagent with immediate dialysis into storage buffer (25 mM Tris–HCl, pH 7.0, 10 mM NaCl) in the dark at 4°C. Derivatized PKR RBD concentration was determined by the Bio-Rad protein assay with BSA as the protein standard. Modified proteins were stored at –20°C. The extent of TFPAM-3 conjugation with the unique cysteine of the PKR RBD was analyzed via electrospray ionization mass spectrometry. Mass spectrometer experiments were performed on a Micromass (Manchester, UK) Quattro II triple quadrupole mass spectrometer. A fraction of the conjugation reaction (75 µl) was supplemented with 75 µl of deionized water and dialyzed into a 50:50 acetonitrile:water, 0.1% formic acid mixture. PKR RBD was infused into the mass spectrometer and molecular weights were calculated by MassLynx software to an error of 0.01%. Myoglobin was used as a reference to calibrate the mass spectrometry which had a calculated/observed mass of 16 951.499/ 16 951 Da. All experimental molecular weights were compared to calculated molecular weights (Compute pI/Mw tool; www.expasy.ch/tools/pi_tool.html ).
RBD/RNA photocrosslinking
RNA complexes with the PKR RBD modified with TFPAM-3 were formed by incubating protein (6 µM final concentration) with 5′-end-labeled RNA at room temperature for 7 min in 25 mM Tris–HCl (pH 7.0), 10 mM NaCl (19,20). Protein–RNA complexes were UV irradiated for 18 min in the tip of a 1.7 ml polypropylene microcentrifuge tube at room temperature using a 312 nm transilluminator (Fisher) at 1.5 cm distance. A time course of UV irradiation demonstrated that an 18 min UV exposure resulted in maximum crosslinking efficiency. Samples were denatured in SDS sample buffer (50 mM Tris–HCl, pH 6.8, 0.25% SDS, 0.1 M DTT, 30% glycerol) by heating to 85°C for 5 min (21). Crosslinked RNA/PKR RBD complexes were separated from free RNA on a 17% (79:1 acrylamide/bisacrylamide) SDS–PAGE gel. Crosslinking efficiencies were calculated by quantifying the band intensities and dividing the amount of RNA crosslinked to total RNA. Proteinase K treatment was performed as follows. Following the 18 min UV irradiation, 6 µl of a 5× proteinase K buffer (250 mM Tris–HCl, pH 7.5, 250 mM NaCl, 25 mM EDTA, 2.5% SDS) was added to a 20 µl sample along with 1 µl of 10% SDS and 3 µl of a 1 mg/ml proteinase K enzyme solution (30 µl final volume). The sample was incubated for 30 min at 37°C. After completion of the incubation, the sample was denatured in SDS sample buffer by heating to 85°C for 5 min and run on a 17% (79:1 acrylamide/bisacrylamide) SDS–PAGE gel.
RESULTS
PKR (C121V, C135V) is an RNA-activated kinase
The RBD of PKR is localized in the first 184 amino acids of the protein and this domain can be expressed independently to high levels in bacteria and easily purified (18). We wished to use site-specific modification of this protein to introduce functional groups that would aid in further defining structural features of the complexes formed with various RNA ligands. Selective modification of cysteine residues with thiol-specific reagents is a useful method to accomplish this goal. However, there are two cysteine residues found naturally in the first 184 amino acids of PKR (C121 and C135). To determine if these two cysteines are required for RNA activation of the kinase, site-directed mutagenesis was used to alter these residues. When the autophosphorylation activity of the bacterially expressed FLAG epitope-tagged PKR (C121V, C135V) double mutant was assayed in vitro, it was found to be stimulated by the addition of dsRNA [poly(I)·poly(C)], as previously seen for the wild-type protein (Fig. 2A) (22). The RNA activation profile is bell shaped, as expected, as high concentrations of activating RNA have been shown to inhibit PKR (Fig. 2B) (22). Therefore, the two cysteines found naturally in the PKR RBD are not essential for RNA-regulated kinase activity.
Figure 2.
