

Abstract
Chimeric antigen receptor (CAR) T-cell therapy has shown promise in treating hematological cancers, but resistance is common, and efficacy is limited in solid tumors. We found that CAR T-cells autonomously propagate epigenetically-programmed type I interferon signaling through chronic stimulation, which hampers antitumor function. EGR2 transcriptional regulator knockout not only blocks this type I interferon-mediated inhibitory program, but also independently expands early memory CAR T-cells with improved efficacy against liquid and solid tumors. The protective effect of EGR2 deletion in CAR T-cells against chronic antigen-induced exhaustion can be overridden by interferon-β exposure, suggesting that EGR2 ablation suppresses dysfunction by inhibiting type I interferon signaling. Finally, a refined EGR2 gene signature is a biomarker for type I interferon-associated CAR T-cell failure and shorter patient survival. These findings connect prolonged CAR T-cell activation with deleterious immunoinflammatory signaling and point to an EGR2-type I interferon axis as a therapeutically amenable biologic system.
Keywords: CAR T-cell therapy, type I interferon, EGR2, memory differentiation, dysfunction, exhaustion, resistance, solid tumors, biomarker
Introduction
The recent success of immunotherapy illustrated by durable clinical responses following treatment with chimeric antigen receptor (CAR) T-cells in hematologic malignancies highlights the ability of adaptive immunity to eliminate cancer in an antigen-specific manner. Despite high rates of response in certain indications, resistance is common. Although these therapies are beneficial to some patients, most subjects who initially respond will eventually relapse, leaving a very large unmet need. Further, the efficacy of CAR T-cells in solid tumors has been limited to date for reasons that are multifactorial, including T-cell-intrinsic fitness (1,2). Thus, it is essential to enhance currently available CAR T-cell products and extend this transformational approach to treat more cancer types.
Optimal CAR T-cell therapy depends upon rigorous clonal expansion following engagement of the synthetic antigen receptor (2,3). In addition to activation and costimulatory signals, induction of an effective antitumor CAR T-cell response requires cytokine signaling (‘Signal 3’) (4). This is essential for clonal proliferation, elicitation of effector functions as well as memory formation and often tracks with favorable immune response parameters (5). A prominent example is type I interferon (IFN) cascade induction, which potentiates T-cell activation and robust downstream antitumor activity (6). In contrast, persistent type I IFN elaboration within the tumor immune microenvironment (TIME) has important pro-survival advantages for cancer cells and orchestrates T-cell immunosuppression (7,8). These opposing effects establish a regulatory relationship to precisely control the magnitude of both adaptive and innate immune antitumor immunity. However, the underlying mechanisms for T-cell autonomous mobilization of the type I IFN pathway and the nature of its paradoxical immunopathologic effects in the setting of CAR T-cell therapy remain poorly defined.
We demonstrate that chronic CAR stimulation, which recapitulates features of exhaustion, results in autonomously propagated type I IFN signaling. While early induction of type I IFN signaling may promote optimal CAR T-cell priming (6), continuous tumor antigen exposure or antigen-independent tonic CAR triggering coordinates an EGR2-dependent epigenomic resistance program in CAR T-cells, characterized by expression of an aberrant downstream type I IFN-stimulated gene regulatory pathway. Knocking out EGR2 not only interferes with this type I IFN-mediated inhibitory network, but also independently enhances early memory differentiation of tumor-targeted T-cells. Clinically, the presence of EGR2-mediated type I interferon-associated dysfunction, as identified by a refined transcriptomic signature, predicts CAR T-cell therapy success and patient survival. Accordingly, we also show that deleting EGR2 in exhausted CLL patient T-cells improves the in vivo potency of CD19-directed CAR T-cells. Thus, our results identify a unique T-cell-intrinsic immunoinflammatory resistance mechanism and additionally reveal EGR2 as a negative regulator of human memory T-cell differentiation in cancer.
Results
Type I IFN pathway upregulation is a feature of antigen-driven CAR T-cell dysfunction.
Prolonged CAR activation is known to drive CAR T-cell dysfunction (9–11), but the molecular underpinnings of T-cell-intrinsic resistance in the setting of chronic antigen exposure have not been fully characterized. We therefore deployed an in vitro ‘stress test’ model of CAR T-cell dysfunction driven by continuous exposure to cognate antigen (Figure 1A) that faithfully recapitulates defined features of T-cell exhaustion, including transcriptional profiles overlapping with those of hypofunctional tumor-infiltrating lymphocytes in patients (12,13). During repetitive antigen stimulation, CAR T-cells gradually lose proliferative potential, which is accompanied by memory T-cell attrition (Figure 1B–D). Further, chronically stimulated CAR T-cells exhibit impaired effector cytokine production and increased proportions of CD8+ T-cells co-expressing TIM3 and LAG3 inhibitory receptors (Figure 1E–G), which indicates progressive dysfunction. In line with these observations, chronic CAR activation elevates expression of a battery of T-cell suppressive genes and associated pathway signatures previously observed in exhausted antigen-specific CD4+ and CD8+ T-cell subsets as well as CAR T-cells (Figure 1H and 1I) (11). Notably, the type I IFN pathway is one of the most significantly upregulated gene signatures in dysfunctional CAR T-cells in this model (Figure 1J and 1K).
Figure 1. Chronically-stimulated CAR T-cells exhibit type I IFN pathway upregulation.
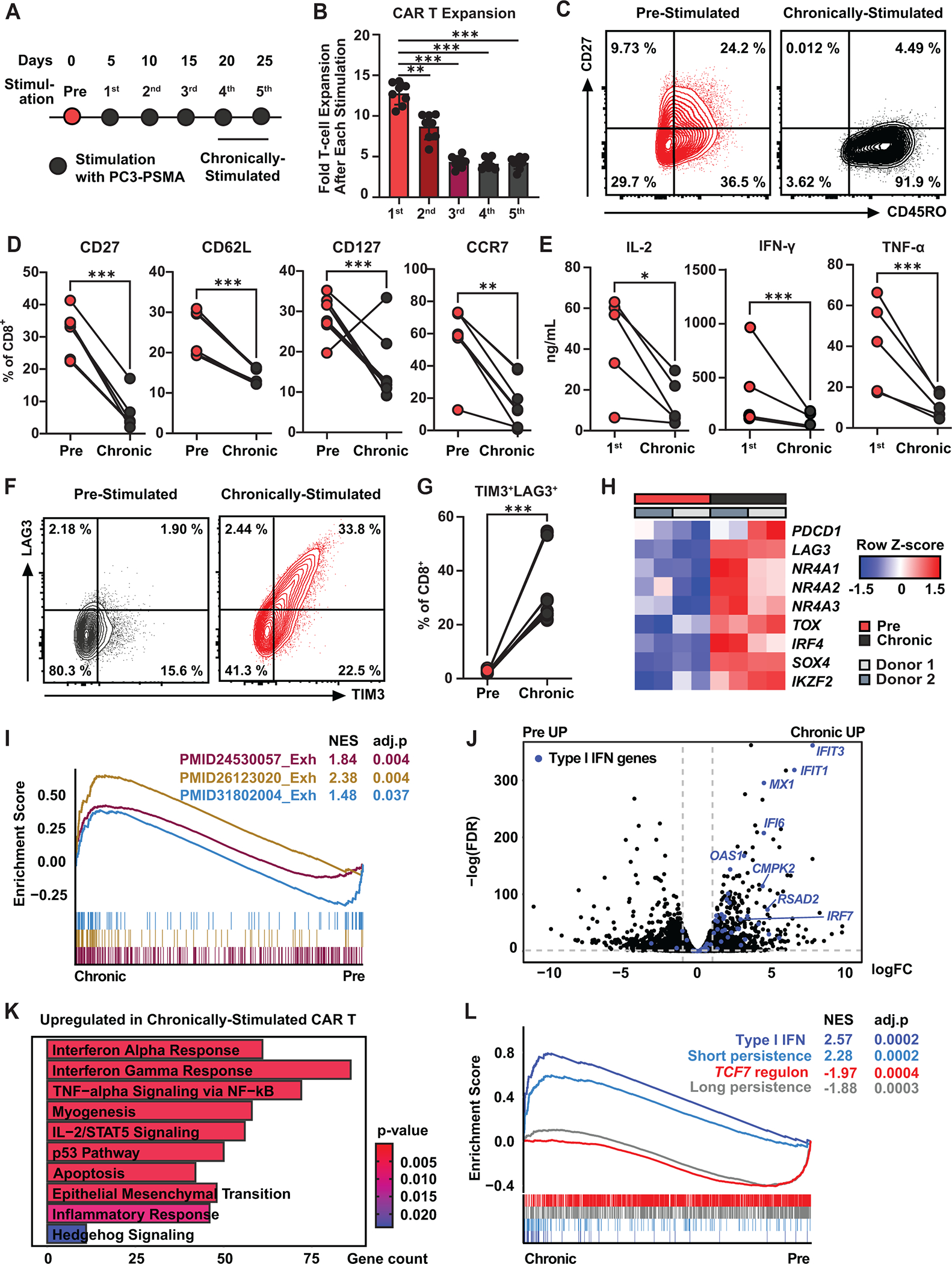
A, Schematic representation of the in vitro “stress test” used to induce CAR T-cell dysfunction. Briefly, PSMA CAR T-cells were challenged with PC3 prostate tumor cells expressing PSMA every five days at an effector to target (E:T) ratio of 3:1. B, Magnitude of CAR T-cell expansion after each antigen stimulation. Data indicate mean ± S.D. from n = 8 individual healthy donors (one-way ANOVA). C, Representative flow cytometry plots showing frequencies of CD8+ CAR T-cells expressing CD27 and CD45RO. D, Proportions of pre- and chronically stimulated CAR T-cells expressing early memory markers (CD27, CD127, and CCR7: n = 8 using four different healthy donors, CD62L: n = 6 using two healthy donors; ratio paired t-test). E, Cytokine production after the first and chronic antigen stimulation time points (n = 5 using four healthy donors; ratio paired t-test). F, Representative flow cytometry plots showing percentages of CAR T-cells expressing TIM3 and LAG3 inhibitory receptors. G, Frequency of TIM3+LAG3+ CD8 CAR T-cells. (H) Heatmap showing expression levels of exhaustion-related genes. I, Gene set enrichment analysis (GSEA) of exhaustion pathways. J, Volcano plot showing differentially expressed genes between pre- and chronically stimulated CAR T-cells. Type I IFN genes are highlighted in blue. K, Top pathways upregulated in chronically stimulated CAR T-cells. L, GSEA analysis using gene sets associated with the type I IFN pathway, TCF7 regulon, and CAR T-cell persistence. *P < 0.05, *P < 0.01, ***P < 0.001, ns: not significant.
In pediatric acute lymphoblastic leukemia (ALL) patients at the time of T-cell collection, enrichment of an IRF7-regulated type I IFN pathway and the TCF7 regulon (governs the formation and maintenance of memory T-cells) predict poor and long-term CAR T-cell persistence, respectively (14). Failure in treating leukemia is commonly associated with a lack of CAR T-cell persistence, while clinical response correlates with robust proliferation and persistence. Accordingly, during the in vitro “stress test,” chronically stimulated CAR T-cells also downregulate the TCF7 regulon and upregulate type I IFN pathway gene signatures (Figure 1L) (14). These collective results indicate that type I IFN pathway upregulation is a defining characteristic of CAR T-cell dysfunction driven by chronic tumor antigen challenge.
Type I IFN and EGR2 pathways are associated with CAR T-cell-intrinsic resistance.