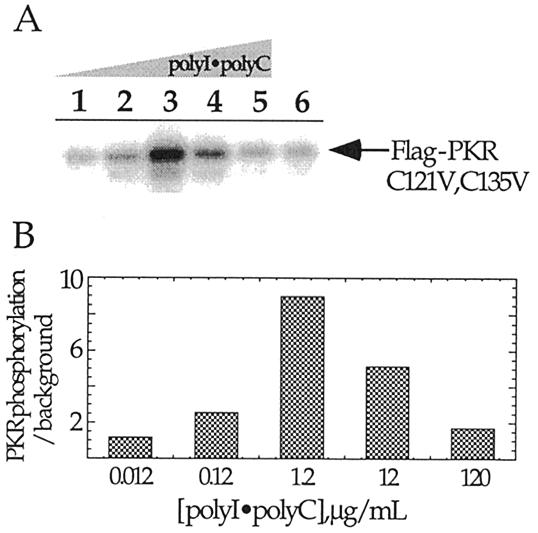
In vitro activation of PKR (C121V, C135V). (A) Shown is a storage phosphor autoradiogram of a 10% SDS–polyacrylamide gel used to resolve PKR (C121V, C135V) autophosphorylated in response to various added concentrations of poly(I)·poly(C). Lanes 1–6, 0.012, 0.12, 1.2, 12, 120 and 0 µg/ml, respectively. (B) Plot of relative extents of PKR (C121V, C135V) autophosphorylation in the presence of different poly(I)·poly(C) concentrations.
A single cysteine mutant of the PKR RBD binds RNA with an affinity similar to wild type
To produce proteins for site-specific modification, mutants of the 20 kDa PKR RBD were generated for overexpression in bacteria. Each of these proteins carried the C121V and C135V mutations. In addition, unique cysteines were reintroduced at various positions (E13C, E29C, S33C and S59C) for further modification. These proteins were overexpressed in bacteria as GST fusion proteins and purified on glutathione–Sepharose. The GST domain was removed from each mutant by specific proteolysis with thrombin.
To assess the RNA binding properties of these proteins, gel mobility shift assays were performed using a 92 nt RNA ligand recently identified as a PKR activator from a sequence-randomized RNA library (Fig. 3A) (8). This RNA was chosen for our initial studies because it binds to the PKR RBD with high affinity to form a single, saturatable complex (8). Proteins that have dsRBMs, including PKR, often form multiple complexes with RNA ligands, presumably due to the existence of multiple binding sites of similar affinity on the RNA. The fact that this RNA forms a single complex under our experimental conditions simplifies data interpretation. Furthermore, like several viral RNA ligands of PKR, this RNA is a single-stranded, highly structured ligand which binds PKR via imperfect duplex regions (8). The binding affinity of the RBD E29C mutant for this RNA (Kd = 0.6 ± 0.1 µM) was found to be nearly identical to that of the wild-type PKR RBD (Kd = 0.5 ± 0.1 µM) (Fig. 3B and C). Similar results were observed for each of the other mutants except S59C, which did not bind the RNA. This observation is consistent with results reported by Williams et al. for an S59A mutant, which also was found to be deficient in RNA binding (23). Furthermore, the crystal structure of the Xlrbpa–RNA complex indicates that the residue of Xlrbpa in a position analogous to S59 of PKR (S162) is in direct contact with the RNA phosphodiester backbone (17).
Figure 3.