Next, we sought to identify transcription factors enriched in dysfunctional CAR T-cells by examining expression levels of genes encoding regulators associated with exhaustion and anergy (e.g., NR4A, TOX, IKZF, EGR, DGK families, ID3, SOX4, IRF4, BATF, FLI1, MAF, ZBED2, CBLB, PRDM1). We found that early growth response 2 (EGR2), which was previously reported to regulate T-cell anergy and the stability of TCF1+ progenitor cells (15,16), is the most differentially upregulated transcription factor gene in chronically stimulated CAR T-cells (Figure 2A and 2B). Together with EGR2, chronic antigen exposure increases the expression of genes that are co-regulated with EGR2 such as NR4A1, NR4A2, NR4A3, BHLHE40, EGR3, CCL1, CD80, SLAMF8, IRF5, CCR1, CCR5, CCL3 (Figure 2C, Table S1). Assay for transposase-accessible chromatin (ATAC) sequencing analysis reveals that chronic CAR activation increases chromatin accessibility at the EGR2 locus (Figure 2D). Moreover, EGR2 binding motifs are enriched in dysfunctional CAR T-cells and observed in 4.2% of differentially accessible peaks between baseline and chronically stimulated CAR T-cells (Figure 2E). Our analysis revealed overlaps between regions of differential peaks in ATAC-seq and EGR2 binding motifs at T-cell exhaustion genes (PDCD1, CD38, SOX4), type I IFN genes (MX2, PTMS, SPATS2L) and genes involved in memory T-cell differentiation (CD28, KLF2, CXCR4) (Figure 2F–2G, S1A–S1C). These overlaps were found in regions near the transcription start site of a subset of exhaustion-associated and type I IFN genes, suggesting that EGR2 may play a role in the transcriptional and epigenetic regulation of genes involved in T-cell dysfunction and type I IFN signaling during chronic CAR T-cell stimulation. However, further studies are needed to fully understand the specific regulatory functions of these regions. In support of this hypothesis, upregulation of the EGR2 gene signature precedes that of the type I IFN cascade during repetitive tumor challenge, and both pathways are highly expressed following several rounds of antigen exposure (Figure 2H).
Figure 2. Type I interferon and EGR2 pathways are associated with resistance to CAR T-cells.

A, Transcription factor-encoding genes related to T-cell dysfunction differentially expressed between pre- and chronically-stimulated PSMA CAR T-cells are highlighted in red. B, Expression of EGR2 in pre- and chronically-stimulated CAR T-cells. C, Enrichment of an EGR2 gene signature (EGR2 ARCHS4 co-expression gene set) in chronically-activated CAR T-cells. D, ATACseq tracks of differential chromatin accessibility within a region of the EGR2 gene region in unstimulated versus chronically stimulated CAR T-cells. The differentially accessible region is boxed. E, An EGR2 binding motif is observed in 4.2% of differentially accessible peaks. F, ATACseq tracks of PDCD1 and G, MX2. Differentially accessible peaks colocalized with EGR2 binding motif are highlighted in light blue (F and G). H, EGR2 and type I IFN signature scores during serial antigen restimulation of PSMA CAR T-cells. I, EGR2 and type I IFN gene signature scores measured during expansion of naïve CD8+ T-cells expressing a GD2 CAR with high level of tonic signaling compared to a non-tonically signaling CD19 CAR. CAR T-cells were generated using T-cells from a healthy donor. (GSE136891). J, Expression levels of EGR2 and an EGR2 gene signature in clinically favorable (naïve/stem cell-like) and unfavorable (effector memory/effector) baseline/apheresis T-cell populations in ALL. K, Top transcription factor (TF) co-expression signatures overexpressed in CD19 CAR T-cell products of non-responding CLL patients. L, EGR2 and type I IFN gene signature scores measured in stimulated CD19 CAR T-cell products from CLL patients. (CR: complete response, PRTD: very good partial response, PR: conventional partial response, NR: no response) *P < 0.05, *P < 0.01, ***P < 0.001, ns: not significant. M, Type I IFN and EGR2 module scores in PSMA CAR T-cells from non-lymphodepleted metastatic castration resistant prostate cancer patients in association with in vivo CAR T-cell expansion and PSA response.
We subsequently examined whether EGR2- and type I IFN-related pathways are associated with T-cell dysfunction mediated by antigen-independent tonic CAR signaling, which leads to exhaustion and impaired antitumor efficacy (10,11). Although initial robust activation may contribute to the expression of exhaustion-related genes after seven days in culture, T-cells engineered with a tonically signaling GD2 CAR exhibit higher levels of sustained exhaustion gene expression compared to CD19 CAR T-cells at the end of expansion (Figure S1D). These cells also have higher levels of EGR2 and type I IFN gene expression scores compared to T-cells engineered with a non-tonically signaling CD19 CAR, as shown in Figure 2I (GSE136891; (11)). Similarly, central memory GD2 CAR T-cells show increased EGR2 signature expression and gradual upregulation of type I IFN genes in the setting of tonic CAR triggering (Figure S1E). EGR2 and type I IFN gene signature upregulation in this tumor-free system suggests that CAR T-cell autonomous pathway propagation is mediated by CAR signaling, even in the absence of cognate antigen.
We aimed to corroborate that aberrant EGR2-type I IFN pathway upregulation is a feature of failed CAR T-cell therapy. In the pediatric ALL study described above, high frequencies of leukapheresed naïve and early memory T-cells forecasted long-term CAR T-cell persistence, while enrichment of effector memory and effector cells correlated with poor CAR T-cell persistence and eventual relapse (14). Both expression of the EGR2 gene and an EGR2 pathway score are highly enriched in clinically unfavorable baseline effector T-cell subsets, whereas optimal naïve and stem cell-like memory T-cells exhibit low levels of EGR2 expression (Figure 2J). To strength these findings, we then compared transcriptional profiles of CAR T-cell products from responder and non-responder chronic lymphocytic leukemia (CLL) patients, where the majority of subjects exhibited primary CD19 CAR T-cell-intrinsic resistance to therapy (1). The transcription factor signature of EGR2 is significantly elevated in CAR T-cells from non-responders compared to those from responders, while the signatures of TCF7 and KLF2 are upregulated in CAR T-cell products from complete responders (Figure 2K, S1F). Furthermore, EGR2 and type I IFN gene signature scores are positively correlated in unstimulated and stimulated CLL CAR T-cells, whereas products from patients with favorable clinical responses show a low EGR2 signature score (Figure 2L and S1G).
We next investigated whether downregulation of EGR2-type I IFN pathway expression is a correlate of CAR T-cell potency in a solid tumor indication. For this analysis, we examined pre-treatment prostate-specific membrane antigen (PSMA)-directed CAR T-cells from patients with metastatic castration resistant prostate cancer (mCRPC) (17). We separated the subjects into two groups based on lymphodepletion conditioning prior to cell product infusion. Patients 2, 5, and 7 were treated without lymphodepletion and Patient 7 received a higher dose of CAR-T cells compared to Patients 2 and 5. Patient 2, who had the lowest levels of type I IFN and EGR2 signature expression in the CAR T-cell product, showed better expansion and PSA response compared to Patients 5 and 7 (Figure 2M). Similarly, in a lymphodepleted cohort, Patient 9 with reduced type I signature score showed enhanced in vivo CAR T cell potency (Figure S1H). Patients 2 and 5 received the lowest dose of CAR T-cells in this trial. Notably, pre-infusion CAR T-cells from Patient 2, with significantly reduced type I IFN and EGR2 scores, exhibited superior in vivo cell product expansion and PSA response compared to the infusion material of Patient 5, regardless of the conditioning and cell dose. These results suggest that EGR2-type I IFN pathway upregulation is associated with CAR T-cell dysfunction and resistance to therapy in patients.
EGR2 deletion increases early memory differentiation and ameliorates CAR T-cell-intrinsic dysfunction.
To investigate the role of the EGR2 transcriptional regulator in human T-cell fate and function, we knocked out its encoding gene using CRISPR/Cas9 technology (Figure 3A). The effect of EGR2 disruption during chronic CAR T-cell stimulation was conducted using our in vitro “stress test” system. Unlike previous studies showing proliferative impairment of T-cells with Egr2 knockout (15,18), EGR2 ablation enhances the proliferation of human CAR T-cells during repetitive antigen stimulation (Figure 3B and 3C). We observed that the CD8+ CAR-T cell population was enriched during chronic CAR stimulation and the ratio of CD4+ to CD8+ T cells was similar between the AAVS1 KO control group and the EGR2 KO group, indicating that the EGR2 deletion does not significantly affect the balance of CD4+ and CD8+ T cells within the CAR T cell product (Figure S2A). This enhanced CAR T-cell expansion capacity is accompanied by elevated frequencies of early memory T-cells in the EGR2 knockout condition, relative to controls (Figure 3D and 3E). Following chronic antigen stimulation, we also observe lower frequencies of EGR2 knockout CAR T-cells co-expressing inhibitory receptors and the TOX exhaustion-related transcription factor compared to AAVS1 knockout control CAR T-cells (Figure 3F and 3G). Accordingly, EGR2 knockout enhances CAR T-cell cytolytic capacity in association with increased IFNγ, IL-4, and IL-10 production, while decreasing expression of other effector and regulatory cytokines, such as IL-2 and TNFα (Figure 3H, 3I, S2B).
Figure 3. EGR2 knockout CAR T-cells demonstrate an early memory differentiation phenotype and reduced dysfunction.
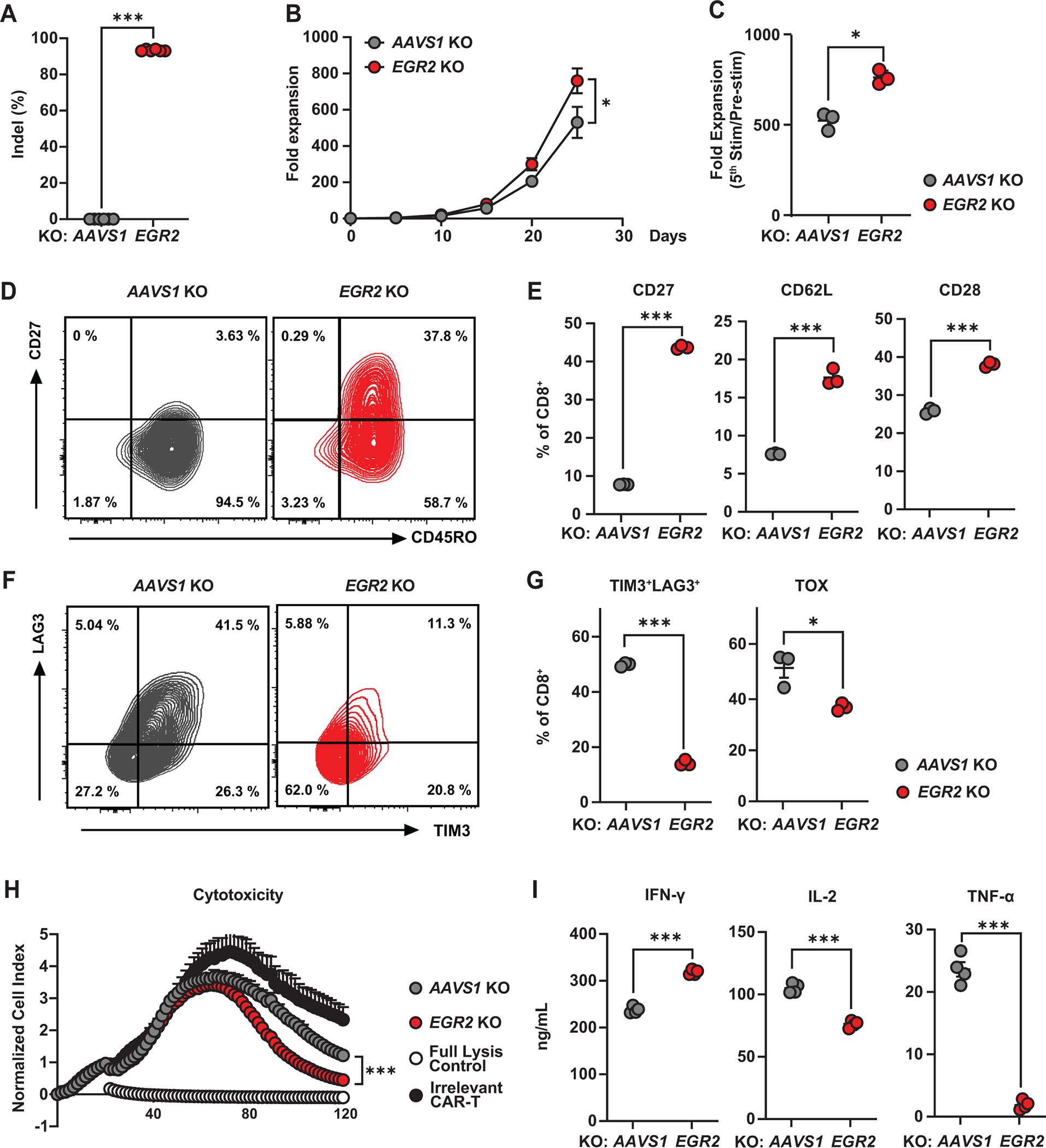
A, CRISPR/Cas9-mediated EGR2 knockout (KO) was confirmed by ICE (Inference of CRISPR Edits) analysis (n = 6). B, C, CAR T-cell expansion capacity during an in vitro stress test. PSMA CAR T-cells were challenged with PC3 cells expressing PSMA every five days at an effector to target (E:T) ratio of 3:1. D, Representative flow cytometry plots showing frequencies of AAVS1 (control) and EGR2-edited CD8+ CAR T-cells expressing CD27 and CD45RO. E, Frequencies of AAVS1 (control) and EGR2 knockout CD8+ CAR T-cells expressing memory markers following two consecutive rounds of antigen stimulation. F, Representative flow cytometric contour plots showing gene-edited CAR T-cells expressing TIM3 and LAG3. G, Exhaustion marker profiles of serially-restimulated CD8+ CAR T-cells following chronic antigen challenges. H, Cytolytic activity of EGR2 knockout CAR T-cells measured using the xCELLigence-based real-time cytotoxicity assay. 20% Tween 20-treated tumor targets were used as a full lysis control. I, Effector cytokines produced by CAR T-cells after 24-hours of antigen stimulation. All in vitro “stress test” experiments were conducted using CAR T-cells manufactured from three different healthy subjects. Figures (B-I) are comprised of representative data from one donor. *P < 0.05, *P < 0.01, ***P < 0.001, ns.: not significant (Student’s t-test).
To determine the effect of EGR2 disruption on different T-cell subsets, we generated CAR T-cells using naïve (CD62L+CD45RO−), central memory (CD62L+CD45RO+), effector memory (CD62L−CD45RO+), and effector (CD62L−CD45RO+) cells (Figure S3A). Our results showed that, similar to CAR T-cells generated from a bulk population, EGR2 disruption led to reduced exhaustion, improved cytotoxicity, and early memory differentiation in all CAR T-cell subsets during chronic CAR stimulation (Figure S3B–D). These findings suggest that EGR2 plays a key role in regulating the fate and function of CAR T-cells, regardless of their differentiation state.
We also investigated whether EGR2 deletion improves CAR T-cell performance with a costimulatory domain different from 4–1BB. We generated EGR2 KO CAR T-cells that target PSMA and have a CD28 costimulatory domain and analyzed CAR T-cell phenotype and function during prolonged CAR stimulation. Similar to the results observed with 4–1BB, EGR2-deficient CD28 CAR T-cells effectively reduced exhaustion and increased cytotoxicity, early memory differentiation, and CAR T-cell expansion (Figure S3E–H).
EGR2 disruption potentiates memory-related transcriptional programing and inhibits type I IFN pathway upregulation.
To examine how EGR2 regulates CAR T-cell memory differentiation and function at high resolution, we isolated CAR T-cells from the in vitro “stress test” after chronic stimulation and conducted single-cell multiome RNA + ATAC sequencing. The clustering of cells on a UMAP plot based on gene expression yields four CD8+ and two CD4+ subclusters (Figure 4A, S4A, S4B). IL7R+ CD4+ T-cells upregulate genes highly expressed by memory T-cells or T helper 2 cells including IL7R, IL17RB, and GATA3, whereas CTLA4+ CD4+ T-cells preferentially express genes associated with T-cell activation and dysfunction such as CTLA4, PRDM1, SOX4, TOX, CD38, and CBLB (Figure 4B, S4C, S4D). KLF2+ CD8+ T-cells are defined by elevated levels of early memory-related genes (KLF2, IL7R, CD28, FOXO1), and CD96+ CD8+ T-cells possess high expression of the CD96 costimulatory receptor gene as well as cell adhesion genes including ITGA1 (Figure 4B, S4E, S4F). GZMA+ CD8+ T-cells and MKI67+ CD8+ T-cells are characterized by elevated expression of cytotoxicity- (GZMA, GZMB, GNLY) and proliferation-related (MKI67, CDK1, CDKN3, G2M cell cycle) genes, respectively (Figure 4B, S4E, S4F, S4G).
Figure 4. EGR2 knockout in CAR T-cells blocks the inhibitory type I IFN transcriptional program and increases memory-related pathways.

A, Uniform manifold approximation and projection (UMAP) plot displaying CD4+ and CD8+ CAR T-cell clusters. Single-cell multiome gene expression experiments were conducted using chronically-stimulated AAVS1 and EGR2 knockout CAR T-cells produced from two healthy donors. B, Bubble plot showing marker gene expression in CD4+ and CD8+ CAR T-cell clusters. C, Frequencies of CAR T-cell clusters in each sample are shown. D, E, Gene signatures scores associated with (D) T-cell stemness, memory, and exhaustion and (E) favorable and unfavorable clinical responses in each CD8+ cluster and CAR T-cell sample. F, Pathways (GO biological processes) differentially regulated in EGR2 knockout CD8+ CAR T-cells. G, Volcano plot showing differentially expressed genes between EGR2 and AAVS1 knockout CD8+ CAR T-cells. Type I IFN genes are highlighted in blue.
We then asked how EGR2 deletion affects the transcriptome during CAR T-cell fate differentiation. Memory-associated genes such as IL7R, BCL2, ZEB1 as well as the quiescence-related genes, FOXP1 and THEMIS (i.e., known to suppress CAR signaling), were upregulated in EGR2 compared to AAVS1 knockout CD8+ CAR T-cells (Table S2) (19–21). Consistent with our flow cytometric analyses (Figure 3D and 3E), EGR2 deletion leads to enrichment of memory-like KLF2+ and CD96+ CD8+ T-cells, while decreasing frequencies of more differentiated CD8+ subclusters such as GZMA+ CD8+ and MKI67+ CD8+ populations (Figure 4C). Unlike CD8+ T-cell clusters, the distribution of CD4+ clusters were comparable between AAVS1 and EGR2 knockout CAR T-cells (Figure 4C). This is likely due to the fact that CD8+ T-cells are preferentially enriched during chronic CAR stimulation, and Egr2 is primarily upregulated in CD4+ T-cells in anergic states, rather than in exhausted T-cells (Figure S2A) (22). We conducted pathway enrichment analysis and found that KLF2+ CD8+ T-cells markedly upregulate multiple gene signatures associated with early T-cell memory differentiation and “stemness”. In contrast, MKI67+ CD8+ and GZMA+ CD8+ T-cells diminished in the EGR2 knockout CAR T-cell pool are highly enriched in T-cell exhaustion pathways, characterized by increased expression of various exhaustion-related genes (CD38, TOX, IKZF2, ENTPD1) compared to memory-like KLF2+ CD8+ T-cells; this is accompanied by low expression of memory-related signatures (Figure 4D, S5A). Gene ontology analysis within various CD8+ clusters in the setting of EGR2 knockout reveals that type I IFN pathways are among the top downregulated pathways across CD8+ subsets, suggesting that the EGR2 ablation blocks the type I IFN signaling cascade independently of the T-cell differentiation state (Figure S5B).
We next sought to identify CD8+ clusters associated with clinical response to CAR T-cell and immune checkpoint blockade therapies. We found that certain CD8+ T-cell clusters are associated with response to CAR T-cell and immune checkpoint blockade therapies. KLF2+ CD8+ T-cells have a gene expression profile highly enriched in CD19 CAR T-cell products from individuals with CLL who had complete responses, as well as T-cells from ALL patients with long-term CAR T-cell persistence (1,14) (Figure 4E, leftmost panel). This same profile is also found in “stem-like” tumor-infiltrating T-cells from patients who respond to immune checkpoint blockade therapy (23). On the other hand, GZMA+ and MKI67+ CD8+ T-cells have a gene expression profile resembling transcriptomic feature of CAR T-cell products from non-responding CLL and lymphoma patients, and neoantigen-specific T-cells from melanoma patients who do not respond to immune checkpoint blockade therapy (Figure 4E, leftmost panel). Our analysis of the effect of EGR2 ablation on CAR T-cell immunophenotype revealed that deletion of EGR2 leads to an increase in pathways associated with T-cell memory, stemness, and favorable response, and a decrease in pathways related to T-cell dysfunction and unfavorable response (Figure 4D, bottom panel and Figure 4E, rightmost panel), further emphasizing the importance of EGR2 in regulating T-cell fate and antitumor activity.
To further understand how EGR2 regulates biological pathways, we conducted gene ontology analysis comparing control AAVS1 and EGR2 knockout CD8+ CAR T-cells. EGR2 deletion significantly upregulates pathways associated with carboxylic acid metabolism, negative regulation of apoptosis, IL-7 signaling, and TCR signaling in naïve T-cells (Figure 4F and S6A–S6C). The type I IFN pathway is one of the most significantly downregulated biological processes in EGR2 knockout CAR T-cells, suggesting that EGR2 potentiates type I IFN signaling (Figure 4F, 4G, S6A–S6C). Similar to CD8+ CAR T-cells, EGR2 deletion leads to significant downregulation of type I IFN pathway-related genes (ISG15, IFI26, IFIT3, IFI6, ISG20, IFIR1, IFITM2, MX2, etc.) and upregulates signatures associated with TCR signaling in naïve CD4+ CAR T-cells (Figure S7A–S7D). Reduced type I IFN signaling is a unique transcriptional feature of the EGR2 KO strategy, as previous T-cell augmentation approaches, such as BRG1/BRM-associated factors (BAF) complex, Gata3, and INO80 complex KO (24), BATF overexpression (25), 4–1BB-based co-stimulation (10), and calibration of CAR activation strength (26) did not result in downregulation of the type I IFN pathway (Figure S8A–F).
EGR2 deletion ameliorates epigenetically-programed CAR T-cell dysfunction.