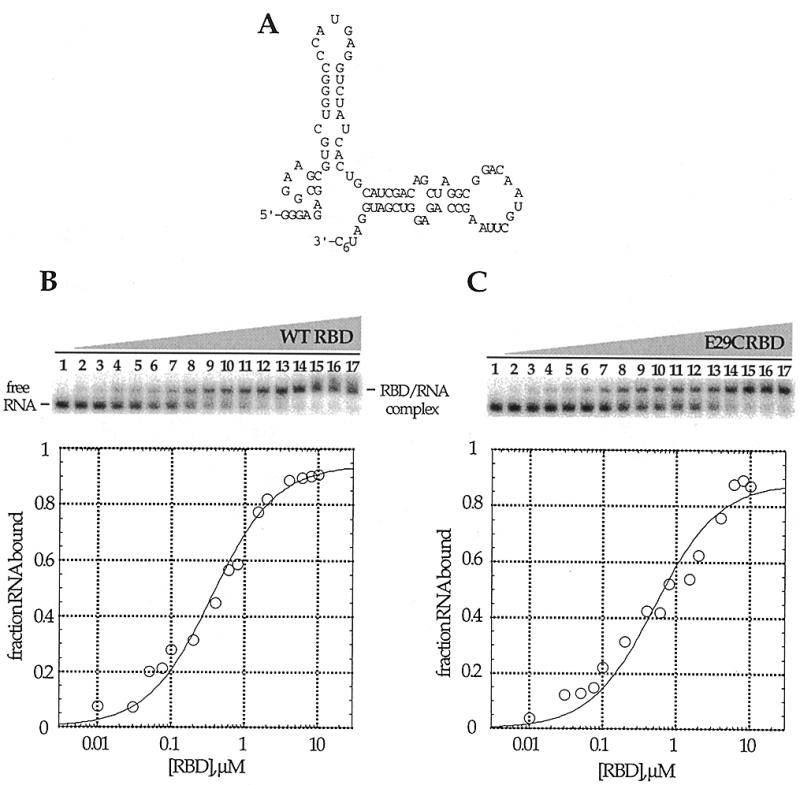
Native gel mobility shifts for the wild-type PKR RBD and PKR RBD (E29C) binding to a 92 nt RNA ligand. (A) Proposed secondary structure of the PKR RNA ligand (8). (B) (Top) Storage phosphor autoradiogram of a representative gel used to separate wild-type PKR RBD-bound from free RNA. Lanes 1–17, 0, 0.01, 0.03, 0.05, 0.075, 0.1, 0.2, 0.4, 0.6, 0.8, 1.0, 1.5, 2.0, 4.0, 6.0, 8.0 and 10.0 µM wild-type PKR RBD added, respectively. (Bottom) Representative plot of fraction RNA bound by wild-type PKR RBD as a function of protein concentration. The data were fitted to the equation: fraction bound = Θ {[RBD]/([RBD] + Kd)} using the least squares method of KaleidaGraph. (C) (Top) Storage phosphor autoradiogram of a representative gel used to separate PKR RBD (E29C)-bound from free RNA. Lanes 1–17, 0, 0.01, 0.03, 0.05, 0.075, 0.1, 0.2, 0.4, 0.6, 0.8, 1.0, 1.5, 2.0, 4.0, 6.0, 8.0 and 10.0 µM PKR RBD (E29C) added, respectively. (Bottom) Representative plot of fraction RNA bound by RBD (E29C) as a function of protein concentration.
Single cysteine mutants of the PKR RBD are stoichiometrically modified with a thiol-selective reagent
Cysteine residues in proteins can react selectively with a variety of modifying reagents such as maleimides, haloacetamides and activated disulfides (see for example 19,21,24). To determine if single cysteine mutants of the PKR RBD could be selectively modified, we carried out modification reactions with the E13C, E29C and S33C mutants with TFPAM-3, which would introduce a photoactivatable crosslinking group at the unique cysteine (Fig. 4) (20). After each reaction, excess modifying reagent was removed by dialysis and the protein product was analyzed by electrospray mass spectrometry. In each case, the mass spectral analysis indicated the presence of singly modified protein with no unmodified or multiply modified species detected (Fig. 5 and Table 1).
Figure 4.
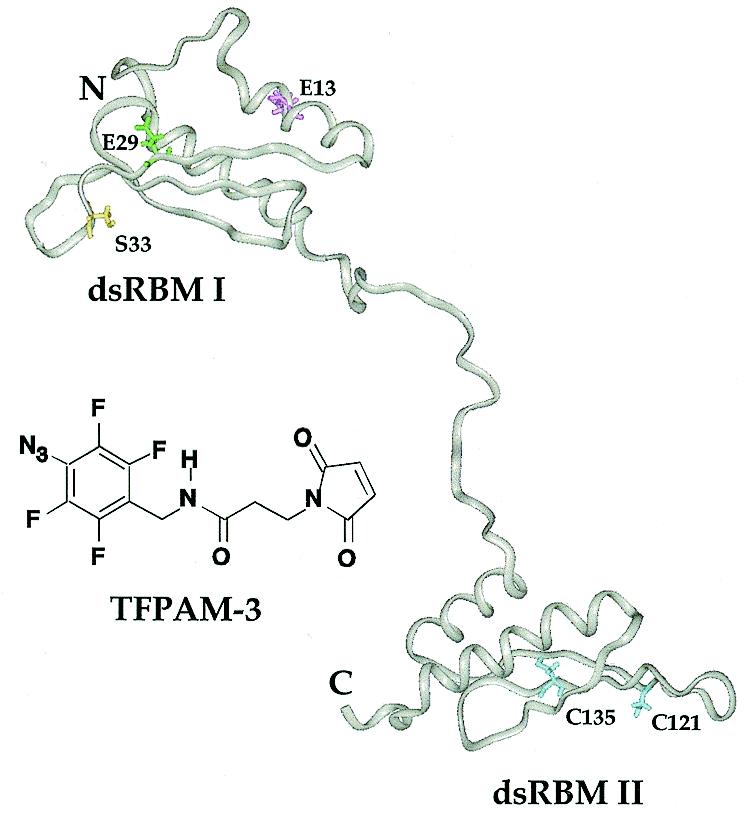
Structure of the PKR RBD (amino acids 1–179) showing the three amino acids selected for mutagenesis along with the structure of the photocrosslinking reagent TFPAM-3 used to modify the different cysteine residues. Using Chem3D (CambridgeSoft), the distance between the reactive maleimide carbon and the putative nitrene was estimated to be ~11 Å. The protein structure shown was generated using Insight II (Biosym) running on a Silicon Graphics O2 workstation and coordinates kindly provided by Qin and co-workers (Lerner Research Institute, The Cleveland Clinic Foundation, Cleveland, OH) (16).
Figure 5.

Electrospray mass spectra of the PKR RBD with a cysteine residue introduced at amino acid position 13. PKR RBD was manually infused on a Micromass Quattro II triple quadrupole mass spectrometer and molecular masses were calculated using MassLynx software. (A) Unmodified PKR RBD (E13C). The minor peak in the spectrum shown occurs at a mass of 21 031. (B) TFPAM-3-modified PKR RBD (E13C).
Table 1. Electrospray mass spectral data for RBD mutants.
| Protein | Molecular weight (Da) | |
|---|---|---|
| Calculateda | Observedb | |
| Wild-type RBDc | 20 664 | 20 664 |
| E13Cd | 20 630 | 20 629 |
| E13C–TFPAM-3 | 21 001 | 21 000 |
| E29C–TFPAM-3 | 21 001 | 21 000 |
| S33C–TFPAM-3 | 21 042 | 21 041 |
aMolecular weights were calculated using the Compute pI/MW tool available at http://www.expasy.ch/tools/pi_tool.html
bSee Materials and Methods for details of the mass spectral analysis.
cWild-type RBD refers to GSEE (human PKR 1–184).
dAll the single cysteine RBD mutants also bear the C121V and C135V mutations.
PKR RBD mutants modified site selectively with a photoactivatable crosslinker bind RNA similarly, but crosslink to different extents
Gel mobility shift assays indicated that each modified protein bound the 92 nt RNA target in a protein concentration-dependent fashion to form a single, saturatable species in a manner similar to the wild-type RBD (Fig. 6A). Upon irradiation of the complexes with UV light, a band of slower mobility was detected in denaturing polyacrylamide gels (Fig. 6B). The maximum yield of the crosslinked species occurred at protein concentrations that saturated the protein–RNA complex observed under non-denaturing conditions (Fig. 6). Formation of this product was dependent on the presence of the TFPAM-3 modification and UV light (Fig. 7A). Also, proteinase K treatment of the irradiation products eliminated the slower moving band, confirming that this was indeed a protein–RNA crosslinked species (Fig. 7A). Importantly, the yield of crosslinked product was dependent on which amino acid in the protein was modified, with the highest efficiency seen with the E29C–TFPAM-3 conjugate (24%) followed by S33C–TFPAM-3 (6%) and E13C–TFPAM-3 (<1%) (Fig. 7B).
Figure 6.