Because EGR2 modulates transcriptional programs associated with memory T-cell differentiation and exhaustion, we investigated how EGR2 deletion epigenetically regulates CAR T-cell fate by integrating ATAC seq data with gene expression profiling at the single-cell level. Analysis of the chromatin landscape shows that memory-like KLF2+ CD8+ T-cells increase accessibility at the FOXO1 locus, while regions corresponding to TOX2, BATF3, ATF3, and IFNG are more open in exhausted-like MKI67+ CD8+ T-cells (Figure S9A). IL7R is one of the most differentially accessible regions in EGR2-deficient CAR T-cells compared to AAVS1 knockout CD8+ CAR T-cells, while EGR2 deletion decreases chromatin accessibility at known loci related to T-cell dysfunction (SOX4, CTLA4) and those associated with type I IFN signaling (MX1, IFIT1, IFIT3) (Figure S9B). These chromatin accessibility profiles are concordant with the above gene expression changes (Tables S2 and S3). We then performed motif enrichment analysis to determine which transcription factors participate in EGR2-mediated transcriptional regulation. Binding motifs for AP-1 and IRF transcription factors, which promote T-cell exhaustion (11,27,28), are enriched in accessible regions of MKI67+ CD8+ CAR T-cells. (Figure 5A and 5B). EGR2 knockout decreases accessibility of these AP-1 and IRF binding motifs, while increasing accessibility at the FOS locus, which encodes an AP-1 transcription factor subunit essential for T-cell effector function (29) (Figure 5B). Notably, transcription factor motifs associated with type I IFN signaling (IRF8, IRF9, STAT1::STAT2, IRF3, and IRF7) showed exaggerated inaccessibility in EGR2 deleted compared to AAVS1 knockout CAR T-cells (Figure 5C). The accessibility of these type I IFN-related transcription motifs was also reduced in the clinically favorable KLF2+ CD8+ subset compared to MKI67+ CD8 T-cells (Figure 5A and B). These data suggest that EGR2 deletion induces remodeling of the exhaustion-associated epigenome of CAR T-cells in association with type I IFN pathway modulation.
Figure 5. EGR2 deletion ameliorates an epigenetic program of CAR T-cell exhaustion that is functionally bypassed by type I IFN exposure.
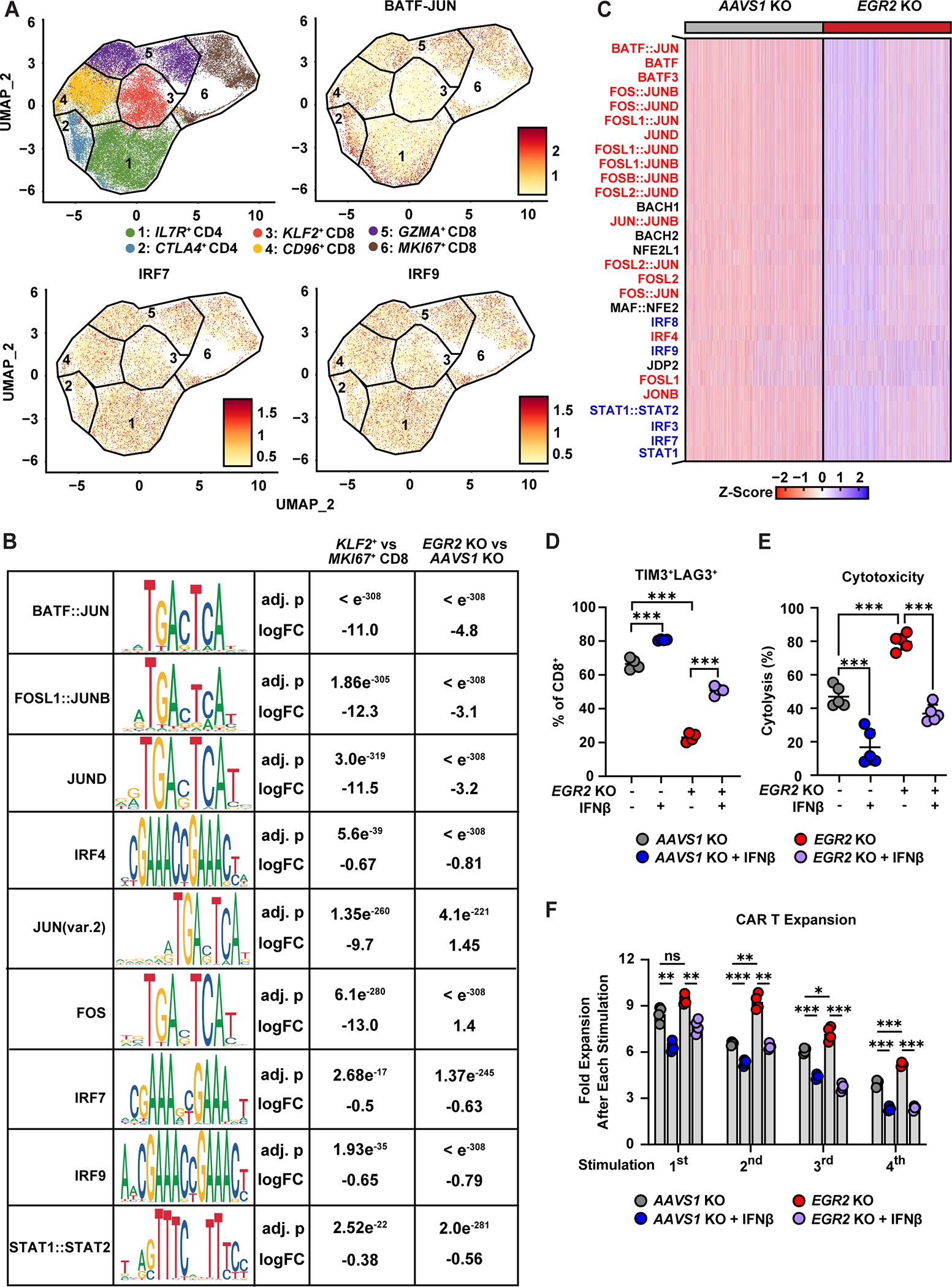
A, UMAP plots showing CD4+ and CD8+ subclusters and chromVAR motif deviation scores. B, Table displaying transcription factor motifs enriched in CD8+subclusters and AAVS1 (control) or EGR2 knockout CAR T-cell samples. C, Heatmap showing top transcription factor motifs inaccessible in EGR2 KO CAR T-cells. Exhaustion-associated AP-1/bZIP motifs and type I IFN-associated IRF/STAT motifs are highlighted in red and blue, respectively. (D-F) PSMA CAR T-cells were serially stimulated with PSMA-expressing PC3 tumor targets in the presence or absence of IFNβ treatment (1ng/mL). Experiments were conducted using CAR T-cells produced from multiple healthy subjects, each represented by a separate dot. D, Frequency of TIM3+LAG3+ CD8+ CAR-T-cells after chronic antigen stimulation (one-way ANOVA, n = 4). E, CAR T-cells were isolated after chronic stimulation and co-cultured with target cancer cells to measure cytolytic capacity using the xCELLigence real-time cytotoxicity assay (one-way ANOVA, n = 5). F, Proliferative capacity of CAR T-cells during the “stress test” (two-way ANOVA, n = 4). Single cell multiome ATACseq experiments were conducted using chronically-stimulated AAVS1 and EGR2 knockout CAR T-cells generated from two different healthy donors. *P < 0.05, **P < 0.01, ***P < 0.001, ns.: not significant.
EGR2 knockout ameliorates CAR T-cell dysfunction through a type I IFN signaling-dependent mechanism.
We next determined whether EGR2-driven CAR T-cell dysfunction is mediated by classical type I IFN signaling through incorporation of recombinant interferon-β (IFNβ) into our in vitro model of chronic CAR T-cell stimulation. Continuous IFNβ exposure during several rounds of antigen challenge significantly reverses the effect of EGR2 knockout on reducing the frequency of CD8+ CAR T-cells co-expressing TIM3 and LAG3 inhibitory receptors (Figure 5D and S9C). Similarly, chronic exposure to IFNβ counteracts the enhancing effect of EGR2 knockout on cytolytic function and proliferative capacity (Figure 5E and 5F). Notably, the enhanced memory differentiation of EGR2 knockout CAR T-cells is not affected by exogenous IFNβ exposure (Figure S9D). Furthermore, we found that addition of a type I IFN receptor-blocking antibody during chronic CAR T cell stimulation had no impact on early memory differentiation (Figure S9E). While IFNAR blockade was able to mitigate exhaustion and improve cytotoxicity in AAVS1 KO control CAR T-cells, it had no additional effect on EGR2 KO CAR T-cells as EGR2 KO alone could significantly reduce type I IFN signaling (Figure S9F–H). These results suggest that EGR2 plays a critical role in regulating CAR T-cell dysfunction through type I IFN signaling, and that its effects on memory differentiation may be independent of this mechanism.
EGR2 knockout improves CAR T-cell antitumor efficacy against blood and solid cancers.
Based on the above results, we interrogated the in vivo antitumor functionality of ERG2 knockout CAR T-cells in xenogeneic mouse models. EGR2 deletion markedly enhances the tumor control capacity of CD19 CAR T-cells in NOD-SCID-γ-chain–/– (NSG) mice engrafted with aggressive NALM-6 B-cell ALL (Figure 6A), which is accompanied by significant prolongation of survival (Figure 6B). In accordance with the results of our in vitro chronic restimulation experiments, the increased potency of ERG2 knockout CAR T-cells is associated with higher proliferative capacity (Figure 6C), early memory differentiation phenotype (Figure 6D), and reduced frequencies of engineered T-cells co-expressing inhibitory receptors (Figure 6E). Additionally, to evaluate the durability of EGR2 KO-mediated enhancement of CAR T-cell antitumor potency, mice that had successfully rejected an initial low dose of NALM-6 cells were rechallenged with a high dose of the leukemia cells (Figure S10A). The mice that had been treated with EGR2 KO CAR T-cells in the first tumor challenge showed improved tumor control and survival after the second challenge. These results further demonstrate the critical role of EGR2 in regulating memory CAR T-cell differentiation and promoting durable antitumor responses (Figure S10B, C).
Figure 6. EGR2 knockout improves CAR T-cell efficacy in multiple liquid and solid tumor in vivo models.

(A-E) NSG mice were intravenously infused with 106 NALM6-CBG cells. On day seven post-tumor injection, AAVS1 and EGR2 knockout CD19 CAR T-cells and negative control, irrelevant PSMA CAR T-cells (pBBz), were adoptively transferred (n = 6–7). A, Longitudinal leukemia burden measured by bioluminescent imaging. Data are representative of two independent experiments. Peripheral blood samples were collected on day twelve post-CAR T-cell injection for immunophenotyping (Mann-Whitney test, n = 6–7). B, Kaplan-Meier curves showing survival of mice following CAR T-cell treatment (Log-rank test). C, Human T-cell counts measured by using counting beads. Frequencies of hCD45+CD8+ cells expressing D, a memory marker (CD27) and E, exhaustion markers (PD1, TIM3, and LAG3). F, NSG mice were treated with 4 × 106 Capan2 cells subcutaneously. On day 38, AAVS1 and EGR2 knockout mesothelin-targeting CAR T-cells were injected intravenously, and tumor growth was monitored over time (Mann-Whitney test, n = 6). Data are representative of two independent experiments. G, NSG mice were subcutaneously injected with 4 × 106 AsPC1 cells. On day 31, the indicated mesothelin-targeting CAR T-cells were injected intravenously, and tumor growth was monitored over time (n = 7). Mice treated with irrelevant CD19 CAR T-cells (19BBz) served as a negative control. Data are representative of two independent experiments. H, On day 37 post-CAR T-cell infusion, tumors were harvested, and CAR T-cells isolated from tumors were reactivated ex vivo. Effector proteins produced by CAR T-cells were measured using flow cytometry (Mann-Whitney test, n = 7). *P < 0.05, *P < 0.01, ***P < 0.001, ns.: not significant.