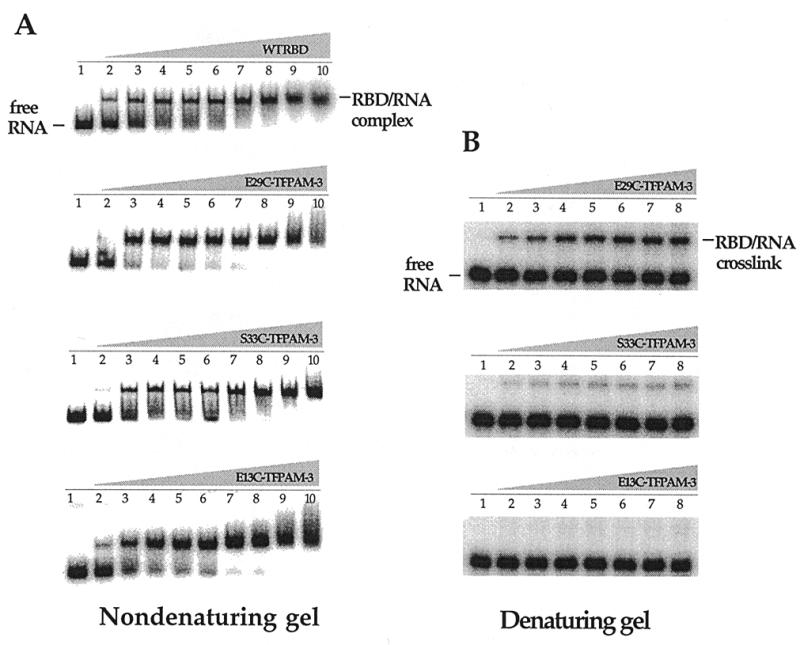
Protein–RNA binding and crosslinking. (A) Native mobility shift experiments of the wild-type PKR RBD and TFPAM-3-modified PKR RBD binding to a 92 nt RNA ligand. Lanes 1–10, 0, 0.005, 0.1, 0.8, 1.0, 2.0, 4.0, 6.0, 12.0 and 20.0 µM wild type or TFPAM-3-modified PKR RBD added, respectively. (B) Denaturing gel experiments resolving the PKR RBD/RNA ligand crosslinked complexes. Lanes 1–8, 0, 0.005, 0.1, 1.0, 2.0, 4.0, 6.0 and 12.0 µM TFPAM-3-modified PKR RBD added, respectively.
Figure 7.
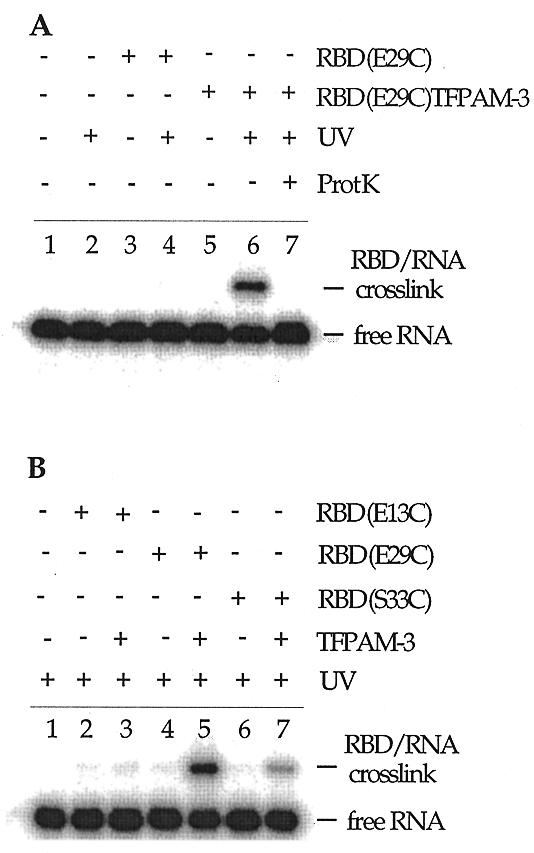
Crosslinking of the PKR RBD to an RNA ligand. See Materials and Methods for photocrosslinking procedures. (A) Characterization of the crosslinked species. Lane 1, no protein added and no irradiation; lane 2, UV irradiation with no protein added; lane 3, 6 µM PKR RBD (E29C) with no UV irradiation; lane 4, PKR RBD (E29C) with 18 min UV irradiation; lane 5, TFPAM-3-modified PKR RBD (E29C) with no UV irradiation; lane 6, TFPAM-3-modified PKR RBD (E29C) with 18 min UV irradiation; lane 7, TFPAM-3-modified PKR RBD (E29C) with 18 min UV irradiation followed by proteinase K treatment. (B) Crosslinking as a function of the amino acid modified. All samples were UV irradiated for 18 min. Lane 1, no protein added; lane 2, PKR RBD (E13C); lane 3, TFPAM-modified PKR RBD (E13C); lane 4, PKR RBD (E29C); lane 5, TFPAM-modified PKR RBD (E29C); lane 6, PKR RBD (S33C); lane 7, TFPAM-modified PKR RBD (S33C).