We then tested this approach in the solid tumor setting first using two different pancreatic cancer xenograft models. The numbers of tumor cells engrafted, and CAR T-cells administered in these systems were carefully titrated so that mice were resistant to unmanipulated CAR T-cell monotherapy. In NSG mice implanted with Capan2 and AsPC1 tumors, EGR2 knockout enhances the in vivo antitumor activity of mesothelin-targeted CAR T-cells and successfully suppresses tumor growth (Figure 6F and 6G). To assess the effector function of tumor-infiltrating CAR T-cells (CAR-TILs), we isolated CAR-TILs from AsPC1 tumors and performed ex vivo stimulation, followed by functional analysis (30). Higher frequencies of EGR2 knockout CAR T-cells produce cytotoxic molecules and effector cytokines in response to activation, compared to AAVS1 knockout controls (Figure 6H). Further, we assessed the efficacy of EGR2-edited anti-PSMA CAR T-cells in an in vivo model of prostate adenocarcinoma. In this fourth in vivo system incorporating a PSMA CAR construct, only EGR2 knockout PSMA CAR T-cells mediated a significant reduction in tumor burden over time (Figure S10D).
The EGR2-targeted transcriptional program in CAR T-cells from patients is predictive of clinical outcome and survival.
We next examined whether a comprehensive EGR2-targeted transcriptional program that emerged from our chronic restimulation model and includes several type I IFN genes can predict CAR T-cell clinical efficacy. First, we explored bulk RNA sequencing data from 33 CLL patient CD19 CAR T-cell infusion products (1). The CAR T-cell products were divided into two groups according to clinical response (i.e., favorable responses: CR and PRTD, unfavorable responses: NR and PR) and genes upregulated or downregulated in chronically-stimulated EGR2 deleted CAR T-cells (Table S2) were used to score EGR2 gene expression, as previously described (31). CAR T-cell products from CR and PRTD are enriched in genes upregulated in EGR2 knockout CAR T-cells, while products from poor responders display signatures downregulated in EGR2 knockout CAR T-cells (Figure 7A and 7B). We then evaluated the relationship between EGR2-targeted gene expression scores and patient survival. Patients infused with CD19 CAR T-cell products exhibiting enrichment of our refined EGR2 knockout molecular signature demonstrate improved overall survival and event-free survival (Figure 7C, S11A and S11B).
Figure 7. A refined EGR2 molecular signature predicts CAR T-cell potency, clinical response and patient survival.

(A-B) EGR2-targeted gene expression scores in A, unstimulated and B, CAR-stimulated CD19 CAR T-cell products from responders (CR/PRTD) and non-responders (NR/PR) in CLL (EGR2 KO UP/DN signatures: genes upregulated or downregulated in EGR2 KO CAR T-cells compared to AAVS1 KO CAR T-cells) (Mann-Whitney test). C, Kaplan–Meier analysis of overall survival (left) and event-free survival (right) stratified by CLL CD19 CAR T cells products with high and low EGR2 KO UP gene expression scores (n = 22 evaluable patients). Correlation between an EGR2-targeted gene expression score and D, peripheral blood CAR T-cell expansion in patients (Pearson correlation), E, frequencies of clinically favorable (CD8+PD1−CD27+) and unfavorable (CD8+PD1+CD27−) subsets in CAR T-cell infusion products (Pearson correlation). (F, G) CD19 CAR T-cells were generated from a CLL non-responder using apheresis material, and their antitumor activity was evaluated in the NALM6 xenograft model. NSG mice were intravenously injected with 105 NALM6-CBG cells. On day 1, the mice received 8 × 105 CAR T-cells with EGR2 KO or a negative control T-cells with a PSMA-targeting CAR (n = 6–7). F, Longitudinal tumor growth. G, Mouse survival.
Our previous work revealed that patients who completely respond to CD19 CAR T-cell therapy display robust peripheral blood CAR T-cell expansion in association with high frequencies of PD1−CD27+ CD8+ T-cells in their engineered infusion products (1). In light of these previous findings, we investigated how the EGR2-targeted transcriptional program correlates with CAR T-cell expansion and frequencies of clinically relevant cell populations. Enrichment of genes elevated in EGR2 knockout CAR T-cells positively correlate with in vivo peripheral blood CAR T-cell expansion as well as the frequency of the clinically favorable PD1−CD27+ CD8+ T-cell subset in patient infusion products (Figure 7D and 7E). In contrast, there is an inverse correlation between this gene signature and the proportion of poorly potent PD1+CD27− CD8+ T-cells (Figure 7E) (1).
We sought to investigate whether the EGR2 knockout strategy could rescue the antitumor efficacy of T-cells from chronic lymphocytic leukemia (CLL) patients who had not responded to CAR T-cell therapy. To do this, we manufactured EGR2 knockout CD19 CAR T-cells using leukapheresis material from a non-responding CLL patient and treated NALM-6 B-ALL-engrafted mice with these cells to assess their in vivo antitumor potency. Our results showed that the EGR2 KO CAR T-cells significantly improved tumor control and mouse survival compared to patient-matched AAVS1 KO control CAR T-cells (Figure 7F and 7G). These findings demonstrate that the EGR2 knockout strategy can be used during CAR T-cell manufacturing to rescue dysfunctional patient T-cells and improve their antitumor efficacy.
To confirm these findings in a different disease indication, we analyzed single-cell RNA sequencing data from a trial of CD19 CAR T-cell therapy in ALL and assessed the association between our EGR2 module score and outcome (32). Pre-infusion CAR T-cells from subjects who had poor clinical responses (i.e., no response due to induction failure or product failure over time leading to eventual relapse with CD19+ leukemia cells) show a significant enrichment of genes downregulated in EGR2 knockout CAR T-cells, compared to highly active products from complete responders who experience durable complete remissions or CD19− leukemia relapse (Figure S11C). These findings support the idea that EGR2 deletion counteracts type I IFN signaling to repress CAR T-cell exhaustion and maintain T-cell memory, which is critical for persistent antitumor responses (Figure S11D).
Discussion
Despite their clinical potential, CAR T-cells do not expand, durably persist, and elicit effective antitumor activity in many patients, including several individuals with hematopoietic and non-hematopoietic malignancies. Our data suggest that the development of CAR T-cell intrinsic resistance is influenced by the duration of type I IFN signaling in the context of persistent and ongoing CAR T-cell stimulation. Several observations reflect the complex biology of type I IFN signaling in CAR T-cell immunotherapy. Zhao and colleagues previously reported that engineering T-cells with a CAR configuration in which 4–1BB ligand is co-expressed rather than incorporating a 4–1BB costimulatory endodomain into the synthetic receptor results in robust activation of the IRF7/IFNβ pathway, especially in CD4+ T-cells; this correlates with vigorous expansion of CAR T-cells and concomitantly enhanced tumor elimination immediately following T-cell infusion (6). In contrast, oncolytic virus-derived type I IFN elaboration was recently shown to hamper adjunctive tumor-targeted adoptive cell therapy through induction of T-cell apoptosis (8). Notably, the amount of type I IFN secretion induced by an oncolytic vesicular stomatitis virus (VSV) encoding murine IFNβ was 2–3 logs higher than that elicited by 4–1BB ligand-co-expressing CAR constructs (6,8). Thus, we reasoned that the beneficial or immunopathological effects of type I IFN signaling on engineered T-cells are determined by the nature of stimulation (i.e., induction in cis versus in trans) and are highly magnitude- and context-dependent.
We found that continuous antigen stimulation promotes functional, transcriptomic and epigenomic features of CAR T-cell dysfunction. Our in vitro “stress test” model captures several hallmarks of progressive exhaustion and unexpectedly identifies the type I IFN pathway as one of the top gene signatures upregulated in chronically-activated CAR T-cells. Notably, continuous type I IFN signaling is implicated in promoting T-cell exhaustion which is marked by co-expression of inhibitory receptors (33,34), diminished effector function (35,36) and reduced survival (37) in chronic viral infection and cancer. However, the underlying mechanism(s) for T-cell intrinsic type I IFN pathway propagation and its effect on CAR T-cell biology are largely unknown. In certain CAR constructs, signaling from the intracellular 4–1BB domain involves TRAF2 (38) to provide CD28-independent costimulatory signals to resting T-cells (39). TRAF2 has been implicated in the activation of type I IFN genes (40) and IFNβ production (41). Although it is possible that the 4–1BB-TRAF2-type I IFN pathway may be operative in the CAR T-cells used in this study, we observe a similar upregulation of the type I IFN pathway in tonically activated GD2 CAR T-cells engineered with a CD28 endodomain that does not elicit co-stimulatory signaling through TRAF2. These findings indicate that independently of type I IFN produced from tumors or other cells within the TIME, CD28- and 4–1BB-based CAR T-cells can autonomously propagate deleterious type I IFN signaling by an alternative mechanism.
A key discovery from our study is that the EGR2 transcription factor bridges persistent antigen-dependent and -independent CAR activation with a late stable type I IFN gene expression program. Excessive type I IFN is induced early in chronic viral infection and drives T-cell exhaustion (42,43). Our findings extend these studies by demonstrating a T cell-intrinsic role for type I IFN signaling and providing insight into EGR2-driven programs of gene expression. We found that the changes in phenotype and function of EGR2 KO CAR T-cells observed in our study are likely a result of modulation of the type I IFN signaling pathway, rather than changes in IFNAR expression. This conclusion is supported by the fact that there is no significant difference in IFNAR expression in T cells between the control and EGR2 KO groups. Upon examination of the chromatin landscape in dysfunctional CAR T-cells, regions where EGR2 motifs colocalize with areas of increased accessibility include type I IFN-stimulated genes, and several transcription factor binding sites associated with type I IFN signaling became inaccessible in EGR2 knockout CAR T-cells. Type I IFN-related transcription factor binding motif accessibility was also reduced in the memory-like KLF2+ CD8+ T-cell population enriched with a known clinically favorable gene signature, compared to less potent MKI67+ CD8+ subsets possessing transcriptional features of dysfunctional T-cells. This suggests that specific dysfunctional CAR T-cell populations that have acquired a type I IFN-related epigenetic imprint may retain the capacity for epigenomic remodeling. A similar phenomenon has been described for a CD8 progenitor population that mediated sustained T-cell-mediated antiviral responses and is programmed by an interplay between type I IFN and the TCF1-Bcl6 axis (44). Future studies are warranted to better define the specific cell populations that undergo epigenetic reprogramming following type I IFN pathway modulation to identify the precise role of EGR2 in remodeling the exhaustion-associated epigenome.