DISCUSSION
Given the diversity of RNA structures that bind PKR and the functional differences in these complexes (kinase active or inactive), simple experiments that can provide structural information on a variety of protein–RNA complexes are of considerable value to understanding the RNA-mediated regulation of this enzyme. We have used site-selective modification of the PKR RBD to generate reagents that can be used to probe PKR–RNA interactions. This method had not been previously applied to the study of PKR (or any dsRBM-containing protein), although there are many examples in the literature of the power of this approach to the study of other protein–RNA complexes (see for example 21,25,26). In this paper, we use photocrosslinking protein conjugates to further characterize the binding of the PKR RBD to a kinase-activating RNA ligand (8).
To ensure that our single cysteine mutants of the PKR RBD had RNA binding properties reflective of the wild-type protein, we ascertained the functional importance of the two cysteines (C121 and C135) found naturally in this domain of the protein. In the dsRBM family of proteins, each of these positions is typically occupied by a hydrophobic residue (13). Therefore, we chose a hydrophobic amino acid (valine) to replace C121 and C135. In fact, valine is found at positions corresponding to C135 in the majority of dsRBM proteins. We found that replacing these residues with valine in the context of the full-length enzyme generated a protein that retained RNA-regulated enzymatic activity, indicating that these cysteines are not required for RNA binding or activation. Furthermore, when the RNA binding affinity of a single cysteine mutant of the RBD (E29C, C121V, C135V) was quantified and compared to that of the wild-type RBD, little difference was observed.
The experiments reported here have focused on functionalization of dsRBM I of the PKR RBD, as mutagenesis studies carried out by others suggested that dsRBM I plays a dominant role in RNA binding (27). Therefore, cysteine was introduced at four different positions in dsRBM I: E13, E29, S33 and S59. These positions were chosen due to their proximity to conserved basic amino acids that had been implicated in the RNA binding of PKR by site-directed mutagenesis (23). Each of these proteins could be expressed to high levels in E.coli as a GST fusion protein, purified to apparent homogeneity and cleaved with thrombin to remove GST. All the mutants except S59C bound to an RNA ligand of PKR with an affinity similar to that observed for the wild-type RBD sequence. Each mutant could be stoichiometrically modified with thiol-specific modifying reagents and these modifications did not appear to decrease the binding affinity.
Using the E13C, E29C and S33C mutants modified at the unique cysteine with the photocrosslinker TFPAM-3, we observed significant differences in RNA crosslinking efficiencies under conditions where each protein was fully bound to the RNA. Upon irradiation of the tetrafluoroaryl azide of TFPAM-3, a reactive nitrene is formed that can insert into C–H bonds of the RNA relatively non-selectively (28). We interpret the differences in crosslinking efficiency observed here to indicate that the crosslinking reagent is held in different positions in the complex, with the E29C azide having greatest access to the RNA and the E13C azide having the poorest. These relative crosslinking efficiencies can be explained if one considers both the NMR structure of the PKR RBD and the Xlrbpa–RNA crystal structure (16,17). If the PKR dsRBM I binds RNA as does Xlrbpa, we can define its RNA-binding surface as the amino acids in positions analogous to those of Xlrbpa that make direct or water-mediated RNA contacts. Figure 8 shows the NMR structure of the PKR dsRBM I with these putative RNA contact residues highlighted in red, along with the E13, E29 and S33 residues also highlighted in color. In this structure, the E29 side chain is found proximal to and oriented in the same general direction as the putative RNA-binding side chains, whereas the E13 side chain is found on the opposite side of the protein, directed away from the RNA-binding surface. Therefore, the relative crosslinking efficiencies observed here are consistent with the hypothesis that the PKR dsRBM I has an RNA-binding surface similar to that of Xlrbpa.
Figure 8.
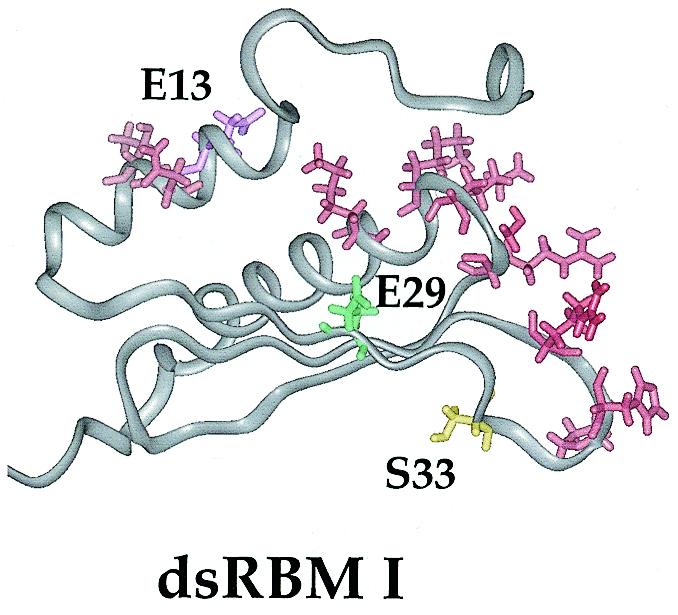
Structure of dsRBM I of the PKR RBD displaying the putative RNA-binding residues (N15, T16, P36, H37, R39, R58, S59, K60, K61 and K64) (16,17). The amino acids selected for modification are also highlighted in color.
Since S33 is in a loop whose structure is not as well defined by NMR as the remainder of the protein, less can be made of its orientation relative to the putative RNA-binding residues shown in Figure 8. However, the fact that crosslinking is observed with the S33C–TFPAM-3 conjugate (6%) suggests that the S33 side chain is indeed near the RNA-binding surface. Interestingly, this serine has been identified as one of 14 autophosphorylation sites found in PKR (4). Given its proximity to the RNA, it is possible that autophosphorylation at S33 affects the RNA binding properties of the enzyme, perhaps by decreasing the overall binding affinity. One could envision this as a mechanism by which an RNA activator is released from the activated enzyme. Additional experiments are required to test this hypothesis.
The results presented here suggest that one could use site-selective modification of the PKR RBD to introduce other diagnostic functional groups to define properties of the protein and protein–RNA complexes. Indeed, a fluorophore as well as a nucleic acid cleaving group have each been introduced at various sites in the PKR RBD using the techniques described here (data not shown). These proteins are currently being used in fluorescent resonance energy transfer experiments to probe factors that control protein oligomerization as well as affinity cleavage experiments to map sites in RNAs proximal to the modified residues. Furthermore, modifications of dsRBM II will lead to a better understanding of its role in RNA binding and its relative location in PKR–RNA complexes.
Acknowledgments
ACKNOWLEDGEMENT
This work was funded by a grant from the National Institutes of Health to P.A.B. (GM-57214).