Our findings also add to the emerging data on the role of EGR2 on AP-1 transcription factor function during T-cell exhaustion. In an elegant study by Wagle and colleagues (15), EGR2 was identified as a key transcriptional and epigenetic regulator of progenitor exhausted cells in a mouse model of chronic LCMV infection. This study corroborates our results in human CAR T-cells showing that EGR2 knockout decreased chromatin accessibility at regions enriched for AP-1/bZIP motifs, accompanied by a concordant transcriptional program of decreased exhaustion (15). However, in LCMV, despite the increase in progenitor exhausted CD8+ T-cells known to play a key role in the maintenance of cell-mediated immunity, the expansion capacity of Egr2 deficient virus-specific CD8+ T-cells induced by checkpoint blockade is significantly impaired, suggesting that EGR2 is required by mouse T-cells for optimal viral control (15). Likewise, other studies done in murine systems demonstrate that Egr2 knockout produces T-cells with a more “effector-like” phenotype possessing a compromised capacity to proliferate and persist (15,18,45). On the contrary, our study reveals that knocking out EGR2 enhances memory CAR T-cell differentiation in association with enhanced antigen-driven expansion. Thus, while murine models have enabled the elucidation of a functional role for EGR2 in T-cell biology, it has become clear that this response and differentiation trajectory varies between rodents and humans (46). Further, we show that EGR2 disruption in CAR T-cells orchestrates a transcriptional network of genes known to direct early memory T-cell differentiation. Our mechanistic studies involving IFNβ incorporation into the “stress test” model suggest that the memory-enhancing effect of EGR2 deficiency is likely independent of the ability of EGR2 to counteract a distinct program of CAR T cell-intrinsic dysfunction involving type I IFN signaling and exhaustion. Specifically, we found that prevention of chronic antigen-induced dysfunction in EGR2-deficient CAR T-cells is mediated by reduced type I interferon signaling, as demonstrated by the ability of recombinant IFNβ exposure to circumvent this effect and the opposite outcome observed with type I interferon receptor blockade. However, the exact mechanism(s) by which EGR2 modifies type I IFN signaling and its separate relationship to the observed memory-enhancing effect requires further study.
We also demonstrate that EGR2 KO can enhance antitumor responses in multiple liquid and solid tumor models, irrespective of the antigen targeted (CD19, mesothelin, PSMA) or CAR construct used for T-cell engineering. While our work provides valuable insights into the role of EGR2 in regulating CAR T-cell dysfunction, it is important to note that the mouse models used in this study have some limitations. For example, cell line-derived xenograft models may not accurately mimic primary tumors and the tumor microenvironment in humans, and our study is further limited by the use of immunodeficient mouse models, which do not fully represent the complex interactions between CAR T cells, the immune system and the tumor microenvironment that occur in humans. Thus, it is important to note that further testing in human patients will be necessary to fully understand the implications of these findings and to establish the short- and long-term clinical utility of the EGR2 KO strategy in enhancing the potency of CAR T-cell therapy. Additional studies are also needed to determine whether this approach can be universally applied to all CARs and whether blocking type I IFN signaling and overcoming suppression of early memory differentiation are the mechanisms by which CAR T-cell-intrinsic EGR2 loss augments functionality in vivo. The findings also raise the prospect for regulatable platforms using clinically relevant pharmacologic inducers of transcription factor degradation to precisely enhance CAR T-cell efficacy resulting from temporal control of EGR2 signaling.
Lastly, we explored clinical correlates for the role of EGR2 in CAR T-cell therapy. Based on our preclinical work, we refined a signature comprising a battery of EGR2-regulated genes gleaned from our in vitro chronic CAR T-cell restimulation model and examined this biomarker in relation to patient response to CD19 CAR T-cell therapy in leukemia. We found that the EGR2 gene score within pre-infusion CAR T-cell products segregates both CLL and ALL patients according to the magnitude of in vivo CAR T-cell expansion and outcome. Moreover, high levels of the EGR2 regulon predict poor survival in CLL patients after CD19 CAR T-cell treatment. This unique molecular signature thus specifies non-obvious aspects of cell physiology associated with clinical response, and long-term may provide a tool for assessing CAR T-cell batch quality prior to administration. In addition, the significant correlation between the EGR2-regulated gene population score and frequencies of highly potent PD1−CD27+ CAR T-cells (1) provides an actionable manufacturing pathway to enrich clinically desirable or deplete undesirable subsets in infusion products. Further studies are needed to compare the efficacy of such an approach to that of EGR2 gene-edited CAR T-cells in cancer.
In summary, our results conceptually link prolonged CAR T-cell stimulation with a T-cell-intrinsic immunoinflammatory resistance mechanism. Genetic modulation of the EGR2-type I IFN signaling may therefore represent an immediately actionable strategy to increase the efficacy of cellular immunotherapy for both hematopoietic and less tractable non-hematopoietic cancers. Our work also underscores the fundamental role of EGR2 in the regulation of human T-cell memory differentiation. These discoveries thus support the development of EGR2 knockout CAR T-cell products for early-phase clinical trials. Beyond the immediate application of this technology to CAR T-cell therapy for cancer, these findings could have broad implications for developing novel cell therapies to treat infectious diseases, autoimmunity, cardiac disease, etc.
Methods
Primary human samples
For CAR T-cell production, peripheral blood mononuclear cells (PBMC) were collected through leukapheresis of healthy donors with approval by the University of Pennsylvania Institutional Review Board. Written informed consent was provided to study subjects according to the Declaration of Helsinki, the International Conference on Harmonization Guidelines for Good Clinical Practice and U.S. Common Rule.
Cell lines
NALM6 B-ALL and PC3 prostate adenocarcinoma cell lines were originally obtained from the American Type Culture Collection (ATCC) and subsequently engineered to express click beetle green luciferase and green fluorescent protein (CBG-GFP) (1,47). PC3-PSMA and Capan2 pancreatic ductal adenocarcinoma cells were provided by Carl H. June (University of Pennsylvania) and maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% streptomycin/penicillin. NALM6 and pancreas adenocarcinoma AsPC1 cells (ATCC) were cultured in Roswell Park Memorial Institute (RPMI) 1640 media with 10% FBS and 1% streptomycin/penicillin (R10 media). HEK 293T cells used for lentivirus production were also procured from the ATCC and cultured in R10 media. We tested all low-passage cell lines used in this study for mycoplasma contamination using a MycoAlert Mycoplasma Detection kit (Lonza), following the manufacturer’s instructions. The University of Arizona Genetics Core performed cell line identity testing within one year of use.
Lentivirus production
Lentiviral vectors encoding CARs were manufactured as previously described (47). In brief, second generation CARs comprised of an anti-CD19 (48), anti-Mesothelin (49), or anti-PSMA (47) single-chain variable fragment (scFv) fused to a CD8α transmembrane domain and 4–1BB as well as CD3ζ intracellular signaling domains were subcloned into the pTRPE lentiviral plasmid. For lentivirus production, HEK293T cells were transfected with transfer and packaging plasmids using Lipofectamine 2000 (Thermo Fisher Scientific). At 24- and 48-hours post-transfection, lentivirus supernatant was harvested from the culture and concentrated using high-speed ultracentrifugation.
Lentiviral transduction and T-cell culture
Healthy donor T-cells were isolated from PBMCs using the Pan T-cell Isolation Kit according to the manufacturer’s instructions (Miltenyi Biotec). To manufacture CAR T-cells from naïve, central memory, effector memory, and effector T-cells, each T-cell subset was magnetically isolated using the REAlease CD62 microbead kit (Miltenyi Biotech) and CD45RO microbeads (Miltenyi Biotech). T-cells were maintained in OpTmizer CTS SFM media (Gibco) supplemented with 5% human AB serum and 100U/mL human IL-2. On day zero, CD3/CD28 Dynabeads CTS™ (Thermo Fisher Scientific) were added to T-cells at a bead to cell ratio of 3:1. After 24-hours of bead-based activation, lentiviral vector encoding an anti-CD19, anti-Mesothelin or anti-PSMA CAR was added to the culture at a multiplicity of infection (MOI) of two. Media was added to cultures every two days and CAR T-cells were maintained at a constant concentration of 5 × 105 cells/mL, followed by harvest and cryopreservation on day nine of the CAR T-cell expansion process.
CRISPR/Cas9-mediated gene editing
On day three post-T-cell activation as described above, CD3/CD28 Dynabeads were magnetically eliminated. T-cells were washed with PBS and electroporated using a P3 primary cell 4D-nucleofector kit (Lonza). 2 × 106 CAR T-cells resuspended in P3 solution were then preincubated with 6μg of TrueCut™ S. pyogenes Cas9 (Invitrogen) and 0.1nmol of chemically-modified tracrRNA and crRNA (Integrated DNA Technologies). T-cells were electroporated using the 4D-Nucleofactor X-Unit (EO-115; Lonza) and cultured in supplemented OpTmizer media as described above. The crRNA sequences used in this study were as follows: AAVS1: 5’-CCATCGTAAGCAAACCTTAG-3’, EGR2: 5’- GGTGCCAGCTGCTACCCAGA-3’. Target gene-editing regions were PCR-amplified using the following primers: EGR2-F1: 5’- GGGTGAGAAGGGAGTCCTGGCTGTATTTTC-3’, EGR2-R1: 5’- AGGATAGTCTGGGATCATTGGGAAGAGACC-3’. The frequency of insertion–deletion mutations (indels) generated by CRISPR/Cas9 editing was determined by ICE (Inference of CRISPR Edits) analysis (50).
Flow cytometry
For CAR T-cell immunophenotyping in in vitro and in vivo experiments, cells were stained with LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (ThermoFisher Scientific), followed by fluorochrome-conjugated antibodies diluted in FACS buffer (PBS + 2% FBS). PSMA CAR expression was detected with an allophycocyanin (APC)-conjugated human PSMA protein (Sino Biological). Surface staining was carried out using the following antibodies: PD1-BV421 (Biolegend #329920), CD45-BV570 (Biolegend #304226), CD8-BV650 (Biolegend #301042), CD8-APC-H7 (BD Biosciences #560179), CD4-BV785 (Biolegend #317442), TIM3-PE (Biolegend #345006), CCR7-PE-CF594 (BD Biosciences #562381), CD62L-PE-Cy5 (Biolegend #304808), LAG3-PE-Cy7(eBioscience #25–2239-42), hCD45-APC (BD Biosciences #340943), murine CD45-PerCP-Cy5.5 (Biolegend #103132), CD127-BV570 (Biolegend #351307), HLA-DR-Alexa Fluor700 (Biolegend #307625), CD25-APC (ebioscience #17–0259-42). For intracellular staining, cells were fixed and permeabilized using the FoxP3 Transcription Factor Staining Buffer Kit (eBioscience) and subsequently stained with following antibodies: IL2-PE-CF594 (BD Biosciences #562384), IFNγ-BV570 (Biolegend #502534), TNFα- Alexa Fluor700 (Biolegend #502928), Perforin-BV421 (Biolegend # 353307), Perforin-APC (Biolegend #308112), GZMB-PE-Cy5.5 (Invitrogen #GRB18), TOX-APC (Miltenyi Biotech #130–107-785). Samples were then acquired on an LSRII Fortessa flow cytometer (BD Biosciences), and analysis was performed using FlowJo software v10.8 (FlowJo, LLC).
In vitro CAR T-cell “stress test”
The proliferative capacity, immunophenotype, transcriptome and effector functions of CAR T-cells were evaluated during in vitro “stress tests” based on chronic tumor antigen stimulation. Briefly, 5 × 105 irradiated PC3-PSMA cells were seeded into 6-well plates at 24-hours prior to CAR T-cell coculture. AAVS1 and EGR2 knockout PSMA-directed CAR T-cells were magnetically isolated using a biotin-goat anti-mouse IgG F(ab)2 fragment (Jackson ImmunoResearch) and anti-biotin beads (Miltenyi Biotech). Following isolation, 1.5 × 106 CAR T-cells were added to PC3-PSMA cell cultures. At 24-hours post-coculture, supernatant was collected for cytokine analysis using the LEGENDplex™ human CD8 panel (Biolegend). CAR T-cell expansion capacity was assessed during the assay using the Luna automated cell counter (Logos Biosystems). Every five days, 1.5 × 106 CAR T-cells were isolated from previous cultures and serially stimulated with new PC3-PSMA cells at an effector to target (E:T) ratio of 3:1. Restimulation assays were carried out for 20–25 days until CAR T-cell proliferation and viability markedly declined in culture. The effect of altering type I IFN signaling on CAR T-cell phenotype and function was assessed by adding 1ng/mL of IFNβ (PeproTech) or 1μg/mL of an IFNAR blocking antibody (Creative Biolabs, Anifrolumab) during in the setting of chronic tumor antigen challenge.
Cytotoxicity assay
The real-time cytolytic capacity of gene-edited CAR T-cells against PC3-PSMA cells was assessed using the xCELLigence system (ACEA Biosciences Inc.). 2 × 104 PC3-PSMA cells were seeded into E-Plate VIEW 96 PET microwell plates on the day preceding CAR T-cell addition. After 24-hours, PSMA CAR T-cells or irrelevant CD19 CAR T-cells (control) were added to microwell plates containing target cells to achieve the desired E:T ratios. A 20% mixture of Tween 20 nonionic detergent (Sigma-Aldrich) was added to separate wells as a full lysis control. Electrical impedance was monitored in 30-minute intervals over six days, and cytotoxicity was assessed by calculating the normalized cell index and percent cytolysis.
Mouse xenograft studies
Mouse experiments were conducted with 6- to 8-week-old NOD/SCID/IL-2Rγ-null (NSG) mice (The Jackson Laboratory) in compliance with a University of Pennsylvania Institutional Animal Care and Use Committee approved protocol.
For the B-cell ALL model, NSG mice were intravenously infused with 106 NALM6-CBG cells. On day seven after tumor injection, AAVS1 or EGR2 knockout CD19 CAR T-cells or a negative control product (i.e., irrelevant PSMA CAR T-cells) were infused. Longitudinal tumor growth was assessed weekly using bioluminescent imaging with a Xenogen IVIS Imaging System (PerkinElmer, Inc., (1)). To measure peak product expansion and assess the immunophenotypes of circulating CAR T-cells, peripheral blood samples were taken on day twelve post-CAR T-cell injection. The absolute number of circulating human T-cells was quantified using 123count eBeads™ (Thermo Fisher Scientific), and the expression of memory as well as exhaustion markers was assessed using flow cytometry. To evaluate the memory function of CAR T-cells, we performed tumor rechallenge experiments. On day 1, 105 NALM6-CBG cells were injected intravenously into the mice, followed by the infusion of 8 × 105 CD19 CAR T-cells. After the leukemia was eradicated by AAVS1 and EGR2 KO CAR T-cells on day 33, the mice were rechallenged with a high dose (5 × 106 cells) of NALM6-CBG cells to assess the ability of the CAR T-cells to mount a secondary response.
To assess the in vivo efficacy of EGR2 knockout CAR T-cells against pancreatic cancer, 4 × 106 Capan2 or AsPC1 cells were premixed with Matrigel (Corning) and subcutaneously injected into the flanks of NSG mice. 3 × 105 mesothelin-targeting CAR T-cells were intravenously injected when tumor volume reached 400–500 mm3. Tumor growth was monitored over time using caliper-based measurements (tumor volume = (length × width2)/2). To evaluate the effector functions of tumor-infiltrating CAR T-cells, tumors were harvested on day thirty-seven post-CAR T-cell infusion and minced with a scalpel, followed by dissociation in 100U/mL collagenase IV (STEMCELL Technologies) and 0.25mg/mL DNase I (STEMCELL Technologies) for 30 minutes at 37°C in 5% CO2. CAR T-cells isolated from tumors were reactivated with 50 ng/mL phorbol 12-myristate 13-acetate 1210 (PMA) and 1ug/mL ionomycin in the presence of 5ug/mL Brefeldin A for 6-hours. Production of effector molecules was measured by intracellular flow cytometry.
For the prostate cancer mouse model, 5 × 106 PC3-PSMA tumor cells were subcutaneously injected to NSG mice. 1 × 105 PSMA CAR T-cells were intravenously infused when the average tumor size reached 400 mm3 and cancer growth was monitored over time by taking caliper measurements.
Single-cell multiome sequencing
AAVS1 and EGR2 knockout PSMA CAR T-cells produced from two different healthy subjects were isolated from the in vitro “stress test” after multiple consecutive rounds of antigen stimulation. Dead cells were removed using the Dead Cell Removal Kit (Miltenyi Biotec), and PSMA CAR-expressing T-cells were enriched by magnetic cell separation using a Biotin-goat anti-mouse IgG F(ab)2 fragment (Jackson ImmunoResearch #115–065-072) and anti-biotin beads (Miltenyi Biotech). CAR T-cells were washed with PBS supplemented with 0.04 % BSA and nuclei were isolated according to the manufacturer’s protocol (10x Genomics). Briefly, cells were incubated with lysis buffer for four minutes, washed three times and resuspended in chilled dilute nuclei buffer (10x Genomics). Absolute cell numbers were enumerated, and viability was determined using the trypan blue dye exclusion method.
Single-cell multiome-seq libraries were prepared using Chromium Single Cell Multiome ATAC and Gene Expression Reagent Kits V1 (10X Genomics) following the manufacturer’s instructions. Isolated nuclei were washed and resuspended in PBS containing 0.04% BSA and ~20,000 cells were loaded per reaction. Sequencing was performed on a Novaseq 6000 (Illumina) at a depth of ~20,000 reads / cell for gene expression and ~10,000 reads / cell for ATAC sequencing.
Reads were aligned to the human reference genome (GRCh38) using Cell Ranger ARC 2.0. Subsequent quality control and downstream analyses were performed using Seurat 4.1.1 and Signac 1.7.0 in R. Low-quality cells were eliminated based on following criteria: (1) a minimum of 1,000 genes and a maximum of 6,000 genes detected per cell, (2) less than 15% of mitochondrial gene counts, (3) a minimum 1,000 and maximum of 25,000 ATAC read counts, (4) a nucleosome signal less than two, and (5) a transcription start site enrichment score greater than two. After quality control, 38,573 cells remained. For gene expression analysis, RNA counts were normalized using SCTransform followed by integration of four CAR T-cell datasets using SelectIntegrationFeatures, PrepSCTIntegration, FindIntegrationAnchors, and IntegrateData functions in Seurat. Data were then scaled using ScaleData, followed by principal-component analysis and graph building using the RunPCA, FindNeighbors, and FindClusters. Differential gene expression analyses between subclusters and samples were conducted using the FindMarkers command. Module scores were then calculated using AddModuleScore to assess the enrichment of gene signatures, and EnrichR pathway analysis was performed via DEenrichRPlot.
For ATACseq data, peaks were called using MACS2 with the CallPeaks function followed by removal of peaks overlapping genomic blacklist regions. We then quantified counts in each peak using FeatureMatrix. Frequency-inverse document frequency (TF-IDF) normalization and dimension reduction were conducted using RunTFIDF, FindTopFeatures, and RunSVD. Per-cell motif activity scores were calculated using chromVAR through the RunChromVAR command and differential motif enrichment analysis was performed using FindMarkers.
Bulk RNAseq analysis
PSMA CAR T-cells that were unstimulated or stimulated one time and four times were isolated from in vitro “stress test” cultures. CD8+ CAR T-cells were magnetically isolated using human CD8 microbeads (Miltenyi Biotec) and total mRNA was extracted with RNA Clean & Concentrator™ kits (Zymo Research). Bulk RNAseq was conducted by Novogene using the Novaseq6000 system (paired-end, 150bp reads) at 40 × 106 reads per sample. Paired-end reads were pseudoaligned to the human genome (GRCh38) using kallisto v0.46.0. Aligned reads were summarized into matrices using tximport 1.24.0.
Differential expression analyses between unstimulated and chronically stimulated CAR T-cells were carried out using the edgeR v3.34.0 and limma v3.48.0 packages. Briefly, filtered RNAseq data was normalized using a Trimmed Mean of M-values method and logarithmically transformed into counts per million (cpm). Differential expression was determined using generalized linear models with edgeR. Gene set enrichment analysis (GSEA) was conducted using the GSEABase and enrichR packages.
Calculation of EGR2 and type I IFN gene signature scores
The EGR2 gene signature was derived from the ARCHS4 Transcription Factors Co-expression library, and the type I IFN gene set was obtained from Gregory et al. ((14); Table S1). UP and DN signatures represent sets of differentially expressed genes expressed between EGR2 and AAVS1 knockout CAR T-cells from single-cell RNA sequencing (threshold: log fold change > 0.25, adjusted P value < 0.01; Table S2). Gene signature scores were calculated as described previously (31). Expression data used in this study for gene signature scoring include bulk RNAseq data from our in vitro “stress test,” our previously published dataset (1), and GSE136891. Using these expression data, transcript counts were filtered and normalized using the edgeR package. The normalized log2 expression of each gene was centered and scaled across the samples, followed by summing all of the scaled expression values of gene sets for each sample.
Bulk ATACseq analysis
Unstimulated and chronically activated CAR T-cells were purified from in vitro “stress tests.” Following dead cell removal, 100,000 CD8+ CAR T-cells per sample were cryopreserved. Library preparation was carried out by Novogene. Briefly, nuclei pellets were resuspended in the Tn5 transposase reaction mixture consisting of transposase and equimolar adapter 1 and adapter 2. The reaction was then incubated at 37°C for 30 minutes. A PCR was subsequently performed to enrich the library. The final libraries were purified using AMPure beads (Beckman Coulter) and library quality was assessed. Paired-end sequencing was performed (150bp reads) on a NovaSeq 6000 instrument (Illumina).
FASTQ files for each sample were trimmed of adapter contamination using cutadapt (https://github.com/marcelm/cutadapt/). Alignments to the hg19 reference genome were performed using Bowtie2 and restricting to properly aligned and paired reads between 10 and 1000 base pairs. Mitochondrial reads were then eliminated (https://github.com/jsh58/harvard/blob/master/removeChrom.py). Files were organized using samtools, and PCR duplicates were removed with Picard. BAM files were indexed with samtools to visualize tracks in the Integrative Genomics Viewer (IGV, Broad Institute). Peak calling was carried out with MACS2 using a false discovery rate (FDR) q-value of 0.01. The R package Diffbind was implemented to remove ENCODE blacklisted regions (https://sites.google.com/site/anshulkundaje/projects/blacklists) and to subsequently identify peaks differentially opened between CAR T-cell samples. The findMotifsGenome script from HOMER was used to map the hg19 genome for occurrences of the EGR2 motif (i.e., derived from ENCODE data accessible via GEO at GSE31477).
Statistical analyses
Normality tests were conducted using Shapiro-Wilk and D’Agostino-Pearson omnibus tests. Statistical tests between two groups were conducted using the Mann Whitney U test, Student’s t-test for unpaired comparisons or the Difference (or Ratio) for Equivalence Test as appropriate. For assessments of three or more groups, the one- or two-way ANOVA test followed by post-hoc tests to correct for multiple comparisons were implemented. Pearson or Spearman correlation tests were used to evaluate relationships between two variables. The log-rank (Mantel-Cox) test was used to analyze the statistical significance of difference for survival analyses. Statistical tests were conducted using Prism 9.4 (GraphPad) and P values < 0.05 were considered significant.
Supplementary Material
Statement of Significance.