REFERENCES
- 1.Farrell P.J., Balkow,B., Hunt,T. and Jackson,R.J. (1977) Cell, 11, 187–200. [DOI] [PubMed] [Google Scholar]
- 2.Jaramillo M.L., Abraham,N. and Bell,J.C. (1995) Cancer Invest., 13, 327–338. [DOI] [PubMed] [Google Scholar]
- 3.Taylor D.R., Lee,S.B., Romano,P.R., Marshak,D.R., Hinnebusch,A.G., Esteban,M. and Mathews,M.B. (1996) Mol. Cell. Biol., 16, 6295–6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X., Herring,C.J., Romano,P.R., Szczepanowska,J., Brzeska,H., Hinnebusch,A.G. and Qin,J. (1998) Anal. Chem., 70, 2050–2059. [DOI] [PubMed] [Google Scholar]
- 5.Chong K.L., Feng,L., Schappert,K., Meurs,E., Donahue,T.F., Friesen,J.D., Hovanessian,A.G. and Williams,B.R.G. (1992) EMBO J., 11, 1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis S. and Watson,J.C. (1996) Proc. Natl Acad. Sci. USA, 93, 508–513.8552671 [Google Scholar]
- 7.Circle D.A., Neel,O.D., Robertson,H.D., Clarke,P.A. and Mathews,M.B. (1997) RNA, 3, 438–448. [PMC free article] [PubMed] [Google Scholar]
- 8.Bevilacqua P.C., George,C.S., Samuel,C.E. and Cech,T.R. (1998) Biochemistry, 37, 6303–6316. [DOI] [PubMed] [Google Scholar]
- 9.Kitajewski J., Schneider,R.J., Safer,B., Munemitsu,S.M., Samuel,C.E., Thimmappaya,B. and Shenk,T. (1986) Cell, 45, 195–200. [DOI] [PubMed] [Google Scholar]
- 10.Burd C.G. and Dreyfuss,G. (1994) Science, 265, 615–621. [DOI] [PubMed] [Google Scholar]
- 11.Bass B.L., Nishikura,K., Keller,W., Seeburg,P.H., Emeson,R.B., O‘Connell,M.A., Samuel,C.E. and Herbert,A. (1997) RNA, 3, 947–949. [PMC free article] [PubMed] [Google Scholar]
- 12.Bycroft M., Gruenert,S., Murzin,A.G. and Proctor,M. (1995) EMBO J., 14, 3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kharrat A., Macias,M.J., Gibson,T.J. and Nilges,M. (1995) EMBO J., 14, 3572–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langland J.O., Kao,P.N. and Jacobs,B.L. (1999) Biochemistry, 38, 6361–6368. [DOI] [PubMed] [Google Scholar]
- 15.Sen G.C. and Ransohoff,R.M. (1993) Adv. Virus Res., 42, 57–102. [DOI] [PubMed] [Google Scholar]
- 16.Nanduri S., Carpick,B.W., Yang,Y., Williams,B.R.G. and Qin,J. (1998) EMBO J., 1, 5458–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryter J.M. and Schultz,S.C. (1998) EMBO J., 17, 7505–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bevilacqua P.C. and Cech,T.R. (1996) Biochemistry, 35, 9983–9994. [DOI] [PubMed] [Google Scholar]
- 19.Pietroni P., Young,M.C., Latham,G.J. and Hippel,P.H.v. (1997) J. Biol. Chem., 272, 31666–31676. [DOI] [PubMed] [Google Scholar]
- 20.Aggeler R., Chicas-Cruz,K., Cai,S.-X., Keana,J.F.W. and Capaldi,R.A. (1992) Biochemistry, 31, 2956–2961. [DOI] [PubMed] [Google Scholar]
- 21.Niranjanakumari S., Stams,T., Crary,S.M., Christianson,D.W. and Fierke,C.A. (1998) Proc. Natl Acad. Sci. USA, 95, 15212–15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galabru J. and Hovanessian,A. (1987) J. Biol. Chem., 262, 15538–15544. [PubMed] [Google Scholar]
- 23.McMillan N.A.J., Carpick,B.W., Hollis,B., Toone,W.M., Zamanian-Daryoush,M. and Williams,B.R.G. (1995) J. Biol. Chem., 270, 2601–2606. [DOI] [PubMed] [Google Scholar]
- 24.Platis I.E., Ermacora,M.R. and Fox,R.O. (1993) Biochemistry, 32, 12761–12767. [DOI] [PubMed] [Google Scholar]
- 25.Culver G.M. and Noller,H.F. (1998) RNA, 4, 1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beck D.L., Stump,W.T. and Hall,K.B. (1998) RNA, 4, 331–339. [PMC free article] [PubMed] [Google Scholar]
- 27.Green S.R., Manche,L. and Mathews,M.B. (1995) Mol. Cell. Biol., 15, 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keana J.F.W. and Cai,S.X. (1990) J. Org. Chem., 55, 3640–3647. [Google Scholar]