To improve CAR T-cell therapy outcomes, modulating molecular determinants of CAR T-cell-intrinsic resistance is crucial. Editing the gene encoding the EGR2 transcriptional regulator renders CAR T-cells impervious to type I interferon pathway-induced dysfunction and improves memory differentiation, thereby addressing major barriers to progress for this emerging class of cancer immunotherapies.
Acknowledgements
The authors acknowledge the Human Immunology Core at the University of Pennsylvania for providing leukocytes and the Hospital of the University of Pennsylvania Apheresis Unit for peripheral blood mononuclear cell collections. We would also like to recognize the contributors who supported development and execution of the trials from which clinical samples were obtained and/or for statistical assistance: Edward Pequignot, Avery Lee Gaymon, Megan Davis, Amy Marshall, Carl H. June, Sophia Ngo, Theresa A. Colligon, Bruce Levine, Whitney Gladney, Anne Chew, Julie Jadlowsky, Diane Frazee, Mary Truran, Elizabeth Veloso, Holly McConville, Jonathan Aguedelo, Samantha Hower, Joan Gilmore, Lester Lledo, Karen Dengel, Vanessa Gonzalez, and Fang Chen. The Clinical Cell and Vaccine Production Facility (CVPF), the Translational and Correlative Studies Laboratory (TCSL), and the Product Development Lab (PDL) from the University of Pennsylvania Center for Cellular Immunotherapies (CCI) are also thanked. This work was supported by the Prostate Cancer Foundation Tactical Award (to AJR, VN, NBH and JAF), F31 CA274961(to CRH), an Alliance for Cancer Gene Therapy Investigator Award in Cell and Gene Therapy for Cancer (to JAF and NBH), a Prostate Cancer Foundation Young Investigator Award (to VN), U54 CA244711 (to JAF), P01 CA214278 (to DLP and JAF), U01 AG066100 (to JAF), a Samuel Waxman Cancer Research Foundation Grant (to JAF), and an ACC P30 Core Grant P30 CA016520–42 (to JAF).
Footnotes
Conflict of Interest Statement: I.Y. Jung, V. Narayan, N.B. Haas, D.L. Siegel, D.L. Porter, S. Guedan, F.D. Bushman and J.A. Fraietta hold patents and other intellectual property in the field of T-cell-based immunotherapy for cancer and have received royalties. The remaining authors declare no potential conflicts of interest related to this work.
Data availability
Raw and analyzed data supporting the conclusions of this manuscript are available at the Gene Expression Omnibus (GEO) under the accession number GSE224194. The data are publicly accessible and can be downloaded for further analysis.
References
- 1.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018;24(5):563–71 doi 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018;558(7709):307–12 doi 10.1038/s41586-018-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah NN, Qin H, Yates B, Su L, Shalabi H, Raffeld M, et al. Clonal expansion of CAR T cells harboring lentivector integration in the CBL gene following anti-CD22 CAR T-cell therapy. Blood Adv 2019;3(15):2317–22 doi 10.1182/bloodadvances.2019000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindner SE, Johnson SM, Brown CE, Wang LD. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci Adv 2020;6(21):eaaz3223 doi 10.1126/sciadv.aaz3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin J, Cheng J, Huang M, Luo H, Zhou J. Fueling chimeric antigen receptor T cells with cytokines. Am J Cancer Res 2020;10(12):4038–55. [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015;28(4):415–28 doi 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gong W, Donnelly CR, Heath BR, Bellile E, Donnelly LA, Taner HF, et al. Cancer-specific type-I interferon receptor signaling promotes cancer stemness and effector CD8+ T-cell exhaustion. Oncoimmunology 2021;10(1):1997385 doi 10.1080/2162402X.2021.1997385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evgin L, Huff AL, Wongthida P, Thompson J, Kottke T, Tonne J, et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat Commun 2020;11(1):3187 doi 10.1038/s41467-020-17011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung IY, Kim YY, Yu HS, Lee M, Kim S, Lee J. CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res 2018;78(16):4692–703 doi 10.1158/0008-5472.CAN-18-0030. [DOI] [PubMed] [Google Scholar]
- 10.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015;21(6):581–90 doi 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019;576(7786):293–300 doi 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019;176(4):775–89 e18 doi 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018;564(7735):268–72 doi 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- 14.Chen GM, Chen C, Das RK, Gao P, Chen CH, Bandyopadhyay S, et al. Integrative Bulk and Single-Cell Profiling of Premanufacture T-cell Populations Reveals Factors Mediating Long-Term Persistence of CAR T-cell Therapy. Cancer Discov 2021;11(9):2186–99 doi 10.1158/2159-8290.CD-20-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagle MV, Vervoort SJ, Kelly MJ, Van Der Byl W, Peters TJ, Martin BP, et al. Antigen-driven EGR2 expression is required for exhausted CD8(+) T cell stability and maintenance. Nat Commun 2021;12(1):2782 doi 10.1038/s41467-021-23044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y, Zha Y, Driessens G, Locke F, Gajewski TF. Transcriptional regulator early growth response gene 2 (Egr2) is required for T cell anergy in vitro and in vivo. J Exp Med 2012;209(12):2157–63 doi 10.1084/jem.20120342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narayan V, Barber-Rotenberg JS, Jung IY, Lacey SF, Rech AJ, Davis MM, et al. PSMA-targeting TGFbeta-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med 2022;28(4):724–34 doi 10.1038/s41591-022-01726-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du N, Kwon H, Li P, West EE, Oh J, Liao W, et al. EGR2 is critical for peripheral naive T-cell differentiation and the T-cell response to influenza. Proc Natl Acad Sci U S A 2014;111(46):16484–9 doi 10.1073/pnas.1417215111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guan T, Dominguez CX, Amezquita RA, Laidlaw BJ, Cheng J, Henao-Mejia J, et al. ZEB1, ZEB2, and the miR-200 family form a counterregulatory network to regulate CD8(+) T cell fates. J Exp Med 2018;215(4):1153–68 doi 10.1084/jem.20171352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei H, Geng J, Shi B, Liu Z, Wang YH, Stevens AC, et al. Cutting Edge: Foxp1 Controls Naive CD8+ T Cell Quiescence by Simultaneously Repressing Key Pathways in Cellular Metabolism and Cell Cycle Progression. J Immunol 2016;196(9):3537–41 doi 10.4049/jimmunol.1501896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun C, Shou P, Du H, Hirabayashi K, Chen Y, Herring LE, et al. THEMIS-SHP1 Recruitment by 4–1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell 2020;37(2):216–25 e6 doi 10.1016/j.ccell.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Y, Zha Y, Spaapen RM, Mathew R, Barr K, Bendelac A, et al. Egr2-dependent gene expression profiling and ChIP-Seq reveal novel biologic targets in T cell anergy. Mol Immunol 2013;55(3–4):283–91 doi 10.1016/j.molimm.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018;175(4):998–1013 e20 doi 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belk JA, Yao W, Ly N, Freitas KA, Chen YT, Shi Q, et al. Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell 2022;40(7):768–86 e7 doi 10.1016/j.ccell.2022.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seo H, Gonzalez-Avalos E, Zhang W, Ramchandani P, Yang C, Lio CJ, et al. BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat Immunol 2021;22(8):983–95 doi 10.1038/s41590-021-00964-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med 2019;25(1):82–8 doi 10.1038/s41591-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, et al. Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 2017;47(6):1129–41 e5 doi 10.1016/j.immuni.2017.11.021. [DOI] [PubMed] [Google Scholar]
- 28.Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol 2014;15(4):373–83 doi 10.1038/ni.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao G, Deng A, Liu H, Ge G, Liu X. Activator protein 1 suppresses antitumor T-cell function via the induction of programmed death 1. Proc Natl Acad Sci U S A 2012;109(38):15419–24 doi 10.1073/pnas.1206370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A 2019;116(25):12410–5 doi 10.1073/pnas.1905675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prinzing B, Zebley CC, Petersen CT, Fan Y, Anido AA, Yi Z, et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med 2021;13(620):eabh0272 doi 10.1126/scitranslmed.abh0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bai Z, Woodhouse S, Zhao Z, Arya R, Govek K, Kim D, et al. Single-cell antigen-specific landscape of CAR T infusion product identifies determinants of CD19-positive relapse in patients with ALL. Sci Adv 2022;8(23):eabj2820 doi 10.1126/sciadv.abj2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1-CD8+ Tumor-Infiltrating T Cells. Immunity 2019;50(1):181–94.e6 doi 10.1016/j.immuni.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sumida TS, Dulberg S, Schupp JC, Lincoln MR, Stillwell HA, Axisa PP, et al. Type I interferon transcriptional network regulates expression of coinhibitory receptors in human T cells. Nat Immunol 2022;23(4):632–42 doi 10.1038/s41590-022-01152-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood 2012;119(1):72–82 doi 10.1182/blood-2011-07-366419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. Journal of Virology 2003;77(8):4911–27 doi 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest 2019;130 doi 10.1172/JCI125423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li G, Boucher JC, Kotani H, Park K, Zhang Y, Shrestha B, et al. 4–1BB enhancement of CAR T function requires NF-kappaB and TRAFs. JCI Insight 2018;3(18) doi 10.1172/jci.insight.121322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saoulli K, Lee SY, Cannons JL, Yeh WC, Santana A, Goldstein MD, et al. CD28-independent, TRAF2-dependent costimulation of resting T cells by 4–1BB ligand. J Exp Med 1998;187(11):1849–62 doi 10.1084/jem.187.11.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sasai M, Tatematsu M, Oshiumi H, Funami K, Matsumoto M, Hatakeyama S, et al. Direct binding of TRAF2 and TRAF6 to TICAM-1/TRIF adaptor participates in activation of the Toll-like receptor 3/4 pathway. Mol Immunol 2010;47(6):1283–91 doi 10.1016/j.molimm.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Shin HH, Lee EA, Kim SJ, Kwon BS, Choi HS. A signal through 4–1BB ligand inhibits receptor for activation of nuclear factor-kappaB ligand (RANKL)-induced osteoclastogenesis by increasing interferon (IFN)-beta production. FEBS Lett 2006;580(6):1601–6 doi 10.1016/j.febslet.2006.01.091. [DOI] [PubMed] [Google Scholar]
- 42.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013;340(6129):207–11 doi 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013;340(6129):202–7 doi 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 2016;1(6) doi 10.1126/sciimmunol.aai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miao T, Symonds ALJ, Singh R, Symonds JD, Ogbe A, Omodho B, et al. Egr2 and 3 control adaptive immune responses by temporally uncoupling expansion from T cell differentiation. J Exp Med 2017;214(6):1787–808 doi 10.1084/jem.20160553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masopust D, Sivula CP, Jameson SC. Of Mice, Dirty Mice, and Men: Using Mice To Understand Human Immunology. J Immunol 2017;199(2):383–8 doi 10.4049/jimmunol.1700453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol Ther 2018;26(7):1855–66 doi 10.1016/j.ymthe.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 2009;17(8):1453–64 doi 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haas AR, Tanyi JL, O’Hara MH, Gladney WL, Lacey SF, Torigian DA, et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol Ther 2019;27(11):1919–29 doi 10.1016/j.ymthe.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Conant D, Hsiau T, Rossi N, Oki J, Maures T, Waite K, et al. Inference of CRISPR Edits from Sanger Trace Data. CRISPR J 2022;5(1):123–30 doi 10.1089/crispr.2021.0113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and analyzed data supporting the conclusions of this manuscript are available at the Gene Expression Omnibus (GEO) under the accession number GSE224194. The data are publicly accessible and can be downloaded for further analysis.